
Wykonały:
Oliwia Jankowska
Klaudia Wrońska
CHOROBY
PRZEWLEKŁE
FENYLOKETONURIA
AKROMEGALIA
PRZYCZYNY CHOROBY
Fenyloketonuria (PKU)
jest najczęstszą
wrodzoną wadą w metabolizmie
aminokwasów. Choroba polega na całkowitym
braku lub znacznym ograniczeniu aktywności
enzymu hydroksylazy fenyloalaninowej (PAH).
Enzym ten jest odpowiedzialny za przemianę
aminokwasu fenyloalaniny w tyrozynę.
Reakcja zachodzi w komórkach wątroby, czyli
w hepatocytach. Źródłem fenyloalaniny są
białka pokarmowe spożywane codziennie wraz
z dietą, w których stanowi ona od 3-7 % ich
masy.
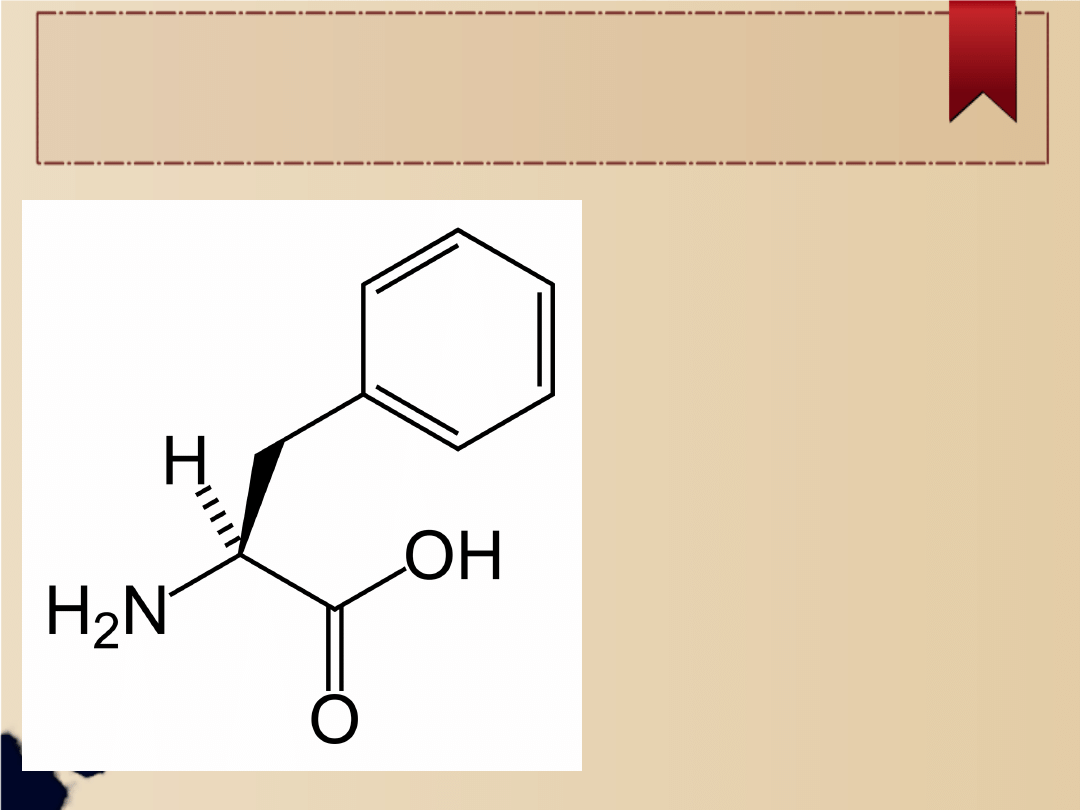
Zaburzenia w
przemianie
fenyloalaniny
powodują wzrost jej
stężenia we krwi,
wraz z którą dostaje
się ona do
ośrodkowego układu
nerwowego (OUN),
gdzie wywołuje
nieodwracalne
uszkodzenia.
Fenyloalanina

