
GENETYKA
NOWOTWORÓW
Lek. Przemysław Łodej
Zakład Genetyki Klinicznej
Uniwersytetu Medycznego w
Lublinie

„Rak zaczyna powstawać wówczas,
gdy komórka wyłamuje się spod
kontroli mechanizmów decydujących
o jej podziałach i lokalizacji”
Robert A. Weinberg

Istota nowotworzenia
W normalnych warunkach w organizmie istnieje
ścisła równowaga pomiędzy tempem podziałów
komórkowych a utratą komórek
W komórkach nowotworowych dochodzi do
zachwiania tej równowagi
Mniejsza liczba komórek ginie niż przybywa w
wyniku podziałów mitotycznych
Wzmożona
proliferacja
skutkuje
dużą
niestabilnością genetyczną , utratą zdolności do
różnicowania, nabyciem zdolności do naciekania
(migracji), oraz kolonizacji (inwazji) obszarów
normalnie zajmowanych przez inne rodzaje komórek

CYKL KOMÓRKOWY

Cykl komórkowy
Cykl komórkowy (cykl podziału komórki)
to
seria
zdarzeń
zachodzących
w
komórce
eukariotycznej, prowadzących do jej podziału.
Ogólnie zdarzenia te można podzielić na 2
okresy:
interfazę -
w trakcie której komórka wzrasta i
gromadzi składniki odżywcze niezbędne do mitozy i
podziału swojego materiału genetycznego (DNA)
fazę mitotyczną (M) -
podczas której komórka
dzieli się na 2 oddzielne komórki, zwane komórkami
potomnymi.
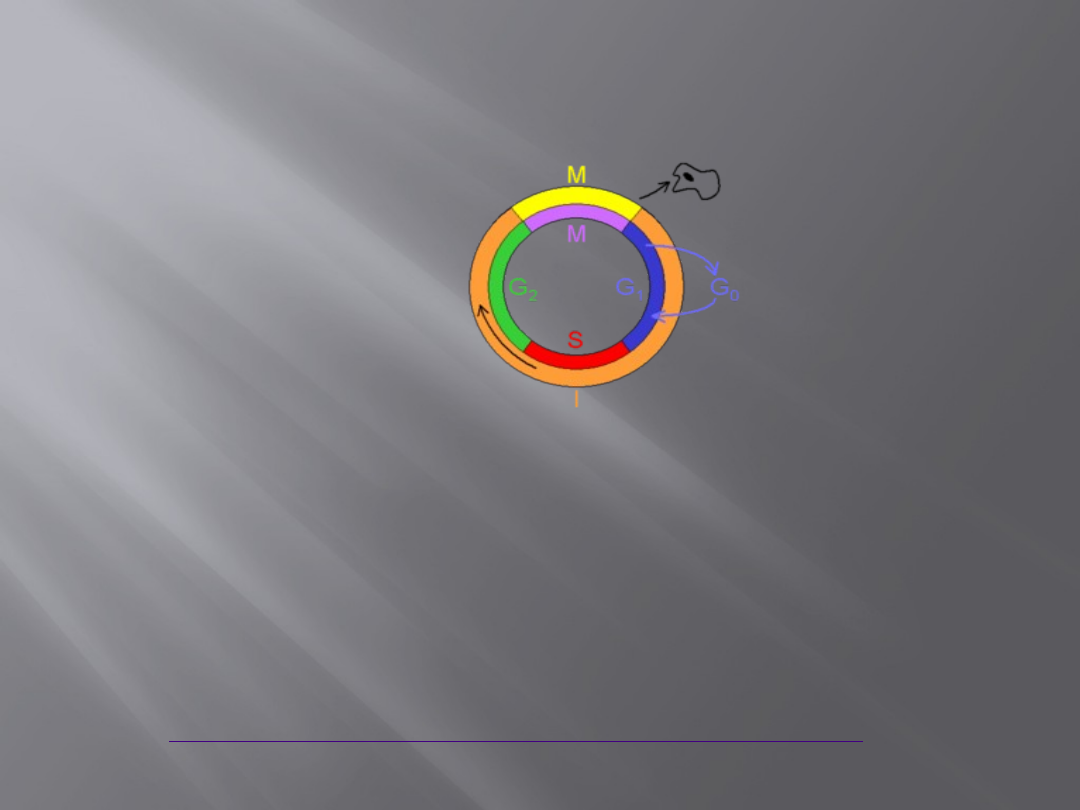
Cykl komórkowy
Etapy cyklu komórkowego:
Interfaza
Mitoza
lub
Mejoza
Diagram nie odzwierciedla stosunków czasu
trwania poszczególnych faz.
http://www.naukowy.pl/encyklopedia/Cykl_kom%C3%B3rkowy
, zmieniony

Cykl komórkowy
Znaczenie cyklu komórkowego:
umożliwia jednokomórkowej zygocie rozwinąć się w dojrzały
organizm
dzięki niemu skóra, włosy, komórki krwi i niektóre inne
narządy wewnętrzne mają możliwość regeneracji
Fazy cyklu komórkowego:
Cykl komórkowy składa się z 4 oddzielnych faz: fazy G
1
, fazy
S, fazy G
2
(zwanych łącznie interfazą) oraz fazy M.
Faza M składa się z kolei z 2 ściśle połączonych ze sobą
procesów: mitozy, w czasie której chromosomy komórki
zostają rozdzielone pomiędzy 2 przyszłe komórki potomne i
cytokinezy, w czasie której dochodzi do podziału cytoplazmy z
uformowaniem odrębnych komórek.

