
Zagadnienia do kartkówki:
1. Podstawy analizy miareczkowej, PK i PR miareczkowania, miareczkowanie bezpo
ś
rednie,
odwrotne, po
ś
rednie.
2. Kompleksy – budowa, nazewnictwo, stała trwało
ś
ci i stała nietrwało
ś
ci; poj
ę
cia: ligand,
jon centralny, liczba koordynacyjna; obliczanie liczby koordynacyjnej.
3. Kompleksometria, kompleksonometria, wska
ź
niki kompleksometryczne.
4. Twardo
ść
wody - definicja,
ź
ródła twardo
ś
ci, rodzaje twardo
ś
ci, jednostki twardo
ś
ci.
5. Metody usuwania twardo
ś
ci wody - reakcje ilustruj
ą
ce ka
ż
d
ą
metod
ę
.
6. Metoda wersenianowa oznaczania twardo
ś
ci wody.
Analiza obj
ę
to
ś
ciowa (miareczkowa) polega na ilo
ś
ciowym oznaczaniu substancji
(analitu) na podstawie dokładnego pomiaru obj
ę
to
ś
ci roztworu dodawanego odczynnika
miareczkuj
ą
cego (titranta) o
ś
ci
ś
le okre
ś
lonym st
ęż
eniu (mianie), odmierzanego za pomoc
ą
biurety, reaguj
ą
cego ilo
ś
ciowo z analitem. Roztwór titranta dodaje si
ę
z biurety stopniowo,
małymi porcjami (miarami), stad nazwa – analiza miareczkowa.
Operacja dodawania titranta z biurety do naczynia zawieraj
ą
cego miareczkowany roztwór
nazywa si
ę
miareczkowaniem. Natomiast roztwór titranta o znanym st
ęż
eniu nosi nazw
ę
roztworu mianowanego.
Metodami miareczkowymi w zale
ż
no
ś
ci od st
ęż
enia titranta i wielko
ś
ci stosowanej biurety,
mo
ż
na oznaczy
ć
zawarto
ść
oznaczanych substancji w zakresie 10
-3
- 10
-1
.
Ze wzgl
ę
du na typ reakcji zachodz
ą
cej podczas miareczkowania, pomi
ę
dzy
oznaczan
ą
substancj
ą
a roztworem titranta, metody miareczkowe zostały podzielone na
cztery grupy:
•
kompleksometri
ę
– opart
ą
na tworzeniu rozpuszczalnych, słabo zdysocjowanych
zwi
ą
zków kompleksowych; najwa
ż
niejszym jej działem jest kompleskonometria,
w której titrantami s
ą
roztwory kompleksonów tworz
ą
cych z metalami kompleksy
chelatowe,
•
miareczkowanie
str
ą
ceniowe
–
oparte
na
reakcjach
wytr
ą
cania
trudno
rozpuszczalnych osadów jonów titranta i substancji oznaczanej,
•
alkacymetri
ę
– opart
ą
na reakcjach zoboj
ę
tnia (kwas – zasada) i obejmuje dwa
działy: alkalimetri
ę
(oznaczanie substancji przez miareczkowanie mianowanym
roztworem zasady oraz acydymetri
ę
(oznaczanie substancji przez miareczkowanie
mianowanym roztworem kwasu),
•
redoksymetri
ę
– opiera si
ę
na reakcjach utlenienia i redukcji; obejmuje dwa działy:
oksydymetri
ę
(oznaczanie
substancji
przez
miareczkowanie
mianowanymi
roztworami utleniaczy) i reduktometri
ę
(oznaczanie substancji przez miareczkowanie
mianowanymi roztworami reduktorów).
W analizie miareczkowej stosuje si
ę
przede wszystkim
ś
rodowisko wodne. Mo
ż
na tak
ż
e
wykorzysta
ć
metody miareczkowe do oznacze
ń
w
ś
rodowiskach niewodnych lub mieszanych.
Punkt miareczkowania, w którym oznaczany składnik przereagował ilo
ś
ciowo i
stechiometrycznie
z
titrantem
nosi
nazw
ę
punktu
równowa
ż
nikowego
(PR)
miareczkowania. Miareczkowanie uznaje si
ę
praktycznie za zako
ń
czone po osi
ą
gni
ę
ciu
punktu ko
ń
cowego (PK), okre
ś
lanego na podstawie widocznych zmian zachodz
ą
cych w
roztworze miareczkowanym (pojawienie si
ę
zabarwienia lub jego zmiana). PK powinien
znajdowa
ć
si
ę
jak najbli
ż
ej PR. Ró
ż
nica obj
ę
to
ś
ci titranta pomi
ę
dzy PR a PK nazywa si
ę
bł
ę
dem systematycznym miareczkowania.
PRACOWNIA nr 5
WST
Ę
P DO ANALIZY MIARECZKOWEJ. KOMPLEKSOMETRIA.
CHEMIA SANITARNA.

2
Ze wzgl
ę
du na sposób prowadzenia miareczkowania wyró
ż
nia si
ę
miareczkowanie
bezpo
ś
rednie i po
ś
rednie. Miareczkowanie bezpo
ś
rednie polega na tym,
ż
e oznaczana
substancja reaguje bezpo
ś
rednio – stechiometrycznie i szybko z dodawanym titrantem.
W tym miareczkowaniu u
ż
ywa si
ę
tylko jednego roztworu mianowanego. Natomiast
miareczkowanie po
ś
rednie polega na dobraniu takiej substancji trzeciej, która reaguj
ą
c
stechiometrycznie i ilo
ś
ciowo z oznaczanym składnikiem tworzy nowy zwi
ą
zek, reaguj
ą
cy
nast
ę
pnie
stechiometrycznie
z
titrantem.
Szczególnym
rodzajem
miareczkowania
po
ś
redniego jest miareczkowanie odwrotne – polega ono na tym,
ż
e do badanego roztworu
dodaje si
ę
odmierzon
ą
ilo
ść
roztworu titranta i w nadmiarze, a nast
ę
pnie nadmiar tego
odczynnika odmiareczkowuje si
ę
innym odpowiednio dobranym roztworem titranta II.
Potrzebne s
ą
wiec dwa roztwory mianowane. Taki typ miareczkowania stosuje si
ę
w
przypadku gdy reakcja przebiega wolno lub gdy trudno jest dobra
ć
odpowiedni wska
ź
nik do
miareczkowania bezpo
ś
redniego.
Technika pipetowania roztworów
Such
ą
od zewn
ą
trz pipet
ę
zanurza si
ę
w cieczy na tak
ą
gł
ę
boko
ść
, aby podczas
pobierania roztworu nie zassa
ć
powietrza. Nast
ę
pnie pobiera si
ę
roztwór trzymaj
ą
c pionowo
pipet
ę
. Po ustaleniu si
ę
dolnego menisku na wysoko
ś
ci kreski na szyjce pipety (oko powinno
by
ć
na wysoko
ś
ci kreski) dotyka si
ę
ko
ń
cem pipety suchego pomocniczego naczynia
szklanego w celu usuni
ę
cia zwisaj
ą
cej kropli i przenosi pipet
ę
nad przygotowane naczynie.
Trzymaj
ą
c pionowo pipet
ę
opró
ż
nia si
ę
jej zawarto
ść
do nachylonego naczynia. Ko
ń
cówka
pipety powinna dotyka
ć
ś
ciank
ę
naczynia. Nie wolno wydmuchiwa
ć
ani wytrz
ą
sa
ć
pozostało
ś
ci roztworu z pipety. Nie powinno si
ę
równie
ż
dotyka
ć
ko
ń
cem pipety cieczy
znajduj
ą
cej si
ę
w naczyniu.
Technika miareczkowania
Dokładnie umyt
ą
biuret
ę
, tak aby woda spływała z niej równomiernie nie pozostawiaj
ą
c
kropel, przepłukuje si
ę
2 – 3 krotnie niewielkimi ilo
ś
ciami roztworu mianowanego i umieszcza
si
ę
w statywie. Do biurety wlewa si
ę
bezpo
ś
rednio z butelki roztwór mianowany powy
ż
ej
kreski zerowej. Nast
ę
pnie nale
ż
y usun
ąć
z ko
ń
cówki biurety powietrze, odkr
ę
caj
ą
c kran tak,
aby z biurety wypłyn
ą
ł mocny strumie
ń
titranta. Pozostawione w biurecie powietrze mo
ż
e by
ć
przyczyn
ą
du
ż
ych bł
ę
dów podczas miareczkowania. Je
ż
eli na ko
ń
cówce biurety pozostanie
jeszcze kropla roztworu, nale
ż
y j
ą
usun
ąć
przez dotkni
ę
cie do suchej
ś
ciany zlewki.
Nast
ę
pnie pod biuret
ę
podstawia si
ę
naczynie (najcz
ęś
ciej kolba sto
ż
kowa) z
miareczkowanym roztworem. Palcami lewej r
ę
ki otwiera si
ę
kurek biurety a praw
ą
r
ę
k
ą
trzyma
si
ę
kolb
ę
sto
ż
kow
ą
jednocze
ś
nie mieszaj
ą
c ruchem wirowym zawarto
ść
kolby. Roztwór
mianowany spuszcza si
ę
ostro
ż
nie z biurety, a w miar
ę
zbli
ż
ania si
ę
do PK miareczkowania
coraz wolniej, dozuj
ą
c go kropla po kropli. Ostatni
ą
kropl
ę
zbiera si
ę
poprzez dotkni
ę
cie
wewn
ę
trzn
ą
powierzchni
ą
kolby, któr
ą
nast
ę
pnie spłukuje si
ę
wod
ą
z tryskawki.

3
Poziom roztworu w biurecie po zako
ń
czeniu miareczkowania nale
ż
y odczytywa
ć
zawsze po
upływie stałego czasu od pocz
ą
tku miareczkowania, np. 1 – 2 minuty, aby zmniejszy
ć
bł
ą
d
spływu. Cało
ść
miareczkowania nale
ż
y przeprowadzi
ć
przy jednorazowym napełnieniu
biurety. Ponowne napełnianie biurety w trakcie miareczkowania zmniejsza dokładno
ść
oznaczenia. Ka
ż
de miareczkowanie nale
ż
y rozpoczyna
ć
od poziomu zerowego. Unika si
ę
w
ten sposób pomyłek w odczytach obj
ę
to
ś
ci. Miareczkowanie badanego roztworu nale
ż
y
wykona
ć
2 – 3 krotnie, a otrzymane wyniki nie powinny ró
ż
ni
ć
si
ę
o wi
ę
cej ni
ż
0,05 – 0,15 ml.
Po sko
ń
czonym miareczkowaniu dopełnia si
ę
biuret
ę
roztworem titranta i nakrywa czyst
ą
,
such
ą
probówk
ą
. Biureta powinna by
ć
stale napełniona mianowanym roztworem lub wod
ą
destylowan
ą
. W razie niepewno
ś
ci czy PK miareczkowania został ju
ż
osi
ą
gni
ę
ty nale
ż
y
odczyta
ć
i zapisa
ć
poziom roztworu w biurecie, a nast
ę
pnie doda
ć
jeszcze jedn
ą
kropl
ę
titranta i obserwowa
ć
czy nast
ą
piła zmiana barwy. Miareczkowanie nale
ż
y prowadzi
ć
w
miejscu dobrze o
ś
wietlonym, lecz zabezpieczonym przed bezpo
ś
rednim działaniem promieni
słonecznych. Po naczyniem z miareczkowanym roztworem nale
ż
y umie
ś
ci
ć
arkusz białego
papieru, bibuł
ę
do s
ą
czenia lub płytk
ę
porcelanow
ą
, co ułatwia obserwowanie barwy
roztworu. W miar
ę
mo
ż
liwo
ś
ci nale
ż
y miareczkowa
ć
przy
ś
wietle dziennym.
Zwi
ą
zki kompleksowe s
ą
grup
ą
zwi
ą
zków zło
ż
on
ą
z rdzenia (atomu centralnego) i ligandów
- przykoordynowanych przez atom centralny jonów lub cz
ą
steczek elektrooboj
ę
tnych.
Atomem centralnym jest zazwyczaj atom lub jon os strukturze elektronowej umo
ż
liwiaj
ą
cej
przyj
ę
cie par elektronowych i utworzenie wi
ą
zania koordynacyjnego, czyli jest akceptorem
elektronów. Ligandy natomiast pełni
ą
rol
ę
donorów elektronów.
Rodzaje kompleksów:
a) ze wzgl
ę
du na rodzaj ligandów wyró
ż
nia si
ę
kompleksy:
- proste – kompleksy utworzone z ligandów jednofunkcyjnych, czyli takich, które zajmuj
ą
w
wewn
ę
trznej sferze koordynacyjnej atomu centralnego tylko jedno miejsce, np. [Fe(SCN)]
2+
,
rozpuszczaj
ą
si
ę
w wodzie
- chelatowe – kompleksy utworzone z ligandów wielofunkcyjnych, czyli takich, które zajmuj
ą
w
wewn
ę
trznej sferze koordynacyjnej atomu centralnego dwa lub wi
ę
cej miejsc, rozpuszczaj
ą
si
ę
w niepolarnych rozpuszczalnikach organicznych np. tetrachlorek w
ę
gla
b) ze wzgl
ę
du na ilo
ść
atomów centralnych wyró
ż
nia si
ę
kompleksy:
- jednordzeniowe – takie, które zawieraj
ą
jeden atom centralny
- wielordzeniowe – takie, które zawieraj
ą
dwa lub wi
ę
cej atomów centralnych
c) ze wzgl
ę
du na szybko
ść
tworzenia si
ę
i dysocjowania kompleksów wyró
ż
nia si
ę
kompleksy:

4
- labilne – czyli takie, w których równowaga pomi
ę
dzy składnikami kompleksów a utworzonym
kompleksem ustala si
ę
bardzo szybko, np. kompleksy niklu (II) z jonem cyjankowym
- bierne – czyli, takie w których ustalanie to przebiega bardzo wolno i stan równowagi zostaje
osi
ą
gni
ę
ty nieraz w ci
ą
gu wielu godzin, np. kompleksy chromu (III) z EDTA.
Liczba jednopozycyjnych ligandów przył
ą
czonych do jonu centralnego nosi nazw
ę
liczby
koordynacyjnej (LK).
Trwało
ść
zwi
ą
zków kompleksowych opisuj
ą
wielko
ś
ci: stała trwało
ś
ci i stała nietrwało
ś
ci.
- stała trwało
ś
ci
β
– stała równowagi tworzenia zwi
ą
zku kompleksowego wyra
ż
aj
ą
ca si
ę
stosunkiem st
ęż
enia kompleksu do iloczynu st
ęż
e
ń
jonów tworz
ą
cych dany kompleks w
stanie równowagi, np. dla reakcji:
2
2
2
]
][
[
]
)
(
[
)
(
2
−
+
−
−
−
+
=
→
+
CN
Ag
CN
Ag
CN
Ag
CN
Ag
β
- stała nietrwało
ś
ci K – stała równowagi reakcji przeciwnej czyli reakcji dysocjacji
kompleksu:
]
)
(
[
]
][
[
2
2
−
−
+
=
CN
Ag
CN
Ag
K
Kompleksy trwałe posiadaj
ą
du
żą
warto
ść
stałej trwało
ś
ci (powy
ż
ej 10
-7
) oraz mał
ą
warto
ść
stałej nietrwało
ś
ci.
Wska
ź
niki w analizie miareczkowej s
ą
substancjami, które ułatwiaj
ą
obserwacj
ę
zmian
fizycznych roztworu w pobli
ż
u PR, a tym samym okre
ś
lenia PK. Typowe efekty u
ż
ycia
wska
ź
nika polegaj
ą
na pojawieniu si
ę
, zmianie lub zaniku barwy, zm
ę
tnienia. Substancja
b
ę
d
ą
ca wska
ź
nikiem zawiera zazwyczaj układ nienasycony – grup
ę
chromoforowi decyduj
ą
c
ą
o barwie wska
ź
nika, np. grupa azowa. Dodatkowo obecno
ść
w cz
ą
steczce wska
ź
nika
pewnych grup pogł
ę
bia intensywno
ść
barwy, np. -OH
-
, -NH
2
, -NR
2
. Takie podstawniki nosz
ą
nazw
ę
grup auksochromowych.
Wska
ź
niki kompleksometryczne
Do wyznaczania PK w miareczkowaniu kompleksometrycznym najcz
ęś
ciej u
ż
ywa si
ę
metalowska
ź
ników. W niektórych miareczkowaniach kompleksometrycznych u
ż
ywa si
ę
tak
ż
e wska
ź
ników redoks. Metalowska
ź
niki to zwi
ą
zki organiczne, które podczas
miareczkowania tworz
ą
z oznaczanym kationem metalu barwny kompleks o warunkowej
stałej trwało
ś
ci ni
ż
szej od stałej trwało
ś
ci oznaczanego kationu z titrantem. Po dodaniu do
miareczkowanego roztworu wska
ź
nika tworzy on z oznaczanym kationem metalu barwny
kompleks. Wprowadzany podczas miareczkowania roztwór kompleksonu (titranta), w PK
wypiera całkowicie kationy metalu z mniej trwałego kompleksu metal- wska
ź
nik tworz
ą
c
trwalszy kompleks metal-titrant, w tym momencie roztwór przyjmuje zabarwienie wolnego
wska
ź
nika.

