
1
Odsalanie roztworu białka metodą chromatografii sita molekularnego
Cel ćwiczenia
Ć
wiczenie ma na celu poznanie podstaw teoretycznych metody chromatograficznej sita
molekularnego. Ponadto studenci zapoznają się praktycznie z frakcjonowaniem związków w
oparciu o ich różnice w masach cząsteczkowych metodą sączenia na żelu Sephadex.
Wprowadzenie
Chromatografia
to fizykochemiczna metoda rozdzielania mieszanin, których składniki
ulegają zróżnicowanemu podziałowi pomiędzy dwie fazy, fazę ruchomą i stacjonarną. Jeżeli
fazą ruchomą jest gaz, to chromatografia nosi nazwę gazowej; gdy ciecz, wówczas nazywana
jest cieczową; w przypadku fazy ruchomej będącej płynem w stanie nadkrytycznym mówi się
o chromatografii nadkrytycznej. Fazą stacjonarną natomiast może być ciało stałe bądź ciecz
osadzona na stałym nośniku. Faza stacjonarna może być umieszczona w kolumnie
(chromatografia kolumnowa) lub na płaszczyźnie (chromatografia planarna: bibułowa lub
cienkowarstwowa).
Faza ruchoma przepływając przez fazę stacjonarną powoduje migrację poszczególnych
składników mieszaniny. Składniki te przemieszczają się z różną szybkością wynikającą z ich
odmiennego powinowactwa do adsorbenta, odmiennego swoistego powinowactwa do ligandu,
z różnic w ich masie cząsteczkowej, z różnic w wypadkowym ładunku, bądź z różnej
rozpuszczalności w określonych warunkach rozdziału. Stąd, w
zależności od przewagi
czynników działających różnicująco na rozdzielane substancje można wyróżnić 5 zasadniczych
metod chromatograficznych: chromatografię adsorpcyjną, powinowactwa, sita molekularnego,
jonowymienną i podziałową.
Chromatografia
sita molekularnego. Jest to jedna z metod chromatograficznych, w
której rozdział mieszaniny opiera się na różnej szybkości migracji poszczególnych składników
przez złoże utworzone z granulek żelu o porach określonej wielkości. Szybkość ta uzależniona
jest od masy cząsteczkowej rozdzielanych związków.
W klasycznej chromatografii niskociśnieniowej (faza ruchoma przepompowywana pod
ciśnieniem zazwyczaj 0.001-0.01MPa), najczęściej rolę sita pełnią odpowiednio usieciowane
polimery, które po umieszczeniu w wodzie pęcznieją tworząc żel, z którego formuje się
kolumnę
.
Są to głównie żele dekstranowe, agarozowe, poliakryloamidowe. W chromatografii
HPLC (HPLC - high pressure liquid chromatography, wysokociśnieniowa chromatografia
cieczowa), w której faza ruchoma przepompowywana jest pod ciśnieniem zazwyczaj 5-10
MPa, stosuje się natomiast złoża odporne na wysokie ciśnienia hydrostatyczne, np.: żele
polistyrenowe, odpowiednio spreparowane żele krzemionkowe czy granulki z porowatego
szkła. Żel dekstranowy to jeden z najpowszechniej stosowanych sit molekularnych,
występujący pod nazwą handlową jako Sephadex (wprowadzony przez firmę Pharmacia,
obecnie GE Healthcare). Żel ten zbudowany jest z łańcuchów α-D-glukozy połączonych
między sobą mostkami poprzecznymi z epichlorohydryny. Od liczby tych mostków zależy
wielkość porów, a od niej z kolei zdolność rozdzielcza żelu, czyli zakres mas cząsteczkowych
związków, które wnikają do wnętrza granulek żelu i są tam selektywnie zatrzymywane.
Sefadeksy są oznaczone liczbami charakteryzującymi zdolność wiązania wody, np.
Sephadex
2
G-100 wiąże ok. 10ml wody na gram suchego preparatu. Poszczególne rodzaje sefadeksów
charakteryzują się różną, ściśle określoną wielkością porów, dostosowaną do rozdziału cząstek
różnej wielkości (Tabela 1).