DZIEDZICZENIE
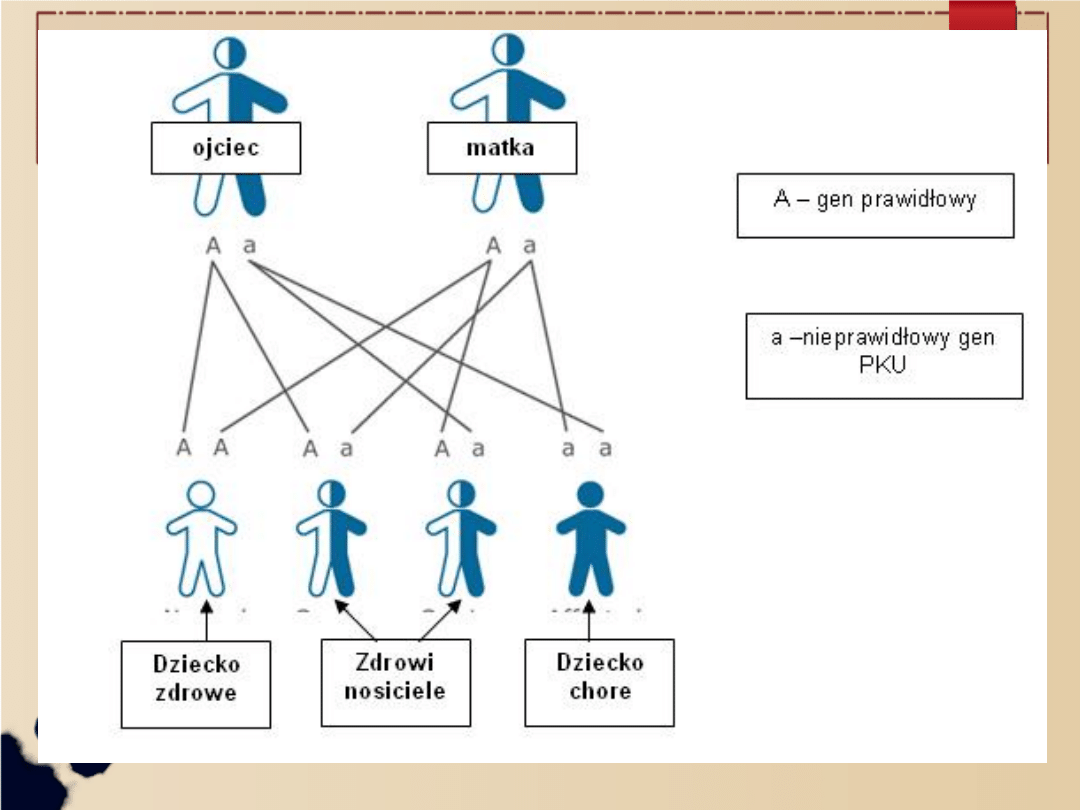
Jest to choroba o podłożu genetycznym,
przekazywana dziecku przez oboje z rodziców.
Dziedziczy się w sposób autosomalny
recesywny co oznacza, że chore dziecko musi
otrzymać po 1 nieprawidłowym genie
wywołującym PKU od każdego z rodziców.
Do pełnego wystąpienia choroby potrzebne są
dwa „chore” geny. Rodzice są najczęściej tylko
bezobjawowymi nosicielami nieprawidłowego
genu i zwykle nie zdają sobie sprawy z tego,
że mogą go przekazać swojemu potomstwu.

OBJAWY
Opóźnienie rozwoju psychoruchowego -
stwierdzane w okresie niemowlęcym często
stanowi pierwszy objaw sugerujący chorobę.
Objawami nieleczonej choroby są:
•
pogłębiające się zaburzenia neurologiczne z
napadami padaczkowymi
•
znacznego stopnia upośledzenie rozwoju
umysłowego i motorycznego
U większości pacjentów upośledzenie umysłowe
odpowiada wartościom charakterystycznym dla
opóźnienia w stopniu głębokim (iloraz
inteligencji 20-40). Różnice parametrów
rozwoju psychicznego są spowodowane
najprawdopodobniej indywidualnie
zróżnicowanym deficytem enzymatycznym
hydroksylazy fenyloalaninowej.

•
zaburzenia chodu, postawy
•
hipotonia mięśniowa
•
zesztywnienie stawów
•
nadpobudliwość z niekontrolowanymi napadami złości
•
napady agresji wobec innych oraz wobec siebie - te ostatnie
mogą doprowadzić do dotkliwych samookaleczeń
•
zachowania destrukcyjne
•
zaburzenia snu
•
deficyty uwagi i zaburzenia koncentracji
•
zachowania autystyczne, m.in. lęk wobec osób obcych i
wobec zmiany otoczenia
•
psychozy
INNE
OBJAWY

CZĘSTOŚĆ
WYSTĘPOWANIA
Średnia częstość występowania
fenyloketonurii wynosi około 1 na
15000 urodzeń, ale pomiędzy różnymi
populacjami występują różnice;
przykładowo w Irlandii choroba
występuje w 1 przypadku na 4500
urodzeń, a w mniej niż jednym
przypadku na 100 000 urodzeń wśród
ludności Finlandii.

ROZPOZNAWANIE
Przy wczesnym rozpoznaniu
choroby (najlepiej zaraz po
urodzeniu) i odpowiednim
leczeniu można zapobiec
wystąpieniu objawów choroby.
Powszechne stosowanie u
noworodków w Polsce i wielu
innych krajach, w trzecim
dniu po urodzeniu prostego
screeningowego badania krwi
(test przesiewowy) umożliwia
wczesne rozpoznanie
fenyloketonurii. Screening w
kierunku fenyloketonurii był
pierwszą, zastosowaną na
szeroką skalę próbą
przeciwdziałania schorzeniu
uwarunkowanemu
genetycznie.
pobieranie krwi z piętki
noworodka w ramach
noworodkowych badań
przesiewowych

METODY LECZENIA
Leczenie PKU jest złożone. Wymaga regularnego
pobierania próbek krwi, notowania danych
dotyczących spożytej żywności, przestrzegania
bardzo restrykcyjnej diety oraz regularnych i
częstych wizyt w poradniach dla chorych na PKU.
Metaboliczną kontrolę choroby można uzyskać w
wyniku leczenia dietetycznego, które polega na
stosowaniu odpowiednio przygotowanych
preparatów białkozastępczych oraz modyfikowanych
produktów o małej zawartości białka, przy
dostarczeniu niezbędnych ilości Phe poprzez
dodanie niewielkich ilości naturalnego białka.

Badania genotypu
U wszystkich osób chorych na PKU należy
przeprowadzić analizę mutacji oraz oznaczyć
genotyp, co ułatwia ustalenie wstępnego
rozpoznania, poradnictwo genetyczne i
leczenie, dalszą kontrolę chorego oraz ustalenie
długoterminowego rokowania. Należy stworzyć
dodatkowe laboratoria, w których będzie można
przeprowadzać oznaczenia genotypu.
W przyszłości może się okazać, że optymalne
leczenie może wymagać identyfikacji mutacji.
Informacje dotyczące częstości występowania
mutacji mogą być również przydatne do
wyliczenia częstości występowania
poszczególnych alleli oraz zapadalności na PKU.