5
Metalowska
ź
niki mo
ż
na podzieli
ć
na trzy grupy
- grupa I - s
ą
to zwi
ą
zki praktycznie bezbarwne, np. kwas salicylowy czy jodek potasu lub
tiomocznik. Zwi
ą
zki te reaguj
ą
z kationami tworz
ą
c barwne kompleksy, np. kwas salicylowy
reaguj
ą
c z jonami
ż
elaza (III) tworzy kompleksy, których barwa zale
ż
y od pH; w
ś
rodowisku
bardzo kwa
ś
nym powstaje fioletowy kompleks, przy pH 4 pomara
ń
czowoczerwony, a przy
pH=9 jasno
ż
ółty
- grupa II - s
ą
to zwi
ą
zki które reaguj
ą
c z kationem powoduj
ą
zm
ę
tnienie, np. kwas
szczawiowy dla jonów wapnia, lub tworz
ą
zabarwione, nierozpuszczalne lub koloidalne laki
(np. galocyjanina dla galu)
- grupa III - czyli tzw. wska
ź
niki metalochromowe, s
ą
to barwniki organiczne zdolne do
tworzenia kompleksów z metalami, przy czym reakcji towarzyszy zmiana zabarwienia, np.:
mureksyd, czer
ń
eriochromowa T, kalces.
W przypadku wska
ź
ników redoks stosowanych w miareczkowaniu kompleksometrycznym ich
zasada działania polega na zmianie st
ęż
enia formy utlenionej b
ą
d
ź
zredukowanej danego
układu redoks obecnego w miareczkowanym roztworze w wyniku zwi
ą
zania jedenej z tych
form w trwały kompleks. Wynikiem tego jest odpowiednia warto
ść
potencjału redoks układu w
PR pozwalaj
ą
ca na utlenienie lub redukcj
ę
wska
ź
nika. Przykładem takiego miareczkowania
jest oznaczanie Fe(III) wobec wska
ź
nika bł
ę
kitu wariaminowego.
Twardo
ść
wody - jest to cecha wody, spowodowan
ą
obecno
ś
ci
ą
w niej jonów wapnia,
magnezu,
ż
elaza, glinu, manganu oraz kationów metali ci
ęż
kich. Poniewa
ż
w wodach
naturalnych sole wapnia i magnezu wyst
ę
puj
ą
w najwi
ę
kszych st
ęż
eniach, przyjmuje si
ę
ż
e
twardo
ść
wody pochodzi głownie od nich. St
ą
d wyró
ż
nia si
ę
twardo
ść
wapniow
ą
i
magnezow
ą
.
Twardo
ść
wody naturalnej powodowana przez w
ę
glany, wodorow
ę
glany i wodorotlenki
wapnia i magnezu okre
ś
la si
ę
mianem twardo
ś
ci w
ę
glanowej (twardo
ść
nietrwała,
przemijaj
ą
ca). Natomiast chlorki, siarczany, azotany wapnia i magnezu odpowiadaj
ą
za
twardo
ść
niew
ę
glanow
ą
wody (trwał
ą
). Suma twardo
ś
ci w
ę
glanowej i niew
ę
glanowej to
twardo
ść
ogólna wody. Podobnie suma twardo
ś
ci wapniowej i magnezowej to twardo
ść
ogólna wody.
Twardo
ść
wody mo
ż
na wyra
ż
a
ć
w ró
ż
nych jednostkach. Podstawowe to mmol/dm
3
i
mval/dm
3
(miligramorównowa
ż
nik zwi
ą
zków powoduj
ą
cych twardo
ść
w 1 dm
3
wody). Jednak
w technologii preparowania wody do celów przemysłowych najcz
ęś
ciej twardo
ść
wody
wyra
ż
ana jest za pomoc
ą
stopni twardo
ś
ci (niemieckich
0
n, francuskich
0
f i angielskich
0
a). W
Ameryce twardo
ść
wody podawana jest natomiast w ppm. W Polsce oprócz jednostek
podstawowych twardo
ś
ci wody zgodnych z układem SI u
ż
ywa si
ę
stopni niemieckich.
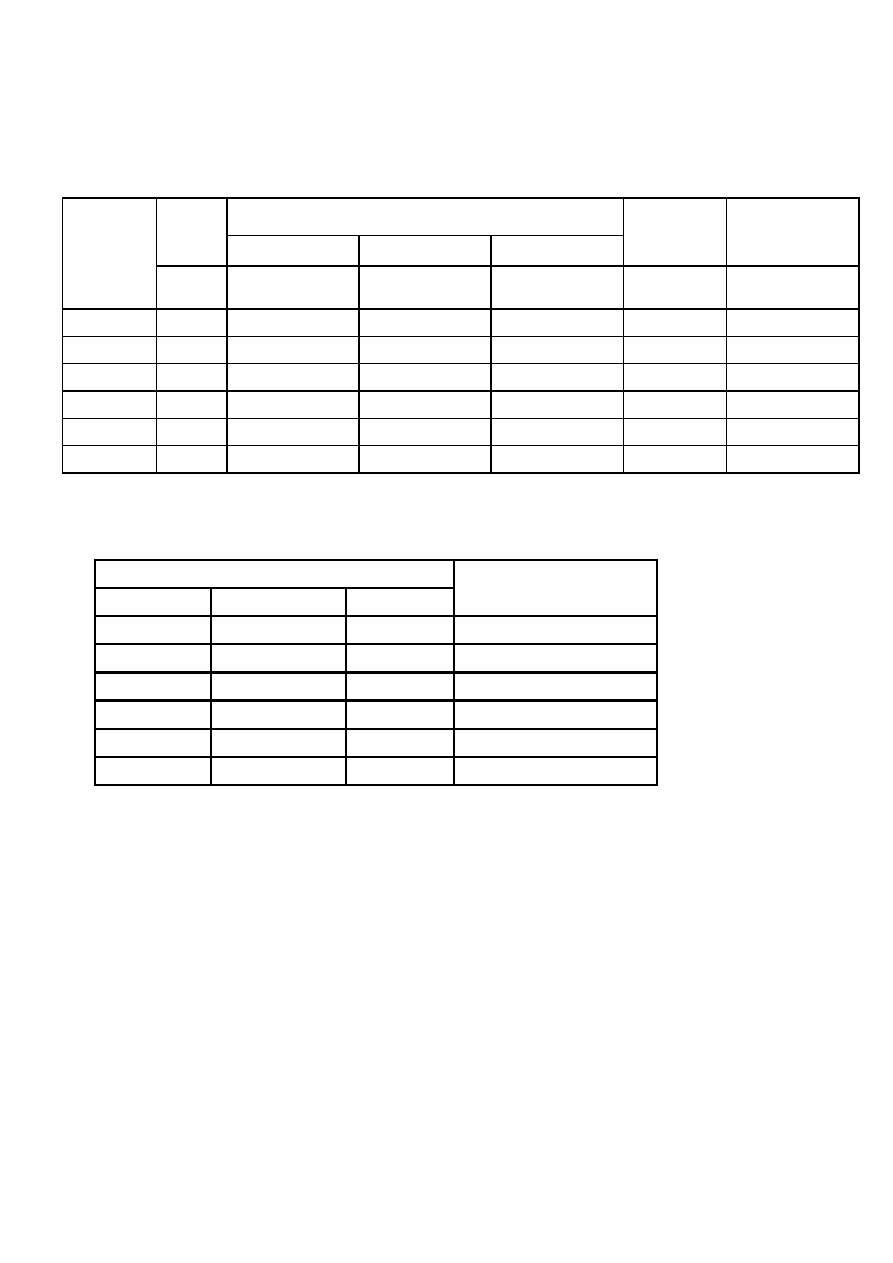
Zale
ż
no
ść
pomi
ę
dzy wymienionymi jednostkami twardo
ś
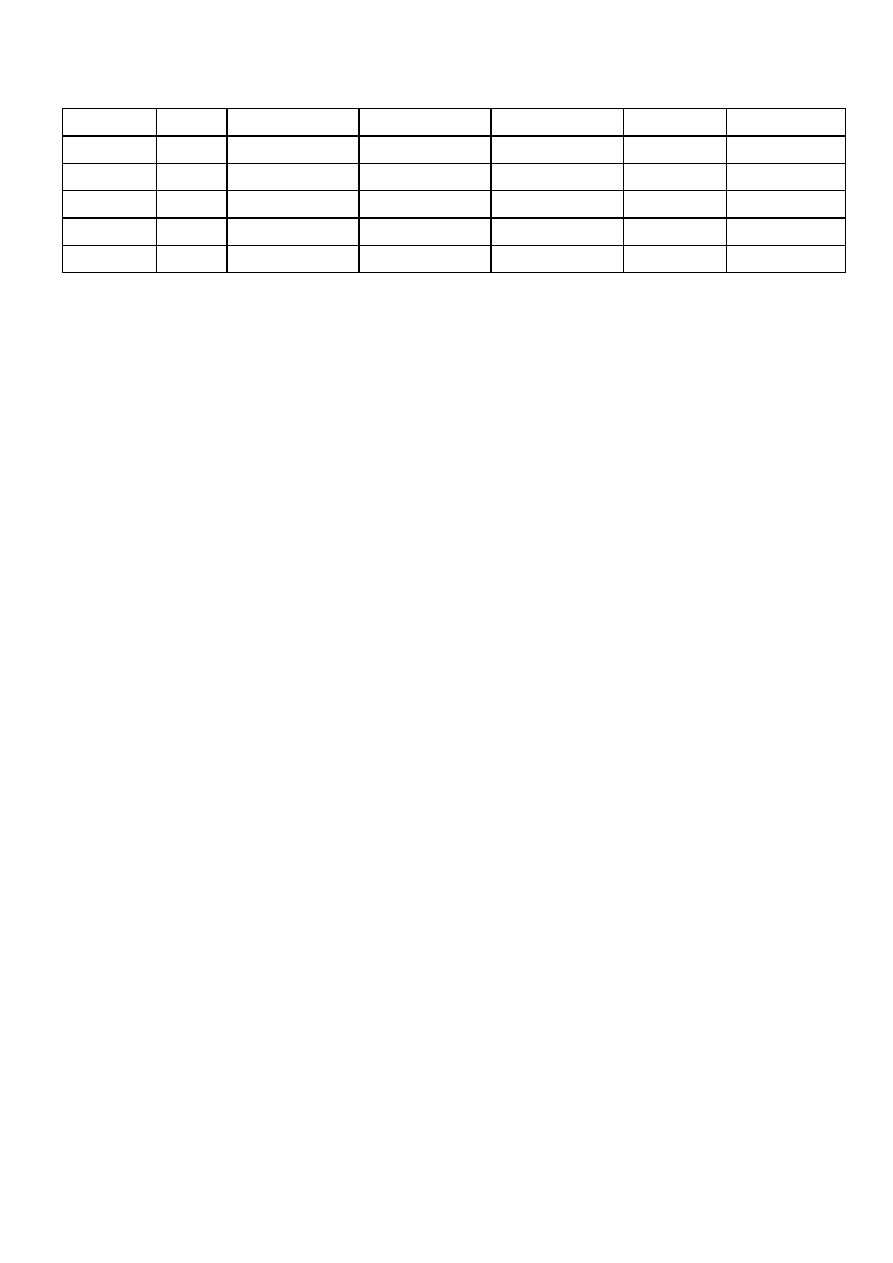
ci wody podano w tabeli nr 8.
Tabela nr 8. Współczynniki przeliczeniowe jednostek twardo
ś
ci wody.
Stopnie twardości
mval/dm
3
niemiecki [n
0
]
francuski [f
0
]
angielski [a
0
]
ppm
mmol/dm
3
Jednostka
twardości
wody
i jej
oznaczenie
28 mg CaO
lub 50 mg
CaCO
3
/dm
3
10 mg CaO/dm
3
10 mg CaCO
3
/dm
3
14,3 mg CaCO
3
/dm
3
1mg CaCO
3
/dm
3
100 mg CaCO
3
/dm
3
6
1 mval/dm
3
1,00
2,8
5,0
3,50
50,0
0,50
1
0
niemiecki
0,357
1,00
1,79
1,25
17,8
0,18
1
0
francuski
0,200
0,56
1,00
0,70
10
0,10
1
0
angielski
0,286
0,80
1,43
1,00
14,3
0,14
ppm
0,02
0,056
0,1
0,07
1
0,01
mmol/dm
3
2
5,6
10,0
7,02
100,0
1,00
Wody naturalne charakteryzuj
ą
si
ę
ró
ż
n
ą
twardo
ś
ci
ą
. Wody opadowe s
ą
bardzo
mi
ę
kkie, a ich twardo
ść
jest bliska zeru. W wodach słodkich przewa
ż
a twardo
ść
wapniowa.
Wody powierzchniowe, zwłaszcza potoków górskich s
ą
na ogół mi
ę
kkie. Wi
ę
ksz
ą
twardo
ść
wykazuj
ą
wody podziemne. Bardzo mi
ę
kkie wody maj
ą
posmak mdły, albo s
ą
bez smaku.
W przeciwie
ń
stwie do nich wody twarde o du
ż
ej zawarto
ś
ci wodorow
ę
glanów wapnia i
magnezu s
ą
smaczne, ale przy du
ż
ej koncentracji soli magnezu s
ą
ju
ż
gorzkie. Typowa
twardo
ść
wody wodoci
ą
gowej wynosi ok. 10 °n. Ze wzgl
ę
dów higienicznych twardo
ść
wody
nie ma istotnego znaczenia. Twarda woda mo
ż
e powodowa
ć
podra
ż
nienie skóry.
W przypadku du
ż
ych st
ęż
e
ń
chlorku i siarczanu magnezu, konsumpcja takiej wody
mo
ż
e skutkowa
ć
pojawieniem si
ę
krótkotrwałych biegunek. Twardo
ść
wody ma za to istotne
znaczenie przy ocenie jej przydatno
ś
ci do celów przemysłowych. W wielu bran
ż
ach
wymagana jest woda mi
ę
kka - woda zasilaj
ą
ca kotły parowe, woda do chłodzenia urz
ą
dze
ń
,
woda w sieciach ciepłowniczych. Mi
ę
kkiej wody wymagaj
ą
przemysł tekstylny, włókienniczy,
galwanizernie, przemysł spo
ż
ywczy. Obecne w twardej wodzie sole wapnia i magnezu mog
ą
powodowa
ć
wtr
ą
canie si
ę
trudno rozpuszczalnych osadów zwanych kamieniem kotłowym
oraz mog
ą
zwi
ę
ksza
ć
wła
ś
ciwo
ś
ci korozyjne wody.
Twardo
ść
wody przeznaczonej do spo
ż
ycia przez ludzi powinna si
ę
zawiera
ć
w przedziale od
60 do 500 mg CaCO
3
/dm
3
.
Metody usuwania twardo
ś
ci wody
Ogólnie metody zmi
ę
kczania wody mo
ż
na podzieli
ć
na cztery rodzaje: destylacja, metody
termiczne, metody chemiczne.
Destylacja
Destylacja daje idealne zmi
ę
kczanie, poniewa
ż
pozbawia wod
ę
wszystkich soli. Jednak
ż
e w
przemy
ś
le nie ma zastosowania ze wzgl
ę
du na wysokie koszty energii cieplnej. Jedynie w
technice cieplnej wykorzystuje si
ę
kondensaty, czyli skropliny z urz
ą
dze
ń
grzewczych. Po
odgazowaniu i odolejeniu kondensaty wody
ś
wie
ż
ej zawraca si
ę
do kotłów.
Metoda termiczna
Pod wpływem podwy
ż
szonej temperatury, ju
ż
powy
ż
ej 40
o
C, nast
ę
puje termiczny rozkład
wodorow
ę
glanów wapnia i magnezu zgodnie z reakcjami:
Ca(HCO
3
)
2
→
CaCO
3
↓
+ CO
2
↑
+ H
2
O
2 Mg(HCO
3
)
2
→
Mg
2
CO
3
(OH)
2
↓
+ 3 CO
2
↑
+ H
2
O
↓
H
2
O
2 Mg(OH)
2
+ CO
2
↑

