
Farmacja weterynaryjna
Ć
wiczenie 2

Rodzaje leków
1. Leki oryginalne
2. Leki odtwórcze (generyczne)
3. Leki o ugruntowanym stosowaniu
(well establish use)
4. Leki homeopatyczne
Leki oryginalne
- Nowa molekuła
- Nowe wskazanie
Produkt leczniczy oryginalny –
produkt leczniczy, wprowadzony do stosowania
w lecznictwie na podstawie pełnej dokumentacji
bada
ń
chemicznych, biologicznych,
farmaceutycznych, farmakologicznych,
toksykologicznych i klinicznych.
Procedura droga i długotrwała.

Leki odtwórcze (generyczne)
Posiadaj
ą
t
ę
sam
ą
, chemicznie zdefiniowan
ą
substancj
ę
aktywn
ą
, w tym samym st
ęż
eniu oraz takie
samo dawkowanie i wskazania co lek oryginalny.
Mog
ą
ró
ż
ni
ć
si
ę
składem substancji pomocniczych.
Najprostsza, najszybsza i najta
ń
sza droga rejestracji
nowych leków po wyga
ś
ni
ę
ciu patentu i wył
ą
czno
ś
ci
danych.
Lek odtwórczy musi wykazywa
ć
równowa
ż
no
ść
biologiczn
ą
lub farmaceutyczn
ą
w zakresie 80-120%
w odniesieniu do leku referencyjnego.

Leki o ugruntowanym stosowaniu
(well establish use)
Leki, w których substancja aktywna znana jest od
ponad 10 lat i istnieje pełna i ugruntowana wiedza na
temat jej działania i bezpiecze
ń
stwa.
Niekiedy nale
ż
y wykona
ć
dodatkowe badanie
w przypadku braku bada
ń
literaturowych. Szczególnie
dotycz
ą
cych okresu karencji.

Leki homeopatyczne
Produkt homeopatyczny –
produkt leczniczy przygotowany z ró
ż
nych składników
lub ich mieszanin, zwanych surowcami homeopatycznymi,
zgodnie z procedur
ą
homeopatyczn
ą
opisan
ą
w odpowiednich farmakopeach uznawanych w pa
ń
stwach
członkowskich Unii Europejskiej.
Najcz
ęś
ciej powy
ż
ej rozcie
ń
czenia 10D.
(10D oznacza,
ż
e 10-krotnie powtórzono procedur
ę
rozcie
ń
czania 1:10,
a np. 20C,
ż
e 20-krotnie powtórzono procedur
ę
rozcie
ń
czania 1:100
Rozcie
ń
czenie Korsakova (K) – rodzaj rozie
ń
czenia 1:100,
np. 200K – 200-krotnie powtórzone rozcie
ń
czenie
Korsakova)
Skuteczno
ść
?

Równowa
ż
no
ść
farmaceutyczna
i biologiczna leków

Lek referencyjny
Lek u
ż
ywany w badaniach biorównowa
ż
no
ś
ci,
do którego jest porównywany lek badany.
Lekiem referencyjnym jest najcz
ęś
ciej lek
oryginalny, w przypadku braku leku
oryginalnego – lek o ugruntowanym
stosowaniu

Dost
ę
pno
ść
farmaceutyczna
Ilo
ść
substancji leczniczej uwolnionej
w okre
ś
lonym czasie z postaci leku
oraz szybko
ść
z jak
ą
ten proces zachodzi.

Test uwalniania leku in vitro
(dissolution test)
Jest odzwierciedleniem dost
ę
pno
ś
ci
farmaceutycznej.
Dotyczy stałych postaci leków.
Wykonywany jest zgodnie z zaleceniami
Farmakopei.

Równowa
ż
no
ść
farmaceutyczna
Dwa leki s
ą
równowa
ż
ne farmaceutycznie je
ś
li
posiadaj
ą
- ten sam skład ilo
ś
ciowy i jako
ś
ciowy
substancji czynnej,
- t
ę
sam
ą
posta
ć
leku (tabletka, kapsułka itp.),
- przeznaczone s
ą
do podawania t
ą
sam
ą
drog
ą

Dost
ę
pno
ść
biologiczna
(Biodost
ę
pno
ść
) – F, BA
Ilo
ść
leku przedostaj
ą
ca si
ę
z miejsca
podania do krwi
(% wchłoni
ę
tej dawki leku).
Po podaniu donaczyniowym
biodost
ę
pno
ść
= 100 %