Jak radzić sobie z
fenyloketonurią w
szkole?
Wcześniejsza rozmowa z nauczycielami,
zapoznanie ich z chorobą dziecka.
Zapoznanie klasy z tą chorobą
Jedzenie przekąsek niskobiałkowych oraz
picie preparatu PKU
Choroba ta nie przeszkadza w wysiłku
fizycznym

Pomysły na posiłki do
szkoły:
kanapki z pieczywa niskobiałkowego np. z warzywami
ser niskobiałkowy z warzywami,
makaron lub ryż niskobiałkowy w pojemniku, który
pozwoli utrzymać ciepłą temperaturę
owoce,
niskobiałkowe przekąski, np. niskobiałkowe chipsy, budyń
lub pucharki z owocami.
Być może dziecko będzie wolało jeść obiad w szkole z kolegami.
Zapytaj, jaką żywność oferuje szkolna stołówka i czy będzie to
możliwe.

AKROMEGALIA
Jest rzadką chorobą spowodowaną zwiększonym
wydzielaniem hormonu wzrostu
(GH, somatotropina) przez
hormonalnie czynnego gruczolaka komórek
kwasochłonnych przedniego płata przysadki mózgowej.
Akromegalia występuje u osób dorosłych, u których
skończony został proces wzrastania kości, a nasady kości
długich uległy mineralizacji i zrośnięciu. U dzieci i
młodzieży nadmiar hormonu wzrostu powoduje gigantyzm,
odróżniający się od akromegalii nadmiernym wzrostem
kości długich.
Choroba rozwija się powoli i bez leczenia postępuje.
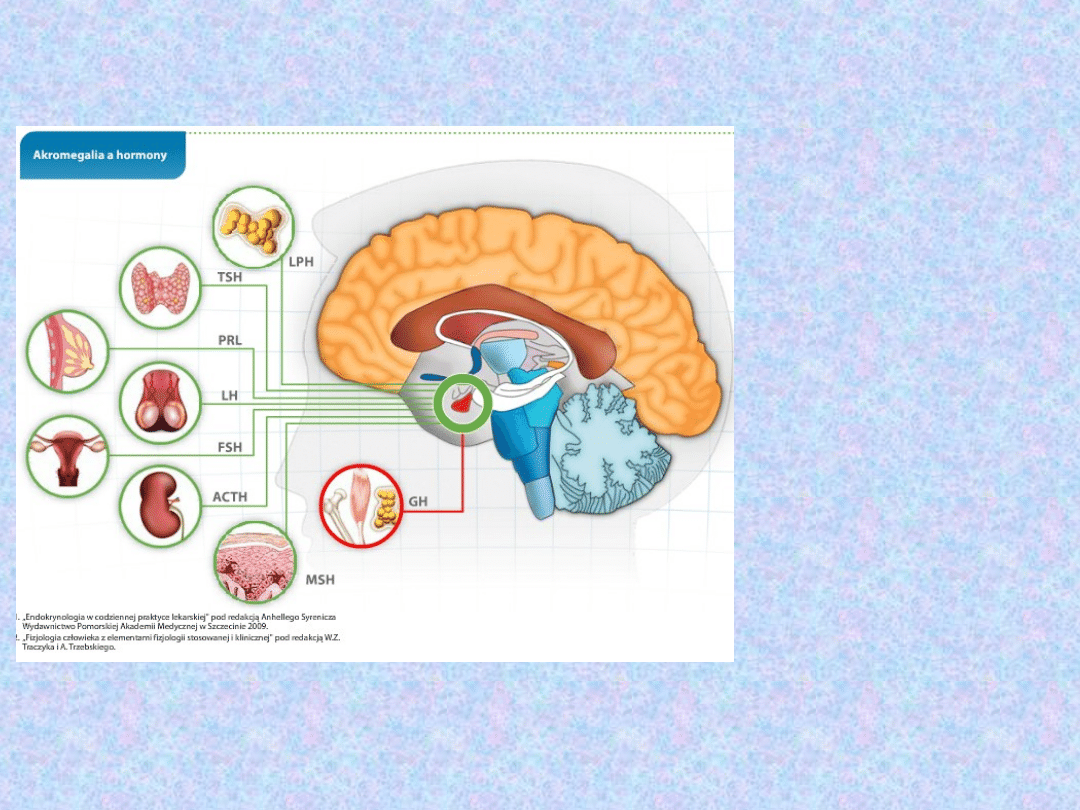
PRZYSADKA
LPH – lipotropina
(aktywuje procesy
rozkładu tłuszczu)
TSH – tyreotropina
(pobudza komórki
nabłonkowe tarczycy)
PRL – prolaktyna
(zapoczątkowuje i
podtrzymuje
wydzielanie mleka)
LH – lutropina
(wydzielanie
progesteronu i
prawidłowy przebieg
cyklu
menstruacyjnego)
FSH – folitropina
(wzrost i procesy
dojrzewania
pęcherzyka
jajnikowego)
ACTH –
kortykotropina
(nadnercza)
MSH – melanotropina
(wzrost syntezy
melaniny)

