
Co to jest jednostka
ruchowa?

Choroby nerwowo-
mięśniowe
Uszkodzenie jednostki ruchowej
(obwodowy neuron ruchowy,
płytka nerwowo-mięśniowa,
włókna mięśniowe)

Choroby nerwowo-
mięśniowe
• Choroby pierwotnie mięśniowe
(miopatie)
• Procesy uszkadzające płytkę nerwowo-
mięśniową
• Procesy neurogenne toczące się w
mięśniu wtórnie do uszkodzenia
obwodowego neuronu ruchowego
(komórki rogu przedniego lub nerwu
obwodowego)

Choroby nerwowo-
mięśniowe
• Zanik mięśniowy
• Niedowład (miopatie –dosiebny,
neurogenny –odsiebny)
• Wiotkość
• Osłabienie lub zniesienie odruchów
(rzadko w miopatiach)
• Objawy miotonii w
– Dystrofia miotoniczna
– Miotonia wrodzona
– Paramiotonia wrodzona
– Porażenie okresowe hiperkalemiczne

Osłabienie i zanik mięśni
dystalnych
• Dystrofia miotoniczna
• Dystalna dystrofia mięśniowa
• Wtrętowe zapalenie mięśni
• Miastenia (rzadko)

Mięśnie twarzy i mięśnie
oczne
• Dystrofia miotoniczna
• Dystrofia oczna
• Dystrofia twarzowo-łopatkowo-
ramieniowa
• Cytopatia/miopatia
mitochondrialna
• Miastenia

Osłabienie mięśni
zginaczy szyi
• Zapalenie wielomięśniowe
• Miastenia
• Dystrofia miotoniczna
• Niedobór kwaśnej maltazy
• Choroba neuronu ruchowego

Niewydolność oddechowa
• Zapalenie wielomięśniowe
• Miastenia
• Miopatia nemalinowa
• Niedobór kwaśnej maltazy
• Choroba neuronu ruchowego
• Zespół Guillain-Barre

Rzekomy przerost mięśni
• Dystrofia Duchenne’a
• Dystrofia Beckera
• Miotonia wrodzona
• Neuropatie obwodowe (bardzo
rzadko)

Ból mięśni
• W spoczynku:
– Miopatie zapalne
– Ostre miopatie z mioglobinurią
– Niektóre miopatie polekowe
– Miopatie związane z zaburzenia
hormonalnymi (niedoczynność tarczycy,
choroba Addisona, zespół Cushinga)
• Po wysiłku:
– Zapalenie wielomięśniowe
– Miopatie metaboliczne

Miopatie - diagnostyka
• Poziom CK, AspAT, aldolza, LDH
• Mioglobinuria
• EMG
– Potencjał wielofazowy
– Obniżenie amplitudy
– Skrócenie długości potencjału
• Biopsja mięśnia - badanie histopatologiczne:
klasyczne i biochemiczne- tłuszcz,
glikogen,
enzymy
• MRI mięśni

Miopatie
• Dystrofie mięśniowe
• Zespoły miotoniczne
• Miopatie wrodzone
• Miopatie metaboliczne
• Miopatie nabyte (w tym zapalne)

Miopatie nabyte
• Hormonalne
(nadczynność i niedoczynność tarczycy,
akromegalia, zesp.Cushinga, niedoczynność przytarczyc, ch.
Addisona)
• Toksyczne i polekowe
(bólowe:
alkohol, amfetamina,
betablokery, bezfibrat,lit, penicylamina, fenytoina, salbutamol,
winkrystyna, teofilina, zidowudyna i in.: bezbólowe: amiodaron,
sterydy, tyroksyna)
• W przebigu chorób narządowych
• W przebiegu choroby nowotworowej
(zapalenie wielomięśniowe i skórno-mięśniowe)
• Zapalne
(zapalenie wielomięśniowe i skórno-mięśniowe,
wirusowe-Coxsackie,grypa, HIV)
• Autoimmunologiczne
(zapalenie wielomięśniowe i
skórno-mięśniowe w Vasculitis, SLE, z. Sjogrena)
• Urazowe

