
PATOFIZJOLOGIA
UKŁADU NERWOWEGO
dr n. med. Arkadiusz Styszyński
Katedra i Zakład Patofizjologii
Uniwersytet Medyczny
im. Karola Marcinkowskiego w Poznaniu

Przyczyny zaburzeń układu nerwowego
• Defekty genetyczne
• Choroby degeneracyjne
• Guzy
• Uszkodzenie mechaniczne
(uraz)
• Krwawienie
• Niedokrwienie
• Zaburzenia metaboliczne
– Hipoglikemia
– Hiperglikemia
– Mocznica
– Niewydolność wątroby
– Zaburzenia hormonalne itp.
• Zaburzenia elektrolitowe
• Leki
• Toksyny
– Metale ciężkie
– Alkohol
• Promieniowanie
• Zapalenie i infekcja
– Wirusy
– Bakterie
– Priony
– Choroby autoimmunologiczne

Uszkodzenie efektorów
obwodowych
Prowadzi do zaburzenia określonych funkcji
• Ogniskowego (pojedyncze mięśnie)
• Uogólnionego (cały układ mięśniowy)
Może skutkować
• nadaktywnością
– Niezamierzone skurcze mięśniowe
– Nieprawidłowa aktywność receptorów czuciowych fałszywa
percepcja czuciowa
• Deficyty czynnościowe (porażenie mięśni lub deficyty
czuciowe)
Percepcja czuciowa, zwłaszcza wzrok i słuch, może być
zaburzona, jeśli aparat przewodzący jest uszkodzony, nawet
przy prawidłowo działających receptorach czuciowych

Przerwanie przewodzenia w nerwie
obwodowym
• Zakłóca sygnały przewodzone w tym nerwie
• Różne typy włókiem (zmielinizowane i niezmielinizowane)
mogą być dotknięte w różnym stopniu
• Całkowite przerwanie przewodzenia w nerwie
– Porażenie wiotkie
– Utrata czucia
– Utrata regulacji autonomicznej
w zakresie unerwienia zajętego nerwu
• Uszkodzenie nerwu rdzeniowego wpływa na odpowidający
mu dermatom
• Rozpoznanie uszkodzenia nerwu wymaga wiedzy na temat
obszaru unerwienia poszczególnych nerwów i dermatomów

Uszkodzenie OUN
Uszkodzenia rdzenia kręgowego
prowadzą do
• Utraty czucia
• Utraty funkcji autonomicznych
• Porażenia wiotkiego lub
spastycznego
Uszkodzenia struktur
nadrdzeniowych prowadzą do
• Deficytów lub nieprawidłowych
pobudzeń ograniczonych co do
funkcji i obszaru ciała (np. w
określonych obszarach kory
czuciowej)
• Częściej powodują złożone
zaburzenia czucia, funkcji
motorycznych i regulacji
autonomicznej
• Upośledzenie czynności
integracyjnych, takich jak pamięć,
emocje, czynności poznawcze
mogą wystąpić w przebiegu wilu
chorób

Nieprawidłowości komórek
nerwowych
•
Gęstość receptorów może ulec zmniejszeniu (down-regulation)
•
Przewodzenie wewnątrzkomórkowe może być zablokowane
– Zablokowanie białka G przez toksynę krztuśca
•
Zaburzenia kanałów jonowych
– Zablokowanie przez leki
– Aktywność zmieniona przez Ca
2+
, Mg
2+
, or H
+
– Wpływ na potencjał błonowy może być zmieniony przez nieprawidłowy gradient jonów –
wzrost lub spadek wewnątrz- lub zewnątrzkomórkowego stężenia K+
•
Transport aksonalny, wytwarzanie, składowanie, uwalnianie i inaktywacja
neurotransmiterów może być zaburzona
– Leki
– Defekty genetyczne
Zaburzenia funkcjonalne – odwracalne, o ile czynnik sprawczy przestaje działać
Uszkodzenia mogą prowadzić do nieodwracalnego zniszczenia neuronów
•
Śmierć komórki na skutek bezpośredniego uszkodzenia - martwica (niedobór
energii, uszkodzenie mechaniczne)
•
Programowana śmierć komórki - apoptoza
U osób dorosłych neurony nie mogą się odnawiać zniszczenie neuronu powoduje
nieodwracalne upośledzenie czynności; inne neurony mogą częściowo przejąć funkcję
martwej komórki

Bariera krew-mózg
Prawidłowa bariera krew-mózg uniemożliwia
przeniknięcie większości substancji i chroni przed
wejściem patogenów i komórek
immunokompetentnych
Szkodliwe substancje muszą przejść przez barierę
krew-mózg jeśli mają wpłynąć na neuronu w OUN
Pewne toksyny (np. krztuśca i jadu kiełbasianego)
osiągają neurony w rdzeniu kręgowym na drodze
wstecznego transportu aksonalnego poprzez
nerwy obwodowe
Niektóre wirusy również osiągają OUN na tej drodze

Przerwanie aksonu
• Obwodowa część aksonu obumiera – zwyrodnienie Wallera
• Aksony neuronów ośrodkowych nie odrastają – uszkodzony neuron umiera
przez apoptozę
– Brak czynnika wzrostu nerwu (nerve growth factor - NGF), który normalnie jest
wydzielany przez unerwioną komórkę postsynaptyczną i w ten sposób poprzez
akson utrzymuje przy życiu komórkę presynaptyczną
• Przerwanie wstecznego transportu aksonalnego w nieuszkodzonym
neuronie również prowadzi do jego śmierci
• Proksymalny „kikut” neuronu obwodowego może odrosnąć
– Makrofagi migrują do nerwu obwodowego, poprzez produkcję interleukiny 1
pobudzają komórki Schanna do produkcji NGF
• Brak unerwienia prowadzi do śmierci komórki docelowej (transneuronalna
degeneracja)
• Niekiedy następuje również śmierć komórki unerwiającej uszkodzoną
komórkę (wsteczna transneuronalna degeneracja)

Demielinizacja
• Przyczyny:
– zwyrodnieniowe
– toksyczne
– zapalne
– niedobór witaminy B6 i B12
• Oporność błony komórkowej zmniejsza się a pojemność elektryczna
zwiększa w segmentach międzywęzłowych wyższe natężenie jest
konieczne aby zmienić polaryzację segmentu międzywęzłowego
dochodzi do znaczej utraty natężenia natężenie generowane w R1 jest
niedostateczne aby zdepolaryzować R2 do potencjału progowego
przewodnictwo jest przerwane
• Niewielkie uszkodzenia segmentu międzywęzłowego spowolnienie
przewodnictwa (nie może przeskakiwać pomiędzy węzłami)
spowolnienie może być niejednakowe w różnych włóknach może
wystąpić rozproszenie czasowe sygnału
• Uszkodzone miejsce samo może wyzwolić potencjał czynnościowy
pobudzenia przeskakuje pomiędzy sąsiednimi włóknami nerwowymi
przewodnictwo może wystąpić wstecznie

