
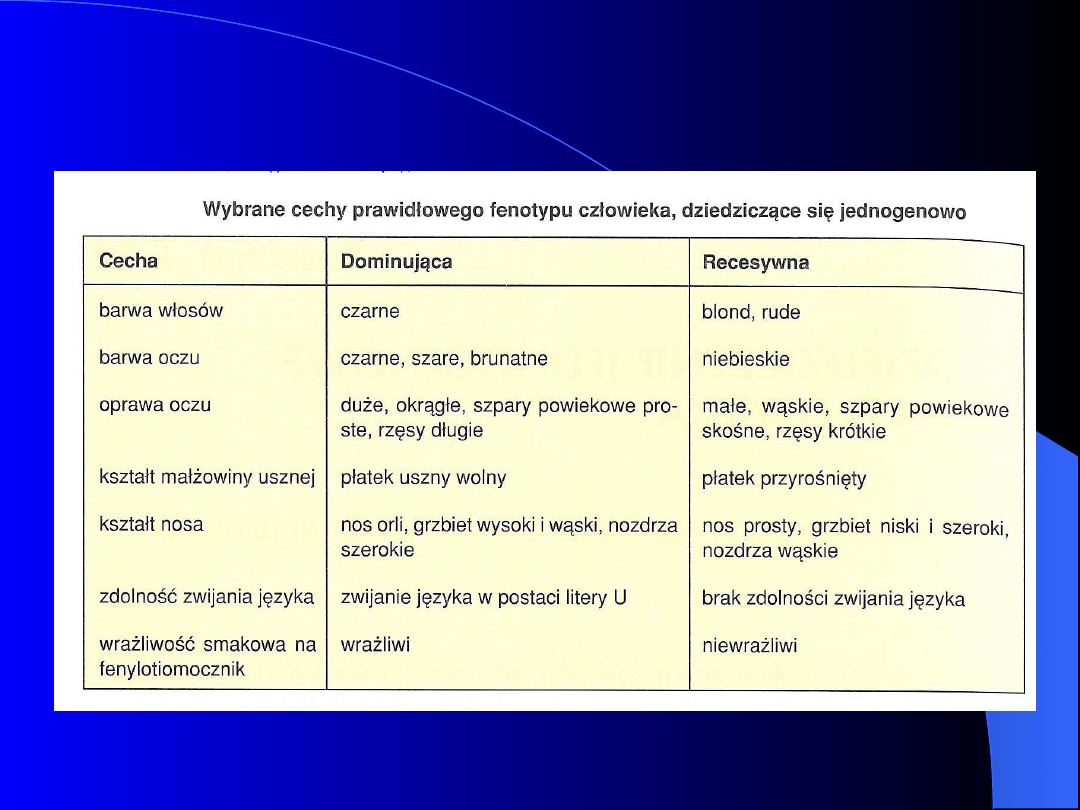
Dziedziczenie
mendlowskie
prawidłowych i
patologicznych cech
człowieka

Ogólne cechy dziedziczenia
Ogólne cechy dziedziczenia
autosomalnego dominującego u
autosomalnego dominującego u
ludzi
ludzi
Cecha (choroba) jest przekazywana z
pokolenia na pokolenie „pionowo”.
Niektóre choroby monogenowe ujawniają
się w późnym wieku , np. choroba
Huntingtona.
Występowanie
chorób
autosomalnych
dominujących może być wynikiem mutacji
de novo o czym decyduje głównie wiek ojca.
Choroba występuje z tą samą częstością u
obu płci.
Nasilenie objawów choroby (zmienność
cechy) może zależeć od płci chorego rodzica
przekazującego cechę.

Obecność lub brak cech klinicznych
oraz ich nasilenie zależy od:
stopnia penetracji patologicznego
genu (pełna penetracja – 100% lub 1;
niepełna penetracja – poniżej 100 %
lub <1),
zmiennej ekspresji genu (chorzy w tej
samej
rodzinie
wykazują
różne
nasilenie objawów choroby),
wieku probanda (penetracja zależna
od wieku).

Geny dominujące wykazują czasem
niepełną penetrację, stąd zjawisko
wyciszania
typowych
objawów
choroby aż do ich zupełnego
zaniku. W wyniku tego zjawiska
może dojść do dziedziczenia z
przeskokiem
pokoleniowym.
Chorują np. dziadkowie i wnuki,
podczas gdy rodzice są zdrowi.

W przypadku kiedy dany gen w populacji
osobników o tym samym genotypie przejawia
się w ten sposób, że badana cecha wykształca
się z różnym nasileniem w fenotypie, to mówi
się o nim, że wykazuje niepełną
ekspresywność.
Jeżeli wśród osobników o tym genotypie tylko
część wykazuje cechę wywołaną posiadanym
genem, a pozostałe osobniki mimo posiadania
tego genu nie przejawiają efektu
fenotypowego, to mówi się, że gen wykazuje
niepełną penetrację.

Jeden gen może wpływać
jednocześnie na powstanie bardzo
różnych cech organizmu. Zjawisko
to określamy jako plejotropowe
działanie genu.

Przykłady chorób dziedziczących się
Przykłady chorób dziedziczących się
autosomalnie dominująco
autosomalnie dominująco
Achondroplazja
(chondrdystrofia, karłowatośc
chondrodystroficzna)
Mała zmienność ekspresji, pełna penetracja
Częstość występowania 1 : 15 000 i 1:77 000
Gen – locus 4p 16.3
Gen receptora czynnika wzrostu
fibroblastów (Fibroblast Growth Factor
Receptor 3 – FGFR3)
Mutacja w nukleotydzie 1138 genu FGFR3:
–
tranzycja G A lub transwersja G C

Achondroplazja
Objawy:
- niski wzrost
- skrócenie kończyn, mikromelia
- szpotawe kolana,
- nadmierna lordoza lędźwiowa,
- duża głowa z wypukłym czołem i
zapadniętą nasadą nosa,

Zespół Marfana (arachnodaktylia)
Częstość występowania 1: 10 000
Gen FBN1 (gen fibrylliny) – locus
15q21.1
Wysoki stopień penetracji i zmienna
ekspresja
Fibryllina – białko o masie 350 kD, jest
głównym składnikiem
zewnątrzkomórkowych mikrofibrylli
Jest to defekt tkanki mezenchymalnej
powodujący zmiany w układzie kostno-
stawowym, w układzie krążenia i w
gałkach ocznych

Zespół Marfana
Objawy:
- smukła sylwetka, wysoki wzrost,
- nadmiernie długie palce rąk i stóp,
- „kurza” lub „lejkowata” klatka
piersiowa,
- nadmiernie elastyczna skóra,
- wady wrodzone serca, tętniaki
aorty, wypadanie zastawki mitralnej,
- krótkowzroczność,

Nerwiakowłókniakowatość
(Neurofibromatosis – NF, choroba
Recklinghausena)
Dwie postaci choroby: NF-1 i NF-2
Gen NF-1 (locus – 17q11.2) – pełna
penetracja, zmienna ekspresja
Produkt genu – białko neurofibromina
(obniżony poziom sprzyja rozwojowi
nowotworów)
Częstość 1:3500
Gen NF-2 (locus – 22q12.2) – pełna penetracja
Produkt genu – merlina (białko cytoszkieletu)
Częstość 1:35 000 – 1:40 000

Nerwiakowłókniakowatość
Objawy
mutacja genu NF-1:
- zmiany barwnikowe na skórze (café au lait) -
we wczesnym dzieciństwie
- w okresie dojrzewania rozwijają się liczne
guzki wywodzące się z nerwów obwodowych
(włókniaki, nerwiakowłókniaki, glejaki nerwu
wzrokowego - zwykle łagodne),
- często niedorozwój umysłowy i padaczka,
mutacja genu NF-2:
- nerwiaki nerwu słuchowego,
- oponiaki rdzenia,
- zmętnienie soczewek,
- pierwsze objawy w okresie dojrzewania lub
w drugiej dekadzie życia,

Siatkówczak (Retinoblastoma)
Nowotwór gałki ocznej
Postać sporadyczna i dziedziczna
(rodzinna)
Częstość występowania 1:20 000
Gen Rb – locus 13q 14.1
Przyczyną choroby jest mikrodelecja
regionu 13q 14.1 lub mutacja genu Rb
zlokalizowanego w tym regionie.
Gen Rb wykazuje właściwości
supresorowe, kodowane przez niego białko
wiąże czynnik transkrypcyjny ,
odgrywający rolę w cyklu komórkowym.