Tabela 1. Charakterystyka kilku rodzajów sefadeksów
typ
zdolność wiązania H
2
O
w ml/g s.m. preparatu
Ś
redni rozmiar „oczek”
sita (mesh)
Zdolność
rozdzielcza
cząsteczek w Da
G-25
2,5
2500
100-5000
G-50
5,0
15000
500-10000
G-100
10,0
65000
1000-100000
G-200
20,0
150000
1000-200000
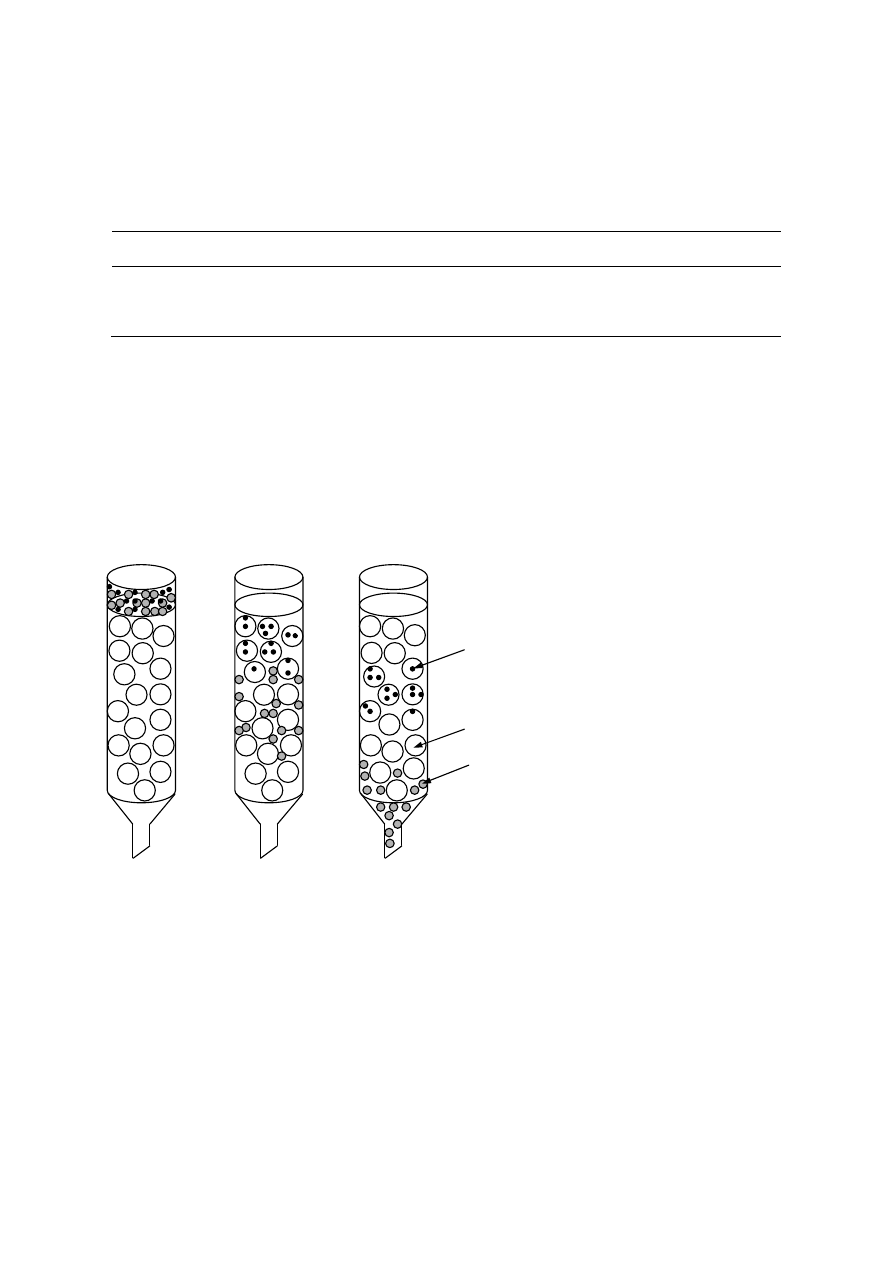
W miarę rozdzielania na kolumnie z sefadeksu, mieszaniny złożonej z związków
różniących się masą cząsteczkową, będzie zachodził następujący proces (Rys.1). Duże
cząsteczki (większe od wielkości porów) nie są w stanie wnikać w pory granulek żelu ze
względu na swój rozmiar, wobec czego będą szybko przemieszczać się przez wolne
przestrzenie między granulkami i wędrować w dół kolumny wraz z czołem rozpuszczalnika.