Dystrofie mięśniowe
Dystrofia Duchenne’a (sprzężona z płcią,
recesywna)
Dystrofia Becker’a (sprzężona z płcią, recesywna)
Dystrofia twarzowo-łopatkowo-ramienna
(autosomalna dominująca)
Dystrofia Emery-Dreifusa (sprzężona z płcią,
recesywna)
Dystrofia łopatkowo-strzałkowa (autosomalna
dominująca)
Dystrofia oczno-gardzielowa (autosomalna
dominująca)
Dystrofia oczna
Dystrofia kończynowo-obręczowa
Dystrofia miotoniczna (autosomalna dominująca)
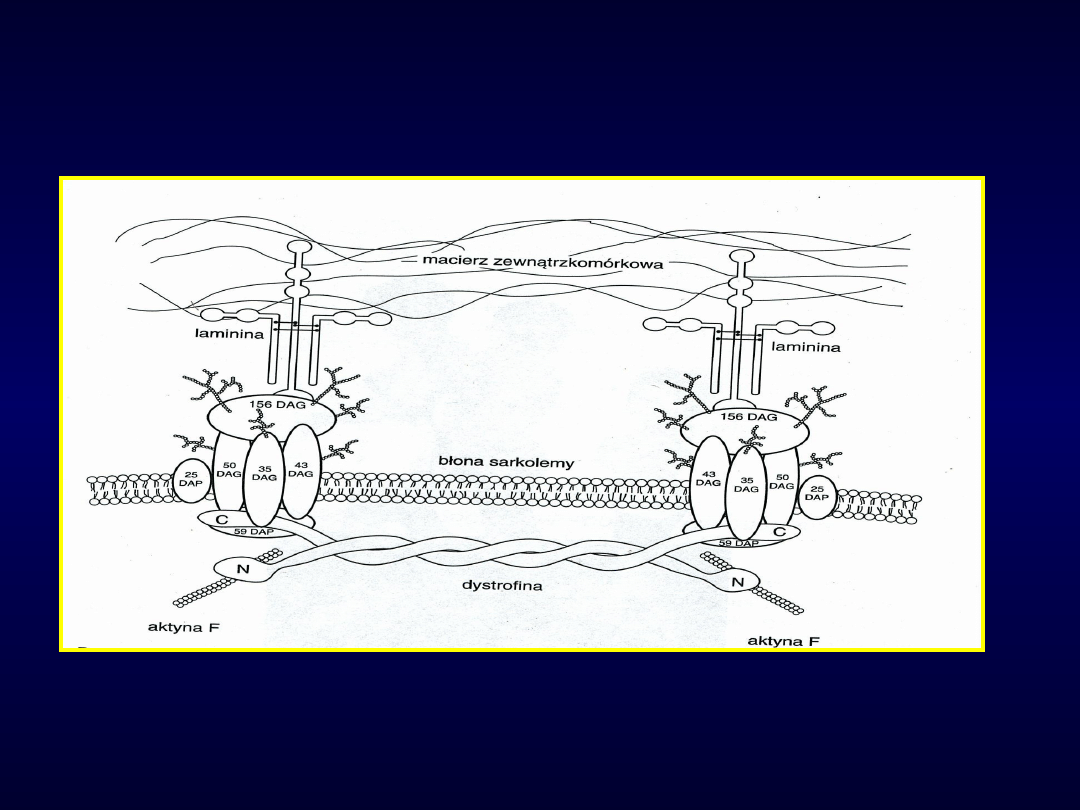
Kompleks dystrofinowo-
glikoproteinowy
DAP-dystrophin associated
protein,
DAG- -dystrophin associated
glycoprotein
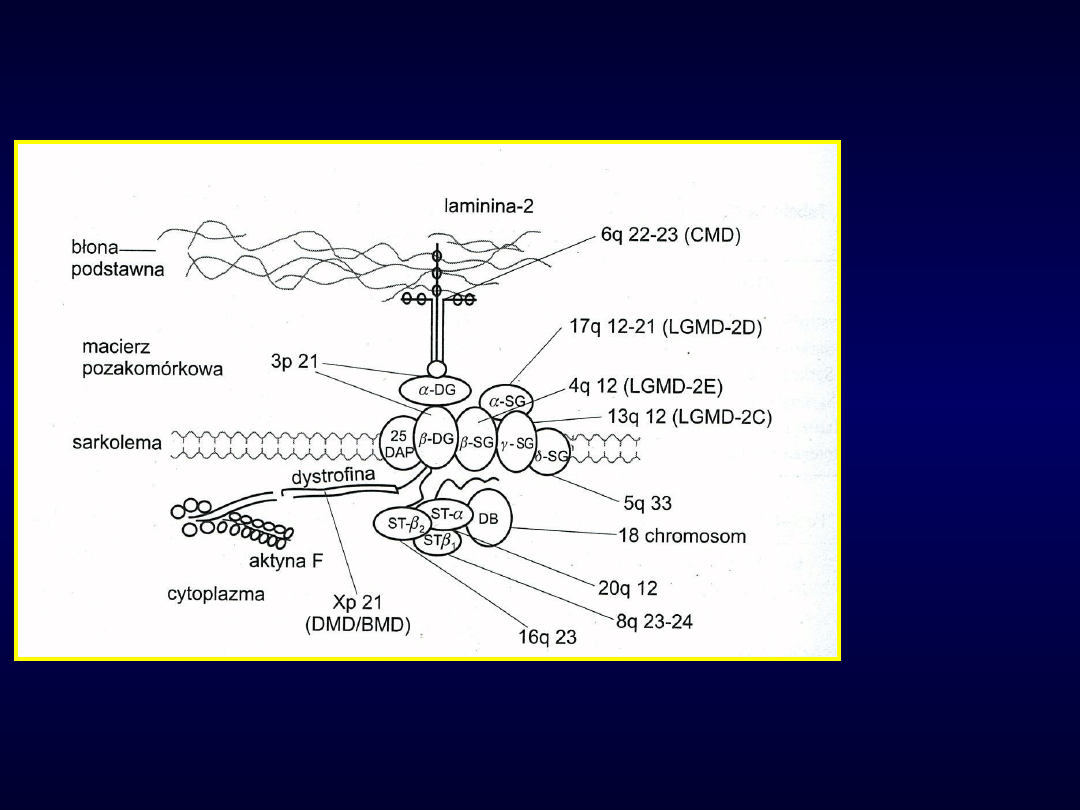
Kompleks dystrofinowo-
glikoproteinowy
DMB/BMD-
Duchenne/
Becker,
LGMD-
kończynow
o-
obręczowa,
CMD-
wrodzona
SG-sarkoglikan, ST-syntrofina, DB-dystobrewina,
DG-dystroglikan, DAP-dystrophin associated protein