Stwardnienie rozsiane
• Choroba autoimmunologiczna która może zostać wyzwolona
infekcją wirusową
• Występują zapalne ogniska demielinizacyjne
• Typowe cechy stwardnienia rozsuanego to czasowo niezwiązane
występowanie zupełnie różnych ubytków nerwowych,
spowodowane zajęciem różnych obszarów mózgu
• Niektóre zmiany mogą częściowo ustąpić jeśli miejscowy stan
zapalny wycofa się, a nerw ulegnie remielinizacji (warunek:
nieuszkodzony akson)
• Przykład
– Początkowo całkowicie odwracalna utrata wzroku z powodu
uszkodzenia nerwu wzrokowego
– Częśniowo odwracalna utrata czucia przy uszkodzeniu dróg czuciowych
w rdzeniu kręgowym
– Ataksja pojawia się przy uszkodzeniu móżdżku

Zaburzenia transmisji nerwowo-mięśniowej
• Anestetyki miejscowe hamują kanały sodowe
sterowane potencjałem w neuronach przerwanie
transmisji nerwowej do płytki motorycznej
• Kanały wapniowe mogą być zablokowane przez
przeciwciała
• Toksyna jadu kiełbasianego inaktywuje synaptobrewinę
– białko odpowiedzialne za wiązanie pęcherzyków
zawierających ACh z błoną komórkową
• Receptory dla ACh mogą być zablokowane przez
przeciwciała przyspieszające internalizację i rozkład
receptorów
• Receptory mogą być zablokowane przez kurarę która
kompetencyjne hamuje wiązanie ACh z receptorem

Zaburzenia transmisji nerwowo-mięśniowej
• Sukcynylocholina powoduje ciągłą stymulację receptora
ciągła depolaryzacja błony postsynaptycznej inaktywacja
postsynaptycznych kanałów Na+ zablokowanie transmisji
nerwowo-mięśniowej
• Inhibitory acetylocholinestareazy (fizostygmina)
zwiększenie dostępności ACh w szczelinie synaptycznej
zwiększenie transmisji nerwowo-mięśniowej
• Wysokie dawki inhibitorów acetylocholinesterazy
wysokie stężenie ACh ciągła depolaryzacja błony
postsynaptycznej inaktywacja kanałów Na+
hamowanie transmisji nerwowo-mięśniowej
• Mg2+ i hemicholina hamowanie wychwytu zwrotnego
choliny przez zakończenie nerwowe

Miastenia gravis (nużliwość mięśni)
• Porażenie mięśni na skutek zablokowanie transmisji nerwowo-
mięśniowej
• Spowodowane przeciwciałami przeciwko receptorom dla ACh w
błonie postsynaptycznej przyspieszenie rozkładu receptora
– Zakażenie wirusami o strukturze podobnej do receptora dla ACh
– Pacjenci z łagodnymi guzami grasicy
– Tworzenie takich przeciwciał częściej występuje u osób, u których
dochodzi do ekspresji określonych podtypów (DR3 i DQw2) układu
zgodności tkankowej (MHC klasy II)
• Powtarzana stymulacja nerwu ruchowego początkowo wywoła
prawidłowo zsumowany potencjał czynnościowy mięśnia
amlituda będzie się zmniejszać w efekcie stopniowo narastającego
„zmęczenia” transmisji nerwowo-mięśniowej

Pseudomiasteniczny zespół
Lamberta i Eatona
• Pacjenci z rakiem drobnokomórkowym płuc
• Kanały Ca
2+
w błonie komórek guza uwrażliwiają
układ odpornościowy i stymulują tworzenie
przeciwciał reagujących z kanałem Ca
2+
błony
presynaptycznej
• Hamowanie kanałów Ca
2+
zsumowany
potencjał czynnościowy jest początkowo niski
powtarzana stymulacja zwiększa ilość Ca
2+
które
ulegają akumulacji w zakończeniu nerwowym
potencjał czynnościowy ulega normalizacji

Choroby jednostki ruchowej
• Uszkodzenie ciał komórek motoneuronów α
– Wirus polio
– Rdzeniowy zanik mięśni – choroba degeneracyjna o nieznanej przyczynie
– Stwardnienie zanikowe boczne (ALS) – defekt genetyczny transportu
aksonalnego
• Uszkodzenie aksonów
– Choroby z autoagresji (np. ch. Guillain-Barre)
– Niedobór witaminy B1 i B12
– Cukrzyca
– Zatrucia (np. ołów, alkohol)
– Defekty genetyczne (np. zespół Charcot–Maire–Tooth)
• Uszkodzenie mięśni
– Choroby z autoagresji (np. zapalenie skórno-mięśniowe)
– Defekty genetyczne - miotonia lub dystrofia

Choroby jednostki ruchowej
• Uszkodzenie jednostki mięśniowej powoduje porażenie zajętych mięśni
• W pierwotnej martwicy motoneuronów αtypowo pojawiają się
fascykulacje (efekt synchronicznej stymulacji i skurczu włókien
mięśniowych jednostki ruchowej)
• Uszkodzenie nerwu obwodowego zmniejszające grubość osłonki
mielinowej skutkuje zmniejszeniem szybkości przewodnictwa nerwowego
• Składowe czuciowe nerwu zostają również dotknięte zaburzeniem
nieprawidłowe funkcje czuciowe (parestezje)
• Pierwotne uszkodzenie mięśni fibrylacje mięśniowe
nieskoordynowane skurcze pojedynczych włókiem mięśniowych
(obserwowane w elektromiografii – EMG)

Dystrofie mięśniowe
Heterogenna grupa
zaburzeń genetycznych
prowadzących do
postępującego zaniku
mięśni i zaburzeń
przewodnictwa w sercu
Klasyfikacja:
• Dystrofinopatie
• Laminopatie
Dystrofina – białko
cytoszkieletu wiążące
aktynę, istotna składowa
kompleksy dystrofiono-
glikoproteinowego (DGC)
Rola DGC
• Połączenie pomiędzy
macierzą
zewnątrzkomórkową i
cytoszkieletem wewnątrz
sarkolemmy
• Ochrona komórek
mięśniowych przez
uszkodzeniem
indukowanym skurczem