7
Metoda termicznego zmi
ę
kczania ma zastosowanie w zakładach przemysłowych
dysponuj
ą
cych du
żą
ilo
ś
ci
ą
pary wymagaj
ą
cej kondensacji. Metoda termiczna nie mo
ż
e
całkowicie usun
ąć
twardo
ś
ci w
ę
glanowej. Pozostaje zawsze resztkowa twardo
ść
wynikaj
ą
ca
z pewnej, niewielkiej zreszt
ą
i zale
ż
nej od temperatury, rozpuszczalno
ś
ci w
ę
glanów wapnia i
magnezu oraz wodorotlenku magnezu. Ogólnie rzecz bior
ą
c metoda termiczna nadaje si
ę
do
zmi
ę
kczania wód o twardo
ś
ci wył
ą
cznie w
ę
glanowej, lub o bardzo znacznej przewadze tej
warto
ś
ci nad niew
ę
glanow
ą
, a tak
ż
e do wst
ę
pnego zmi
ę
kczania przed innymi zabiegami
uzdatniania wody.
Metody chemiczne
Metody chemiczne polegaj
ą
na str
ą
ceniu na drodze chemicznej nierozpuszczalnych
osadów lub wi
ą
zania w kompleksowe zwi
ą
zki jonów wapnia i magnezu przy pomocy ró
ż
nych
reagentów jak np. wodorotlenku wapna (wapno), wodorow
ę
glanu sodu (soda), wodorotlenku
sodu (soda kaustyczna), fosforanów, soli baru i in.
a) Dekarbonizacja wapnem.
Najcz
ęś
ciej stosowan
ą
metod
ą
zmi
ę
kczania jest dekarbonizacja wapnem. Jest prosta, tania i
polega na wytr
ą
caniu składników twardo
ś
ci w
ę
glanowej zgodnie z rekcjami:
Ca(HCO
3
)
2
+ Ca(OH)
2
→
2 CaCO
3
↓
+ 2 H
2
O
Mg(HCO
3
)
2
+ Ca(OH)
2
→
2 CaCO
3
↓
+ Mg(OH)
2
↓
+ 2 H
2
O
Wodorow
ę
glan magnezu, który pocz
ą
tkowo daje z Ca(OH)
2
zasadowy w
ę
glan, reaguje z
nadmiarem wodorotlenku, tworz
ą
c trudniej rozpuszczalny wodorotlenek magnezu. Je
ś
li w
wodzie znajduj
ą
si
ę
inne zwi
ą
zki magnezu np. MgCl
2
lub MgSO
4
tworz
ą
ce twardo
ść
magnezow
ą
– wówczas nast
ę
puje zamiana na twardo
ść
niew
ę
glanow
ą
wapniow
ą
– a
równowa
ż
na ilo
ść
Mg(OH)
2
str
ą
ca si
ę
:
MgSO
4
+ Ca(OH)
2
→
Mg(OH)
2
↓
+ CaSO
4
Z wapnem reaguj
ą
te
ż
inne składniki wody, takie jak dwutlenek w
ę
gla oraz
wodorotlenek
ż
elaza(II):
4 Fe(HCO
3
)
2
+ 8 + Ca(OH)
2
+ O
2
→
4 Fe(OH)
3
↓
+ 8 CaCO
3
↓
+ 6 H
2
O
CO
2
+ Ca(OH)
2
→
CaCO
3
↓
+ H
2
O
Jako dekarbonizatora u
ż
ywa si
ę
wody wapiennej (klarowny roztwór nasycony Ca(OH)
2
) lub
mleka wapiennego (zawiesina Ca(OH)
2
w wodzie). Woda wapienna jest dogodna ze wzgl
ę
du na
łatwo
ść
dokładnego dozowania, ale posiada t
ą
wad
ę
,
ż
e wymaga pojemników o wielkiej
obj
ę
to
ś
ci, poniewa
ż
nasycony roztwór Ca(OH)
2
ma bardzo małe st
ęż
enie (rozpuszczalno
ść
w
wodzie w 20
o
C wynosi zaledwie 1 ,1 g Ca(OH)
2
/l).
Tote
ż
przewa
ż
nie stosuje si
ę
do dekarbonizacji mleko wapienne. Ka
ż
d
ą
z tych cieczy
przyrz
ą
dza si
ę
mieszaj
ą
c wapno ze znaczn
ą
ilo
ś
ci
ą
wody w du
ż
ym pojemniku (sytniku), a
nast
ę
pnie dozuj
ą
c do reaktora, w którym zachodzi str
ą
canie i usuwanie osadu.
Proces dekarbonizacji przebiega z ró
ż
n
ą
szybko
ś
ci
ą
, zale
ż
nie od warunków. Na zimno zachodzi
powoli - w ci
ą
gu 3
÷
6 godzin; w temperaturze bliskiej 100 °C czas skraca si
ę
do około 10 minut.
Na szybko
ść
procesu wpływaj
ą
te
ż
inne warunki: twardo
ść
wody poddawanej dekarbonizacji,
zawarto
ść
w niej zwi
ą
zków organicznych, sposób mieszania wody z. reagentem oraz obecno
ść
masy kontaktowej w postaci drobnego piasku, lub innych substancji drobno rozkruszonych, np.
marmur, kwarc i in. Masa kontaktowa przy
ś
piesza proces powstawania w
ę
glanu wapnia,
dostarczaj
ą
c zarodków dla krystalizacji str
ą
caj
ą
cego si
ę
CaCO
3
.
Poniewa
ż
dla krystalizacji CaCO
3
na ziarnach masy kontaktowej najkorzystniejsza jest
temperatura w granicach 20
÷
30°C, wobec tego proces dekarbonizacji prowadzi si
ę
w tym
zakresie temperatur, zapewniaj
ą
c wła
ś
ciwe mieszanie i utrzymuj
ą
c cz
ęść
str
ą
caj
ą
cego si
ę
osadu wraz z mas
ą
kontaktow
ą
w stanie zawiesiny w dolnej cz
ęś
ci reaktora.

8
b) Dekarbonizacja kwasem solnym.
Ten sposób dekarbonizacji ma zastosowanie w uzdatnianiu wody, do chłodzenia - i w
technice nosi nazw
ą
"szczepienia kwasem". Zabieg szczepienia sprowadza si
ę
do zmiany
twardo
ś
ci w
ę
glanowej na niew
ę
glanow
ą
, co wprawdzie przeciwdziała tworzeniu si
ę
kamienia
kotłowego, ale zwi
ę
ksza własno
ś
ci korozyjne wody.
c) Zmi
ę
kczanie sod
ą
.
Ten sposób dawniej do
ść
rozpowszechniony ust
ę
puje ostatnio miejsca metodom bardziej
nowoczesnym. Polega na dodawaniu sody Na
2
CO
3
, która reaguje z solami wapnia i magnezu
zgodnie z równaniami reakcji:
CaSO
4
+ Na
2
CO
3
→
CaCO
3
↓
+ Na
2
SO
4
Ca(HCO
3
)
2
+ Na
2
CO
3
→
CaCO
3
↓
+ 2 NaHCO
3
MgSO
4
+ Na
2
CO
3
→
MgCO
3
+ Na
2
SO
4
Mg(HCO
3
)
2
+ Na
2
CO
3
→
MgCO
3
+ 2 NaHCO
3
Jakkolwiek z powy
ż
szych reakcji wynika,
ż
e soda usuwa oba rodzaje twardo
ś
ci, to jednak
sposób ten ma zastosowanie-do zmi
ę
kczania wód posiadaj
ą
cych prawie wył
ą
cznie twardo
ść
niew
ę
glanow
ą
i bardzo niewielk
ą
twardo
ść
magnezow
ą
. Szczególnie istotny jest warunek
małej zawarto
ś
ci soli Mg, co ma uzasadnienie w tym,
ż
e MgCO
3
, ma do
ść
dobra
rozpuszczalno
ść
i mo
ż
e si
ę
str
ą
ca
ć
tylko cz
ęś
ciowo jako sól zasadowa Mg
2
(OH)
2
CO
3
,
powstaj
ą
ca w wyniku wtórnie zachodz
ą
cej hydrolizy. Natomiast rozpuszczona cz
ęść
MgCO
3
jest str
ą
cona dopiero w kotle, gdzie nadmiar sody u
ż
ytej do zmi
ę
kczania w wysokiej temperaturze
ulega hydrolizie zgodnie z reakcj
ą
:
Na
2
CO
3
+ H
2
0
→
2 NaOH + CO
2
↑
a powstaj
ą
cy ług sodowy str
ą
ca nierozpuszczalny wodorotlenek magnezowy Mg(OH)
2
. Aby nie
dopu
ś
ci
ć
do str
ą
cania si
ę
tego osadu w kotle stosuje si
ę
zwykle zabieg zawracania wody
kotłowej do reaktora, w którym odbywa si
ę
zmi
ę
kczanie wody surowej. Obecny w wodzie
kotłowej NaOH str
ą
ca wówczas sole magnezowe, które razem z innymi osadami oddziela si
ę
na filtrach. Zmi
ę
kczanie wody sod
ą
pozwala uzyska
ć
spadek twardo
ś
ci nawet poni
ż
ej 1 st. tw.
d) Zmi
ę
kczanie wapnem i sod
ą
(metoda "wapno-soda")
Ta metoda nale
żą
ca dawniej do najbardziej rozpowszechnionych polega na jednoczesnym
usuwaniu twardo
ś
ci w
ę
glanowej i niew
ę
glanowej przez dodawanie wody wapiennej lub mleka
wapiennego i 10
÷
20% roztworu sody, przy czym:
Ca(OH)
2
usuwa: twardo
ść
w
ę
glanow
ą
wszystkie sole magnezowe
rozpuszczony dwutlenek w
ę
gla
Na
2
CO
3
usuwa: twardo
ść
niew
ę
glanow
ą
.
Znane z poprzednich metod reakcje zachodz
ą
w zasadzie szybko, natomiast powolny jest
wzrost powstaj
ą
cych kryształów CaCO
3
, i zawiesiny Mg(OH)
2
, który głównie determinuje
szybko
ść
procesu zmi
ę
kczania. Tote
ż
stosuje si
ę
nast
ę
puj
ą
ce warunki:
- podgrzewanie wody do 70
÷
90°C,
- zwi
ę
kszenie nadmiaru odczynników do 10
÷
20%;
- dodatek masy kontaktowej (CaCO
3
, kwarc).
Wy
ż
sza temperatura sprzyja zwi
ę
kszeniu szybko
ś
ci procesu oraz osi
ą
ganiu lepszych
wyników zmi
ę
kczania (mniejsza twardo
ść
szcz
ą
tkowa).
Dodatek masy kontaktowej - np. sproszkowanego kamienia wapiennego znacznie skraca
czas procesu zachodz
ą
cego w reaktorze. Masa kontaktowa mo
ż
e skróci
ć
czas reakcji z 2
godzin do 30 minut nawet be
ż
podgrzewania. Działanie masy kontaktowej polega na
przy
ś
pieszeniu wzrostu powstaj
ą
cego osadu.

9
Czynnikiem, który ujemnie wpływa na wyniki zmi
ę
kczania s
ą
substancje organiczne koloidowe
(głównie zwi
ą
zki humusowe), które opó
ź
niaj
ą
reakcj
ą
i powoduj
ą
m
ę
tno
ść
wody uchodz
ą
cej z
reaktora i nawet mog
ą
przechodzi
ć
przez filtr.
e) Zmi
ę
kczanie wody wodorotlenkiem sodowym (ługiem).
Zmi
ę
kczanie ługiem sodowym wody zawieraj
ą
cej oba rodzaje twardo
ś
ci sprowadza si
ą
do
działania 2 czynników: NaOH i tworz
ą
cego si
ę
wtórnie Na
2
CO
3
. według ni
ż
ej podanych równa
ń
reakcji. Wodorotlenek sodowy usuwa twardo
ść
wapniow
ą
w
ę
glanow
ą
, twardo
ść
magnezow
ą
i
dwutlenek w
ę
gla rozpuszczony w wodzie. Natomiast wtórnie powstaj
ą
ca soda likwiduje
twardo
ść
wapniow
ą
niew
ę
glanow
ą
. Wida
ć
to z reakcji:
Ca(HCO
3
)
2
+ 2 NaOH
→
CaCO
3
↓
+ Na
2
CO
3
+ 2 H
2
O
Mg(HCO
3
)
2
+ 4 NaOH
→
Mg(OH)
2
↓
+ 2 Na
2
CO
3
+ 2 H
2
O
MgSO4 + 2 NaOH
→
Mg(OH)
2
↓
+ Na
2
SO
4
CO
2
+ 2 NaOH
→
Na
2
CO
3
+ H
2
O
CaSO
4
+ Na
2
CO
3
→
CaCO
3
↓
+ Na
2
SO
4
Jak wida
ć
z bilansu wszystkich, tych reakcji, zmi
ę
kczanie ługiem sodowym nadaje si
ą
do
tych wód, w których twardo
ść
w
ę
glanowa jest w przybli
ż
eniu równa niew
ę
glanowej. Je
ś
li
twardo
ść
w
ę
glanowa przewy
ż
sza niew
ę
glanow
ą
— wówczas w wodzie zmi
ę
kczonej zostaje
pewien nadmiar utworzonego w reakcjach Na
2
CO
3
. W niektórych przypadkach do ługu dodaje si
ę
sod
ę
- mianowicie wtedy, gdy wtórnie utworzony Na
2
CO
3
nie wystarcza do zlikwidowania
twardo
ś
ci niew
ę
glanowej.
Czasami stosuje si
ę
zmi
ę
kczanie ługiem z dodatkiem taniego wapna, które zast
ę
puje
cz
ęść
kosztownego NaOH. W takim wypadku nale
ż
y obliczy
ć
ilo
ść
NaOH tak, aby ilo
ść
utworzonego wtórnie Na
2
CO
3
wystarczyła do usuni
ę
cia twardo
ś
ci niew
ę
glanowej. Tym
sposobem mo
ż
na zmi
ę
kcza
ć
wody o du
ż
ej twardo
ś
ci w
ę
glanowej i małej niew
ę
glanowej.
Metoda zmi
ę
kczania ługiem jest kosztowna, ale dogodna, poniewa
ż
przyrz
ą
dzanie
roztworu NaOH jest łatwe i nie wymaga dodatkowych urz
ą
dze
ń
. Łatwe jest te
ż
ś
cisłe dozowanie
ługu.
f) Zmi
ę
kczanie wody fosforanami.
Metoda fosforanowa polega na str
ą
caniu praktycznie nierozpuszczalnych fosforanów
wapnia i magnezu ( ich iloczyny rozpuszczalno
ś
ci s
ą
wielokrotnie mniejsze od CaCO
3
), dzi
ę
ki
czemu osi
ą
galna jest bardzo mała twardo
ść
szcz
ą
tkowa.
Do zmi
ę
kczania stosuje si
ę
zwykle fosforan trójsodowy, Na
3
PO
4
·10H
2
O, który reaguje
z solami wapnia i magnezu zgodnie z równaniami reakcji:
3 Ca(HCO
3
)
2
+ 2 Na
3
PO
4
→
Ca
3
(PO
4
)
2
↓
+ 6 NaHCO
3
3 Mg(HCO
3
)
2
+ 2 Na
3
PO
4
→
Mg
3
(PO
4
)
2
↓
+ 6 NaHCO
3
3 CaSO
4
+ 2 Na
3
PO
4
→
Ca
3
(PO
4
)
2
↓
+ 3 Na
2
SO
4
3 MgCl
2
+ 2 Na
3
PO
4
→
Mg
3
(PO
4
)
2
↓
+ 6 NaCl
Powstaj
ą
cy w reakcjach wodorow
ę
glan sodowy w wy
ż
szych temperaturach rozkłada
si
ę
z utworzeniem w
ę
glanu sodowego oboj
ę
tnego, który mo
ż
e likwidowa
ć
twardo
ść
niew
ę
glanow
ą
, zmniejszaj
ą
c w ten sposób zu
ż
ycie fosforanu. Rozkład termiczny NaHCO
3
zachodzi wg reakcji:
6 NaHCO
3
→
3 Na
2
CO
3
+ 3 CO
2
+ 3 H
2
O
Metoda fosforanowa ma wiele zalet. Zapewnia 2
÷
3 krotnie wi
ę
ksz
ą
szybko
ść
procesu
zmi
ę
kczania ni
ż
np. metoda wapno-soda; nie wymaga te
ż
bardzo
ś
cisłego dozowania
fosforanu, którego nadmiar nie tylko nie szkodzi, ale nawet jest po
żą
dany. Fosforany bowiem
nie koroduj
ą
blachy
ż
elaznej, a w wodzie zapobiegaj
ą
tworzenia si
ę
kamienia kotłowego,