Inne parametry farmakokinetyczne:
AUC – pole pod krzyw
ą
zale
ż
no
ś
ci
st
ęż
enie leku/czas
C
max
- st
ęż
enie maksymalne – najwy
ż
sze
obserwowane st
ęż
enie leku we krwi
t
max
- czas wyst
ą
pienia st
ęż
enia
maksymalnego
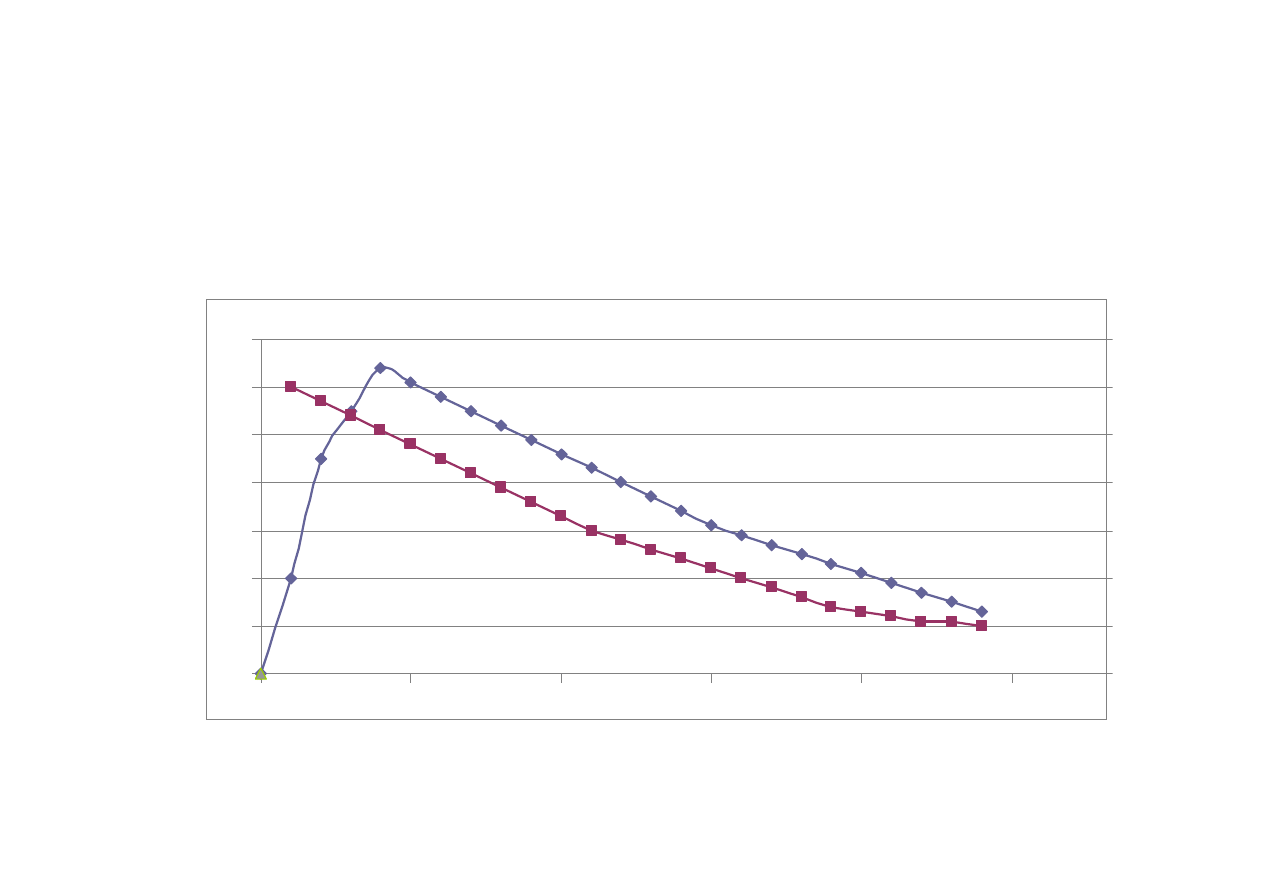
Krzywa st
ęż
enie/czas
st
ęż
enie
czas
0
1
2
3
4
5
6
7
0
5
10
15
20
25

Biorównowa
ż
no
ść
Dwa leki s
ą
równowa
ż
ne biologicznie je
ś
li:
- s
ą
równowa
ż
ne farmaceutycznie (czyli posiadaj
ą
ten
sam skład ilo
ś
ciowy i jako
ś
ciowy substancji czynnej,
t
ę
sam
ą
posta
ć
leku i przeznaczone s
ą
do podawania
t
ą
sam
ą
drog
ą
)
- ich parametry farmakokinetyczne, m.in. biodost
ę
pno
ść
,
po podaniu w tej samej dawce, t
ą
sam
ą
drog
ą
, nie
ró
ż
ni
ą
si
ę
istotnie (80-120 %).
Mo
ż
na wi
ę
c oczekiwa
ć
identycznej
(lub wystarczaj
ą
co zbli
ż
onej) skuteczno
ś
ci
terapeutycznej i bezpiecze
ń
stwa stosowania.
Taka sama ilo
ść
tej samej substancji w tym
samym czasie powoduje taki sam efekt.

Biorównowa
ż
no
ść
Iloraz pola pod krzyw
ą
AUC po podaniu leku
odtwórczego w stosunku do AUC leku
referencyjnego, podanego w tej samej dawce,
t
ą
sam
ą
drog
ą
.
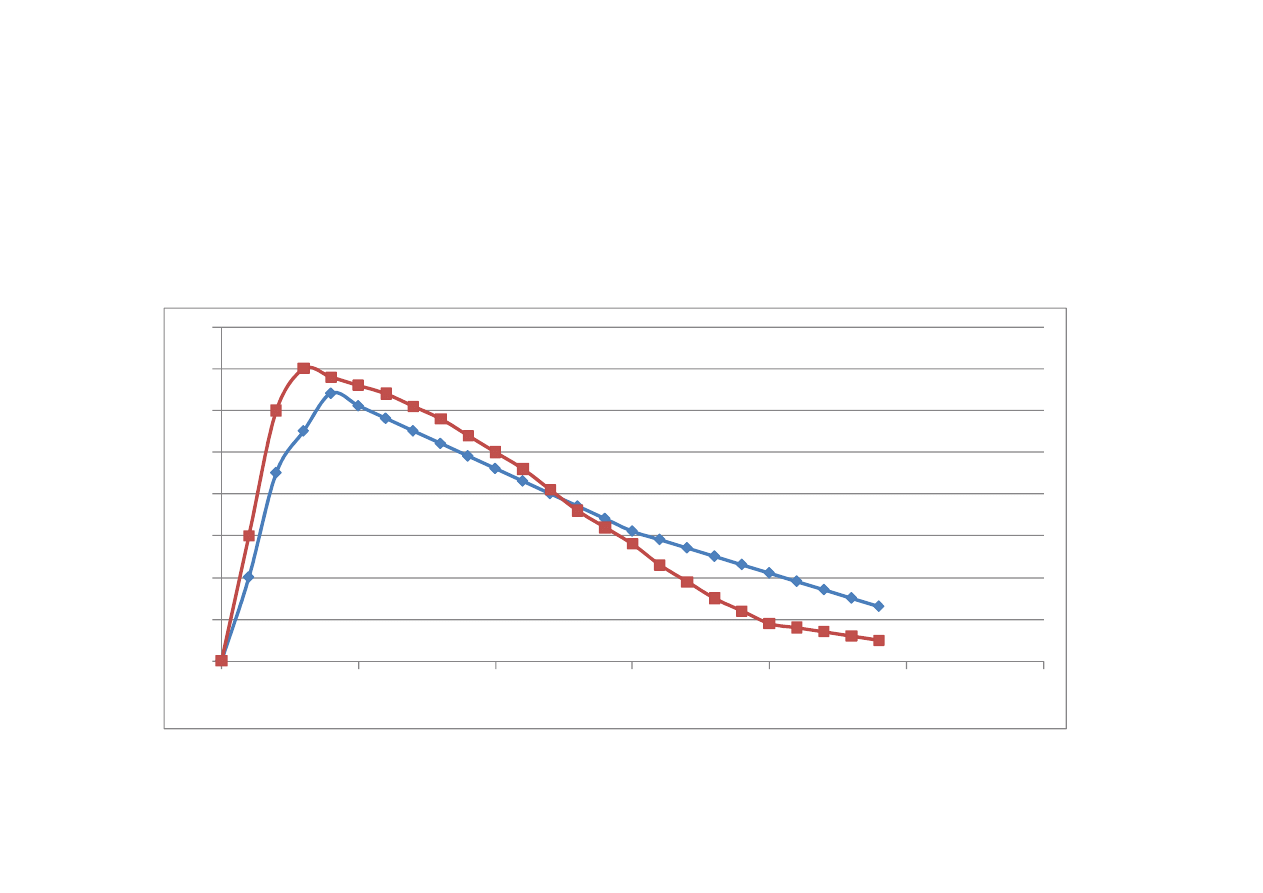
Biorównowa
ż
no
ść
st
ęż
enie
czas
0
1
2
3
4
5
6
7
8
0
5
10
15
20
25
30

