
Ćwiczenie nr 13
ANALIZA ILOŚCIOWA. KOLORYMETRIA.
KOLORYMETRYCZNE OZNACZANIE
ŻELAZA I FOSFORANÓW.
WPROWADZENIE
ŻELAZO I JEGO STOPY
Ż e l a z o jest najbardziej rozpowszechnionym w skorupie ziemskiej metalem ciężkim, jednak ze
względu na dużą reaktywność chemiczną nic występuje w postaci czystej. W technice żelazo spotykane
jest jako stal (zawartość C < 2% wag.) i żeliwo (zawartość C > 2% wag.).
Charakterystyczne dla żelaza, w związkach jest występowanie na dwóch stopniach utlenienia (II, III)
i łatwość zmiany elektrowartościowości Fe
2 +
<=* Fe
3 +
w roztworach wodnych. Żelazo jest odporne na
działanie wody wapiennej, jak również zimnych ługów, co ma duże znaczenie z punktu widzenia
zbrojenia betonu (żelbet).
Węglan żelazawy (II), występujący w skałach jako syderyt, ulega rozpuszczeniu pod wpływem wody
zawierającej C0
2
, z utworzeniem łatwo rozpuszczalnego kwaśnego węglanu, analogicznie do przypadku
rozpuszczania węglanu wapnia
FeCO
3
+ CO
2
+ H
2
O -> Fe(HCO
3
)
2
.
Gdy na roztwór Fe(HC0
3
)
2
działa tlen z powietrza, wytrąca się Fe(OH)
3
, co powoduje powstanie
brunatnych osadów w rowach melioracyjnych, zbiornikach itp. Powoduje to również kolmatację złóż
wodonośnych oraz zatykanie się przewodów wodociągowych
4Fe(HCO
3
)
2
+ O
2
+ 2H
2
O ->4Fe(OH)
3
+ 8CO
2
.
Farby mineralne (pigmenty) o barwach brązowej do czerwonej, odporne na działanie cementu,
światła i innych czynników klimatycznych, zawierają tlenek żelazowy (III). Występuje on w różnych
naturalnych farbach, jak ochra (produkt wietrzenia rud żelazistych) czy umbra (glina zawierająca sole
żelaza i manganu); natomiast czerwień angielska („czerwień żelazowa") jest dokładnie zmielonym —
syntetycznie otrzymanym — tlenkiem żelazowym (Fe
2
0
3
). Obecność Fe
2
0
3
jest również przyczyną
zabarwienia ceramiki budowlanej, jeśli:
- < 1% Fe
2
0
3
i dużo A1
2
0
3
; kolor biały,
- < 2% Fe
2
0
3
i dużo A1
2
0
3
; kolor żółty,
- < 4% Fe
2
0
3
, mało A1
2
0
3
, dużo CaO; kolor żółty,
- 4^-7% Fe
2
0
3
, mało A1
2
0
3
, mało CaO; kolor czerwony. Surówkę żelaza otrzymuje się przez
redukcję w wielkich piecach rud
tlenkowych, jak hematyt Fe
2
0
3
, magnetyt Fe
3
0
4
lub limonit Fe
4
0
3
(OH)
6
:
Fe
2
0
3
+ 3C^2Fe + 3CO, Fe
2
0
3
+ 3CO -»• 2Fe + 3C0
2
, C +
C0
2
^2CO.
Rudy siarczkowe, jak pirotyn FeS lub piryt FeS
2
, muszą zostać uprzednio wypalone na Fe
2
0
3
.
Wyprodukowaniu 1 tony żeliwa towarzyszy powstanie 1 tony żużla — produktu odpadowego,
będącego wynikiem reakcji dodatku CaC0
3
z zanieczyszczeniami znajdującymi się w rudach:
2CaC0
3
+ Si0
2
-* Ca
2
Si0
4
+ 2C0
2
.
składnik skały płonnej
Żużel zawiera: 36-45% CaO, 30-40% Si0
2
, 6-10% A1
2
0
3
, a także FeO, MnO. Odpad ten jest
domielany jako dodatek do cementów hutniczych.
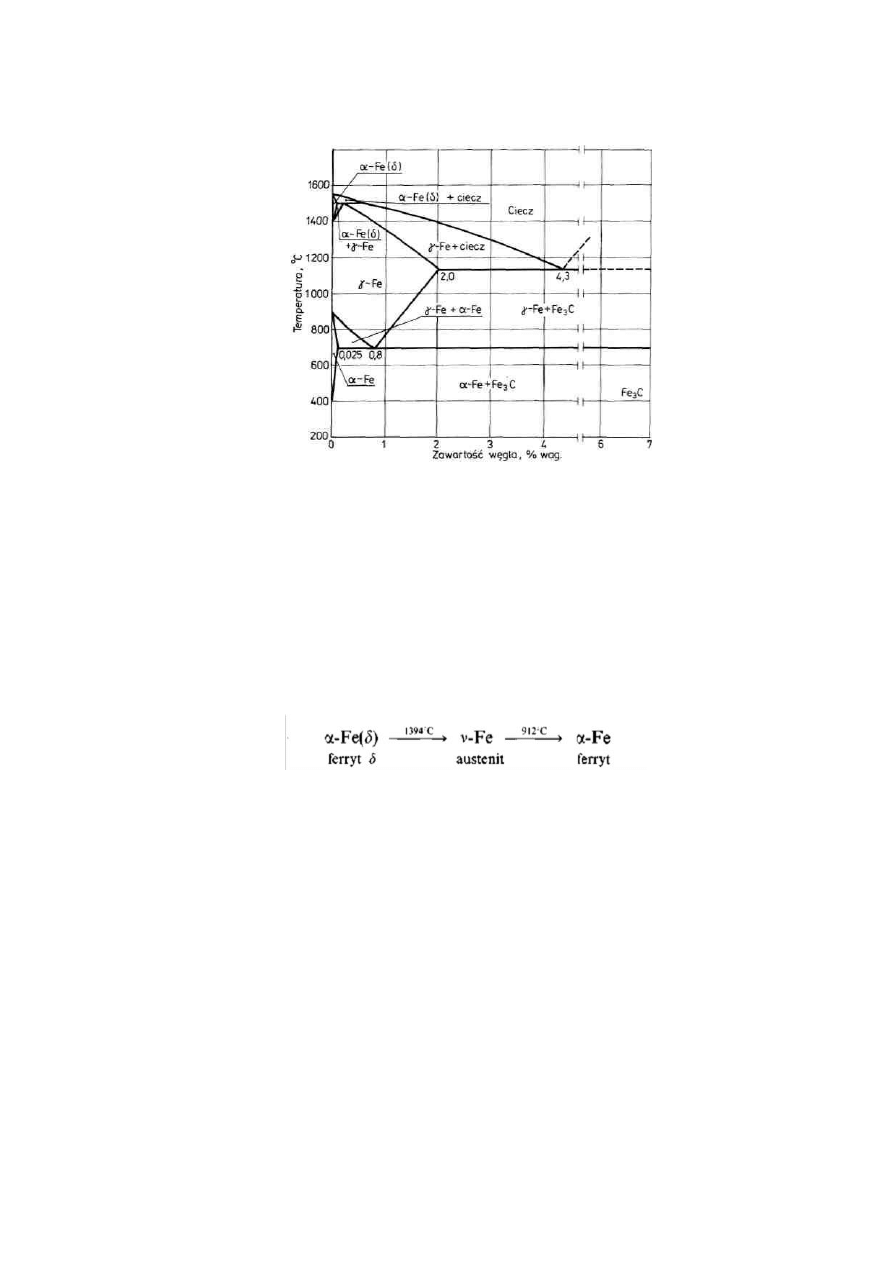
Rys. 1. Układ dwuskładnikowy żelazo (Fe) — cementyt (Fe
3
C)
W technice największe znaczenie mają stopy żelaza z węglem (rys. 1.), występujące w
postaci stali (stop obrabiany plastycznie) oraz żeli (stop odlewniczy). Stal w postaci odlanej
nosi nazwę staliwa.