Miotonia
Sztywność mięśni spowodowana nieprawidłowościami
błony mięśniowej nadreaktywność błony
komórkowej włókien mięśni szkieletowych
Przyczyna – mutacje genów kodujących kanały sodowe i
chlorkowe błony komórek mięśni szkieletowych
Patofizjologia objawów:
• Sztywne mięśnie seria potencjałów czynnościowych
komórek mięśnia zostaje wyzwolona pojedynczą
stymulacją nerwu
• Słabość mięśni niedostateczna odpowiedź kanałów
sodowych włókien mięśniowych na skutek ich
inaktywacji wcześniejszą nadmierną stymulacją
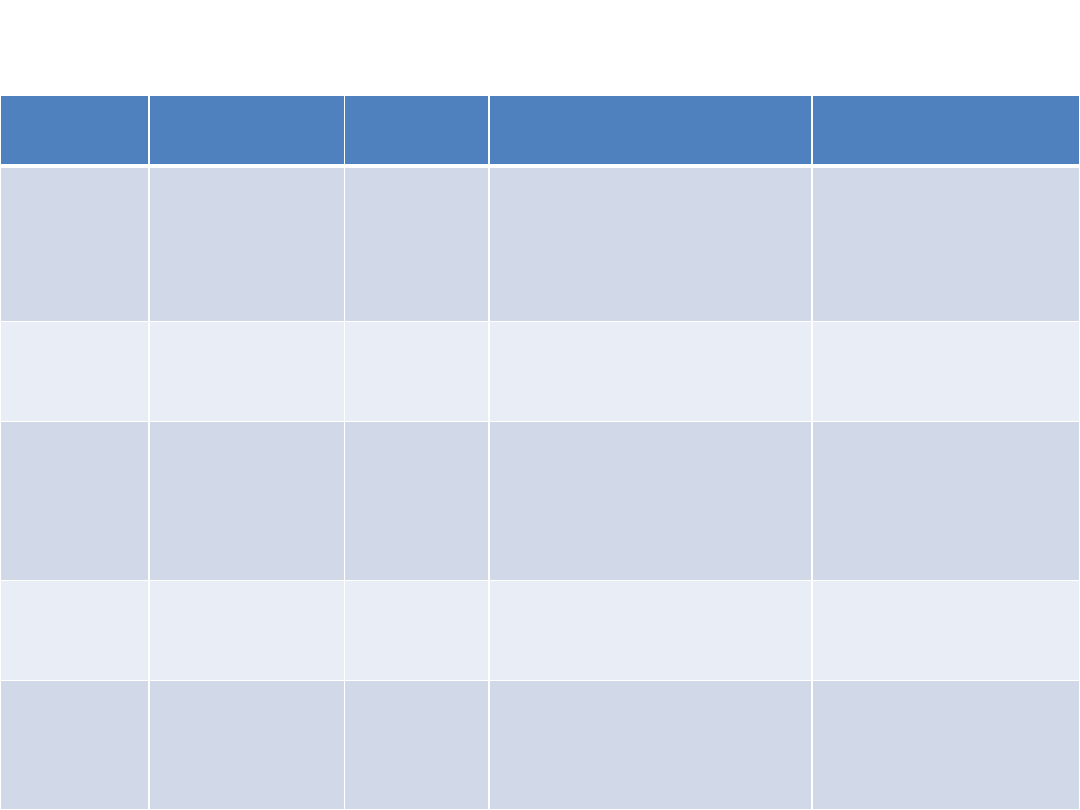
Dystrofie mięśniowe
Choroba
Zaburzenie
genetyczne
Wiek
wystąpienia
Obraz kliniczny
Szybkość postępu
DM
Duchenne’a
Brak dystrofiny
3
Słabość mięśni pośladkowych i
okolicy lędźwiowej wszystkich
grup mięśnowych
Szybka, śmierć na przełomie
2. i 3. dekady życia z powodu
niewydolności oddechowej
lub krążenia
DM Beckera
Zaburzenie domeny
dystrofiny wiążącej
aktynę
5 - 15
Mniej nasilony niż w DMD
Powolniejsza niż w DMD,
śmierć w średnim wieku
DM Emery-
Dreifussa
Emeryna
zlokalizowana na
błonie jądrowej
komórek
mięśniowych
Dzieciństwo
lub
dojrzewanie
Wcześnie występujące skurcze
mięśni, dotyczą proksymalnych
mięśni kończyn górnych i
dystalnych mięśni kończyn dolnych;
arytmie całkowity blok serca
Powolna, zwykle dożywają
średniego wieku
Postać oczno-
gardłowa
Białko
poliadenylowe 2
Piąta dekada
Ptoza, dysfagia, słabość i zanik
języka, mięśni twarzy i
proksymalnej cz. kończyn górnych
Postępująca
Dystrofia
miotoniczna
Kinaza białkowa
miotoninowa
Wiek dorosły
Słabość mięśni i dystrofia,
miotonia, zaćma, zanik jąder,
łysienie czołowe, zaburzenie
przewodnictwa w sercu
Powolny postęp

Uszkodzenia zstępujących dróg ruchowych
• Uszkodzenie torebki wewnętrznej (np. krwawienie lub niedokrwienie
obszaru zaopatrywanego przez tętnicę środkową mózgu) przerwanie
przewodzenia impulsu w zstępujących drogach korowych (droga
piramidowa i inne połączenia kory ruchowej) zmniejszona aktywność
drogi piramidowej, czerwienno-rdzeniowej; mniej uszkodzona droga
przedsionkowo-rdzeniowa i siatkowo-rdzeniowa (silniejszy pozakorowy
wpływ móżdżku) nadmierna aktywność prostowników w nodze i
zginaczy w ręce
• Początkowy szok rdzeniowy w związku z utratą nadrdzeniowego
unerwienia motoneuronów α porażenie wiotkie mięśni, brak odruchów
(arefleksia) częściowe odnerwienie motoneuronów α, γ i
interneuronów stopniowy wzrost wrażliwości tych neuronów
zakończenia neuronów neuronów nadrdzeniowych zostają zastąpione
synapsami z neuronami rdzeniowymi silniejszy wpływ odruchów na
aktywność motoneuronów α hiperrefleksja spastyczność

Uszkodzenia zstępujących dróg ruchowych
• Utrata funkcji dróg zstępujących aktywność
motoneuronów α pod zwiększających się wpływem
wrzecion mięśniowych i narządów Golgiego
rozciąganie wrzecion mięśniowych stymuluje
motoneurony α tego samego mięśnia poprzez odruch
monosynaptyczny silny skurcz przy rozciąganiu
• Odpowiedź wrzecion mięśniowy ma charakter fazowy
przy powolnym rozciąganiu ich aktywność powoli
maleje wpływ narządu Golgiego zaczyna dominować
przy rozciąganiu mięśnia jego skurcz jest hamowany
poprzez hamowanie interneuronu przy powolnym
rozciąganiu mięsień ulegnie nagłemu zwiotczeniu po
poczatkowym wzroście napięcia (objaw scyzoryka)

Uszkodzenia zstępujących dróg ruchowych
• Dominacja mięśni rozciągających prowadzi do
prostowania palucha przy drażnieniu podeszwy stopy
zamiast prawidłowego zgięcia grzbietowego – dowód
na uszkodzenie drogi piramidowej
• Uszkodzenie jądra czerwiennego (np. niedokrwienie
śródmózgowia lub w chorobie Wilsona) drżenie
grubofaliste (jądro czerwienne wytłumia oscylacje
wynikające z ujemnego sprzężenia zwrotnego w
kontroli motoneuronów α)
• Uszkodzenia jądra przedsionkowego zaburzenia
równowagi z zawrotami głowy, oczopląsem i
nudnościami