Dystrofia Duchenne’a
• 2/100.000
• sprzężona z płcią, recesywna – chorują chłopcy,
b. rzadko dziewczynki (translokacja
autosomalna) i zesp. Turnera
• delecja genu dla dystrofiny, zmiana ramki
odczytu
• ujawnia się ok. 2-3-4 roku życia
• Zajęcie obręczy miednicy potem barkowej
• Objaw Gowersa
• Przykurcze 8-9 r.ż.
• Zniekształcenie klatki piersiowej –niewydolność
oddechowa, zaburzenia krążenia
• EKG, ECHO serca –kardiomiopatia
rozstrzeniowa
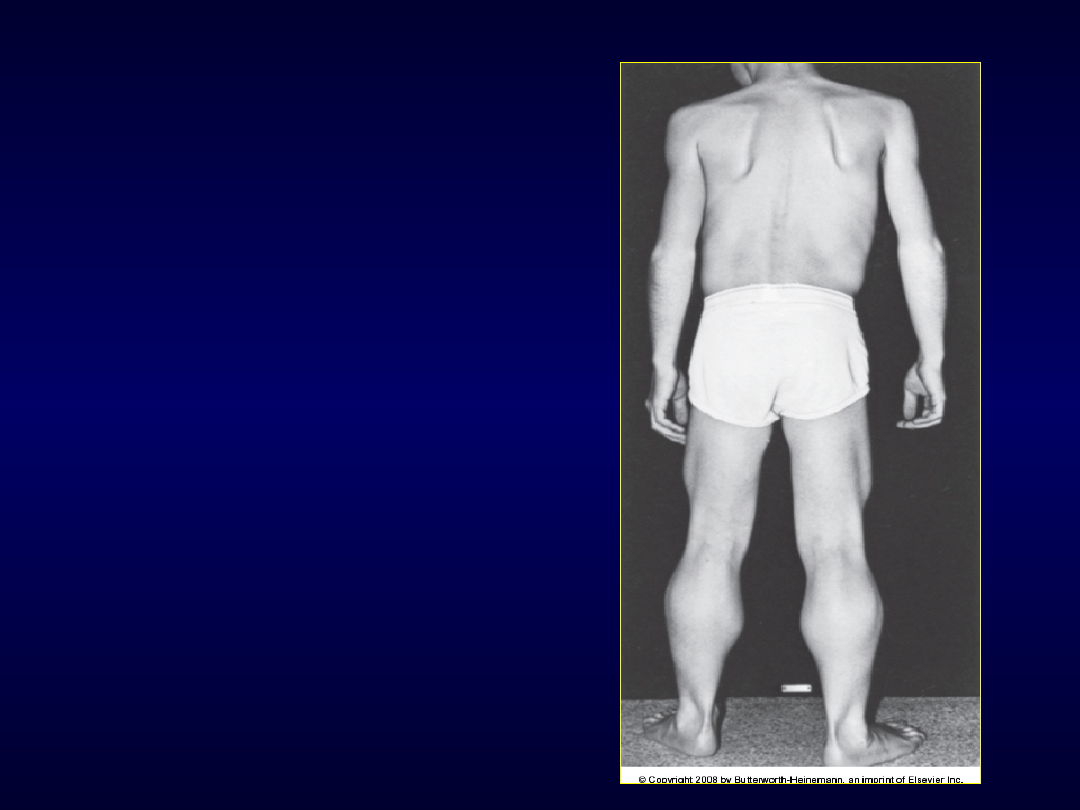
Dystrofia
Duchenne’
a

Dystrofia Becker’a
• 3-6/100.000
• sprzężona z płcią, recesywna – chorują
chłopcy
• delecja genu dla dystrofiny, bez zmiany
ramki odczytu
• ujawnia się ok. 10 roku życia
• Zajęcie obręczy miednicy potem barkowej
• Objaw Gowersa
• Skurcze mięśniowe u nastolatków
• EKG, ECHO serca – rzadko kardiomiopatia
rozstrzeniowa

Dystrofia twarzowo-
łopatkowo-ramienna
• autosomalna dominująca
• Chr.4
• 7-17 r.ż.
• Najpierw mm. twarzy (brak
gwizdania, „usta tapira”)
• Obręcz barkowa (lekarz)
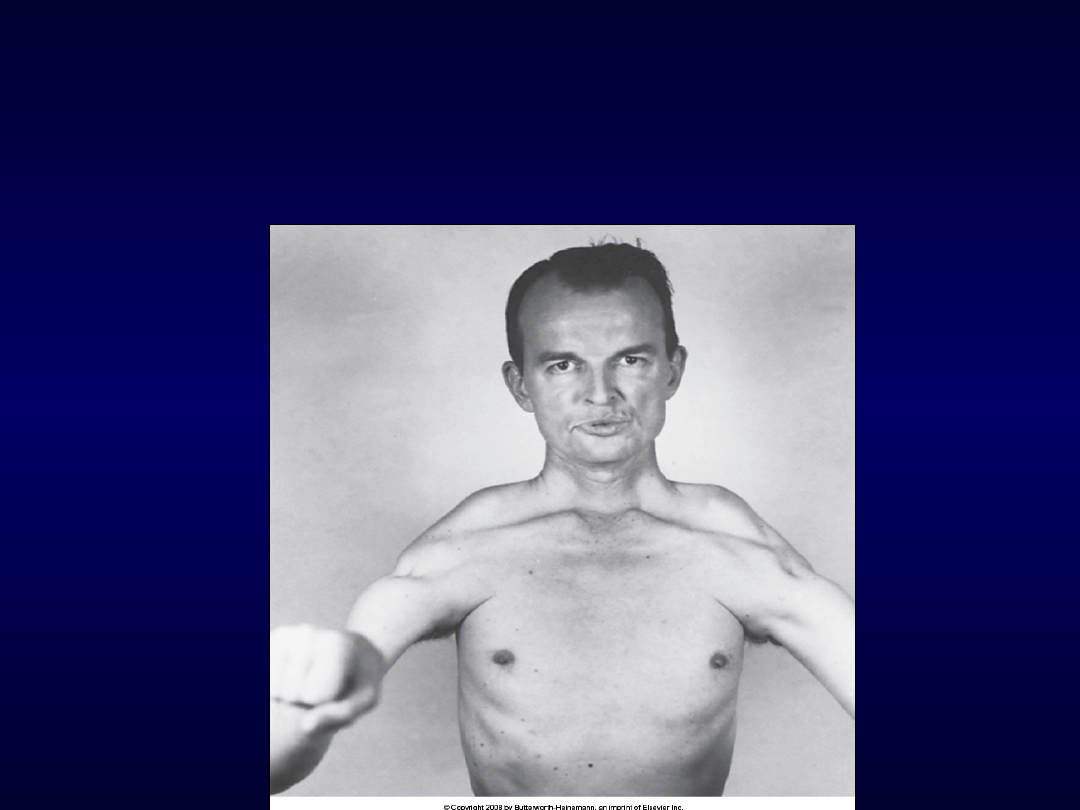
Dystrofia twarzowo-
łopatkowo-ramienna