Stopione w wielkim piecu żeliwo ulega nawęgleniu, w wyniku czego powstaje cementyt 3 Fe +
C -* Fe
3
C oraz roztwory stałe węgla w żelazo (rozpuszczenie węgla w sieci krystalicznej żelaza).
Podczas studzenia czystego żelaza zachodzą następujące przemiany fazowe: w temperaturze
1538°C z fazy ciekłej krystalizuje wysokotemperaturowa odmiana ferrytu oc-Fe(<S), a
następnie:
Odmiany te różnią się właściwościami: y-Fe rozpuszcza węgiel, a pozostałe odmiany rozpuszczają go
tylko w niewielkim stopniu.
Podczas wytopu stal ulega utlenieniu, co powoduje pogorszenie jej właściwości
wytrzymałościowych — stal nieuspokojona. Stal poddana odtlenieniu ma lepsze i bardziej stabilne
właściwości mechaniczne — stal uspokojona.
Właściwości stali zależą od zawartości węgla, zawartości składników stopowych, a także od
zastosowanej obróbki mechanicznej (np. walcowanie na zimno) i cieplnej (wyżarzanie, ulepszanie
cieplne, tzn. hartowanie i odpuszczanie, itp.). Podział gatunków stali na grupy jest zależny od przyjętej
podstawy klasyfikacji (np. skład chemiczny, struktura po schłodzeniu). Dla praktyki przemysłowej
przyjęto podział na stale węglowe i stopowe oraz ze względu na główne i szczegółowe zastosowanie.
W budownictwie stosuje się przede wszystkim stale konstrukcyjne (węglowe lub stopowe). Mają one
niską zawartość węgla (0,05^-0,65%) i często są ulepszane cieplnie. Właściwości stali można
modyfikować przez odpowiednie dodatki stopowe; np. dodatek Cr i Ni poprawia odporność na korozję, a
dodatek W i V podnosi odporność na działanie wysokich temperatur oraz ścieranie. Wzrost zawartości
węgla i pierwiastków stopowych pogarsza spawalność stali. Podstawowym wyróżnikiem stali są:
wymagana wytrzymałość oraz przeznaczenie. Stale k o n s t r u k c y j n e w ę g l o w e zwykłej jakości
oznaczane są literami St i liczbą porządkową lub liczbą oznaczającą wymaganą wytrzymałość; np. St41
oznacza stal o wymaganej wytrzymałości na rozciąganie R
m
= 41 kG/mm
2
(ok. 400 MPa). Stale wyższej
jakości oznaczane są maksymalną lub średnią zawartością węgla (np. 35 oznacza stal o maksymalnej

zawartości C = 0,35°o wag.). Stale węglowe o szczególnym przeznaczeniu, o dodatkowych wymaganiach
lub zwiększonej zawartości danego pierwiastka oznacza się dodatkowymi literami na końcu oznaczenia
(np. J — odporna na starzenie, P — stale dla kolejnictwa, S — stale do spawania. Cu — zwiększona
zawartość miedzi). Wyjątkowo oznacza się dodatkową literą przed oznaczeniem: B — stal na blachy, D
— stal na druty (np. D85 oznacza stal na druty o średniej zawartości C = 0,85% wag.). Stale s t o p o w e
oznacza się dwucyfrową liczbą, określającą średnią zawartość węgla w setnych częściach procentu, oraz
literami umownymi, oznaczającymi pierwiastki stopowe, z dodaniem liczby określającej ich zawartość,
jeśli przekracza ona średnio 1,5%. Umowne znaki składników stopowych to: A-azot, B-bor, F-wanad, G-
mangan. H-chrom, J-aluminium, K-kobalt, M-molibden, N-nikiel, S-krzem. T-tytan, W-wolfram, Cu-
miedź, Nb-niob (np. 35HGS oznacza stal o zawartości C = 0,35% wag. oraz zawartości pierwiastków
stopowych chromu, manganu i krzemu w ilości nie przekraczającej 1,5%). Stale wysokostopowe, odporne
na korozję, oznaczane są w odmienny sposób. 0 na początku symbolu oznacza zawartość węgla poniżej
0,08%. a 00 — zawartość węgla poniżej 0,03% (np. 00H18N10 oznacza stal o zawartości węgla poniżej
0,03% oraz zawartości dodatków: 18% chromu i 10% niklu).
W budownictwie najczęściej wykorzystuje się następujące wyroby hutnicze ze stali
konstrukcyjnych: pręty zbrojeniowe, pręty ogólnego przeznaczenia, taśmy, kształtowniki, blachy, druty i
rury.
Korozja metali
Proces korozji metali jest równoznaczny z niszczeniem ich użytecznych właściwości. W wyniku
korozji metal ulega utlenieniu - przyjmuje elektrowartościowość dodatnią, a produktami tego procesu są
głównie tlenki i wodorotlenki.