OBJAWY
AKROMEGALII
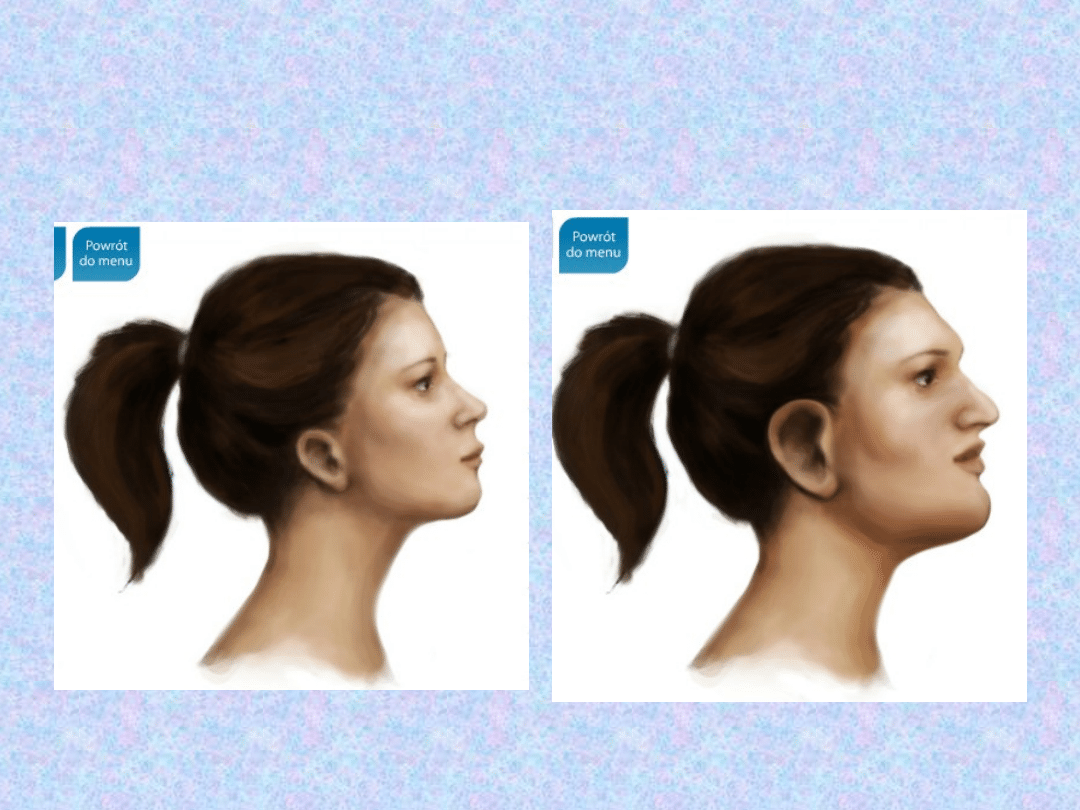
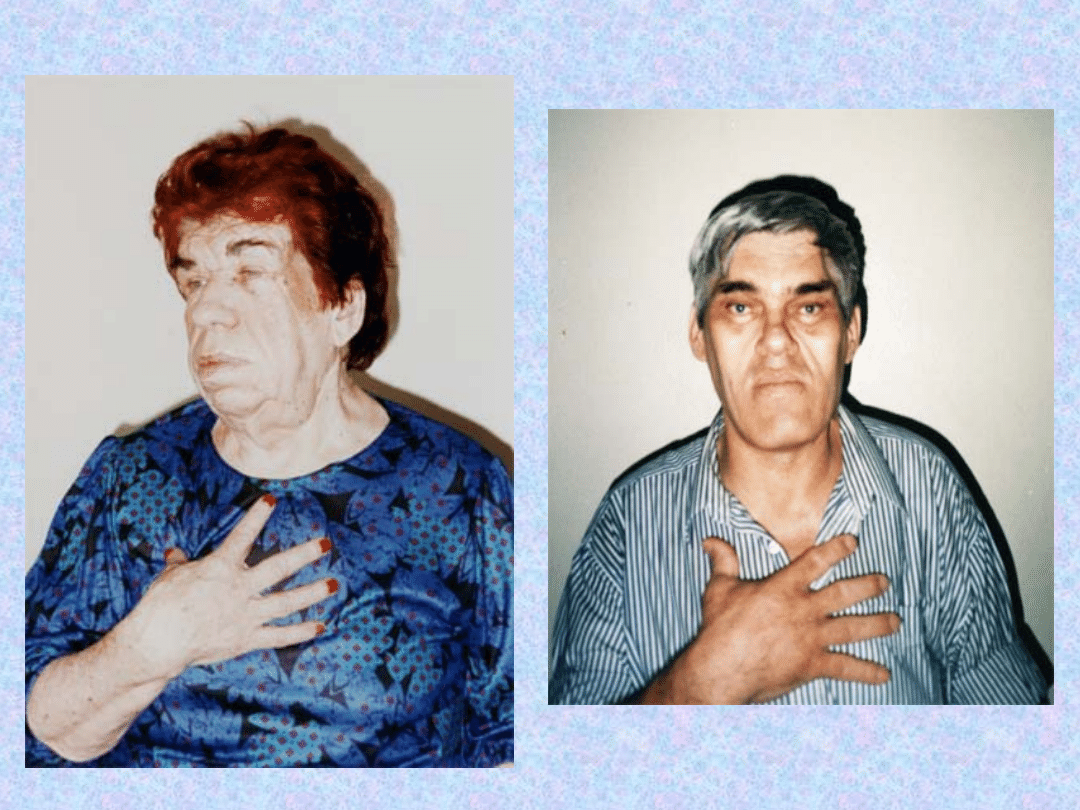
TWARZ
– bardzo
charakterystyczne są zmiany
rysów twarzy:
- powiększenie łuków
brwiowych,
- guzów czołowych,
- nosa,
- małżowin usznych,
- pogrubienie warg.
Zmiany w wyglądzie są
konsekwencja przerostu kośćca
i rozrostu chrząstek.