Cykl komórkowy
Aktywacja kolejnej fazy jest zależna od właściwego
postępu i ukończenia poprzedzającej ją fazy.
Komórka, która czasowo i w sposób odwracalny
zatrzymała swoje podziały, to komórka w fazie
spoczynkowej, zwanej fazą G
0
.
FAZA M
faza M jest dość krótka, trwa około 1 godzinę
następuje w niej podział komórki (mitoza,
ewentualnie mejoza)
obejmuje podział jądra (kariokinezę) i podział
cytoplazmy (cytokinezę)

Cykl komórkowy
INTERFAZA
Po fazie M każda z komórek potomnych zaczyna interfazę
nowego cyklu komórkowego.
FAZA G
1
(G – ang. gap – przerwa)
pierwsza faza interfazy
zaczyna się od końca fazy M poprzedniego cyklu i trwa
do początku syntezy DNA (fazy S)
podjęcie na nowo zwolnionych w fazie M procesów
biosyntezy w komórce
synteza różnych enzymów potrzebnych głównie do
replikacji DNA w fazie S
czas trwania fazy G
1
jest znacznie zróżnicowany, trwa od
kilku do kilkunastu godzin

Cykl komórkowy
FAZA S (ang. synthesis – synteza)
rozpoczyna się wraz z rozpoczęciem syntezy DNA
po jej zakończeniu wszystkie chromosomy są
zreplikowane, tzn. każdy chromosom ma 2
siostrzane chromatydy
ilość DNA w komórce zostaje podwojona, mimo że
ploidalność komórki pozostaje ta sama
tempo syntezy RNA i białek w tej fazie jest niskie
produkcja histonów
czas trwania tej fazy jest zazwyczaj względnie
stały w komórkach tego samego gatunku, u
ssaków trwa około 7 godzin

Cykl komórkowy
FAZA G
2
trwa od zakończenia replikacji DNA do
rozpoczęcia mitozy
ponownie znacząco zwiększa się synteza białek,
głównie tubuliny, celem wytworzenia mikrotubul
–
składnika
wrzeciona
podziałowego
niezbędnego w procesie mitozy
FAZA G
0
faza postmitotyczna, dotyczy komórek w fazie
spoczynku, jak i komórek starzejących się

Cykl komórkowy
FAZA G
0
niedzielące się komórki u organizmów eukariotycznych
generalnie wchodzą w fazę G
0
z fazy G
1
i mogą pozostawać
w tej fazie spoczynkowej przez długi okres, możliwe że i na
zawsze (np. neurony)
starzenie się komórki jest stanem, który występuje w
odpowiedzi na uszkodzenie lub zniszczenie DNA, które
mogłoby uczynić potomstwo komórki niezdolnym do życia.
Jest
to
często
biochemiczna
alternatywa
dla
samozniszczenia tak uszkodzonej komórki przez apoptozę
niektóre typy komórek w dojrzałym organizmie, np. komórki
miąższowe wątroby i nerek, wchodzą w fazę G
0
w sposób na
wpół trwały i mogą zostać pobudzone do ponownych
podziałów w bardzo szczególnych okolicznościach
inne komórki, np. komórki nabłonkowe, kontynuują dzielenie
się przez okres całego życia organizmu

Regulacja cyklu komórkowego
Nadzór nad prawidłowym przebiegiem cyklu jest
wynikiem istnienia wielu punktów kontrolnych (tzw.
checkpoints).
Szczególne znaczenie mają 2 punkty – noszą one
nazwę punktów przejścia (punktów restrykcyjnych).
Znajdują się one na granicy faz G1 i S oraz G2 i M.
Przebieg
cyklu
komórkowego
może
zostać
zatrzymany zarówno w punktach kontrolnych, jak i
restrykcyjnych. Przejście przez punkt kontrolny nie
oznacza wejścia w kolejną fazę cyklu, ale przejście
przez punkt restrykcyjny oznacza bezwzględne
rozpoczęcie kolejnej fazy cyklu komórkowego.
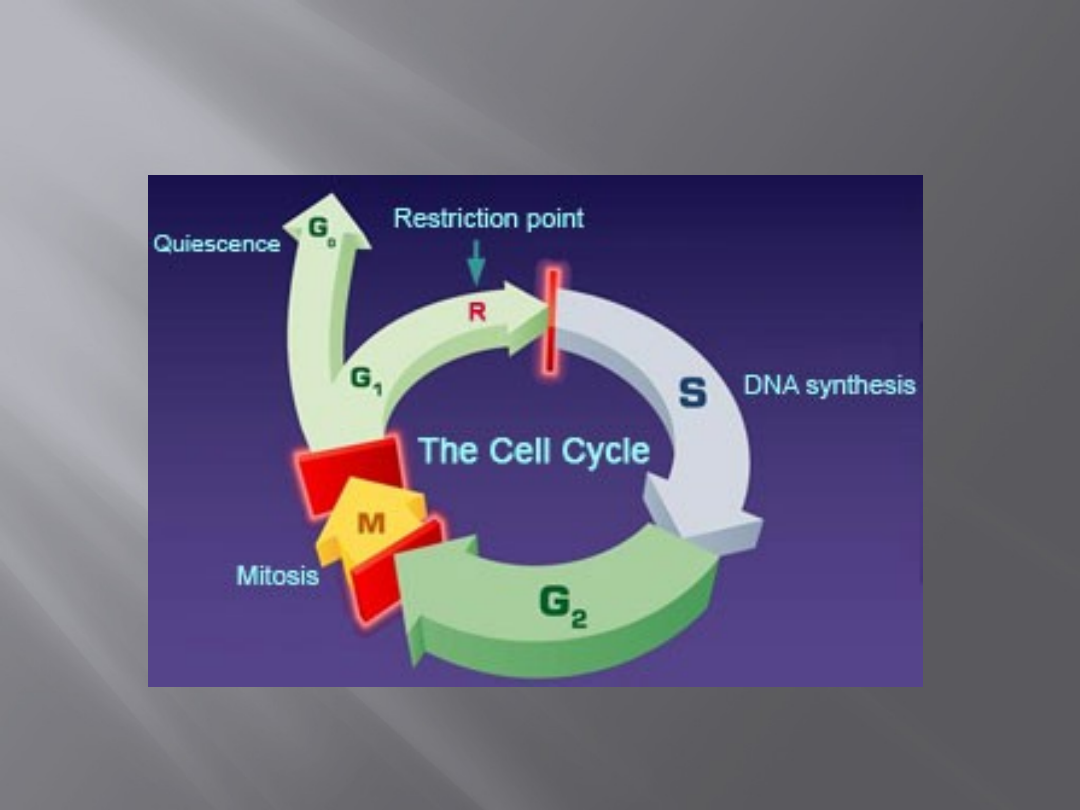
Regulacja cyklu komórkowego
http://sydney.edu.au/wmi/cellcycle/cellcycle.htm