Jądra podstawy mózgu
• Ciało prążkowane
– Jądro ogoniaste
– skorupa
• Część wewnętrzna i zewnętrzna gałki bladej
• Jądro podwzgórzowe
• Istota czarna
– Część siatkowata [p. r.]
– Część zbita [p. c.]
Funkcja: kontrola czynności ruchowych wraz z móżdżkiem
korą ruchową, drogami korowo-rdzeniowymi i jądrami
ruchowymi z pniu mózgu

Jądra podstawy
Neurony prążkowia są aktywowane za pośrednictwem kwasu
glutaminowego przez neurony kory. Połączenia pomiędzy jądrami
podstawy obejmują głównie przekaźnictwo hamujące zależne od
kwasu γ-aminomasłowego (GABA).
Jądra podstawy mają hamujący wpływ na wzgórze poprzez neurony
GABA-ergiczne wewnętrznej cz. gałki bladej i istoty czarnej (p.r.).
Neurony te są aktywowane przez kwas glutaminowy pochodzący z
neuronów jądra podwzgórzowego. Neurony prążkowia są częściowo
aktywowane, częściowo hamowane przez dopaminę z istoty
czarnej (p.c.) oraz aktywowane przez neurony dopaminergiczne.
Zaburzenie równowagi pomiędzy wpływami aktywującymi i
hamującymi ma szkodliwy wpływ na funkcje motoryczne: zbyt silna
inhibicja skutkuje hipokinezą, zbyt słaba hiperkinezą.

Choroba Parkinsona - mechanizm
•
Choroba istoty czarnej (p. c.) która, poprzez drogi dopaminergiczne wpływa na
GABA-ergiczne komórki ciała prążkowanego
•
Przyczyny
– Wrodzona predyspozycja prowadząca do degeneracji neuronów istoty czarnej w średnim
wieku
– Uraz (np. u bokserów)
– Zapalenie (encephalitis)
– Upośledzenie krążenia (miażdżyca)
– Guzy
– Zatrucia, zwłaszcza CO, mangan, 1-metylo-4-fenylo-1,2,3,6-tetrahydropyridina[MPTP] –
substytut heroiny
•
Uszkodzenie komórek głównie poprzez apaptozę (nadtlenki)
•
Uszkodzenie ponad 70% neuronów istoty czarnej (p. c.) objawy choroby
•
Utrata komórek istoty czarnej (p.c.) zmniejsza unerwienie dopaminergiczne
prążkowia odhamowanie komórek glutaminergicznych w jądrze
podwzgórzowym zwiększona aktywacja wewnętrznej części gałki bladej i części
siatkowatej istoty czarnej nadmierne hamowanie wzgórza (przekaźnictwo
GABA)

Choroba Parkinsona - objawy
• Hamowanie wzgórza ogranicza ruchy zależne od woli
• Hipokinezja – trudność w rozpoczęciu ruchu
• Rigor – zwiększenie napięcia mięśni
• Drżenie spoczynkowe (4-8/sek) naprzemienne ruchy rąk i palcó
(„liczenie pieniędzy”)
• hipokinezja zgarbiona postawa, nieznacznie ugięte przedramiona
i nogi
• Sztywna mimika
• Mikrografia
• Cicha, monotonna mowa
• Zwiększona produkcja śliny, depresja, otępienie utrata neuronów
jądra szwu, miejsca sinawego lub nerwu błędnego

Choroba Parkinsona - leczenie
Cel – zwiększenie produkcji dopaminy w neuronach istoty czarnej i prążkowia
•
L-dopa, prekursor dopaminy (która nie przechodzi przez barierę krew-mózg)
•
Pochodne amfetaminy
– Pobudzają uwalnianie dopaminy
– Hamują wychwyt zwrotny dopaminy
– Zwiększają stężenie dopaminy w szczelinie synaptycznej
•
Inhibitory monoaminooksydazy (MAO) – opóźnienie rozkładu dopaminy
•
Efekt dopaminy może być imitowny przez leki dopamino-podobne
•
Próby przeszczepienia komórek produkujących dopaminę do prążkowia
•
Hamowanie neuronów cholinergicznych w prążkowiu (które stymulują neurony
prawidłowo hamowane przez dopaminę)
•
Antagoniści kwasu glutaminowego i uszkodzenie jądra podwzgórzowego lub
wewnętrznej części gałki bladej powoduje odhamowanie wzgórza poprawa
obrazu klinicznego choroby
•
Opóźnienie śmierci apoptotycznej neuronów istoty czarnej i prążkowia poprzez leki
antyoksydacyjne i czynniki wzrostu

Hiperkinezja
•
Pląsawica to najczęstsza choroba hiperkinetyczna jąder podstawy00
•
Pląsawica Huntingtona
– Dziedziczna
– Ujawnia się w 4. lub 5. dekadzie życia
– Prowadzi do nieodwracalnego postępującego zniszczenia neuronów prążkowia
– Odpowiedzialny gen na ramieniu krótkim 4. chromosomu
– Defekty genetyczne skutkują zwiększeniem zawartości białka (hungtingtyny) nie podlegającego
rozkładowi
– Śmierć komórek jest przyspieszona przez pobudzający wpływ kwasu glutaminowego
aktywującego neurony przezkanał wapniowy nadmierny napływ Ca2+ do neuronów
•
Pląsawica Sydenhama
– Odwracalne uszkodzenie neuronów prążkowia
– Odkładanie kompleksów immunologicznych w przebiegu gorączki reumatycznej
– Zwykle u dzieci
•
Inne przyczyny: niedokrwienie, guz, zapalenie
•
Efekt uszkodzenia neuronów prążkowia
– Nasilone hamowanie neuronów jądra podwzgórzowego (prawidłowo aktywują hneurony
hamujące w istocie czarnej [p. r.]) odhamowanie komórek wzgórza nagłe, błędne ,
niezależne od woli ruchy

Ból
•
Nocyceptory w skórze i w narządach wewnętrznych są stymulowane intensywnymi
bodźcami nieuszkadzającymi (rozciąganie, temperatura) i urazem tkanek
•
Martwe komórki uwalniają K+ i białka wewnątrzkomórkowe
– Wzrost zewnątrzkomórkowego K+ depolaryzacja nocyceptorów
– Białka i napływające mikroogranizmy zapalenie
•
Uwalnianie czynników stymulujących ból
•
Substancje uwrażliwiające hyperalgesia lub allodynia:
– leukotrieny
– prostaglandyna E2
– histamina
•
Uszkodzenie tkanek krzepniecie krwi uwolnienie bradykininy i serotoniny
•
Okluzja naczyń niedokrwienie zewnątrzkomórkowe gromadzenie K+ i H+
akywacja uwrażliwionych nocyceptorów
•
Histamina, bradykinina, prostaglandyna E2 zwiększają przepuszczalność ścian
naczyń obrzęk miejscowy wzrost ciścienia tkankowego pobudzenie
nocyceptorówPobudzane nocyceptory uwalniają substancję P i peptyd zależny od
genu kalcytoniny (CGRP) zwiększenie reakcji zapalnej, wazodylatacja,
zwiększenie przepuszczalności ścian naczyń
•
Skurcz naczyń wywołany przez serotoninę z następczym ich rozszerzeniem
napady migreny (nawracający silny ból głowy, zwykle jednostronny związany z
zaburzeniami neurologicznymi z powodu nieprawidłowości wazomotorycznych
mózgu)