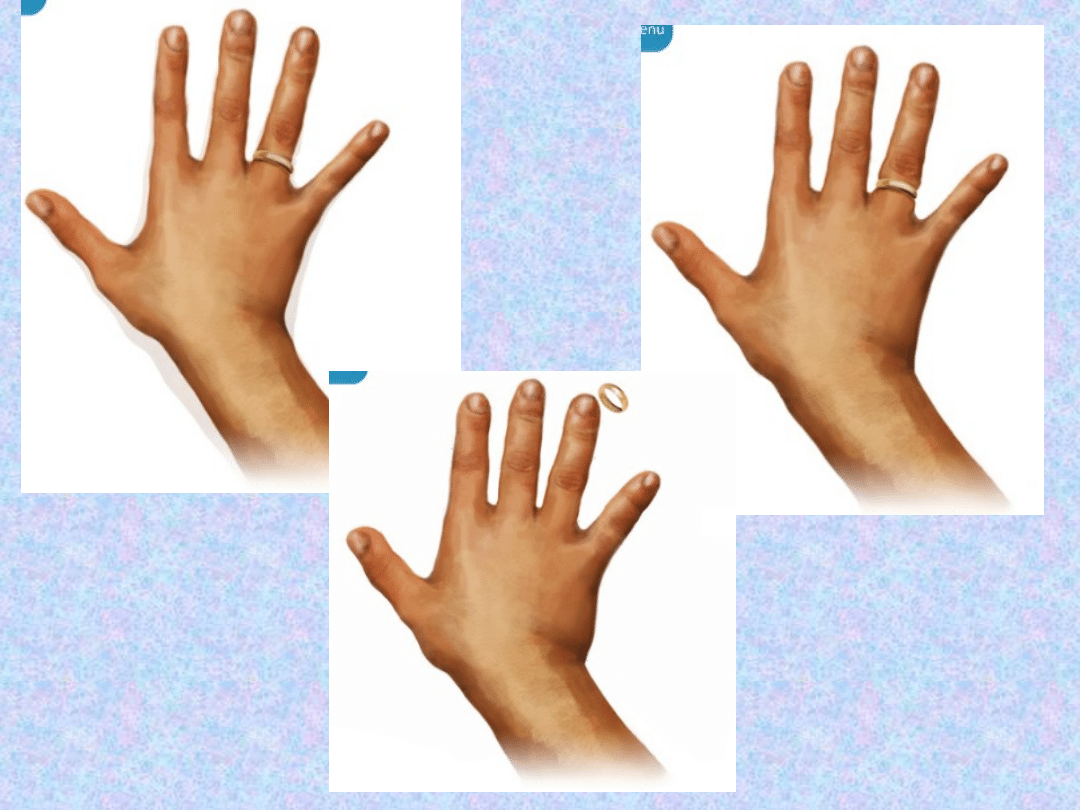
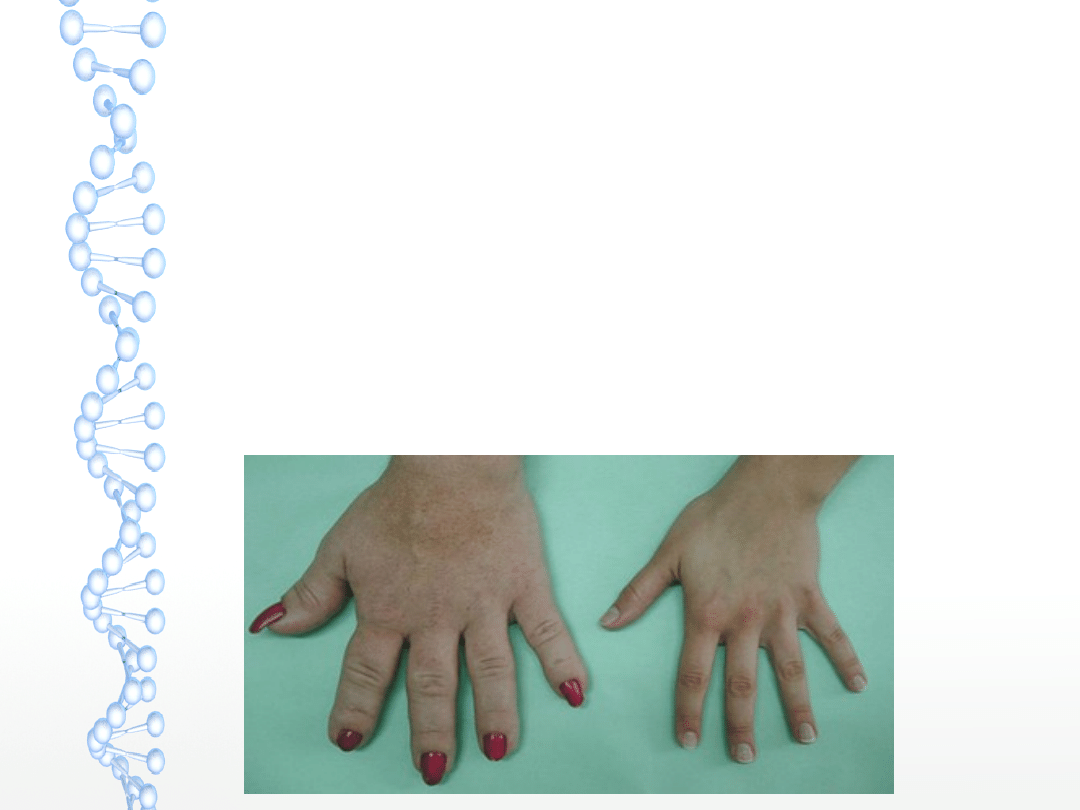
RĘCE
– do typowych objawów należą
powiększenie między innymi rąk (tzw.
„ręce i nogi niedźwiedzie”).
W konsekwencji trzeba zmieniać
rozmiary rękawiczek i pierścionków.
Wzrost jest głównie warunkowany
obrzękiem tkanek miękkich –
zwiększenie szerokości palców i dłoni.
Występuje również zespół cieśni
nadgarstka.