Zagadnienia prawne dotycz
ą
ce rejestracji
1. Rejestracja leków weterynaryjnych:
- European Medicines Agency - EMA - Londyn, (Europejska
Agencja Leków)
- Urz
ą
d Rejestracji Produktów Leczniczych, Wyrobów
Medycznych i Produktów Biobójczych (URPLWMiPB) –
Warszawa
2. Akty prawne:
- Dyrektywa EU 2004/28/WE
- EudraLex
- Ustawa Prawo Farmaceutyczne
- Rozporz
ą
dzenia dotycz
ą
ce prawa farmaceutycznego
3. Rodzaje rejestracji leków

Leki obecne na rynku EU
• Rynek leków
nie jest
wspólny
• Na rynkach krajowych mog
ą
znajdowa
ć
si
ę
tylko leki
dopuszczone do obrotu w danym pa
ń
stwie członkowskim
za pomoc
ą
odpowiednich procedur
• Tylko na własne potrzeby mo
ż
na przywozi
ć
z zagranicy leki, które nie s
ą
dopuszczone do obrotu w
danym pa
ń
stwie członkowskim – max. 5 najmniejszych
opakowa
ń
(NIE DOTYCZY
ś
rodków odurzaj
ą
cych i substancji
psychotropowych oraz leków weterynaryjnych
przeznaczonych dla zwierz
ą
t „producentów
ż
ywno
ś
ci”)
• Import równoległy
Rodzaje rejestracji:
- centralna – wniosek o rejestracj
ę
do EMA, umo
ż
liwia rejestracj
ę
leku
od razu we wszystkich krajach UE
- krajowa (narodowa) – umo
ż
liwia rejestracj
ę
leku danym kraju,
w Polsce przez URPLWMiPB
- w kilku krajach:
• procedura wzajemnego uznawania – MRP –
kiedy produkt leczniczy jest ju
ż
zarejestrowany w jednym kraju UE,
a rejestracja w innym kraju mo
ż
liwa jest przez uznanie w mniejszym
w wi
ę
kszym zakresie wcze
ś
niejszej dokumentacji z mo
ż
liwym
zgłaszaniem do niej uwag
• procedura zdecentralizowana - DCP –
rejestracja równoległa w kilku krajach UE; kiedy produkt leczniczy
nie jest jeszcze zarejestrowany w
ż
adnym kraju UE, wtedy jedno
z pa
ń
stw przyjmuje na siebie obowi
ą
zek koordynacji rejestracji
i wst
ę
pnej oceny dokumentacji, a pozostałe pa
ń
stwa, w których lek
ma by
ć
zarejestrowany, do niej si
ę
odnosz
ą
(zgłaszanie uwag)

Rozporz
ą
dzenie Ministra Zdrowia
z dnia 18 stycznia 2010 r.
w sprawie szczegółowego sposobu
przedstawiania dokumentacji doł
ą
czanej
do wniosku o dopuszczenie do obrotu
produktu leczniczego weterynaryjnego

SPOSÓB PRZEDSTAWIANIA DOKUMENTACJI
W PRZYPADKU PRODUKTÓW LECZNICZYCH
WETERYNARYJNYCH
CZE
ŚĆ
I - STRESZCZENIE DOKUMENTACJI
CZ
ĘŚĆ
II – DOKUMENTACJA CHEMICZNA, FARMACEUTYCZNA
I BIOLOGICZNA
CZ
ĘŚĆ
III – DOKUMENTACJA TOKSYKOLOGICZNA
I FARMAKOLOGICZNA, W TYM DOKUMENTACJA
POZOSTAŁO
Ś
CI ORAZ EKOTOKSYCZNO
Ś
CI
CZ
ĘŚĆ
IV – DOKUMENTACJA PRZEDKLINICZNA I KLINICZNA

Cz
ęść
I - Streszczenie dokumentacji
Cz
ęść
I A – Wniosek o dopuszczenie do obrotu oraz
dokumenty uzupełniaj
ą
ce doł
ą
czane do wniosku
zgodnie z wykazem zawartym w formularzu
tego wniosku
Cz
ęść
I B – Charakterystyka produktu leczniczego
weterynaryjnego, ulotka, oznakowanie opakowa
ń
Cz
ęść
I C – Raporty ekspertów
Cz
ęść
I C. 1 – Raport eksperta na temat dokumentacji
chemicznej, farmaceutycznej i biologicznej
Cz
ęść
I C. 2 – Raport eksperta na temat dokumentacji
toksykologicznej i farmakologicznej, w tym
dokumentacji pozostało
ś
ci oraz ekotoksyczno
ś
ci
Cz
ęść
I C. 3 – Raport eksperta na temat dokumentacji
przedklinicznej i klinicznej

Cz
ęść
II - Dokumentacja chemiczna, farmaceutyczna
i biologiczna dla substancji czynnych
pochodzenia chemicznego
Cz
ęść
II A – Skład produktu leczniczego weterynaryjnego
i proponowany sposób opakowania
Cz
ęść
II B – Opis metody wytwarzania
Cz
ęść
II C – Kontrola surowców u
ż
ytych do wytwarzania
produktu leczniczego weterynaryjnego
Cz
ęść
II D – Szczególne
ś
rodki podj
ę
te w celu ochrony przed
przenoszeniem si
ę
g
ą
bczastych encefalopatii zwierz
ą
t
Cz
ęść
II E – Badania kontrolne produktów po
ś
rednich
Cz
ęść
II F – Badania kontrolne produktu ko
ń
cowego
Cz
ęść
II G – Badania stabilno
ś
ci
Cz
ęść
II H - Dane dotycz
ą
ce oceny ryzyka dla
ś
rodowiska
naturalnego w przypadku produktów leczniczych
zawieraj
ą
cych organizm genetycznie zmodyfikowany
(GMO) lub składaj
ą
cych si
ę
z GMO
Cz
ęść
Q – Inne dane