Mechanizm procesu korozyjnego i jego przebieg zależy od rodzaju środowiska. Rozróżnia się przy
tym dwa główne typy korozji: chemiczną i elektrochemiczną.
Korozja chemiczna - jest to proces niszczenia metalu zachodzący w środowisku
nieelektrolitycznym. Obejmuje reakcje chemiczne przebiegające między metalem, a suchym gazem lub
cieczą nie przewodzącą prądu elektrycznego (nieelektrolitem). Do tego typu korozji należy, np.:
- korozja elementów grzejnych pieców elektrycznych,
- korozja zbiorników, instalacji i reaktorów chemicznych powodowana działaniem: gazów (tj. H2S, H2,
CO, CO2, CI2, NH3), przegrzanej pary wodnej, ciekłych nieelektrolitów (np. ropy naftowej, stopionej
siarki, substancji organicznych),
- korozja elementów turbin, np. łopatek, stykających się z gorącymi gazami spalinowymi.
Spośród różnych rodzajów korozji chemicznej największe straty w gospodarce wyrządzone są
poprzez korozję gazową, tj. utlenianie metali w wysokiej temperaturze, w atmosferze suchych gazów.
Towarzyszy temu ciągłe tworzenie się i narastanie na powierzchni metali warstwy stałego produktu
reakcji tzw. zgorzeliny. Warstwa ta oddziela metal od środowiska korozyjnego i dalszy przebieg
utleniania uzależniony jest od dyfuzji metalu i utleniacza. Ponieważ w praktyce korozja gazowa zachodzi
najczęściej w środowisku powietrza, czynnikiem utleniającym jest tlen, a produktami korozji,
tworzącymi zgorzelinę, głównie tlenki metali.
Proces wysokotemperaturowego utleniania metali w powietrzu składa się z dwóch procesów
cząstkowych:
I - utlenianie metalu na granicy faz metal / tlenek: Me —» Me2+ + 2 e
II - redukcja tlenu na granicy faz tlenek / gaz:
0.5 O2 + 2 e —> 0
2
~
sumarycznie:
Me + 0.5 O2 -» MeO
Narastająca warstwa tlenkowa spełnia następujące funkcje:
- jest przewodnikiem jonów i elektronów,
- jest elektrodą, przy której następuje redukcja tlenu,
- jest barierą dyfuzyjną dla jonów i elektronów.
Proces narastania zgorzeliny na metalach i ich stopach przebiega bardzo różnie w zależności od
rodzaju metalu, temperatury, składu chemicznego środowiska itp. Poznanie tych zależności jest bardzo
istotne, gdyż umożliwia określenie odporności korozyjnej metali pracujących w wysokich temperaturach.
Odporność korozyjna jest tym większa, im bardziej warstewka zgorzeliny na powierzchni metalu pełni
funkcję ochronną, tj. gdy:

- zgorzelina jest zwarta, tzn. nie występują w niej szczeliny i pory ułatwiające dyfuzję utleniacza;
- zgorzelina wykazuje dobrą przyczepność do metalicznego podłoża; szybkość dyfuzji metalu i
utleniacza przez zgorzelinę jest mała;
- temperatura topnienia zgorzeliny jest wysoka.
Gdy powierzchnia metalu pokryta zostanie bardzo cienką (nie przekraczającą 0.1 μm) zwartą
warstewką tlenku chroniącego metal przed dostępem tlenu, mówimy, że powierzchnia metalu uległa
pasywacji. Na tym polega właśnie większa, w porównaniu z innymi metalami, odporność aluminium na
korozję, wykazującego również duże powinowactwo do tlenu. Bardzo szybko powstająca pierwotna
warstewka tlenku glinu jest tak zwarta i trwała, że całkowicie zabezpiecza znajdujący się pod nią metal
przed dalszą korozją. Podobnie jest w przypadku chromu i tytanu, natomiast na czystym żelazie
warstewka tlenku łatwo pęka (zwłaszcza przy większej grubości), co umożliwia dopływ tlenu do
powierzchni metalu i jego dalszą korozję. Cenną zaletą chromu jest to, że wprowadzony jako dodatek
stopowy do żelaza przekazuje mu niejako swoją zdolność ulegania pasywacji (stal nierdzewna zawiera
powyżej 12 % chromu). Należy jednak zaznaczyć, że warstwa pasywna wobec tlenu nie zawsze jest
odporna na działanie innych czynników chemicznych, które mogą z nią reagować niwelując jej działanie
ochronne.
Korozja elektrochemiczna -jest to proces niszczenia metalu przebiegający w roztworach
elektrolitów. Wiąże się z powstawaniem ogniw galwanicznych oraz przepływem elektronów lub jonów
przez granicę faz metal - elektrolit.
Mechanizm korozji elektrochemicznej jest analogiczny do działania ogniwa galwanicznego. Ogniwo
takie składa się z dwóch elektrod (ujemnej - anody i dodatniej - katody) zanurzonych w roztworze
elektrolitu. Elektrodami mogą być różne lub dwa takie same metale pod tym jednak warunkiem, że
między nimi występuje różnica potencjałów. Warunek ten jest spełniony, gdy np. metale znajdują się w
środowiskach różniących się temperaturą, natlenieniem, stężeniem roztworów. Środowisko korozyjne
może stanowić dowolny elektrolit, np. woda morska, wodociągowa, roztwory chemikaliów, a także
wilgotne powietrze oraz gleba. Elektrody w ogniwie muszą być zwarte, tzn. połączone przewodem
metalicznym lub bezpośrednio stykające się ze sobą. W takim ogniwie galwanicznym lub ogniwie
korozyjnym energia chemiczna ulega zmianie na elektryczną, w wyniku czego płynie prąd elektryczny.
Proces korozji elektrochemicznej składa się z procesu anodowego, katodowego oraz procesu
przepływu elektryczności.
Proces anodowy (korozja) polega na przechodzeniu metalu do roztworu w postaci utlenionych
jonów zgodnie z reakcją:
Me -» Me
n+
+ n e (np. utlenianie żelaza: Fe -> Fe
2
+ + 2 e);
Proces katodowy polega na pochłanianiu elektronów przez tzw. depolaryzatory, którymi mogą być
atomy, cząsteczki lub jony roztworu ulegające redukcji na katodzie (katoda nie ulega korozji). W
przypadku, gdy redukcji ulegają jony wodorowe, mówimy o depolaryzacji wodorowej, a gdy redukcji
ulega tlen
- mamy do czynienia z depolaryzacją tlenową.