Regulacja cyklu komórkowego
Wykrycie nieprawidłowości powoduje zatrzymanie cyklu
w danej fazie i próbę naprawy. Jeśli defekty okażą się
zbyt poważne, dochodzi do uruchomienia procesów
apoptozy. Jednak jeśli z jakiegoś powodu nie nastąpi
uruchomienie
procesów
programowanej
śmierci,
komórka wejdzie w kolejną fazę cyklu komórkowego z
uszkodzeniami. Grozi to pojawieniem się mutacji w
materiale genetycznym i jest zwykle pierwszym krokiem
w kierunku powstania komórki nowotworowej.
W regulacji cyklu komórkowego istotną rolę odgrywają
produkty białkowe dwóch genów supresorowych: TP53 i
RB1. Białka te kontrolują bezpośrednio oba punkty
restrykcyjne
cyklu
komórkowego.
Są
więc
odpowiedzialne za przejście komórki do fazy syntezy
DNA i fazy podziału.

Białko TP53
Gen TP53 (tumor protein 53) jest zlokalizowany w
chromosomie 17p13.1.
Składa się z 11 eksonów, pierwszy ekson jest niekodujący.
Większość mutacji genu występuje w obrębie tzw.
regionów o wysokiej homologii międzygatunkowej HCD
(jest ich 5). Są to głównie mutacje punktowe lub delecje
fragmentu genu. Występują głównie w eksonach 5-8. W
obszarach HCD można wyróżnić ponadto 4-5 tzw.
gorących miejsc (hot spot region – HSR), w których
najczęściej dochodzi do mutacji.
Gen TP53 jest najczęściej zmienionym genem w
nowotworach człowieka.
Produktem tego genu jest białko TP53 zbudowane z 393
aminokwasów.

Białko TP53
Białko TP53 jest czynnikiem transkrypcyjnym dla ponad
30 różnych genów, również dla hamujących cykl
komórkowy (hamuje kinazy CDK, aktywuje białko RB),
uczestniczących w systemach naprawy DNA (np. białko
GADD45), a także w procesach apoptozy (np. białko BAX –
indukuje apoptozę).
Biologicznie
aktywną
formą
białka
TP53
jest
ufosforylowany homotetramer i w tej postaci jest ono
głównym regulatorem cyklu komórkowego. Spełnia ono
rolę sensora stresu w komórce. Jest sensorem uszkodzeń
w DNA, dlatego nazywane jest „strażnikiem genomu”.
Regulatorem funkcji białka TP53 jest białko MDM2.
reguluje ono stężenie wolnego TP53, tworząc z nim
kompleks i tym samym uniemożliwiając oddziaływanie z
DNA, a więc aktywację TP53-zależnych genów.

Białko TP53
Mechanizm działania białka TP53 w punkcie restrykcyjnym G1/S: w
cytoplazmie rośnie ilość białka TP53, w przypadku uszkodzeń DNA
białko to zaczyna przedostawać się do jądra komórkowego, indukuje
ekspresję genu p21
Cip
, którego produkt jest inhibitorem kinaz, co
prowadzi do zatrzymania cyklu komórkowego. Jednocześnie białko
TP53 aktywuje geny systemów naprawy DNA. Jeśli uszkodzenia
zostaną naprawione, blok zostanie zwolniony i cykl komórkowy jest
wznawiany. W przypadku braku możliwości naprawy uszkodzeń
białko TP53 aktywuje geny apoptozy.
Mechanizm działania białka TP53 w punkcie restrykcyjnym G2/M: w
przypadku poreplikacyjnych uszkodzeń DNA stężenie białka TP53
rośnie. Hamuje to transkrypcję genu cykliny B, co skutkuje brakiem
możliwości tworzenia kompleksu kinaza CDC2/cyklina B. Dochodzi
do indukcji kinaz ATM i ATR, które aktywują kinazy z rodziny CHEK.
Represja transkrypcji genu cykliny B i brak defosforylacji kinazy
CDC2 prowadzą do zahamowania cyklu komórkowego.

Białko TP53
Skutki mutacji genu TP53:
Obniżenie poziomu prawidłowego białka TP53
prowadzi do przedwczesnego zwolnienia bloku
proliferacyjnego.
Rozpoczyna się runda replikacji DNA.
W przypadku niezakończenia procesu naprawy
uszkodzonego DNA w komórce pojawiają się
mutacje.
Utrata kontroli nad proliferacją komórki powoduje
gromadzenie
się
komórek
genetycznie
niestabilnych.
Efektem jest szybka selekcja stransformowanych
nowotworowo komórek.

Białko RB1
Należy do rodziny białek transportujących (białek
kieszeniowych) – wiążą inne białka za pomocą tzw.
kieszeni.
Gen RB1 ma wielkość około 180 kpz, zlokalizowany jest w
chromosomie 13q14. Składa się z 27 eksonów. W
genomie występuje w pojedynczej kopii. Koduje białko o
wielkości 928 aminokwasów.
Mutacje występują głównie w eksonach 13-17, 24 i 27.
Większość to mutacje nonsensowne i przesunięcia ramki
odczytu, prowadzące do przedwczesnej terminacji białka.
Białko RB1 reguluje przejście przez punkty restrykcyjne
G1/S i G2/M. Regulacja wynika ze zdolności do wiązania i
uwalniania
białek
będących
regulatorami
cyklu
komórkowego.