Cz
ęść
III – Dokumentacja toksykologiczna
i farmakologiczna, w tym dokumentacja
pozostało
ś
ci i ekotoksyczno
ś
ci
Cz
ęść
III A – Dokumentacja dotycz
ą
ca bezpiecze
ń
stwa
Cz
ęść
III A.1 – Opis produktu leczniczego wet.
Cze
ść
III A.2 – Badania farmakologiczne
Cz
ęść
III A.3 – Badania toksykologiczne
Cz
ęść
III A.4 – Inne badania
Cz
ęść
III A.5 – Bezpiecze
ń
stwo osoby podaj
ą
cej
produkt leczniczy weterynaryjny
Cz
ęść
III A.6 – Ocena wpływu na
ś
rodowisko
Wnioski
Cz
ęść
III B – Dokumentacja dotycz
ą
ca pozostało
ś
ci
Cz
ęść
III B.1 – Opis produktu leczniczego wet.
Cz
ęść
III B.2 – Badania pozostało
ś
ci
Cz
ęść
III B.3 – Metoda (-y) analityczna (-e)
Wnioski

Cz
ęść
IV – Dokumentacja przedkliniczna i kliniczna
Cz
ęść
IV A – Dokumentacja przedkliniczna
Cz
ęść
IV A 1 – Farmakodynamika
Cz
ęść
IV A 2 – Farmakokinetyka
Cz
ęść
IV A 3 – Badania tolerancji u docelowych
gatunków zwierz
ą
t
Cz
ęść
IV A 4 – Dane z zakresu oporno
ś
ci
Cze
ść
IV B – Dokumentacja kliniczna

Atrybuty leku
- Opakowanie
- Etykieta i ulotka
- Seria
- Data wa
ż
no
ś
ci
Rozporz
ą
dzenie Ministra Zdrowia
z dnia 29 pa
ź
dziernika 2009 r.
w sprawie wymaga
ń
dotycz
ą
cych oznakowania
opakowa
ń
produktu leczniczego
weterynaryjnego i tre
ś
ci ulotki

Rozporz
ą
dzenie Ministra Zdrowia
z dnia 12 grudnia 2002 r.
w sprawie podmiotów uprawnionych
do zakupu produktów leczniczych
w hurtowni farmaceutycznej
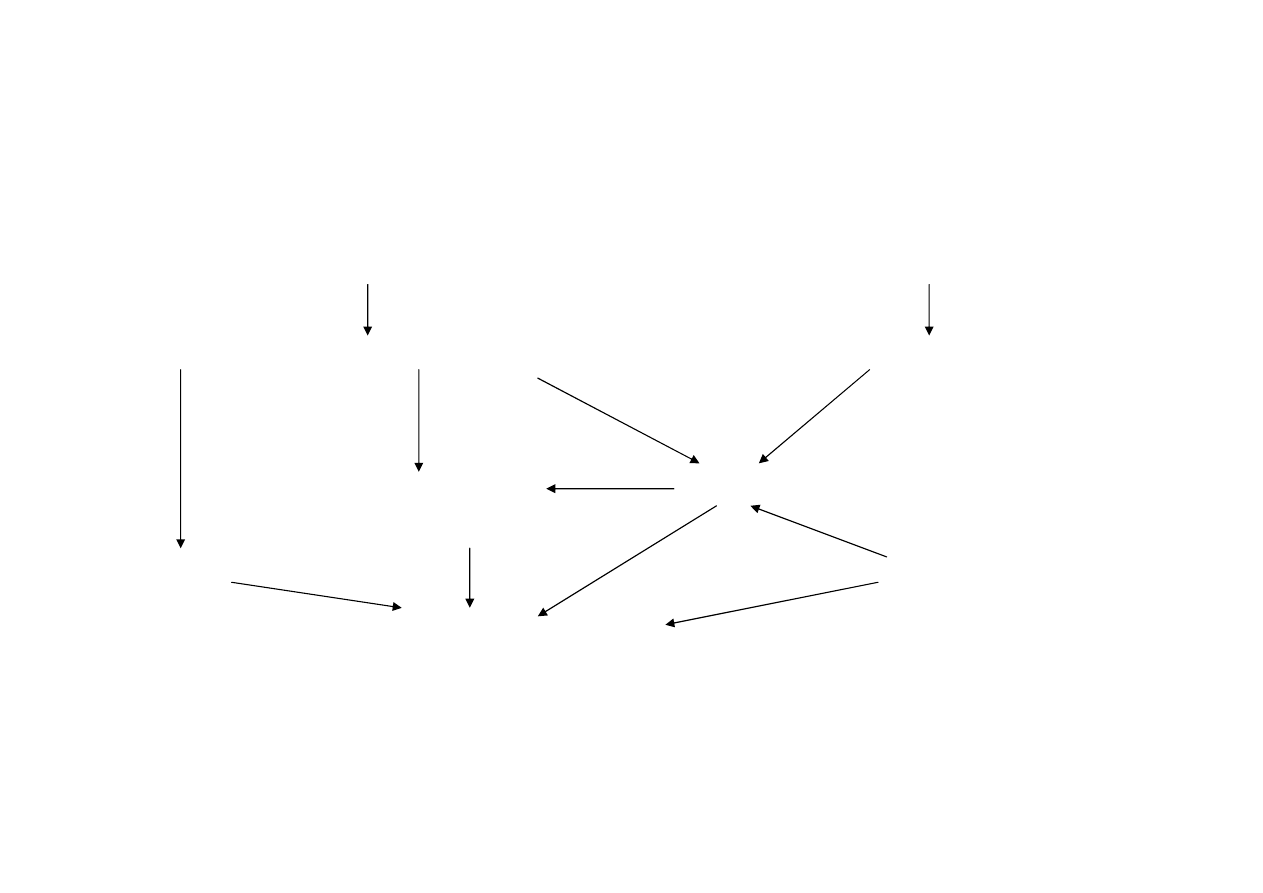
Obrót lekami u
ż
ywanymi w lecznictwie
weterynaryjnym
Producent/imp. leków weterynaryjnych
Producent/imp. leków ludzkich
Hurtownia leków weterynaryjnych
Hurtownia leków „ludzkich”
Mieszalnia
Lekarz weterynarii
pasz leczniczych
Sklepy
Apteka ogólnodost
ę
pna
Wła
ś
ciciel zwierz
ę
cia