Przykładem reakcji katodowych w środowisku kwaśnym są:
- redukcja jonów wodorowych:
2 H
+
+2 e -» 2 H —> H
2
- redukcja rozpuszczonego tlenu:
O
2
+4H++4e—»2 H
2
O
a w środowisku obojętnym lub zasadowym:
- redukcja rozpuszczonego tlenu:
O
2
+ 2 H
2
O + 4 e -» 4 OH
-
Proces przepływu elektryczności w metalu odbywa się poprzez ruch elektronów od obszarów
anodowych do katodowych, natomiast w roztworze przez ruch kationów od obszarów katodowych do
anodowych.
W roztworze elektrolitu, podczas zetknięcia się produktów reakcji anodowych (jonów metali) z
produktami reakcji katodowych (np. z grupami wodorotlenowymi) może dojść do wtórnych reakcji, np.:
Fe
2+
+ 2 OH- -> Fe(OH)
2
Powstały wodorotlenek żelaza(II) jest trudno rozpuszczalny, więc wytrąca się z roztworu. Jeżeli
proces przebiega w obecności rozpuszczonego tlenu z powietrza, następuje reakcja utleniania do
wodorotlenku żelaza(III):

4 Fe(OH)
2
+ O
2
+ 2 H
2
0 -> 4 Fe(OH)
3
Ponieważ wodorotlenek żelaza(II) jest nietrwały, rozpada się tworząc uwodniony tlenek Fe203 • H2O
o barwie czerwonobrunatnej. Gdy ilość tlenu jest za mała do całkowitego utlenienia Fe(OH)2, tworzy się
magnetyt o wzorze Fe3C>4 :
6 Fe(OH)
2
+ O
2
-> 4 H
2
O + 2 Fe
3
O
4
• H
2
O (zielony)
Fe
3
0
4
■ H2O -> H2O + Fe
3
04 (czarny)
Tworzenie się uwodnionych tlenków żelaza na powierzchni żelaza lub stali nazywa się potocznie
rdzewieniem, zaś wymienione wyżej produkty korozji -rdzą.
Ogniwa galwaniczne tworzą się na każdym korodującym metalu. Przyczyny powstawania ogniw
korozyjnych są różne i wynikają zarówno z właściwości metalu (czynniki wewnętrzne), jak i środowiska
elektrolitycznego (czynniki zewnętrzne).
Do wewnętrznych czynników wpływających na szybkość i charakter procesu korozyjnego
zaliczamy: rodzaj metalu, jego skład chemiczny, strukturę, stan powierzchni, obróbkę cieplną i
mechaniczną, obecność naprężeń własnych itp. Największą odporność na korozję wykazują metale o
dużym stopniu czystości. Jednak uzyskanie bardzo czystych metali jest trudne i wiąże się z wysokimi
kosztami. Poza tym wysoka czystość metalu nie gwarantuje dobrych właściwości wytrzymałościowych,
np. czyste żelazo jest miękkie i nie nadaje się do celów konstrukcyjnych. Stopy metali w zależności od
wzajemnej rozpuszczalności mogą być jednofazowe lub wielofazowe. Stopy wielofazowe charakteryzują
się niejednorodnością składu i struktury, co prowadzi do tworzenia się na ich powierzchni mikroogniw
korozyjnych w przypadku zetknięcia się z elektrolitem. Do stopów wielofazowych należą stale węglowe,
mosiądze (o zawartości Zn powyżej 37 %) lub stale chromoniklowe. Struktura większości stopów metali
zależy m.in. od ich obróbki cieplnej, którą stosuje się w celu nadania materiałowi żądanych właściwości
wytrzymałościowych (np. hartowanie stali zwiększa wytrzymałość na rozciąganie, wzrasta twardość i
sprężystość). Podczas obróbki cieplnej oraz w trakcie przeróbki plastycznej metali (kucie, walcowanie), a
także podczas spawania, zgrzewania itp. powstają w metalu naprężenia własne. Ich obecność jest
niekorzystna, gdyż może prowadzić do pękania korozyjnego. Obróbka mechaniczna, np. skrawanie metali,
nie powoduje wzrostu naprężeń własnych, decyduje jednak o gładkości powierzchni. Im wyższa gładkość,
tym większa odporność metalu na korozję.
Zewnętrzne czynniki korozji są to te czynniki związane ze środowiskiem korozyjnym, które
wpływają na szybkość i charakter korozji. Zaliczamy do nich rodzaj i skład środowiska, temperaturę,
ciśnienie, szybkość przepływu elektrolitu itp. Przykładowo, obecność jonów wodorowych w środowisku
korozyjnym ma duży wpływ na przebieg korozji metali. Zmiana wartości ich stężenia, a więc zmiana pH
środowiska powoduje, zmianę szybkości korozji różnych metali. Metale szlachetne, tj. platyna, złoto są
odporne na korozję zarówno w kwasach, jak i w zasadach. Szybkość korozji tych metali nie zależy od pH
roztworu. Metale amfoteryczne, np. aluminium lub cynk, które w kwasach tworzą jony Al
3+
, Zn
2+
, a w
zasadach jony AIO
2
, ZnO
2
korodują z różną szybkością w zależności od pH środowiska. Zależność ta ma
przebieg paraboliczny. Minimalna korozja dla aluminium występuje przy pH ~ 6.5, a dla cynku gdy pH ~
11.5. Przy każdej zmianie wartości pH (wzrost lub spadek) następuje zwiększenie
szybkości korozji. Metale, tj. nikiel, miedź, kobalt, chrom, mangan, kadm, magnez tworzą tlenki
rozpuszczalne w kwasach, a nierozpuszczalne w zasadach. Dlatego szybkość korozji tych metali w
środowisku alkalicznym maleje. Zależność szybkości korozji żelaza od pH środowiska jest następująca:
- pH < 4 - silna korozja żelaza z wydzieleniem wodoru oraz rozpuszczonych produktów korozji;
- pH = 4+9 - szybkość korozji nie zależy od pH, lecz jedynie od szybkości dyfuzji tlenu do powierzchni
żelaza;
- pH = 9^-12 - spadek szybkości korozji spowodowany mniejszą rozpuszczalnością produktów korozji;
- pH > 12 - ponowne zwiększenie szybkości korozji w wyniku tworzenia się rozpuszczalnych żelazinów.