Białko RB1
Represja transkrypcji następuje po utworzeniu kompleksu
pRB/E2F/DP/HDAC (E2F – rodzina czynników transkrypcyjnych;
DP – białka stabilizujące; HDAC – rodzina deacetylaz
histonowych).
Związanie
HDAC
prowadzi
do
wzrostu
upakowania
chromatyny i czyni ją niedostępną dla czynników
transkrypcyjnych.
Głównym mechanizmem odpowiedzialnym za tworzenie i
destrukcję kompleksu jest proces fosforylacji białka RB, która
jest dokonywana przez kompleksy kinaza CDK/cyklina. W
fazie G1 jest to kompleks CDK4 lub 6/cyklina D, w późnej fazie
G1 kompleks CDK2/cyklina E, w fazie S CDK2/cyklina A.
Przy wejściu w fazę G1 białko RB zostaje częściowo
zdefosforylowane i w tej postaci ma ono zdolność do
tworzenia kompleksu z E2F i HDAC.

Białko RB1
Fosforylacja końca C RB1 pod koniec fazy G1
powoduje
uwalnianie
czynników
transkrypcyjnych E2F i HDAC, które aktywują
odpowiednie geny odpowiedzialne za zwolnienie
bloku G1/S i rozpoczęcie syntezy DNA.
Przez całą fazę S i G2 białko RB1 pozostaje w
pełni ufosforylowane.
Pod koniec fazy G2 aż do anafazy fazy M białko
RB1 ulega częściowej defosforylacji, co prowadzi
do tworzenia komleksu RB1/E2F/DP/HDAC. W tej
postaci kompleks utrzymuje się aż do fazy G1.
Cykl rozpoczyna się od nowa.

Białko RB1
Białko RB1 wykazuje zdolność do
wiązania się z wirusowymi onkogenami.
Stwierdzono tworzenie kompleksów z
białkiem E1A adenowirusów, antygenem
T wirusa SV40, białkiem E7 wirusa HPV
16.
Utworzenie takiego kompleksu hamuje
zdolność regulacyjną RB w wyniku
aktywacji procesów transkrypcyjnych.

Etapy karcynogenezy
Przekształcenie się komórki prawidłowej w nowotworową
nazywamy
transformacją
nowotworową
(karcynogenezą)
.
Etapy transformacji nowotworowej:
1.
Preinicjacja
– ekspozycja na karcynogeny fizyczne (np.
promieniowanie jonizujące), chemiczne, biologiczne
(głównie
wirusy,
toksyny
bakteryjne,
toksyny
pasożytów, zaburzenia hormonalne, np. nadmiar
estrogenów). Trwa całe życie. Podatność zależy od
osobniczej zmienności genetycznej, głównie od
polimorfizmu
genów
kodujących
systemy
detoksykacyjne (np. geny oporności wielolekowej –
MDR) oraz genów kodujących systemy naprawcze (jeśli
już dojdzie do uszkodzenia materiału genetycznego).

Etapy karcynogenezy
2.
Inicjacja:
Rozpoczyna się, gdy wystąpi pierwsza mutacja.
Kolejne
mutacje
mogą
być
następstwem
pierwszej, mogą też powstawać spontanicznie.
Ich gromadzeniu sprzyja ciągła ekspozycja na
kancerogeny.
Mutacje odpowiedzialne za przejście komórki
prawidłowej
w
nowotworową
tworzą
tor
mutacyjny
.
Nagromadzenie mutacji prowadzi do transformacji
nowotworowej.
Etap inicjacji trwa od kilku do 20-30 lat.

Etapy karcynogenezy
3.
Promocja:
Dochodzi
do
niej,
gdy
zawiodą
wszystkie
mechanizmy
zabezpieczające komórkę.
Wzrost aktywności mitotycznej.
Narastanie mutacji.
Zmiany strukturalne w chromosomach – translokacje, delecje,
duplikacje, inwersje. Możliwe zmiany liczby chromosomów –
aneuploidie.
Powstawanie subklonów komórek nowotworowych, często nie
wykazujących podobieństwa do komórek, z których się wywodzą.
Większość jest eliminowana na drodze presji selekcyjnej.
Powstaje ograniczony rozrost nowotworowy – nowotwór in situ
(
Carcinoma in situ
), złożony z kilku subklonów komórek i liczący 10
6
-
10
7
komórek. Otaczają one naczynie krwionośne, które dostarcza
substancji odżywczych.
Na tym etapie guz nie wytwarza jeszcze naczyń krwionośnych,
nabywa jednak zdolności do migracji.
Etap trwa zwykle poniżej kilku lat.

Etapy karcynogenezy
4.
Progresja:
Etap ten rozpoczyna się w momencie
inicjacji neoangiogenezy.
Narastają zmiany w genomie.
Trwa presja selekcyjna. Prowadzi ona do
powstania
klonów
komórek,
które
nabywają
zdolności
do
swobodnej
migracji i przemieszczania się w inne
regiony organizmu – zdolność do
przerzutowania.
Trwa od kilku miesięcy do kilku lat.

Onkogeny
W organizmie większość komórek znajduje się
w fazie spoczynkowej – w fazie G0 cyklu
komórkowego.
Komórki
poddane
działaniu
czynników
stymulujących je do wzrostu wchodzą w cykl
podziałowy.
Proliferacją steruje klasa genów zwanych
protoonkogenami
.
Dotychczas
opisano
ponad
500
protoonkogenów.
Białka
kodowane
przez
protoonkogeny
dzielimy na 3 grupy.

Onkogeny
Klasy białek kodowane przez protoonkogeny:
Białka regulatory cyklu komórkowego
– czynniki
wzrostu (np. PDGF, FGF), receptory dla tych
czynników,
kinazy
białkowe
tyrozyny
i
seryny/treoniny, białka wiążące GTP, czynniki
transkrypcyjne.
Białka uczestniczące w procesie apoptozy
– białka
błony mitochondrialnej i jądrowej (kodowane przez
geny
BCL-2
i
BAX),
białka
błonowe
zewnątrzkomórkowe i ich ligandy (kodowane przez
geny FAS/APO1 i FAS L).
Różne inne białka
, np. białka tworzące kanały
jonowe.