Zagadnienia prawne dotycz
ą
ce obrotu
leków
1. Rejestracja w Izbie Lekarsko-Weterynaryjnej
zakładu leczniczego dla zwierz
ą
t.
2. Rejestracje w Woj. Inspektoracie
Farmaceutycznym i uzyskanie pozwolenia na obrót
wybranymi lekami psychotropowymi.
3. Posiadanie warunków do przechowywania leków
weterynaryjnych – strefy temperaturowe.
4. Prowadzenie ewidencji stosowania i zu
ż
ycia
leków, w tym oddzielnej ewidencji dotycz
ą
cej leków
psychotropowych.

Rozporz
ą
dzenie
Ministra Rolnictwa i Rozwoju Wsi
z dnia 17 pa
ź
dziernika 2008 r.
w sprawie sposobu prowadzenia dokumentacji
obrotu detalicznego produktami leczniczymi
weterynaryjnymi i wzoru tej dokumentacji

Rozporz
ą
dzenie
Ministra Rolnictwa i Rozwoju Wsi
z dnia 29 wrze
ś
nia 2011 r.
w sprawie zakresu i sposobu prowadzenia
dokumentacji lekarsko-weterynaryjnej
i ewidencji leczenia zwierz
ą
t oraz wzorów
tej dokumentacji i ewidencji

Lekarz weterynarii prowadzi dokumentacj
ę
lekarsko-
weterynaryjn
ą
:
- dla zwierz
ą
t producentów
ż
ywno
ś
ci –
papierowa ksi
ąż
ka leczenia zwierz
ą
t (numerowane,
samokopiuj
ą
ce si
ę
strony), po dokonaniu wpisu lekarz
wet. pozostawia oryginał strony posiadaczowi
zwierz
ę
cia
- dla zwierz
ą
t towarzysz
ą
cych –
papierowa lub elektroniczna ksi
ąż
ka leczenia zwierz
ą
t
Posiadacz zwierz
ą
t gospodarskich prowadzi ewidencj
ę
leczenia zwierz
ą
t, któr
ą
stanowi
ą
chronologicznie
uło
ż
one oryginały stron ksi
ąż
ki leczenia zwierz
ą
t
producentów
ż
ywno
ś
ci.
W ww. rozporz
ą
dzeniu wzory ksi
ąż
ek leczenia zwierz
ą
t.

Rozporz
ą
dzenia Ministra Zdrowia
z dnia 6 lutego 2012 r.
w sprawie preparatów zawieraj
ą
cych
ś
rodki odurzaj
ą
ce
lub substancje psychotropowe, które mog
ą
by
ć
posiadane w celach medycznych oraz stosowane do
bada
ń
klinicznych, po uzyskaniu zgody wojewódzkiego
inspektora farmaceutycznego

Weterynaryjne leki psychotropowe
Grupa II-P - ketamina,
Grupa III-P - pentobarbital
Obowi
ą
zek
ś
cisłego ewidencjonowania.


Odpowiedzialno
ść
karna lekarza
Ust. O przeciwdziałaniu narkomanii
Art. 56. 1. Kto, wbrew przepisom art. 33-35 i 37, wprowadza do obrotu
ś
rodki
odurzaj
ą
ce, substancje psychotropowe lub słom
ę
makow
ą
albo uczestniczy w
takim obrocie,
podlega grzywnie i karze pozbawienia wolno
ś
ci od 6
miesi
ę
cy do lat 8.
Art. 67. Kto, wbrew przepisom ustawy, rozporz
ą
dzenia 273/2004 lub
rozporz
ą
dzenia 111/2005, nie dopełnia obowi
ą
zku prowadzenia ewidencji
wytwarzania, przetwarzania, przerobu
ś
rodków odurzaj
ą
cych, substancji
psychotropowych lub prekursorów i obrotu nimi albo w inny sposób narusza
przepisy okre
ś
laj
ą
ce zasady stosowania
ś
rodków, substancji lub prekursorów i
obrotu nimi,
podlega karze grzywny.

Ustawa Prawo Farmaceutyczne:
Art. 124a. 1. Kto wprowadza do obrotu lub stosuje niewpisane do Rejestru
Produktów Leczniczych Dopuszczonych do Obrotu na terytorium
Rzeczypospolitej Polskiej, o którym mowa w art. 28, produkty lecznicze
weterynaryjne,
podlega grzywnie albo karze ograniczenia wolno
ś
ci albo
pozbawienia wolno
ś
ci do lat 2.
2. Tej samej karze podlega osoba odpowiedzialna za zwierz
ę
ta, która
dopuszcza do stosowania u zwierz
ą
t produkty lecznicze weterynaryjne
niedopuszczone do obrotu.
Art. 132a. Kto wprowadza do obrotu lub stosuje w praktyce weterynaryjnej
nieprzetworzone surowce farmaceutyczne,
podlega grzywnie albo karze pozbawienia wolno
ś
ci do lat 3 albo obu tym
karom ł
ą
cznie.
Art. 132b. Kto nie posiada dokumentów nabycia i stosowania u zwierz
ą
t, z
których lub od których tkanki i produkty s
ą
przeznaczone do spo
ż
ycia przez
ludzi, produktu leczniczego weterynaryjnego posiadaj
ą
cego wła
ś
ciwo
ś
ci
anaboliczne, przeciwbakteryjne, przeciwpaso
ż
ytnicze, przeciwzapalne,
hormonalne i psychotropowe
podlega grzywnie albo karze pozbawienia
wolno
ś
ci do lat 2 albo obu tym karom ł
ą
cznie.