Obecność rozpuszczonych soli wywiera różny wpływ na przebieg korozji w zależności od ich
chemicznego charakteru. Sole metali alkalicznych, np. NaCl, KCl, KI, Na
2
SO
4
obecne w roztworze
obojętnym powodują początkowo wzrost szybkości korozji wraz ze wzrostem stężenia soli aż do
osiągnięcia wartości maksymalnej, a następnie jej zmniejszanie. Sole ulegające hydrolizie z odczynem
kwaśnym, np. AICl
3
, FeCl
2
, NiS04 przyśpieszają proces korozji w zależności od pH roztworu. Sole
ulegające hydrolizie z odczynem alkalicznym o pH > 10 hamują korozję żelaza i stali. Do tych soli należą,
np. Na
2
SiO
3
, Na
2
C03, Na
3
P0
4
.
Czynnikiem powodującym wzrost szybkości korozji jest podwyższona temperatura, np. szybkość

korozji stali w kwasie HC1 (z depolaryzacją wodorową) wzrasta dwukrotnie przy wzroście temperatury
kwasu o 10°C. Często jednak zależność ta ma charakter bardziej złożony, szczególnie, gdy korozja
przebiega z depolaryzacją tlenową. Do takich przypadków zaliczamy korozję stali w środowisku wód
naturalnych.
Na przebieg korozji elektrochemicznej wpływają także warunki eksploatacji konstrukcji
metalowych. W zależności od nich rozróżniamy następujące rodzaje korozji:
- atmosferyczna - jest to korozja metali zachodząca w atmosferze wilgotnego powietrza;
- ziemna - spowodowana jest agresywnym działaniem gleby;
- morska - zachodzi w wyniku agresywnego działania środowiska morskiego, tj. atmosfery i wody
morskiej;
- elektrolityczna (elektrokorozja) - spowodowana prądami elektrycznymi pochodzącymi ze źródeł
zewnętrznych (tzw. prądami błądzącymi);
- biologiczna - zachodzi w wyniku zmian powstałych w środowisku pod wpływem działania
organizmów żywych, np. bakterii, glonów, pleśni, grzybów.
Konstrukcje metalowe mogą korodować w różny sposób. W zależności od rodzaju zniszczenia
korozyjnego rozróżnia się:
- korozję równomierną - jest najmniej niebezpieczna, gdyż wpływa na obniżenie wytrzymałości
konstrukcji tylko poprzez zmniejszenie przekroju poprzecznego materiałów. Zachodzi z jednakową
szybkością na całej powierzchni metalu (np. korozja atmosferyczna blachy stalowej);
- korozję wżerową - zwaną również korozją punktową, gdyż zachodzi w określonych miejscach na
powierzchni metalu. Rozwija się na niewielkich obszarach, lecz powoduje duże zniszczenia w głębi
materiału wskutek powstawania tzw. wżerów wpływających na obniżenie wytrzymałości materiału jak i
konstrukcji. Ten typ korozji obserwuje się w stalach zanurzonych w roztworach zawierających
agresywne jony chlorkowe, np. w wodzie morskiej;
- korozję międzykrystaliczną - rozwija się ona wzdłuż granic ziaren metalu lub stopu. Tworzą się wówczas
wewnętrzne ogniwa korozyjne, w których granice ziaren ulegają rozpuszczeniu, a tym samym zostaje
naruszona struktura metalu. Jest to najniebezpieczniejszy typ korozji, gdyż przy minimalnych zmianach
wyglądu zewnętrznego następuje znaczne pogorszenie właściwości mechanicznych metalu.
W celu zmniejszenia strat spowodowanych niszczącym działaniem procesu korozji opracowano wiele
metod zapobiegania lub hamowania przebiegu tego niepożądanego zjawiska. Najistotniejszymi metodami
ochrony antykorozyjnej są:
- dobór odpowiedniego metalu lub stopu do budowy urządzeń oraz odpowiednia ich obróbka, gdyż
zanieczyszczenia materiału oraz niejednorodność jego powierzchni sprzyjają korozji;
- ochrona elektrochemiczna, która polega na podłączeniu konstrukcji metalowej do ujemnego bieguna
źródła prądu stałego o niewielkim napięciu (1-^2 V) lub połączeniu jej z metalem bardziej
elektroujemnym, który stanowi anodę w utworzonym w ten sposób ogniwie. Metal ten jest tak zwanym
protektorem, a metoda ochrony - metodą protektorową. Zarówno przy zastosowaniu prądu
zewnętrznego, jak i w metodzie protektorowej chroniony metal pełni rolę katody, na której zachodzą
wyłącznie procesy redukcji (nie zachodzi proces korozji);
-
ochrona za pomocą inhibitorów korozji, tj. substancji, które dodane w niewielkiej ilości do środowiska
agresywnego znacznie zmniejszają szybkość korozji. Mechanizm ich działania antykorozyjnego zależy
od rodzaju zastosowanego inhibitora. Inhibitory utleniające powodują powstanie cienkiej warstewki
tlenków na metalu zapobiegającej jego korozji. Inhibitory organiczne absorbują się na powierzchni
metalu izolując go od agresywnego środowiska. Inhibitory lotne charakteryzuje duża prężność par, które
osadzają się na powierzchni chronionej zabezpieczając ją przed korozją; stosowanie powłok
ochronnych - polega na pokrywaniu elementów chronionych powłokami metalowymi lub
niemetalowymi. Powłoka wykonana z metalu o niższym potencjale od metalu chronionego oprócz
izolacji od tlenu i wilgoci zapewnia ochronę protektorową, gdyż sama jako anoda ulega korozji.
Powłoka katodowa wykonana jest z metalu o wyższym potencjale elektrodowym niż element chroniony.
Jej działanie trwa tak długo, jak długo powłoka jest szczelna. Zadaniem powłok niemetalicznych,
organicznych i nieorganicznych jest izolowanie powierzchni metalu od dostępu tlenu i wilgoci.
Stosowane w tym celu farby i lakiery służą zarazem do dekoracji chronionych powierzchni.
