
Opracowanie pytań na kolokwium
wykładowe z „Analizy
instrumentalnej”
Z
AGADNIENIA
OGÓLNE
:
Wymień znane metody instrumentalne oparte na zjawiskach fizykochemicznych.
1.
Wydzielanie elektrolityczne – elektrograwimetria, kulometria, miareczkowanie kulometryczne.
2. Przepływ prądu między elektrodami – polarografia, woltamperometria, amperometria, miareczkowanie
amperometryczne.
3. Zmiana potencjału elektrody wskaźnikowej – potencjometria, miareczkowanie potencjometryczne.
4.
Przewodnictwo elektryczne roztworów – konduktometria, miareczkowanie konduktometryczne,
oscylometria, miareczkowanie oscylometryczne.
5. Promieniowanie α, β, γ powstające w wyniku reakcji jądrowych – metody radiometryczne.
6. Zmiana masy ogrzewanej próbki – termo grawimetria (TG).
7. Efekty cieplne (związane ze zmianą masy) – termiczna analiza różnicowa (DTA).
Wymień znane metody instrumentalne oparte na zjawiskach fizycznych.
1. Absorpcja promieniowania – spektrofotometria absorpcyjna cząsteczkowa (UV, VIS, IR), spektometria
absorpcyjna atomowa (ASA), absorpcja promieni rentgenowskich, magnetyczny rezonans jądrowy
(NMR), elektronowy rezonans paramagnetyczny (EPR, ESR).
2. Rozproszenie i absorpcja – turbidymetria.
3. Rozproszenie promieniowania – nefelometria, dyfrakcja promieni rentgenowskich.
4.
Odbicie światła – reflektometria.
5. Załamanie światła – refraktometria, interferometria.
6. Skręcanie płaszczyzny polaryzacji światła spolaryzowanego – polarymetria.
7.
Emisja promieniowania – fotometria płomieniowa, spektrografia i spektrometria emisyjna,
fluorescencja rentgenowska, fluorescencja atomowa, spektrofluorymetria.
8. Strumień cząstek naładowanych w polu magnetycznym oróżnym stosunku m/z (masy do ładunku) –
spektrometria mas (MS).
9. Strumień elektronów lub jonów o różnej energii – spektrometria elektronów i jonów.
10. Efekty cieplne (bez zmian masy) – termiczna analiza różnicowa (DTA).
Metoda krzywej wzorcowej.
Metoda wykorzystywana do ilościowych oznaczeń spektrofotometrycznych.
Krzywa wzorcowa jest graficzną zależnością absorbancji (A) od stężenia substancji wzorcowej (c). Wykonanie
wykres pozwala na bezpośredni odczytanie szukanych stężeń na podstawie zmierzonych wartości absorbancji
oznaczanych próbek. Prostoliniowy przebieg prostej świadczy o spełnieniu prawa Beera.
W celu wykreślenia krzywej wzorcowej przygotowuje się 5 – 8 roztworów wzorcowych tej samej substancji o
coraz większych stężeniach i mierzy ich absorbancję. Otrzymana krzywa wzorcowa nie może być
wykorzystywana jako wykres uniwersalny, gdyż zmiana warunków pracy i zmiana temperatury powodują
przesunięcie krzywej lub zmianę jej kąta nachylenia.
Krzywa wzorcowa może przechodzić przez układ współrzędnych ale nie musi:
1
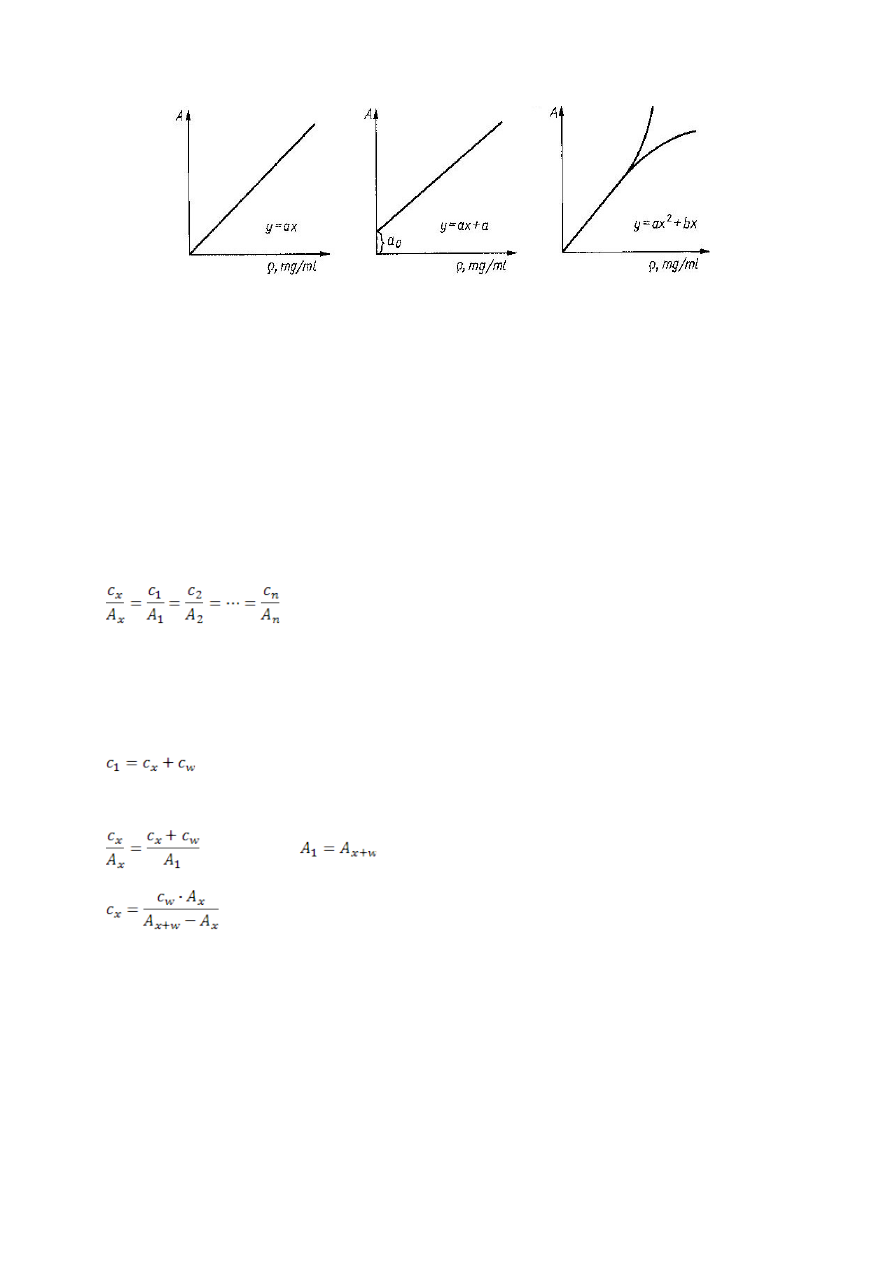
Otrzymana prosta opisana jest równaniem A = m * c
c – stężenie, m – współczynnik kierunkowy.
Metoda krzywej wzorcowej jest najczęściej stosowana podczas wykonywania wielokrotnych analiz roztworów o
podobnym składzie chemicznym. Metoda daje dobre wyniki, jeżeli w badanym zakresie stężeń spełnione jest
prawo Beera.
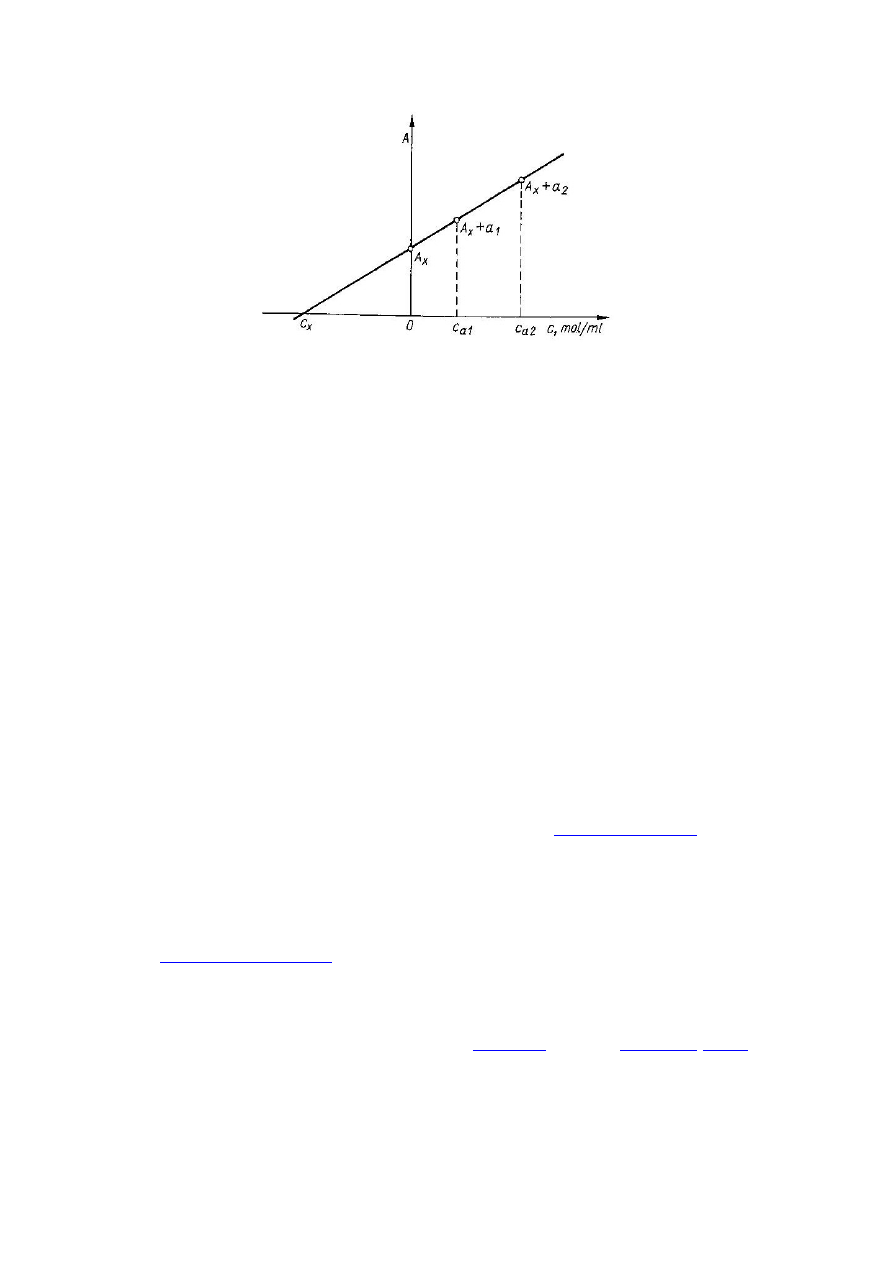
Metoda dodawania wzorca.
Stosowana podczas wykonywania pojedynczych analiz lub dla uniknięcia błędów wynikających z
niejednakowego składu roztworu oznaczanego i wzorca. Najczęściej stosowane dla oznaczenia ilości śladowych
obok dużych stężeń substancji towarzyszących. Substancja w badanych przedziale stężeń powinna spełniać
prawo Beera.
Podstawą tej metody jest zależność, którą graficznie można udowodnić twierdzeniem Talesa:
c
x
– stężenie badanej substancji w badanym roztworze, A
x
– sygnał analityczny badanego roztworu, c
1
(c
n
) –
stężenie badanej substancji w roztworze próbki zawierającej jeden dodatek wzorca (n dodatków wzorca), A
1
(A
n
)
– sygnał analityczny roztworu badanego zawierającego jeden dodatek wzorca (n dodatków wzorca).
Po dodaniu jednego dodatku wzorca (c
w
):
czyli:
Należy przygotować badany roztwór o stężeniu dwukrotnie większym niż potrzebny do analizy.
Z tego roztworu przygotowuje się dwa roztwory do pomiarów. Pierwszy przez dwukrotne rozcieńczenie
badanego roztworu, drugi przez takie samo rozcieńczenie, ale po dodaniu do niego określonej ilości
oznaczanego pierwiastka. Następnie mierzy się absorbancję obu roztworów i oblicza stężenie w roztworze
badanym.
Oznaczenia można również dokonać w sposób graficzny:
2
Słowniczek:
Absorpcja – proces polegający na wnikaniu cząsteczek, atomów lub jonów do wnętrza innej substancji
tworzącej dowolną fazę ciągłą - (gazu, cieczy, ciała stałego itp.) Absorpcji nie należy mylić z adsorpcją, która
jest zjawiskiem powierzchniowym. Absorpcja, adsorpcja i wymiana jonowa są wspólnie nazywane procesami
sorpcji.
Elektrograwimetria (analiza elektrograwimetryczna) – metoda analizy ilościowej, polegająca na wagowym
oznaczeniu substancji wydzielonej uprzednio na elektrodzie.
Kulometria – zespół metod elektrochemicznych, opartych na zastosowaniu praw elektrolizy Faradaya,
określających zależność pomiędzy ilością przepływającego przez obwód ładunku a ilością substancji ulegającej
elektrolizie. Pomiar ładunku elektrycznego prowadzi się za pomocą kulometru.
Polarografia – część woltamperometrii, elektrochemiczna metoda analityczna polegająca na przyłożeniu
liniowo wzrastającego potencjału elektrycznego do kroplowej elektrody rtęciowej będącej elektrodą pracującą z
cyklicznie zmieniającą się w trakcie pomiaru powierzchnią i rejestracji natężenia prądu płynącego przez nią.
Wartość natężenia prądu jest proporcjonalna do stężenia obecnej w roztworze substancji ulegającej utlenieniu
lub redukcji. Krzywa zależności natężenia prądu od liniowo rosnącego potencjału, rejestrowana za pomocą
aparatu zwanego polarografem, w postaci tzw. krzywej polarograficznej pozwala zidentyfikować substancję
badaną i określić jej stężenie.
Woltamperometria – analiza woltamperometryczna, dział chemicznej
. Jej podstawą
jest pomiar zależności natężenie prądu - potencjał w układzie elektrod pracującej i odniesienia (niekiedy
występuje dodatkowo trzecia elektroda pomocnicza) zanurzonych w roztworze badanym zawierającym
oznaczaną substancję (depolaryzator) i elektrolit podstawowy. Elektroda porównawcza (odniesienia) jest
niepolaryzowana (np. elektroda kalomelowa), natomiast elektroda pracująca jest polaryzowaną obojętną, i może
to być:
1.
(ker)
2. inna elektroda rtęciowa
3. mikroelektroda stała
Pomiary wykonywane z zastosowaniem ker są nazywane
, choć
często pojęcie to jest utożsamiane z woltamperometrią.
Amperometria – zespół metod elektrochemicznych, obejmuje pomiary zmian natężenia prądu elektrycznego
wywołane przebiegiem reakcji elektrodowej, której ulega substancja elektroaktywna w warunkach stałej
(wymuszonej) różnicy potencjałów.
3

Amperometryczne miareczkowanie (miareczkowanie polarymetryczne) – metoda chemicznej analizy
instrumentalnej polegająca na pomiarze prądu granicznego płynącego w obwodzie polarograficznym (elektroda
kroplowa rtęciowa) podczas miareczkowania oznaczanej substancji. Metodę tą stosuje się do oznaczeń opartych
na reakcjach strąceniowych, kompleksowania i utleniająco – redukujących.
– metoda wykorzystująca zależność między aktywnością oznaczanego jonu w roztworze, a
potencjałem elektrycznym elektrody. Praktycznie wyznacza się stężenie oznaczanego składnika na podstawie
SEM ogniwa utworzonego z elektrody wzorcowej i pomiarowej (dla roztworów rozcieńczonych). Zależność tę
opisuje najogólniej równanie
Miareczkowanie potencjometryczne – metoda miareczkowa polegająca na pomiarze zmian SEM ogniwa
złożonego z elektrody wskaźnikowej i elektrody odniesienia w funkcji objętości dodanego titranta.
Konduktometria – metoda elektroanalityczna oparta na pomiarze przewodności elektrolitów, zmieniającej się
wraz ze zmianą stężenia roztworów. Na podstawie wyników pomiaru przewodności określa się stężenie
badanego roztworu w odniesieniu do danych wzorcowych. Inną mozliwością zastosowania tej metody jest
miareczkowanie konduktometryczne, gdzie na podstawie zmiany przewodności ustala się punkt końcowy
miareczkowania.
Miareczkowanie konduktometryczne - w chemii analitycznej chemiczna technika miareczkowa polegająca na
pomiarze zmian przewodnictwa elektrycznego analizowanego roztworu w trakcie stopniowego dodawania do
niego odczynnika miareczkującego.