Dystrofia Emery-Dreifusa
• sprzężona z płcią, recesywna, wiele
mutacji
• Białko emeryna
• 3-6 r.ż.
• Niedowład mm. dosiebnych kk.g. i
odsiebnych kk.d
• Chodzenie na palcach
• Zaburzenia przewodnictwa sercowego
blok przedsionkowo-komorowy

Dystrofia łopatkowo-
strzałkowa
• autosomalna dominująca?,
sprzężona z płcią?
• Odmiana dystrofii twarzowo-
łopatkowo-ramiennej lub dystrofii
Emery-Dreifusa

Dystrofia kończynowo-
obręczowa
• Mieszana grupa miopatii
• Wspólne: zanik mięśni dosiebnych
obręczy miednicznej, mniej
obręczy barkowej
• Chód kaczkowaty

Dystrofia oczno-
gardzielowa
• autosomalna dominująca
• 30-40 r.ż
• Niedowład mm. zewn. gałki ocznej
(bez podwójnego widzenia)
• Opadnięcie powiek
• Czasem dysfagia, osłabienie mm.
twarzy
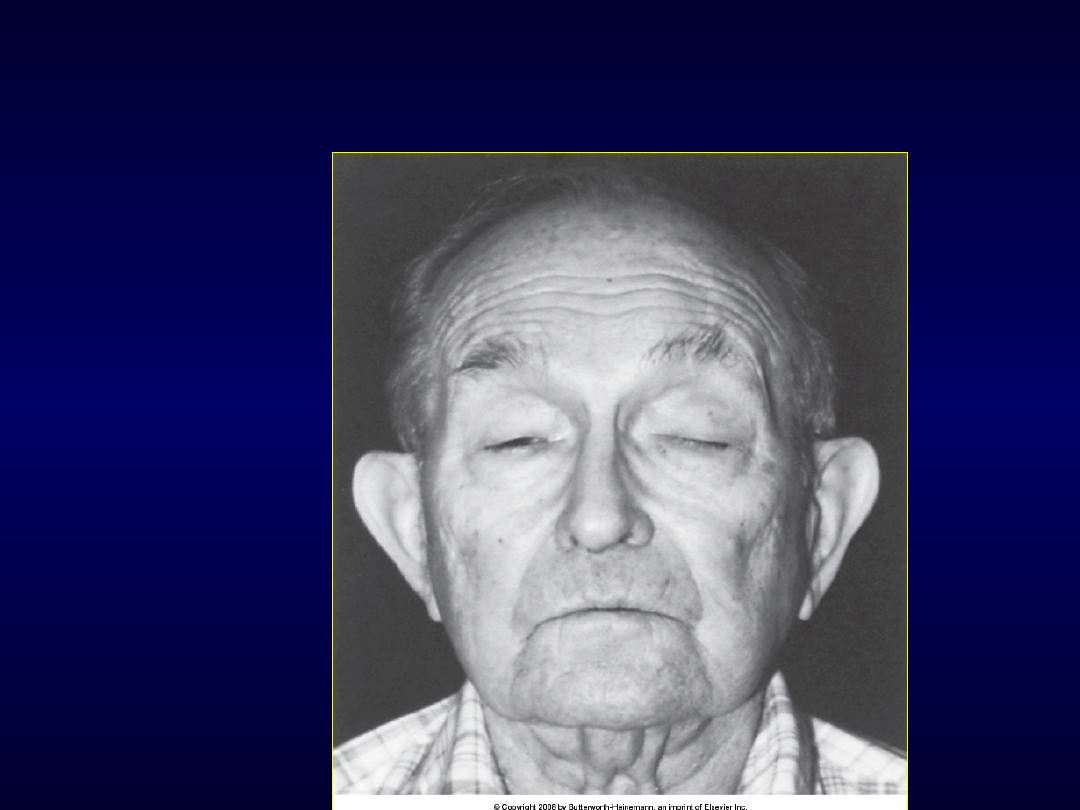
Dystrofia oczno-
gardzielowa

Dystrofia oczna
• Dystrofia oczno-gardzielowa lub
• Zesp. Kerns-Sayre
– Miopatia mitochondrialna (uszkodzenie
genów łańcucha oddechowego)
– Dziedziczenie „od matki”
– Początek w dzieciństwie
– Postępująca oftalmoplegia zewnętrzna
– Retinitis pigmentosa
– „zaburzenia mitochondrialne” w in. tkankach–
może być spastyczność i wzmożenie odruchów
głębokich, ataksja móżdżkowa
– Zaburzenia przewodnictwa sercowego

Dystrofia miotoniczna
• autosomalna dominująca, Chr 19, 14/100.000
urodzeń
• Mutacja dynamiczna (kinaza proteinowa dystonii
miotonicznej) gł młodzi dorośli
• Objaw miotonii
• Zaniki dystalne mm. kończyn (gł. ręki)
• Zaniki mm. twarzy języka, gardła, opadanie powiek
(gł. bez zajęcia mm. gałkoruchowych)
• Nasilenie obj. w niskich temp.
• Dot. wszystkich mięśni (dyzartria, dysfagia) w tym m.
Sercowego
• Zaćma, Łysienie czołowe, przerost kk. Czaszki, zab.
hormonalne

Dystrofia miotoniczna

Miotonia wrodzona
• miotonia Thomsena -AD, u niemowląt;
• miotonia Beckera -AR, u dzieci starszych
– Objawy:
•Miotonia nasilająca się w niskich
temp.
•Poprawa pod wpływem ciepła i
wysiłku
•Przerost mięśni

Leczenie miotonii
• Fenytoina 100-200 mg / 2xdz
• Acetazolamid (paramiotonia)
• Prokainamid 250-500mg/ 4xdz

Paramiotonia
• AD,
• kanał Na+
• Przed 10 r.ż
• Objawy: Napady miotonii,
osłabienia mięśni, prowokowane
niską temp., hiperkalemią i beta-
blokera

