1
Choroby nerwowo-mięśniowe
Klinika Neurologii
2008
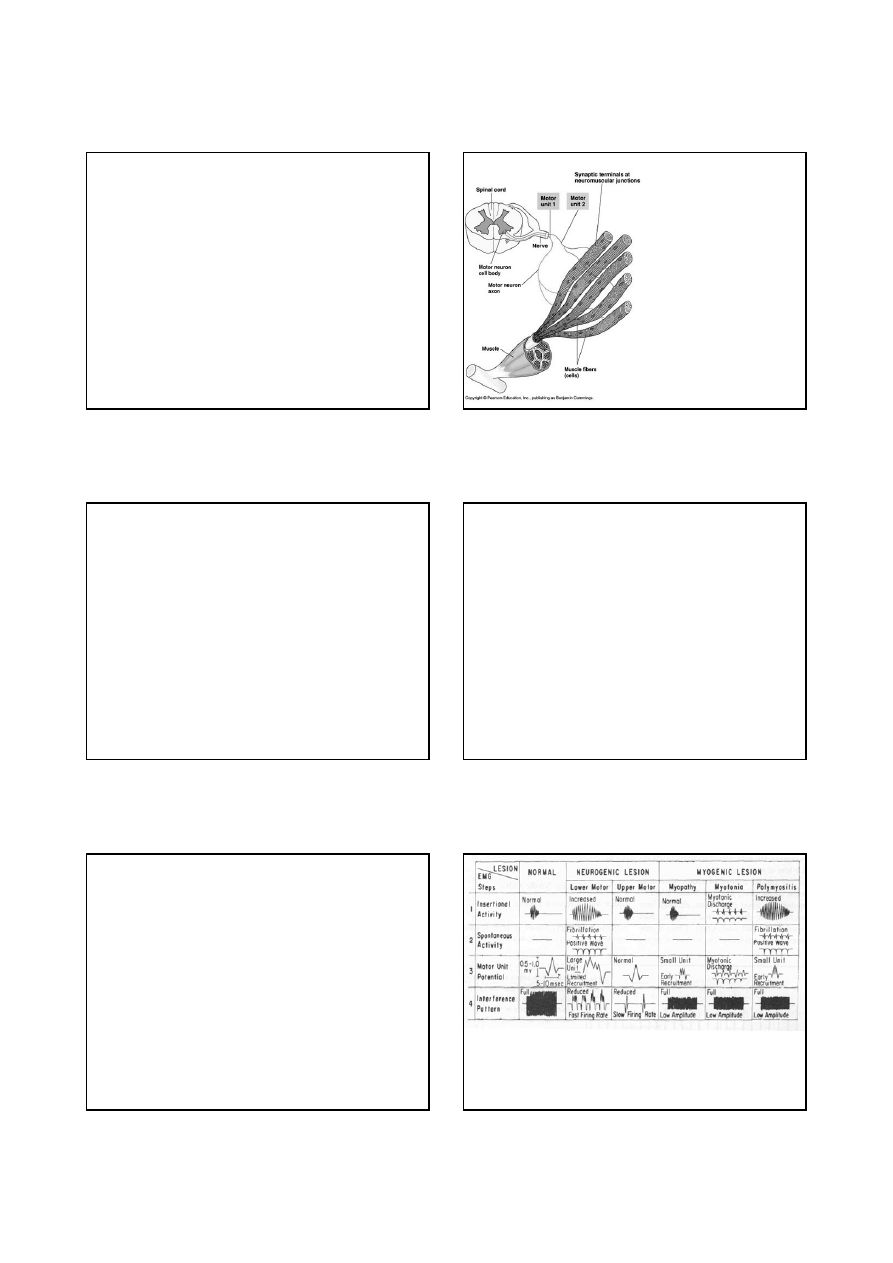
Jednostka ruchowa:
Pojedynczy
motoneuron wraz ze
wszystkimi
unerwianymi przez
niego włóknami
mięśniowymi
Choroby nerwowo-mięśniowe - definicja
Choroby spowodowane strukturalnym uszkodzeniem
lub czynnościowym zaburzeniem w tzw. jednostkach
ruchowych, na które składają się:
– komórki ruchowe rogów przednich
– ich wypustki osiowe
– płytka nerwowo-mięśniowa
– włókna mięśniowe
Proces chorobowy może dotyczyć jednostki ruchowej
w całości lub tylko jej części mięśniowej
Choroby nerwowo-mięśniowe - podział
Zależny od poziomu uszkodzenia jednostki
ruchowej:
Choroby obwodowego neuronu
ruchowego
Choroby płytki nerwowo-mięśniowej
Choroby pierwotnie mięśniowe (miopatie)
Wspólne cechy
Proces chorobowy dotyczy jednostki ruchowej
Podobny obraz kliniczny:
- osłabienie mięśni
- zanik mięśni
- wiotkość
Metody badawcze i diagnostyczne:
– Elektromiografia
– Badanie histopatologiczne (biopsja nerwu i mięśnia)
– Swoiste badania biochemiczne
Porównanie cech uszkodzenia neurogennego i
miogennego w EMG

2
Ogólna klasyfikacja (wg Hausmanowej)
Uszkodzenie neuronu ruchowego
(na poziomie komórki rogu przedniego):
– Zanik rdzeniowy mięśni
– Stwardnienie boczne zanikowe
Uszkodzenie neuronu ruchowego
(na poziomie nerwu obwodowego) – neuropatie
Uszkodzenie złącza nerwowo mięśniowego:
– Miastenia
– Zespoły miasteniczne
Uszkodzenie mięśnia (miopatie):
– Dystrofie mięśniowe
– Zespoły miotoniczne
– Wrodzone defekty strukturalne i metaboliczne
– Miopatie nabyte
Miopatie - objawy kliniczne
niedowład (zwykle bardziej nasilony
proksymalnie)
zanik
przerost (prawdziwy lub rzekomy)
przykurcze
nużliwość (męczliwość) – niedowład
pojawiający się lub nasilający się po
wysiłku i zmniejszający się po odpoczynku
ból (samoistny, uciskowy)
Miopatie - objawy kliniczne
kurcze mięśni
miotonia
(przedłużony skurcz mięśni po ruchu
dowolnym, trudności z rozluźnieniem
mięśni)
mioglobinuria
miokimie
sztywność mięśni
brak lub osłabienie odruchów głębokich
Choroby mięśni - miopatie
Pierwotne:
– dystrofie mięśniowe
postępujące
– zapalenie
wielomięśniowe
– miopatie wrodzone
– dystrofia miotoniczna
Wtórne:
– endokrynne
– nowotworowe
– toksyczne
– w kolagenozach
– objawowe zapalenie
mięśni (np. włośnica)
Dystrofie mięśniowe
Postępujące pierwotne choroby mięśni
Uwarunkowane genetycznie
Postępujące zmiany zwyrodnieniowe
mięśnia
Objawy wynikają z postępującego
osłabienia mięśni szkieletowych (niekiedy
również mięśnia serca i mięśni gładkich)
Dystrofia mięśniowa Duchenne’a
Najczęstsza i najcięższa (klasyczna) postać
dystrofii mięśni opisana po raz pierwszy w
połowie XIX w.