Choroba Huntingtona (Huntington
Disease HD)
Częstość występowania 4 –7 : 100 000
Pełna zależna od wieku penetracja
Gen HD – locus 4p 16.3
Białko – huntingtina
Mutacja dynamiczna: niestabilność
trójnukleotydowych sekwencji powtarzalnych -
(CAG)
n
na końcu 5’ genu kodującego huntingtinę
Osoby zdrowe: 10 –20 powtórzeń CAG
30 – 35 powtórzeń stan przedmutacyjny
Osoby chore: >36 powtórzeń
Antycypacja – ostrzejszy przebieg choroby w
następujących po sobie pokoleniach oraz
występowanie choroby w coraz młodszym wieku
Antycypacja w HD jest mocniej wyrażona, jeżeli
zmutowany gen przekazywany jest przez ojca.

Choroba Huntingtona
Objawy:
- początek choroby zwykle w czwartej
dekadzie życia,
- zmiany neuropatologiczne – selektywne
obumieranie komórek jądra ogoniastego,
- zaburzenia hiperkinetyczne
(przypominające taniec)
- zaburzenia mowy,
- postepująca utrata aktywności
umysłowej,
- charłactwo fizyczne,

Ogólne cechy dziedziczenia
Ogólne cechy dziedziczenia
autosomalnego recesywnego u ludzi:
autosomalnego recesywnego u ludzi:
Choroby o tym typie dziedziczenia
występują głównie u rodzeństwa (poziome
przekazywanie cechy).
Cecha (choroba) ujawnia się tylko u
homozygot
recesywnych
(rodzice
są
heterozygotami
pod
względem
zmutowanego genu).
Częstość
występowania
chorób
autosomalnych
recesywnych
(w
tym
rzadkich) jest zwiększona w małżeństwach
spokrewnionych.

Choroby
dziedziczone
autosomalnie
recesywnie najczęściej są wynikiem mutacji
genów
strukturalnych,
kontrolujących
syntezę
białek
enzymatycznych,
co
prowadzi do zaburzeń metabolicznych
ustroju, a przez to do zaburzeń jego
procesów życiowych. Większość bloków
metabolicznych dziedziczy się autosomalnie
recesywnie.
W
niektórych
chorobach
recesywnych
istnieje
możliwość
pośredniego wykrywania heterozygot –
nosicieli zmutowanego genu, np. metoda
obciążania fenyloalaniną w fenyloketonurii.

Fenyloketonuria
Częstość występowania w populacjach
europejskich średnio 1: 10 000
Locus genu – 12q 24.1
Brak hydroksylazy fenyloalaninowej
(4-monooksygenazy fenyloalaninowej)
Wykrywanie :
test Guthriego – wykrywanie we krwi
zwiększonego stężenia fenyloalaniny
test z FeCl
3
– wykrywanie w moczu kwasu
fenylopirogronowego,
oznaczanie poziomu fenyloalaniny i tyrozyny w
surowicy krwi i ich metabolitów w moczu –
metody enzymatyczne, chromatograficzne i
fluorymetryczne.

Fenyloketonuria
Objawy:
- jasne włosy i jasna karnacja,
- uporczywe wymioty,
- „mysi” zapach moczu,
- wzrost napięcia mięśniowego,
- kiwanie tułowia,
- upośledzenie umysłowe,
Zastosowanie diety pozbawionej
fenyloalaniny umożliwia rozwój
intelektualny zbliżony do normalnego
.

Albinizm (bielactwo uogólnione)
–
częstość średnio 1: 10 000
–
blok metaboliczny przemiany
tyrozyny
–
mutacja genu kontrolującego syntezę
monooksygenazy monofenolowej i
oksydazy katecholowej (tyrozynaza)
–
zahamowanie syntezy melanin w
melanocytach naskórka, cebulek
włosowych, tęczówce i siatkówce

Albinizm
Objawy:
- skóra różowoczerwona,
- włosy białe,
- tęczówki nibieskie lub różowe
(„czerwone źrenice”),
- światłowstręt, zmniejszona
ostrość wzroku,

Alkaptonuria
- częstość 1 : 200 000
- brak 1,2-dioksygenazy
homogentyzynianowej
- kwas homogentyzynowy wydalany z
moczem
Objawy:
- ochronoza – ciemnienie chrząstek,
ścięgien i więzadeł,
- zmiany zapalne i zwyrodnieniowe stawów,
- ciemnienie moczu po zetknięciu z
powietrzem,

Choroba Gauchera
niedobór beta – glukozydazy,
rozkładającej glukocerebrozyd do
glukozy i ceramidu
Choroba Niemanna-Picka
(sfingomielinoza)
niedobór sfingomielinazy, rozkładającej
sfingomielinę do ceramidu i fosfocholiny
Choroba Tay-Sachsa
niedobór N-acetyloheksozaminidazy,
odszczepiającej N-acetylogalaktozaminę
od glikolipidu

Mukowiscydoza (zwłóknienie
torbielowate trzustki, cystic fibrosis –
CF)
Częstość :
- Europa 1 :2500
- rasa czarna 1 : 17 000
- rasa żółta 1 : 90 000
Gen CFTR (Cystic Fibrosis
Transmembrane Regulator gene) – locus
7q 31–q32
Wielkość genu 250 kb, 27 eksonów

Mukowiscydoza
Zidentyfikowano ponad 500 różnych
mutacji w genie CFTR
Białko CFTR – błonowy regulator
przewodnictwa specyficzny dla
mukowiscydozy
Charakterystyczny objaw: słony pot
Podwyższone stężenie Na+ i Cl- w
pocie
Na+ > 60 mmol/l (n. 20-25mmol/l)
Cl- .70 mmol/l (n. 16-19 mmol/l)

Mukowiscydoza
Objawy:
- słony pot,
- u noworodków : niedrożność
smółkowa, powiększenie brzucha,
wymioty, brak smółki,
- nawracające infekcje dróg
oddechowych,
- zmiany oskrzelowo-płucne (w 90%
przypadków są przyczyną śmierci)
- zaburzenia procesów trawienia,
- dysfunkcja gruczołów wydzielniczych,
- wrodzony brak nasieniowodów,

Niedokrwistość
sierpowatokrwinkowa
Gen HBB – locus 11p 15.15, koduje
beta-globinę
Hemoglobina S (HbS)
Mutacja punktowa w genie HBB –
zamiana tripletu GAG (kwas
glutaminowy) na GUG (walina)
Heterozygoty HbS/HbA – większa
odporność na zarażenie zarodźcem
malarii
Homozygoty (HbS/HbS) – ciężka
postać niedokrwistości hemolitycznej

Niedokrwistość
sierpowatokrwinkowa
Objawy:
- sierpowaty kształt erytrocytów,
- krwinki łatwiej ulegają hemolizie,
- podwyższona lepkość krwi,
skłonność do tworzenia
zakrzepów,
- niewydolność krążenia,
uszkodzenie wiele organów,

Ogólne cechy dziedziczenia dominującego
Ogólne cechy dziedziczenia dominującego
sprzężonego z chromosomem X
sprzężonego z chromosomem X
Dominujący tor dziedziczenia chorób
sprzężonych z chromosomem X należy
do rzadkości i można go podejrzewać
gdy:
chory mężczyzna ma wyłącznie chore
córki i wyłącznie zdrowych synów,
chore
kobiety
heterozygoty
przekazują
cechę
50%
swego
potomstwa, niezależnie od płci,

Ogólne
cechy
dziedziczenia
dominującego
sprzężonego
z
chromosomem X
chore
kobiety
homozygoty
przekazują
cechę
wszystkim
swoim dzieciom,
w potomstwie chorej kobiety
(heterozygoty)
i
zdrowego
mężczyzny 50% synów i 50%
córek będzie chorych.