Miareczkowanie konduktometryczne przeprowadzane jest zwykle w układzie kwas-zasada. O przewodnictwie
układu kwas-zasada decydują głównie bardzo ruchliwe jony hydroniowe, a zatem jest ono funkcją pH układu.
Przy miareczkowaniu słabych kwasów, nie zauważa się początkowego spadku przewodnictwa, gdyż od razu
powstaje mocny elektrolit. Pomimo tego po osiągnięciu punktu równoważnikowego, stężenie jonów
hydroksylowych i ciągłe zwiększanie się sumy stężeń wszystkich jonów, powoduje powstanie wyraźnego
załamania w punkcie zobojętnienia na krzywej miareczkowania. Przy miareczkowaniu mieszaniny mocny –
słaby kwas, najpierw zostaje zneutralizowany mocny kwas.
Oscylometria – jedna z niespecyficznych elektrochemicznych metod analitycznych, polega na pośrednim
pomiarze admitancji lub impedancji naczynia pomiarowego w zależności od stężenia roztworu. Brak jest reakcji
elektrodowych, a elektrody stanowią okładki kondensatora
Metody radiometryczne – chemiczne metody analityczne, które są oparte na pomiarach energii
α, β lub γ wysyłanego w czasie przemian
, które mogą być spontaniczne lub
wymuszone. Reakcje wymuszone przebiegają, gdy w wyniku zderzeń i
energię w postaci pędu i masy utworzy się układ w stanie wzbudzonym, który rozpada się na produkty.
Radiometryczne techniki analityczne:
•
Metoda rozcieńczeń izotopowych
•
Termograwimetria (TG) – w metodzie tej dokonuje się pomiarów masy próbki w funkcji temperatury lub
czasu. Próbka jest ogrzewana w kontrolowanej atmosferze.
Termiczna analiza różnicowa (DTA) – metoda polegająca na rejestracji różnicy temperatur między substancją
badaną i substancją odniesienia względem czasu lub temperatury, jako dwu próbek znajdujących się w
identycznych warunkach w środowisku ogrzewanym lub chłodzonym w sposób kontrolowany. Rezultatem
pomiaru jest krzywa termicznej analizy różnicowej (krzywa DTA). Na krzywej tej różnica temperatur .T
odkładana jest na osi rzędnych, a na osi odciętych temperatura lub czas wzrastające od lewej ku prawej. Na
krzywej DTA wyróżnia się odcinki określane jako linia podstawowa. Oznaczają one przedziały temperatur w
których w próbce nie zachodzą procesy związane z pochłanianiem lub wydzielaniem ciepła. W momencie
reakcji lub przemiany fazowej linia
4

podstawowa przechodzi w pik. Jest to część krzywej DTA, gdzie odchyla się ona od linii podstawowej, a
następnie do niej wraca. Pik endotermiczny powstaje wówczas, gdy temperatura próbki badanej jest niższa niż
wzorcowa, zaś egzotermiczny pojawia się wtedy, gdy temperatura próbki badanej wzrasta powyżej temperatury
próbki wzorcowej.
Spektroskopia UV – spektroskopia świetlna, w której widmo powstaje na skutek przejścia lub odbicia się
światła ultrafioletowego przez analizowaną próbkę.
W chemii organicznej absorpcyjna spektroskopia UV jest stosowana do wykrywania w związkach chemicznych
grup zawierających sprzężone wiązania wielokrotne węgiel-heteroatom lub węgiel-węgiel, występujące w
alkenach, arenach i wielu związkach heterocyklicznych. Związki zawierające tego typu ugrupowania posiadają
bowiem zdolność do absorpcji światła UV. W absorpcyjnych widmach UV, w odróżnieniu od widm w
podczerwieni, występują zwykle bardzo szerokie piki absorpcyjne, których maksimum i kształt jest jednak
charakterystyczny dla danych grup funkcyjnych.
W technologii materiałowej spektroskopia UV umożliwia wstępne ustalenie przydatności materiałów jako np.
filtrów UV, czy też przewodników prądu elektrycznego, a także zbadanie niektórych własności ich powierzchni.
Spektroskopia UV-VIS – to zespół technik spektroskopowych, w których wykorzystuje się promieniowanie
elektromagnetyczne leżące w zakresie światła widzialnego oraz bliskiego ultrafioletu i bliskiej podczerwieni
(długość fali od 200 nm do 1100 nm).
Spektroskopia UV-VIS jest rutynowo stosowana w ilościowej analizie roztworów jonów metali przejściowych i
złożonych związków organicznych. Urządzeniem służącym do badań za pomocą tej techniki jest spektrofotometr
UV/VIS.
Spektroskopia IR – rodzaj spektroskopii, w której stosuje się promieniowanie podczerwone. Najpowszechniej
stosowaną techniką IR jest absorpcyjna spektroskopia IR, służąca do otrzymywania widm oscylacyjnych (choć
w zakresie dalekiej podczerwieni obserwuje się także przejścia rotacyjne). Przy pomocy spektroskopii IR można
ustalić jakie grupy funkcyjne obecne są w analizowanym związku.
Spektroskopia w podczerwieni pozwala na analizę zarówno struktury cząsteczek jak i ich oddziaływania z
otoczeniem. Jest to jedna z podstawowych metod stosowanych w badaniu wiązań wodorowych. Metodą
komplementarną do spektroskopii IR jest spektroskopia Ramana.
Atomowa Spektrometria Absorpcyjna (ASA) – technika analityczna pozwalająca na oznaczanie pierwiastków
(przede wszystkim metali) w próbkach ciekłych, stałych i gazowych. Zasada pomiaru opiera się na zjawisku
absorpcji promieniowania o specyficznej długości fali przez wolne atomy metali.
Procedura pomiarowa (pomijając przygotowanie próbki) polega na wprowadzeniu próbki do aparatu, atomizacji,
pomiarze absorbancji i obliczeniu na jej podstawie stężenia. ASA jest metodą wymagającą wykonania krzywej
wzorcowej przed przystąpieniem do pomiarów. Niezbędne jest również posiadanie odpowiedniej lampy dla
każdego oznaczanego pierwiastka.
Spektroskopia Magnetycznego Rezonansu Jądrowego (NMR) – jedna z najczęściej stosowanych obecnie
technik spektroskopowych w chemii i medycynie.
Spektroskopia ta polega na wzbudzaniu spinów jądrowych znajdujących się w zewnętrznym polu
magnetycznym poprzez szybkie zmiany pola magnetycznego, a następnie rejestrację promieniowania
elektromagnetycznego powstającego na skutek zjawisk relaksacji, gdzie przez relaksację rozumiemy powrót
układu spinów jądrowych do stanu równowagi termodynamicznej. NMR jest zatem jedną ze spektroskopii
absorpcyjnych.
Spektroskopia Elektronowego Rezonansu Paramagnetycznego (EPS) – jest techniką pozwalającą na
wykrycie związków posiadających niesparowane elektrony, czyli będące wolnymi rodnikami. Z powodu tego, iż
większość stabilnych cząsteczek nie posiada wolnych elektronów, technika ta jest rzadziej używana niż
spektroskopia NMR.
5

Podstawowe fizyczne założenia techniki są analogiczne do tych wykorzystywanych w spektroskopii NMR, ale
badane są spiny elektronów, a nie spiny jąder atomowych. Z powodu różnic w masie pomiędzy jądrami a
elektronami, w technice EPR używane są słabsze pola magnetyczne i wyższe częstotliwości promieniowania
mikrofalowego niż w spektroskopii NMR. Dla elektronów, rezonans paramagnetyczny w polu magnetycznym o
wartości ok. 0,3 tesli zachodzi przy częstotliwości ok. 10
Spektroskopia EPR jest wykorzystywana m.in. w fizyce ciała stałego do identyfikacji wolnych rodników, w
chemii do badań przebiegu reakcji oraz w biologii i medycynie do śledzenia znaczników spinowych.
Ponieważ wolne rodniki są bardzo reaktywne, nie występują one w układach biologicznych w wysokich
stężeniach. Aby badać układy biologiczne zaprojektowano małoreaktywne molekuły mogące wiązać się do
specyficznych miejsc w komórce czy białku, pozwala to na otrzymanie informacji o otoczeniu danej próbki
spinowej.
Turbidymetria – jedna z metod spektrofotometrycznych w chemii analitycznej; służy do pomiaru mętności
zawiesin. Istota metody jest analogiczna, jak w przypadku innych metod spektrofotometrycznych i opiera się na
pomiarze relacji pomiędzy ilością światła emitowanego przez źródło, a ilością światła docierającą do detektora
spektrofotometru, po przejściu przez komórkę (kuwetę) z badaną próbką. Relacja ta zależy głównie od stężenia
cząstek zawiesiny, na których zachodzi dyspersja światła.
Turbidymetria należy do metod analitycznych o stosunkowo niewielkiej precyzji. Bywa wykorzystywana m.in.
w konstrukcji bioreaktorów, gdzie służy do badania ilości utworzonej biomasy mikroorganizmów (jest to tzw.
turbidostat).
Nefelometria – metoda analizy stężenia roztworu na podstawie pomiaru natężenia światła rozproszonego przez
zawiesinę, wykorzystująca efekt Tyndalla.
Wiązka światła przechodząc przez roztwór koloidalny pod określonym kątem względem wiązki padającej, staje
się widoczna w postaci tzw. Stożka Tyndalla. Na tej podstawie oznacza się stężenie tej zawiesiny lub rozmiary
tworzących ją cząstek.
Nefelometrię wykorzystuje się w medycynie. Jest jedną z metod analizy instrumentalnej.
Refraktometria – instrumentalna metoda analityczna wykorzystująca pomiary współczynników załamania
światła badanych roztworów. Na tej podstawie wnioskuje się o stężeniu oznaczanych substancji oraz o strukturze
związków
chemicznych
Refraktometria jest stosowana najczęściej do oznaczania związków organicznych. Pomiary w refraktometrii
wykonywane są za pomocą refraktometrów. Do najbardziej popularnych refraktometrów należą: Pulfricha,
Abbego i refraktometr zanurzeniowy.
Interferometria – technika wykorzystująca zjawisko interferencji fal elektromagnetycznych (światła, fal
radiowych) do pomiarów, np. długości fali, pomiarów kątowych gwiazd, kontroli jakości elementów i układów
optycznych.
Interferometria znajduje też zastosowanie w mechanice eksperymentalnej (interferometria moiré, holografia, czy
interferometria plamkowa). Techniki te wykorzystuje się zasadniczo do pomiarów pola przemieszczeń i kształtu
obiektów, choć dzięki informacjom uzyskanym z pomiarów pola przemieszczeń można, za pomocą
numerycznego różniczkowania, w łatwy sposób wyznaczyć odkształcenia badanego obiektu.
Pierwszym etapem każdej z powyższych metod jest oświetlenie powierzchni badanego obiektu wiązką fali
nośnej. Fala nośna jest znanym sygnałem, który zostanie zmodyfikowany na skutek zmian powierzchni obiektu.
W przypadku interferometrii holograficznej uzyskany, charakterystyczny rozkład prążkowy jest wynikiem
interferencji fali nośnej odbitej od zmienionego, na przykład zdeformowanego, obiektu z falą odbitą od obiektu
pierwotnego, przy czym jako fale nośną stosuje się spójne, monochromatyczne światło laserowe. Prążki zwane
są często liniami izoteicznymi (ang. isoteic lines) lub liniami stałego przemieszczenia, ponieważ każdy prążek
znajduje się nad tymi punktami obiektu, które przemieściły się o tę samą wielkość.
6

Polarymetria – technika analityczna polegająca na pomiarze stężenia substancji optycznie czynnej na podstawie
wielkości kąta skręcenia płaszczyzny polaryzacji światła.
Jest to możliwe dzięki temu, że wielkość kąta skręcenia dla danej substancji jest proporcjonalna do jej stężenia w
roztworze. Przez polarymetrię rozumie się też często oznaczanie kąta skręcenia płaszczyzny polaryzacji czystych
związków chemicznych. Oprócz oznaczania stężeń związków czynnych optycznie, technika ta umożliwia także
pomiar tzw. czystości optycznej enancjomerów.
Fotometria płomieniowa – metoda analityczna oparta na pomiarze promieniowania emitowanego przez
odpowiednio wzbudzoną próbkę. Jako źródło wzbudzenia stosuje się w niej płomień palnika, do którego
wprowadza się badaną substancję, zwykle w postaci rozpylonego roztworu. Badany roztwór jest przy użyciu
sprężonego powietrza zasysany z naczynka i rozpylany do płomienia gazowego. Atomy spalanej substancji
emitują charakterystyczne widmo. Światło płomienia przechodzi przez układ optyczny z filtrem
przepuszczającym jedynie widmo badanego pierwiastka i trafia na fotoogniwo. Powstały w fotoogniwie prąd
elektryczny jest miarą ilości badanej substancji.
Fotometrię płomieniową stosuje się do naturalnych materiałów ciekłych, takich jak wody różnego pochodzenia
lub ścieki. W hydrochemii używa się jej przede wszystkim do pomiaru stężenia potasowców i wapniowców
(sód, potas, wapń, magnez, stront i in.) Ponadto ta technika analityczna ma duże znaczenie m.in. w biochemii i
medycynie (badanie moczu, surowicy krwi i tkanek), w geologii i mineralogii (analiza rud i minerałów), w
agrochemii (badanie gleb, analiza nawozów i minerałów roślinnych).
Fluorescencja rentgenowska – metoda analiz chemicznych, polega na pobudzaniu rentgenowskiego
promieniowania charakterystycznego danego materiału poprzez umieszczenie go w strumieniu
wysokoenergetycznych fotonów (kwantów gamma lub promieni rentgenowskich z lampy rentgenowskiej).
Energia padających fotonów musi być wyższa od energii analizowanego promieniowania charakterystycznego.
Do rejestracji promieniowania fluorescencji rentgenowskiej stosuje się obecnie spektrometry rentgenowskie z
detektorami półprzewodnikowymi. Odpowiednia kalibracja spektrometru pozwala przejść od obserwowanych
natężeń linii widmowych promieniowania charakterystycznego do koncentracji pierwiastków w badanym
materiale.
Spektrometria mas (MS) – uniwersalna technika analityczna, zaliczana do metod spektroskopowych, której
podstawą jest pomiar stosunku masy do jej ładunku elektrycznego (m/z). Pierwszy spektrometr mas został
zbudowany przez J. J. Thompsona w 1911 roku.
Współcześnie istnieje wiele odmian tej techniki, z których każda posiada inne zastosowanie i wymaga
stosowania aparatów o innej konstrukcji. Wszystkie te techniki są jednak oparte na jonizacji cząsteczek lub
atomów, a następnie detekcji liczby i stosunku masy do ładunku (m/z) powstających jonów. Wyniki działania
spektrometru mas są przedstawiane w postaci tzw. widma masowego.
Spektrometria mas służy do:
•
identyfikacji związków chemicznych i ich mieszanin,
•
ustalania struktury związków chemicznych,
•
ustalania ich składu pierwiastkowego,
•
ustalania składu izotopowego analizowanych substancji, co m.in. umożliwia określenie ich źródła
pochodzenia
•
precyzyjnego ustalania składu złożonych mieszanin związków o wysokich masach molowych
M
ETODY
OPTYCZNE
:
Zdefiniować następujące pojęcia: absorbancja, transmitancja, molowy współczynnik absorpcji.
7

Absorbancja – logarytm stosunku natężenia promieniowania padającego (I
0
) do natężenia promieniowania
wychodzącego (I
t
).
Absorbancję definiuje się również jako logarytm odwrotności transmitancji.
Transmitancja – stosunek natężenia promieniowania wychodzącego z danego ośrodka (I
t
) do natężenia
promieniowania padającego (I
0
). Wykazuje jaka część promieniowania padającego została przepuszczona przez
roztwór (w %).
Molowy współczynnik absorpcji
Wywodzi się z prawa Lamberta – Beera, które zakłada, że stopień absorpcji i rozpraszania światła jest
proporcjonalny do grubości warstwy i jej właściwości optycznych.
Absorbancja jest funkcją liczby cząsteczek absorbujących (dla jednej substancji absorbującej).
A – współczynnik absorbancji. Stężenia substancji absorbującej można obliczyć jako stężeni molowe, wtedy:
Gdzie ε jest molowym współczynnikiem absorbancji
.
Prawa absorpcji i odstępstwa od tych praw.
Pierwsze prawo Lamberta
Dotyczy zależności pomiędzy natężeniem światła padającego i natężeniem światła przechodzącego. Uzależnia
wielkość pochłaniania światła od natężenia światła padającego.