ZASADA OZNACZANIA ŻELAZA METODĄ Z RODANKIEM
Jony żelaza trójwartościowego w środowisku kwaśnym dają z roztworem rodanku
krwistoczerwone zabarwienie, którego intensywność jest proporcjonalna do stężenia żelaza w badanej
próbce. Przed oznaczaniem całą ilość żelaza należy przeprowadzić w formę trójwartościową. W
przypadku oznaczania żelaza ogólnego, całą ilość żelaza obecnego w badanej wodzie należy
przeprowadzić w formę trójwartościową utleniając jony żelazawe Fe
2+
nadtlenkiem wodoru H
2
O
2
(wodą
utlenioną). W trakcie oznaczania zachodzą reakcje:
2 Fe
2+
+H
2
O
2
+ 2H
+
→ 2 Fe
3+
+ 2H
2
O
Fe
3+
+ n SCN
-
→ Fe (SCN)
n
3-n
W oznaczaniu przeszkadzają:
— fluorki i fosforany oraz inne związki tworzące połączenia kompleksowe z trójwartościowym
żelazem, w środowisku kwaśnym,
— srebro, kadm, rtęć i antymon tworzące związki kompleksowe z rodankiem i zmniejszające
intensywność zabarwienia rodanku żelazowego,
— duża zawartość związków organicznych oraz wysoka barwa i mętność wody.
Metoda pozwala na oznaczenie żelaza od 0,01 do 0,2 mg w próbce, co przy użyciu próbki o objętości 100
cm
3
stanowi 0,l-0,2 mg/dm
3
.
Jeżeli oznaczenie nie może być wykonane w ciągu kilku godzin od pobrania, próbkę należy utrwalić
przez dodanie 2 cm
3
kwasu solnego (c.wł. 1,18) na 1 dm
3
wody.
Wykonanie oznaczenia
Do cylindrów Nesslera odmierzyć po 50 cm
3
badanych prób. Dodać 5 cm
3
HC1 (1+1) i 0,5 cm
3
nadtlenku wodoru. Próbę dokładnie wymieszać. Po upływie 5 minut dodać 2,5 cm
3
roztworu
rodanku. Dokładnie wymieszać i porównać ze skalą wzorców.
Zawartość żelaza ogólnego obliczyć według wzoru:
x
=
v
a 1000
×
[mg Fe
og
/dm
3
]
w którym:
a - ilość żelaza w próbce odczytana ze skali wzorców, mg Fe;
v - objętość próby wody użyta do badań, cm
3
.
W celu oznaczenia w badanej próbie zawartości żelaza trójwartościowego należy
wykonać identyczne oznaczenie, lecz bez dodawania nadtlenku wodoru. Zawartość żelaza
dwuwartościowego należy wyliczyć z różnicy:
Fe
2+
= Fe
og
- Fe
3+
[mg Fe
2+
/dm
3
]
wykorzystując wyniki wykonanych wcześniej oznaczeń.
WPROWADZENIE DO ABSORPCJOMETRII.
Większość oznaczeń analitycznych w analizie instrumentalnej (ilościowej) oparta jest na metodach
kolorymetrycznych. Zasada tych metod polega na reakcjach oznaczanych związków z dodanymi
odczynnikami, w wyniku których powstają reakcje barwne. W zależności od stężenia substancji badanej
zmienia się natężenie zabarwienia roztworu. W analizie kolorymetrycznej wykorzystuje się własności
zabarwionych roztworów, polegające na pochłanianiu (absorpcji) przechodzącego przez nie
promieniowania. Jeżeli na badaną próbkę wody skierujemy wiązkę światła, wówczas część
promieniowania ulegnie odbiciu lub rozproszeniu, część pochłonięciu, a część przechodzi przez warstwę
wody. Zależności ilościowe tych wiązek promieniowania opisują prawa absorpcji.
Prawa absorpcji
I Prawo absorpcji (prawo Lamberta)
Wiązka promieniowania monochromatycznego po przejściu przez jednorodny ośrodek absorbujący
o grubości b ulega osłabieniu według równania:
I = I
0
e
-kb
w którym I
0
oznacza natężenie wiązki promieniowania monochromatycznego padającego na jednorodny
ośrodek absorbujący, I— natężenie promieniowania po przejściu przez ośrodek absorbujący, b —
grubość warstwy absorbującej, k — współczynnik absorpcji, e — podstawę logarytmów naturalnych.
Stąd:
ln
I
I
0
= kb =A lub A= log
I
I
0
ab
gdzie: a = 0,4343 k, a A — zdolność pochłaniania promieniowania zwana absorbancją.
I prawo absorpcji można zatem sformułować w sposób następujący: Jeśli wiązka promieniowania
monochromatycznego przechodzi przez jednorodny ośrodek absorbujący absorbancja jest
proporcjonalna do grubości warstwy absorbującej,
Inną wielkością stosowaną do określania absorpcji promieniowania jest transmitancja T określana
jako:
A= log
T
1
Transmitancję podajemy najczęściej w procentach, stąd:
% T = 100
0
I
I
= 100 T lub A=log
T
%
100
II Prawo absorpcji (prawo Lamberta — Beera)
Prawo to dotyczy absorpcji promieniowania przez roztwory i można je sformułować w następujący
sposób: jeśli współczynnik absorpcji rozpuszczalnika jest równy zeru, to wiązka promieniowania
monochromatycznego, po przejściu przez jednorodny roztwór substancji absorbującej o stężeniu c,
ulega osłabieniu według równania:
I = I
0
e
-kbc
Stąd, po przekształceniach jak wyżej, można zapisać:
A= log
I
I
0
abc
Prawo to można sformułować w sposób następujący: Jeżeli współczynnik absorpcji rozpuszczalnika jest
równy zeru, to absorbancja wiązki promieniowania monochromatycznego przechodzącej przez
jednorodny roztwór jest wprost proporcjonalna do stężenia roztworu c i do grubości warstwy
absorbującej b
III Prawo absorpcji (prawo addytywności absorpcji)
Absorbancją roztworu wieloskładnikowego równa się sumie absorbancji poszczególnych składników
A = A
1
+A
2
+...A
n
gdzie: A
t
, A
2
, ..., A„ są to absorbancje poszczególnych składników
W równaniu:
A = a c b
wielkość a jest właściwym współczynnikiem absorpcji, gdy stężenie wyrażamy w kg/dm
3
lub g/cm
3
.
Natomiast, gdy stężenie c wyrażamy w mol/dm
3
, równanie to przybiera postać:
A = ε c b
gdzie
ε
jest to molowy współczynnik absorpcji, a jego wymiar podawany jest dwojako:
[dm
3
• mol
-1
-•cm
-1
] lub w jednostkach SI [m
2
• mol
-1
].