Wrodzona hipoplazja skóry
(Incontinentia pigmenti – zespół
Blocha i Sulzbergera)
Częstość 1: 75 000
Locus genu – Xq 27-q28
Chorują głównie dziewczęta
Dla płodów męskich – zespół
letalny

Wrodzona hipoplazja skóry
Objawy:
- dla płodów męskich – zespół letalny,
- po urodzeniu na skórze dziecka
pojawiają się plamy rumieniowe,
zawierające male pęcherzyki,
- naturalny tatuaż skóry,
- zez,
- wady układu kostnego i serca,
- u 50% chorych – niedorozwój
umysłowy, porażenia, napady
drgawek,

Zespół Retta
Częstość :
Europa 1: 15 000
USA 1 : 20 000
Locus genu – Xp 21.3
Dla płci męskiej - letalny

Zespół Retta
Objawy:
- zaburzenia psychoruchowe –
pojawiają się między 6 a 18
miesiącem życia,
- głębokie upośledzenie umysłowe,
- zaburzenia neurologiczne
(padaczka, spastyczność),
- liczne zaburzenia w rozwoju
fizycznym,

Zespół łamliwego chromosomu X
(s. Zespół Martina-Bella, fragile X
syndrom)
Częstość:
Mężczyźni – 1: 1250
Kobiety – 1 : 2000
Uszkodzenie funkc ji genu FMR1 (Fragile
X Mental Retardation)
Locus genu – Xq 27.3 (37kb, 17 eksonów)
Niestabilne sekwencje (CGG)
n
Osoby zdrowe 6 – 52 powtórzeń CGG
Nosiciele bezobjawowi 52 – 200
Osoby chore 230 – 1500 (pełna mutacja)

Zespół łamliwego chromosomuX
Produkt genu FMR1 – białko FMRP – 64
aminokwasy, należy do białek
wiążących RNA.
Kobiety z pełna mutacją – upośledzenie
umysłowe w stopniu lekkim (20-30%)
lub umiarkowanym (1-2%)
Mężczyźni z pełną mutacją ciężki
stopień upośledzenia (max. IQ = 31)

Zespół łamliwego chromosomu X
Objawy:
u noworodków i niemowląt płci męskiej:
- niska waga urodzeniowa,
- mały obwód głowy,
- zwiększona objętość jąder,
- duże małżowiny uszne,
- hipotonia mięśniowa,
u dzieci:
- autyzm, zaburzenia mowy,
- opóźnienie rozwoju psychoruchowego,
u dorosłego mężczyzny:
- deformacje twarzoczaszki (wydatne guzy czołowe,
duże i odstające małżowiny uszne, duża żuchwa,
wysokie podniebienie, hiperteloryzm),
- powiększenie jąder, bladoniebieskie tęczówki,
zniekształcenia kręgosłupa, padaczka, encefalopatia,

Zespół łamliwego chromosomu X
Objawy
- u 1/3 kobiet z pełną mutacją genu
FMR1 występują:
- zmiany w obrębie twarzoczaszki,
upośledzenie umysłowe średniego
stopnia ( IQ 24-41),
- spowolnienie ruchowe,
- trudności wymowy,
- problemy z koncentracją,

Ogólne cechy dziedziczenia recesywnego
Ogólne cechy dziedziczenia recesywnego
sprzężonego z chromosomem X
sprzężonego z chromosomem X
O recesywnym sposobie dziedziczenia
chorób sprzężonych z chromosomem
X mówimy wówczas gdy:
choroba występuje znacznie częściej
u mężczyzn (hemizygota) niż u
kobiet,
kobiety chorują tylko w układzie
homozygotycznym dla zmutowanej
pary alleli,

u kobiet heterozygot (nosicielek
zmutowanego genu) choroba nie
występuje,
chory
mężczyzna
nigdy
nie
przekazuje choroby (cechy) synom, a
wszystkie córki chorego mężczyzny
są nosicielkami zmutowanego genu,
istnieje 50% prawdopodobieństwa,
że
heterozygotyczne
kobiety
przekażą zmutowany gen zarówno
synom, jak i córkom.

Dystrofia mięśniowa Duchenne’a
(DMD)
Częstość u chłopców – 1 : 3500
Locus genu – Xp 21.2 (2,5 mln par zasad,
79 eksonów)
Sekwencje niekodujące stanowią 99,4%
genu.
Brak dystrofiny w mięśniach, zgon w
drugiej dekadzie życia.
Dystrofina – białko strukturalne
zlokalizowane po stronie cytoplazmatycznej
błony komórkowej.
Choroba ujawnia się przed 5 rokiem życia

Dystrofia mięśniowa typu Duchenne’a
Objawy:
- symetryczny zanik mięśni obręczy
miednicy i barkowej,
- „kaczy” chód,
- trudności przy wchodzeniu po
schodach i wstawaniu z pozycji
leżącej,
- przerost łydek,
- zwiększona aktywność w surowicy
krwi kinazy kreatynowej, aldolazy i
dehydrogenazy mleczanowej,

Dystrofia mięśniowa Beckera
(BMD)
Częstość – 1 : 20 000
Nie jest letalna.
Mniejsza ilość dystrofiny (3-10%)
lub zmieniona dystrofina w
mięśniach.
DMD i BMD to alleliczne formy tej
samej choroby.

Dystrofia mięśniowa typu Beckera
Objawy:
- pojawiają się w drugiej dekadzie
życia,
- zanik mięśni kończyn najpierw
dolnych, a potem górnych,
- przebieg łagodniejszy, ale
prowadzi do inwalidztwa,

Ślepota na barwy
Choruje od 5 do 9% mężczyzn
Locus genu – Xq 28
Protanopia - ślepota na barwę czerwoną
Deuteranopia - ślepota na barwę zieloną
Tritanopia – ślepota na barwę niebieską
Niedowidzenie barw:
- protanomalia
- deuteranomalia
- tritanomalia
Gen odpowiedzialny za percepcję barwy
niebieskiej zajduje się na chromosomie 7.

Hemofilia A i hemofilia B
Częstość występowania hemofilii A
wynosi od 1 :10 000 do 1 : 20 000
Locus genu Xq 28 (186 kb)
Niedobór lub brak VIII czynnika
krzepnięcia krwi
Częstość występowania hemofilii B
(choroba Christmasa) wynosi 1 : 30 000
Locus genu – Xq 27.1-q 27.2
Brak czynnika IX krzepnięcia krwi

Hemofilia
Objawy:
- w ciężkiej postaci: samoistne wylewy
dostawowe prowadzące do inwalidztwa
(artropatia dostawowa)
- w umiarkowanej postaci: krwawienia
występują po urazach, wylewy
śródstawowe są mniej ciężkie i występują
rzadziej,
- w postaci łagodnej wylewy występują
tylko po znacznych urazach lub po
operacjach,
- czas krzepnięcia krwi znacznie
wydłużony,

Dziedziczenie grup krwi

UKŁAD GRUPOWY ABO
UKŁAD GRUPOWY ABO
został po raz pierwszy opisany w 1901 roku przez
Landsteinera
zawiera antygeny A i B, zwane substancjami
grupowymi, na podstawie których wyróżnia się
cztery podstawowe grupy krwi : A, B, AB, O
antygen A posiada wiele odmian, z których
najważniejsze są odmiany A
1
, A
2
i dlatego wyróżnia
się następujące grupy krwi : A
1
, A
2
, B , A
1
B , A
2
B ,
O
antygeny znajdują się w błonie erytrocytów, oraz na
powierzchni pozostałych komórek z wyjątkiem
komórek układu nerwowego
antygeny pojawiają się w 6 tygodniu życia
płodowego , ale do ich pełnej ekspresji dochodzi w 6
– 18 miesięcy po urodzeniu


Przeciwciała skierowane przeciw antygenom A i B
stanowią naturalny składnik ludzkiego osocza poza
tym obecne są w płynach ustrojowych i
wydzielinach.
Są to tzw.
izoaglutyniny
– należą do klasy Ig M, a
ich wytwarzanie rozpoczyna się zaraz po
urodzeniu i do 3 – 6 miesiąca życia ich stężenie
jest niskie. Największe miano przeciwciał
obserwuje się w 5 – 10 roku życia ( z wiekiem
stopniowo maleje ).
Odpornościowe przeciwciała anty –A i anty – B
są
wytwarzane głównie w następstwie immunizacji
kobiet przez krwinki płodu, które posiadają
antygeny nieobecne u matki lub po przetoczeniu
niezgodnej grupowo krwi. Należą one do klasy Ig
M i Ig G.