Jeżeli światło monochromatyczne o początkowym natężeniu I
0
przechodzi przez roztwór to natężenie światła
zmniejsza się w miarę przechodzenia światła przez poszczególne warstwy. Zatem jeśli umownie podzielimy
roztwór na kilka warstw tej samej grubości, to różnice pomiędzy natężeniami światła wchodzącego do danej
warstwy i wychodzącego z niej będą się zmniejszać w kierunku przechodzenia światła. Jednak w każdej
warstwie stosunek zmniejszania się natężenia światła do natężenia światła wychodzącego będzie stały. Zatem
można ogólnie zapisać, że:
Po przekształceniu:
8

Według pierwszego prawa Lamberta stosunek światła monochromatycznego po przejściu przez ośrodek
optycznie jednorodny jest proporcjonalny do natężenia światła padającego.
Prawo Bouguera – Lamberta
Określa związek pomiędzy absorpcją a grubością ośrodka absorbującego. Jeżeli warstwa absorbująca o grubości
l składa się z nieskończenie małych warstewek dl, z których każda zmniejsza natężenie padającego
promieniowania o Di, to względne zmniejszanie natężenia dI/I jest proporcjonalne do grubości warstwy
absorbującej:
I – natężenie światła wchodzącego do danej warstwy, k – wartość stała. Minus, bo w miarę zwiększania grubości
warstwy natężenie światła wychodzącego zmniejsza się.
Wynika z tego, że natężenie światła przechodzącego przez warstwę absorbującą zmniejsza się wykładniczo wraz
z liniowym zwiększaniem się grubości warstwy.
Prawo Beera
Podaje zależność pomiędzy absorbancją a stężeniem substancji absorbującej w
roztworze o stałej grubości warstwy.
Prawo Bouguera – Lamberta – Beera – Watlera
9
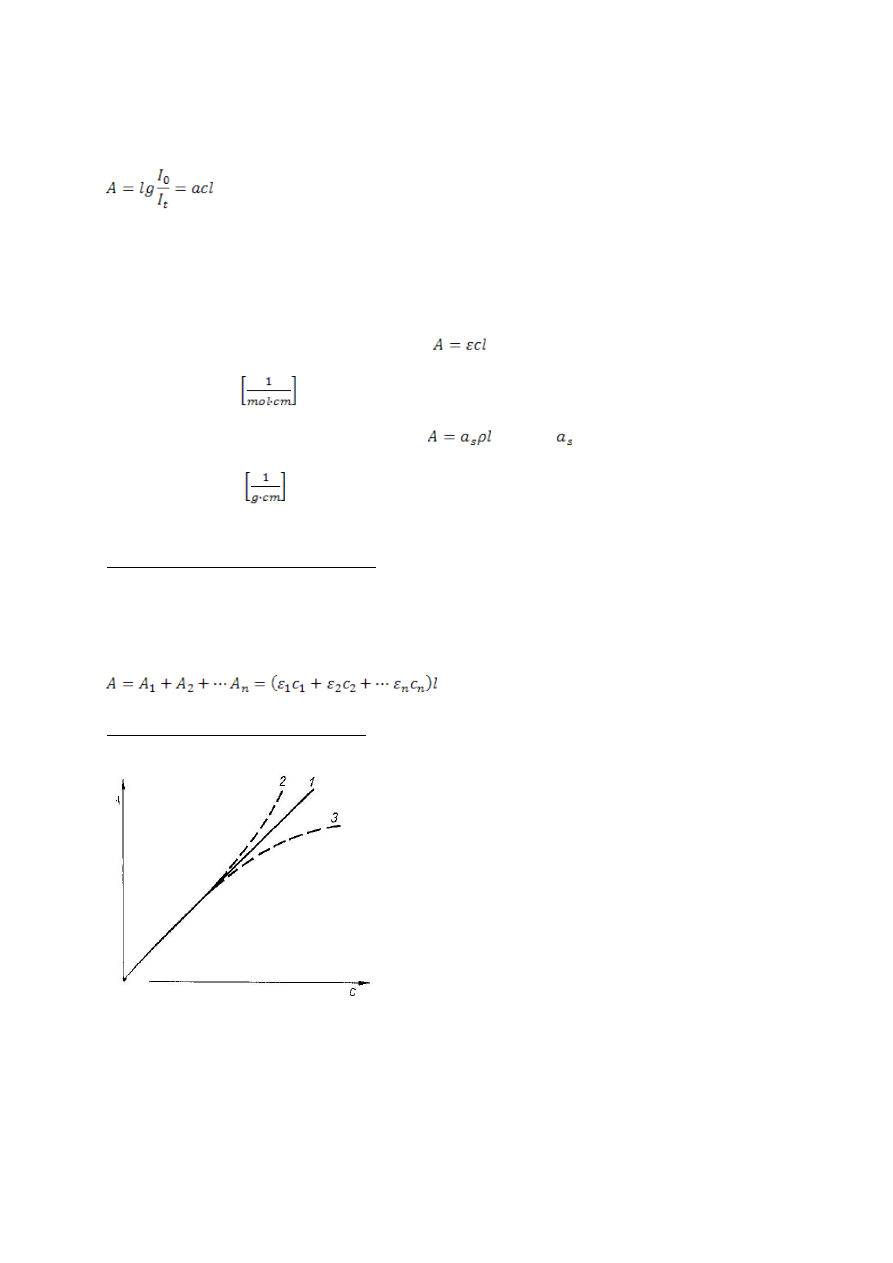
Określa zależność absorbancji od grubości warstwy absorbującej i stężenia
roztworu:
A – absorbancja, c – stężenia roztworu, l – grubość warstwy absorbującej [cm], a –
współczynnik absorpcji.
Stężenie absorbującej substancji można wyrazić na dwa sposoby:
•
jako stężenie molowe c:
, gdzie ε – molowy współczynnik
absorpcji
•
jako stężenie masowe ρ:
, gdzie - współczynnik absorpcji
właściwej
Prawo addytywności absorbancji
Dla przypadków, kiedy w roztworze znajduje się więcej niż jedna substancji
absorbująca. Wtedy absorbancja całkowita jest sumą absorbancji poszczególnych
składników:
Odstępstwa od prawa absorpcji
Wykres przedstawia zależność absorbancji od
stężenia. Dla substancji spełniających prawo
Beera wykres przedstawia linię prostą
przechodzącą przez początek układu
współrzędnych (1), której tanges kąta
nachylenia jest równy iloczynowi cl. Krzywa 2
wykazuje dodatnie odchylenie od prawa
Beera, krzywa 3 – odchylenie ujemne.
Odstępstwa od prawa Beera mogą być
spowodowane przyczynami chemicznymi i
fizycznymi. Jeżeli kształt widma absorpcyjnego
danej substancji zmienia się wraz ze zmianą stężenia substancji w roztworze, to
występują odstępstwa chemiczne. Zachodzi wtedy oddziaływanie cząsteczek
substancji ze sobą lub z cząsteczkami rozpuszczalnika. Reakcje dysocjacji
powodują ujemne odchylenie od prawa Beera. Najważniejsza przyczyną fizyczną
odstępstw od prawa Beera jest niemonochromatyczność promieniowania. Z
reguły stanowią odchylenia ujemne.
10

Precyzja i czułość oznaczeń spektrofotometrycznych.
Czułość metody oznaczania jest to najmniejsze oznaczalne stężenie pierwiastka
(najmniejsza oznaczalna ilość), które można określić za pomocą danej metody.
Można ją również definiować jako najmniejszą różnicę stężeń lub zawartości
składnika oznaczanego, którą można określić za pomocą danej metody. Zatem
czułość metody jest decydującym kryterium wyboru sposobu oznaczenia.
Liczbową miarą czułości metod spektrofotometrycznych jest molowy
współczynnik absorpcji:
Współczynnik nie zależy od stężenia. Zależy natomiast od długości fali światła
padającego i rodzaju substancji absorbującej światło.
Za czułe uważa się metody o wartości współczynników absorpcji ε > 10000. Gdy
ε < 1000 metodę uważa się za mało czułą.
Precyzja metody jest miarą zgodności otrzymywanych wyników, charakteryzuje
więc powtarzalność metody. Zależny ona od zakresu oznaczanych zawartości i od
stosowanej techniki w pomiarach absorbancji precyzja zależy od mierzonych
wartości.
Bardzo
małe
stężenie
substancji
barwnej
w roztworze są oznaczane z dużym błędem. Gdyż przepuszczalność roztworu
badanego jest podobna do przepuszczalności roztworu odniesienia i najczęściej
bliska 100%. W przypadku intensywnie zabarwionych roztworów tylko mała
część promieniowania przechodzi przez roztwór, co powoduje zwiększenie błędów
pomiaru. Aby uniknąć błędów należy zróżniczkować równanie Beera
i przedstawić na wykresie jako dA/A.
Schemat budowy spektrofotometru. Omówić krótko rolę każdego
elementu. Przykłady części składowych.
11
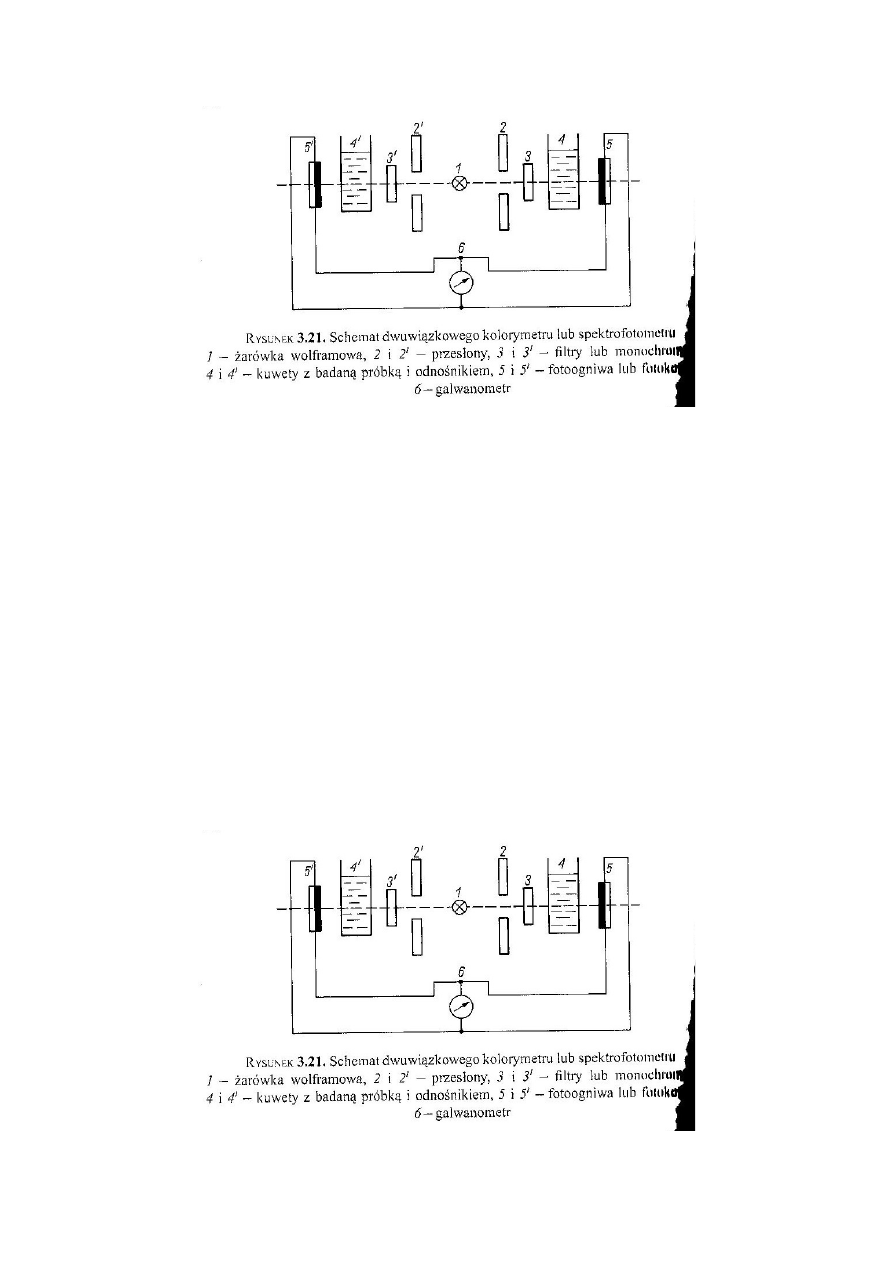
Źródłem promieniowania w zakresie widzialnym jest lampa z elektrycznie
żarzonym włóknem wolframowym, która dostarcza wiązkę światła „białego” o
ciągłym widmie i odpowiednim natężeniu. Regulacja wiązki odbywa się za
pomocą przesłony irysowej lub regulowanej szczeliny. Następnie dochodzi do
monochromatyzacji wiązki, czyli wydzieleniu ze światła złożonego
promieniowania
o określonej długości fali. Do monochromatyzacji służą filtry świetlne, pryzmaty
czy siatki dyfrakcyjne. Tak dobrana wiązka światła przechodzi przez próbkę
badanego roztworu. Następnym elementem budowy jest detektor, którego
zadaniem jest efekt fotoelektryczny, czyli zamiana energii świetlnej na energię
elektryczną. Pozwala to na bezpośredni pomiar natężenia promieniowania.
Detektorem może być: fotoogniwo, fotokomórka, fotopowielacz elektronowy,
fotodioda. Ostatnim elementem jest rejestrator. Jest nim najczęściej
galwanometr.
Schemat ogólny spektrofotometru. Stosowane źródła promieniowania.
12

Źródłami promieniowania są lampy: wodorowa lub deuterowa dla zakresu UV,
wolframowa lub halogenowa dla zakresu VIS.
Monochromatory i detektory stosowane w spektrofotometrii UV/VIS.
Monochromator w spektrofotometrach składa się z dwóch szczelin: wejściowej i
wyjściowej oraz urządzenia monochromatyzującego. Zadaniem szczeliny
wejściowej jest regulacja natężenia wiązki promieniowania pochodzącej od
źródła promieniowania. Szczelina wyjściowa pozwala na wyodrębnienie z widma
wiązki promieniowania o wybranej długości fali i określonej szerokości
spektralnej. Szerokość szczeliny wyjściowej wywiera również wpływ na zdolność
rozdzielczą monochromatora.
Do urządzeń monochromatyzujących zalicza się pryzmaty i siatki dyfrakcyjne.
Proces polega na rozszczepieniu wiązki światła białego wskutek: załamania
światła na granicy dwóch ośrodków w monochromaty zatorach pryzmatycznych;
lub ugięcia promieniowania na wąskich szczelinach i jego interferencji w
monochromaty zatorach siatkowych.
Pryzmat:
Promieniowanie padające na pryzmat ulega dwukrotnie załamaniu na ściankach
pryzmatu ustawionych pod kątem łamiącym, w wyniku czego ulega ono
rozszczepieniu na widmo. Z widma tego wyodrębnia się w wąskiej szczelinie
wyjściowej wiązkę promieniowania o wybranej długości fali i określonej szerokości
spektralnej. Pryzmaty, w zależności od zakresu stosowania mogą być wykonane
ze szkła lub kwarcu. Pryzmaty szklane wykorzystuje się w zakresie widzialnym
(dają lepsze rozszczepienie promieniowania niż pryzmaty kwarcowe). Pryzmaty
kwarcowe są stosowane dla zakresu nadfioletowego.
Siatka dyfrakcyjna:
Jest to wypolerowana płytka szklana lub metalowa z dużą liczbą równoległych rys
położonych blisko siebie. Zasada działania oparta jest na zjawisku interferencji
promieni ugiętych podczas przechodzenia przez wąskie szczeliny (szerokość
mniejsza niż długość fali padającej). Przechodząc przez siatkę dyfrakcyjną wiązka
promieniowania monochromatycznego ulega rozszczepieniu na wiele wiązek,
które odchylają się od kierunku padania pod kątami zależnymi od odległości
pomiędzy wyrysowanymi liniami i od długości fali promieniowania padającego.
Siatki dyfrakcyjne można podzielić na płaskie, odbiciowe i holograficzne.