STOPY
– jak w przypadku rąk (w
konsekwencji zmiana obuwia.
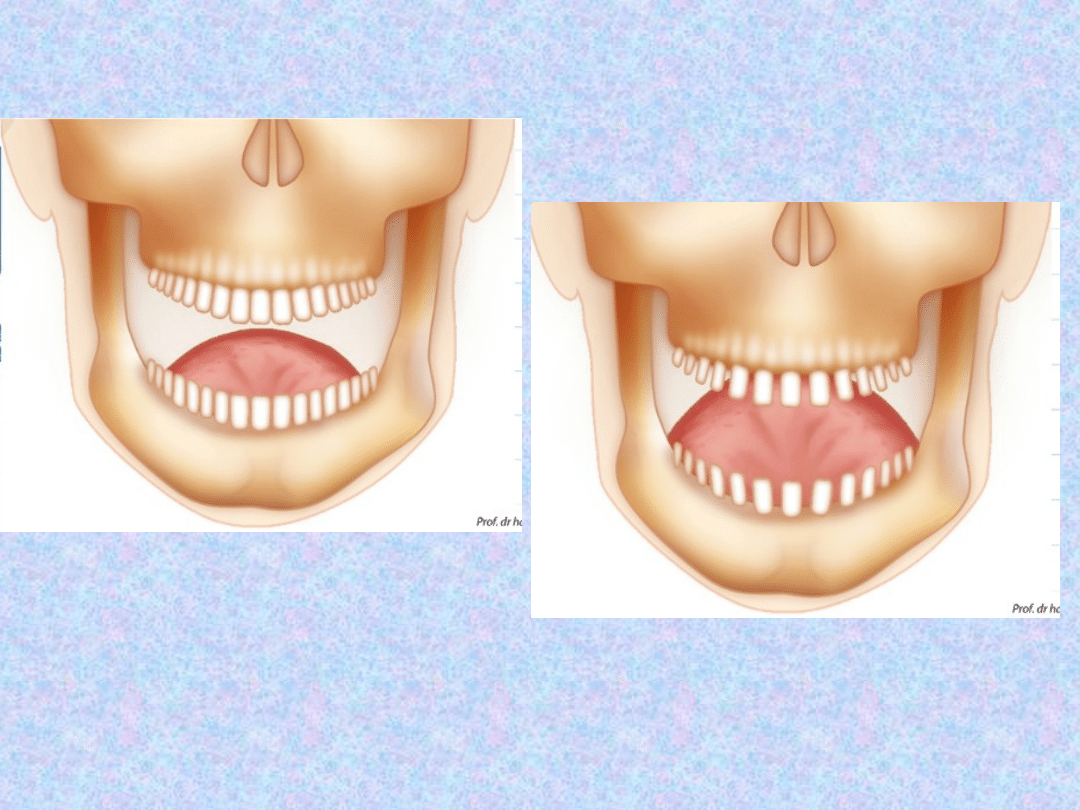
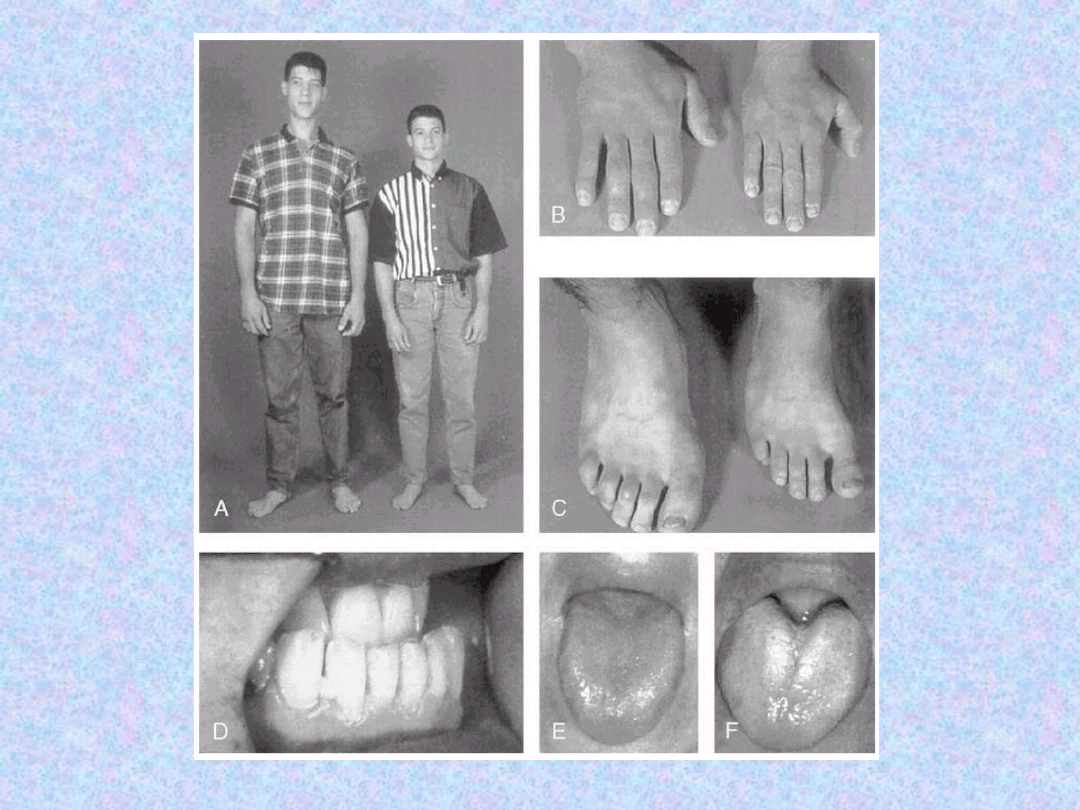
JAMA USTNA
– powiększenie
języka w skutek przerostu mięśni,
pogrubienie i powiększenie warg,
czy rzadziej przerost dziąseł.
Następuje również powiększenie
żuchwy (powiększenie przerw
między zębami -> wypadanie).