Ból odniesiony
• Impulsacja z narządów i powierzchni skóry podlega konwergencji na
tych samych neuronach w obrębie rdzenia kręgowego
• Pobudzenie nocyceptorów w narządach wewnętrznych wzbudza
odczucie bólu na tych obszarach skóry których unerwienie tworzy
połączenia w obrębie tego samego segmentu rdzenia
– Np. w zawale ból promieniuje do lewego barku i ramienia
• Ból prjekcyjny jest spowodowany stymulacją nerwu (np. nerw
łokciowy w bruździe łokciowej) odczucie bólu ulega projekcji do
obszaru unerwienie tego nerwu
– Bóle fantomowe amputowanej kończyny
– Neuralgia –ciągłą, nieprawidłowa stymulacja nerwy lub korzenia
tylnego prowadzi do przewlekłego bólu w zakresie unerwienia

Leczenie bólu
• Aktywacja receptorów bólu może zostać zahamowana poprzez
schłodzenie uszkodzonego obszaru ciała i inhibitory syntezy
prostaglandyn
• Przewodzenie bólu w nerwie obwodowym może zostać
zahamowane poprzez jego schłodzenie i przez blokery kanałów Na+
(anestetyki miejscowe)
• Transmisja poprzez wzgórze może zostać zahamowana przez środki
znieczulenia ogólnego i alkohol
• Próbuje się przerwać przewodzenie bólu przez chirurgiczne
przecięcie nerwu
• Elektroakupunktura i przezskórna stymulacja nerwu oddziałują
poprzez aktywację zstępujących dróg hamujących
• Receptory opioidowe są stymulowane przez morfinę i jej pochodne
• Endogenne mechanizmy hamujące ból mogą zostać uruchomione
dzięki psychoterapii.

Padaczka – rodzaje napadów
• Napad padaczkowy jest wyzwalany przez spontanicznym,
zsynchronizowanym pobudzeniem dużej liczby neuronów, co
skutkuje miejscową lub uogólnioną aktywacją czynności
– ruchowych (drgawki)
– czuciowych (wrażenia sensoryczne)
– autonomicznych (np. ślinienie)
– złożonych (poznawczych, emocjonalnych)
• Napad częściowy – napad padaczki występujący lokalnie (np. lewy
zakręt przedśrodkowy w obszarze kontrolującym prawą stopę)
• Napad jacksonowski – napad rozprzestrzenia się z obszaru lokalnego
na cały zakręt przedśrodkowy (kloniczny skurcz prawej nogi
rozprzestrzenia się na całą prawą połowę ciała); nie zawsze
związany z utratą przytomności
• Napad częściowy z wtórnym uogólnieniem – napad przenosi się na
drugą stronę ciała, pacjent traci przytomność
• Napady pierwotnie uogólnione – zawsze związane z utratą
przytomności
• Niektóre napady mogą prowadzić jedynie do utraty przytomności

Padaczka
•
Zjawisko wyzwalające – napadowa depolaryzacja pojedynczych neuronów
•
Aktywacja kanałów Ca2+ napływający Ca2+ nieswoiste kanały kationowe masywna
depolaryzacja otwarcie kanałów K+ i Cl- aktywowanych Ca2+ napad padaczkowy ma
miejsce, gdy dostateczna liczba neuronów została pobudzona
•
Przyczyny lub czynniki sprzyjające padaczce:
– Zaburzenia genetyczne (kanały K+ i inne)
– Zaburzenia rozwojowe mózgu
– Uraz mózgu (blizna glejowa)
– Guz
– Krwawienie
– Ropnie
– Zatrucie (np. alkohol)
– Zapalenie
– Gorączka
– Obrzęk lub obkurczenie komórek
– Hipoglikemia
– Hipomagnezemia
– Hipokalcemia
– Brak snu
– Niedokrwienie lub hipoksja
– Powtarzające się bodźce (np. błyskające światło)
– Hiperwentylacja hipokapnia skurcz naczyń mózgowych niedotlenienie mózgu
– Wyższe występowanie napadów padaczkowych u kobiet w ciąży

Padaczka
Mechanizmy komórkowe rozprzestrzeniania się pobudzenia:
•
Dendryty komórek piramidowych zawierają kanały Ca2+ sterowane potencjałem otwierające się
podczas depolaryzacji
•
Przy uszkodzeniu neuronów większa ekspresja kanałów Ca2+ hamowanych przez Mg2+;
hipomagnezemia sprzyja aktywności tych kanałów
•
Zwiększone zewnątrzkomórkowe stężenie K+ zmniejsza wypływ K+ przez kanały K+ wpływ
depolaryzujący sprzyja aktywacji kanałów Ca2+
•
Dendryty komórek piramidowych są depolaryzowane przez glutaminian z synaps pobudzających
glutaminian działa na kanał kationowy nieprzepuszczalny dla Ca2+ (kanał AMPA) i na przepuszczalny
dla Ca2+ (kanał NMDA – normalnie blokowany przez Mg2+) depolaryzacja wyzwalana aktywacją
kanału AMPA znosi blokadę przez Mg2+ (współdziałanie dwóch kanałów) niedobór Mg2+ i
depolaryzacja sprzyjają aktywacji kanału NMDA
•
Potencjał błonowy neuronów jest normalnie podtrzymywany przez kanały K+; warunkiem do tego
jest prawidłowy gradient K+ poprzez błonę komórkową; gradient ten jest utrzymywany przez
Na+/K+-ATPazę niedobór energii (z powodu niedoboru tlenu lub hipoglikemii) upośledza Na+/K+-
ATPazę sprzyja depolaryzacji komórek
•
Prawidłowo depolaryzacja jest zmniejszana przez neurony hamujące, które aktywują kanały K+ lub
Cl- poprzez GABA; jest on tworzony przez dekarboksylazę glutaminianową, enzym wymagający
pirydoksyny (wit. B6) jako kofaktora niedobór wit. B6 albo zmniejszone powinowactwo enzymu
do wit. B6 (defekt genetyczny) sprzyja występowaniu padaczki
•
Hiperpolaryzacja neuronów wzgórza może zwiększać gotowość kanałów Ca2+ typu T do aktywacji,
sprzyjając występowaniu napadów nieświadomości