Miareczkowanie spektrofotometryczne
Miareczkowanie to należy do grupy metod analizy objętościowej, gdzie wyznacza
się PK poprzez pomiar absorbancji roztworu. Dzięki tej metodzie można śledzić
prawie wszystkie reakcje chemiczne. Metodę stosuje się wtedy, gdy zmiana
13
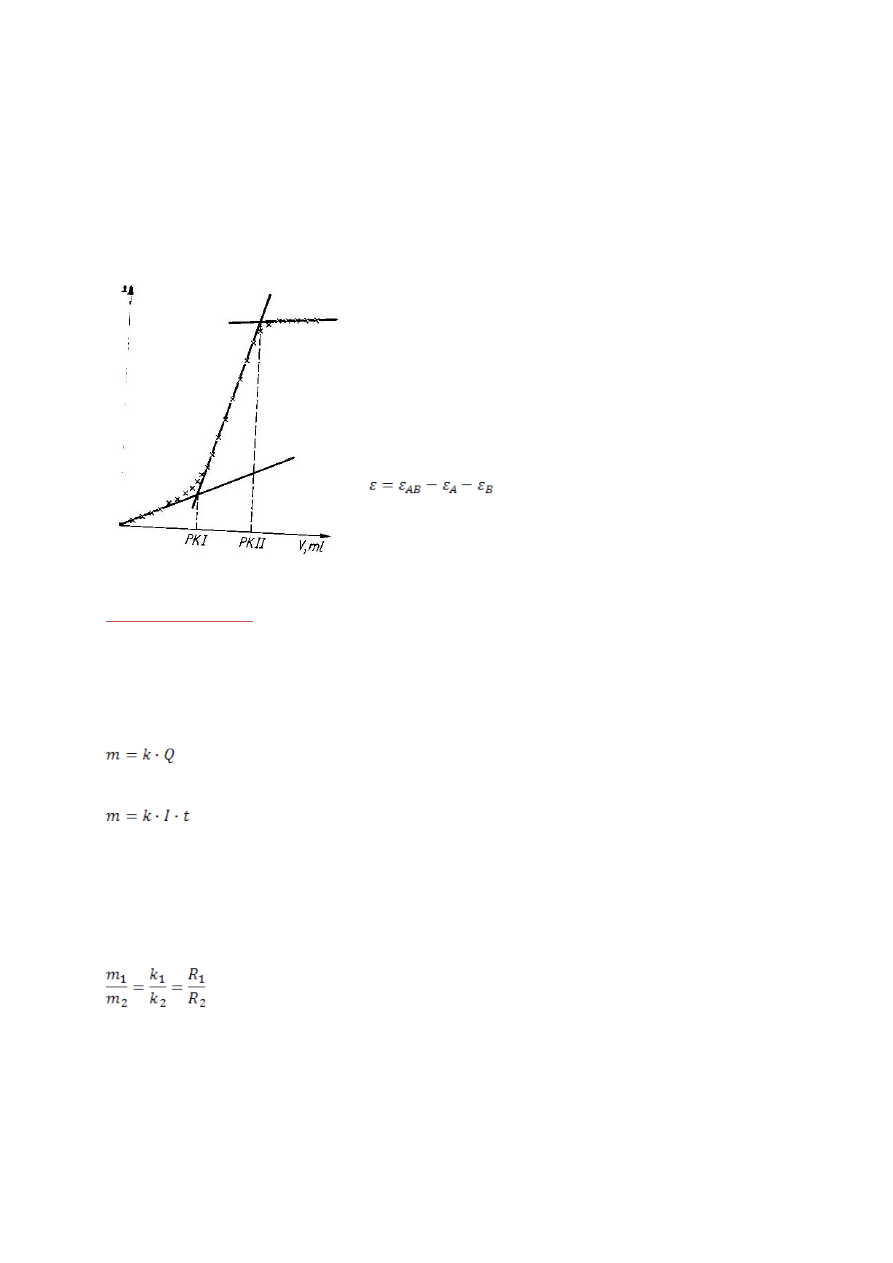
barwy podczas miareczkowania wizualnego nie jest dostatecznie wyraźne w
punkcie końcowym lub też przebiega stopniowo.
Metoda miareczkowania spektrofotometrycznego polega na kolejnych pomiarach
absorbancji analizowanego roztworu podczas miareczkowania przy wcześniej
wyznaczonej długości fali. Jeżeli badany roztwór spełnia prawo Beera to proces
miareczkowania można przedstawić w następujący sposób:
Na rysunku widoczne są dwa załamania
odpowiadające dwóm punktom końcowym (PKI i
PKII) dla różnych związków. Przebieg krzywej
miareczkowania przed punktem końcowym i po
nim zależy od wartości molowych współczynników
absorpcji składnika oznaczanego (ε
A
), reagenta
(ε
B
) oraz produktu (ε
AB
):
E
LEKTROCHEMIA
:
Prawa Faradaya.
Pierwsze prawo Faradaya – masa substancji wydzielonej podczas elektrolizy jest
proporcjonalna do ładunku, który przepłynął przez elektrolit.
Drugie prawo Faradaya - stosunek mas m
1
oraz m
2
substancji wydzielonych na
elektrodach podczas przepływu jednakowych ładunków elektrycznych jest równy
stosunkowi ich równoważników elektrochemicznych k
1
oraz k
2
i stosunkowi ich
mas równoważnikowych R
1
oraz R
2
.
Techniki elektrograwimetryczne.
Techniki elektrograwimetryczne można podzielić na:
14
•
elektrograwimetrię klasyczną – dwie elektrody przy stałej gęstości prądu
(prąd stały).nadaje się przede wszystkim do analizy ilościowej
pojedynczych substancji – zwłaszcza metali wydzielających się na
elektrodzie;
•
elektrograwimetrię z kontrolowanym potencjałem elektrody czynnej –
elektroliza prowadzona w warunkach gdy potencjał elektrody pracującej
ma wartość stałą. Elektroliza zachodzi przy stałym potencjale i stałym
natężeniu (podczas wydzielania się substancji potencjał, natężenie
zmniejsza się w wyniku czego należy zwiększać napięcie prądu). metoda
wykorzystywana do oznaczania metali o zbliżonych do siebie potencjałach
wydzielania;
•
elektrograwimetrię wewnętrzną – proces wydzielania substancji na
elektrodzie przebiega samorzutnie w wyniku zwarcia elektrod
przewodnikiem bez doprowadzania prądu zewnętrznego w ogniwie
zbudowanego z dwóch elektrod o odpowiednio dobranych potencjałach.
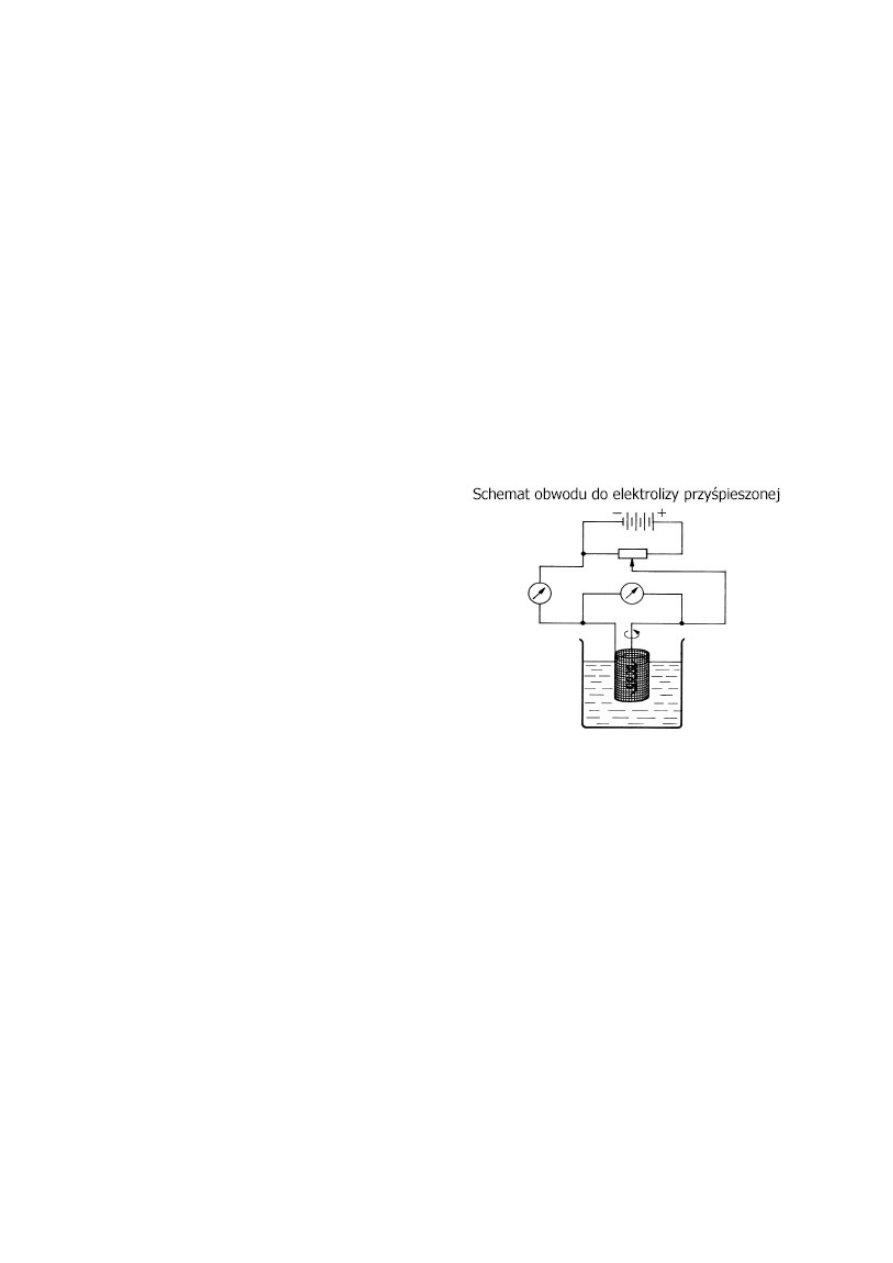
Co to jest elektroliza przyspieszona? Co to są bufory potencjału?
Stosuje się mieszanie i zwiększanie temperatury.
Bufory potencjału stosuje się aby wydzielała się tylko
jedna substancja z mieszaniny związków. Utrzymuje
on określoną wartość potencjału i nie pozwala na
osiągnięcie potencjału potrzebnego do wydzielenia
drugiej.
Wpływ gęstości prądowej na postać wydzielonego osadu.
Postać wydzielonego osadu zależy od gęstości prądowej (szybkości procesu).
Wartość ta nie może być zbyt mała ani zbyt duża. Gęstość prądu dobiera się
empirycznie, kierując się zasadą, że wydzielony osad powinien być zwarty i
mocno osadzony na elektrodzie. Gdy gęstość prądowa będzie zbyt duża osad
będzie przyjmował postać gąbczastą – z wolnymi przestrzeniami.
Potencjał elektrody. Wzór Nernsta. Rodzaje elektrod.
Potencjał elektrody - siła elektromotoryczna ogniwa zbudowanego z ogniwa
badanego, zawierającego jony o jednostkowej aktywności, oraz elektrody
wodorowej, której potencjał przyjmuje się za równy 0 we wszystkich
temperaturach, aby było możliwe określenie potencjału badanej elektrody. Jeśli
badana elektroda jest anodą, to jej potencjał jest ujemny, jeśli natomiast jest
katodą to jej potencjał jest dodatni.
Wzór Nernsta – stanowi podstawową zależność elektrochemiczną wyrażającą
równowagowy potencjał elektrody względem jej potencjału standardowego i
15
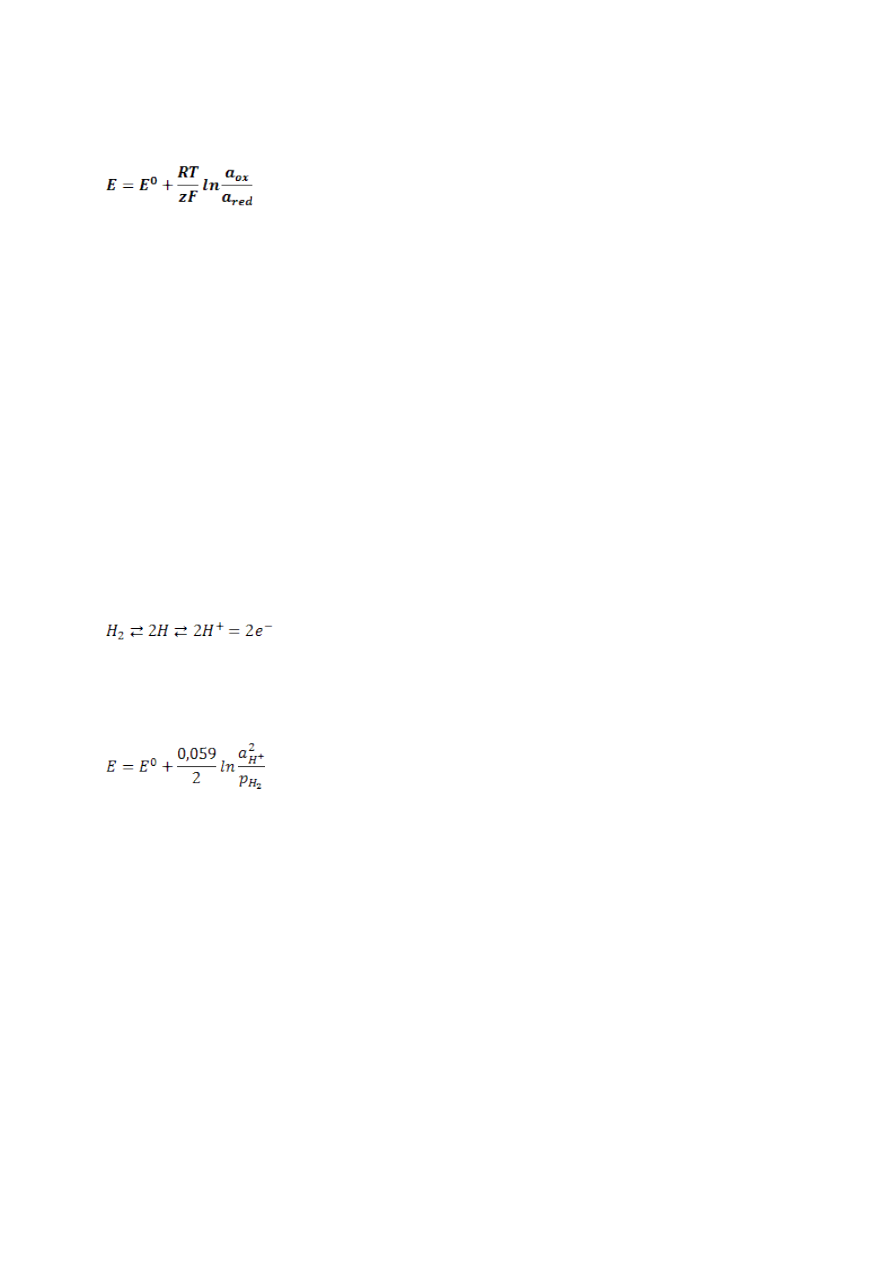
stężenia
substancji
biorących
udział
w procesie elektrodowym.
Rodzaje elektrod:
Istnieją trzy rodzaje elektrod. Pierwsze dwa rodzaje to anoda i katoda. Anoda to
ta
z elektrod, która przyjmuje ładunek ujemny lub wysyła dodatni, zaś katoda to
elektroda wysyłająca ładunek ujemny lub przyjmująca dodatni. Ładunek
elektryczny przepływający między anodą i katodą może przybierać formę
wolnych elektronów lub jonów. Trzecim rodzajem są elektrody oddziałujące na
przestrzeń swoim potencjałem.
Elektroda wodorowa. Normalna elektroda wodorowa.
Elektroda wodorowa jest elektrodą odwracalną względem kationu, a zatem
elektrodą pierwszego rzędu. Elektrodę wodorową stanowi platynowa blaszka
pokryta czernią platynową, zanurzona częściowo lub całkowicie w roztworze
zawierającym jony H
+
, omywana gazowym wodorem. Na powierzchni elektrody
ustala się wtedy równowaga:
Zgodnie z równaniem Nernsta potencjał elektrody wodorowej w temperaturze
25°C można wyrazić wzorem:
Kiedy ciśnienie cząstkowe wodoru wynosi 1atm, a aktywność jonów wodorowych
równa jest 1 to mamy do czynienia z normalną elektrodą wodorową, w której E =
E
0
(E
0
umownie przyjęta jako zero). Normalną elektrodę wodorową stosuje się
jako wzorcowy układ , za pomocą którego określa się potencjał względny
wybranej elektrody.
Podać znane podziały elektrod. Dla każdej grupy elektrod podać
przykłady.