Choroba dziedziczona recesywnie w
sprzężeniu z chromosomem X.
Mutacja genu na krótkim ramieniu
chromosomu X w miejscu 21 (Xp21)
1 przypadek na 5000 urodzonych chłopców,
chorobowość – 2/100.000
3
Dystrofia mięśniowa Duchenne’a
Początek choroby: 2 – 4 rok życia
Postępujące symetryczne osłabienie i zanik mięśni
początkowo obręczy biodrowej, potem barkowej,
potem innych
Przerost prawdziwy lub rzekomy mięśni łydek,
czasem innych mięśni
Zaburzenia chodu (chód kaczkowaty), nadmierna
lordoza lędźwiowa, chód na palcach, upadki,
trudność we wstawaniu z pozycji leżącej (objaw
Gowersa – wspinania się po sobie) i wchodzenia na
schody
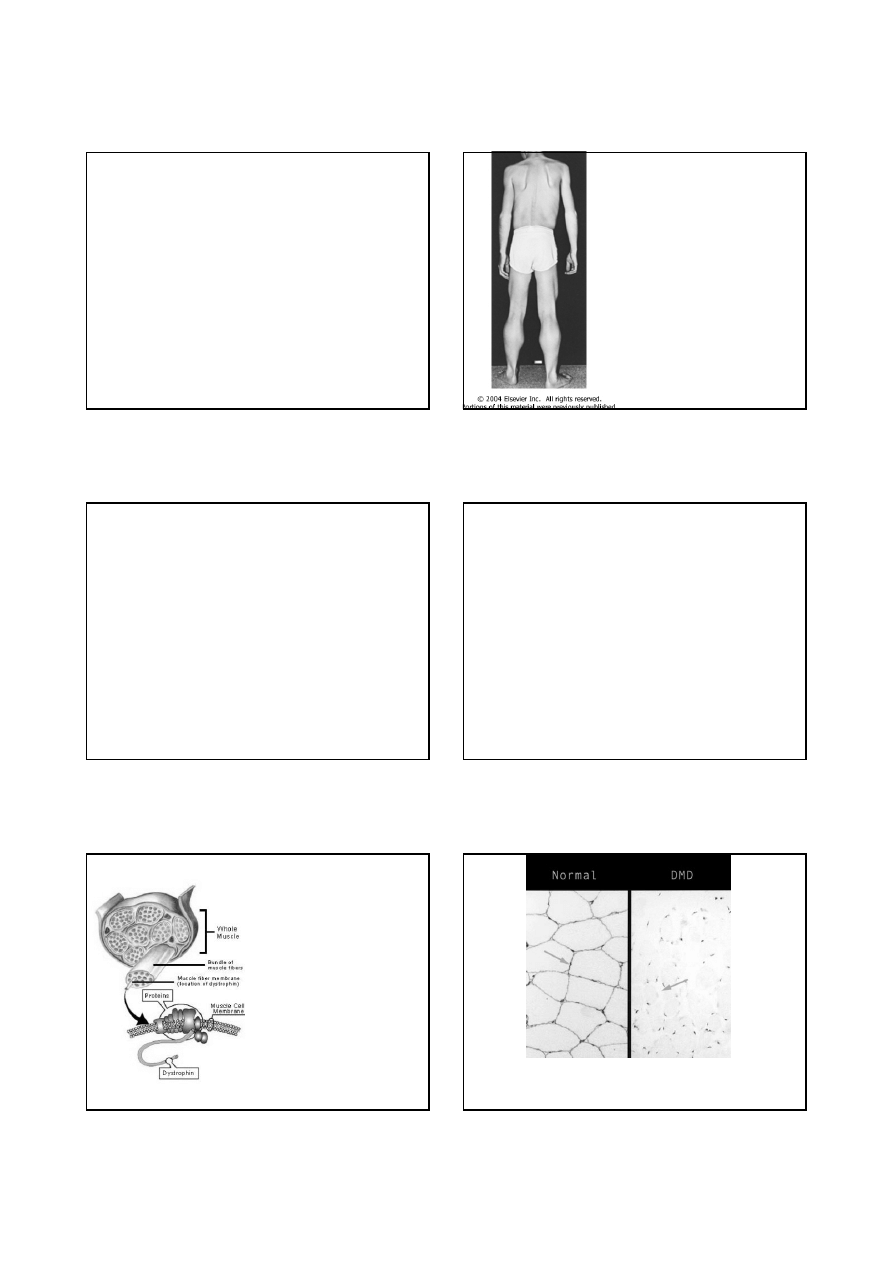
Przerost mięśni łydek u
chorego na dystrofię
Duchenne’a
Dystrofia mięśniowa Duchenne’a
Wychudzenie lub otyłość
Później (8-9 rok życia) przykurcze, głównie ścięgna
Achillesa, potem innych mięśni i ścięgien,
postępujące skrzywienie kręgosłupa
Ok. 9-12 r.ż. Poruszanie się na wózku inwalidzkim
Ok. 20 r.ż. niewydolność oddechowa
30-50% chorych upośledzenie umysłowe, niezależne
od postępu choroby
Dystrofia mięśniowa Duchenne’a
Patomechanizm:
brak białka – dystrofiny
Badania laboratoryjne:
– EMG (zapis miogenny)
– Znaczny wzrost aktywności kinazy kreatynowej (CK)
– Wykrycie delecji w genie dystrofiny w badaniu
genetycznym
– Brak lub niedobór dystrofiny mięśniowej (badanie
immunofluorescencyjne)
– EKG: zaburzenia rytmu
– USG serca: kardiomiopatia rozstrzeniowa lub
przerostowa
Umiejscowienie
dystrofiny w
obrębie włókna
mięśniowego
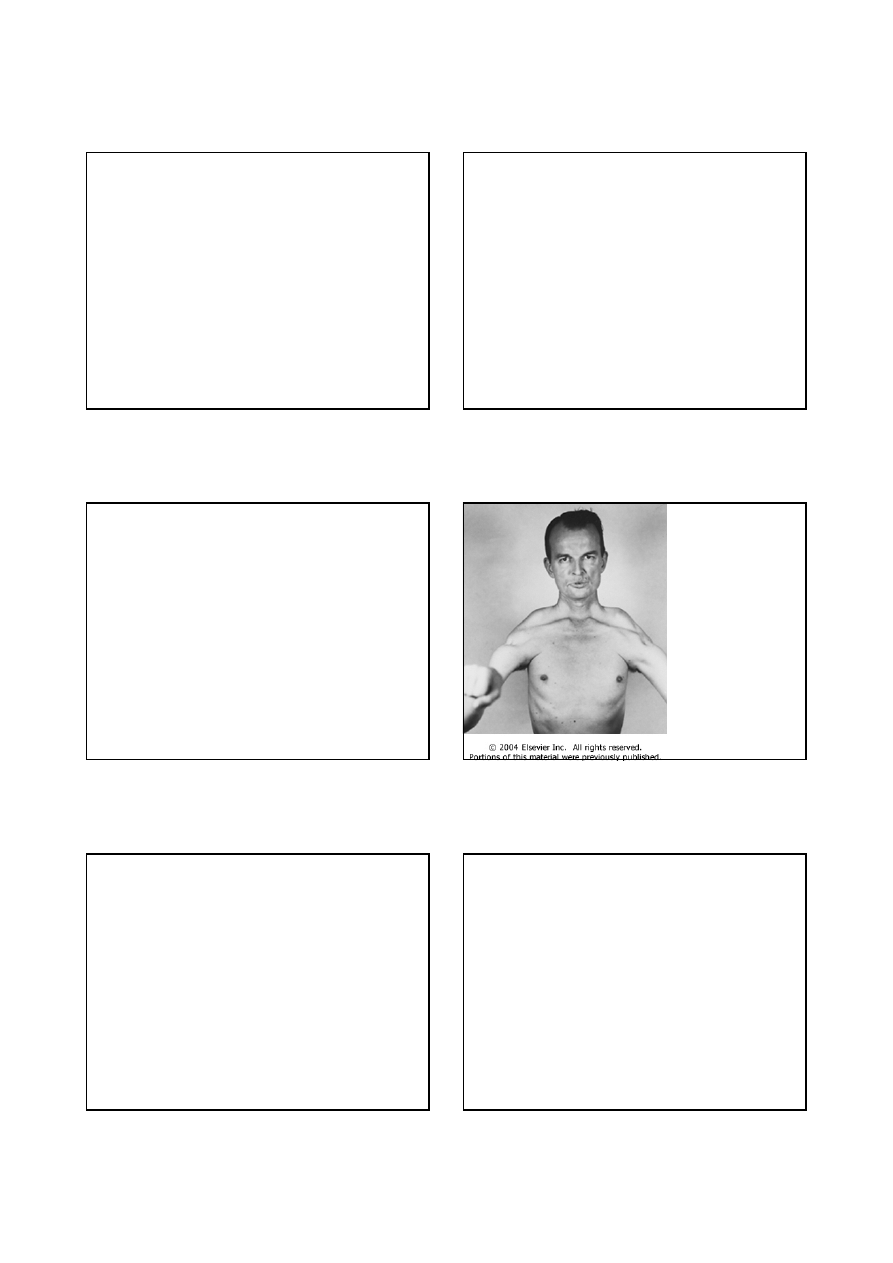
Dystrofina (badanie immunohistochemiczne) w
mięśniu prawidłowym i jej brak w mięśniu