10
który mo
ż
e powstawa
ć
w postaci krzemianu z resztkowej twardo
ś
ci szcz
ą
tkowej w obecno
ś
ci
SiO
2
. Jest to mo
ż
liwe dzi
ę
ki temu,
ż
e na np. Ca
3
(PO
4
)
2
jest trudniej rozpuszczalny od CaSiO
3
i
zgodnie z ogóln
ą
reguł
ą
str
ą
cania osadów mo
ż
e nie tylko wytr
ą
ca
ć
si
ę
w pierwszej kolejno
ś
ci,
ale nawet rozkłada
ć
krzemian wapniowy ju
ż
istniej
ą
cy zgodnie z reakcj
ą
:
3 CaSiO
3
+ 2 Na
3
PO
4
→
Ca
3
(PO
4
)
2
↓
+ 3 NaSiO
3
w której tworzy si
ę
rozpuszczalny krzemian sodowy.
Podobnie zachowuj
ą
si
ę
inne składniki kamienia kotłowego np. CaSO
4
, ulegaj
ą
rozkładowi pod wpływem fosforanów na tej samej zasadzie.
Mimo przytoczonych tu zalet metoda fosforanowa nie ma zastosowania do
zmi
ę
kczania wody surowej - jest na to za kosztowna. Natomiast nadaje si
ę
do usuwania
twardo
ś
ci resztkowej, pozostałej po zmi
ę
kczaniu wst
ę
pnym innymi metodami. Cz
ę
sto te
ż
u
ż
ywa si
ę
fosforanów do likwidowania minimalnej twardo
ś
ci resztkowej w kondensatach
zasilaj
ą
cych wysokopr
ęż
ne kotły.
Najcz
ęś
ciej w metodzie fosforanowej u
ż
ywa si
ę
ortofosforanu trójsodowego
znajduj
ą
cego si
ę
w handlu w .postaci soli uwodnionej Na
3
PO
4
·10H
2
O. Mo
ż
na stosowa
ć
równie
ż
ta
ń
sze wodorofosforany NaH
2
PO
4
i Na
2
HPO
4
, a nawet kwas fosforowy H
3
PO
4
- ale
pod warunkiem,
ż
e w wodzie znajduj
ą
si
ę
składniki alkaliczne (NaOH, Na
2
CO
3
), które
zoboj
ę
tniaj
ą
je.
g) Wymiana jonowa.
W zale
ż
no
ś
ci od wymaganego stopnia zmniejszenia twardo
ś
ci wody oraz rodzaju
usuwanej twardo
ś
ci stosuje si
ę
wymian
ę
jonow
ą
:
- w cyklu wodorowym na kationitach słabo kwa
ś
nych – usuwanie twardo
ś
ci w
ę
glanowej;
- w cyklu sodowym lub wodorowym na kationitach silnie kwa
ś
nych -usuwanie twardo
ś
ci
w
ę
glanowej i niew
ę
glanowej;
- w cyklu wodorowym i cyklu sodowym – usuwanie twardo
ś
ci w
ę
glanowej i niew
ę
glanowej;
- w cyklu wodorowym (kationit słabo kwa
ś
ny – dekarbonizacja + kationit silnie kwa
ś
ny –
usuwanie twardo
ś
ci w
ę
glanowej i niew
ę
glanowej);
- dekarbonizacja i dekationizacja na kationicie silnie kwa
ś
nym pracuj
ą
cym w cyklu sodowym i
anionicie zasadowym pracuj
ą
cym w cyklu chlorkowym.
Kationity słabo kwa
ś
ne wodorowe wymieniaj
ą
kationy zwi
ą
zane z resztami kwasowymi
słabych kwasów, st
ą
d zapewniaj
ą
rozkład wodorow
ę
glanów, a wi
ę
c usuwanie twardo
ś
ci
w
ę
glanowej zgodnie z reakcjami:
2KtH + Ca(HCO
3
)
2
→
Kt
2
Ca + 2H
2
O + 2CO
2
2KtH + Mg(HCO
3
)
2
→
Kt
2
Mg + 2H
2
O + 2CO
2
KtH + NaHCO
3
→
KtNa + H
2
O + CO
2
Kationity te, po regeneracji du
ż
ym nadmiarem regeneranta (najcz
ęś
ciej HCl), mog
ą
wymienia
ć
równie
ż
cz
ęść
kationów w zwi
ą
zanych z resztami kwasowymi silnych kwasów
według reakcji:
2KtH + CaCl
2
→
Kt
2
Ca + 2HCl
Woda po jonitowej dekarbonizacji zawiera CO
2
i pozbawiona jest twardo
ś
ci
w
ę
glanowej. W wyniku wymiany jonowej usuwana jest równie
ż
cz
ęść
twardo
ś
ci
niew
ę
glanowej, a w wodzie obecne s
ą
kwasy mineralne. Kationity słabo kwa
ś
ne pracuj
ą
ce w
cyklu wodorowym stosowane s
ą
wi
ę
c do zmi
ę
kczania wody maj
ą
cej du
żą
twardo
ść
w
ę
glanow
ą
i zawieraj
ą
cej NaHCO
3
. W celu usuni
ę
cia CO
2
z wody stosuje si
ę
odgazowywacze.
Kationity silnie kwa
ś
ne pracuj
ą
ce w cyklu sodowym lub wodorowym zapewniaj
ą
prawie
całkowit
ą
wymian
ę
jonów Ca
2+
i Mg
2+
zwi
ą
zanych z anionami silnych i słabych kwasów, a wi
ę
c
usuwanie zarówno twardo
ś
ci w
ę
glanowej, jak i niew
ę
glanowej.

11
Wymiana w cyklu sodowym:
2KtNa + Ca(HCO
3
)
2
→
Kt
2
Ca + 2NaHCO
3
2KtNa + Mg(HCO
3
)
2
→
Kt
2
Mg + 2NaHCO
3
2KtNa + CaCl
2
→
Kt
2
Ca + 2NaCl
2KtNa + MgSO
4
→
Kt
2
Mg + Na
2
SO
4
w cyklu wodorowym
2KtH + Ca(HCO
3
)
2
→
Kt
2
Ca + 2H
2
O + 2CO
2
2KtH + Mg(HCO
3
)
2
→
Kt
2
Mg + 2H
2
O + 2CO
2
2KtH + CaCl
2
→
Kt
2
Ca + 2HCl
2KtH + MgSO
4
→
Kt
2
Mg + H
2
SO
4
W wyniku wymiany jonowej w cyklu sodowym lub wodorowym woda pozbawiona jest
wszystkich jonów Ca
2+
i Mg
2+
oraz innych kationów w tym radionukidów i metali ci
ęż
kich.
Układ zawieraj
ą
cy kationity pracuj
ą
ce w cyklu wodorowym (słaby lub silny) i sodowym
(silny) zapewnia całkowite usuni
ę
cie twardo
ś
ci ogólnej, cz
ęś
ciowe odsolenie wody oraz
neutralizacj
ę
kwasów mineralnych.
Oznaczanie twardo
ś
ci wody metod
ą
wersenianow
ą
Metoda wersenianowa oznaczania twardo
ś
ci wody polega na kompleksometrycznym
miareczkowaniu próbki wody wersenianem dwusodowy (EDTA) wobec barwnika czerni
eriochromowej T w
ś
rodowisku alkalicznym (pH=10). Barwnik ten tworzy z kationami wapnia i
magnezu zwi
ą
zki kompleksowe o barwie czerwono-fioletowej. Poniewa
ż
zwi
ą
zki te s
ą
mniej
trwałe ni
ż
zwi
ą
zki wapnia i magnezu z wersenianem, w trakcie miareczkowania jony wapnia i
magnezu uwalniaj
ą
czer
ń
eriochromow
ą
T, co prowadzi do zmiany zabarwienia roztworu z
czerwonofiolotewego na niebieskie. W oznaczaniu przeszkadzaj
ą
jony baru, cynku, kadmu,
kobaltu, manganu, miedzi, niklu, ołowiu, strontu,
ż
elaza oraz wysoka m
ę
tno
ść
i substancje
organiczne. Wi
ę
kszo
ść
z wymienionych jonów eliminuje si
ę
poprzez stosowanie odczynnika
maskuj
ą
cego: siarczku sodu oraz chlorowodorku hydroksyloaminy NH
2
OH*HCl.
Literatura
1. Buczkowski R., Wybrane zagadnienia proekologiczne w chemii, Wydawnictwo
Uniwersytetu M. Kopernika w Toruniu, Toru
ń
2002.
2.
Cyga
ń
ski A., Chemiczne metody analizy ilo
ś
ciowej, WNT Warszawa 1999.
3.
Hermanowicz W., Dojlido J., Do
ż
a
ń
ska W., Kosiorowski B., Zerze J., Fizyczno-
chemiczne badanie wody i
ś
cieków, Arkady, Warszawa 1999.
4. Kiedry
ń
ska L., Papciak D., Granops M., Chemia sanitarna, Wydawnictwo SGGW,
Warszawa 2006.
5. Kowal A.,
Ś
widerska-Bró
ż
M., Oczyszczanie wody, Wydawnictwo PWN, Warszawa
2008.
6. Krzechowska M., Podstawy chemii ogólnej i
ś
rodowiska przyrodniczego.
Ć
wiczenia
laboratoryjne, Oficyna Wydawnicza Politechniki Warszawskiej, Warszawa 2007.
7. Persona A. (red.), Chemia analityczna. Podstawy klasycznej analizy ilo
ś
ciowej.
Wydawnictwo Medyk, Warszawa 2007.
8. Pokojska U., Przewodnik metodyczny do analizy wód, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
1999.
9. Szłyk E., Kurzawa M., Szydłowska-Czerniak A., Jastrz
ę
bska A., Ilo
ś
ciowa analiza
chemiczna. Metody wagowe i miareczkowe, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
2005.
10. Szmal S., Lipiec T., Chemia analityczna z elementami analizy instrumentalnej,
PZWL, 1997.
12
Celem
ć
wiczenia jest zapoznanie z podstawami oznacze
ń
kompleksometrycznych na
przykładzie oznacze
ń
kompleksonometrycznych.
Zakres
ć
wiczenia obejmuje wykonanie oznaczenia twardo
ś
ci ogólnej i wapniowej badanej
wody metod
ą
wersenianow
ą
.
Uwaga: roztworów mianowanych pobranych z butelki do biurety lub zlewki a nie
wykorzystanych do analizy, NIE WOLNO wlewa
ć
z powrotem do butelki, w której s
ą
przechowywane.
A. Oznaczanie twardo
ś
ci ogólnej wody.
Zasada metody
Do próbki badanej wody wprowadza si
ę
roztwór wersenianu dwusodowego EDTA, który
wi
ąż
e kompleksometrycznie kationy wapnia i magnezu. Miareczkowanie przeprowadza si
ę
w
obecno
ś
ci wska
ź
nika – czerni eriochromowej T.
Odczynniki: bufor amonowy, 1% roztwór chlorowodorku hydroksyloaminy, 5% Na
2
S,
0,025 N EDTA, czer
ń
eriochromowa T
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipet
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- do ka
ż
dej kolby doda
ć
po 5 ml roztworu buforu amonowego, 2 ml 1%-ego roztworu
chlorowodorku hydroksyloaminy, 1 ml 5%-ego roztworu siarczku sodu Na
2
S,
- do ka
ż
dej kolby wsypa
ć
(bezpo
ś
rednio przed miareczkowaniem) szczypt
ę
czerni
eriochromowej T,
- miareczkowa
ć
roztworem wersenianu dwusodowego EDTA do zmiany zabarwienia
z czerwonofioetowego na niebieskie,
po dodaniu czerni eriochromowej T po miareczkowaniu EDTA
Ć
wiczenie nr 3
KOMPLEKSOMETRIA. CHEMIA SANITARNA.
OZNACZANIE TWARDO
Ś
CI WODY - instrukcja.
13
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu EDTA i obliczy
ć
twardo
ść
ogóln
ą
według
poni
ż
szego wzoru:
T
og
=
pr
EDTA
V
V
n *
*
2000
[mval/dm
3
]
gdzie: V
EDTA
– obj
ę
to
ść
zu
ż
ytego roztworu EDTA, ml
n - miano EDTA, n = 0,025 N
V
pr
– obj
ę
to
ść
próbki wody u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
B. Oznaczanie twardo
ś
ci wapniowej wody.
Zasada metody
Metoda polega na zmiareczkowaniu badanego roztworu zawieraj
ą
cego jony wapnia, przy
pH 9-10 mianowanym roztworem wersenianu dwusodowego w obecno
ś
ci wska
ź
nika –
kalcesu. Kompleks chelatowy wap
ń
- kalces o barwie czerwonej zostaje zast
ą
piony przez
bardziej trwały kompleks wap
ń
– wersenianu dwusodowy i roztwór przybiera barw
ę
niebiesk
ą
.
Z ilo
ś
ci EDTA zu
ż
ytego do zwi
ą
zania jonów wapnia oblicza si
ę
twardo
ść
wapniow
ą
wody.
Odczynniki: 3 N NaOH, 1% roztwór chlorowodorku hydroksyloaminy, 5% Na
2
S,
0,025 N EDTA, kalces
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipet
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- do ka
ż
dej kolby doda
ć
pipet
ą
wielomiarow
ą
po 2 ml 3N roztworu wodorotlenku
sodu NaOH, 2 ml 1 % - ego roztworu chlorowodorku hydroksyloaminy,
1 ml 5% -ego roztworu siarczku sodu Na
2
S,
- do ka
ż
dej kolby wsypa
ć
(bezpo
ś
rednio przed miareczkowaniem) szczypt
ę
kalcesu,
- miareczkowa
ć
roztworem wersenianu dwusodowego EDTA do zmiany zabarwienia
z czerwonofioletowego na niebieskie,
po dodaniu kalcesu po miareczkowaniu EDTA
14
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu EDTA i obliczy
ć
twardo
ść
wapniow
ą
wg
poni
ż
szego wzoru:
T
Ca
=
pr
EDTA
V
V
n *
*
2000
[mval/dm
3
]
gdzie: V
EDTA
– obj
ę
to
ść
zu
ż
ytego roztworu EDTA, ml
n - miano EDTA, n = 0,025 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
C. Oznaczanie twardo
ś
ci magnezowej wody.
- na podstawie oznacze
ń
twardo
ś
ci ogólnej i wapniowej, obliczy
ć
twardo
ść
magnezow
ą
korzystaj
ą
z poni
ż
szej zale
ż
no
ś
ci:
T
Mg
= T
og
– T
Ca
[mval/dm
3
]
Wyniki otrzymane z oznacze
ń
twardo
ś
ci ogólnej i wapniowej wpisa
ć
do tabeli.
Sprawozdanie z
ć
wiczenia powinno zawiera
ć
:
- stron
ę
tytułow
ą
- cel i zakres
ć
wiczenia
- opis wykonania
ć
wiczenia (zasada oznaczenia, odczynniki, szkło, sprz
ę
t, materiały,
wykonanie
ć
wiczenia)
- otrzymane wyniki (tabela podpisana przez prowadz
ą
cego
ć
wiczenia)
- wnioski – odnie
ść
si
ę
do aktualnych przepisów prawnych dotycz
ą
cych jako
ś
ci wody
przeznaczonej do spo
ż
ycia przez ludzi oraz danych zawartych w Tabelach 8 i 9.
15
Tabela nr 8. Współczynniki przeliczeniowe jednostek twardo
ś
ci wody.
Stopnie twardości
mval/dm
3
niemiecki [n
0
]
francuski [f
0
]
angielski [a
0
]
ppm
mmol/dm
3
Jednostka
twardości
wody
i jej
oznaczenie
28 mg CaO
lub 50 mg
CaCO
3
/dm
3
10 mg CaO/dm
3
10 mg CaCO
3
/dm
3
14,3 mg CaCO
3
/dm
3
1mg CaCO
3
/dm
3
100 mg CaCO
3
/dm
3
1 mval/dm
3
1,00
2,8
5,0
3,50
50,0
0,50
1
0
niemiecki
0,357
1,00
1,79
1,25
17,8
0,18
1
0
francuski
0,200
0,56
1,00
0,70
10
0,10
1
0
angielski
0,286
0,80
1,43
1,00
14,3
0,14
ppm
0,02
0,056
0,1
0,07
1
0,01
mmol/dm
3
2
5,6
10,0
7,02
100,0
1,00
Tabela 9. Skala twardo
ś
ci wody.
Twardość ogólna wody
mmol/dm
3
mval/dm
3
n
0
Określenie opisowe
0 - 0,89
0 – 1,72
0 - 5
woda bardzo miękka
0,9 – 1,78
1,73 – 3,57
6 – 10
woda miękka
1,79 – 2,67
3,8 – 5,35
11 – 15
woda o średniej twardości
2,68 – 3,56
5,36 – 7,13
16 – 20
woda o znacznej twardości
3,57 – 5,34
7,14 – 10,70
21 – 30
woda twarda
> 5,34
> 10,71
> 30
woda bardzo twarda