Podział elektrod:
•
ze względu na rolę:
o wskaźnikowe (elektroda srebrowa, platynowa)
o odniesienia – porównawcza (elektroda kalomelowa)
16
•
ze względu na aktywność:
o aktywne
o obojętne
•
ze względu na mechanizm reakcji
o
I rodzaju (elektroda srebrowa, chlorowa)
o
II rodzaju (elektroda kalomelowa, chlorosrebrowa)
o
III rodzaju (układ złożony z metalu, jego trudno rozpuszczalnej soli i
drugiej trudno rozpuszczalnej soli o wspólnym anionie z pierwszą –
ołów, węglan ołowiu, węglan wapnia))
o
redoks (elektroda chinhydrynowa)
o
metaliczne (elektroda antymonowa, bizmutowa)
o
jonoselektywne (elektroda szklana)
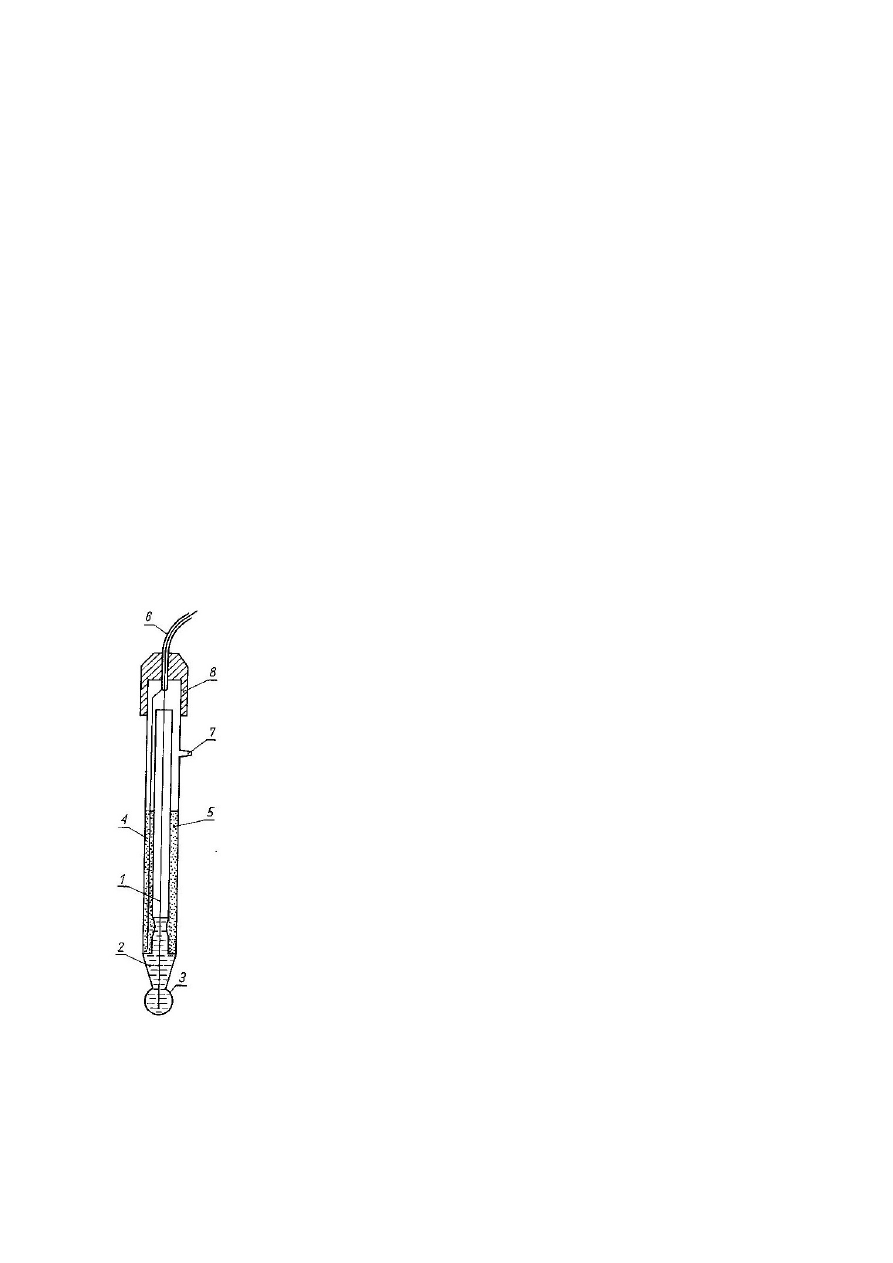
Budowa, działanie i zastosowanie elektrody kombinowanej.
Zestaw dwóch elektrod we wspólnej oprawie.
1 – elektroda wyprowadzająca chlorosrebrowa
2 – roztwór wewnętrzny elektrody wyprowadzającej
3 – membrana szklana
4 – elektroda porównawcza chlorosrebrowa
5 – roztwór wewnętrzny elektrody porównawczej
6 – kabel koncentryczny
7 – wlew roztworu wewnętrznego
8 – korek
Zbudowana jest z elektrody szklanej i elektrody chlorosrebrowej
umieszczonej we wspólnej oprawce. Membrana szklana elektrody
wykonana jest ze specjalnego szkła o małym oporze i dużej
wytrzymałości mechanicznej. Elektroda składa się z części szklanej
(wskaźnikowej) zakończonej kulistą banieczką (membraną), której potencjał
zależy od pH badanego roztworu oraz części odniesienia zakończonej przeponą o
potencjale niezależnym od pH badanego roztworu. Rolę półogniwa odniesienia
pełni
która
zanurzona
jest
17
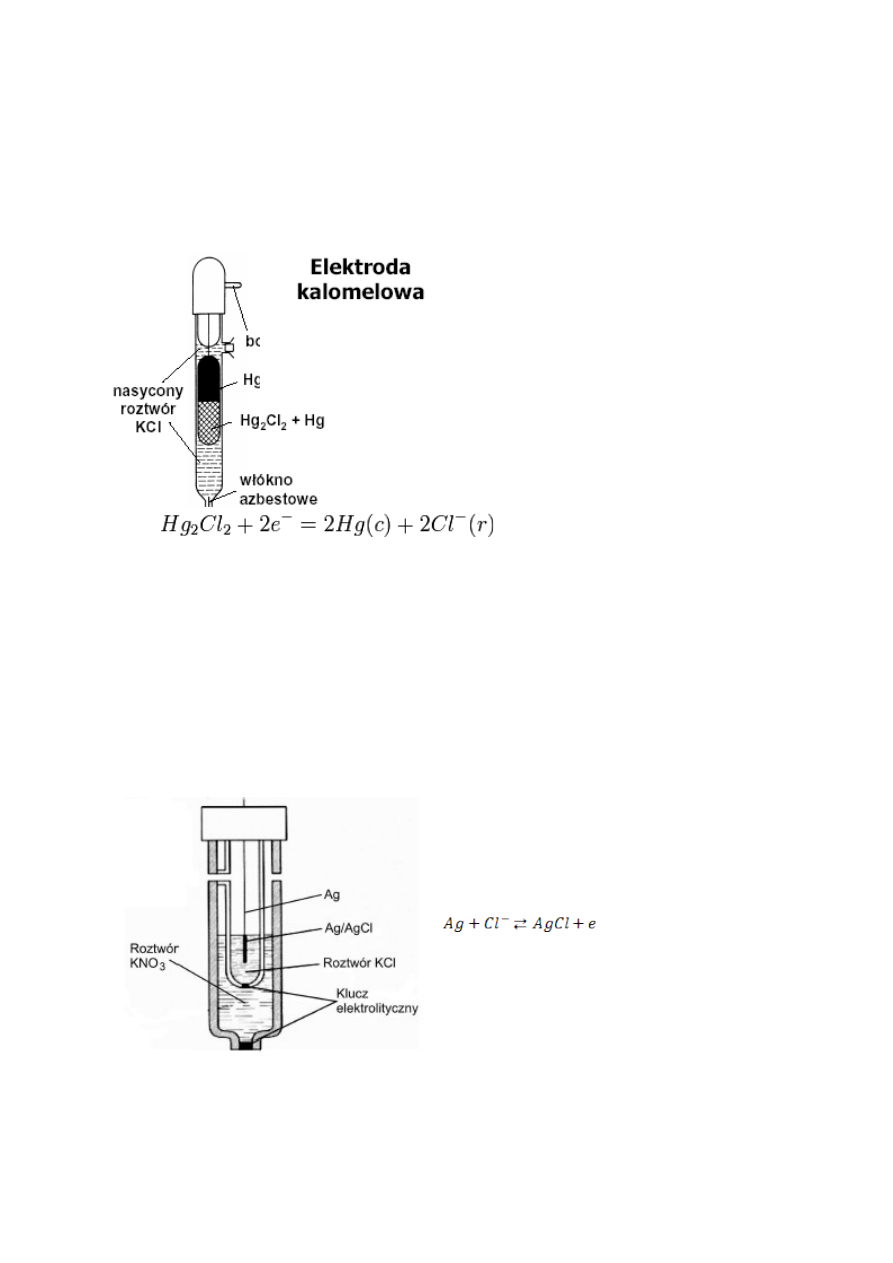
w nasyconym roztworze chlorku potasu. Po zanurzeniu w roztworze jest ogniwem
pomiarowym.
Budowa, działanie i zastosowanie elektrody kalomelowej.
Elektroda drugiego rzędu, którą stanowi
rtęć stykająca się z chlorkiem rtęci(I) (w
celu zabezpieczenia elektrody przed
obecnością Hg
2+
do sporządzenia jej nie
używa się czystego kalomelu, lecz pasty
kalomelowej zawierającej niewielkie
ilości rozdrobnionej rtęci) w roztworze
chlorku potasu (KCl)
Reakcja elektrodowa:
wskazuje, że potencjał takiego półogniwa zależy od stężenia jonów chlorkowych.
Stosując nasycony roztwór jonów Cl- (np. KCl), uzyskuje się półogniwo o stałym
potencjale, który w temperaturze 25°C wynosi E = + 0,2679 V.
Elektroda kalomelowa jest często stosowana w praktyce laboratoryjnej jako
półogniwo odniesienia do pomiaru potencjału innych półogniw, zamiast
niewygodnej w użyciu elektrody wodorowej.
Może być stosowana w temperaturach nie przekraczających 70°C
Budowa, działanie i zastosowanie elektrody chlorosrebrowej.
Jest nią drucik srebrny pokryty solą
trudno rozpuszczalną AgCl, zanurzony w
roztworze zawierającym jony chlorkowe.
Przemiany zachodzące w elektrodzie
można zapisać następująco:
Przejściu atomów srebra w stan jonowy
towarzyszy zanikanie jonów chlorkowych
i odwrotnie. Jest to więc elektroda II
rzędu – odwracalna względem anionu.
18
Naszkicować przebieg krzywej miareczkowania potencjometrycznego
mieszaniny
jodków
i bromków za pomocą roztworu azotanu srebra, zaznaczyć na krzywej
punkty końcowe miareczkowania i podać wzory ogólne na obliczanie
zawartość
masy
jodków
i
bromków
w otrzymanej próbce z wyjaśnieniem stosowanych symboli.
masa jodu = V
jodu
. C
AgNO3
. M
I
. W
masa bromu = (V
bromku
– V
jodu
). C
AgNO3
.
M
Br
. W
C
AgNO3
= stężenie titranu
M
I, Br
= masa molowa jodu, bromu
W =współczynnik kolby i pipety
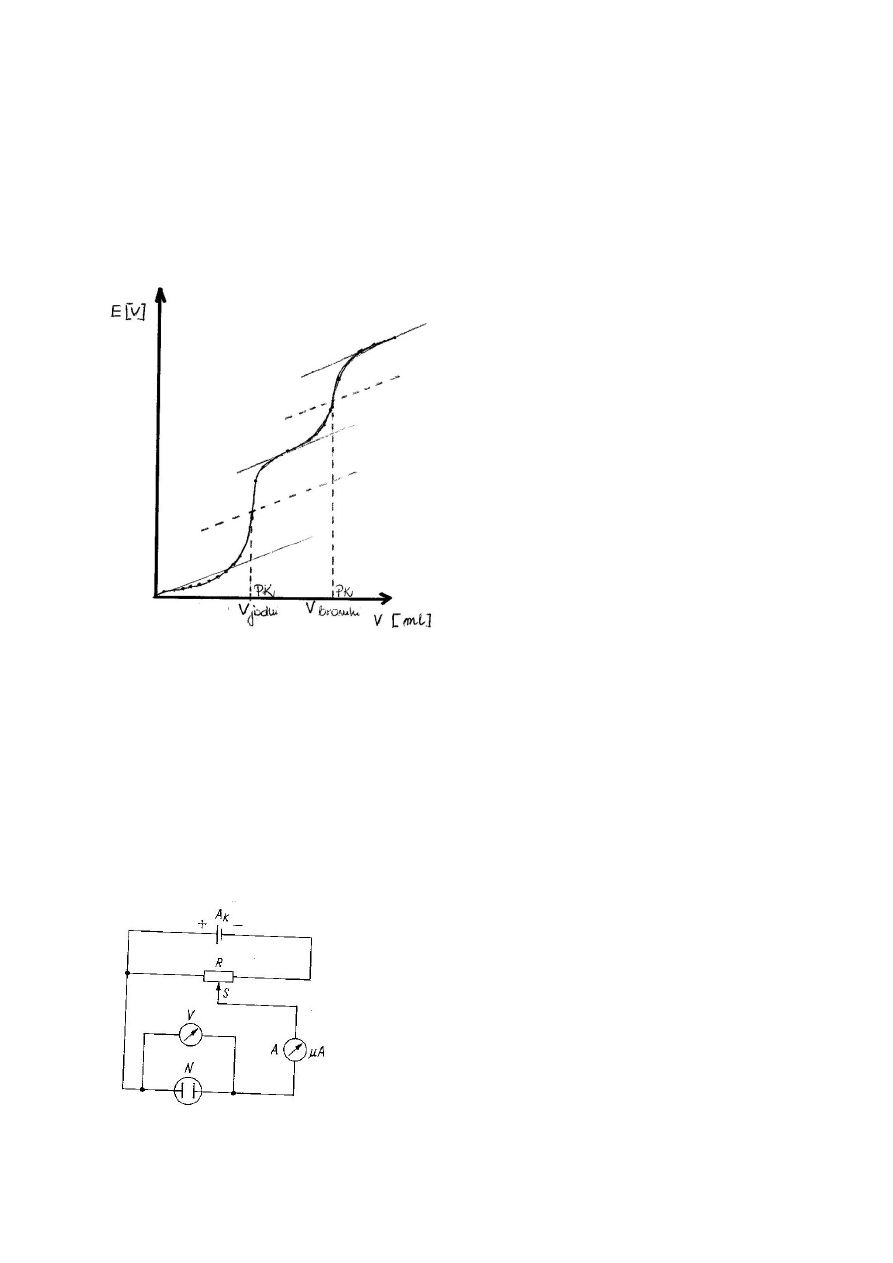
Polarografia stałoprądowa (klasyczna)
W klasycznej polarografii stałoprądowej doprowadza się do kroplowej elektrody
rtęciowej zmieniający się liniowo potencjał mierzony względem odpowiedniej
elektrody porównawczej (elektrody kalomelowej). Natężenie prądu stałego,
płynącego w tych warunkach przez roztwór jest wykreślane w funkcji
przyłożonego napięcia.
Schemat obwodu elektrycznego do oznaczeń polarograficznych:
W analizie polarograficznej zamiast wyznaczać
wartość natężenia prądu dyfuzyjnego, stosuje się
wyznaczenie wysokości fali polarograficznej (mierzy
się ją bezpośrednio z polarogramu w mm), która
jest wprost proporcjonalna do natężenia prądu w
μA.