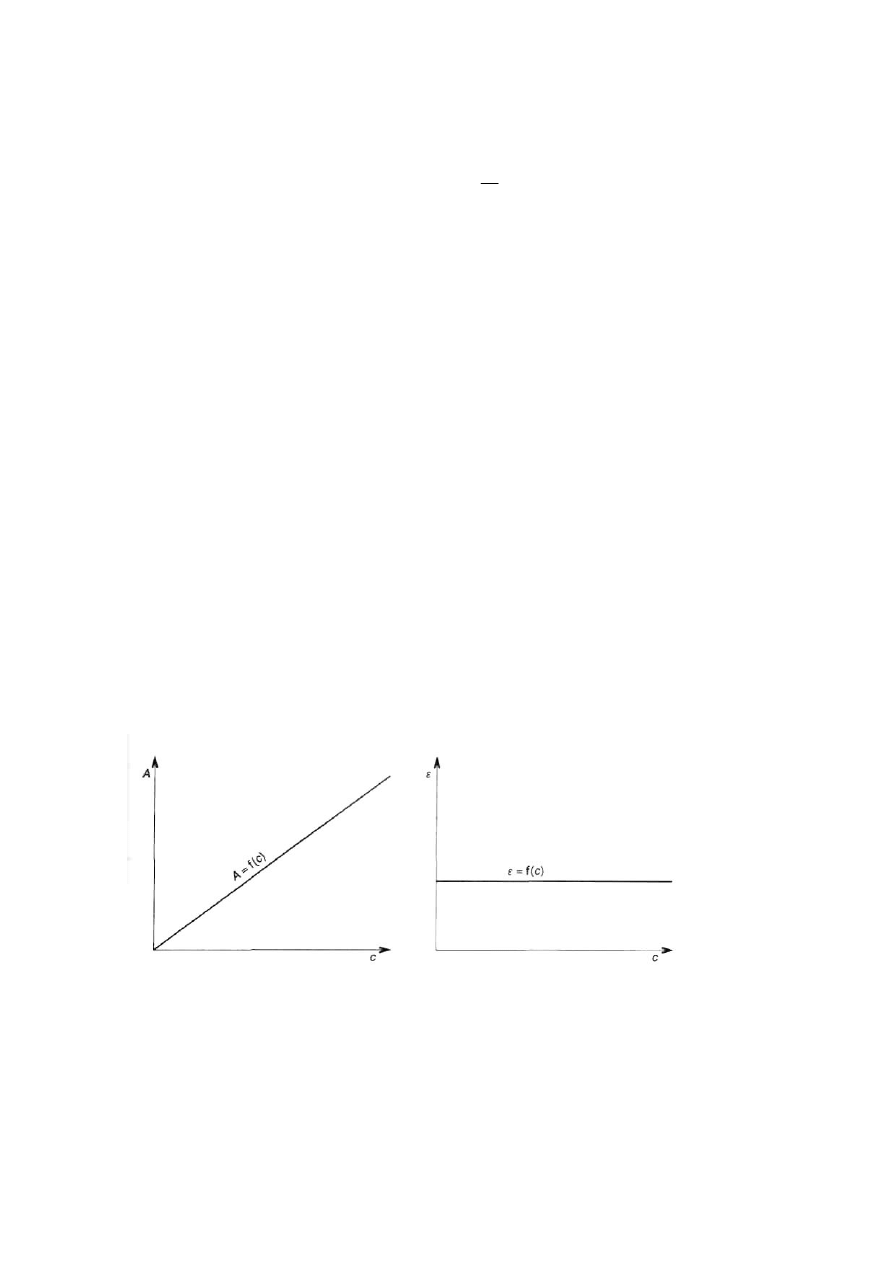
Funkcja A = f(c) jest linią prostą, jeżeli roztwór spełnia II prawo absorpcji. Natomiast
ε
jest wartością
stałą dla danego chromoforu.
Rys. 2. Wykres zależności A = f (c) Rys. 3. Wykres zależności
ε
= f (e)

FOSFORANY
Fosfor w wodach naturalnych może pochodzić z rozkładu związków organicznych roślinnych lub
zwierzęcych, z pól nawożonych nawozami fosforowymi oraz z zanieczyszczeń ściekami przemysłowymi.
Związki fosforu w przyrodzie ulegają podobnym przemianom jak związki azotowe, przy czym
przechodzą one w fosforany, co stanowi ostatnie stadium mineralizacji. Szczególnie intensywnie
przebiega przemiana fosforanowa w wodach powierzchniowych, w których mikroorganizmy asymilują
fosforany, a następnie obumierając opadają na dno, gdzie z kolei następuje mineralizacja. Z tych
względów w wodach powierzchniowych obserwuje się okresowość występowania fosforanów na jesieni,
zimą i wiosną, a zanik w lecie. Ilość fosforanów w czystych wodach powierzchniowych jest nieznaczna.
Wody pochodzące z terenów bogatych w związki humusowe mogą zawierać do 0,25 mg/dm
3
P0
4
.
Fosforany w wodach podziemnych płytkich pochodzą najczęściej z zanieczyszczeń wydalinami lub
nawożonej gleby, a zawartość ich wynosi od 01 do 0,2 mg/dm
3
PO
4
. W wodach podziemnych głębokich
fosforany występują tylko w wyjątkowych przypadkach. Wody naturalne zawierają fosfor zarówno w
postaci związków organicznych, jak i mineralnych — rozpuszczonych i nierozpuszczonych.
Znaczenie związków fosforu przy ocenie wody do picia jest podobne lak azotowych i zazwyczaj
składniki te występują równolegle. Z tych względów przy badaniu wody do picia oznaczanie związków
fosforowych ma drugorzędne znaczenie, tym bardziej że produkty przemiany związków azotowych
występują w większych ilościach. Oznaczanie fosforu w wodach powierzchniowych ma duże znaczenie,
gdyż fosforany stanowią jeden z podstawowych czynników biogennych, powodujących masowy rozwój
glonów i stwierdzenie ich obecności zmusza do śledzenia składu wody i zwalczania zakwitów.
Eutrofizacja naturalnych zbiorników wodnych, w związku z odprowadzaniem do nich takich
substancji biogennych jak związki fosforu i azotu, stanowi wysoce niekorzystne zjawisko w problemie
ochrony wód przed zanieczyszczeniem.
Dużym zagrożeniem dla odbiorników są zwłaszcza znaczne ilości związków fosforu, które dostają się
do odbiorników wraz z detergentami, stosowanymi na coraz większą skalę do potrzeb gospodarstwa
domowego oraz w przemyśle.
Obecność fosforanów w wodzie wodociągowej lub w wodzie przechowywanej przez pewien czas
sprzyja rozwojowi mikroorganizmów i z tych względów fosforany nie są pożądane.