16
POLITECHNIKA LUBELSKA
Wydział In
ż
ynierii
Ś
rodowiska
In
ż
ynieria
Ś
rodowiska
SPRAWOZDANIE
Z
Ć
WICZE
Ń
LABORATORYJNYCH Z CHEMII
Nr
ć
wiczenia
3
Temat
ć
wiczenia
Kompleksometria. Chemia sanitarna.
Oznaczanie twardo
ś
ci wody.
Imi
ę
i nazwisko studenta
……………………………………………
……………………………………………
……………………………………………..
Rok studiów
Semestr
Data
Imi
ę
i nazwisko prowadz
ą
cego
ć
wiczenia
Uwagi prowadz
ą
cego:
17
Kompleksometryczne oznaczanie twardości wody
...................................................
…………………………………
…………………………………
Imię i Nazwisko
Grupa BDi .............
Data.......................
Oznaczanie twardości ogólnej Tw
og
Nr próbki
V
1
EDTA [ml]
Tw
og
[mval/dm
3
]
Tw
og
[mmol/dm
3
]
1
2
3
Tw
og
=
.........................
[mval/dm
3
]
Oznaczanie twardości wapniowej Tw
Ca
Nr próbki
V
1
EDTA [ml]
Tw
Ca
[mval/dm
3
]
Tw
Ca
[mmol/dm
3
]
1
2
3
Tw
Ca
=
..................
[mval/dm
3
]
Oznaczanie twardości magnezowej Tw
Mg
Nr próbki Tw
Mg
= Tw
og
- Tw
Ca
[mval/dm
3
]
Tw
Mg
[mmol/dm
3
]
1
2
3
Tw
Mg
=
.....................
[mval/dm
3
]
gdzie:
Tw
og
=
pr
EDTA
V
V
a
∗
∗
2000
[mval/dm
3
]
Tw
ca
=
pr
EDTA
V
V
a
∗
∗
2000
[mval/dm
3
]
V
EDTA
– objętość zużytego EDTA (ml),
a – miano EDTA, 0,025m,
V
pr
- objętość próbki użytej do analizy (ml)
Uwaga: jako wynik podać średnią arytmetyczną z trzech oznaczeń
18
Zagadnienia do kartkówki:
1. Miareczkowanie str
ą
ceniowe, wska
ź
niki w miareczkowaniu str
ą
ceniowym.
2. Argentometria, merkurometria– definicje.
3.
Ź
ródła chlorków w wodzie.
4. Metody oznaczania chlorków.
5. Argentometryczne oznaczanie chlorków; metoda Mohra i Volharda – opis metod,
reakcje.
6. Iloczyn rozpuszczalno
ś
ci soli trudno rozpuszczalnych – zadania.
Analiza miareczkowa wytr
ą
ceniowa (precypitometria) polega na wydzielaniu
okre
ś
lonej substancji w postaci trudno rozpuszczalnego osadu przy u
ż
yciu mianowanego
roztworu titranta. W celu stwierdzenia ko
ń
ca miareczkowania tj. momentu, gdy
miareczkowany roztwór nie zawiera ju
ż
oznaczanego składnika, stosuje si
ę
odpowiednie
wska
ź
niki, wła
ś
ciwe dla danego oznaczenia. Krzywa miareczkowania wytr
ą
ceniowego jest
zale
ż
no
ś
ci
ą
wykładnika st
ęż
enia jonu wytr
ą
canego od obj
ę
to
ś
ci titrantu.
Miareczkowanie wytr
ą
ceniowe nie stanowi zwartej cało
ś
ci jak np. alkacymetria.
Najwa
ż
niejszym działem precypitometrii jest argentometria. Obejmuje ona oznaczenia
oparte na reakcjach tworzenia si
ę
trudno rozpuszczalnych soli srebra. Rol
ę
titrantu w tej
grup
ę
oznacze
ń
pełni mianowany roztwór azotan(V) srebra. Metodami argentometrycznymi
mo
ż
na oznaczy
ć
jony chlorkowe, jodkowe, bromkowe, rodankowe, fosforany(V), srebro.
Merkurometria - metoda chemicznej analizy obj
ę
to
ś
ciowej, polegaj
ą
ca na dodawaniu
mianowanego roztworu azotanu rt
ę
ci do roztworu substancji badanej w celu jej ilo
ś
ciowego
oznaczenia.
W miareczkowaniu str
ą
ceniowym nie ma wska
ź
ników uniwersalnych. Praktycznie dla
ka
ż
dej metody istnieje specjalny, wła
ś
ciwy dla niej sposób okre
ś
lania PK miareczkowania.
Np. w przypadku oznaczania chlorków metod
ą
Mohra jest nim powstanie brunatno-
czerwonego osadu na tle białej zawiesiny chlorku srebra a w metodzie Volharda oznaczania
chlorków pojawienie si
ę
czerwonego zabarwienia od utworzonego kompleksu Fe(III) z
tiocyjanianem. Najbardziej uniwersalnymi w argentometrii wska
ź
nikami s
ą
obecnie ju
ż
rzadko
stosowane wska
ź
niki adsorpcyjne Fajansa. Zasada działania tych wska
ź
ników polega na
tym,
ż
e powoduj
ą
one zmian
ę
zabarwienia nie w gł
ę
bi roztworu lecz na powierzchni osadu - w
punkcie równowa
ż
nikowym wskutek zmiany ładunku na powierzchni osadu nast
ę
puje
adsorpcja lub desorpcja wska
ź
nika, czemu towarzyszy zmiana zabarwienia lub fluorescencji.
Przykłady wska
ź
ników adsorpcyjnych:
nazwa wskaźnika
titrant
oznaczany jon
zamiana barwy
Cl
-
, Br
-
, SCN
-
żółtozielona → różowa
fluoresceina
Ag
+
J
-
żółtozielona → pomarańczowa
Cl
-
, Br
-
, SCN
-
żółta → niebieska
3,6-dichlorofluorosceina
Ag
+
J
-
żółtozielona → niebieskozielona
Ag
+
Cl
-
,
czerwonofioletowa → różowa
Br
-
, J
-
,
pomarańczowa → żółta
fuksyna
SCN
-
niebieska → różowa
eozyna
Ag
+
Cl
-
pomarańczowa → różowa
PRACOWNIA nr 6
ANALIZA WYTR
Ą
CENIOWA. CHEMIA SANITARNA.
OZNACZANIE ZAWARTO
Ś
CI CHLORKÓW.

19
Chlorki ze wzgl
ę
du na swoj
ą
wysok
ą
rozpuszczalno
ść
w wodzie (wyj
ą
tek AgCl,
Hg
2
Cl
2
, CuCl i PbCl
2
) oraz szerokie rozpowszechnienie w skorupie ziemskiej w postaci
naturalnych pokładów soli, stanowi
ą
podstawowy składnik wód naturalnych. Ich
ź
ródła w
wodach mog
ą
mie
ć
charakter naturalny (geologiczny) oraz antropogeniczny. Chlorki o
geologiczne pochodz
ą
z gruntu, z naturalnych pokładów soli. Natomiast chlorki
antropogeniczne obecne w wodzie
ś
wiadcz
ą
o jej ska
ż
eniu
ś
ciekami, zarówno komunalnymi
jak te
ż
i przemysłowymi. St
ęż
enia chlorków w wodach naturalnych zmieniaj
ą
si
ę
od ilo
ś
ci
ś
ladowych do kilkuset mg/dm
3
. Według wymaga
ń
sanitarno – higienicznych zawarto
ść
chlorków geologicznych wodzie do picia nie powinna przekracza
ć
250 ng/dm
3
, przy czym
warto
ść
zalecana wynosi 25 mg/dm
3
. Chlorki innego pochodzenia czyni
ą
j
ą
niezdatn
ą
do
spo
ż
ycia. Zawarto
ść
chlorków powy
ż
ej 100 mg/dm
3
w pewnych warunkach wzmacnia
wła
ś
ciwo
ś
ci korozyjne wody.
Metody oznaczania chlorków:
- metoda miareczkowania argentometrycznego (metoda Mohra)
- metoda miareczkowania merkurometrycznego
- metoda potencjometryczna
- metoda turbidymetryczna
Metoda miareczkowania merkurometrycznego chlorków polega na miareczkowaniu
roztworu zawieraj
ą
cego chlorki mianowanym roztworem Hg(NO
3
)
2
wobec difenylokarbazonu
jako wska
ź
nika, w
ś
rodowisku o pH=2,3-2,8. Jony chlorkowe reaguj
ą
z jonami Hg
2+
tworz
ą
c
słabo zdysocjowany HgCl
2
. Dodany wska
ź
nik difenylokarbazon tworzy z nadmiarem jonów
Hg
2+
zwi
ą
zek kompleksowy o fioletowym zabarwieniu.
Metoda
turbidymetryczna
oznaczania
chlorków
polega
na
ocenie
zm
ę
tnienia
spowodowanego wytr
ą
ceniem si
ę
koloidalnego osadu AgCl w wyniku reakcji AgNO
3
z jonami
chlorkowymi.
Literatura
1. Buczkowski R., Wybrane zagadnienia proekologiczne w chemii, Wydawnictwo
Uniwersytetu M. Kopernika w Toruniu, Toru
ń
2002.
2.
Cyga
ń
ski A., Chemiczne metody analizy ilo
ś
ciowej, WNT Warszawa 1999.
3.
Hermanowicz W., Dojlido J., Do
ż
a
ń
ska W., Kosiorowski B., Zerze J., Fizyczno-
chemiczne badanie wody i
ś
cieków, Arkady, Warszawa 1999.
4. Kiedry
ń
ska L., Papciak D., Granops M., Chemia sanitarna, Wydawnictwo SGGW,
Warszawa 2006.
5. Krzechowska M., Podstawy chemii ogólnej i
ś
rodowiska przyrodniczego.
Ć
wiczenia
laboratoryjne. Oficyna Wydawnicza Politechniki Warszawskiej, Warszawa 2007.
6. Persona A. (red.), Chemia analityczna. Podstawy klasycznej analizy ilo
ś
ciowej.
Wydawnictwo Medyk, Warszawa 2007.
7. Pokojska U., Przewodnik metodyczny do analizy wód, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
1999.
8. Szłyk E., Kurzawa M., Szydłowska-Czerniak A., Jastrz
ę
bska A., Ilo
ś
ciowa analiza
chemiczna. Metody wagowe i miareczkowe, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
2005.
9. Szmal S., Lipiec T., Chemia analityczna z elementami analizy instrumentalnej
PZWL, 1997.

20
Celem
ć
wiczenia jest zapoznanie z podstawami oznacze
ń
stosowanych w analizie
wytr
ą
ceniowej na przykładzie oznacze
ń
argentometrycznych.
Zakres
ć
wiczenia obejmuje wykonanie oznaczenia zawarto
ś
ci chlorków w badanej wodzie
metod
ą
Mohra i metod
ą
Volharda.
Uwaga: roztworów mianowanych pobranych z butelki do biurety lub zlewki a nie
wykorzystanych do analizy, NIE WOLNO wlewa
ć
z powrotem do butelki, w której s
ą
przechowywane.
A. Oznaczanie chlorków metod
ą
Mohra.
Zasada metody
Metoda ta polega na bezpo
ś
rednim miareczkowaniu roztworu zawieraj
ą
cego jony chlorkowe
mianowanym roztworem azotanu(V) srebra w obecno
ś
ci chromianu(VI) potasu jako
wska
ź
nika w
ś
rodowisku o pH =6,5-10. dy praktycznie cała obecna w roztworze ilo
ść
chlorków wydzieli si
ę
w postaci chlorku srebra (biały serowaty osad), nadmiar roztworu
azotanu srebra wytr
ą
ca chromian(VI) srebra i nast
ę
puje zmiana zabarwienia roztworu z
ż
ółtozielonkawego na czerwonobrunatne, co wskazuje na ko
ń
cowy punkt miareczkowania.
Metody Mohra nie mo
ż
na stosowa
ć
w przypadku gdy roztwór zawiera inne aniony tworz
ą
ce w
ś
rodowisku oboj
ę
tnym trudno rozpuszczalne sole srebra albo kationy, które daj
ą
zwi
ą
zki
trudno rozpuszczalne sole z jonami chromianowymi(VI), lub substancje redukuj
ą
ce azotan
srebra do srebra metalicznego. Dodatkowo w oznaczaniu przeszkadzaj
ą
: barwa roztworu
powy
ż
ej 30 mgPt/dm
3
, m
ę
tno
ść
powy
ż
ej 20 mg/dm
3
,
ż
elazo, siarczki, siarczany(VI),
siarkowodór i fosforany. Metod
ą
Mohra oprócz chlorków mo
ż
na jeszcze oznacza
ć
bromki w
miareczkowaniu bezpo
ś
rednim i jony srebra w miareczkowaniu po
ś
rednim.
Odczynniki: 5%
K
2
CrO
4
, 0,1 N AgNO
3
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- do ka
ż
dej kolby doda
ć
pipet
ą
wielomiarow
ą
po 1 ml 5% -wego roztworu chromianu
(VI) potasu K
2
CrO
4
,
- miareczkowa
ć
0,1 N roztworem AgNO
3
do zmiany zabarwienia na
ż
ółto-pomara
ń
czowe,
po dodaniu K
2
CrO
4
po miareczkowaniu AgNO
3
Ć
wiczenie nr 4
ANALIZA WYTR
Ą
CENIOWA. CHEMIA SANITARNA.
OZNACZANIE ZAWARTO
Ś
CI CHLORKÓW – instrukcja.

21
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu AgNO
3
i obliczy
ć
zawarto
ść
chlorków wg
poni
ż
szego wzoru:
[Cl
-
] =
pr
AgNO
V
V
n
)
3
,
0
(
5
,
35
1000
3
−
∗
∗
∗
[mg/dm
3
]
gdzie: V
AgNO3
– obj
ę
to
ść
zu
ż
ytego roztworu AgNO
3
, ml
n – miano roztworu AgNO
3
, n = 0,1 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
B. Oznaczanie chlorków metod
ą
Volharda.
Zasada metody polega na po
ś
rednim oznaczaniu chlorków. Do zakwaszonego
rozcie
ń
czonym kwasem azotowym(V) roztworu zawieraj
ą
cego chlorki dodaje si
ę
nadmiar
mianowanego roztworu azotanu(V) srebra. Wskutek tego praktycznie cały chlorek wytr
ą
ci si
ę
w postaci AgCl, a w roztworze pozostanie pewien nadmiar AgNO
3
. Ten nadmiar
odmiareczkowuje si
ę
mianowanym roztworem tiocyjanianem amonu lub potasu w obecno
ś
ci
soli
ż
elaza(III) – siarczan(VI) amonu i
ż
elaza(III) lub azotanu(V)
ż
elaza(III), które pełni
ą
rol
ę
wska
ź
ników. Pojawienie si
ę
czerwonego zabarwienia roztworu oznacza, ze cał
ą
ilo
ść
jonów
srebra została odmiareczkowana i utworzył si
ę
kompleks tiocyjanianu z
ż
elazem(III) –
FeSCN
2+
. Metod
ą
Volharda mo
ż
na oznacza
ć
jony srebra wykonuj
ą
c miareczkowanie
bezpo
ś
rednie, oraz jony chlorkowe, bromkowe, jodkowe i rodankowe, prowadz
ą
c
miareczkowanie po
ś
rednie. W oznaczeniach nie przeszkadzaj
ą
aniony fosforanowe(V),
szczawianowe i arsenianowe(V) oraz kationy ulegaj
ą
ce hydrolizie, np. glinu(III).
Odczynniki: HNO
3
(1+1), 0,1 N AgNO
3
, CHCl
3
, FeNH
4
(SO
4
)
2
, 0,05 N KSCN
Szkło: kolby sto
ż
kowe z doszlifowanym korkiem o poj. 500 ml, pipety wielomiarowe,
biureta, cylinder miarowy 100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych z doszlifowanymi korkami o pojemno
ś
ci 500 ml pobra
ć
pipet
ą
jednomiarow
ą
po 100 ml otrzymanej do analizy próbki,
- do ka
ż
dej kolby doda
ć
pipet
ą
wielomiarow
ą
po 5 ml roztworu kwasu azotowego (V)
HNO
3
(1+1) i po 25 ml (odmierzy
ć
pipet
ą
jednomiarow
ą
) roztworu AgNO
3
,
- doda
ć
po 3 ml chloroformu CHCl
3
i po 1 ml 10% -wego roztworu siarczanu amonu-
ż
elaza (III) FeNH
4
(SO
4
)
2
,
- wytrz
ą
sa
ć
przez 3 minuty,

22
- nadmiar jonów Ag
+
odmiareczkowa
ć
0,05 N roztworem rodanku potasu KSCN do
wyst
ą
pienia trwałego brunatno-pomara
ń
czowego zabarwienia,
przed miareczkowaniem KSCN po miareczkowaniu KSCN
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu rodanku potasu KSCN i obliczy
ć
zawarto
ść
chlorków wg poni
ż
szego wzoru:
[Cl
-
] =
pr
KSCN
AgNO
V
V
n
V
n
)
(
5
,
35
1000
2
1
3
∗
−
∗
∗
∗
[mg/dm
3
]
gdzie: V
AgNO3
– obj
ę
to
ść
zu
ż
ytego roztworu AgNO
3
, ml
n
1
– miano roztworu AgNO
3
, n
1
= 0,1 N
V
KSCN
– obj
ę
to
ść
zu
ż
ytego roztworu KSCN, ml
n
2
– miano roztworu KSCN, n
2
= 0,05 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
Wyniki otrzymane z oznacze
ń
zawarto
ś
ci chlorków metodami Mohra i Volharda wpisa
ć
do
tabeli.
Sprawozdanie z
ć
wiczenia powinno zawiera
ć
:
- stron
ę
tytułow
ą
- cel i zakres
ć
wiczenia
- opis wykonania
ć
wiczenia (zasada oznaczenia, odczynniki, szkło, sprz
ę
t, materiały,
wykonanie
ć
wiczenia)
- otrzymane wyniki (tabela podpisana przez prowadz
ą
cego
ć
wiczenia)
- wnioski – odnie
ść
si
ę
do aktualnych przepisów prawnych dotycz
ą
cych jako
ś
ci wody
przeznaczonej do spo
ż
ycia przez ludzi.