chorego na dystrofię Duchenne’a
4
Dystrofia mięśniowa Duchenne’a
Profilaktyka: poradnictwo genetyczne i
diagnostyka prenatalna (wykrywanie
nosicielek)
Leczenie:
– Wyłącznie objawowe
– Kortykosteroidy (prednizon) opóźniają
unieruchomienie chorego
– Środki wzmacniające, dieta bogatobiałkowa
– Rehabilitacja
– Chirurgiczne leczenie skrzywienia kręgosłupa i
zniekształceń kończyn
Dystrofia mięśniowa Beckera
Patomechanizm i dziedziczenie jak w
dystrofii Duchenne’a
Ze względu na duży niedobór (a nie brak)
dystrofiny, objawy kliniczne występują
później i rozwijają się wolniej
Diagnostyka jak w dystrofii Duchenne’a
Dystrofia twarzowo-łopatkowo-
ramieniowa
Dziedziczenie autosomalnie dominujące, penetracja
prawie pełna, duża kliniczna zmienność
wewnątrzrodzinna
Częstość: 1/20.000 urodzeń
Początek: 7 -17 rok życia: osłabienie dosiebnych
mięśni obręczy barkowej
Osłabienie mięśni twarzy (niemożność gwizdania,
poprzeczny uśmiech), odstające łopatki
Objawy często asymetryczne
Chory na
dystrofię
twarzowo-
łopatkowo-
ramieniową
Dystrofia twarzowo-łopatkowo-
ramieniowa
Przebieg bardzo różny, od poronnego przez
stacjonarny do bardzo ciężkiego, prowadzącego
do unieruchomienia
Rozpoznanie trudne
- CK tylko nieznacznie zwiększona lub w normie,
badanie
- EMG w jakościowej ocenie może dawać obraz
zmian neurogennych
- biopsja mięśniowa nie kwalifikuje zmian do
grupy dystrofii
Dystrofia miotoniczna
Najczęstsza dystrofia mięśniowa u osób
dorosłych
Dziedziczy się jako cecha autosomalnie
dominująca z prawie 100% penetracją.
Mutacja genu – zbyt duża liczba powtórzeń CTG
(powyżej 50) →
nieprawidłowa kinaza
proteinowa
. Antycypacja.