Cząsteczki mniejsze (mniejsze od wielkości porów) będą wnikać do wnętrza porów i ich
prędkość migracji będzie znacznie mniejsza. Im cząsteczka mniejsza tym głębiej będzie
penetrować pory granulek, tym wolniej będzie migrować przez złoże i tym później pojawi się
ona w eluacie od cząsteczki większej od niej.
.
Rys 1. Przepływ przez kolumnę wypełnioną sefadeksem związków nisko- i
wysokocząsteczkowych.
Rozdział związków na sefadeksie można przyrównać do chromatografii podziałowej,
przyjmując, że objętość całkowita złoża w kolumnie (V
c
) jest sumą objętości rozpuszczalnika
znajdującego się między ziarnami żelu, niezwiązanego z żelem (V
o
)
; objętości rozpuszczalnika
wypełniającego ziarna żelu, związanego z żelem (V
i
); oraz objętości matrycy żelu – (Vg):
V
c
= V
o
+ V
i
+ Vg
Rozpuszczalnik przemieszczający się pomiędzy granulkami żelu można więc traktować
jako fazę ruchomą, a zalegający w porach granulek żelu – jako fazę stacjonarną. Objętość
związki
wielkocząsteczkowe
związki
niskocząsteczkowe
granulka żelu
3
zerową kolumny, V
o
, wyznacza się stosując najczęściej błękit dekstranowy – związek barwny o
wysokiej masie cząsteczkowej, wędrujący w kolumnie z czołem rozpuszczalnika.
Odsalanie na złożu Sephadex.
Chromatografia sita molekularnego znalazła
zastosowanie m.in. do: rozdzielania mieszanin na kolumnie; odsalania substancji
wysokocząsteczkowych, np. białek, od związków niskocząsteczkowych; wyznaczania
przybliżonych
mas
cząsteczkowych;
zagęszczania
roztworów
substancji
wysokocząsteczkowych. Na ćwiczeniu studenci zapoznają się z procesem odsalania białka od
azotanu potasu, który przeprowadzony zostanie na złożu Sephadex G-25. W doświadczeniu
tym, jako rozpuszczalnik zastosowano wodę, która stanowi zarówno fazę stacjonarną, jak i
fazę ruchomą. W takich warunkach duże cząsteczki białka, niezdolne do wnikania do wnętrza
ż
elu, wędrują z czołem rozpuszczalnika. Pojawienie się białka w eluacie (czyli objętość
elucyjna V
e
) odpowiada równocześnie objętości zerowej kolumny (V
o
):
V
e
białka = V
o
Składniki soli natomiast wnikają bez trudu do rozpuszczalnika wypełniającego pory
sefadeksu, swobodnie dyfundują w porach granulek żelu i dlatego wędrują znacznie wolniej.