Onkogeny
Mutacje protoonkogenów prowadzą do powstania
onkogenu
.
Onkogeny to zmutowane/ zmienione protoonkogeny.
Mutacja protoonkogenu prowadzi do zaburzeń w transmisji
sygnałów wzrostu.
Najczęściej
zmutowanym
onkogenem
w
komórkach
nowotworowych jest
RAS
. Jest to grupa 3 genów (Ha-RAS, Ki-
RAS i N-RAS), każdy umiejscowiony jest w innym
chromosomie i koduje białko p21 o aktywności kinazy
białkowej. Białko to przenosi sygnał z błony komórkowej do
wnętrza komórki i zapoczątkowuje kaskadę fosforylacji.
Prowadzi to do aktywacji genów kodujących czynniki
transkrypcyjne, np. MYC, MYB, FOS czy JUN. Konsekwencją
jest aktywacja czynników transkrypcyjnych, które z kolei
uruchamiają geny, których produkty odpowiadają za proces
proliferacji i różnicowania komórek.

Onkogeny
Cechą charakterystyczną komórek nowotworowych
jest zdolność do dzielenia się bez ograniczeń.
Komórki nowotworowe są albo poddane stałej
stymulacji przez czynniki wzrostu albo utraciły
kontrolę nad procesem przekazywania sygnałów do
wzrostu.
Jest to efekt mutacji protoonkogenów. Mutacje te
prowadzą
do
niekontrolowanej
aktywacji
protoonkogenów.
Skutkiem jest stymulacja proliferacji, niezależnie od
sygnałów docierających do komórki z jej otoczenia.

Geny supresorowe
Komórki nowotworowe cechują się utratą zdolności do
rozpoznawania sygnałów antywzrostowych.
Produkty genów supresorowych mają za zadanie
powstrzymywać komórki przed proliferacją i utrzymywać
je w fazie spoczynkowej cyklu komórkowego, a tym
samym zapobiegać transformacji nowotworowej.
Opisano ponad 30 genów supresorowych.
W nowotworach obserwuje się zniesienie funkcji obu alleli
genu supresorowego. Jedna z kopii zostaje uszkodzona
np. w drodze mutacji punktowej, druga wskutek utraty
regionu genomu, w którym znajduje się dany gen
supresorowy – tzw. utrata heterozygotyczności (LOH).
Utrata aktywności supresorowej prawie zawsze prowadzi
do transformacji nowotworowej.

Geny supresorowe
Produkty genów supresorowych pełnią w prawidłowej
komórce podstawowe funkcje, np. są elementami
struktur komórkowych, odpowiadają za kontakty
międzykomórkowe,
są
inhibitorami
aktywacji
(fosforylacji) białek, kontorlują przebieg cyklu
komórkowego i różnicowania.
Do genów supresorowych należą m.in.:
TP53 (czynnik
transkrypcyjny, kontroler prawidłowej
proliferacji, „strażnik genomu”),
RB1 (czynnik transkrypcyjny, kontroler proliferacji),
BRCA1 (aktywator transkrypcji, element systemu naprawy
dwuniciowych pęknięć DNA, uczestniczy w remodelowaniu
chromatyny),
BRCA2
(aktywator
transkrypcji,
ma
aktywność
deacetylotransferazy histonów).

Geny stabilizacyjne
Kolejną cechą transformacji nowotworowej są zaburzenia w
systemach naprawy DNA.
Prawidłowa komórka dysponuje wieloma systemami
naprawy materiału genetycznego. Mutacja w takiej
komórce zdarza się co sekundę.
Geny stabilizacyjne (MMR – mismatch repair) stanowią
jeden z elementów systemu naprawy. Są to geny MSH2,
MSH3, MSH6, MLH1, PMS1 i PMS2.
Mutacja któregokolwiek z tych genów powoduje, że
zmieniony jego produkt nie rozpoznaje lub rozpoznaje w
ograniczonym zakresie źle sparowane zasady, co prowadzi
do narastania liczby mutacji rozproszonych po całym
genomie, także w onkogenach i genach supresorowych, a
nawet w kolejnych genach stabilizacyjnych.

Geny stabilizacyjne
Powoduje
to
lawinowe
narastanie
niestabilności genetycznej komórek i
sprzyja transformacji nowotworowej.
Mutacje dziedziczne genów układu MMR
(głównie MSH2 i MMLH1) odpowiadają za
rozwój dziedzicznego zespołu raka jelita
grubego niezwiązanego z polipowatością
(HNPCC). W zespole tym drugim co do
częstości
nowotworem
jest
rak
endometrium.

Nabywanie nieograniczonego
potencjału podziałowego
W prawidłowej komórce istnieje równowaga pomiędzy
czynnikami
wzrostu
(protoonkogenami)
i
czynnikami
antywzrostowymi (genami supresorowymi).
Skutkiem zaburzenia tej równowagi są zmiany w potencjale
podziałowym.
Prawidłowe
komórki
mają
ograniczony
potencjał
proliferacyjny, czyli mogą się podzielić pewną ilość razy,
ściśle określoną dla danej komórki, zwaną liczbą Hayflicka. Po
przekroczeniu tej liczby komórka wkracza na drogę apoptozy.
W prawidłowej komórce z każdym kolejnym podziałem
dochodzi do skracania telomerów (polimerazy DNA nie mają
zdolności syntezy opóźnionej nici DNA do samego końca nici
prowadzącej – matrycy). Telomery to krótkie sekwencje
obecne na końcu nici DNA, powtarzające się wiele tysięcy
razy.

Nabywanie nieograniczonego
potencjału podziałowego
Skracanie się telomerów przy każdym cyklu komórkowym
jest rodzajem zegara biologicznego komórki.
Proces biologicznego starzenia się komórki jest związany
z osiągnięciem pewnej krytycznej długości telomerów.
Sugeruje się, że krytyczna jest długość telomeru w
chromosomie 17.
Osiągnięcie krytycznej długości telomerów jest sygnałem
do uruchomienia genów odpowiedzialnych za śmierć
komórki.
Zmniejszenie długości telomerów poniżej 1,5 kpz
prowadzi do destabilizacji struktury chromosomów.
Prowadzi to do pojawienia się aberracji chromosomowych.