Wyznaczanie okresu karencji
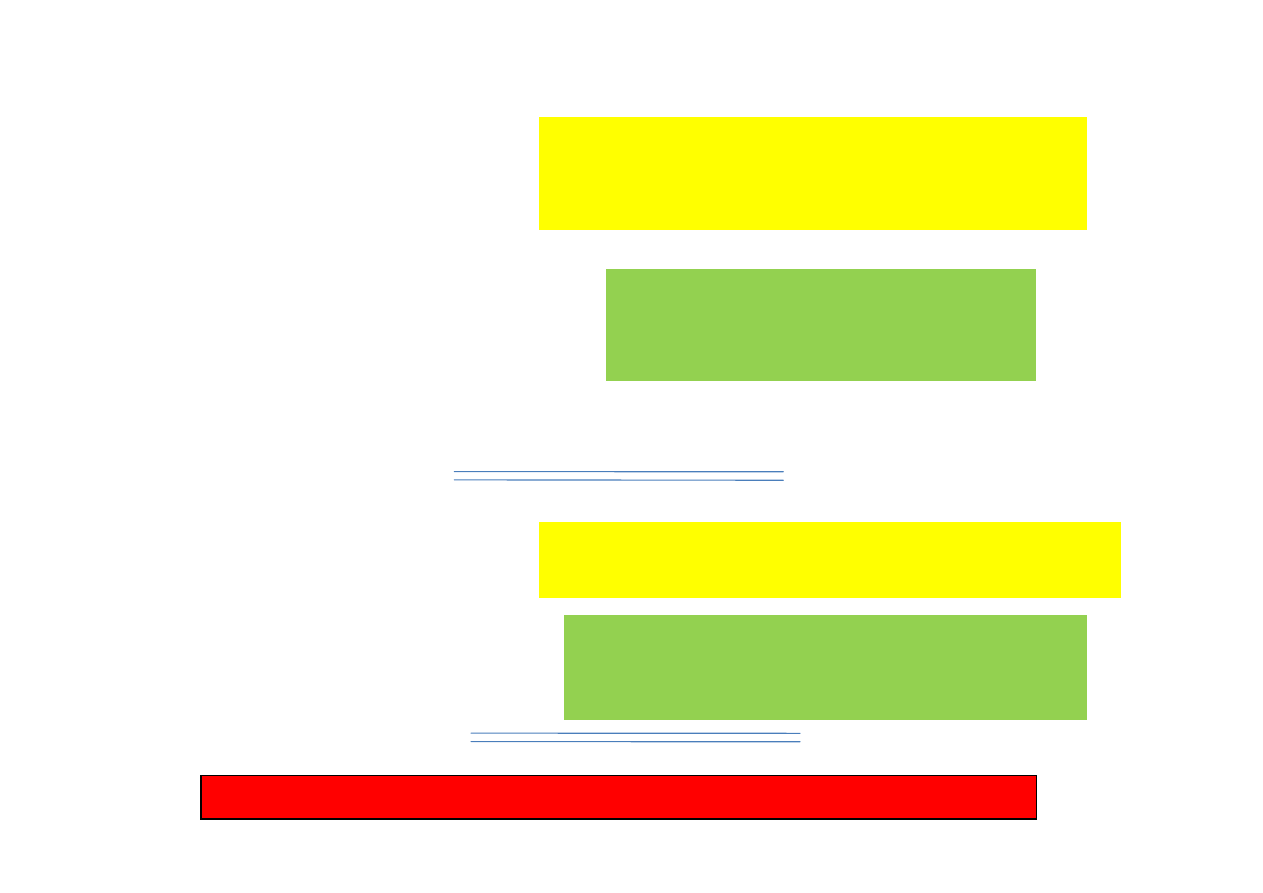
NOAEL
ADI
MRL
(pol. NDP)
Okres
karencji
Producent leku oryginalnego (sponsor)
wprowadzaj
ą
cy lek na rynek
(Badania toksykometryczne lub mikrobiologiczne)
Europejska Agencja Leków
EMA (CVMP)
(Ustalenie wysoko
ś
ci MRL)
Podmiot rejestruj
ą
cy lek (generyczny)
(Badania pozostało
ś
ci i propozycja okresu karencji)
Pa
ń
stwowa komisja rejestruj
ą
ca leki
(Ustalenie okresu karencji dla rejestrowanego
produktu leczniczego)
Lekarz weterynarii - nadzór nad przestrzeganiem okresu karencji

NOAEL (NOEL)
(No Observed Adverse Effect Level)
Maksymalne st
ęż
enie/dawka substancji,
przy której nie obserwuje si
ę ż
adnych
działa
ń
niepo
żą
danych po długotrwałym
stosowaniu.

Badania toksykologiczne nad NOAEL wykonywane s
ą
na zwierz
ę
tach
do
ś
wiadczalnych in vivo i na hodowlach komórkowych (in vitro) i na ogół
obejmuj
ą
one pomiary czynników charakteryzuj
ą
cych:
1) toksyczno
ść
ostr
ą
(DL
50
);
2) genotoksyczno
ść
(mutagenno
ść
, klastogenno
ść
);
3) podprzewlekł
ą
toksyczno
ść
doustn
ą
;
4) przewlekł
ą
toksyczno
ść
doustna/rakotwórczo
ść
;
5) toksyczny wpływ na rozrodczo
ść
wł
ą
cznie z teratogenno
ś
ci
ą
; oraz
6) inne badania.
W przypadku jakiegokolwiek powodu do obaw przeprowadza si
ę
dalsze
badania dostarczaj
ą
ce dodatkowych informacji niezb
ę
dnych do oceny
nieszkodliwo
ś
ci substancji czynnej i jej pozostało
ś
ci.
Na podstawie wyników wspomnianych bada
ń
nale
ż
y ustali
ć
NOAEL
toksykologiczny (akceptowany przez gremia mi
ę
dzynarodowe) – Jecfa
(eksperci FAO, WHO), EMA, FDA (USA)

ADI (Acceptable Daily Intake)
Dopuszczalne dzienne spo
ż
ycie
okre
ś
la maksymaln
ą
ilo
ść
substancji, która -
zgodnie z aktualnym stanem wiedzy - mo
ż
e by
ć
przez człowieka pobierana z
ż
ywno
ś
ci
ą
codziennie
przez całe
ż
ycie prawdopodobnie bez negatywnych
skutków dla zdrowia.
ADI jest okre
ś
lany nie tylko dla pozostało
ś
ci leków
weterynaryjnych, ale równie
ż
m.in. dla wi
ę
kszo
ś
ci
dodatków do
ż
ywno
ś
ci, pozostało
ś
ci pestycydów.
Wska
ź
nik ADI jest najcz
ęś
ciej podawany w
µ
g/kg,
mg/kg na 1 dzie
ń
i dotyczy ł
ą
cznego pobrania
substancji ró
ż
nymi drogami (z
ż
ywno
ś
ci
ą
,
powietrzem, lekami, kosmetykami, etc.).