Choroba Alzheimera
• Najczęstsza przyczyna otępienia w podeszłym wieku (ok. 70%), której
sprzyja predyspozycja genetyczna
• Choroba nie jest genetycznie jednorodna
• W rodzinach z chorobą Alzheimera znaleziono defekty na chromosomach
1, 12, 14, 19 lub 21
• Uszkodzony gen na chromosomie 19 koduje apolipoproteinę E (ApoE4)
• Odpowiedzialny gen na chromosomie 21 dla białka (prekursora β-
amyloidu), które może zostać rozłożone do małych peptydów amyloidu
peptydy gromadzą się w fibryle białkowe o długości 7-10 nm fibryle
amyloidu mogą tworzyć agregaty (o rozmiarze 10 do kilkuset um – płytki
starcze) płyki zawierają zniszczone dendryty i aksony z nieprawidłowymi
neurofibrylami wewnątrzkomórkowymi – nietypowe elementy
cytoszkieletu, co poprzedza śmierć neuronów
• Pewne mutacje genu prekursora β-amyloidu sprzyjają tworzeniu płytek
starczych
• Toksyny, które mogą przenikać do mózgu poprzez nerwy węchowe mogą
wywołać chorobę
• Depozyty amyloidu występują również w trisomii 21 (zespole Downa) co
prowadzi do otępienia

Choroba Alzheimera
• Fibryle β-amyloidu mogą reagować z receptorami na powierzchni
komórek
– Receptory dla produktów zaawansowanej glikacji (RAGE)
– Receptor zmiatacza wolnych rodników (RA)
• Tworzenie rodników tlenowych depolaryzacja błony komórkowej
zwiększenie wewnątrzkomórkowego stężenia Ca2+ rodniki
tlenowe i Ca2+ promują śmierć komórki
• W komórkach mikrogleju aktywacja RAGE i RA pobudza tworzenie i
uwalnianie NO, prostaglandyn, cytokin, TNF-α, TGF-β1, b-FGF
zapalenie uszkadzające neurony
• Zwiększone stężenie substancji osmotycznej, inozytolu powoduje
zaburzenie regulacki objętości komórek
• Śmierć neuronów jest przyspieszana brakiem NGF lub receptorów
dla NGF i może zostać spowolniona przez NGF

Choroba Alzheimera
•
Neurony cholinergiczne jądra Meynerta, w hipokampie i w korze śródwęchowej są
szczególnie dotknięte śmiercią komórkową; śmierć komórek zachodzi również w
innych obszarach: płatach czołowych, przedniej cz. płatów skroniowych, płatach
ciemieniowych, korze węchowej, podwzgórzu, miejscu sinawym, jądrach szwu
•
Śmierci neuronów towarzyszy zmniejszona produkcja i wydzielanie
neurotransmiterów
– Acetylocholina
– Acetylotransferaza cholinowa (enzym uczestniczący w syntezie acetylocholiny)
– Inne neurotransmitery (noradrenalina, serotonina, somatotropina, neuropeptyd Y, substancja
P, kortykoliberyna)
•
Utrata funkcji mózgowych
– Choroba przebiega podstępnie – niewielkie deficyty pamięci, zaniedbanie wyglądu i higieny,
okresy splątania, podejmowanie błędnych decyzji
– Progresja – niepamięć wsteczna, pogorszenie pamięci odległej, pamięci proceduralnej
– Uszkodzenia w układzie limbicznym – niepokój i letarg
– Zaburzenia ruchowe (zaburzenia mowy, nieprawidłowe napięcie mięśni, ataksja, hiperkinezja,
mioklonie)

Depresja
• Występowanie rodzinne
• Depresja przedzielona fazami manii (choroba dwubiegunowa) albo sama
depresja (choroba jednobiegunowa)
• Zmniejszona dostępność noradrenaliny i serotoniny w mózgu
• Noradrenalina jest tworzona w neuronach miejsca sinawego o pokrywy
aksony z pokrywy łączą się z podwzgórzem, przednią częścią przysadki
mózgowej, pniem mózgu i rdzeniem kręgowym
aksony miejsca sinawego dochodzą do rdzenia, podwzgórza, wzgórza,
układu limbicznego i kory
• Wydzielanie i oddziaływanie noradrenaliny w zakończeniu nerwowym
może zostać zmniejszone poprzez:
– Synteza noradrenaliny z tyrozyny poprzez DOPA może zostać zmniejszona przez
inhibitory enzymów (np. metyltyrozynę)
– Wychwyt noradrenaliny do magazynów presynaptycznych poże zostać
zahamowany (np. rezerpina)
– Noradrenalina może zostać wyparta z receptorów (np. fenoksybenzamina,
fentolamina)

Depresja
Stężenie noradrenaliny w synapsie może się zwiększyć
• Inhibitory monoaminooksydazy A (MAO-A) specyficznej dla
noradrenaliny i serotoniny (np. trancylpromina,
moklobemid) mogą spowolnić ich rozkład w zakończeniu
presynaptycznym
• Substancje hamujące metylotransferazę katecholową
(COMT; np. tropolon) spowalniają rozkład noradrenaliny
• Pochodne amfetaminy zwiększają stężenie synaptyczne
noradrenaliny, dopaminy i serotoniny poprzez hamowanie
ich transportu
• Dezypramina hamuje wychwyt zwrotny noradrenaliny
• Receptory mogą być pobudzane przez agonistów (np.
klonidyna)

Depresja
• Serotonina (5-hydroksytryptamina [5-HT]) jest
tworzona w neuronach jąder szwu, które dochodzą do
rdzenia kręgowego, móżdżku, wzgórza, podwrzgórza,
jąder podstawy, ukladu limbicznego i kory mózgu
• Zmniejszona dostępność i działanie serotoniny sprzyja
rozwojowi depresji
– Hamowanie syntezy z tryptofanu (np. chlorofenyloalanina)
– Hamowanie wychwytu w magazynach presynaptycznych
(np. rezerpina)
– Z powodu zwiększongo zużycia serotoniny poprzez
tworzenie nieaktywnj melatoniny (w okresie ciemności, w
szyszynce)

Depresja
Efekt antydepresyjny obserwuje się, gdy zwiększa się działanie serotoniny lub
pobudzane są receptory serotoninergiczne
• Dostępność tryptofanu zwiększa się przy podaży glukozy glukoza
zwiększa wydzielanie insuliny antyproteolityczne i anaboliczne działanie
insuliny zmniejszenie stężenia aminokwasów we krwi niektóre
aminokwasy współzawodniczą z tryptofanem w transporcie przez barierę
krew-mózg brak hamowania zwiększy przyswajanie tryptofanu przez
komórki mózgowe
• Trójcykliczne leki antydepresyjne (np. imipramina, amitryptylina) hamują
wychwyt serotoniny przez magazyny presynaptyczne
• Inhibitory MAO-A zmniejszają rozkład serotoniny
• Ekspozycja na światło hamuje konwersję serotoniny do melatoniny
• Agoniści receptorów serotoninergicznych (np. LSD) mogą je bezpośrednio
pobudzać
• Lit prawdopodobnie wywiera efekt antydepresyjnypoprzez wpływ na
przekaźnictwo wewnątrzkomórkowe

Schizofrenia
Występowanie rodzinne
• Objawy wytwórcze
– Urojenia
– Omamy
– Zachowanie nieakceptowane społecznie
• Objawy ubytkowe
– Brak motywacji
– Spłycenie lub zanik emocji
U niektórych pacjentów dominują objawy pozytywne
(typ I), u innych negatywne (typ II).