układ ABO uwarunkowany jest trzema
allelami, które zajmują to samo locus w
ramieniu długim chromosomu 9 (9q34)
allele I
A
i I
B
są dominujące w stosunku do
allela I
0
(i) i kodominujące względem
siebie, co prowadzi
do powstania sześciu różnych genotypów
dziedziczenie układu ABO odbywa się
według praw Mendla


Reguły dziedziczenia grup krwi układu AB0
Reguły dziedziczenia grup krwi układu AB0
Z praw dziedziczenia głównych grup
krwi wynikają pewne prawidłowości
pomiędzy fenotypami występującymi u
rodziców
oraz
fenotypami
ich
potomstwa. Jeżeli:
oboje rodzice mają grupę krwi A (I
A
I
A
lub I
A
i ), to żadne z ich dzieci nie może
mieć grupy krwi B oraz AB; jeżeli oboje
rodzice mają grupę krwi B (I
B
I
B
lub I
B
i),
to żadne z ich potomstwa nie może
mieć grupy krwi A oraz AB;

Reguły dziedziczenia grup krwi układu AB0
Reguły dziedziczenia grup krwi układu AB0
jedno z rodziców ma grupę krwi 0 (ii),
to żadne z ich dzieci nie może mieć
grupy AB;
jedno z rodziców ma grupę krwi AB
(I
A
I
B
), to żadne z ich potomstwa nie
może mieć grupy krwi 0;
jedno z rodziców ma grupę krwi A (I
A
i),
a drugie grupę krwi B (I
B
i), to u ich
potomstwa mogą wystąpić wszystkie
grupy krwi (A, B, AB i 0).

Antygeny grupowe układu AB0 mogą występować w
3 różnych postaciach: jako wielocukry, glikoproteiny
lub glikolipidy.
Mocz – oligocukry
Płyny biologiczne – dominują glikoproteiny
Błony komórkowe – najczęściej glikolipidy
Powierzchnia krwinek czerwonych – 80% antygenów
układu AB0 jest związanych z glikoproteinami,
pozostałe 20% występuje w połączeniu z
glikolipidami błony erytrocytu.

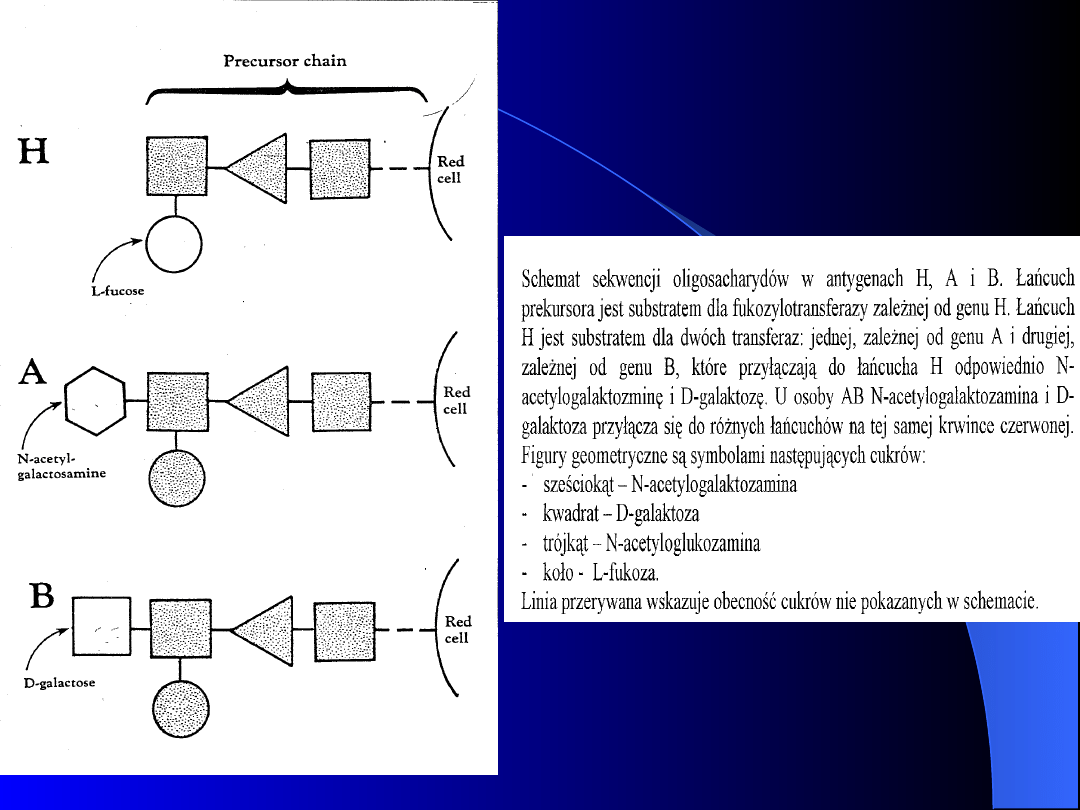
Geny A i B kodują swoiste transferazy (1,3-N-
acetylo-galaktozaminotransferaza i 1,3-
galaktozylotransferaza), które przyłączają
końcowe cukry do łańcucha polisacharydowego
zwanego łańcuchem H lub prekursorowym.
Swoistość antygenu warunkuje cukier
zajmujący ostatnią pozycję łańcucha
prekursorowego (N - acetylogalaktozamina =
antygen A; D - galaktoza = antygen B)

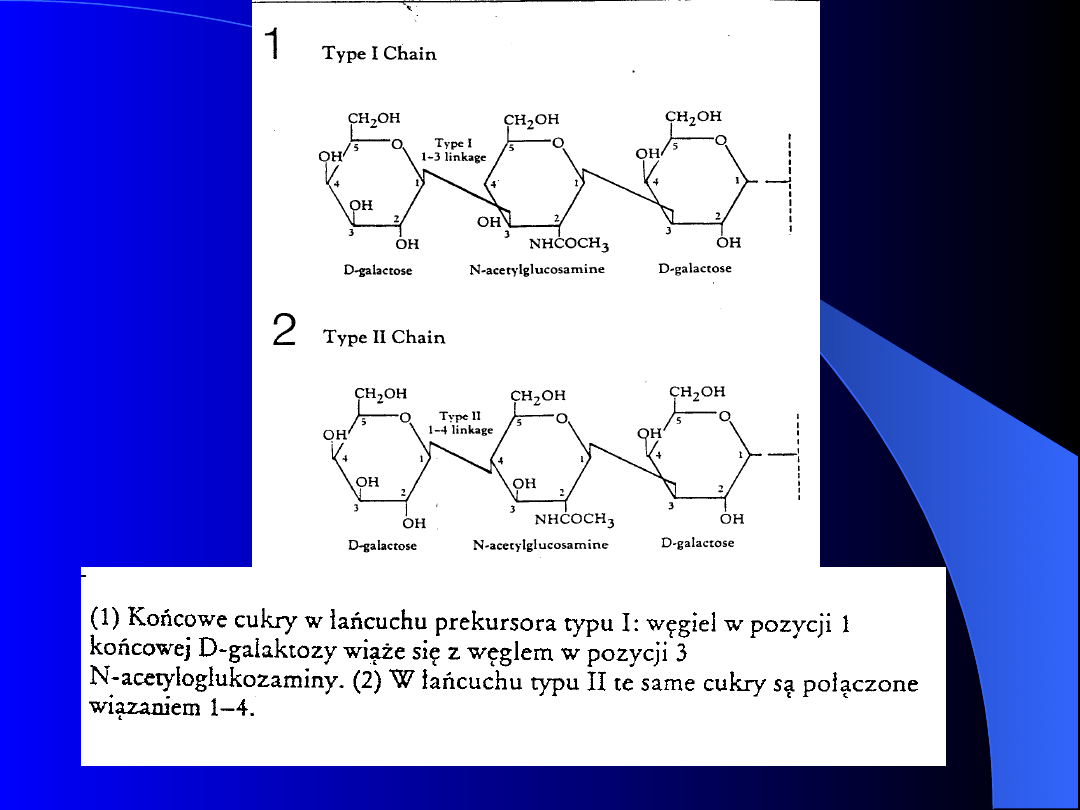
- Łańcuchy prekursorowe są dwóch
rodzajów i różnią się tylko wiązaniem
między węglem terminalnej galaktozy i
węglem przedostatniego cukru, którym jest
N-acetyloglukozamina.
- Łańcuchy będące integralnym składnikiem
erytrocytów są głównie typu II, podczas gdy
łańcuchy obecne w płynach ustrojowych są
typu I.