Ich objętość elucyjna będzie więc sumą objętości rozpuszczalnika znajdującego się między
ziarnami żelu (V
o
)
i objętości rozpuszczalnika wypełniającego ziarna żelu (V
i
):
V
e
soli = V
o
+ V
i
Efekt procesu odsalania sprawdzony zostanie poprzez detekcję rozdzielanych składników
mieszaniny w eluacie z kolumny. Na ćwiczeniach, białko będzie wykrywane metodą
Lowry’ego i wsp. (1951) lub metodą spektrofotometryczną (Layne, 1957) (obie metody
opisane w ćwiczeniu: „Fotometryczne oznaczanie zawartości białka”). Jony azotanowe
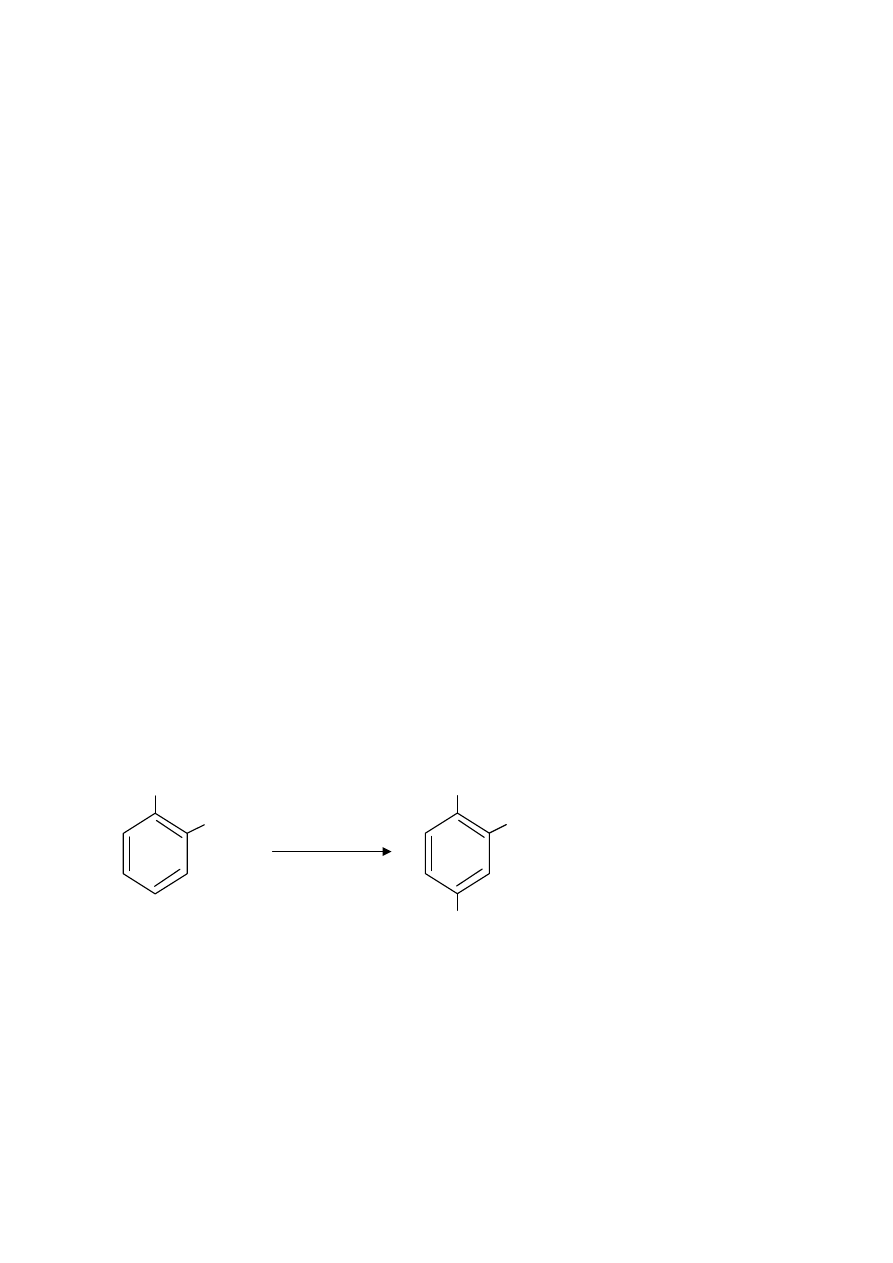
oznaczane będą metodą Cataldo i wsp. (1975), która polega na nitrowaniu kwasu salicylowego
w środowisku silnie kwaśnym (Rys. 2). Powstający kwas nitrosalicylowy w środowisku
zasadowym (po zmianie pH) przyjmuje zabarwienie żółte, którego intensywność jest
wprostproporcjonalna do stężenia NO
3
-
i mierzy się ją przy 420 nm.