Ogólny wzór na obliczenie wartości ADI to
NOEL
ADI = -------------------------------- mg x kg m.c.
-1
x doba
-1
współczynnik bezpieczeństwa
Współczynnik bezpieczeństwa ma charakter uznaniowy, jest
najczęściej liczbą 10, 100, 500 lub 1000. Na jego przyjęcie ma
wpływ szereg czynników np. różnica toksyczności danego
ksenobiotyku u zwierząt i człowieka, zróżnicowanie reakcji w
populacji, działania odległe ksenobiotyku i inne.
Współczynnik ten, dla poszczególnych związków przyjmowany jest
od lat przez Wspólny Komitet Ekspertów WHO i FAO d/s
Dodatków Żywnościowych - JECFA (Joint FAO/WHO Expert
Committee on Food Additives)

Dla antybiotyków przeciwbakteryjnych okre
ś
la si
ę
ADI na podstawie
MIC
50
dla najwra
ż
liwszego szczepu flory bakteryjnej człowieka
Np. ADI dla florfenikolu wyliczono z wzoru
MIC
50
(
µµµµ
g/ml) x masa zawarto
ś
ci jelit (g)
ADI = -----------------------------------------------------------------------------------------
dost
ę
pna cz
ęść
dawki x wsp. bezpiecze
ń
stwa x masa ciała człowieka
Przyj
ę
to
ż
e warto
ść
MIC
50
dla Fusobacterium wynosi 0,36
µµµµ
g/ml,
masa zawarto
ś
ci jelit – 150 g,
dost
ę
pna ilo
ść
podanego leku wynosi 0,3 (reszta ulega wchłoni
ę
ciu),
współczynnik bezpiecze
ń
stwa za 1 i mas
ę
ciała człowieka za 60 kg.

MRL (Maximum Residue Level)
Maksymalny Limit Pozostało
ś
ci
Najwy
ż
sze dopuszczalne st
ęż
enie pozostało
ś
ci
leku lub jego metabolitów w tkankach
i produktach pochodzenia zwierz
ę
cego
przeznaczonych do spo
ż
ycia przez człowieka

MRL wylicza si
ę
o dane liczbowe opisuj
ą
ce teoretyczne dzienne
spo
ż
ycie przez ludzi (g tkanek lub produktów)
Ssaki Ptaki
Ryby Inne
Mi
ęś
nie 300 300
300
(*)
W
ą
troba 100 100
—
Nerki 50 10
—
Tłuszcz 50 (**) 90 (***)
—
+Mleko 1 500 —
—
+Jaja —
100 —
+Miód
20
(*) Mi
ęś
nie i skóra w naturalnych proporcjach.
(**) W przypadku
ś
wi
ń
50 g tłuszczu i skóry w naturalnych proporcjach.
(***) Tłuszcz i skóra w naturalnych proporcjach
Definicje stosowane przy wyznaczaniu NDP
i-j Poszczególne tkanki/produkty (w
ą
troba, nerki, mi
ęś
nie, skóra + tłuszcz, mleko, jaja, miód) w ró
ż
nych okresach
NDPi-j Najwy
ż
szy dopuszczalny poziom pozostało
ś
ci w tkankach/produktach (mg substancji znacznikowej kg-1)
Qti-j Dzienne spo
ż
ycie przez ludzi poszczególnych tkanek/produktów (kg) ustalone za pomoc
ą
tabeli 1 lub jej poprawionej
wersji
TRCi-j Ł
ą
czne st
ęż
enie pozostało
ś
ci w poszczególnych tkankach/produktach (mg kg-1)
MRCi-j St
ęż
enie pozostało
ś
ci znacznikowej w poszczególnych tkankach/produktach (mg kg-1)
RMTRi-j Stosunek MRCi-j do TRCi-j w odniesieniu do poszczególnych tkanek/produktów
DITRi-j Pobranie z diet
ą
w odniesieniu do poszczególnych tkanek/produktów, wyliczone z ł
ą
cznych pozostało
ś
ci (mg)
DITRi-j = Qti-j x TRCi-j
DITRNDPi-j Pobranie z diet
ą
wyliczone z NDP (mg) poszczególnych tkanek/produktów
DITRNDPi-j = Qti-j x NDPi-j x RMTRi-j

NAJWY
Ż
SZE DOPUSZCZALNE ST
ĘŻ
ENIA
POZOSTAŁO
Ś
CI (MRL)
• Ustala CVMP agenda EMA w Londynie.
• Opiera si
ę
na analizie ryzyka niebezpiecze
ń
stwa
szkodliwego oddziaływania substancji aktywnej lub
pomocniczej leku na człowieka w przypadku jej
konsumpcji.
• Do jego ustalenia koniecznych jest szereg bada
ń
toksykometrycznych.
• Dotyczy leków stosowanych u zwierz
ą
t, z których
otrzymujemy
ż
ywno
ść
• Leki zgrupowane w 4 aneksach

Aneksy
Aneks I – substancje o ustalonym MRL
Aneks II – substancje dopuszczone do
stosowania bez konieczno
ś
ci
wyznaczania MRL
Aneks III – substancje z czasowo ustalonym MRL,
warunkowo dopuszczone do
stosowania u zwierz
ą
t producentów
ż
ywno
ś
ci do okre
ś
lonego terminu
Aneks IV – leki z nieustalonym MRL

Leków z Aneksu IV
NIE WOLNO
stosowa
ć
u zwierz
ą
t
producentów
ż
ywno
ś
ci
U zwierz
ą
t producentów
ż
ywno
ś
ci
mo
ż
na stosowa
ć
wył
ą
cznie leki
z Aneksów I, II i III

Aneks IV
• Aristolochia spp.
• Chloramfenikol
• Chloroform
• Chloropromazyna
• Kolchicyna
• Dapson
• Dimetridazol
• Metronidazol
• Nitrofurany
• Ronidazol