Nieprawidłowo duże mRNA zakłóca składanie
(splicing) pre-mRNA białek ważnych dla
funkcjonowania komórek mięśniowych

5
Dystrofia miotoniczna
Objawy kliniczne:
- osłabienie i zanik mięśni
kończyn i twarzy
- miotonia
- zaćma
- łysina czołowa
- zanik jąder
- zaburzenia osobowości
- nadmierna senność z
bezdechami
-zaburzenia rytmu
Dystrofia miotoniczna -
postępowanie
Leczenie wyłącznie objawowe
Zmniejszanie miotonii – fenytoina,
meksyletyna
Monitorowanie i leczenie zaburzeń rytmu
Wczesne wykrycie zaćmy
Możliwa jest diagnostyka genetyczna (w
tym prenatalna)
Miopatie zapalne
zapalenie wielomięśniowe
lub skórno-mięśniowe
- idiopatyczne
- towarzyszące nowotworom
- towarzyszące kolagenozom
wtrętowe zapalenie mięśni
Idiopatyczne zapalenie wielomięśniowe
– objawy kliniczne
Symetryczne osłabienie mięśni głównie
proksymalnych
Osłabienie mięśni szyi, gardła (dyzartria,
dysfagia)
Bóle samoistne i uciskowe mięśni
Szybka progresja
Spontaniczne remisje i nawroty
Długo zachowane odruchy kolanowe
10% ma zapalną kardiomiopatię
Zapalenie wielomięśniowe
-badania diagnostyczne
Enzymy mięśniowe: CPK, aldolaza
EMG
Wycinek mięśnia
Badania immunologiczne: przeciwciała
przeciwcytoplazmatyczne (antysyntetazowe),
przeciwciała przeciwjądrowe
Zapalenie wielomięśniowe i zapalenie
skórno-mięśniowe
- leczenie
prednizon (80-100 mg/d)
dożylne immunoglobuliny
- w dawce 0,3-0,4g/kg/dobę
- co miesiąc trzydniowe kolejne wlewy
przez okres 3-6 miesięcy
inne leki immunosupresyjne
6
Miopatie mitochondrialne
Patomechanizm związany z zaburzeniami czynności
mitochondriów
Dziedziczone często po linii matczynej (mutacje
mitochondrialnego DNA)
Objawy wynikające z uszkodzenia mięśni, mózgu, innych
tkanek
- postępująca oftalmoplegia zewnętrzna
(bez podwójnego widzenia)
- opadanie powiek
- inne objawy miopatii
- w zależności od mutacji: kardiomiopatia, napady
padaczkowe, udary mózgu
Leki, które mogą powodować lub
nasilać miopatię
statyny
fibraty
glikokortykosteroidy
penicylamina
zydowudyna
chlorochina
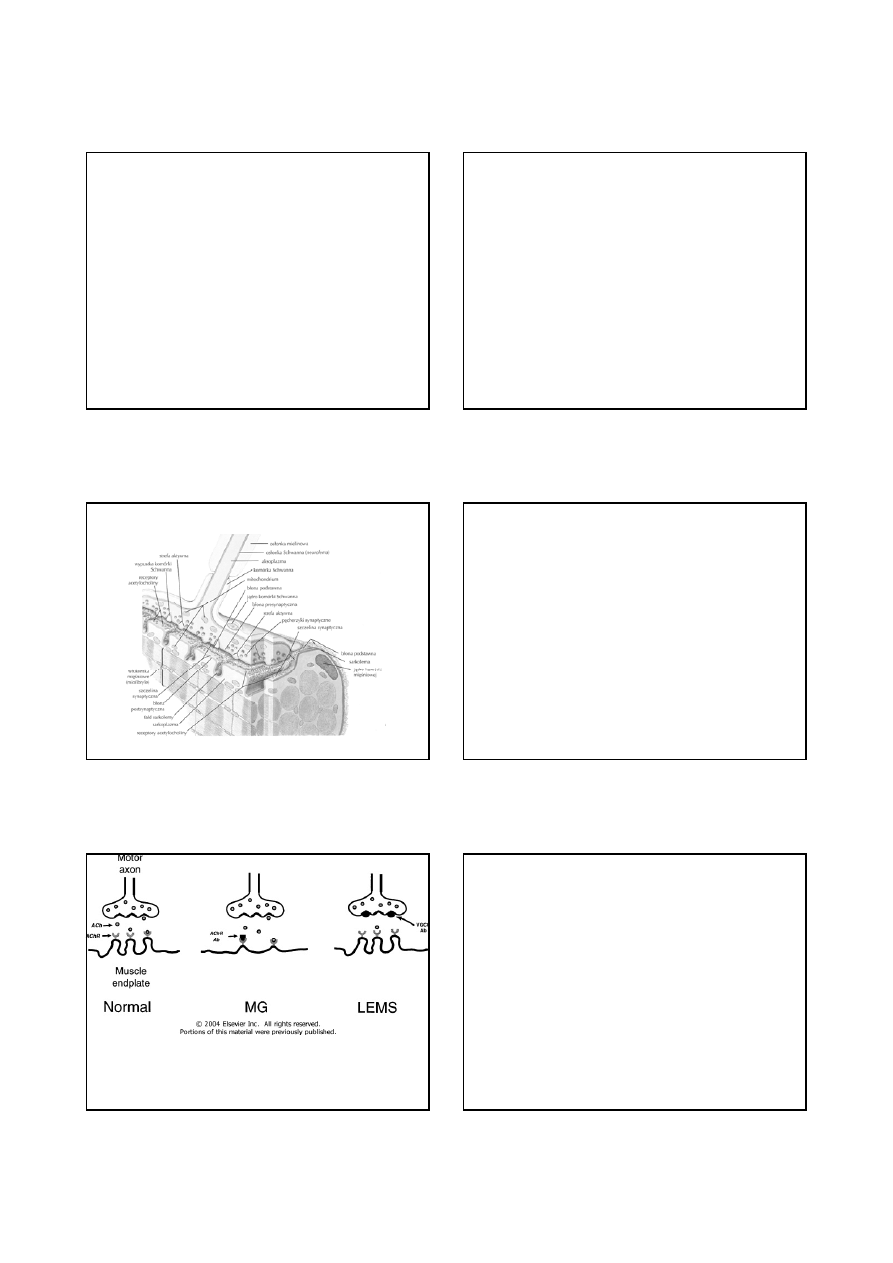
Choroby złącza nerwowo-mięśniowego
Schemat złącza nerwowo-mięśniowego
Miastenia rzekomoporaźna
Myasthenia gravis,
choroba Erba-Goldflama
Chorobowość: 5-12/100.000, zapadalność:
0,2-0,4/100.000/rok
Początek choroby: 18-30 rok życia (kobiety 2-
3-krotnie częściej) lub 45-50 rok życia. Może
występować u dzieci i u ludzi starych
Miastenia rodzinna 1-2%
Lokalizacja zaburzenia transmisji nerwowo-mięśniowej w
miastenii (MG) i w zespole miastenicznym Lamberta-Eatona
(LEMS)
Miastenia - objawy
Główny objaw: męczliwość zależna od
wysiłku fizycznego, ustępująca po
odpoczynku, bardziej nasilona wieczorem
Przebieg: możliwe krótkotrwałe rzuty i
remisje, nasilenie w czasie infekcji,
przemęczenia, stresu emocjonalnego, w
wysokiej temperaturze, w bólu, po lekach,
w ciąży, w czasie miesiączki
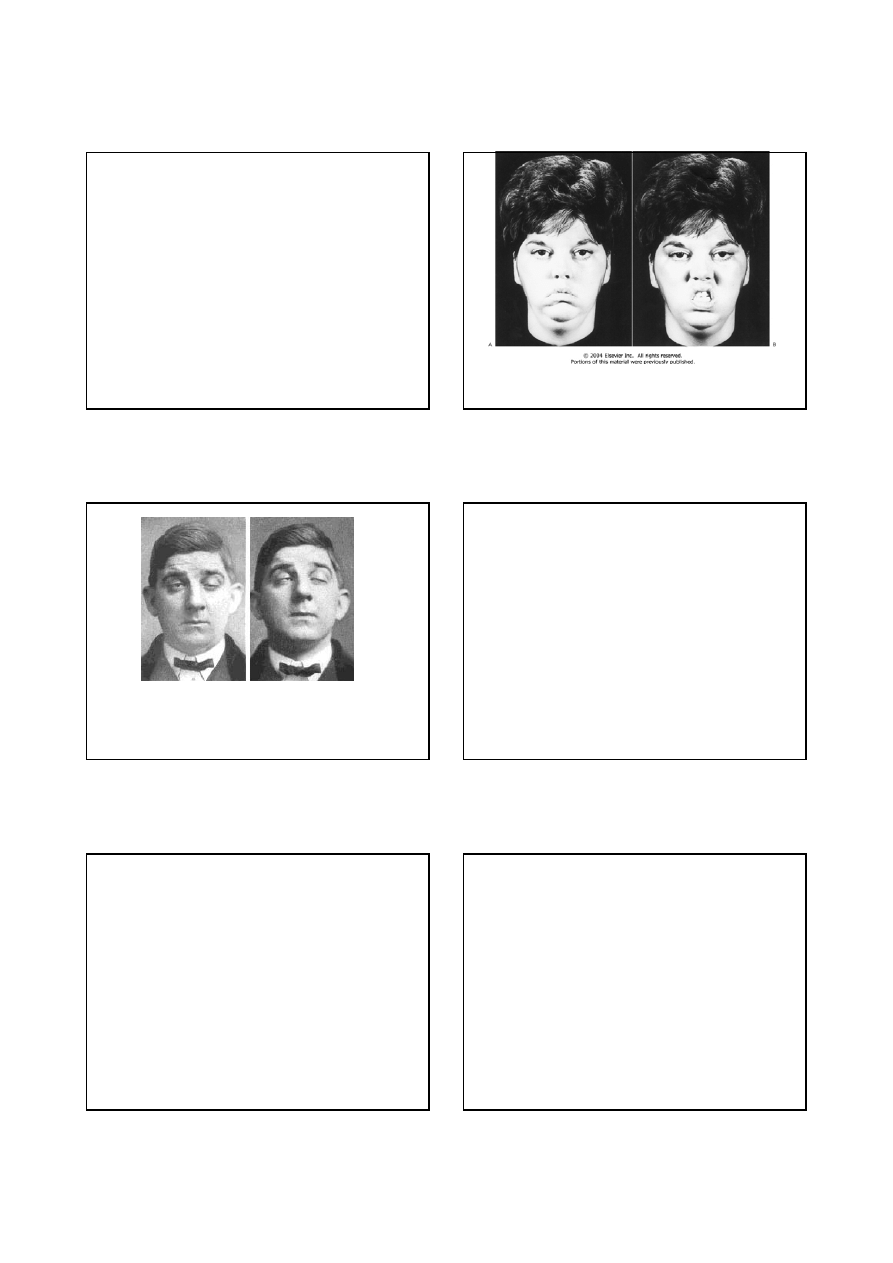
7
Miastenia - objawy
Pierwsze objawy dotyczą mięśni ocznych (60%)
(podwójne widzenie, opadanie powiek)
potem gardła i języka (zaburzenia mowy,
połykania), kończyn, mięśni oddechowych
(duszność, niewydolność oddechowa),
czasem po wielu latach, inne mięśnie (twarzy –
uśmiech Giocondy, żuchwy - opadanie)
Kliniczny podział miastenii:
– Postać oczna
– Postać uogólniona: łagodna, podostra, ostra
Mimika twarzy u chorej na miastenię
Opadanie powiek i niedowład mięśni twarzy u
chorego na miastenię
Miastenia - objawy
Postać łagodna:
– Objawy głównie oczne rozwijają się
powoli z rzutami i remisjami, następnie
dołączają się objawy opuszkowe i
kończynowe
– nie ma zaburzeń oddechowych
– chorzy dobrze reagują na objawowe
leczenie inhibitorami esterazy
cholinowej
Miastenia - objawy
Postać podostra:
– Objawy rozwijają się ostrzej
– dotyczą mięśni opuszkowych, ocznych i
kończynowych bez zaburzeń oddechowych
– odpowiedź na leczenie nie jest pełna, po kilku
miesiącach leczenia reakcja na leki jest słaba lub
żadna
Postać ostra:
– Objawy występują nagle, z ciężkimi objawami
opuszkowymi i niewydolnością oddechową
– Może rozwinąć się z postaci podostrej lub
łagodnej (rzadziej)
Miastenia - objawy
Przełom miasteniczny:
– Ostro narastające osłabienie mięśni, w tym
opuszkowych i oddechowych
Przełom cholinergiczny:
– Ostro narastające osłabienie mięśni, w tym
opuszkowych i oddechowych, w wyniku
przedawkowania inhibitorów esterazy
cholinowej, z towarzyszącymi objawami
cholinergicznymi: zamazane widzenie, wąskie
źrenice, ślinotok, przyspieszone tętno,
biegunka, lęk, itp.