Rys. 2. Rekacja nitrowania kwasu salicylowego
Odczynniki
1. Sephadex G-25 drobnoziarnisty (fine).
2. 0.25-proc. roztwór białka (albumina wołu frakcja V) w 2.5 mM azotanie potasu
OH
COOH
OH
COOH
NO
2
HNO
3
, H
2
SO
4
- H
2
O
kwas salicylowy
kwas nitrosalicylowy

4
3. Odczynnik miedziowy: a) 2-proc. roztwór węglanu sodowego w 0,1-molowym
wodorotlenku sodowym, b) 2-proc. wodny roztwór winianu sodowo-potasowego, c) 1-
proc. wodny roztwór CuSO
4
· 5 H
2
O; przed użyciem w cylindrze miarowym na 100 ml
zmieszać 98 ml roztworu a) z 1 ml roztworu b) i 1 ml roztworu c).
4. Odczynnik Folina, powszechnie dostępny w handlu (rozcieńczony 1:1) wodą.
5. 5-proc. roztwór kwasu salicylowego w stężonym kwasie siarkowym
6. 4 M wodorotlenek sodu
Wykonanie
Studenci pracując w zespołach 2-osobowych oddzielają białko od soli azotanowej na
złożu Sephadex G-25. Na zajęciach 4-godzinnych, studenci oznaczają zawartość białka w
roztworze wyjściowym nanoszonym na kolumnę oraz w eluacie metodą Lowry’ego, natomiast
na zajęciach 3-godzinnych białko wykrywane jest metodą spektrofotometryczną tylko w
zebranych frakcjach. Studenci na zajęciach 3- i 4-godzinnych w zebranych frakcjach
wykrywają także jony azotanowe.
1. Sporządzanie kolumny. Kolumnę formuje się z uprzednio napęczniałego wodą żelu
Sephadex G-25 (1). Rurkę chromatograficzną zawierającą przy wylocie zwitek z waty szklanej
umocować pionowo w statywie i napełnić jednorazowo żelem do wysokości ok. 20 cm. Na
powierzchni żelu ułożyć delikatnie krążek z bibuły filtracyjnej w celu zabezpieczenia kolumny
przed uszkodzeniem. Następnie odkręcić zacisk, aby płyn wyciekał z szybkością 15–20 kropli
na minutę i przemywać kolumnę 2–3-krotną objętością wody.
2. Rozdział na kolumnie (studenci wykonują parami). Wyskalować 20 małych
probówek na 1 ml i jedną probówkę na 7 ml. Z przygotowanej kolumny wypełnionej
sefadeksem odkręcić zacisk
i odsączyć płyn znad żelu. Gdy warstwa buforu nad żelem będzie
wynosiła ok. 2 mm zakręcić zacisk kolumny, nanieść ostrożnie pipetą (aby nie naruszyć żelu) 1
ml roztworu białkowego (2) i pod wylot kolumny podstawić probówkę wyskalowaną na 7 ml,
po czym odkręcić zacisk. Po prawie całkowitym wniknięciu naniesionego roztworu w żel
również ostrożnie nanieść na kolumnę 1 ml wody w celu wmycia białka i soli do wnętrza żelu.
Po wniknięciu i tej porcji czynność powtórzyć jeszcze raz. Następnie kolumnę nad żelem
napełnić ostrożnie wodą i utrzymywać jej objętość na względnie stałym poziomie przez cały
czas prowadzenia rozdziału. Po zebraniu pierwszych 7 ml podstawić kolejne wyskalowane na
1 ml probówki, zbierając do nich następne frakcje po 1 ml. W sumie zebrać 20 frakcji, po czym
zamknąć wylot kolumny za pomocą ściskacza.