Nabywanie nieograniczonego
potencjału podziałowego
Zmiany wywołane utratą telomerów mogą być
zniesione przez onkogenne białka wirusowe, np.
antygen T wirusa SV40, białko E1A adenowirusa,
białka E6 i E7 brodawczaka ludzkiego.
Pojawienie się aberracji chromosomowych prowadzi
do
uruchomienia
mechanizmu
obronnego
–
mianowicie niektóre chromosomy dzięki procesom
rekombinacyjnym łączą się ze sobą koniec z końcem,
tworząc tzw. Asocjacje telomeryczne. Sprzyja to
powstawaniu wtórnych aberracji chromosomowych.
Tworzenie
asocjacji
telomerycznych
umożliwia
komórce przetrwanie kryzysu nazwanego stanem M2.
Komórki, które przetrwają stan M2 stają się
nieśmiertelne.

Nabywanie nieograniczonego
potencjału podziałowego
Jedną
z
cech
komórek
nowotworowych
jest
nieśmiertelność, czyli zdolność do nieograniczonych
podziałów.
W komórkach tych procesy kontroli podziałów są
wyłączone lub poważnie uszkodzone, np. mutacja
onkogenu RAS czy genu supresorowego TP53.
Cechą
charakterystyczną
komórek
większości
nowotworów jest wysoka aktywność enzymu telomerazy –
odpowiadającego za odbudowę telomerów. W normalnych
komórkach (poza macierzystymi i częściowo limfocytami)
praktycznie nie wykrywa się aktywności tego enzymu.
Telomeraza zapobiega osiągnięciu krytycznej długości
telomerów przez komórkę nowotworową, a tym samym
zapewnia jej nieśmiertelność.

Transformacja nowotworowa a
apoptoza
Codziennie organizm traci około 10
12
komórek. Na ich miejsce na drodze
podziału powstają nowe komórki. Śmierć
ta jest ściśle zaprogramowana, zachodzi
na
drodze
apoptozy,
pozwala
na
eliminację
komórek
starych
i
uszkodzonych, a także już niepotrzebnych.
Apoptoza może być indukowana na drodze
dwóch mechanizmów:
Aktywacji przez czynniki wewnętrzne
Aktywacji przez czynniki zewnętrzne

Transformacja nowotworowa a
apoptoza
Aktywacja przez czynniki wewnętrzne – prowadzi do
uszkodzenia mitochondriów, głównie powodowana przez
produkty białkowe genów z rodziny BCL2 – antyapoptotyczne
(np. BCL2, BCL-X
L
) i proapoptotyczne (np. BAX, BAK).
Skierowanie na drogę proliferacji lub apoptozy zależy od
ilościowych zależności pomiędzy produktami tych genów –
np.
przewaga
białek
proapoptotycznych
nad
antyapoptotycznymi prowadzi do apoptozy.
Aktywacja przez czynniki zewnętrzne – polega na aktywacji
receptorów powierzchniowych, takich jak FAS/APO1 (CD95),
receptora dla IGFR1 (insulinopodobnego czynnika wzrostu),
czy receptora dla TNFR1 (czynnika wzrostu nowotworów).
Przekazywany sygnał prowadzi do aktywacji prokaspazy 8,
która przekształca się w kaspazę 8 i aktywuje inne kaspazy.
Kaspazy te dokonują trawienia ważnych dla życia komórki
białek.

Transformacja nowotworowa a
apoptoza
Cechą charakterystyczną komórek nowotworowych
jest zanik zdolności do apoptozy.
Najczęstszą przyczyną jest uszkodzenie genu TP53.
zmutowane białko nie jest w stanie zatrzymać cyklu
komórkowego i uruchomić systemów naprawy DNA.
Nie może też aktywować genów proapoptotycznych,
takich jak BAX czy FAS/APO1.
W konsekwencji komórki nowotworowe tracą zdolność
do umierania. Stają się nieśmiertelne.
Kolejne rundy replikacyjne powodują powstawanie
kolejnych populacji komórek z różnymi mutacjami i
aberracjami chromosomowymi. Prowadzi to do
szybkiej selekcji komórek o fenotypie nowotworowym.

Nabywanie zdolności do
unaczynienia guza
W dorosłym organizmie proces angiogenezy praktycznie
nie zachodzi (poza gojeniem ran).
Proces angiogenezy jest kontrolowany przez równowagę
czynników proangiogennych i antyangiogennych.
Czynniki proangiogenne to: czynnik wzrostu śródbłonka
naczyń (VEGF), czynniki wzrostu fibroblastów (FGF),
czynnik wzrostu nowotworów (TGF), interleukina 8.
Czynniki antyangiogenne to: angiostatyna, fragment
plazminogenu, endostatyna, trombospondyna, tkankowe
inhibitory metaloproteaz (TIMP), IL-1, IL-6, IL-10, IL-12.
Przesunięcie równowagi w kierunku angiogenezy
indukuje proces neoangiogenezy – tworzenia nowych
naczyń krwionośnych w guzie nowotworowym.

Nabywanie zdolności do
unaczynienia guza
Neoangiogeneza może być aktywowana poprzez mutację
w genie TP53. Prawidłowe białko TP53 aktywuje czynnik
antyangiogenny – trombospondynę. Spadek poziomu
białka TP53 powoduje spadek trombospondyny, co
przesuwa równowagę na korzyść angiogenezy.
Innym przykładem pobudzenia neoangiogenezy jest
wzrost
stężenia
VEGF,
spowodowany
wzrostem
aktywności genu VEGF na skutek oddziaływań ze strony
zrębu komórkowego. W obecności VEGF komórki
nabłonka naczyń włosowatych sąsiadujących z rozrostem
nowotworowym zaczynają się dzielić mitotycznie i
formują sieć naczyń przenikających guz. Powstające
naczynia wydzielają czynniki wzrostu, które stymulują
komórki guza do dalszych podziałów.