23
POLITECHNIKA LUBELSKA
Wydział In
ż
ynierii
Ś
rodowiska
In
ż
ynieria
Ś
rodowiska
SPRAWOZDANIE
Z
Ć
WICZE
Ń
LABORATORYJNYCH Z CHEMII
Nr
ć
wiczenia
4
Temat
ć
wiczenia
Analiza wytr
ą
ceniowa. Chemia sanitarna.
Oznaczanie chlorków metod
ą
Mohra i Volharda.
Imi
ę
i nazwisko studenta
……………………………………………
……………………………………………
……………………………………………
Rok studiów
Semestr
Data
Imi
ę
i nazwisko prowadz
ą
cego
ć
wiczenia
Uwagi prowadz
ą
cego:

24
Argentometryczne oznaczanie chlorków
...................................................
…………………………………
…………………………………
Imię i Nazwisko
Grupa BDi .............
Data.......................
Oznaczanie chlorków metodą Mohra
Nr próbki
V
1
AgNO
3
[ml]
[Cl
-
]
[mg/dm
3
]
1
2
3
[Cl
-
] =
.........................
[mg/dm
3
]
Oznaczanie chlorków metodą Volharda
Nr próbki
V
2
KSCN [ml]
[Cl
-
]
[mg/dm
3
]
1
2
3
[Cl
-
] =
.........................
[mg/dm
3
]
gdzie:
[Cl
-
] =
pr
V
V
a
)
3
,
0
(
5
,
35
1000
1
−
∗
∗
∗
[mgl/dm
3
] - metoda Mohra
[Cl
-
] =
pr
V
V
n
V
n
)
(
5
,
35
1000
2
2
1
1
∗
−
∗
∗
∗
[mg/dm
3
] - metoda Volharda
V
1
– objętość roztworu AgNO
3
(ml),
V
2
– objętość roztworu KSCN (ml),
a – miano AgNO
3
– 0,1n
n
1
– normalność roztworu AgNO
3
– 0,1 n,
n
2
– normalność roztworu KSCN – 0,05 n,
V
pr
- objętość próbki użytej do analizy (ml)
Uwaga: jako wynik podać średnią arytmetyczną z trzech oznaczeń

25
Zagadnienia do kartkówki:
1. Alkacymetria (podział metod alkacymetrycznych), wska
ź
niki alkacymetryczne.
2. Kwasowo
ść
– definicja,
ź
ródła, rodzaje, jednostki, metody oznaczania.
3. Zasadowo
ść
– definicja,
ź
ródła, rodzaje, jednostki, metody oznaczania.
4. Zale
ż
no
ść
mi
ę
dzy zasadowo
ś
ci
ą
, charakterem chem. zwi
ą
zków zawartych w wodzie i pH.
5. Definicja pH, jednostka pH, skala pH, wska
ź
niki pH – przykłady. pH słabych i mocnych
elektrolitów – zadania.
Alkacymetria obejmuje grup
ę
metod analizy obj
ę
to
ś
ciowej opartych na reakcji
zoboj
ę
tniania. Metody alkacymetryczne stosuje si
ę
do oznaczania kwasów i zasad
nieorganicznych i organicznych.
Alkacymetria obejmuje dwa działy:
- alkalimetri
ę
– oznaczanie substancji przez miareczkowanie mianowanym roztworem
zasady
- acydymetri
ę
– oznaczanie substancji przez miareczkowanie mianowanym roztworem
kwasu.
Wska
ź
niki pH – czyli indykatory pH, słu
żą
do wyznaczania PK miareczkowania
alkacymetrycznego i okre
ś
lania pH roztworu. S
ą
to zwi
ą
zki chemiczne – najcz
ęś
ciej słabe
kwasy lub zasady organiczne (stosowane w postaci roztworów wodnych lub alkoholowych),
zmieniaj
ą
ce barw
ę
w okre
ś
lonym zakresie pH roztworu. W roztworach wodnych tworz
ą
one
układy sprz
ęż
one kwas –zasada. Je
ż
eli tylko jedna forma w układzie jest zabarwiona, co
wynika z obecno
ś
ci grupy chromoforowej, wówczas mówi si
ę
o wska
ź
nikach
jednobarwnych (np. fenoloftaleina). Je
ż
eli dwie formy układu maj
ą
ró
ż
ne zabarwienie, wtedy
s
ą
to wska
ź
niki dwubarwne (np. oran
ż
metylowy). Wyra
ź
n
ą
zmian
ę
barwy wska
ź
nika
dwubarwnego zauwa
ż
a si
ę
, gdy około 10% indykatora przejdzie w w posta
ć
inaczej
zabarwion
ą
. Całkowita zmiana barwy wska
ź
nika wyst
ę
puje w zakresie dwóch jednostek pH.
Zakres pH, w którym wyst
ę
puje przej
ś
ciowe zabarwienie wska
ź
nika nazywa si
ę
zakresem
zmiany barwy wska
ź
nika. W przypadku wska
ź
ników jednobarwnych widoczna jest tylko jedna
barwna posta
ć
wska
ź
nika.
W przypadku niektórych wska
ź
ników trudno jest zauwa
ż
y
ć
zmian
ę
barwy, dlatego
stosuje si
ę
wska
ź
niki mieszane, czyli mieszaniny dwóch odpowiednio dobranych wska
ź
ników
kwasowo-zasadowych lub mieszanin
ę
jednego wska
ź
nika z oboj
ę
tnym barwnikiem, np. oran
ż
metylowy mo
ż
na zmiesza
ć
z bł
ę
kitem metylowym. Taka mieszanina ma barw
ę
fioletow
ą
przy
pH=3 a zielon
ą
przy pH=5.
Najcz
ęś
ciej stosowane wska
ź
niki pH:
- oran
ż
metylowy
- fenoloftaleina
- bł
ę
kit tymolowy
- lakmus
PRACOWNIA nr 7
ALKACYMETRIA. CHEMIA SANITARNA.
OZNACZANIE ZASADOWO
Ś
CI i KWASOWOSCI WODY.

26
Tabela 11. Zakresy zmiany barwy najcz
ęś
ciej stosowanych wska
ź
ników pH.
Wskaźnik
zakres pH
zmiana barwy
fiolet metylowy
0,1 – 3,2
żółta – fioletowa
oranż metylowy
3,1 – 4,4
czerwona pomarańczowa – żółta
czerwień metylowa
4,2 – 6,3
czerwona - pomarańczowa – żółta
lakmus
5,2 – 8,0
czerwona - różowa - niebieska
błękit bromotymolowy
6,0 – 7,6
żółta – zielona – niebieska
fenoloftaleina
0,0 - 8,2
8,2 – 12,0
> 12,0
bezbarwna
różowa
bezbarwna
żółcień alizarynowa G
10,0 – 12,0
żółta – pomarańczowa - czerwona
pH za pomoc
ą
wska
ź
ników nale
ż
y mierzy
ć
tylko w rozcie
ń
czonych roztworach
wodnych. Do okre
ś
lania pH roztworu stosuje si
ę
tak
ż
e wska
ź
niki w postaci papierków
(pasków) wska
ź
nikowych, którymi s
ą
wysuszone kawałki bibuły filtracyjnej nas
ą
czone
uprzednio roztworem wska
ź
nika. W laboratoriach znalazły tak
ż
e zastosowanie papierki
uniwersalne. S
ą
to kawałki bibuły nas
ą
czone mieszanin
ą
ró
ż
nych wska
ź
ników i zmieniaj
ą
ce
barw
ę
(kilkakrotnie) ze zmian
ą
pH
ś
rodowiska, od mocno kwa
ś
nego do mocno zasadowego
(z dokładno
ś
ci
ą
do 0,5 jednostki pH). Dokładne pH roztworu mo
ż
na zmierzy
ć
za pomoc
ą
urz
ą
dzenia zwanego pehametrem.
Warto
ś
ci pH roztworów znanych z
ż
ycia codziennego:
- krew – 7,35-7,45
- mleko – 6,5
- sok
ż
oł
ą
dkowy – 1,0 – 2,0
- ocet – 3,0
- Coca cola – 2,3 – 3,0
Kwasowo
ść
wody naturalnej jest to zdolno
ść
do zoboj
ę
tniania silnych zasad
mineralnych lub w
ę
glanów wobec umownych wska
ź
ników. Kwasowo
ść
wód naturalnych jest
wywołana głównie obecno
ś
ci
ą
wolnego CO
2
(rozpuszczonego), a tak
ż
e obecno
ś
ci
ą
słabych
kwasów organicznych (np. kwasów humusowych) oraz produktów hydrolizy koagulantów
(sole glinu i
ż
elaza). Kwasowo
ść
wody jest niepo
żą
dana w przypadku wykorzystania jej do
celów przemysłowych. Je
ż
eli pH wody jest mniejsze od 4,6 wyst
ę
puje wówczas tzw.
kwasowo
ść
mineralna wody (wywołana mocnymi kwasami) w woda wykazuje silne
wła
ś
ciwo
ś
ci korozyjne wobec betonu i
ż
elaza. Ten rodzaj kwasowo
ś
ci bardzo rzadko
wyst
ę
puje w wodach powierzchniowych. W zakresie pH = 4,6 – 8,3 oznacza si
ę
kwasowo
ść
ogóln
ą
wody (wywołan
ą
przez sum
ę
kwasów mocnych i słabych) współistniej
ą
c
ą
z
zasadowo
ś
ci
ą
ogóln
ą
. Kwasowo
ść
wody oznacza si
ę
za pomoc
ą
miareczkowania
potencjometrycznego
lub
miareczkowania
roztworem
NaOH
lub
Na
2
CO
3
wobec
odpowiedniego wska
ź
nika. W przypadku kwasowo
ś
ci mineralnej jest nim oran
ż
metylowy a
dla kwasowo
ś
ci ogólnej – fenoloftaleina. W oznaczaniu kwasowo
ś
ci przeszkadzaj
ą
sole
ż
elaza w st
ęż
eniu powy
ż
ej 1 mg/dm
3
oraz twardo
ść
w
ę
glanowa powy
ż
ej 3,6 mval/dm
3
.
Czynniki przeszkadzaj
ą
ce usuwa si
ę
poprzez dodanie przed miareczkowaniem do
analizowanej próbki kilku kropel roztworu winianu potasu u sodu. Jednostk
ą
kwasowo
ś
ci s
ą
mmol/dm
3
, a tak
ż
e mval/dm
3
.
Kwasowo
ść
wody do picia ma bezpo
ś
rednie znaczenie jedynie wówczas gdy jest
wywołana przez kwasy mineralne. W czystych wodach naturalnych istnieje naturalny układ
buforowy (wodorow
ę
glany i kwas w
ę
glanowy), którego zadaniem jest utrzymywanie
odpowiedniego pH wody po wprowadzeniu niewielkich ilo
ś
ci silnych kwasów.
Zdolno
ść
wody do zoboj
ę
tniania mocnych kwasów mineralnych w okre
ś
lonych
warunkach nosi nazw
ę
zasadowo
ś
ci wody. Wła
ś
ciwo
ść
t
ę
nadaj
ą
wodzie obecne w niej
27
wodorow
ę
glany, w
ę
glany oraz rzadziej wodorotlenki, borany, ortokrzemiany i ortofosforany.
Parametr ten jest wa
ż
ny w analizie wód technologicznych (kotłowych, chłodniczych) i
naturalnych (przy ocenie ich agresywno
ś
ci wzgl
ę
dem betonu). W ocenie higienicznej
zasadowo
ść
ma znaczenie drugorz
ę
dne. Oznaczanie zasadowo
ś
ci polega na okre
ś
leniu
zawarto
ś
ci zwi
ą
zków reaguj
ą
cych w wodzie zasadowo wobec odpowiedniego wska
ź
nika.
Miareczkowanie kwasem solnym wodorotlenków, w
ę
glanów i wodorow
ę
glanów wapnia,
magnezu, sodu i potasu zawartych w wodzie, pozwala oznaczy
ć
ich sumaryczne st
ęż
enie
zwane zasadowo
ś
ci
ą
ogóln
ą
. Miareczkuj
ą
c te zwi
ą
zki 0,1M HCl wobec oran
ż
u metylowego
do pH=4,5, okre
ś
la si
ę
zasadowo
ść
ogóln
ą
wody (M, m lub Z
m
). Natomiast zasadowo
ść
mineralna (F, p lub Z
p
) pochodzi od wolnych jonów OH
-
z dysocjacji zasad oraz od jonów OH
-
powstaj
ą
cych w wyniku hydrolizy w
ę
glanów i oznacza si
ę
j
ą
przez miareczkowanie 0,1M HCl
wobec fenoloftaleiny przy pH=8,3. W zale
ż
no
ś
ci od uzyskanych wyników miareczkowania
próby wody kwasem solnym wobec fenoloftaleiny i oran
ż
u metylowego, mo
ż
na okre
ś
li
ć
udziały jonów OH
-
, HCO
3
2-
, CO
3
2-
(mmol/dm
3
) w zasadowo
ś
ci ogólnej w sposób podany
w Tabeli 10.
Tabela 10. Współistnienie jonów OH
-
, CO
3
2-
, HCO
3
-
w wodach przy pH < 9.
Zasadowość spowodowana obecnością jonu (mval/dm
3
)
zależność pomiędzy
zasadowością p i m
OH
-
CO
32-
HCO
3-
p=0, m>0
0
0
m
2p<m
0
p
m - 2p
2p=m
0
p
0
2p>m
2p - m
m – p
0
p=m
m
0
0
Zale
ż
nie od zwi
ą
zku, który nadaje wodzie charakter zasadowy, przy badaniach specjalnych
rozró
ż
nia si
ę
:
- zasadowo
ść
ogóln
ą
Z
m
(OH
-
, CO
3
2-
, HCO
3
-
)
- zasadowo
ść
z fenoloftalein
ą
Z
p
(OH
-
, CO
3
2-
)
- zasadowo
ść
w
ę
glanow
ą
(CO
3
2-
)
- zasadowo
ść
wodorow
ę
glanow
ą
(HCO
3
-
)
- zasadowo
ść
wodorotlenow
ą
(OH
-
).
Zasadowo
ść
wody z punktu oceny sanitarnej nie ma wi
ę
kszego znaczenia, natomiast ma
istotne znaczenie w przypadku wód do celów przemysłowych.
Literatura
1. Buczkowski R., Wybrane zagadnienia proekologiczne w chemii, Wydawnictwo
Uniwersytetu M. Kopernika w Toruniu, Toru
ń
2002.
2.
Cyga
ń
ski A., Chemiczne metody analizy ilo
ś
ciowej, WNT Warszawa 1999.
3.
Hermanowicz W., Dojlido J., Do
ż
a
ń
ska W., Kosiorowski B., Zerze J., Fizyczno-
chemiczne badanie wody i
ś
cieków, Arkady, Warszawa 1999.
4. Kiedry
ń
ska L., Papciak D., Granops M., Chemia sanitarna, Wydawnictwo SGGW,
Warszawa 2006.
5. Krzechowska M., Podstawy chemii ogólnej i
ś
rodowiska przyrodniczego.
Ć
wiczenia
laboratoryjne. Oficyna Wydawnicza Politechniki Warszawskiej, Warszawa 2007.
6. Pokojska U., Przewodnik metodyczny do analizy wód, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
1999.
7. Szłyk E., Kurzawa M., Szydłowska-Czerniak A., Jastrz
ę
bska A., Ilo
ś
ciowa analiza
chemiczna. Metody wagowe i miareczkowe, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
2005.
8. Szmal S., Lipiec T., Chemia analityczna z elementami analizy instrumentalnej
PZWL, 1997.