3. Oznaczanie zawartości białka w próbie wyjściowej i w frakcjach wycieku z kolumny
metodą Lowry’ego
. W celu oznaczenia zawartości białka w roztworze wyjściowym należy
przenieść dokładnie 1 ml tego roztworu do kolby miarowej na 50 ml, kolbę dopełnić wodą do
kreski i dobrze wymieszać. Do dwóch probówek (próby pełne) pobrać po 0,5 ml roztworu z
kolbki, a do próbówki trzeciej (próba kontrolna) zamiast roztworu białka odmierzyć 0,5 ml
wody. Do wszystkich probówek dodać po 2,5 ml odczynnika miedziowego (3), wymieszać i po
10 min dodać, intensywnie wstrząsając probówką, po 0,25 ml odczynnika Folina (4). Po 30
min mierzyć intensywność zabarwienia w kolorymetrze przy długości fali 750 nm, wobec
próby kontrolnej. Uzyskaną wartość absorbancji (A) przeliczyć na µg białka, wykorzystując w
tym celu krzywą wzorcową (ew. własną, wykonaną na ćwiczeniu „Fotometryczne oznaczanie
zawartości białka”).
W celu oznaczenia zawartości białka w kolejnych frakcjach wycieku z kolumny należy
pobrać po 0,1 ml z frakcji od 1 do 11, uzupełnić wodą do 0,5 ml i postępować dalej jak opisano
przy oznaczeniu białka w próbie wyjściowej.
5
4. Wykrywanie jonu azotanowego. Do 14 probówek pobrać kolejno po 0.1 ml roztworu
z frakcji od 5 do 18, do każdej dodać po 0,2 ml kwasu salicylowego (5) (UWAGA! Ostrożnie
ż
rące!), wymieszać i pozostawić w temperaturze pokojowej przez 20 minut. Po tym czasie
dodać ostrożnie (!) 2,2 ml 4 M wodorotlenku sodowego (6), wymieszać, schłodzić (!) i
odczytać absorbancję przy długości fali 420 nm, wobec próby odczynnikowej zawierającej 0,1
ml wody dejonizowanej zamiast roztworu frakcji.
5. Wykrywanie białka metodą spektrofotometryczną. Zawartość frakcji od 1 do 11 (po
odebraniu 0.1 ml na oznaczenie jonu azotanowego) przenosić kolejno do kuwety i mierzyć
absorbancję przy długości fali 280 nm, wobec wody dejonizowanej.
Opracowanie wyników
Zajęcia 4-godzinne
W sprawozdaniu należy podać wyniki pomiarów zawartości białka w roztworze wyjściowym i
w eluacie, oraz obliczyć procent odzyskanego białka w stosunku do naniesionego na kolumnę.
Ponadto obliczyć objętość zerową kolumny, na podstawie objętości potrzebnej do
wyeluowania maksymalnego stężenia białka, oraz objętość elucyjną dla jonów azotanowych,
na podstawie objętości eluatu, w której jony azotanowe osiągają maksymalne stężenie.
Wykreślić krzywe elucji obu składników mieszaniny, odkładając na osi odciętych kolejne
numery frakcji a na osi rzędnych odczytane dla tych frakcji wartości absorbancji.
Zajęcia 3-godzinne
W sprawozdaniu należy podać wyniki odczytów absorbancji dla każdej frakcji eluatu
przy 280 nm (białko) i 420 nm (jony azotanowe). Ponadto wykreślić krzywe elucji obu
składników mieszaniny, odkładając na osi odciętych kolejne numery frakcji a na osi rzędnych
odczytane dla tych frakcji wartości absorbancji.
Przykładowe obliczenia (zajęcia 4-godzinne)
a. ilość białka naniesionego na kolumnę
Absorbancja
1
Absorbancja
2
Ś
rednia absorbancja Ilość białka w µg odczytana z krzywej
wzorcowej dla średniej absorbancji
0.175
0.157
0.166
30
Schemat rozcieńczenia roztworu białka przed oznaczeniem:
1 ml 50 ml
0.5 ml
Ś
rednia absorbancja dla 0.5 ml rozcieńczonego roztworu – 0.166, ilość białka w 0.5 ml
rozcieńczonego roztworu – 30 µg.