Okres karencji
Czas, jaki musi upłyn
ąć
od ostatniego podania
leku do uboju zwierz
ę
cia, a w przypadku mleka,
jaj lub miodu, do momentu rozpocz
ę
cia
pozyskiwania tych produktów do celów
spo
ż
ywczych.
Czas ten musi upłyn
ąć
, aby tkanki zw. oraz
inne produkty poch. zwierz
ę
cego nie zawierały
pozostało
ś
ci leku w ilo
ś
ci przekraczaj
ą
cej
maksymalny limit pozostało
ś
ci (MRL)

Postacie leków
Proszki
Granulaty
Tabletki
Kapsułki
Systemy terapeutyczne
Aerozole lecznicze
Ma
ś
ci
Czopki oraz inne postacie leków doodbytniczych i dopochwowych
Mydła
Mazidła
Syropy, eliksiry, miody
Roztwory lecznicze
Leki do oczu
Leki pozajelitowe
Postacie leków ro
ś
linnych
Postacie leków homeopatycznych
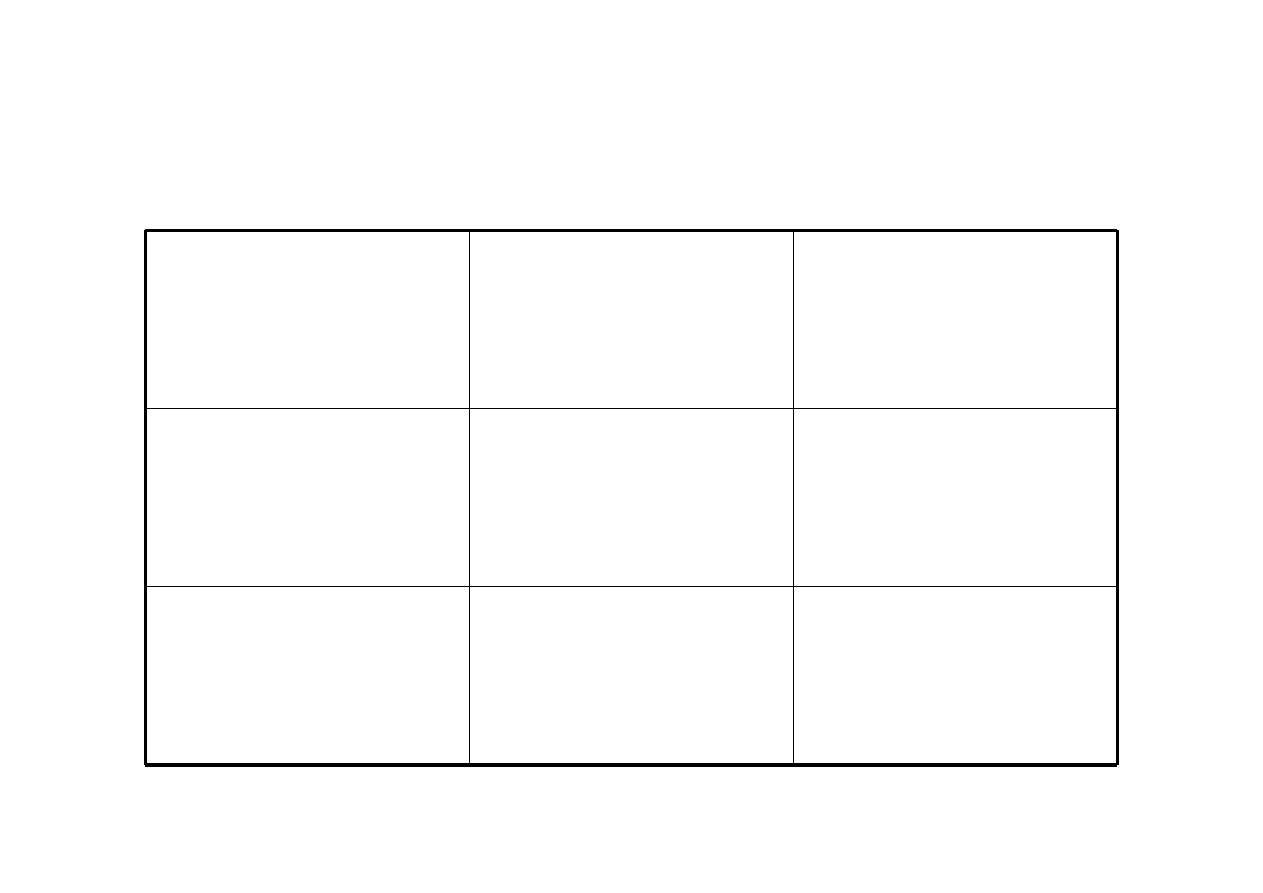
Biofarmaceutyczny system klasyfikacji leków wg. FDA
Rozpuszczalno
ść
wodzie wysoka
Rozpuszczalno
ść
wodzie niska
Przenikalno
ść
przez błony
wysoka
Klasa I
Klasa II
Przenikalno
ść
przez błony
niska
Klasa III
Klasa IV

Substancje pomocnicze
Substancje, które s
ą
stosowane w procesie
technologii produkcji leku, pomagaj
ą
w uzyskaniu odpowiedniej postaci leku oraz
poprawiaj
ą
jego trwało
ść
, działanie i wygl
ą
d.
Substancje pomocnicze nie mog
ą
w zastosowanych ilo
ś
ciach wywiera
ć
własnego
działania farmakologicznego ani działania
dra
ż
ni
ą
cego, ani wpływa
ć
negatywnie na
trwało
ść
postaci leku i dost
ę
pno
ść
biologiczn
ą
substancji leczniczej.
Wyszukiwarka
Podobne podstrony:
Farmacja cw 1 2012
Farmacja cw 3 2012
Farmacja cw 4 2012
Farmacja cw 1 2012
NOTATKI ĆW 3 2012
PNOP cw 2012 06 23
Farmacja cw 4
Matematyka II (Ćw) 2012 06 01
Instrukcja laboratorium ETP ćw 2 2012
Prawo cywilne ćw.8 2012-02-20, Prawo Cywilne
Ćw 4 2012 Przepływ w stopniu turbiny
Farmacja cw 1 id 168164 Nieznany
Prawo cywilne ćw.9 2012-02-27, Prawo Cywilne
Prawo cywilne ćw.7 2012-02-13, Prawo Cywilne
więcej podobnych podstron