28
Celem
ć
wiczenia jest zapoznanie z podstawami miareczkowania alkacymetrycznego.
Zakres
ć
wiczenia obejmuje wykonanie oznaczenia zasadowo
ś
ci ogólnej i mineralnej oraz
kwasowo
ś
ci ogólnej i mineralnej badanej wody.
Uwaga: roztworów mianowanych pobranych z butelki do biurety lub zlewki a nie
wykorzystanych do analizy, NIE WOLNO wlewa
ć
z powrotem do butelki, w której s
ą
przechowywane.
A. Oznaczanie zasadowo
ś
ci mineralnej P i zasadowo
ś
ci ogólnej M wody.
Zasada metody
Oznaczanie zasadowo
ś
ci mineralnej Z
p
polega na zoboj
ę
tnianiu zawartych w wodzie
zwi
ą
zków zasadowych mianowanym roztworem HCl do pH=8,3, przy u
ż
yciu wska
ź
nika –
fenoloftaleiny. Miareczkowanie kwasem prowadzi si
ę
do zaniku ró
ż
owego zabarwienia próbki.
Natomiast oznaczanie zasadowo
ś
ci ogólnej Z
m
polega na miareczkowaniu badanej wody
mianowanym roztworem HCl do pH=4,5, przy u
ż
yciu wska
ź
nika – oran
ż
u metylowego.
Miareczkowanie kwasem prowadzi si
ę
do zmiany zabarwienia próbki wody z
ż
ółtego na
ż
ółtoczerwone.
Odczynniki: 0,1 N HCl, fenoloftaleina, oran
ż
metylowy
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- doda
ć
3 - 5 kropli fenoloftaleiny do uzyskania malinowego zabarwienia,
- miareczkowa
ć
0,1 N roztworem kwasu solnego HCl do odbarwienia
miareczkowanego roztworu,
po dodaniu fenoloftaleiny po miareczkowaniu HCl
Uwaga: je
ż
eli woda nie zabarwi si
ę
na ró
ż
owo, oznacza to,
ż
e ten rodzaj zasadowo
ś
ci
w badanej wodzie nie wyst
ę
puje, czyli Z
p
= 0 mval/dm
3
Ć
wiczenie nr 5
ALKACYMETRIA. CHEMIA SANITARNA.
OZNACZANIE ZASADOWO
Ś
CI i KWASOWOSCI WODY - instrukcja.
29
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu HCl,
- doda
ć
2 – 3 krople oran
ż
u metylowego i miareczkowa
ć
tym samym kwasem do
zmiany zabarwienia z
ż
ółtej na cebulkow
ą
,
po dodaniu oran
ż
u metylowego po miareczkowaniu HCl
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu HCl i obliczy
ć
zasadowo
ść
M i P ze wzoru:
P
=
pr
V
V
n
1
1000
∗
∗
[mval/dm
3
]
M
=
pr
V
V
n
2
1000
∗
∗
[mval/dm
3
]
gdzie: V
1
– obj
ę
to
ść
zu
ż
ytego roztworu 0,1 N HCl na zmiareczkowania próbki wobec
fenoloftaleiny, ml
V
2
– obj
ę
to
ść
zu
ż
ytego roztworu 0,1 N HCl na zmiareczkowania próbki wobec
oran
ż
u metylowego, ml
n – normalno
ść
roztworu HCl - 0,1 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
B. Oznaczanie kwasowo
ś
ci ogólnej K
og
wody.
Zasada metody polega na zoboj
ę
tnieniu mocnych i słabych kwasów zawartych w badanej
wodzie (do pH=8,3) mianowanym roztworem NaOH w obecno
ś
ci wska
ź
nika – fenoloftaleiny.
Miareczkowanie prowadzi si
ę
do powstania ró
ż
owego zabarwienia roztworu.
Odczynniki: 0,1 N NaOH, fenoloftaleina, 33% sól Seignetta
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 2 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- doda
ć
po 3 – 5 kropli fenoloftaleiny i 4 krople 33% -wego roztworu winianu potasu i
sodu (soli Seignetta),
- miareczkowa
ć
0,1 N roztworem NaOH do uzyskania lekko ró
ż
owej barwy roztworu,
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu NaOH i obliczy
ć
kwasowo
ść
ogóln
ą
wg wzoru:

30
K
Og
=
pr
V
V
a
1
1000
∗
∗
[mval/dm
3
]
gdzie: V
1
– obj
ę
to
ść
zu
ż
ytego roztworu 0,1 N NaOH na zmiareczkowania próbki, ml
a - normalno
ść
wodorotlenku sodu NaOH - 0,1 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 2 oznacze
ń
C. Oznaczanie kwasowo
ś
ci mineralnej K
m
wody.
Zasada metody polega na
zoboj
ę
tnieniu mocnych kwasów zawartych w badanej wodzie (do
pH=4,5) mianowanym roztworem NaOH w obecno
ś
ci metylooran
ż
u do uzyskania
ż
ółtopomara
ń
czowego zabarwienia.
Odczynniki: 0,1 N NaOH, oran
ż
metylowy, 33% sól Seignetta
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 2 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- doda
ć
po 3 – 5 kropli oran
ż
u metylowego,
- miareczkowa
ć
0,1 N roztworem NaOH do zmiany barwy z czerwonego na
ż
ółto -
pomara
ń
czowe,
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu NaOH i obliczy
ć
kwasowo
ść
mineraln
ą
wg wzoru:
K
m
=
pr
V
V
a
1
1000
∗
∗
[mval/dm
3
]
gdzie: V
1
– obj
ę
to
ść
zu
ż
ytego roztworu 0,1 N NaOH na zmiareczkowania próbki, ml
a – normalno
ść
NaOH - 0,1N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 2 oznacze
ń
Wyniki otrzymane z oznacze
ń
zasadowo
ś
ci i kwasowo
ś
ci wpisa
ć
do tabeli.

31
Sprawozdanie z
ć
wiczenia powinno zawiera
ć
:
- stron
ę
tytułow
ą
- cel i zakres
ć
wiczenia
- opis wykonania
ć
wiczenia (zasada oznaczenia, odczynniki, szkło, sprz
ę
t, materiały,
wykonanie
ć
wiczenia)
- otrzymane wyniki (tabela podpisana przez prowadz
ą
cego
ć
wiczenia)
- wnioski – odnie
ść
si
ę
do aktualnych przepisów prawnych dotycz
ą
cych jako
ś
ci wody
przeznaczonej do spo
ż
ycia przez ludzi.

32
POLITECHNIKA LUBELSKA
Wydział In
ż
ynierii
Ś
rodowiska
In
ż
ynieria
Ś
rodowiska
SPRAWOZDANIE
Z
Ć
WICZE
Ń
LABORATORYJNYCH Z CHEMII
Nr
ć
wiczenia
5
Temat
ć
wiczenia
Alkacymetria. Chemia sanitarna.
Oznaczanie zasadowo
ś
ci i kwasowo
ś
ci wody.
Imi
ę
i nazwisko studenta
……………………………………………
……………………………………………
……………………………………………
Rok studiów
Semestr
Data
Imi
ę
i nazwisko prowadz
ą
cego
ć
wiczenia
Uwagi prowadz
ą
cego:

33
Oznaczanie zasadowości i kwasowości wody
...................................................
..................................................
..................................................
Imię i Nazwisko
Grupa BDi .............
Data.......................
Oznaczanie zasadowości mineralnej P
Nr próbki
V
1
HCl [ml]
P [mval/dm
3
]
1
2
3
P
=
.........................
[mval/dm
3
]
Oznaczanie zasadowości ogólnej M
Nr próbki
V
2
HCl [ml]
M
[mval/dm
3
]
1
2
3
M =
..................
[mval/dm
3
]
Oznaczanie kwasowości ogólnej K
Og
Nr próbki
V
1
NaOH [ml]
K
Og
[mval/dm
3
]
1
2
3
K
Og
=
.....................
[mval/dm
3
]
Oznaczanie kwasowości mineralnej K
m
Nr próbki
V
2
NaOH [ml]
K
m
[mval/dm
3
]
1
2
3
K
m
=
.....................
[mval/dm
3
]
gdzie:
P
=
pr
V
V
n
1
1000
∗
∗
[mval/dm
3
]
M
=
pr
V
V
V
n
)
(
1000
2
1
+
∗
∗
[mval/dm
3
]
K
Og
=
pr
V
V
a
3
1000
∗
∗
[mval/dm
3
] K
m
=
pr
V
V
a
4
1000
∗
∗
[mval/dm
3
]
V
1
- objętość HCl zużytego na zmiareczkowanie próbki wobec fenoloftaleiny (ml), przy
oznaczaniu zasadowości P,
V
2
– objętość HCl zużytego na zmiareczkowanie próbki wobec oranżu metylowego (ml), przy
oznaczaniu zasadowości M,
V
3,
V
4
– objętość NaOH (ml), przy oznaczaniu kwasowości,
n – normalność – 0,1 n,
a – normalność roztworu NaOH – 0,1 n,
V
pr
- objętość próbki użytej do analizy (ml)

34
Zagadnienia do kartkówki:
1. Reakcje utlenienia i redukcji, definicja utleniacza i reduktora, stopnie utlenienia
pierwiastków w zwi
ą
zkach chemicznych, zasady ich wyznaczania.
2. Uzgadnianie reakcji redox.
3. Podstawy metod analitycznych opartych na reakcjach redox, miareczkowanie
redoksometryczne, jodometria (definicja).
4.
Ź
ródła tlenu w wodzie, cel oznaczania zawarto
ś
ci O
2
w wodzie, zale
ż
no
ść
mi
ę
dzy
zawarto
ś
ci
ą
tlenu w wodzie a czysto
ś
ci
ą
wód.
5. Stopnie utlenienia chromu w zwi
ą
zkach chemicznych, podstawowe zwi
ą
zki
chromu oraz ich charakter redoks,
ź
ródła chromu (VI) w wodzie i
ś
ciekach.
6. Metoda jodometryczna oznaczania Cr(VI) – reakcje.
Reakcjami redoks, czyli reakcjami utleniania i redukcji nazywa si
ę
reakcje chemiczne
przebiegaj
ą
ce ze zmian
ą
stopnia utlenienia reaguj
ą
cych ze sob
ą
cz
ą
steczek, atomów lub
jonów. Przyczyn
ą
zmiany stopnia utlenienia substratów reakcji jest zmiana liczby elektronów
zwi
ą
zanych z atomem lub jonem, który uległ utlenieniu lub redukcji.
Utlenianiem (dezelektronacj
ą
) nazywa si
ę
wszystkie procesy chemiczne, w których
cz
ą
steczki, atomy lub jony trac
ą
elektrony, czyli wzrasta ich stopie
ń
utlenienia. Natomiast w
przypadku procesów redukcji (elektronacji) nast
ę
puje przył
ą
czanie elektronów i w
konsekwencji obni
ż
enie warto
ś
ci stopnia utlenienia reaguj
ą
cych cz
ą
steczek, atomów lub
jonów.
W rzeczywisto
ś
ci oba procesy s
ą
ze sob
ą
sprz
ęż
one. Procesowi utleniania zawsze
towarzyszy proces redukcji i dlatego nie istniej
ą
procesy, które byłyby tylko reakcjami
utleniania lub tylko reakcjami redukcji.
Substrat reakcji redoks oddaj
ą
cy elektrony, a wi
ę
c zawieraj
ą
cy pierwiastek, w którym
ro
ś
nie stopie
ń
utlenienia, nazywa si
ę
reduktorem (elektronatorem), poniewa
ż
dostarcza
elektronów innemu reagentowi. Analogicznie utleniacz (dezelektronator) to substrat
pobieraj
ą
cy elektrony, czyli zawieraj
ą
cy pierwiastek, którego stopie
ń
utlenienia maleje.
Cz
ą
steczki powstaj
ą
ce w rezultacie podwy
ż
szenia stopnia utlenienia nazywa si
ę
form
ą
utlenion
ą
, natomiast cz
ą
steczki tworz
ą
ce si
ę
w wyniku obni
ż
enia stopnia utlenienia nosz
ą
nazw
ę
formy zredukowanej.
Funkcj
ę
utleniacza w reakcjach redoks mog
ą
pełni
ć
pierwiastki na najwy
ż
szych
stopniach utlenienia, np.:
- jony metali, które przył
ą
czaj
ą
c elektrony przejd
ą
w atomy lub jony na ni
ż
szym
stopniu utlenienia, np.: manganiany(VII), dichromiany(VI), sole cyny(IV), sole
ż
elaza(III), jony metali szlachetnych (Ag
+
, Cu
2+
),
- atomy niemetali wyst
ę
puj
ą
ce w stanie wolnym lub w zwi
ą
zkach na dodatnich
stopniach utlenienia, np.: tlen, ozon, siarka, fluorowce, kwas azotowy(V), kwas
chlorowy(VII), st
ęż
ony kwas siarkowy(VI), H
2
O
2
.
Funkcj
ę
reduktora w reakcjach redoks mog
ą
pełni
ć
substancje zawieraj
ą
ce
pierwiastki na mo
ż
liwie najni
ż
szych stopniach utlenienia, czyli:
- atomy metali w stanie wolnym lub jony metali na ni
ż
szych stopniach, np.: aktywne
metale I i II grupy, sole cyny(II),
ż
elaza(II),
- atomy niemetali wyst
ę
puj
ą
ce w stanie wolnym lub w zwi
ą
zkach na ni
ż
szych
stopniach utlenienia, np.: wodór, w
ę
giel, fluorowce, siarczany(IV), azotany(III).
Własno
ś
ci utleniaj
ą
co-redukuj
ą
ce danej substancji zale
żą
nie tylko od jej charakteru
chemicznego, ale tak
ż
e od
ś
rodowiska reakcji i obecno
ś
ci innych substancji o wła
ś
ciwo
ś
ciach
utleniaj
ą
co-redukuj
ą
cych. Substancje zawieraj
ą
ce pierwiastki na po
ś
rednich stopniach
utlenienia mog
ą
w obecno
ś
ci silnego reduktora wykazywa
ć
własno
ś
ci utleniaj
ą
ce, a w
obecno
ś
ci silnego utleniacza własno
ś
ci redukuj
ą
ce. Takie substancje nosz
ą
nazw
ę
amfoterów redoks.
PRACOWNIA nr 8
REDOKSOMETRIA. CHEMIA SANITARNA.
OZNACZANIE TLENU ROZPUSZCZONEGO i CHROMU(VI).

35
Znajomo
ść
stopni utlenienia pozwala nie tylko okre
ś
li
ć
tendencj
ę
do utleniania lub
redukcji, ale tak
ż
e ułatwia ocen
ę
niektórych wła
ś
ciwo
ś
ci chemicznych substancji, np.: moc
elektrolitów oraz skłonno
ść
do tworzenia jonów. Wraz ze wzrostem stopnia utleniania
pierwiastka wchodz
ą
cego w skład danego zwi
ą
zku obserwuje si
ę
:
- wzrost wła
ś
ciwo
ś
ci utleniaj
ą
cych
- wzrost mocy kwasów tlenowych,
- wzrost tendencji do tworzenia anionów,
- spadek wła
ś
ciwo
ś
ci redukuj
ą
cych,
- spadek mocy wodorotlenków,
- wzrost tendencji do tworzenia anionów.
Mo
ż
na wiec oczekiwa
ć
, ze je
ś
li pierwiastek grupy przej
ś
ciowej wyst
ę
puje na kilku stopniach
utlenienia, to jego tlenek na najni
ż
szym stopniu utlenienia powinien wykazywa
ć
wła
ś
ciwo
ś
ci
zasadowe, tlenki na stopniach po
ś
rednich – amfoteryczne, a na najwy
ż
szym – kwasowe.
Takie stwierdzenie jest prawdziwe m.in. dla zwi
ą
zków chromu i manganu.
Stopie
ń
utlenienia pierwiastka wchodz
ą
cego skład okre
ś
lonej substancji jest
poj
ę
ciem umownym, definiowanym jako liczba dodatnich lub ujemnych ładunków
elementarnych, jakie przypisane zostałyby atomom tego pierwiastka, gdyby cz
ą
steczki tej
substancji miały budow
ę
jonow
ą
. Stopie
ń
utlenienia wyznacza si
ę
na podstawie poni
ż
szych
reguł:
1. Atomy pierwiastków w stanie wolnym maj
ą
stopie
ń
utlenienia równy zero,
niezale
ż
nie od zło
ż
ono
ś
ci ich budowy, np.: Na, H
2
, S
8
.
2. Suma algebraiczna stopni utlenienia wszystkich atomów wchodz
ą
cych w skład
oboj
ę
tnej cz
ą
steczki jest równa zero.
3. Suma stopni utlenienia pierwiastków tworz
ą
cych jon jest równa ładunkowi tego
jonu.
4. Fluor we wszystkich poł
ą
czeniach ma stopie
ń
utlenienia -1.
5. Metale przyjmuj
ą
w zwi
ą
zkach dodatnie stopnie utlenienia, przy czym litowce maj
ą
zawsze stopie
ń
utlenienia +1, a berylowce +2.
6. Wodór w zwi
ą
zkach ma stopie
ń
utlenienia + 1, z wyj
ą
tkiem wodorków metali I i II
grupy głównej, np.: NaH, CaH
2
, w których stopie
ń
utlenienia wodoru wynosi -1.
7. Tlen w zwi
ą
zkach przyjmuje stopie
ń
utlenienia -2, z wyj
ą
tkiem nadtlenków, np.:
H
2
O
2
, BaO
2
, w których stopie
ń
utlenienia tlenu wynosi -l, podtlenków, np.: KO
2
, w
których stopie
ń
utlenienia tlenu wynosi -0,5; fluorku tlenu OF
2
, w którym stopie
ń
utlenienia tlen wynosi +2.
W niektórych przypadkach stopie
ń
utlenienia mo
ż
e mie
ć
warto
ść
ułamkow
ą
, np. w
jonie tetrationowym S
4
0
6
2-
stopie
ń
utlenienia siarki wynosi 5/2. Całkowity stopie
ń
utlenienia dla
siarki wynosi +10 i jest to
ś
redni stopie
ń
utlenienia, który nie podaje rozmieszczenia
elektronów w cz
ą
steczce, ale mo
ż
e by
ć
wykorzystywany do bilansowania reakcji redoks.
Dla wi
ę
kszo
ś
ci pierwiastków najwy
ż
szy dodatni stopie
ń
utlenienia równy jest liczbie
elektronów na powłoce walencyjnej atomu, a najni
ż
szy, ujemny - odpowiada liczbie
elektronów brakuj
ą
cych do wypełnienia tej powłoki.
Przy ustalaniu stopni utlenienia w cz
ą
steczkach zwi
ą
zków organicznych analizuje si
ę
ka
ż
dy atom w
ę
gla oddzielnie i korzysta z nast
ę
puj
ą
cej zasady: suma stopni utlenienia atomu
w
ę
gla i zwi
ą
zanych z nim podstawników nie b
ę
d
ą
cych atomami w
ę
gla jest równa zero, np.:
-4 +1 -3 +1 -2 +1 -2 +1 -2 +1 -3 +1 +3 - 2 -2 +1 -3 +1 +1 -2 +1
C H
4
, C H
3
C H
2
CH
2
O H , C H
3
C O O
H, C H
3
C O H
Stopnia utlenienia nie mo
ż
na uto
ż
samia
ć
z warto
ś
ciowo
ś
ci
ą
, np.: w kwasie
szczawiowym H
2
C
2
O
4
stopie
ń
utlenienia w
ę
gla wynosi +3, podczas gdy atom w
ę
gla tworzy 4
wi
ą
zania. Zatem stopie
ń
utlenienia, cho
ć
cz
ę
sto nie ma fizycznego sensu, jest przydatny do
ustalania współczynników w równaniach redoks.