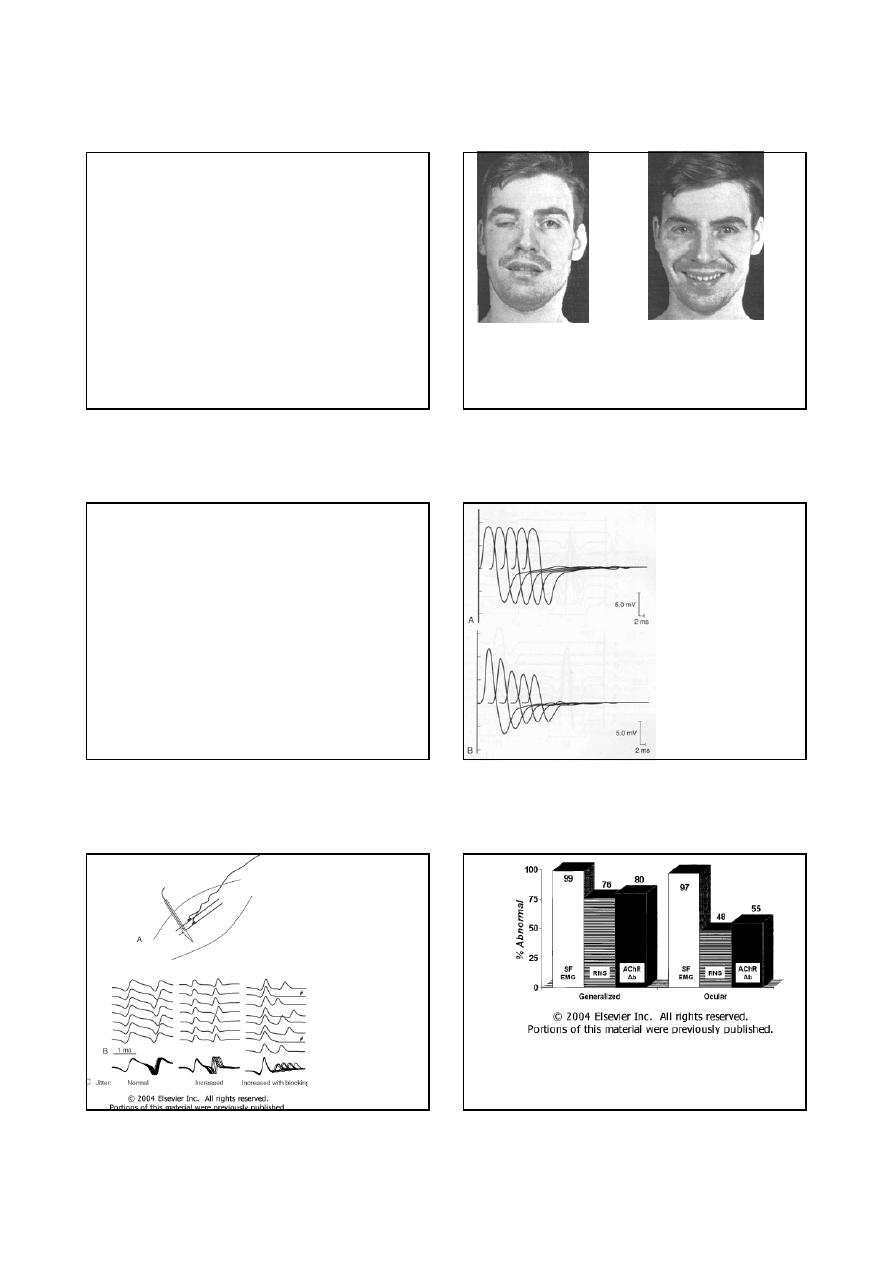
8
Miastenia - rozpoznanie
Wywiad:
- opadanie powiek, podwójne widzenie, trudności
w połykaniu, w mówieniu, osłabienie kończyn
- dolegliwości mają zmienne nasilenie, większe
wieczorem, po wysiłku, w stresie, podczas infekcji
Badanie neurologiczne:
apokamnoza – narastające osłabienie mięśni po
powtarzanych ruchach
-
wielokrotne zaciskanie i otwieranie powiek
-
głośne liczenie bez przerwy do 60
-
utrzymywanie gałek ocznych w skrajnym położeniu
Farmakologiczna próba nużliwości
Pacjent przed i po podaniu edrofonium (Tensilon)
krótkotrwała (minuty) poprawa w zakresie osłabienia
mięśni, powodowana dożylnym podaniem
krótkodziałającego inhibitora acetylocholinesterazy
Miastenia - rozpoznanie
Diagnostyka elektrofizjologiczna
– Elektrostymulacyjna próba nużliwości
– Elektromiografia pojedynczego włókna
mięśniowego
Badania immunologiczne
– Przeciwciała przeciwko receptorom Ach
(85-90% chorych w miastenii uogólnionej,
50-60% chorych w miastenii ocznej)
Ocena grasicy (tomografia śródpiersia)
– Grasiczak u 15-25% chorych
Elektrofizjologiczna
próba nużliwości:
Potencjały ruchowe
powstające wskutek
powtarzanej
stymulacji nerwu
(2 Hz):
A. osoba zdrowa
B. chory na
miastenię
Elektromiografia
pojedynczego
włókna
mięśniowego
(SFEMG) i jej
nieprawidłowości
spotykane w
przebiegu
miastenii
Czułość badań pomocniczych w wykrywaniu
miastenii uogólnionej i ocznej

9
Miastenia rzekomoporaźna
75% chorych ma przetrwałą grasicę
15% chorych ma grasiczaka
3-5% chorych ma choroby tarczycy
3% reumatoidalne zapalenie stawów
2% toczeń układowy
Miastenia rzekomoporaźna i zespół
miasteniczny
Miastenia – zaburzenia transmisji
nerwowo-mięśniowej w wyniku bloku
postsynaptycznego, wywołanego
zablokowaniem receptorów dla ACh
przez przeciwciała.