19
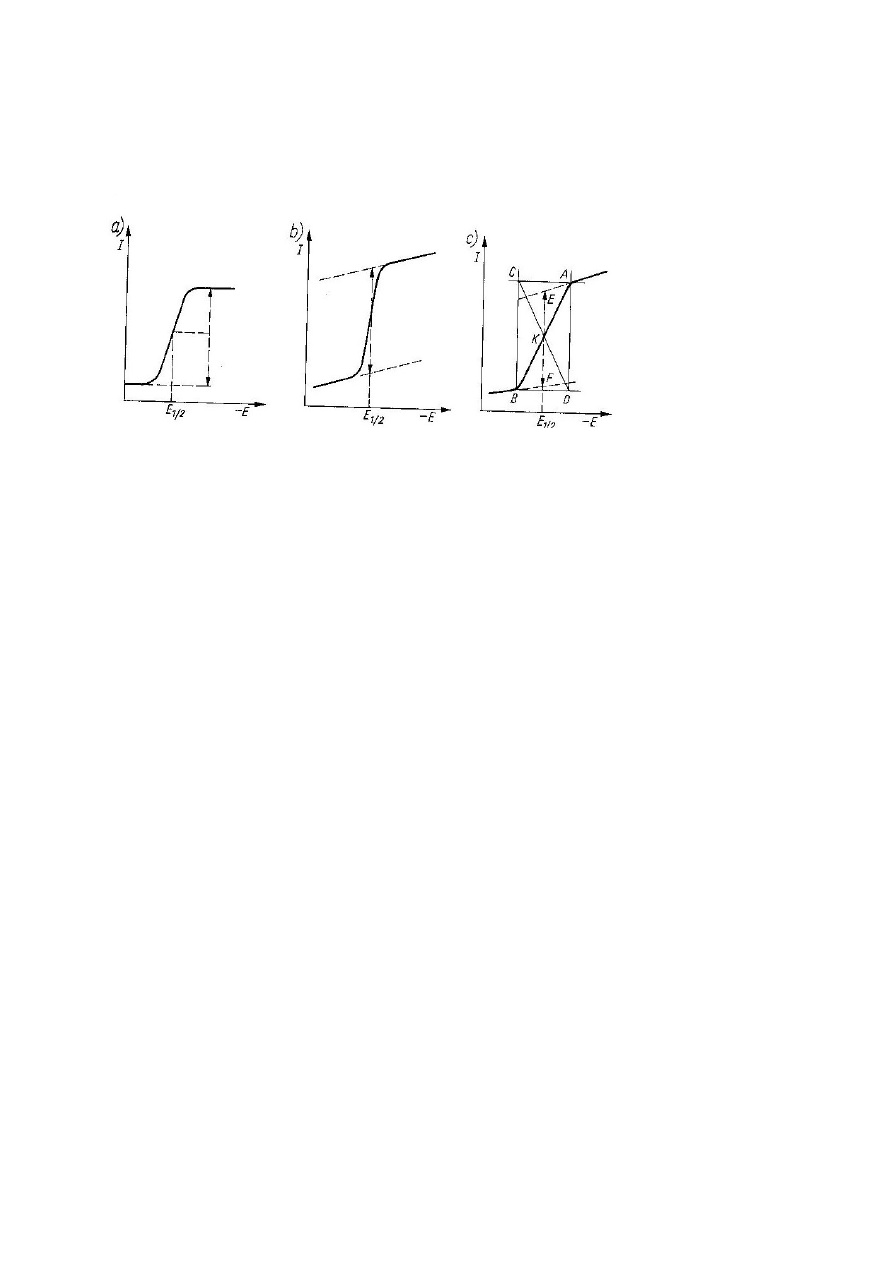
Graficzne metody wyznaczania wysokości fali i potencjału półfali:
Do metod oznaczeń ilościowych w polarografii stałoprądowej zalicza się metodę
krzywej wzorcowej jak i metodę dodawania wzorca.
Polarografia stałoprądowa pozwala na wykrywanie i oznaczanie substancji
elektroaktywnych
o stężeniach 10
-3
– 10
-5
mol/l. stosuje się ją do oznaczeń niewielkich zawartości
składników
w stopach, do oznaczeń zanieczyszczeń w produktach spożywczych i materiałach
biologicznych oraz do oznaczania składników metalicznych w glinkach,
minerałach, materiałach ceramicznych. Analiza polarograficzna obejmuje nie
tylko jony proste, ale i złożone a nawet związki organiczne. Zaletą tej metody jest
możliwość oznaczenia więcej niż jednej substancji w tym samym roztworze.
Metoda jest również przydatna dla oznaczania jonu tego samego pierwiastka ale
na różnym stopniu utlenienia.
Prądy w polarografii
•
szczątkowy – jest sumą prądu dyfuzyjnego spowodowanego niewielką
ilością aktywnych polarograficznie zanieczyszczeń i prądu
pojemnościowego,
•
dyfuzyjny – jego natężenie zależy tylko od szybkości dyfuzji depolaryzatora
z głębi roztworu do powierzchni elektrody kroplowej. Szybkość ta zależy od
stężenia
oznaczanej
substancji
w roztworze,
•
pojemnościowy – występuje dlatego, że układ elektrod tworzy pewnego
rodzaju kondensator. Jego wartość jest bardzo mała. Jest spowodowany
procesem ładowania się podwójnej warstwy elektrycznej na stale
odnawiającej się powierzchni kropli rtęci. Jest niezależny od występowania
prądu dyfuzyjnego,
•
migracyjny – eliminuje się go poprzez użycie w nadmiarze elektrolitu
podstawowego, który przejmuje na siebie całe przewodnictwo,
•
inne prądy:
20
o
kinetyczny
o
katalityczny
o
adsorpcyjny
Rola elektrolitu podstawowego
Elektrolit podstawowy przewodzi prąd. Ponad to spełnia rolę czynnika
zmniejszającego niejednorodność pola elektrycznego w okolicy KER, która
wywołuje
prądy
powodujące
zakłócenia
w odczycie fali polarograficznej.
Stężenie elektrolitu podstawowego powinno być 100 – 1000 – krotnie większe od
stężenia substancji oznaczanej. Elektrolit musi być substancją nie depolaryzującą
KER w jak największym zakresie (nie przeszkadzać w odczycie fali
polarograficznej). Powinien to być mocny elektrolit nie wchodzący
w reakcje z rtęcią ani oznaczaną substancją.
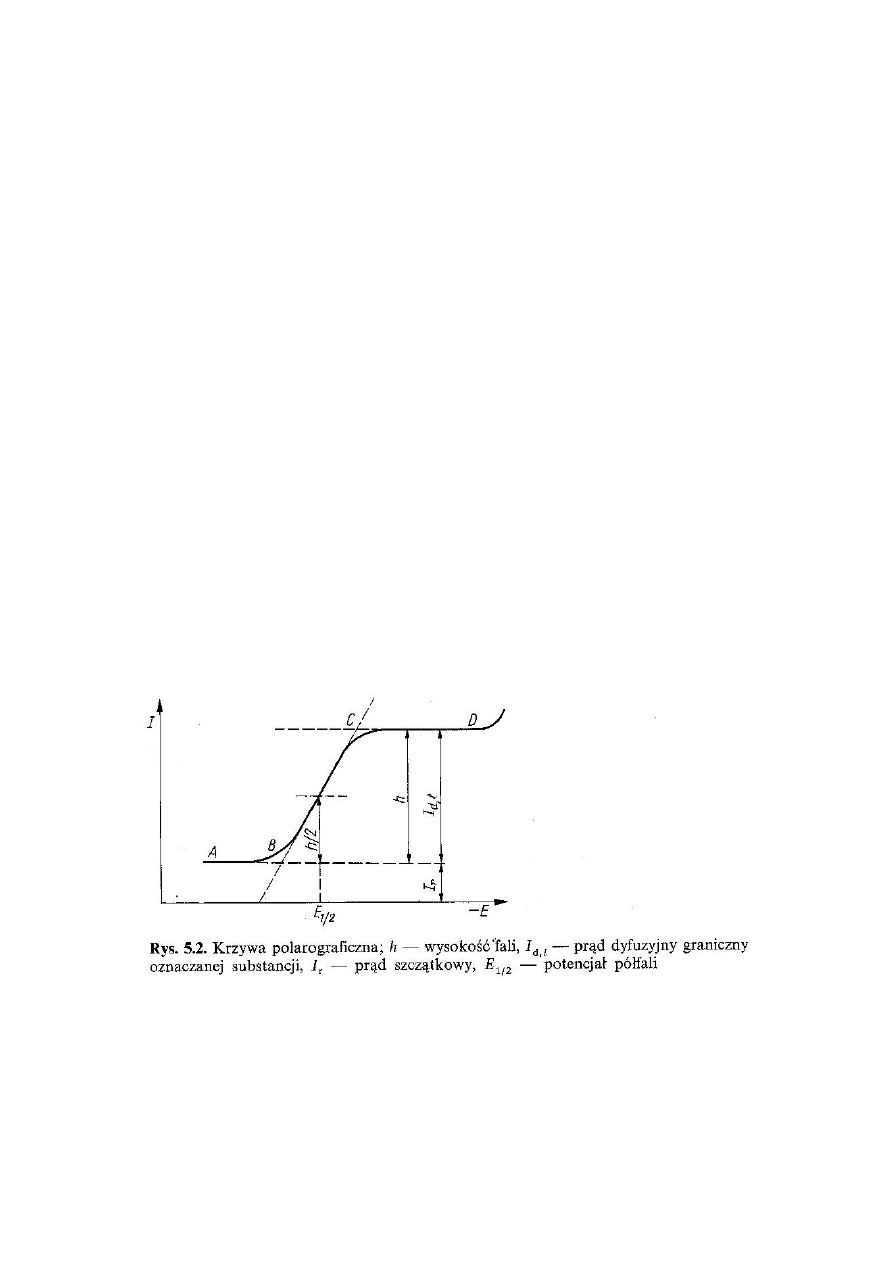
Krzywa polarograficzna
Krzywa składa się z trzech części AB, BC i CD.
Odcinek AB – jest prawie równoległy do osi OX i mimo wzrostu napięcia,
natężenie prądu jest bardzo małe. Prąd ten jest nazywany szczątkowym;
spowodowany jest głównie obecnością elektroaktywnych zanieczyszczeń i
prądem pojemnościowym. Ta część wykresu odpowiada polaryzacji KER.
21

Punkt B – odpowiada potencjałowi wydzielania. Od tego punktu następuje
znaczny wzrost natężenia prądu i krzywa ulega zagięciu. Trwa normalna
elektroliza, podczas której na KER redukują się jony. Pomiędzy różnica stężeń
jonów między cienką warstwą roztworu przy elektrodzie a pozostałą częścią
roztworu. W wyniku tej różnicy następuje dyfuzja i płynie prąd dyfuzyjny. Którego
obrazem jest stroma część krzywej polarograficznej (odcinek BC).
Punkt C – jest granicznym prądem dyfuzyjnym, prąd dyfuzyjny osiąga wartość
maksymalną, na którą nie ma wpływu dalsze zwiększanie napięcia (odcinek CD).
Interpretacja krzywej polarograficznej. Na podstawie jakich danych z tej
krzywej możemy wyciągnąć wnioski jakościowe i ilościowe?
Fala polarograficzna – odcinek BC – odpowiada określonej reakcji elektrodowej.
Jeżeli potencjał jest większy niż w punkcie D, to zaczyna zachodzić reakcja
elektrodowa innego składnika niż oznaczamy.
Wysokość fali h jest równa natężeniu granicznego prądu dyfuzyjnego substancji
oznaczanej i jest proporcjonalna do stężenia tej substancji.
22

NMR:
Definicja przesunięcia chemicznego. Jakie czynniki wpływają na jego
wartość?
Przesunięcie chemiczne, δ – wielkość związana z ekranowaniem jąder atomów w
cząsteczkach przez otaczające je elektrony. W praktyce przez przesunięcie
chemiczne rozumie się przesunięcie chemiczne względne, podane w stosunku do
odpowiedniego wzorca.
Przesunięcie chemiczne względne – różnica w położeniu sygnału absorpcji
określonego
protonu
w odniesieniu do położenia sygnału protonu wzorca.
Przesunięcie chemiczne podaje się w jednostkach ppm (część na milion) i wyraża
się wzorem:
Sygnał wzorca umieszcza się w punkcie zero.
Przesunięcie chemiczne zależy od czynników:
•
zewnętrznych:
o temperatura,
o rodzaj rozpuszczalnika,
o stężenia badanego roztworu.
•
wewnętrznych:
o rozkładu gęstości elektronowej w otoczeniu protonu
o wielkości wtórnego pola magnetycznego powstającego w wyniku ruchu
elektronów wokół innych jąder w cząsteczce.
Opisać metodę określania rzędowości atomów techniką
13
C NMR.
Technika DEPT – rejestrowanie jedynie atomów węgla, które sąsiadują z
protonami. Metoda DEPT pozwala na określanie rzędowości atomów węgla.
Na początku otrzymujemy widmo
13
C{
1
H}:
23
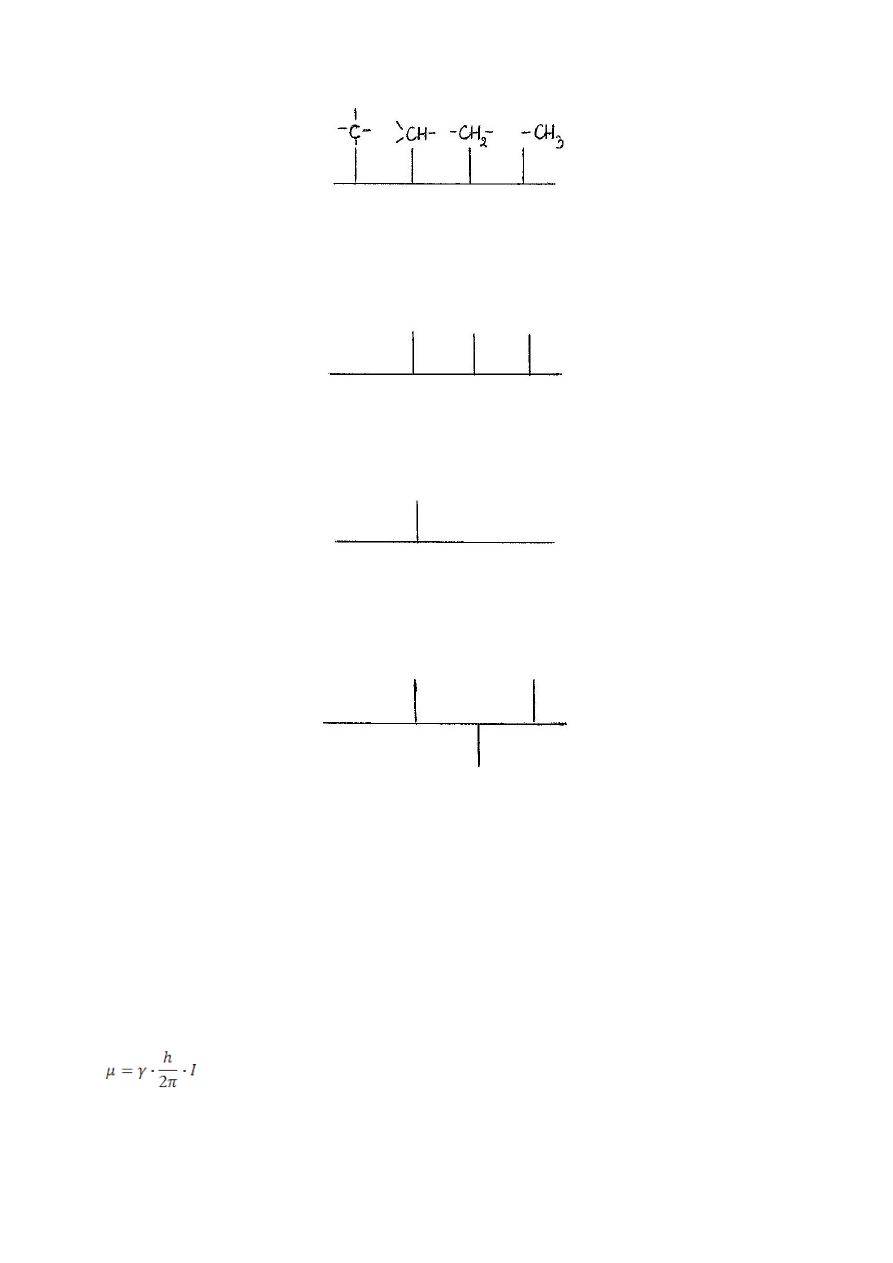
Przedstawia ono atomy węgla pierwszo-, drugo-, trzecio- i czwartorzędowe.
Otrzymując taki widmo nie wiadomo który atom ma jaką rzędowość.
W tym celu przeprowadza się badanie techniką DEPT. Po naświetlaniu DEPT 45
(45 – kąt o jaki wychylamy atomy) otrzymujemy następujące widmo:
Widmo nie zawiera atomów czwartorzędowych. Zatem wiemy już który sygnał
pierwszego widma należał do atomu czwartorzędowego.
Następnie używamy naświetlania DEPT 90:
Otrzymane widmo przedstawia tylko jeden sygnał należący do trzeciorzędowego
atomu węgla.
Naświetlanie DEPT 135:
Na otrzymanym widmie można zaobserwować sygnały pochodzące od atomów
węgla pierwszo-, drugo-, i trzeciorzędowych. Drugorzędowe są skierowane do
dołu.
Jakie czynniki charakteryzujące jądro atomowe są istotne dla zjawiska
magnetycznego rezonansu jądrowego?
W metodzie NMR wykorzystuje się jądra o właściwościach magnetycznych, czyli
takich, których liczba kwantowa spinowa I jest różna od zera, np.
1
H,
13
C,
31
P.
I = n *1/2 ,gdzie n = 0, 1, 2,3, …..