Ilość białka w 50 ml rozcieńczonego roztworu (w kolbce) – 3000 µg (30 µg - 0.5 ml
x µg - 50 ml
x = 3000 µg)
Do kolbki, przed dopełnieniem wodą do kreski, wlano 1 ml roztworu białka nanoszonego na
kolumnę. Ilość białka w kolbce przed rozcieńczeniem (w 1ml) i po rozcieńczeniu wodą (w 25

6
ml) jest taka sama, zmienia się tylko stężenie roztworu. Stąd ilość białka w 1 ml roztworu
nanoszonego na kolumnę równa jest 3000 µg.
b. ilość białka w wycieku z kolumny (odzyskanego z kolumny)
Na podstawie absorbancji otrzymanych dla poszczególnych frakcji (1-11) odczytać z krzywej
wzorcowej µg białka. Zsumować odczytane dla poszczególnych frakcji µg białka, np. suma
równa jest 280 µg. Uwzględnić, że do oznaczenia białka pobrano tylko 1/10 objętości każdej
frakcji, stąd: 280 µg x 10 = 2800 µg. Ilość białka odzyskanego z kolumny (suma µg białka z
poszczególnych frakcji) = 2800 µg.
c. Procent odzyskanego białka w stosunku do naniesionego na kolumnę
3000 µg – 100 %
2800 µg – x %
x = 93,3 %
Pytania
1. Na jakiej zasadzie opiera się rozdział związków metodą chromatografii sita molekularnego?
2. Jakie zastosowania w biochemii może mieć Sephadex?
3. Jak zbudowany jest żel Sephadex?
4. Co to jest i od czego zależy zdolność rozdzielcza żelu Sephadex?
5. W jaki sposób można się przekonać o
efekcie rozdziału mieszaniny na złożu Sephadex?
6. W jakiej kolejności wypływają z kolumny wypełnionej żelem Sephadex G-25 i G-100 jon
amonowy, peptyd protamina (2–3 kDa) i pepsyna (35 kDa)?
Literatura
1. Cataldo D.A., Haroon M., Schrader L.E., Youngs V.L. (1975). Rapid colorimetric
determination of nitrate in plant tissue by nitration of salicylic acid. Commun. Soil Sci.
Plant Anal. 6, 71-80.
2. Layne E. (1957). Spectrophotometric and turbidimetric methods for measuring
proteins. Methods in Enzymology 3, 447-455.
3. Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. (1951). Protein measurement
with the Folin phenol reagent. J. Biol. Chem. 193, 265-275.
Wyszukiwarka
Podobne podstrony:
Odsalanie roztworu białka metoda chromatografii sita molekularnego
Na czym polega metoda chromatografii bibułowej, kosmetologia, chemia kosmetyczna
Całkowity potencjał antyoksydacyjny wyznaczony metodą chromatograficzną niektórych ziół i napoi alko
Oznaczanie składu metodami chromatografii gazowej 1
Identyfikacja cukrów metodą chromatografii bibułowej
Wyznaczanie stężenia roztworów koloidalnych metodą nefelometryczną (2), FIZYKA-sprawozdania
Sita molekularne typJ
Całkowity potencjał antyoksydacyjny wyznaczony metodą chromatograficzną niektórych ziół i napoi a
Analiza gazów energetycznych metodą chromatografii gazowej
Oznaczenie roztworu zasady metodą miareczkowania alkacymetrycznego, Studia, Semestr II, Chemia, Labo
Ćw. 5 Rozdział metodą chromatografii bibułowej - sprawozdanie, Chemia ogólna i nieorganiczna
Oznaczanie składu metodami chromatografii gazowej, AKADEMIA GÓRNICZO - HUTNICZA
Sita molekularne typJ
Ilościowe oznaczanie stężenia białka metodą Lowry
OZNACZANIE BIALKA metoda biuretowa BIOL (1)
więcej podobnych podstron