Porażenie okresowe
• Hipokalemiczne: AD, kanał Ca, napady
wywoływane jedzeniem bogatym w
węglowodany, zimnem, ciężkie porażenie,
bez miotonii, podanie K usuwa objawy,
przerwy między napadami –tyg./mc., przed
10 r.ż
• Hiperkalemiczne: AD, kanał Na, napady
wywołane podaniem K, podanie glukozy
znosi objawy, miotonia, przerwy między
napadami – może nie być, przed 10 r.ż

Miopatie wrodzone
• Typu „central core”, AD,- histopatol.: włókna
mięśniowe- ogniska odmiennej barwliwości –
skupienie miofilamentów; nieprawidłowości
kostne (wydrążone stopy, skrzywienie
kręgosłupa, dzieci zaczynają chodzić po 2 r.ż,
przebieg łagodny
• Nitkowata (nemalinowa), AR lub AD,
histopatol.: włókna mięśniowe- struktury
nitkowate „rods”, dyzmorfizm kostny, zajęcie
także mm. Twarzy i gałkoruchowych
• Miotubularne, z chr.X lub AD lub AR,
histopatol.: włókna mięśniowe- łańcuchy jąder,
uogólniona wiotkość, niewydolność oddechowa
zgon po kilku tyg./mc ż. (respirator)

Miopatie metaboliczne
• Niedobor kwaśnej maltazy, AR,
gromadzenie glikogenu w wątrobie, sercu,
oun, mięśniach, zgon w dzieciństwie
• Niedobór fosforylazy mięśniowej (Ch.
McArdle’a), AR, początek w dzieciństwie,
charakter niepostępujący, brak zaniku
mięśni, charakterystyczne zmęczenie,
osłabienie i sztywność zwłaszcza po
wysiłku

Miopatie zapalne
• Zapalenie wielomięśniowe i skórno-mięśniowe
–
Każdy wiek
–
Początek: ostry, podostry i przewlekły
–
Rozlane, symetryczne osłabienie proksymalnych mięśni
kk
–
(obj. Skórne rumień na twarzy, tułowiu, wyprostne pow.
kk)
–
Wys. OB., wys. Enz. Mięśń., EMG, biopsja
–
Leczenie:
• sterydy długotrwale 80 mg prednisolonu
• Immunosupresja: azatiopryna, metotreksat, cyklofosfamid,
chlorambucyl, cyklosporyna A
• IgG i.v.
• Wtrętowe zapalenie mięśni
–
Po 50 r.ż., leczenie bez ewidentnego efektu
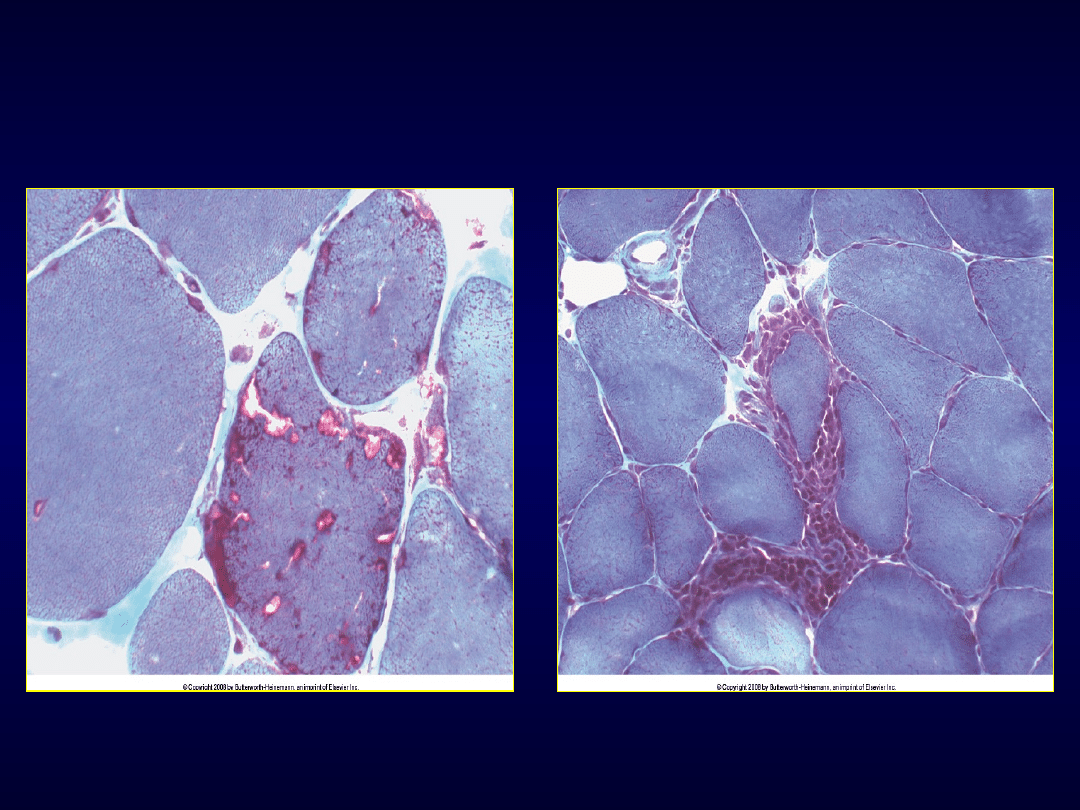
Miopatie zapalne
Wtrętowe zapalenie mięśni zapalenie wielomięśniowe

Choroby nerwowo-
mięśniowe
• Choroby pierwotnie mięśniowe
(miopatie)
• Procesy uszkadzające płytkę nerwowo-
mięśniową
• Procesy neurogenne toczące się w
mięśniu wtórnie do uszkodzenia
obwodowego neuronu ruchowego
(komórki rogu przedniego lub nerwu
obwodowego)