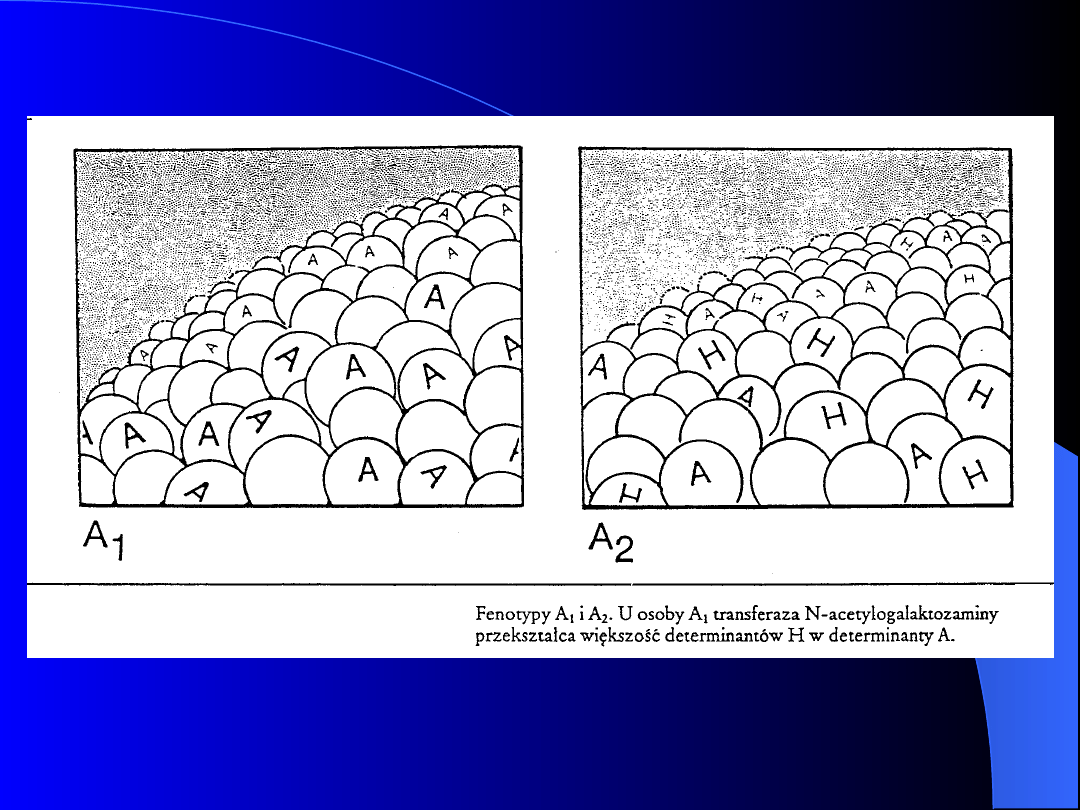
Około 80% osób posiadających grupę
krwi A należy do odmiany A
1
, natomiast
pozostałe 20% osób do A
2
. Transferaza
produkowana przez genA
2
różni się od
produkowanej przez gen A
1
niższą
efektywnością w przekształcaniu
łańcucha H w A. Końcowy cukier w
obu odmianach jest ten sam.

NIEZGODNOŚĆ W UKŁADZIE ABO
NIEZGODNOŚĆ W UKŁADZIE ABO
MIĘDZY MATKĄ A PŁODEM
MIĘDZY MATKĄ A PŁODEM
matka ma grupę A, a dziecko B
matka ma grupę B, a dziecko A
matka ma grupę O, a dziecko A lub B
Niezgodność występuje zatem, gdy w osoczu matki
znajdują się przeciwciała anty – A lub anty – B,
natomiast krwinki płodu zawierają odpowiedni
antygen odziedziczony po ojcu.
Istnieje możliwość wystąpienia choroby
hemolitycznej noworodków (w pierwszej dobie
pojawia się narastająca żółtaczka spowodowana
wzrostem poziomu bilirubiny po rozpadzie
erytrocytów)

Niezgodność w układzie ABO dotyczy ok. 20%
ciąż
Naturalne przeciwciała są klasy IgM, nie
przechodzą przez łożysko, przechodzą natomiast
przeciwciała odpornościowe należące do klasy IgG
Choroba hemolityczna występuje często u dzieci z
pierwszej ciąży
GRUPY KRWI A ZAPADALNOŚĆ NA CHOROBY
- ludzie z grupą krwi A częściej chorują na raka
żołądka i dróg rodnych, anemię złośliwą, cukrzycę
- ludzie z grupą krwi O na chorobę wrzodową
żołądka i dwunastnicy

oprócz alleli A i B istnieje gen H nie
sprzężony z genami locus ABO
gen H koduje enzym fukozylotransferazę
przenoszący fukozę do „terminalnej”
galaktozy (Gal T) substancji prekursorowej,
w efekcie tego powstaje substancja grupowa
H, która jest prekursorem antygenów A i B

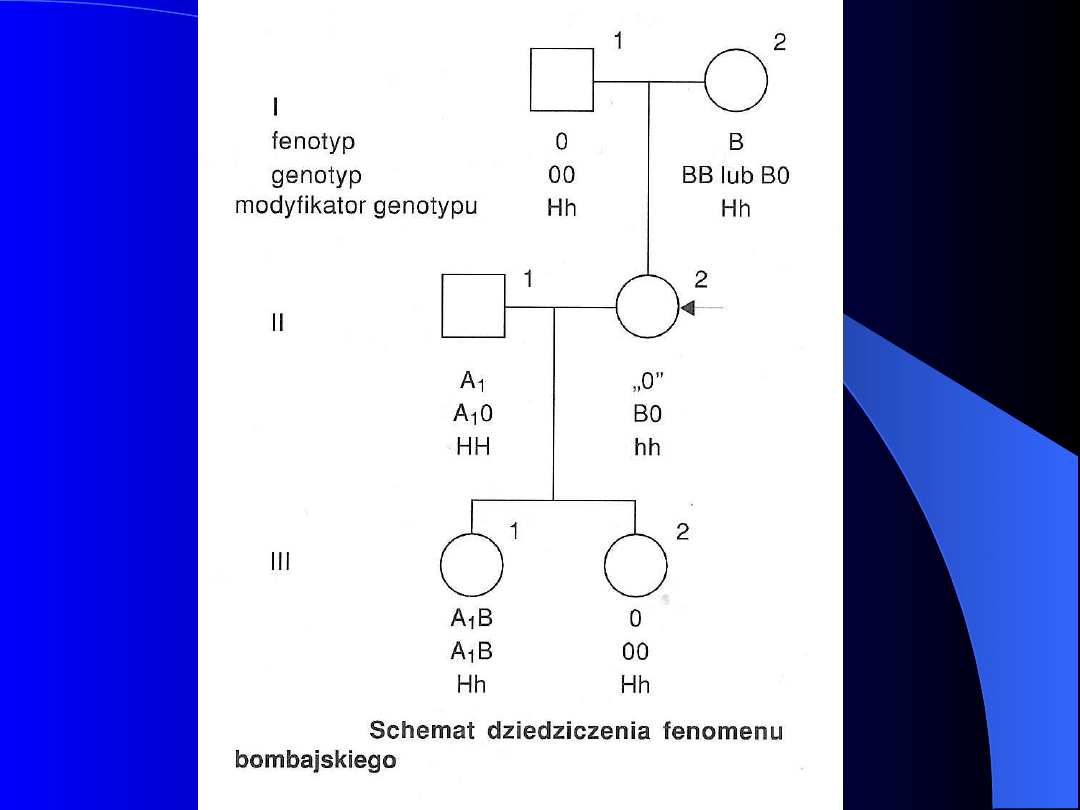
FENOMEN BOMBAJSKI
FENOMEN BOMBAJSKI
Osobnicy z niezwykle rzadką grupą krwi
Bombay nie posiadają genu H (locus na
chromosomie 19).
Ich genotyp określa się hh a fenotyp O
hA
,
O
hB
,
O
hAB ,
gdzie litery A, B, AB oznaczają grupę
krwi, której ekspresja jest przytłumiona przez
genotyp hh
U homozygoty hh brak jest fukozylotransferazy
(przy prawidłowym stężeniu pozostałych
transferaz), nie dochodzi do syntezy łańcucha
prekursorowego H i antygeny A i B nie mogą
być syntetyzowane.