Fosforany w ilościach normalnie występujących w wodzie do picia nie są szkodliwe dla zdrowia. W
ostatnich czasach stosuje się dodawanie fosforanów do wody w celu zwalczania korozji oraz
zapobiegania wytrącaniu się niektórych związków, jak np. żelaza lub wapnia. Ilości fosforanów
dodawane w tym celu wahają się w granicach od 0,25 do 1,0 mg/dm
3
PO4. W wyniku stosowania
nowych metod uzdatniania wody oraz stosowania na coraz większą skalę syntetycznych środków
powierzchniowoczynnych, dostają się do wód polifosforany, które hydrolizując tworzą ortofosforany.
Dopuszczalną ilość fosforanów w wodach regulują przepisy prawne
ł)
. Fosforany należy oznaczać
możliwie szybko po pobraniu próbki; jeżeli jest to niemożliwe, próbkę należy utrwalić przez dodanie 40
mg HgCl
2
/dm
3
. Nie należy stosować do utrwalania kwasu lub chloroformu. Wszystkie formy fosforu,
występujące w wodzie, oznacza się w postaci ortofosforanów po uprzedniej zamianie w tę postać
różnymi metodami podanymi przy badaniu ścieków. Przy oznaczaniu fosforanów rozpuszczalnych
należy przesączyć próbkę na miejscu pobrania.
Do oznaczania fosforanów w wodzie najczęściej stosuje się metodę kolorymetryczną z molibdenianem
amonowym i chlorkiem cynawym jako reduktorem. Mogą być również stosowane inne metody
kolorymetryczne.
OZNACZANIE FOSFORANÓW METODĄ MOLIBDENOWĄ
Zasada oznaczania polega na tworzeniu się w roztworze kwaśnym kwasu fosforomolibdenowego
H
7
(P)MoO
2
(O
4
)
6
o żółtym zabarwieniu, który ulega redukcji pod wpływem chlorku cynawego, tworząc
związek kompleksowy — błękit molibdenowy — o intensywnym niebieskim zabarwieniu. Intensywność
zabarwienia jest proporcjonalna do zawartości fosforanów. Oznacza się ją wizualnie lub
fotokolorymetrycznie. Dodatkowe zastosowanie ekstrakcji fosforanów z badanej próbki pozwala na
zwiększenie czułości oznaczania i zmniejsza wpływ czynników przeszkadzających. Minimalne stężenie
wynosi ok. 0,01 mg/dm
3
P0
4
.
W oznaczeniu przeszkadzają: krzemionka w postaci jonowej w stężeniu powyżej 25 mg/dm
3
,
arseniany, mętność, barwa, znaczne ilości chlorków, azotyny, Fe
3+
powyżej 1 mg/dm
3
, Fe
2+
powyżej 100
mg/dm
3
, związki organiczne. Bardzo alkaliczne lub bardzo kwaśne wody należy zobojętnić wobec

fenoloftaleiny. Wpływ krzemionki eliminuje się przez rozcieńczenie próbki. Wpływ żelaza można usunąć
przez odpowiednie rozcieńczenie próbki lub dodanie równoważnej ilości 0,1 m roztworu wersenianu.
Przy dużych ilościach chlorków powstaje błękitnozielone zabarwienie, które się kompensuje porównując
zabarwienie próbki z wzorcem zawierającym chlorki o takim samym stężeniu. Przy stężeniu azotynów do
25 mg/dm
3
należy dodać do próbki 0,1 g kwasu aminosulfonowego. Mętność usuwa się przez
odwirowanie próbki lub przesączenie. Związki organiczne, barwę, arseniany, w zależności od rodzaju
oznaczanych fosforanów, eliminuje się na drodze mineralizacji próbki lub przez odpowiednie
rozcieńczenie.
Wykonanie oznaczenia
Wykreślenie krzywej wzorcowej.
Wzorce należy przygotować dopełniając kolbki miarowe do kreski. Następnie dodać —
mieszając dokładnie za każdym razem — 2 cm
3
roztworu molibdenianu amonowego i 0,5 cm
3
roztworu chlorku cynawego. Następnie dokonać pomiaru absorbancji prób spektrofotometrem
Spekol 10 przy długości fali 690 nm stosując jako odnośnik wzorzec z wodą destylowaną i
wykreślić krzywą wzorcową, odkładając na osi odciętych stężenia fosforanów (PO
4
) we
wzorcach, a na osi rzędnych odpowiednie wartości absorbancji.
Wykonanie oznaczenia w badanych próbach.
Badane próby umieszczone w kolbkach miarowych dopełnić wodą destylowaną do kreski.
Następnie postępować identycznie jak podczas przygotowywania próbek do wykreślenia
krzywej wzorcowej. Po wykonaniu pomiaru absorbancji badanych prób dokonać odczytu
wartości stężeń fosforanów z wykreślonej wcześniej krzywej wzorcowej.
Obliczanie i podawanie wyników. Zawartość fosforanów obliczyć i podać wg wzoru:
x
=
v
a 1000
×
[mg/dm
3
PO
4
]
gdzie: a — ilość fosforanów odczytana z krzywej, wzorcowej, mg;
v— objętość próbki wziętej do badania, cm
3
.
Jeżeli wynik trzeba podać jako P, należy otrzymaną wartość podzielić przez 3,06.
Wyszukiwarka
Podobne podstrony:
Cwiczenie nr 10 Analiza ilościowa Alkacymetria Oznacznie weglanow i wodoroweglanow
Cwiczenie nr 11 Analiza ilościowa (miareczkowa) Oznacznie Ca 2 , Mg 2 Twardosc
Drgania Ćwiczenie nr 13, Politechnika Lubelska, Studia, semestr 5, Sem V, Sprawozdania, Laborka, Lab
Cwiczenia nr 13 RPiS id 124686 Nieznany
Kolorymetr oznaczanie Fe id 241 Nieznany
Cwiczenia nr 13 (z 14) id 98681 Nieznany
ćwiczenia nr 13 Rozwój emocji i potrzeb, Matczak rozwój społeczny, Matczak „Rozwój społeczny&r
Cwiczenie nr 13 Szablony i praca zespolowa id 9
ćwiczenia nr 13, Rozwoj cw 13 - Kepinski
Ćwiczenie nr 13(1)
Ćwiczenie nr 13, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwicz
Zeszyt Ćwiczeń nr 13
Cwiczenie nr 13
cwiczenie nr 13
ćwiczenie nr 13
więcej podobnych podstron