Procesy uszkadzające
płytkę nerwowo-
mięśniową
• Wrodzone
– Defekt syntezy ACh
– Niedobór AChE (acetylocholinoesterazy)
płytkowej
– Niedobór receptorów Ach (AChR)
– Zespół wolnego kanału receptora ACh
• Nabyte
– Toksyczne i polekowe (jad
kiełbasiany,pestycydy, penicylamina)
– Immunologiczne (miastenia, zespół Lamberta-
Eatona, leczenie penicylaminą)
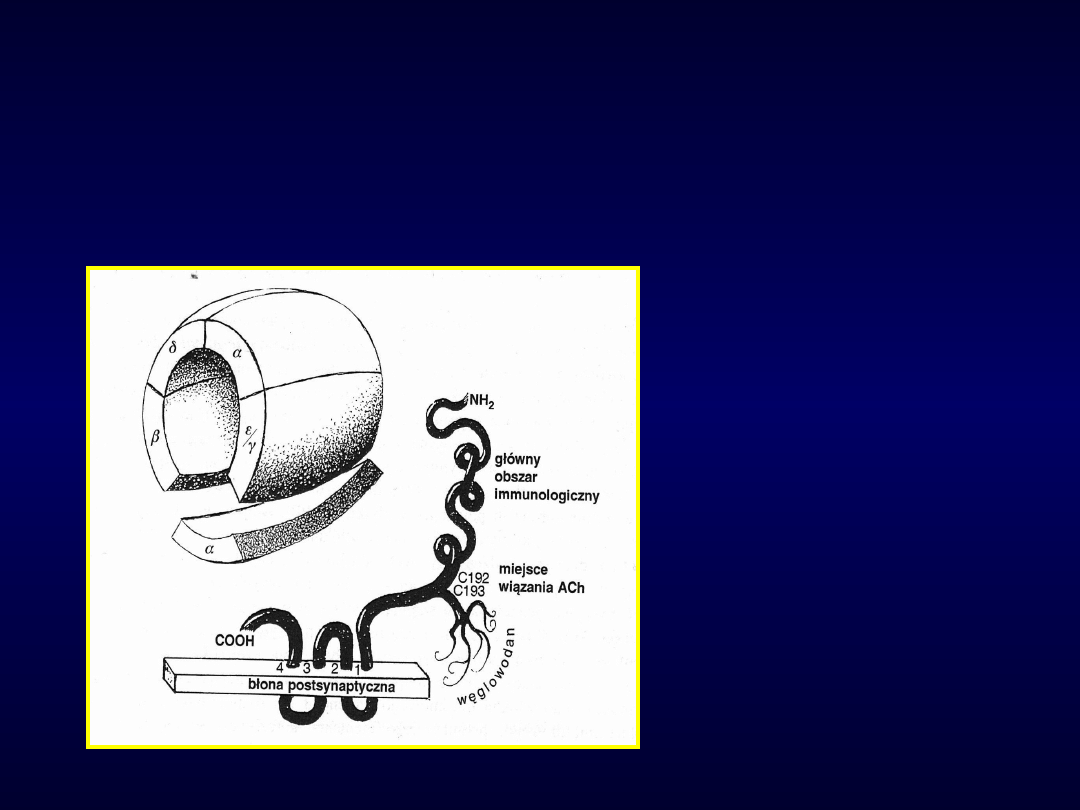
Receptor nikotynowy
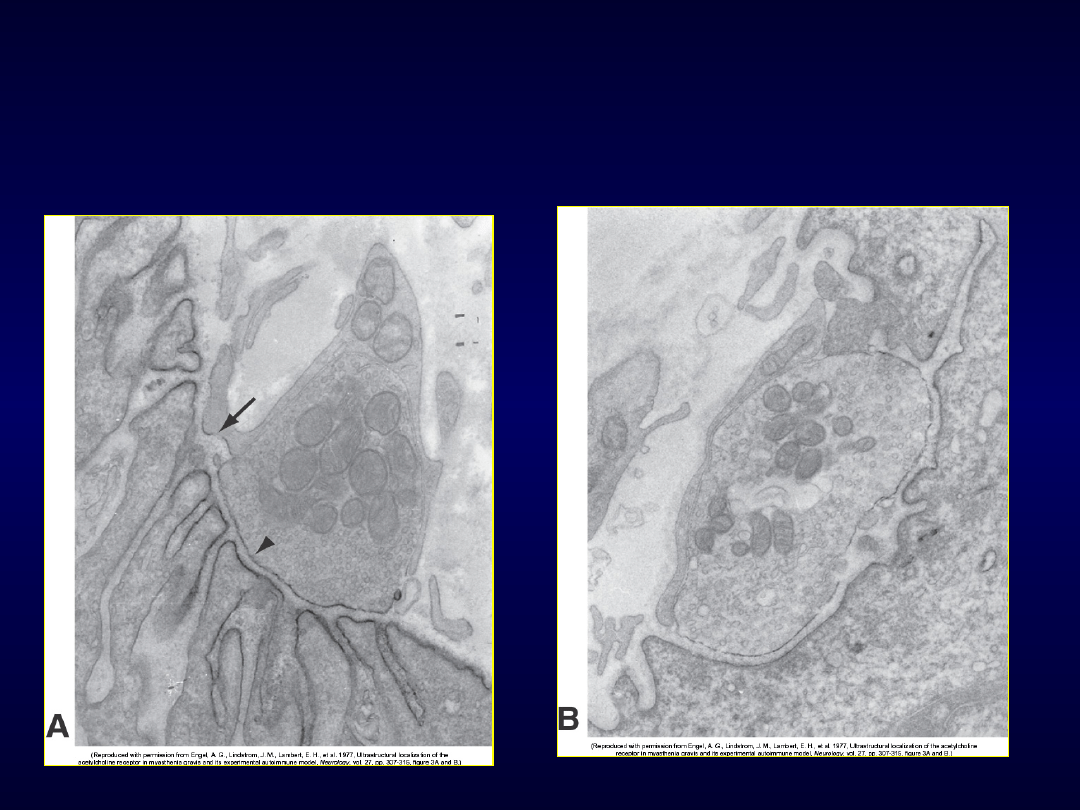
AChR

Miastenia
• Przeciwciała przeciwko receptorowi
nikotynowemu ACh lub kinazy tyrozynowej
mięśni MUSK
• 5/100.000
• Kobiety: mężczyźni 2:1
• Zachorowania każdy wiek – najczęściej 20-
30 r.ż. (kobiety), 50-60r.ż. (mężczyźni)
• Apokamnoza
• Mięśnie: gałkoruchowe, twarzy, objawy
opuszkowe, mięśnie kk i oddechowe
• 10% - grasiczak
• Miastenia przejściowa noworodków

Miastenia
• Diagnostyka:
– Apokamnoza
– Test inhibitorami AChE Tensilonem (10 mg) lub
prostygminą (1-1,5mg) obj. Niepożądane muskarynowe
bradycardia i spadek RR – atropina 0,6mg
– Próba miasteniczna
– Przeciwciała
• Lecznie
– tymektomia
– Inhibitory AChE Mestinon, Mytelasa
– sterydy
– Immunosupresja azatiopryna
– Plazmaferezy
– IgG i.v.