Zespół miasteniczny występuje w
przebiegu innej choroby lub jest
spowodowany środkami powodującymi
zaburzenia transmisji nerwowo-
mięśniowej w części presynaptycznej
płytki nerwowo-mięśniowej.
Zespół miasteniczny
Nadczynność tarczycy
Niedoczynność tarczycy
Zespół miasteniczny polekowy
– Penicylamina (Cuprenil) w leczeniu choroby
reumatycznej, choroby Wilsona
Zespół Eatona-Lamberta
Zespół Eatona-Lamberta
Często u chorych z rakiem drobno-komórkowym
płuc
Głównie osłabienie proksymalne kończyn dolnych
Bóle mięśni
Brak odruchów kolanowych i skokowych
Objawy autonomiczne: suchość w jamie ustnej,
hipotonia ortostatyczna, zaburzenia potliwość
Słaba reakcja na środki przeciw-cholinesterazowe
Zjawisko torowania przy szybkiej (50 Hz)
stymulacji w przebiegu zespołu miastenicznego
Lamberta-Eatona
Miastenia rzekomoporaźna
– różnicowanie
Nerwica (20-40% pacjentów)
Stwardnienie rozsiane
Postać oczna miastenii – cukrzyca,
zmiany zapalne, zatrucie jadem
kiełbasianym
Postać opuszkowa miastenii – guzy
pnia, tętniaki tylnego kręgu
unaczynienia, zmiany zapalne w pniu

10
Miastenia – różnicowanie z miopatami
Miopatia mitochondrialna – nie ma rzutów
i remisji, choroba postępuje powoli, dotyczy
mięśni gałkoruchowych, a nie powoduje
dwojenia obrazów, mogą być zajęte mięśnie
opuszkowe i kończynowe (
EMG,wycinek)
Dystrofia mięśniowa postać oczna i oczno-
gardłowa
(CPK, EMG, wycinek)
Miastenia – różnicowanie z miopatami
Porażenie okresowe rodzinne – osłabienie
mięśni okresowe, nigdy nie są zajęte nerwy
czaszkowe
(poziom K, EMG)
Zapalenie wielomięśniowe – obraz może
być podobny
(CPK, EMG, próba stymulacji)
Miastenia – różnicowanie z
neurogennym uszkodzeniem mięśni
Stwardnienie zanikowe boczne – objawy
narastają, są objawy uszkodzenia
obwodowego i ośrodkowego uszkodzenia
układu nerwowego
(EMG)
Rdzeniowy zanik mięśni - objawy
narastają bardzo powoli, nie ma zajęcia
mięśni unerwionych przez nerwy czaszkowe
(EMG, wycinek)
Miastenia – różnicowanie z
neurogennym uszkodzeniem mięśni
Zapalenie wielonerwowe Guillaina-Barrego
(p
ł
yn mózgowy-rdzeniowy, EMG)
Polineuropatia w przebiegu porfirii
(metabolizm porfirin)
Miastenia - profilaktyka
Unikanie nadmiernego wysiłku fizycznego, stresu, infekcji
Ostrożność w zlecaniu szczepień
Unikanie leków, które zaburzają przekaźnictwo nerwowo-
mięśniowe:
- Leki blokujące złącze nerwowo-mięśniowe: kurara,
chinina
- Antybiotyki: aminiglikozydy, tetracykliny, ampicylina,
erytromycyny, klindamycyna
- Leki stabilizujące błonę komórkową:
chinina, prokainamid, lignokaina, fenytoina, beta-blokery
- Sole litu
- chlorpromazyna
Miastenia - leczenie
Inhibitory cholinesterazy:
– pirydostygmina (Mestinon, tabl., amp., syrop)
– ambenonium (Mytelase, tabl.)
– neostygmina (Polstigminum, tabl., amp.)
Tymektomia
Kortykosteroidy
Leki immunosupresyjne
- azatiopryna
- cyklosporyna
- cyklofosfamid
Plazmafereza
Immunoglobuliny

11
Miastenia – tymektomia
wskazania
– Obecność grasiczaka
– Obecność przetrwałej grasicy u chorych z postacią
uogólnioną lub opuszkową, którzy nie reagują dobrze
na leczenie farmakologiczne
zalety
– ustąpienie objawów u 30-40%
– poprawa u 30-40%
– nieobecność odległych objawów niepożądanych
– poprawa najczęściej w okresie 1-2 lat, może być do 10
lat
Miastenia – leczenie
immunosupresyjne