Gen h jest genem amorficznym.
Fenotyp hh określa się jako Bombay, bardziej
prawidłową nazwą jest 0
h
lub ABH
null
.
Krwinki osób hh nie są aglutynowane przez
żadną z wzorcowych surowic (anty-A, anty-B
i anty A+B), natomiast w ich surowicy
stwierdza się przeciwciała anty-A, anty-B i
anty-H. Te ostatnie aglutynują krwinki grupy
0.

Antygeny A, B i H mogą być również obecne w
płynach ustrojowych. Wydzielanie substancji
A, B i H jest kontrolowane przez parę alleli Se
i se zwanych genami wydzielania (sekrecji) .
Możliwe są trzy genotypy SeSe, Sese i sese i
dwa fenotypy. Tylko homozygoty recesywne są
tzw. niewydzielaczami = gen se jest
amorficzny.
Wydzielacze (SeSe lub Sese) stanowią około
80% osób rasy białej

Rola genu Se
gen Se kontroluje produkcję fukozylotransferazy,
która przyłącza fukozę do łańcucha prekursorowego
typu I - powstaje łańcuch H typu I
geny Se i H to dwa odrębne geny, które kodują różne
fukozylotransferazy
transferaza zależna od genu H używa jako substratu
łańcucha prekursorowego typu II.

Osoby
hhsese
– brak łańcuchów typu II i typu I.
Fenotyp tych osób, to
klasyczny Bombay
(niewydzielacz Bombay).
Osoby
hhSeSe lub hhSese
- łańcuchy H typu I są
obecne w ich płynach ustrojowych, brak H dotyczy
tylko krwinek czerwonych. Fenotyp to
para - Bombay
(wydzielacz Bombay).
W zależności od tego, który z genów – A czy B został
odziedziczony, osoby para-Bombay poza substancją
H wydzielają substancję A lub B.

Układ grupowy Rh
Antygeny pojawiają się w 6 tygodniu życia płodowego i
występują tylko na krwinkach czerwonych
Dziedziczy się niezależnie od układu ABO
Przeciwciała układu Rh mają charakter odpornościowy,
należą do klasy IgG i mogą przechodzić przez łożysko
Przeciwciała powstają w wyniku przetaczania krwi Rh
(+) osobom Rh (-) lub w przypadku immunizacji matki
Rh(-) antygenem płodu Rh(+)

Zgodnie z teorią Fishera-Race’a antygeny
układu Rh są determinowane przez trzy
pary genów allelomorficznych, które
zajmują blisko leżące i sprzężone loci na
chromosomie 1 (1p36)
W każdym z loci znajduje się jeden z pary
alleli: D/d, C/c, E/e. Występujące genotypy
stanowią kombinację 2 z 8 możliwych
zestawów: cde, Cde, CDe, CDE, cDE, cDe,
cdE, CdE.
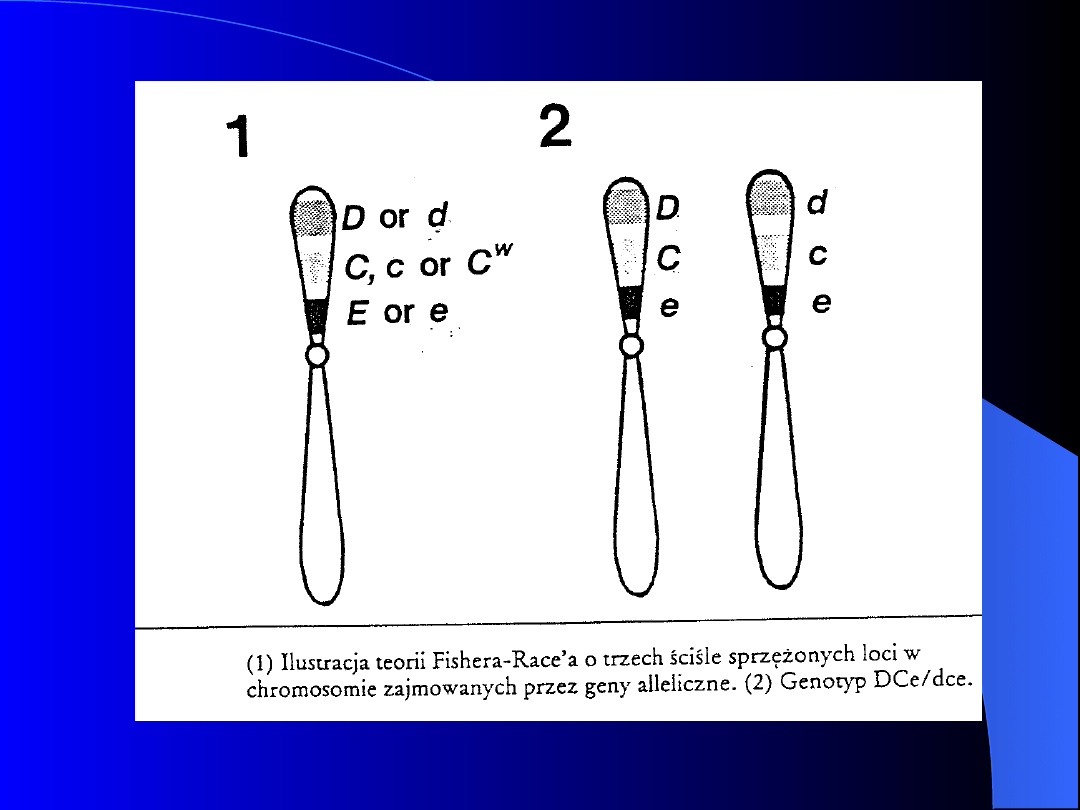


W praktyce największe znaczenie ma antygen
D, ponieważ odznacza się znaczną mocą
pobudzającą do wytwarzania przeciwciał
Dominacja allelu D nad allelem d jest
całkowita, co powoduje, że powstaje tylko
antygen D
Dominacja alleli C i E nad allelami c i e nie
jest zupełna - powstają antygeny Cc i Ee
W zależności od obecności antygenu D na
erytrocytach wyróżnia się osoby : z fenotypem
Rh(+) i genotypem DD lub Dd, oraz z
fenotypem Rh(-) o genotypie dd

KONFLIKT SEROLOGICZNY W
KONFLIKT SEROLOGICZNY W
UKŁADZIE Rh
UKŁADZIE Rh
Jest następstwem reakcji immunologicznej jaka
zachodzi między antygenami krwinek czerwonych
płodu a przeciwciałami anty-Rh organizmu matki
matka Rh(-), płód Rh(+)
- krwinki płodu dostają się do krążenia matki i stymulują
powstanie przeciwciał anty-Rh (klasy IgG)
- organizm matki jest zdolny do odpowiedzi
immunologicznej
- w krążeniu matki jest wysoki poziom przeciwciał anty-
Rh,
które przechodząc przez łożysko niszczą erytrocyty
płodu

W celu zapobiegania konfliktu serologicznego
u noworodków każdej nieuczulonej kobiecie
Rh(-), która rodzi dziecko Rh(+) należy
podawać gamma-globulinę anty-Rh.
Przeciwciała te reagują z erytrocytami płodu
które przedostały się do organizmu matki.
Immunoglobulina anty-RhD powinna być
stosowana przed porodem, bowiem mimo
prawidłowego podawania jej po porodzie 1-
2% Rh ujemnych kobiet uodparnia się.