Nabywanie zdolności do
tworzenia nacieków i przerzutów
Zdolność pierwotnego nowotworu do tworzenia przerzutów
jest jedną z głównych przyczyn niepowodzeń w leczeniu.
W prawidłowych tkankach komórki ściśle przylegają do
siebie i są zakotwiczone w zrębie pozakomórkowym (ECM).
Odpowiadają za to białka – kadheryny E i integryny.
Komórki nowotworowe charakteryzują się częściową utratą
właściwości adhezyjnych. Powoduje to rozluźnienie
oddziaływań międzykomórkowych i przylegania do
macierzy zewnątrzkomórkowej.
Dodatkowo
komórki
nowotworowe
mogą
nabywać
zdolności degradacji błony podstawnej.
Komórki takie mogą zostać uwolnione z nowotworu
pierwotnego, następnie mogą spenetrować śródbłonek
naczynia krwionośnego lub chłonnego i dostać się do jego
światła.

Nabywanie zdolności do
tworzenia nacieków i przerzutów
Komórka następnie wędruje z prądem krwi lub
chłonki do innych rejonów organizmu. Na skutek
mechanicznego spowolnienia w świetle naczynia
komórka nowotworowa przylega do śródbłonka,
degraduje go i opuszcza naczynie.
Po zlokalizowaniu się w innym rejonie organizmu
i w obecności sprzyjających warunków (czynniki
wzrostu, substancje odżywcze) komórka zaczyna
się dzielić i tworzyć przerzut.
Spośród całej populacji komórek pierwotnego
nowotworu tylko nieliczne mają zdolność do
przerzutowania.

Nabywanie zdolności do
tworzenia nacieków i przerzutów
Inwazyjność nowotworu jest regulowana
przez równowagę produktów genowych.
Geny proinwazyjne to m.in. geny, których
produkty odpowiadają za adhezję komórek,
geny enzymów hydrolizujących błonę
podstawną i macierz zewnątrzkomórkową –
białka zwiększające zdolność do migracji.
Geny hamujące inwazję to m.in. geny
regulujące
oddziaływania
między
komórkami i kodujące inhibitory enzymów
hydrolitycznych.

Nabywanie zdolności do
tworzenia nacieków i przerzutów
Rodzina genów NME (NM23) – składa się z dwóch genów: NME1 i
NME2. W komórkach przerzutujących stwierdzono obniżenie poziomu
białek kodowanych przez te geny. Powoduje to zwiększenie
ruchomości komórek.
Kadheryna E – jest cząsteczką adhezyjną, ma zdolność wiązania
innych białek. Wiąże się z innymi kadherynami i integrynami, tworzy
sieć powiązań międzykomórkowych. Tą drogą za pomocą kinaz FAK i
SRC przekazywany jest drugi sygnał proliferacyjny (pierwszy
pochodzi z cyklu komórkowego). Hamowanie przekazywania sygnału
proliferacyjnego przez białka adhezyjne za pomocą ww. kinaz nosi
nazwę inhibicji kontaktowej. Komórki nowotworowe są zazwyczaj
pozbawione inhibicji kontaktowej z powodu spadku ilości kadheryny
E i mutacji w genach kodujących kinazy FAK i SRC. Prowadzi to do
zwiększenia zdolności do migracji komórek.
Kolagenaza IV – enzym degradujący kolagen IV występujący w błonie
podstawnej i macierzy pozakomórkowej. W komórkach zdolnych do
tworzenia przerzutów stwierdza się wzmożoną ekspresję genu
kodującego kolagenazę IV i wzrost stężenia tego enzymu.