Schizofrenia
• Zmniejszony przepływ krwi i zużycie glukozy głównie w
korze przeczołowej
• W typie II zmniejszenie liczby neuronów
• Nieprawidłowa migracja neuronów podczas rozwoju
mózgu
• Atrofia dendrytów komórek piramidowych w korze
przedczołowej i zakręcie obręczy – dendryty te
zawierają synapsy glutaminergiczne zaburzenie
transmisji glutaminergicznej
• Tworzenie GABA i liczba neuronów GABAergicznych
jest zmniejszona zmniejszone hamowanie komórek
piramidowych

Schizofrenia
Szczególna rola dopaminy:
• Nadmierana dostępność dopaminy lub jej
agonistów może prowadzić do objawów
schizofrenii
• Inhibitory receptora D2 skutecznie leczą
objawy schizofrenii
• Zmniejszenie receptorów D2 stwierdzono w
korze przedczołowej, co koreluje z
negatywnymi objawami schizofrenii

Schizofrenia
Niektóre sybstancja zwiększają wydzielanie
dopaminy
Leki przeciwparkinsonowskie:
• L-dopa
• Inhibitory MAO
Kokaina pobudza uwalnianie dopaminy w
szczelinie synaptycznej
Amfetamina hamuje wychwyt dopaminy w
zakończeniu nerwowym

Schizofrenia
Leki antydopaminergiczne zmniejszają nasilenie objawów:
• Niektóre substancje (np. fenotiazyny, haloperidol)
wypierają dopaminę z jej receptorów
• Rezerpina zmniejsza uwalnianie dopaminy do szczeliny
synaptycznej
• Długotrwałe stosowanie antagonistów dopaminy może
prowadzić do „późnych dyskinezji” na skutek ich
działania na prążkowie
• Możliwe, że serotonina również odgrywa rolę w
wytwarzaniu objawów schizofrenii – nadmierne
działąnie serotoniny może wywołać omamy, wiele
leków przeciwpsychotycznych blokuje receptor 5-HT2

Zespół Hornera
• Opadanie powieki
• Zwężenie źrenicy
• Brak potliwości
• Przekrwienie zajętej części
twarzy
• Brak pigmetacji tęczówki (w
postaci wrodzonej)
Skutek przerwania drogi
współczulnej biegnącej od
podwzgórza do oka
• Uszkodzenie ośrodkowe
– Niedokrwienie pnia mózgu
– Jamistość rdzenia
– Guz mózgu
• Uszkodzenia obwodowe
– Guz Pancoasta
– Adenopatia szyjna
– Obrażenia szyi i czaszki
– Rozwarstwienie aorty lub
tętnicy szyjnej
– Tętnik części piersiowej aorty

Zespół wieloukładowy
Trzy składowe:
• Zanik oliwkowo-mostowo-
móżdżkowy
• Zwyrodnienie prążkowiowo-
czarne
• Zespół Shy’a i Dragera
Mechanizm: gromadzenie alfa-
synukleiny w komórkach
oligedendrogleju
Objawy:
• Niedociśnienie
ortostatyczne
• Zatrzymanie moczu
• Zaparcia
• Ataksja
• Sztywność
• Niestabilność postawy

PATOFIZJOLOGIA BÓLU

Nocyceptory
• Wolne zakończenia nerwowe cienkich włókien
z osłonką mielinową (Aδ) lub bez osłonki (C)
• Reagują wybiórczo na bodźce uszkadzające:
– Mechaniczne
– Chemiczne
– Termiczne
• Bradykinina i histamina uwalniane z
uszkodzonych komórek pobudzają
nocyceptory
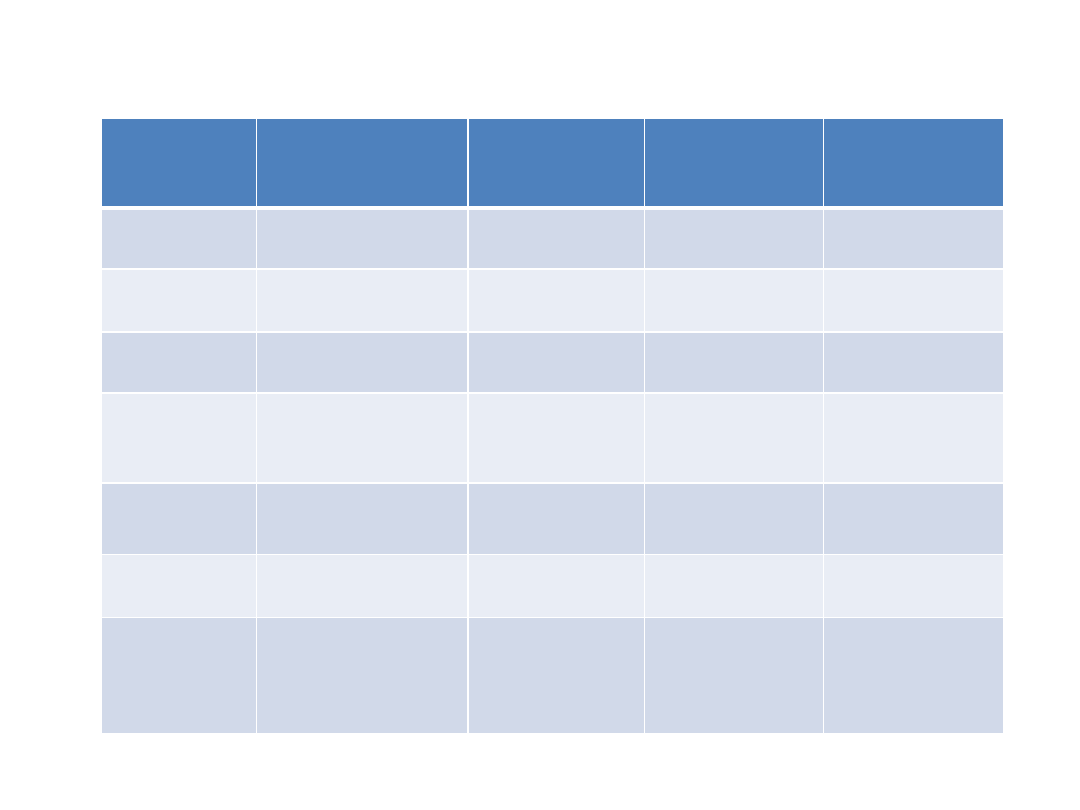
Endogenne czynniki aktywujące nocyceptory
Substancja
Źródło
Enzym
zaangażowany w
produkcję
Wpływ na
pierwszorzędowy
neuron czuciowy
Działanie
farmakologiczne
Potas
Uszkodzone komórki
Aktywacja
Serotonina
Płytki
Hydroksylaza
tryptofanu
Aktywacja
Bradykinina
Kininogen osoczowy
Kalikreina
Aktywacja
Histamina
Komórki tuczne
Aktywacja
Antagoniści
receptora H1
(difenhydramina)
Prostaglandyny Kwas arachidonowy /
uszkodzone komórki
Cyklooksygenaza
Uwrażliwienie
NLPZ (aspiryna,
ibuprofen)
Leukotrieny
Kwas arachidonowy /
uszkodzone komórki
Lipooksygenaza
Uwrażliwienie
Substancja P
Pierwszorzędowy
neuron czuciowy
Uwrażliwienie
Agoniści
receptorów
opioidowych
(morfina)