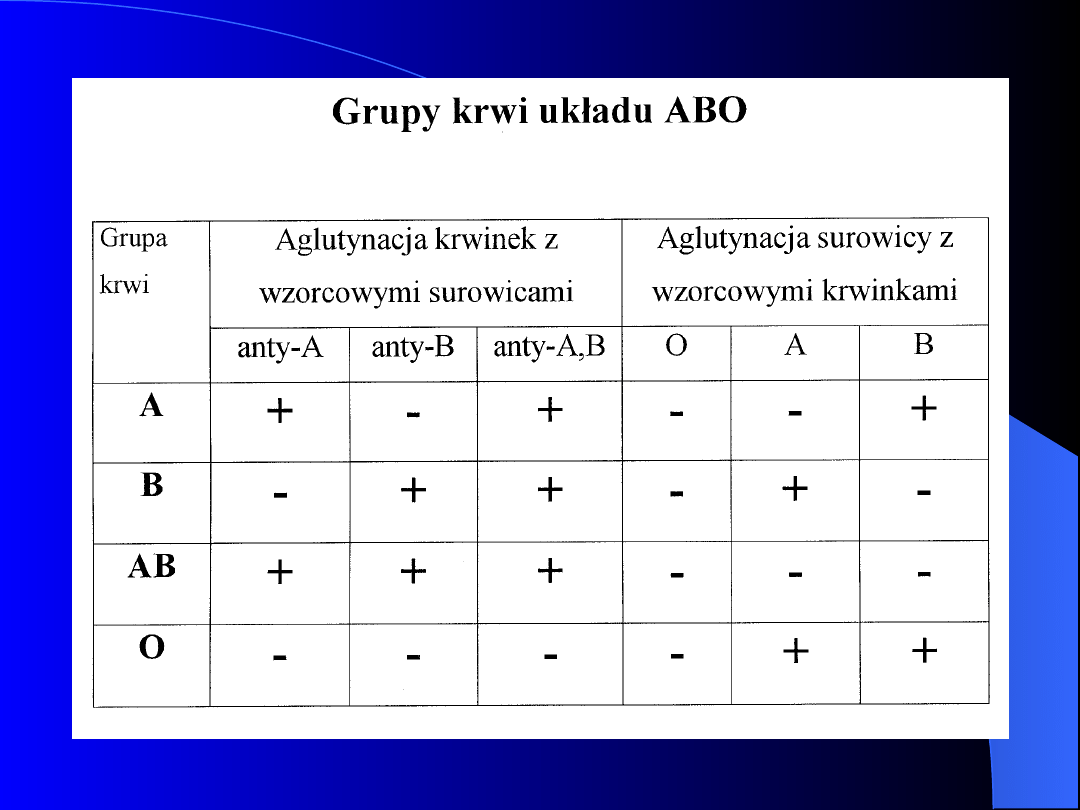
GRUPY KRWI A TRANSFUZJA
GRUPY KRWI A TRANSFUZJA
Największe znaczenie kliniczne mają układy
ABO i Rh, przetaczana krew musi być zgodna
w zakresie antygenów układu ABO oraz w
zakresie antygenu D układu Rh
Przed transfuzją wykonuje się próbę krzyżową
pomiędzy krwią biorcy i dawcy (aglutynacja
wyklucza transfuzję)
W celu określenia grupy krwi należy u
badanego przeprowadzić zarówno reakcję
aglutynacji jego krwinek z surowicami
wzorcowymi, jak i pomiędzy jego surowicą a
krwinkami wzorcowymi

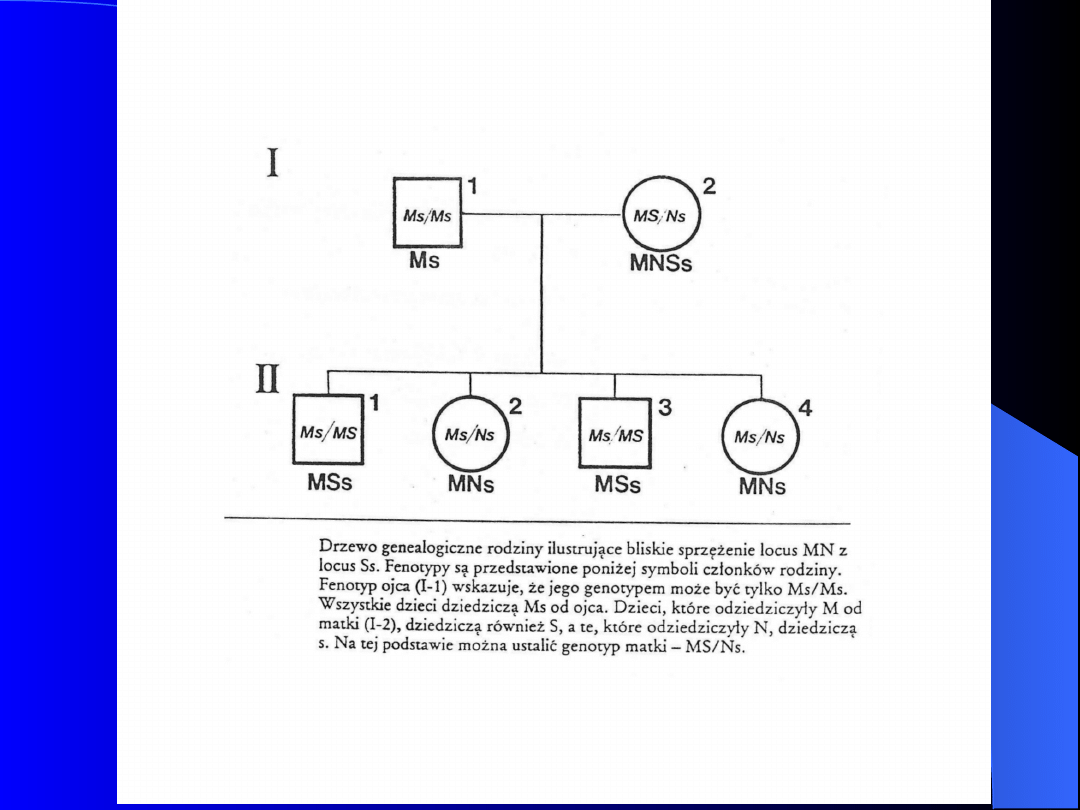
Układ grupowy MNSs
Nośnikami antygenów M, N, S i s są glikoproteiny. Ponieważ
większość cukrów stanowi kwas sjalowy, związki te się określa jako
sjaloglikoproteiny.
Glikoforyna A - jest nośnikiem determinantów M i N; Glikoforyna
B - jest nośnikiem determinantów S i s.
Antygeny oznaczone M i N dziedziczone są niezależnie od
antygenów A i B
W osoczu ludzkim brak jest naturalnych przeciwciał anty-M i anty-
N
Locus alleli M i N - chromosom 4

Antygeny M i N występują w krwinkach w
różnych kombinacjach (trzy typy
serologiczne: M, N i MN)
Dziedziczenie antygenów M i N
uwarunkowane jest kodominującą parą
alleli L
M
i L
N
fenotyp M N MN
genotyp L
M
L
M
L
N
L
N
L
M
L
N

W 1947 Walsh i Montgomery odkryli
antygeny S i s, które warunkują trzy
serologiczne grupy krwi: S, Ss i ss
Antygeny M i N oraz S i s stanowią jeden
układ grupowy dziedziczony na zasadzie
dwóch par genów allelomorficznych,
sprzężonych ze sobą
Pojawienie się antygenów MNSs warunkują
cztery allele L
MS
L
Ms
L
NS
L
Ns

Każdy allel koduje jednocześnie
antygeny z układu MN i Ss
Przeciwciała odpornościowe anty-M i
anty-N należą do klasy IgM. Antygeny
tego układu bardzo rzadko powodują
immunizację
Aglutyniny anty-S,s są klasy – IgG
Badanie antygenów MN stosuje się w
antropologii kryminalistyce, w
określaniu ojcostwa