Immunosupresja wskazana przy braku poprawy
po inhibitorach acetylocholinesterazy:
– kortykosteroidy
– azatiopryna
– cyklospryna
Miastenia oczna
Słabo reaguje na inhibitory cholinesterazy
Tymektomia niezalecana
Dobra reakcja na immunosupresję
– mniejsze dawki prednizonu (0,5 mg/kg)
– cyklofosfamid
Miastenia u kobiet ciężarnych
Ciąża w zróżnicowany sposób wpływa na
nasilenie objawów
objawy mogą zaostrzyć się kilka miesięcy
po porodzie
inihibitory cholinesterazy doustne są
bezpieczne
inihibitory cholinesterazy dożylne są
przeciwwskazane (poronienie,
przedwczesny poród)
Choroby neuronu ruchowego
Uszkodzenie górnego i dolnego neuronu
ruchowego
- stwardnienie boczne zanikowe
Uszkodzenie dolnego neuronu ruchowego
- rdzeniowy zanik mięśni
Uszkodzenie górnego neuronu ruchowego
- pierwotne stwardnienie boczne
Stwardnienie boczne zanikowe
pierwotnie postępująca choroba neurodegeneracyjna o
nieustalonej przyczynie (rzadko rodzinnie)
nieodwracalne uszkodzenie neuronów ruchowych w korze
ruchowej, w pniu mózgowym i rdzeniu kręgowym
zwykle w wieku średnim lub starszym
Powoli postępują objawy uszkodzenia:
dolnego neuronu ruchowego (niedowład, zanik mięśni,
fascykulacje, zespół opuszkowy)
górnego neuronu ruchowego (niedowład, wygórowane odruchy
głębokie, objaw Babińskiego, zespół rzekomoopuszkowy)
Zwykle nie występują zaburzenia czucia ani zwieraczy, rzadko
zaburzenia poznawcze
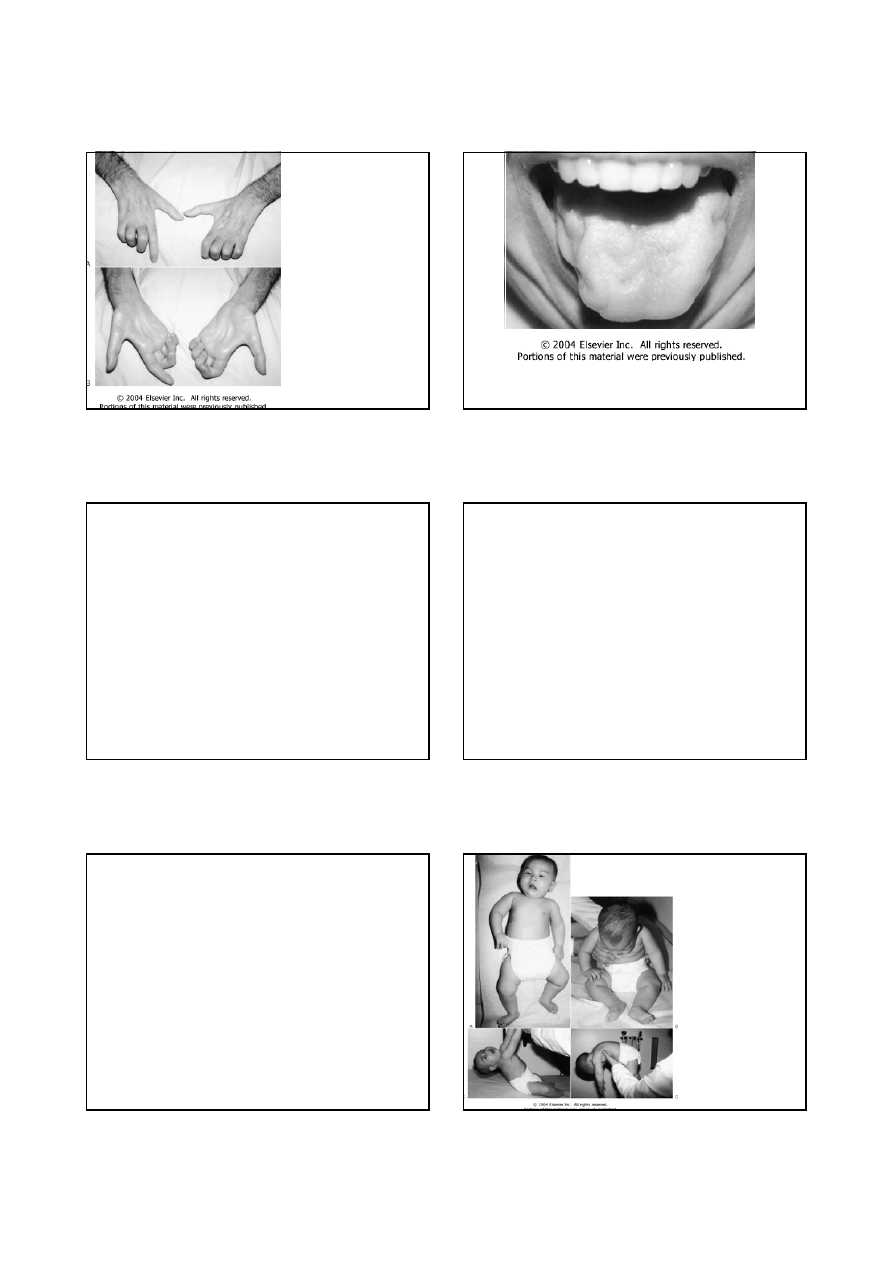
12
Zanik mięśni rąk
u chorego na
stwardnienie
boczne zanikowe
Zanik mięśni języka w przebiegu stwardnienia bocznego zanikowego
Stwardnienie boczne zanikowe
- diagnostyka
EMG
W różnicowaniu wykorzystuje się m.in.:
- RM rdzenia szyjnego
- badanie płynu mózgowo-rdzeniowego
- próby nużliwości
- badanie przewodnictwa nerwowego
Stwardnienie boczne zanikowe - postępowanie
Choroba postępuje, prowadząc do śmierci w ciągu 3-
5 lat
Leczenie quasiprzyczynowe: riluzol (antagonista
NMDA)
Leczenie objawowe i paliatywne:
- przezskórna gastrostomia
- nieinwazyjna wentylacja
- leczenie nietrzymania afektu (amitryptylina)
- rehabilitacja
Kwestie związane z terminalnym okresem choroby
Rdzeniowy zanik mięśni (SMA)
genetycznie uwarunkowane zwyrodnienie komórek
ruchowych rogów przednich rdzenia
mutacja genu SMN (survival motor neuron) na
chromosomie 5
SMA I-IV (różnią się początkiem wystąpienia choroby i
czasem jej trwania)
postępujące objawy z dolnego neuronu ruchowego:
niedowład, zanik, fascykulacje, wiotkie napięcie
zajęcie mięśni opuszkowych, oddechowych
bez zaburzeń czucia, zaburzeń zwieraczy czy funkcji
intelektualnych
diagnostyka: EMG, badanie genetyczne, biopsja mięśnia
leczenie: rehabilitacyjne
Uogólniona
wiotkość
mięśni w
przebiegu
rdzeniowego
zaniku mięśni
(SMA I)
Wyszukiwarka
Podobne podstrony:
Choroby nerwowo-mięsniowe(1), fizjoterapia
metody oceny AUN, choroby nerwowo-mięśniowe
Choroby nerwowo miesniowe
Choroby nerwowo mięśniowe
Choroby nerwowo mięśniowe
choroby nerwowo mięśniowe
Choroby nerwowo mięśniowe
Choroby nerwowo miesniowe
choroby nerwowo-miesniowe, choroby i ich leczenie
Choroby nerwowo mięśniowe
Wytyczne postępowania w chorobach nerwowo - mięśniowych , Neurologia1
choroby nerwowo-mięśniowe, neurologia
choroby nerwowo miesniowe, Neurologia(1)
Choroby nerwowo miesniowe (2)
05 Choroby nerwowo miesniowe
więcej podobnych podstron