Moment magnetyczny :
24
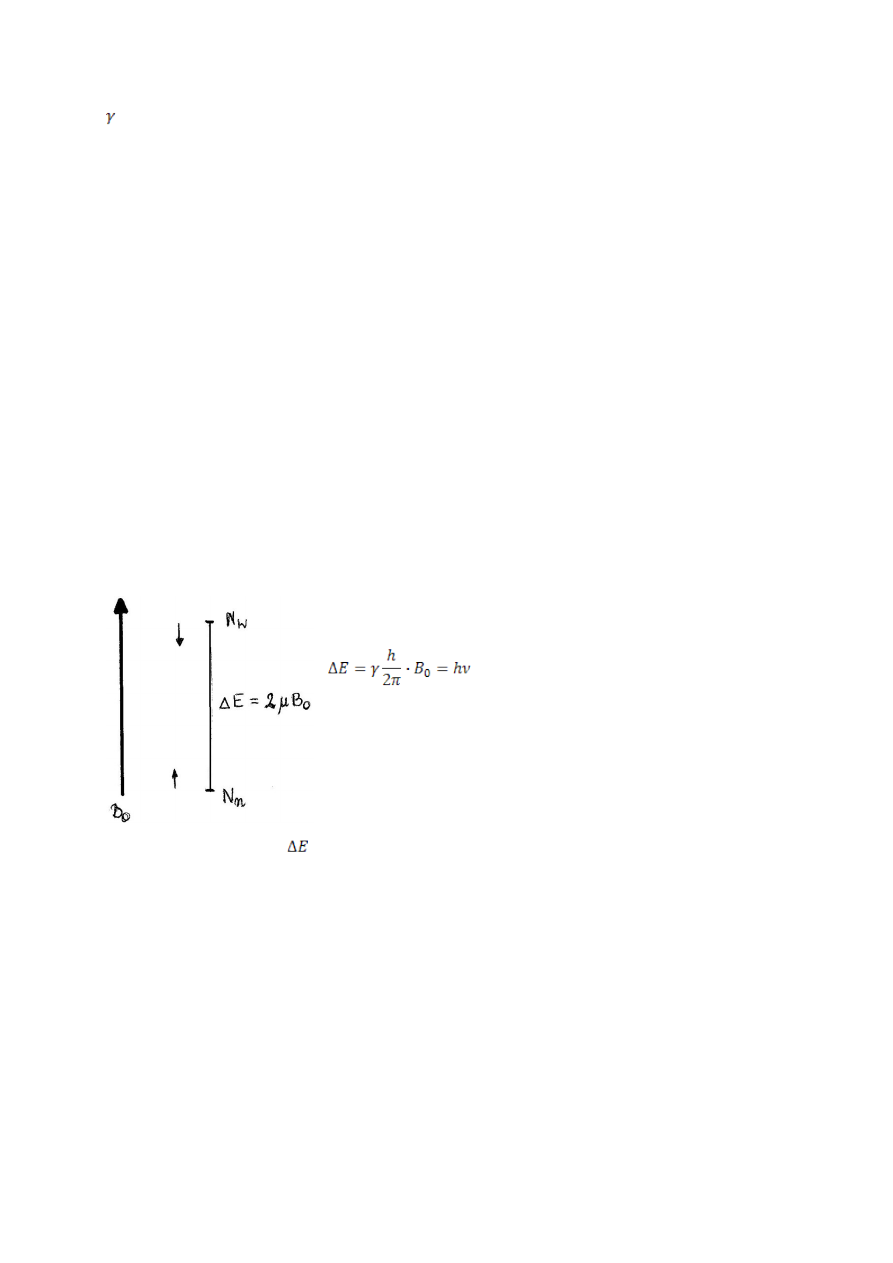
- współczynnik magnetogiryczny, charakterystyczny dla każdego jądra
atomowego, jest miarą oddziaływania danego jądra atomowego w polu
magnetycznym.
Opisać metody rejestracji widm metodą NMR.
Widmo NMR można otrzymać poprzez umieszczenie próbki substancji o
właściwościach magnetycznych (I różne od zera) w silnym polu magnetycznym i
działaniu na nią promieniowaniem o częstości radiowej.
Do otrzymania widma stosuje się spektrometry składające się z:
•
elektromagnesów nadprzewodnościowych,
•
nadajnika promieniowania o częstotliwości radiowej,
•
sondy z próbką,
•
detektora – odbiornika promieniowania o częstości radiowej,
•
rejestratora, komputera.
Do wytwarzania jednorodnego pola magnetycznego stosuje się chłodzone helem
elektromagnesy nadprzewodnościowe.
Im większe jest natężenie pola magnetycznego tym większa jest różnica
poziomów energetycznych ustawienia równoległego i antyrównoległego spinów
jądrowych w zewnętrznym polu magnetycznym:
Zwiększenie wartości
powoduje zwiększenie liczby jąder na niższym poziomie
energetycznym, dzięki czemu zwiększa się czułość oraz otrzymuje się lepszą
jakość widm.
Próbkę znajdującą się w cienkościennej rurce umieszcza się w sondzie, w
szczelinie magnesu. Sonda składa się z uchwytu próbki, rotora umożliwiającego
ruch wirowy i cewki nadawczo – odbiorczej.
25
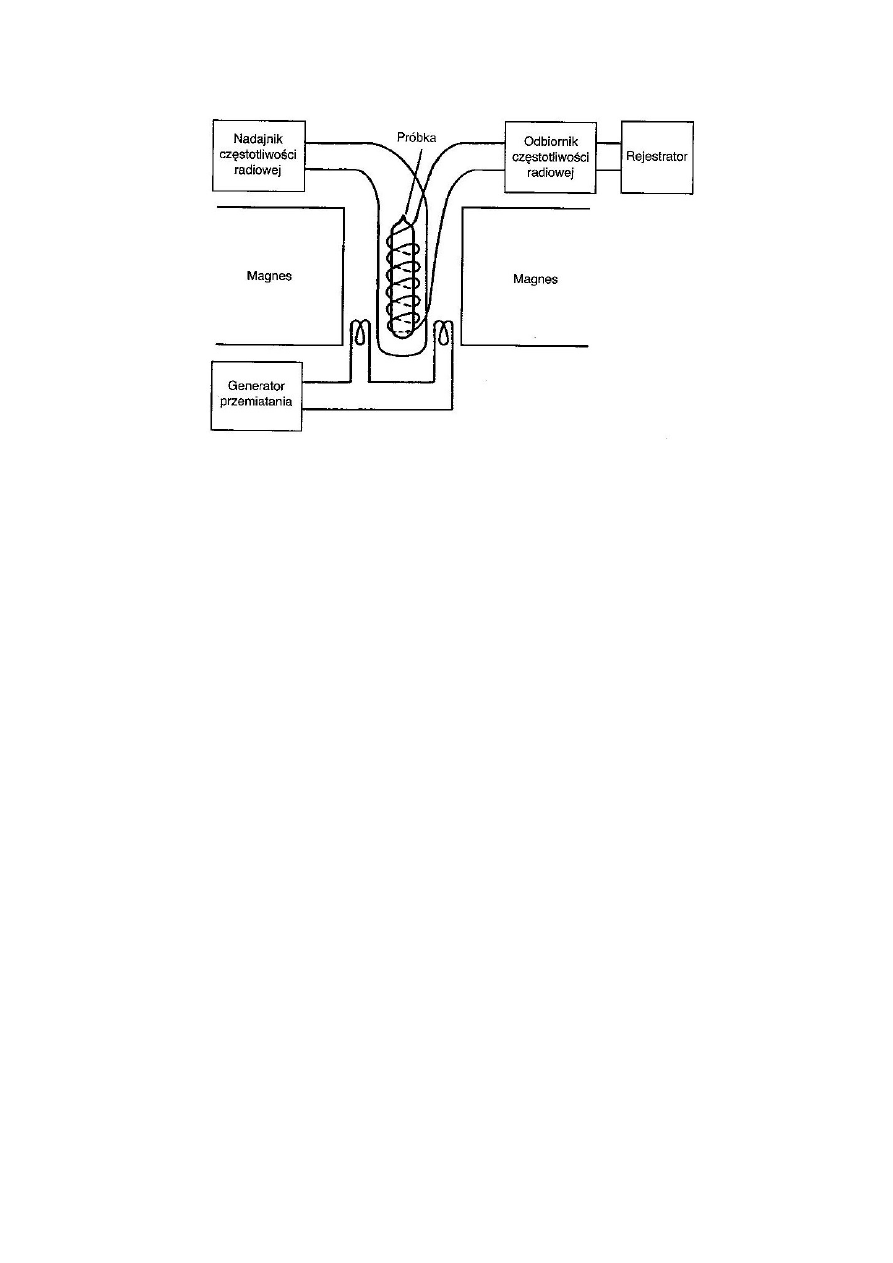
Schemat spektrometru NMR
Badaną próbkę rozpuszcza się w rozpuszczalniku, który nie daje widma w
zakresie, w którym otrzymuje się widma
1
H NMR. Rozpuszczalniki muszą być
deuterowane lub nie zawierać atomów wodoru.
Częstość rezonansową próbki mierzy się względem sygnału rezonansowego
wzorca, którym najczęściej jest TMS (tetrametylosilan). Sygnał wzorca jest ostrą
pojedynczą linią umieszczaną w punkcie zerowym na skali przesunięć
chemicznych.
Pojęcie stałej sprzężenia. Zależność wicynalnej stałej sprzężenia od
geometrii cząsteczki.
Stała sprzężenia, J – odległość pomiędzy sąsiednimi składowymi rozszczepionego
sygnału. Wielkość mierzona w Hz, zwykle nie przekracza 20Hz. Wartość
niezależna od natężenia pola magnetycznego.
O wielkości sprzężenia decydują:
•
współczynniki magnetogiryczne,
•
odległość protonów mierzona liczbą wiązań chemicznych (
n
J),
•
rodzaju sprzęgających się jąder,
•
hybrydyzacji sprzęgających się jąder,
•
geometrii cząsteczki (zależy od kąta dwuściennego dla rozprzęgania przez
trzy wiązania).
Sprzężenia obserwuje się dla jąder o spinach różnych od zera.
Stałą sprzężenia wylicza się ze wzoru :
26
J [Hz] = (σ
1
– σ
2
)*γ
podst.
γ
podst.
– częstość spektrometru.
Zasadnicze elementy spektrometru NMR. Zasada działania spektrometru
NMR.
1. rotor
2. płaszcz próżniowy
3. próbka
4. ciekły azot
5. ciekły hel
6. magnes nadprzewodzący
A
BSORPCYJNA
SPEKTROMETRIA
ATOMOWA
:
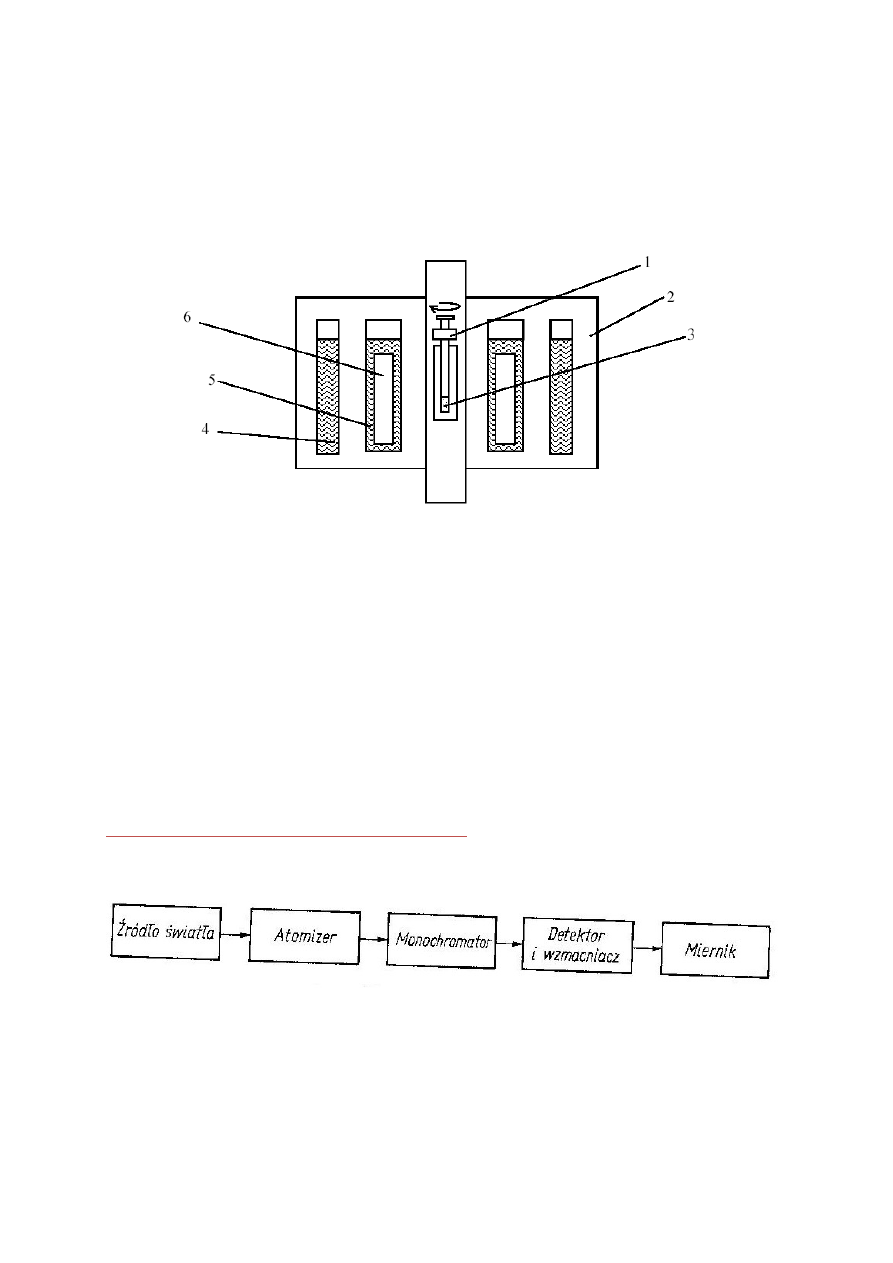
Schemat blokowy aparatu do absorpcyjnej spektrometrii atomowej.
Promieniowanie ze źródła emitującego widmo liniowe, charakterystyczne dla
oznaczanego pierwiastka, przechodzi przez atomizator i pada na szczelinę
monochromatora, który oddziela linię rezonansową od pozostałych, a następnie w
detektorze zostaje przetworzone na sygnał elektryczny, mierzony na mierniku.
27
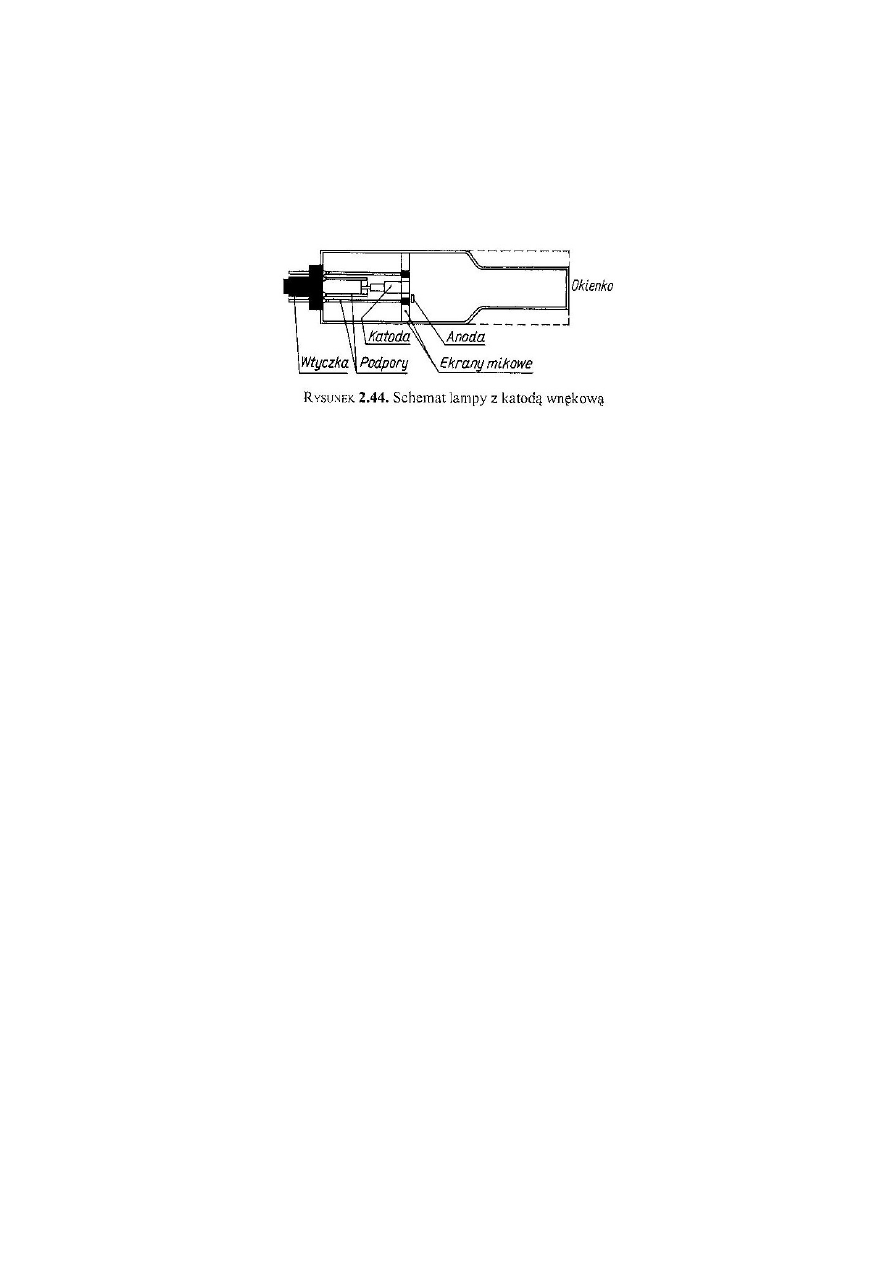
Źródła promieniowania w metodzie AAS
Aby uzyskać dużą czułość i precyzję pomiaru, źródło promieniowania używane w
spektrometrach absorpcji atomowej powinno emitować promieniowanie stabilne
o
możliwie
dużym
natężeniu
i z wąskim konturem linii emitowanych.