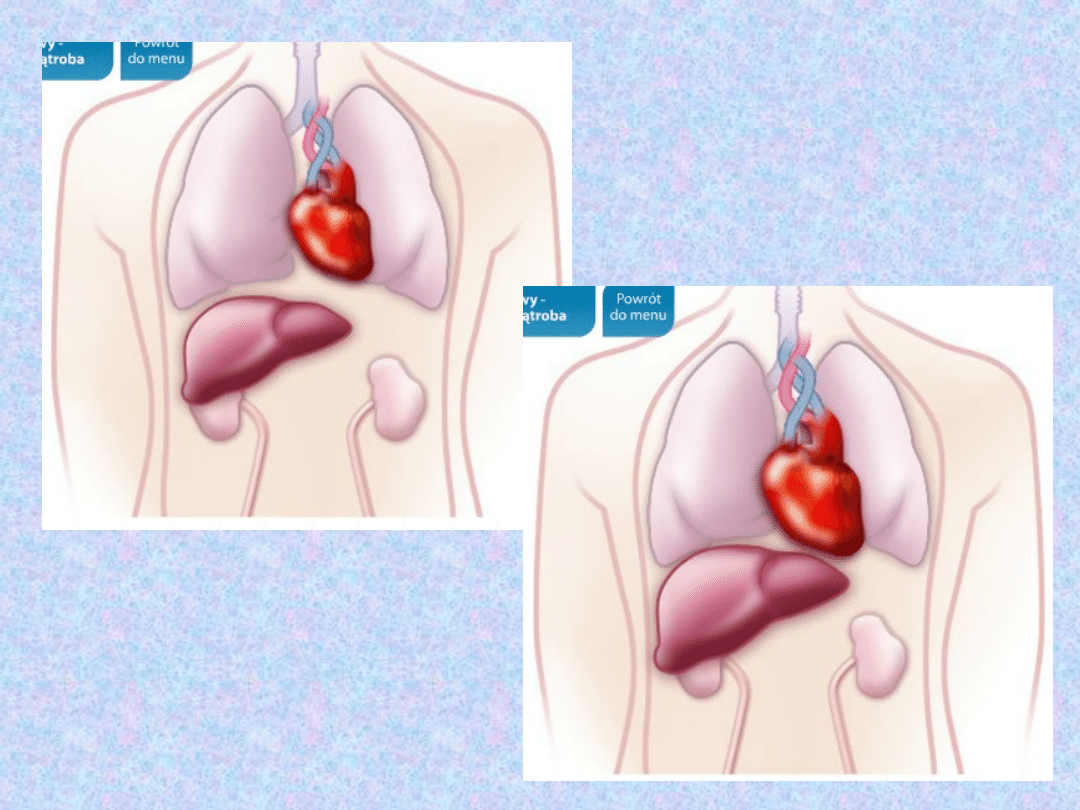
SERCE I WĄTROBA
– chorzy z
niewyrównaną akromegalią żyją 10-20 lat
krócej!
Choroby układu sercowo- naczyniowego
takie jak kardiomiopatia przerostowa,
nadciśnienie tętnicze, czy zaburzenia
rytmu serca będące główną przyczyną
śmiertelności występują u 90% chorych z
długotrwałą, nieleczoną akromegalią i są
w ok. 60% przyczyna umieralności.
W większości przypadków dochodzi do
przerostu innych narządów wewnętrznych
(zwłaszcza wątroby).
W nieleczonej akromegalii zwiększa
się ryzyko wystąpienie innych
nowotworów.

..INNE OBJAWY
• przerost i deformacja kości, deformacje
stawów, bóle stawów i kości,
• wzmożona potliwość (zlewne poty),
• pogrubienie skóry, nadmierne
owłosienie,
• cukrzyca z insulinoopornością,
• bóle głowy,
• zaburzenia czynności wzroku
(ograniczone pola widzenia),
• zaburzenia miesiączkowania,
brak miesiączki, mlekotok, zaburzenia
potencji, obniżone libido,
• kamica nerkowa,
• zaparcia,