Przełomy
• Przełom miasteniczny
– osłabienie mięśni – niewydolność oddechowa
– objawy opuszkowe
• Przełom cholinergiczny
(przedawkowanie inhibitorów AChE)
– osłabienie mięśni – niewydolność oddechowa
– objawy opuszkowe
– wąskie źrenice,
– zab. Akomodacji,
– tachycardia,
– poty,
– wymioty,
– biegunka,
– zaleganie wydzieliny w oskrzelach,
– lęk, kurcze mięśni
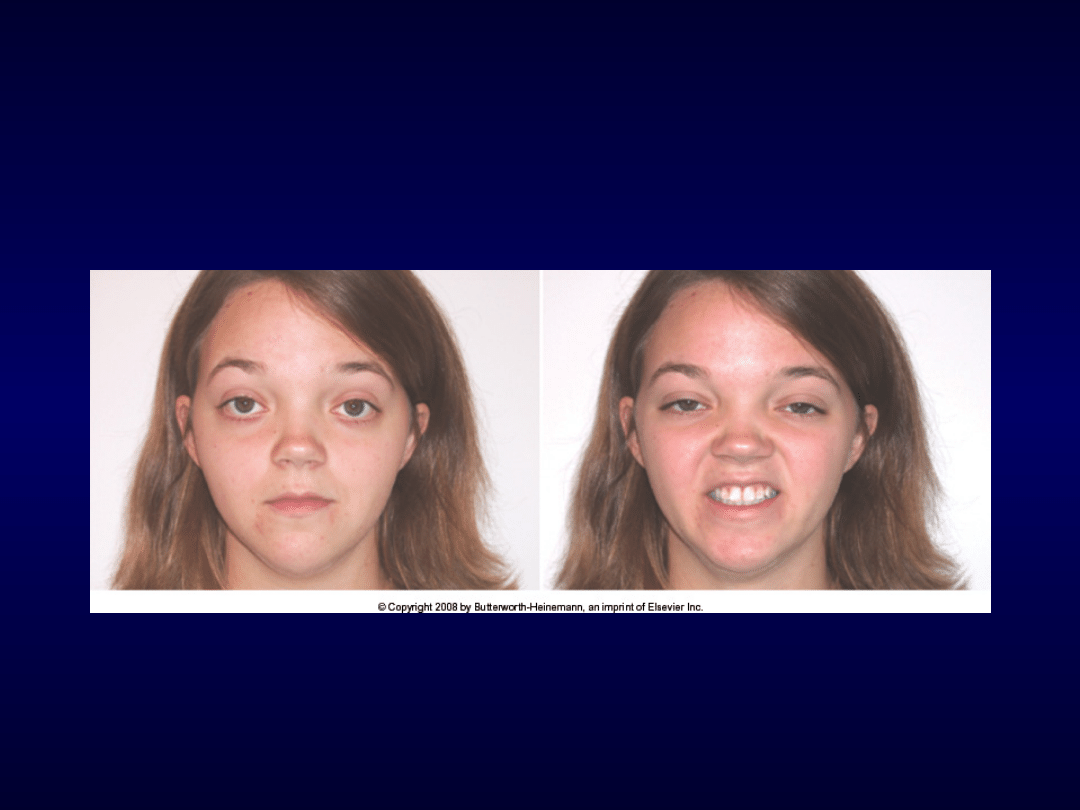
Miastenia