Lampa składa się ze szklanego lub kwarcowego cylindra, wypełnionego neonem
lub argonem pod ciśnieniem kilku hPa, w który są wtopione anoda i katoda.
Anodę stanowi zwykle wolframowy pręcik, a katodę wydrążony walec glinowy,
wyłożony wewnątrz warstwą metalu, którego promieniowanie chcemy otrzymać.
Po doprowadzeniu do elektrod odpowiedniego napięcia zachodzą wyładowania
i następuje jonizacja gazu. Jony rozładowując się na katodzie wybijają atomy
metalu z katody. Wybite atomy zderzają się z jonami gazu i ulegają wzbudzeniu,
w wyniku czego emitują charakterystyczne promieniowanie. Cylindryczny kształt
katody powoduje, że emitowane promieniowanie jest dobrze zogniskowane.
Ekrany mikowe zapobiegają rozprzestrzenianiu się wyładowań na zewnątrz
katody.
Zaletą lampy z katodą wnękową jest duża intensywność. Mogą być w niej
wzbudzane wszystkie pierwiastki w dowolnym stanie skupienia.
Atomizery w metodzie AAS
Atomizery w metodzie ASS można podzielić na:
•
płomieniowe – oznaczany pierwiastek występuje zwykle w postaci związku
chemicznego, z którego należy wydzielić go w postaci wolnych atomów.
Najczęściej używanym atomizerem jest płomień. Związek z oznaczanym
pierwiastkiem jest wprowadzany do płomienia w postaci małych kropel, co
pozwala na otrzymanie dużego stężenia atomów. O najczęściej
stosowanych płomieni należą: płomień powietrze – acetylen, podtlenek
azotu – acetylen (dla pierwiastków tworzących trudno dysocjujące tlenki) i
powietrze – metan (dla metali łatwo ulegających jonizacji). Jednak
używanie płomienia jako atomizera powoduje otrzymywanie niskich
wydajności atomizacji, powoduje absorpcję promieniowania przez gazy
płomienia, niejednorodność oraz interferencje fizyczne i chemiczne.
28

•
bezpłomieniowe – do atomizacji elektrotermicznej wykorzystuje się
atomizery typu kuwety grafitowej. Urządzenie to jest ogrzewane
elektrycznie w 4 etapach. Etap 1 polega na odparowaniu próbki. Etap 2 –
mineralizacja próbki. Etap 3 – ponowne odparowanie i atomizacja próbki.
Etap 4 – czyszczenie kuwety z próbki przed następnym cyklem
pomiarowym. Do atomizacji bezpłomieniowej stosuje się również piece
kwarcowe i łódki tantalowe o zbliżonym działaniu.
S
PEKTROMETRIA
MAS
:
Jak działa analizator magnetyczny?
Analizator ten wykorzystuje zjawisko zmiany toru lotu jonów w
. Tor lotu jonów jest zakrzywiany, stopień zakrzywienia lotu zależy
od stosunku masy do ładunku (m/z) i prędkości jonu a także od parametrów pola
magnetycznego. Sektor magnetyczny charakteryzuje się stosunkowo małą
rozdzielczością - mniej niż 5000 thomsonów. Związane jest to głównie z dużymi
różnicami prędkości cząsteczek wpadających do urządzenia. Problem ten
rozwiązuje przez zastosowanie sektora elektrycznego przed sektorem
magnetycznym, w którym cząsteczki są rozpędzane, dzięki czemu różnice
prędkości są mniejsze.
Jak działa analizator elektryczny?
Urządzenie to wykorzystuje zjawisko zmiany toru lotu jonów w
, jest zbudowane z dwóch równoległych, zakrzywionych płyt do
. Jony o jednakowej
mają jednakowe tory lotu w sektorze elektrycznym. Za sektorem elektrycznym
znajduje się szczelina przez którą przelatują tylko jony o określonej energii.
Sektor elektryczny jest stosowany przed sektorami magnetycznymi w
spektrometrach
mas
o podwójnym ogniskowaniu.
Metody jonizacji w spektrometrii mas.
•
jonizacja elektronowa (EI),
•
Jonizacja chemiczna (CI),
29

•
bombardowanie szybkimi atomami (FAB),
•
bombardowanie szybkimi jonami (FIB),
•
jonizacja polem i desorpcja polem (FI i FD),
•
termosprej (TS lub TSI),
•
elektrosprej, elektrorozpylanie (ES lub ESI),
•
jonizacja chemiczna pod ciśnieniem atmosferycznym (APCI),
•
desorpcja promieniowaniem laserowym z użyciem matrycy (MALDI),
•
jonizacja plazmą wzbudzoną indukcyjnie (ICP).
Słowniczek:
Jonizacja elektronami (Electron Ionisation - EI) – jonizacja przy pomocy wiązki
elektronów. Jonizacja odbywa się w próżni. Metoda ta powoduje zwykle
fragmentację badanych cząsteczek. EI charakteryzuje się stosunkowo małą
wydajnością - poniżej 1% cząsteczek ulega jonizacji.
Elektrorozpylanie (Electrospray, ESI) – polega na rozpylaniu cieczy zawierającej
badaną
substancję
z igły, do której przyłożono wysokie napięcie (zwykle 1 - 5 k
) pod ciśnieniem
atmosferycznym. Jest to jedna z łagodnych metod jonizacji - zwykle nie powoduje
fragmentacji badanych cząsteczek. Metoda ta jest bardzo często stosowana w
badaniach nad wielkocząsteczkowymi
Termorozpylanie (Termospray, TE) – jonizacja przez podgrzanie przy pomocy
prądu elektrycznego roztworu zawierającego sól i analizowaną substancję
wewnątrz stalowej
. Gorąca substancja jest rozpylana w komorze
próżniowej z prędkością naddźwiękową.
Jonizacja chemiczna (Chemical Ionisation, CI) – jony wytwarzane są na skutek
zderzeń cząsteczek badanego związku chemicznego z jonami pierwotnymi
obecnymi w źródle jonów. Jest to metoda nie powodująca fragmentacji
cząsteczek (łagodna jonizacja). Jonizacja odbywa się zwykle przy ciśnieniu rzędu
60
Bombardowanie szybkimi atomami (Fast-Atom Bombardment FAB) – polega
na bombardowaniu cząsteczki obojętnymi atomami o wysokiej energii (zwykle 17
lub 70
). Cząsteczki mogą znajdować się w fazie gazowej lub być rozpuszczone
w ciekłej, mało lotnej substancji (matrycy) np.
Bombardowanie jonami (spektrometria mas jonów wtórnych - Secondary Ion
Mass Spectrometry - SIMS) – metoda ta początkowo była stosowana do substancji
przewodzących prąd lub substancji naniesionych na metalowe płytki. Obecnie
metodę SIMS stosuje się z powodzeniem do substancji nie przewodzących prądu.
Istnieje odmiana techniki SIMS, w której badana substancja jest rozpuszczona
w ciekłej matrycy (najczęściej glicerolu). Technika ta jest nazywana czasami
LSIMS (Liquid Secondary Ion Mass Spectrometry) lub FIB (Fast Ion Bombardment).
Desorpcja laserowa (Laser Desorption - LD) – jonizacja następuje przez
naświetlanie próbki silnym
, a zatem bombardującymi cząstkami są
30
Desorpcja laserowa z udziałem matrycy (Matrix Assisted Laser Desorption
Ionisation - MALDI) – stosuje się jonizację laserową, ale z tak dobraną energią
wiązki, aby nie doprowadzać do fragmentacji cząsteczek (łagodna metoda
jonizacji), lecz tylko do ich "wybijania" ze specjalnie przygotowanej matrycy.
Matryca absorbuje energię lasera, która jest później przekazywana do
analizowanych cząsteczek. Metoda ta jest bardzo często stosowana w badaniach
nad
syntetycznymi.
Plazma wzbudzona indukcyjnie (ICP) – jonizowana substancja jest
wprowadzana do
płomienia palnika znajdującego się w
Rura otoczona jest cewką, przez którą przepływa
częstotliwości. Plazma ogrzewa się do temperatury rzędu 10 000
wytworzonym przez prąd płynący w cewce.
Metoda nadaje się doskonale do analizy pierwiastków
Jakie istotne dane o substancji można uzyskać na podstawie analizy
metodą spektrometrii mas?
•
jaka jest masa cząsteczkowa badanego związku,
•
jaki jest skład pierwiastkowy, elementarny (wzór sumaryczny),
•
jaka jest struktura badanego związku (wzór strukturalny),
•
jaki jest skład izotopowy analizowanej substancji,
•
jaki jest precyzyjny skład mieszaniny związków,
•
czy związek jest czysty, czy zawiera domieszki lub zanieczyszczenia.
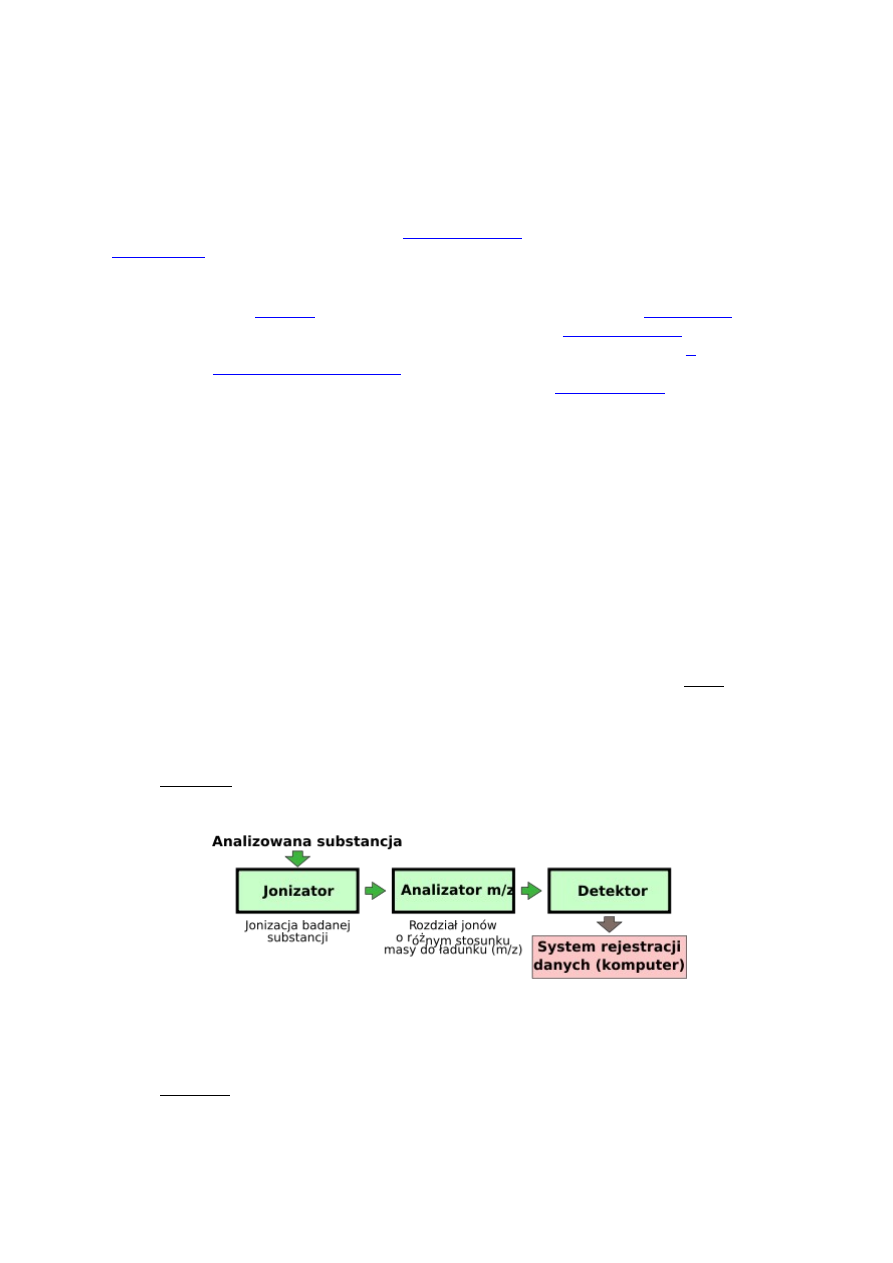
Z jakich podzespołów (bloków) składa się spektrometr mas? Dwa z nich
mają zasadnicze znaczenie – które? Odpowiedź uzasadnić.
•
jonizator
– urządzenie, w którym następuje jonizacja cząsteczek przy
użyciu różnorodnych technik, z których część prowadzi do pękania wiązań
chemicznych na skutek czego dochodzi do ich podziału na mniejsze
fragmenty. Inne techniki powodują tylko naładowanie cząsteczek bez ich
fragmentowania,
•
analizator – w którym wcześniej powstałe jony ulegają rozdziałowi na
podstawie stosunku ich masy do ładunku.
•
detektor
– urządzenie "zliczające" jony napływające z analizatora.
31
Na czym w uproszczeniu, polega metoda spektrometrii mas? Jak może
wyglądać
widmo
masowe
i jakie wielkości mogą w nim występować?
Działanie tradycyjnego spektrometru mas opiera się na odchylaniu strumienia
jonów badanej substancji w polu elektrycznym. Wszystkie cząsteczki analizowane
w spektrometrze mas muszą mieć ładunek elektryczny. Wewnątrz spektrometru
mas panuje próżnia, dzięki czemu ruch jonów nie jest zakłócany przez zderzenia z
cząsteczkami gazów.
Pierwszym przedziałem spektrometru mas jest źródło jonów. Urządzenie to
przeprowadza substancje analizowane w spektrometrze w jony unoszące się w
fazie gazowej. Zjonizowane cząsteczki przechodzą do dalszych przedziałów
spektrometru mas, gdzie formowana jest wiązka jonów. Wiązka ta jest kierowana
do analizatora masy.
Analizator masy rozdziela jony ze względu na stosunek ich masy do ładunku. Jony
kierowane są do detektora, który zamienia w sposób ilościowy sygnał w postaci
prądu jonowego na sygnał elektryczny, który jest rejestrowany przez komputer w
postaci widma stosunku masy do ładunku elektrycznego (nazywanego często
widmem masowym). W widmie takim na osi poziomej odłożone są stosunki mas
do ładunków w thompsonach (1 Th = 1 Dalton / liczbę ładunków elementarnych
jonu), na osi pionowej intensywności (liczba jonów zarejestrowanych przez
spektrometr).
32
Document Outline
Wyszukiwarka
Podobne podstrony:
opracowanie pytań na kolokwium, SOCJOLOGIA UJ, Współczesne teorie socjologiczne
opracowanie pytań na kolowkium bez deskolaryzacji nr 18, opracowanie pytań na kolokwium
ped. ogólna sciaga, opracowanie pytań na kolokwium
MI-PTAKI-OPRACOWANIE PYTAN NA KOLOKWIUM II, Dokumenty(1)
opracowanie pytań na kolowkium- BEZ 2 REFERATÓW, opracowanie pytań na kolokwium
opracowanie pytań na kolowkium, opracowanie pytań na kolokwium
opracowanie pytan na kolokwium trl 1
opracowanie pytań na kolokwium
Opracowania pytań na analizę instrumentalną
Opracowania pytań na analizę instrumentalną
2Opracowanie pytań na kolokwium z analizy instrumentalnej
wykłady, TUW opracowane pytan na egz, 1
notatki z wykładów i opracowane pytania na kolokwium, Ogrodnictwo UP Lbn, mikrobiologia
Analiza matematyczna 2 - opracowane zagadnienia na egzamin, Wykłady - Studia matematyczno-informatyc
opracowania pytań na andrago na podstawie wykładów(1), Pedagogika, Andragogika
więcej podobnych podstron