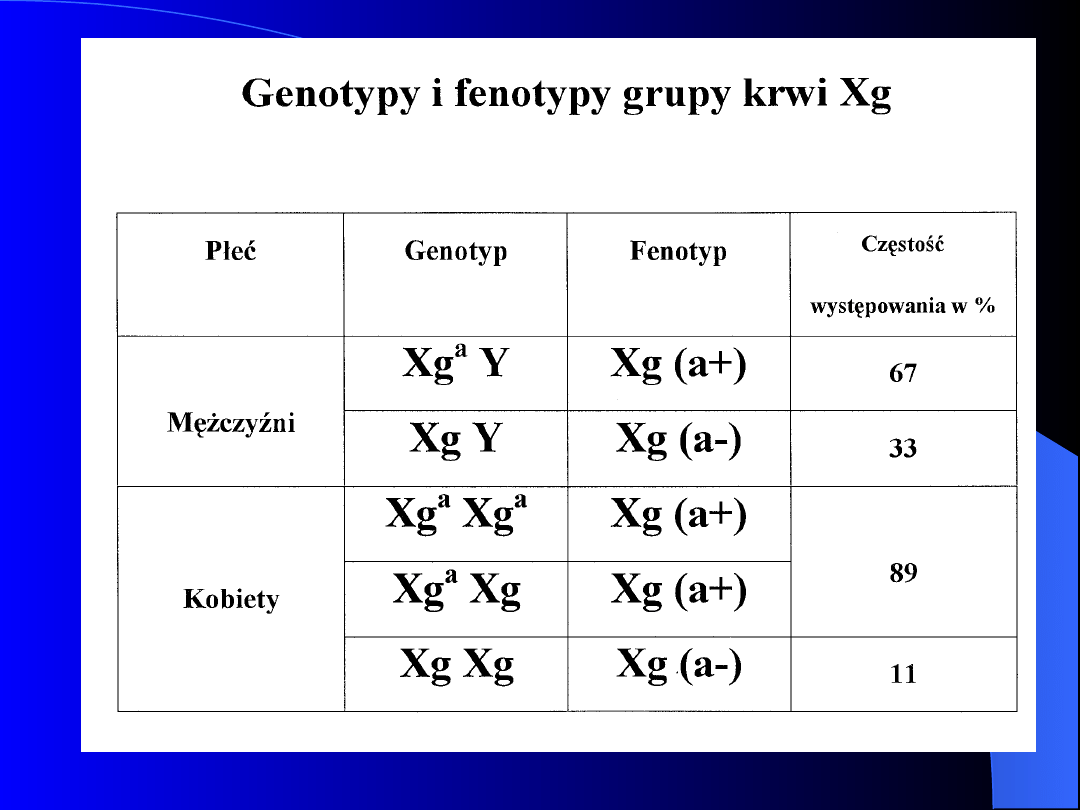
UKŁAD GRUPOWY Xg
W 1962 r odkryto antygen Xg i przeciwciała
skierowane przeciwko temu antygenowi
Gen antygenu Xg zlokalizowany jest na
ramieniu krótkim chromosomu X (Xp22.3)
Dziedziczenie tej cechy jest związane z płcią
Istnieją dwa typy osobników : Xg(a+) i Xg(a-)
Para alleli: Xg
a
i Xg
Ojciec posiadający cechę Xg(a+) nie może
mieć córki z cechą Xg(a-)
Matka Xg(a-) nie może mieć syna z cechą
Xg(a+)

Nabyte odmiany antygenów grupowych krwi
Ekspresja antygenów może mieć miejsce w
następstwie zmniejszonej aktywności lub
pojawienia się egzo- lub endogennych
transferaz.
1. W chorobach rozrostowych krwi i w ciąży
krwinki osób z grupą krwi A lub B mogą
reagować jak erytrocyty osób z grupą 0.
- dzieje się tak na skutek zmniejszenia
aktywności odpowiedniej trasferazy i
odsłonięcia antygenu H
- zmiany te mogą ulec odwróceniu w okresie
remisji nowotworu

2. Niektóre szczepy bakterii (E. coli, Proteus
vulgaris) produkują enzym, deacylazę, która
zmienia N-acetylogalaktozaminę (grupa krwi
A) w galaktozaminę.
- galaktozamina zajmująca końcową pozycję
jest rozpoznawana przez przeciwciała anty-B.
- zakażenia wymienionymi bakteriami mogą
doprowadzić do zmiany fenotypu krwinek
chorego z grupy krwi A na B.

3. Zmiany antygenów grupowych krwi
obserwuje się u chorych po przeszczepieniu
szpiku, jeśli dawca i biorca różnili się w
układzie AB0.
4. Antygeny grupowe krwi wykrywa się na
niektórych komórkach nowotworowych.
-
znane jest zjawisko zwiększonej częstości raka
żołądka i jajnika u osób z grupą krwi A
-
według hipotezy, rozwój nowotworu
posiadającego substancję grupową A hamują
odpowiednie przeciwciała obecne u osób z
grupą B lub 0. Tego mechanizmu obronnego
pozbawieni byliby chorzy z grupą krwi A.

Układ ABO – allele I
A
, I
B
, i
I
A
I
A
= A, I
a
i = A; I
B
I
B
= B, I
B
i = B
I
A
I
B
= AB; ii = O
Układ Rh – allele D i d
(pełna
dominacja)
DD = Rh(+), Dd = Rh(+) i dd = Rh (-)

Układ MNSs – allele
L
MS
L
Ms
L
NS
L
Ns
L
MS
L
MS
= MS, L
MS
L
Ms
= MSs
,
L
Ms
L
Ms
= Ms
L
NS
L
NS
= NS, L
NS
L
Ns
= NSs
,
L
Ns
L
Ns
= Ns
L
MS
L
NS
= MNS, L
MS
L
Ns
= MNSs
,
L
Ms
L
Ns
=
MNs

Układ Kell – allele K i k
(pełna
dominacja)
KK = K(+), Kk = K(+) i kk = K(-)
Układ P – allele P
1
, P
2
i p
P
1
P
1
= P1
P
1
p = P1, P
2
P
2
= P2, P
2
p = P2,
P
1
P
2
= P1P2, pp = p

Układ Lutheran – allele Lu
a
, Lu
b
i lu
Lu
a
Lu
a
= Lu (a+, b-)
Lu
a
lu = Lu (a+, b-)
Lu
b
Lu
b
= Lu (a-, b+)
Lu
b
lu = Lu (a-, b+)
Lu
a
Lu
b
= Lu (a+, b+)
lu lu = Lu (a-, b-)

Układ Lewis – allele Le
a
, Le
b
i le
Le
a
Le
a
= Le (a+, b-)
Le
a
le = Le (a+, b-)
Le
b
Le
b
= Le (a-, b+)
Le
b
le = Le (a-, b+)
Le
a
Le
b
= Le (a+, b+)
le le = Le (a-, b-)

Układ Duffy– allele Fy
a
, Fy
b
i fy
Fy
a
Fy
a
= Fy (a+, b-)
Fy
a
fy = Fy (a+, b-)
Fy
b
Fy
b
= Fy (a-, b+)
Fy
b
fy = Fy (a-, b+)
Fy
a
Fy
b
= Fy (a+, b+)
fy fy = Fy (a-, b-)

Zadania
Zadania
1.
Jeśli matka i dziecko mają grupę
krwi AB, to ojcem dziecka nie
mógł być mężczyzna o grupie
krwi ___?
2.
Jeżeli dziecko ma grupę krwi 0,a
a matka A, to ojciec dziecka mógł
ieć grupę krwi ____ ?

Zadania
Zadania
1.
Jakie jest prawdopodobieństwo
urodzenia się córki o grupie krwi O,
MNSs, Rh (+) jeżeli rodzice mają
grupy krwi A, MNS, Rh(+) i B, Ms,
Rh(-).
2.
Podaj możliwe fenotypy i genotypy
potomstwa rodziców o grupach krwi
O, Rh(+), MS, wydzielacz i AB, Rh(-),
Ms, niewydzielacz.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
Wyszukiwarka
Podobne podstrony:
Ćw 3 Dziedziczenie grup krwi
Cw[1][1] 3 Dziedziczenie grup krwi
Ćw 4 Dziedziczenie grup krwi
Ćw 3 Dziedziczenie grup krwi
GENETYKA i parazyty KOLOKWIUM 20092, Fizjoterapia CM UMK, Biologia medyczna
Dziedziczenie grup krwi
Dziedziczenie grup krwi, Ratownicto Medyczne, Biologia
przysadka cw histologia, II rok, II rok CM UMK, Giełdy, 2 rok, histologia
GENETYKA i parazyty KOLOKWIUM 2009, farmacja cm umk
genetyka i parazyty gieldy, Pielęgniarstwo licencjat cm umk, I rok, Genetyka i parazytologia koła pi
zadania fizyczna ćw. 4, Chemia fizyczna dr Cysewski farmacja CM UMK Bydgoszcz
cw kolo2 chem lek, Chemia leków CM UMK
GENETYKA i parazyty KOLOKWIUM 20092, Fizjoterapia CM UMK, Biologia medyczna
Zawartość cukru we krwi, Pielęgniarstwo licencjat cm umk, III rok, Geriatria i pielęgniarstwo geriat
więcej podobnych podstron