Miastenia apokamnoza
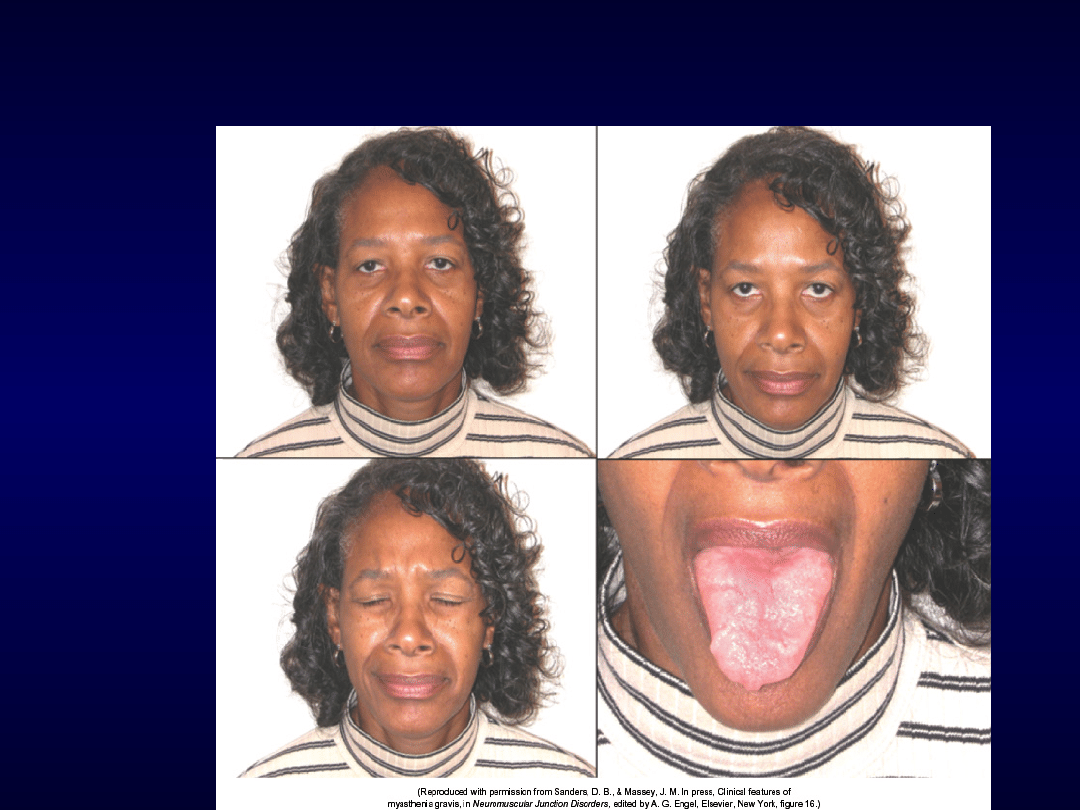
Miastenia „MUSK”
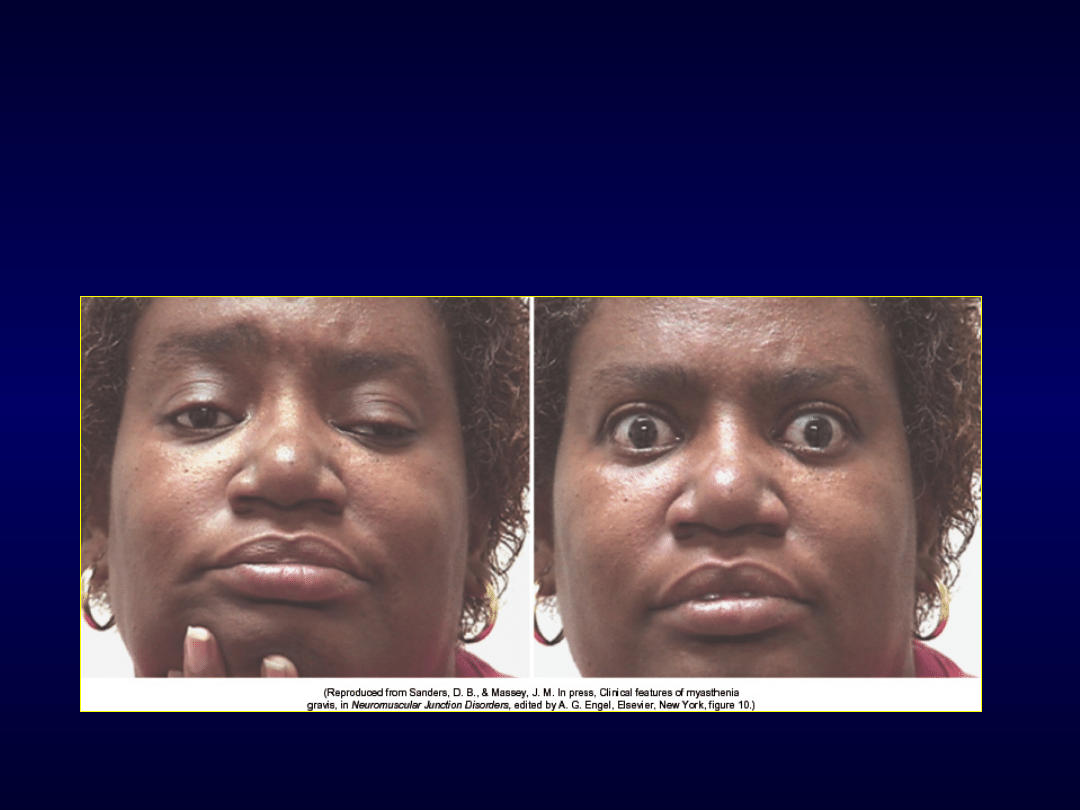
Miastenia - tensilon

Zespoły miasteniczne
wrodzone
• Niedobór AChE płytkowej: AD lub AR (po
urodzeniu obj. oczne, później uogólnienie, bez
reakcji na leki p-miasteniczne)
• Niedobór AChR: AR (noworodki objawy oczne i
opuszkowe, przebieg stacjonarny, leki p-
miasteniczne)
• Zespół wolnego kanału receptora Ach: AD (dzieci
niedowłady i zaniki pasa barkowego, rzadziej obj.
oczne i opuszkowe)
• Dziedziczna miastenia niemowląt: AR (po
urodzeniu objawy opuszkowe, rzadziej oczne, leki
p-miasteniczne)

Zespół Lamberta-Eatona
• 50% nowotwór w tym 80% rak
drobnokomórkowy płuc
• Przeciwciała przeciwko kanałowi Ca
(VGCC- voltage gated calcium channel)
upośledzenie wydzielania ACh
• Osłabienie mm. kkd
• EMG
• Guanidyna 5-10 max.30 mg/kg/dz w
dawkach podzielonych co 4-6h
• Diaminopirydyna (DAP) 5-25 mg 3-4xdz
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
Wyszukiwarka
Podobne podstrony:
Wytyczne postępowania w chorobach nerwowo - mięśniowych , Neurologia1
choroby nerwowo-mięśniowe, neurologia
choroby nerwowo miesniowe, Neurologia(1)
miastenia, Medycyna, Neurologia, 12 choroby nerwowo-miesniowe
Choroby nerwowo-miesniowe, MEDYCYNA - ŚUM Katowice, V ROK, Neurologia, Materiały dodatkowe
Choroby nerwowo-mięsniowe(1), fizjoterapia
metody oceny AUN, choroby nerwowo-mięśniowe
Choroby nerwowo miesniowe
Choroby nerwowo mięśniowe
Choroby nerwowo mięśniowe
choroby nerwowo mięśniowe
Choroby nerwowo mięśniowe
Choroby nerwowo miesniowe
Choroby nerwowo mięśniowe 2008
choroby nerwowo-miesniowe, choroby i ich leczenie
więcej podobnych podstron