Rodzaje czucia bólu
• Ból szybki (początkowy)
– Wyraźne, dobrze zlokalizowane odczucie
– Aktywacja zakończeń bólowych włókiem Aδ
• Ból powolny (późny)
– Nieprecyzyjnie zlokalizowany
– Tępe palące wrażenie
– Aktywacja zakończeń bólowych włókiem C

Drogi docierania bólu do ośrodków
świadomości
• Czucie somatyczne
– Ból szybki
– Ból powolny
• Czucie trzewne
– Ból odniesiony
• Ból projekcyjny

Czucie somatyczne
• Ból szybki
– Potencjały przewodzone przez szybkie włókna
drogą przez szlak rdzeniowo-wzgórzowy
– Małe pola recepcyjne bólu
– Włokna czuciowe mają reprezentację
topograficzną w korze
– Zdolność włókien do kodowania lokalizacji bodźca
wywołującego ból

Czucie somatyczne
• Ból powolny
– Biegnie drogą rozproszoną przez szlak rdzeniowo-
siateczkowo-wzgórzowy
– Kolaterale przechodzą przez twór siatkowaty
pobudzajc reakcję emocjonalną związaną z
odczuwaniem bólu
– Odpowiedzielny na intensywne, nieprzyjemne
odczuwanie powolnego bólu

Czucie trzewne
• Ból odniesiony – zaczyna się w narządach
wewnętrznych i jest odnoszony do miejsc na
skórze
– Włókna bólowe somatyczne i trzewne przebiegają
wspólną drogą do mózgu
– Rozpoznawany jako pochodzący ze skóry (np. ból
wieńcowy)
– Skóra jest reprezentowana topograficznie w korze,
a narządy wewnętrzne nie

Ból projekcyjny
• Wynika z bezpośredniego podrażnienia
włókien na drodze nerwowej
• Mechanizm oznaczonej linii – kodowanie
lokalizacji bólu
• Drażnienie w dowolnym miejscu drogi bólowej
powoduje taką samą percepcję (np. uderzenie
w łokieć, bóle międzyżebrowe)
• Bóle fantomowe – po amputacji kończyn

Odruchy
• Ból szybki
– Wywołuje odruch cofnięcia
– Wywołuje reakcję współczulną
• Wzrost ciśnienia krwi
• Mobilizacja zapasów energetycznych
• Ból powolny
– Nudności
– Obfite poty
– Spadek ciśnienia krwi
– Spadek napięcia mięśni

Ośrodkowy mechanizm czucia bólu
• Czucie bólu przesyłane przez przednio-boczny
kwadrant rdzenia
• Ból przewlekły – odczuwany długo po
zniknięciu bodźca i wyleczeniu uszkodzenia
– Prawdopodobnie w wyniku spontanicznej
aktywności ośrodków bólowych OUN
• Krążenie impulsów po zamkniętych obwodach
• Nadwrażliwość odnerwieniowa – wzrost wrażliwości na
krążący neuroprzekaźnik, kiedy normalne wejście
synaptyczne jest usunięte z neuronu

Znieczulenie podpajęczynówkowe
•
Podanie miejscowego środka znieczulającego do płynu mózgowo-rdzeniowego
otaczającego rdzeń kręgowy
•
Wykonywane poniżej kręgu L2 (nie ma tu rdzenia kręgowego)
•
Stosowane leki:
– Lignokaina – krótkie znieczulenie
– Bupiwakaina – średnia długość znieczulenia
– Tetrakaina – długi czas trwania
•
Mechanizm – blokada kanałów sodowych błony drugorzędowego neuronu drogi
czuciowej brak napływu sodu do wnętrza neuronu neuron nie ulega
depolaryzacji i potencjałowi czynnościowemu ból nie może być przewodzony do
wyższych struktur OUN
•
Miejsce wiązania – podjednostka kanału sodowego w pobliżu wewnętrznej
powierzchni błony komórkowej – lek musi wejść do wnętrza neuronu
•
Drugorzędowe neurony drogi czuciowej są najbardziej podatne na znieczulenie
podpajęczynówkowe – mają mały rozmiar, niezmielinizowane; neurony ruchowe
alfa podatne tylko w wyższych stężeniach – większy rozmiar i zmielinizowane

Znieczulenie nadtwardówkowe
• Podanie anestetyku miejscowego do przestrzeni nadtwardówkowej
• Położona poza kanałem rdzenia kręgowego na jego grzbietowej
powierzchni, zawiera tłuszcz i jest silnie unaczyniona
• Może zostać bezpiecznie przeprowadzona na każdym poziomie
rdzenia
• Miejsce działania – korzenie nerwów rdzeniowych
• Wybór leku
– Bupiwakaina – długie znieczulenie
– Lignokaina – pośredni czas trwana
– Chloroprokaina – krótkie znieczulenie
• Środki wstrzykiwane do przestrzeni nadtwardówkowej mogą
przeniknąć do krwi poprzez bogate sploty żylne w tym obszarze
• Podczas porodu mogą one przeniknąć przez łożysko do krążenia
płodowego i wywrzeć depresyjny wpływ na noworodka
Wyszukiwarka
Podobne podstrony:
Anatomia i patofizjologia układu nerwowego
patofizjologia układu nerwowego
Patofizjologia układu nerwowego 2
Patofizjologia ukladu nerwowego
dp 651 wykl miazdzyca-bez rycin wl 2013 [tryb zgodnosci](czyli 2014.03.04)
Choroby układu nerwowego ppt
patofizjologia układu odpornościowego
SPECYFIKA CHORÓB UKŁADU NERWOWEGO OKRESU ROZWOJOWEGO
Patofizjologia układu dokrewnego
Zakażenia wirusowe układu nerwowego psów i kotów
Patofizjologia układu pokarmowego
Choroby układu nerwowego
2 Patofizjologia układu oddechowegoid 19598 ppt
BUDOWA I FUNKCJA UKŁADU NERWOWEGO, weterynaria, Anatomia
Ocena sprawności funkcjonalnej w chorobach układu nerwowego, Neurologia
Biochemia ukladu nerwowego, Biologia, Biochemia
więcej podobnych podstron