ROZPOZNANIE
Dla rozpoznania czynnej akromegalii
konieczne jest udokumentowanie braku
supresji wydzielania GH poniżej 1,0 µg/l
(ng/ml) po doustnym obciążeniu
glukozą (75 g glukozy) i zwiększonego
wydzielania IGF-1.
Aby móc jednoznacznie określić
przyczynę akromegalii należy wykonać
badanie przysadki w rezonansie
magnetycznym (MRI) z zastosowaniem
kontrastu. U prawie wszystkich
pacjentów z chorobą pochodzenia
przysadkowego stwierdza się
gruczolaka przedniego płata
przysadki, w większości wielkości ≥ 1
cm (makrogruczolak).
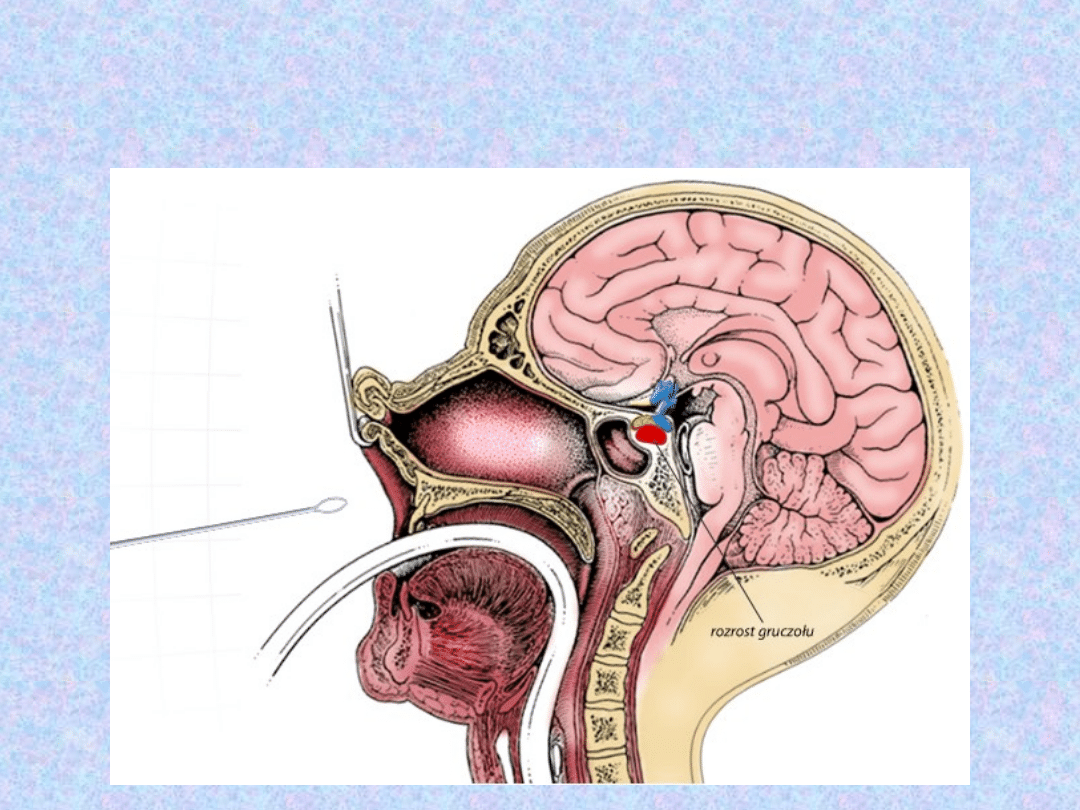
LECZENIE
CHIRURGICZNE-
usunięcie gruczolaka przysadki
mózgowej

LECZENIE
FARMAKOLOGICZNE
Leczenie farmakologiczne
analogami somatostatyny o
przedłużonym działaniu wdraża się,
gdy istnieją przeciwwskazania do
zabiegu lub gdy pacjent nie wyraża
zgody, albo gdy leczenie operacyjne
okazało się nieskuteczne lub guz jest
nieoperacyjny (nacieka nerw
wzrokowy).
RADIOTERAPIA
stosowana jest jako leczenie
uzupełniające w razie niepowodzenia
leczenia chirurgicznego i
farmakologicznego.
W Polsce
, co roku rozpoznajemy
około 200 nowych przypadków
akromegalii. W sumie, w naszym
kraju na tę chorobę cierpi aktualnie
ok. 2,5 tys. osób.
Chorobę obserwuje się dwukrotnie
częściej u kobiet.

BIBLIOGRAFIA
• Akromegalia.pl
• Zdrowie.gazeta.pl
• Medyczny-poznan.pl
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
Wyszukiwarka
Podobne podstrony:
26. Reakcje rodziców na info. o chorobie przewlekłej dziecka, opieka nad dziećmi z chorobą przewlekł
epidemiologia chorób przewlekłych 2012
Choroba przewlekła jako czynnik ryzyka krzywdzenia emocjonalnego dziecka, Dziecko- Metody terapii
choroba przewlekla, Studia, Edukacja i rehabilitacja osób z niepełnosprawnością ruchową, materiały n
Życie dziecka z chorobą przewlekłą
Dziecko z chorobą reumatyczną, Edukacja integracyjna i wlączająca, Metodyka pracy edukacyjno - terap
choroba przewlekła
choroby przewlekłe
20101005 Choroby przewlekłe z uwzględnieniem aspektów opieki położniczej
wyklad 1 Choroba przewlekla w zyciu dziecka i rodzicow
choroby przewlekle studenci iv rok 240512
RYZYKO CHORÓB PRZEWLEKŁYCH A BRAK AKTYWNOŚCI FIZYCZNEJ
poradnictwo w zakresie chorób przewlekłych, poradnictwo w resocjalizacji
Problemy dziecka z choroba przewlekla19
reumetalogia, RZS reum, RZS reumoitedalne zapelanie stawów -gościec przewlekły postępujący ,choroba
pytania - choroby przewlekłe niepelnosprawnośc, Pielęgniarstwo - materiały na studia, Pytania
Jakość życia w chorobach przewlekłych
3a.Choroby przewlekłe okresu rozwojowego cz.1, medyczne różne, pediatria
Detoksykacja w chorobach przewlekłych a, ZDROWIE, Medycyna Naturalna
więcej podobnych podstron