Nabywanie zdolności do
tworzenia nacieków i przerzutów
CD44 – cząsteczka uczestnicząca m.in. w
oddziaływaniach międzykomórkowych i między
komórką a macierzą zewnątrzkomórkową. Komórki
zdolne do przerzutowania wykazują na swojej
powierzchni zmienione cząsteczki CD44 (większe,
znacznie bardziej glikozylowane), które wykazują
zmniejszone zdolności adhezyjne.
Szacuje się, że na 105-106 komórek dostających się
do naczyń przeżywa tylko kilka. Te, które przeżyją,
nie zawsze są w stanie zasiedlić nowe środowisko i
często giną. To tłumaczy powstawanie przerzutów
w późnej fazie nowotworzenia.
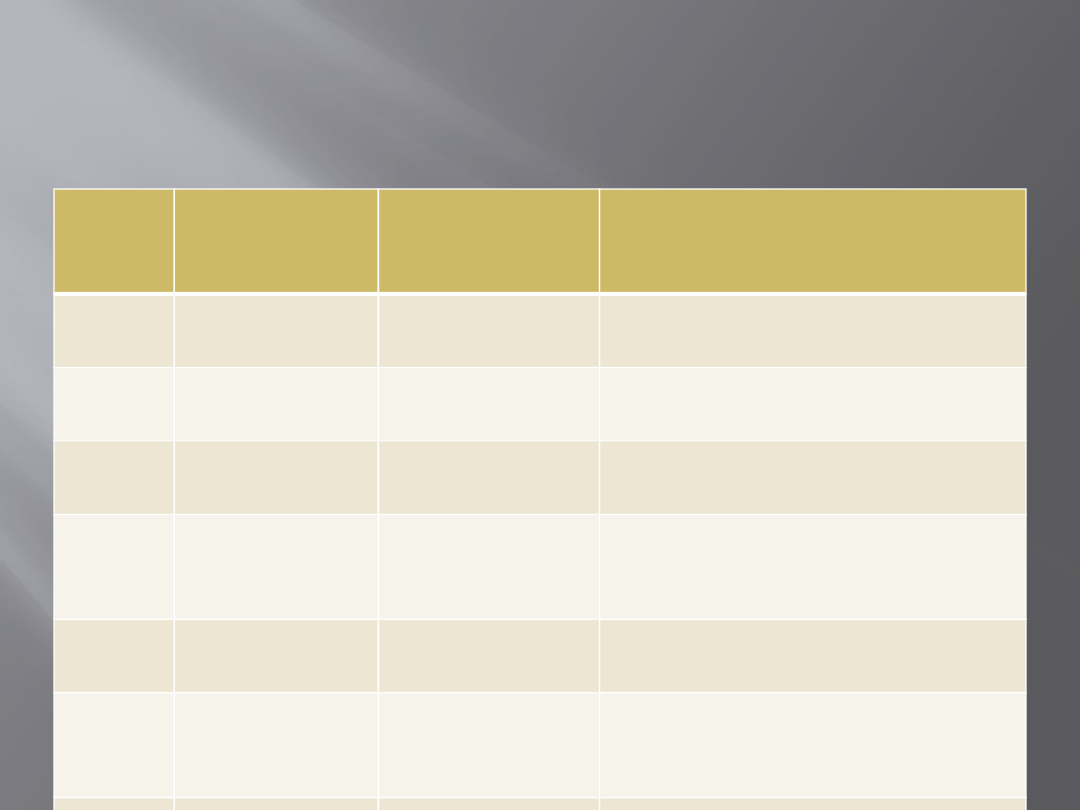
Wybrane geny podatności
zachorowania na nowotwory
rodzinne
Nazw
a
genu
Dziedziczen
ie
Rodzaj genu/
lokalizacja
Rodzaj nowotworu
APC
Autosomalne
dominujące
Supresorowy
5q21
Rak jelita grubego na podłożu
FAP; inne nowotwory
RB1
Autosomalne
dominujące
Supresorowy
13q14
Siatkówczak
VHL
Autosomalne
dominujące
Supresorowy
3p25
Choroba von Hippla-Lindaua;
nowotwory nerek
TP53
Autosomalne
dominujące
Supresorowy
17p13
Zespół Li-Fraumeni –
nowotwory mózgu, sutka,
mięsaki, białaczki
WT1
Autosomalne
dominujące
Supresorowy
11p13
Guz Wilmsa
BRCA1
BRCA2
Autosomalne
dominujące
Supresorowy
17q21.2;
13q12.2
Rak sutka i jajnika; raki
prostaty i sutka u mężczyzn
hMSH2
,
hMLH1
Autosomalne
dominujące
Stabilizacyjny
2p16; 3p21
Rak jelita grubego HNPCC;
raki macicy

Wybrane geny podatności
zachorowania na nowotwory
rodzinne
Nazwa
genu
Dziedziczen
ie
Rodzaj
genu/
lokalizacja
Rodzaj nowotworu
NBS1
Autosomalne
recesywne
Stabilizacyjny
; 8q21.3
Chłoniaki, guzy mózgu w
zespole Nijmegen
(homozygoty)
RET
Autosomalne
dominujące
Protoonkogen
; 10q11
Rak rdzeniasty tarczycy
KIT
Autosomalne
dominujące
Protoonkogen
; 4q12
Guzy stromalne przewodu
pokarmowego

Dziękuję za uwagę
Document Outline
- Slide 1
- Slide 2
- Istota nowotworzenia
- Slide 4
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Cykl komórkowy
- Regulacja cyklu komórkowego
- Regulacja cyklu komórkowego
- Regulacja cyklu komórkowego
- Białko TP53
- Białko TP53
- Białko TP53
- Białko TP53
- Białko RB1
- Białko RB1
- Białko RB1
- Białko RB1
- Etapy karcynogenezy
- Etapy karcynogenezy
- Etapy karcynogenezy
- Etapy karcynogenezy
- Onkogeny
- Onkogeny
- Onkogeny
- Onkogeny
- Geny supresorowe
- Geny supresorowe
- Geny stabilizacyjne
- Geny stabilizacyjne
- Nabywanie nieograniczonego potencjału podziałowego
- Nabywanie nieograniczonego potencjału podziałowego
- Nabywanie nieograniczonego potencjału podziałowego
- Nabywanie nieograniczonego potencjału podziałowego
- Transformacja nowotworowa a apoptoza
- Transformacja nowotworowa a apoptoza
- Transformacja nowotworowa a apoptoza
- Nabywanie zdolności do unaczynienia guza
- Nabywanie zdolności do unaczynienia guza
- Nabywanie zdolności do tworzenia nacieków i przerzutów
- Nabywanie zdolności do tworzenia nacieków i przerzutów
- Nabywanie zdolności do tworzenia nacieków i przerzutów
- Nabywanie zdolności do tworzenia nacieków i przerzutów
- Nabywanie zdolności do tworzenia nacieków i przerzutów
- Wybrane geny podatności zachorowania na nowotwory rodzinne
- Wybrane geny podatności zachorowania na nowotwory rodzinne
- Dziękuję za uwagę
Wyszukiwarka
Podobne podstrony:
Genetyka Nowotworów
Niestabilność genetyczna w nowotworach 1
genetyka nowotworow
genetyka nowotworow
Genetyka nowotworów
Niestabilność genetyczna w nowotworach., Niestabilność chromosomowa:
03 Genetyka w nowotworachid 4362 ppt
Genetyka nowotworów
Niestabilność genetyczna w nowotworach, NIESTABILNOŚĆ GENETYCZNA W NOWOTWORACH
14 Genetyka nowotworow
Niestabilność genetyczna w nowotworach 1
Genetyka nowotworzenia 01
Genetyczne podstawy nowotworów, Biologia medyczna
Diagnostyka predyspozycji genetycznych do chorob nowotworowych
GENETYCZNE PODSTAWY NOWOTWORÓW(1)
GENETYKA KLINICZNA V rok seminarium Nowotwory dziedziczne wprowadzenie Nowotwory jelita grubeg
nowotwory dla studentów, psychologia, Genetyka
IG.4 - Uszkodzenia i naprawa DNA w komórkach nowotworowych, Genetyka, Inżynieria genetyczna
więcej podobnych podstron