36
Typy reakcji redoks.
Reakcj
ę
chemiczn
ą
mo
ż
na sklasyfikowa
ć
jako reakcj
ę
redoks je
ż
eli przemianie
substratów w produkty towarzyszy zmiana stopnia utlenienia pierwiastków.
0 +1-1 +2 -1 0
Zn + 2HCl
→
ZnCl
2
+ H
2
Powy
ż
sza reakcja b
ę
dzie procesem redoks, poniewa
ż
stopie
ń
utlenienia cynku po reakcji (+2)
jest wy
ż
szy od stopnia utlenienia przed reakcj
ą
(0). Równocze
ś
nie ulega obni
ż
eniu stopie
ń
utlenienia wodoru z +1 na 0. Podwy
ż
szenie stopnia utlenienia któregokolwiek pierwiastka
musi spowodowa
ć
obni
ż
enie stopnia utlenienia innego pierwiastka. Wynika to z faktu,
ż
e
suma stopni utlenienia we wszystkich elektrycznie oboj
ę
tnych cz
ą
steczkach jest równa zeru.
W
ś
ród reakcji redoks wyró
ż
niamy tak
ż
e takie, w których nast
ę
puje zmiana stopnia utlenienia
tylko jednego pierwiastka. S
ą
to reakcje:
- dysproporcjonowania (dysmutacji), w których cz
ęść
atomów tego samego pierwiastka
podwy
ż
sza, a cz
ęść
obni
ż
a stopie
ń
utlenienia, np.:
+6 +7 +4
3MnO
4
-
+ 2H
2
O
→
2Mn O
4
-
+ Mn O
2
+ 4OH
-
- synproporcjonacji, w których atomy tego samego pierwiastka z ró
ż
nych stopni
utlenienia przechodz
ą
w jeden, wspólny stopie
ń
utlenienia, np.:
+7 +2 +6
4MnO
4
-
+ Mn
2+
+ 8OH
-
→
5MnO
4
2-
+ 4H
2
O
Redoksometria jest działem analizy miareczkowej opartym na reakcjach utlenienia i
redukcji. Metody oksydymetryczne słu
żą
do bezpo
ś
redniego oznaczania substancji o
charakterze redukuj
ą
cym a stosowany w tych metodach titrant jest odczynnikiem o
wła
ś
ciwo
ś
ciach utleniaj
ą
cych. Do tej grupy metod nalez
ą
: manganometria, bromianometriami,
chromianometria. Metody reduktometryczne słu
żą
do oznaczania substancji o
wła
ś
ciwo
ś
ciach utleniaj
ą
cych a miareczkowanie prowadzi si
ę
titrantem o wła
ś
ciwo
ś
ciach
redukuj
ą
cych.
Zwykle przed miareczkowaniem redoks nale
ż
y przeprowadzi
ć
oznaczan
ą
substancj
ę
w posta
ć
zredukowan
ą
(je
ś
li miareczkuje si
ę
roztworem utleniacza) lub w posta
ć
utlenion
ą
(je
ś
li miareczkuje si
ę
roztworem reduktora). Najcz
ęś
ciej stosuje si
ę
takie roztwory utleniacza i
reduktora, które nast
ę
pnie mo
ż
na łatwo usun
ąć
z roztworu.
Do najwa
ż
niejszych metod oksydymetrycznych nale
żą
: manganometria, cerometria,
chromianometria i bromianometria. W przypadku metod reduktometrycznych mo
ż
na
wymieni
ć
: jodometri
ę
i tytanometria.
Najcz
ęś
ciej stosowan
ą
metod
ą
reduktometryczn
ą
jest jodometria, w której
mianowanym roztworem tiosiarczanu sodu odmiareczkowuje si
ę
jod, wydzielany w
równowa
ż
nej ilo
ś
ci w reakcji oznaczanej substancji utleniaj
ą
cej z jonami jodkowymi.
Oznaczenia jodometryczne mo
ż
na prowadzi
ć
za pomoc
ą
miareczkowania
bezpo
ś
redniego i po
ś
redniego poniewa
ż
potencjał normalny układu redoks J
2
/2J
-
znajduje si
ę
pomi
ę
dzy potencjałami mocnych utleniaczy i mocnych reduktorów.
W miareczkowaniu bezpo
ś
rednim titrantem jest mianowany roztwór jodu. W ten sposób
mo
ż
na oznacza
ć
reduktory, czyli substancje których potencjały utleniaj
ą
ce s
ą
ni
ż
sze od
potencjału układu J
2
/2J
-
. Podczas miareczkowania zachodzi redukcja jodu przez oznaczane
substancje:

37
J
2
+ 2
ĕ
→
2J
-
Metod
ą
bezpo
ś
redniego miareczkowania jodometrycznego mo
ż
na oznacza
ć
jony: S
2-
, SO
3
2-
,
S
2
O
3
2-
, Sn(II), As(III) i Sb(III).
W po
ś
rednim miareczkowaniu jodometrycznym jako titrant wykorzystuje si
ę
mianowany
roztwór Na
2
S
2
O
3
. Za jego pomoc
ą
odmiareczkowuje si
ę
jod wydzielony z KJ przez substancje
oznaczane w ilo
ś
ci równowa
ż
nej do ilo
ś
ci substancji oznaczanej. Tym sposobem oznacza si
ę
utleniacze, tj. substancje o potencjałach utleniaj
ą
cych wi
ę
kszych od potencjału układu J
2
/2J
-
.
Podczas oznaczania jony jodkowe s
ą
utleniane przez oznaczane zwi
ą
zki, a nast
ę
pnie jod
utlenia titrant:
J
2
+ 2
ĕ
→
2J
-
/2
2S
2
O
3
2-
→
S
4
O
6
2-
+ 2
ĕ
/2
J
2
+ 2S
2
O
3
2-
→
S
4
O
6
2-
+ 2J
-
W po
ś
rednim miareczkowaniu jodometrycznym oznaczane s
ą
jony BrO
3
-
, JO
3
-
, CrO
4
2-
,
Cr
2
O
7
2-
, MnO
4
-
, Cl
2
, H
2
O
2
, Cu(II), Fe(III) i Ce(IV).
Wska
ź
nikiem ko
ń
ca miareczkowania w jodometrii jest sam jod, który w wodnym roztworze
jodku, mo
ż
e mie
ć
w zale
ż
no
ś
ci od st
ęż
enia zabarwienie
ż
ółte lub br
ą
zowe. Znacznie
intensywniejsze zabarwienie uzyskuje si
ę
w obecno
ś
ci roztworu skrobi jako wska
ź
nika. W
reakcji jodu zeskrobi
ą
powstaje niebiesko zabarwiony zwi
ą
zek adsorpcyjny.
Tlen rozpuszczony wyst
ę
puje we wszystkich wodach naturalnych stykaj
ą
cych si
ę
z
powietrzem atmosferycznym (wody powierzchniowe i płytkie wody podziemne). Gł
ę
bokie
wody podziemne zawieraj
ą
tlen w niewielkich ilo
ś
ciach lub s
ą
go zupełnie pozbawione. Tlen
rozpuszczony w wodzie pochodzi głównie z powietrza atmosferycznego a tak
ż
e z procesu
fotosyntezy ro
ś
lin wodnych (makrolitów, glonów, fitoplanktonu). Zawarto
ść
tlenu
rozpuszczonego w wodach naturalnych waha si
ę
w zakresie 0 – 14 mg O
2
/dm
3
.
Ilo
ść
rozpuszczonego tlenu podawana w mg/dm
3
nie okre
ś
la nasycenia wody tlenem
w danych warunkach. Parametr ten jest bardzo wa
ż
ny przy okre
ś
laniu stopnia
zanieczyszczenia wód powierzchniowych. Nasycenie wody tlenem podawane jest najcz
ęś
ciej
jako procent nasycenia wody tlenem (N) w danej temperaturze. Definiowany jest jako
stosunek zawarto
ś
ci tlenu rozpuszczonego do maksymalnej zawarto
ś
ci tlenu rozpuszczonego
w wodzie destylowanej w danej temperaturze przy ci
ś
nieniu 1 atm (Tabela 12).
Tabela 12. Rozpuszczalno
ść
tlenu w wodzie destylowanej stykaj
ą
cej si
ę
z powietrzem
o zawarto
ś
ci 20,9% tlenu pod ci
ś
nieniem 1013, 25hPa.
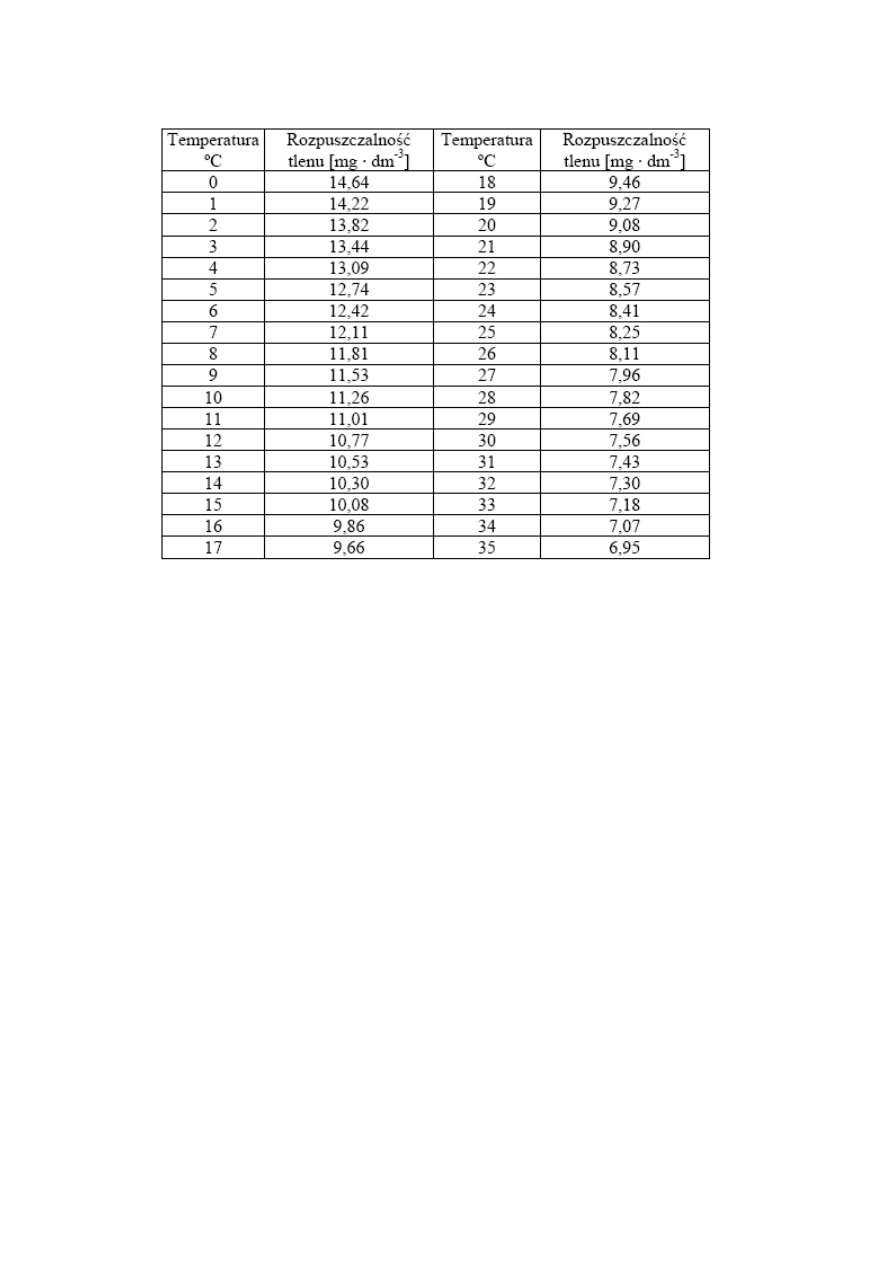
38
Wody powierzchniowe czyste zawieraj
ą
tlen w ilo
ś
ciach odpowiadaj
ą
cych prawie
100 % nasycenia. Wody powierzchniowe zanieczyszczone w nieznacznym stopniu maj
ą
stopie
ń
nasycenia tlenem w granicach 80-95%, natomiast wyra
ź
nie zanieczyszczone
wykazuj
ą
stopie
ń
nasycenia w granicach 40-50%. Je
ż
eli stopie
ń
nasycenia wody tlenem
spadnie poni
ż
ej 30% mo
ż
e to skutkowa
ć
ś
ni
ę
ciem ryb. Przy obni
ż
eniu stopnia nasycenia do
zera ma miejsce deficyt tlenowy i w zbiorniku zaczynaj
ą
dominowa
ć
procesy beztlenowe.
Przyczyn
ą
obni
ż
enia stopnia nasycenia w wodach powierzchniowych jest wprowadzanie
nadmiernych
ilo
ś
ci
zanieczyszcze
ń
,
w
st
ęż
eniach
przekraczaj
ą
cych
zdolno
ść
samooczyszczania danego zbiornika lub cieku wodnego. W wodach powierzchniowych
stoj
ą
cych, zwłaszcza gł
ę
bokich (jeziorach), zawarto
ść
tlenu uwarunkowana jest nie tylko
zawarto
ś
ci
ą
zanieczyszcze
ń
, ale tak
ż
e naturaln
ą
ich stratyfikacj
ą
. Im gł
ę
biej poło
ż
ona
warstwa tym ilo
ść
rozpuszczonego tlenu jest mniejsza. W płytkich powierzchniowych wodach
stoj
ą
cych przy silnym nasłonecznieniu zachodzi zjawisko przesycenia wody tlenem, czyli
stopie
ń
nasycenia wody osi
ą
ga warto
ść
powy
ż
ej 100% na skutek intensywnej fotosyntezy
ro
ś
lin wodnych. Dla takich zbiorników wodnych obserwuje si
ę
tak
ż
e dobow
ą
zmienno
ść
zawarto
ś
ci rozpuszczonego tlenu w wodzie – w ci
ą
gu dnia jest ona bardzo wysoka, natomiast
noc
ą
obni
ż
a si
ę
ze wzgl
ę
du na brak fotosyntezy i zu
ż
ywanie go przez plankton.
W wodach podziemnych obecno
ść
rozpuszczonego tlenu ma zarówno znaczenie dla
oceny jej bezpiecze
ń
stwa sanitarnego jak równie
ż
przy wykorzystaniu tej wody do celów
przemysłowych. Płytkie wody podziemne zawieraj
ą
nieznaczne ilo
ść
tlenu rozpuszczonego,
wody gł
ę
bokie s
ą
go praktycznie pozbawione. Je
ż
eli tlen w nich wyst
ę
puje w wi
ę
kszych
ilo
ś
ciach, mo
ż
e to
ś
wiadczy
ć
o ich zanieczyszczeniu wodami płytkimi (przebicie warstwy
wodono
ś
nej). Przy ocenie wła
ś
ciwo
ś
ci korozyjnych wody podziemnej oprócz st
ęż
enia
rozpuszczonego tlenu nale
ż
y uwzgl
ę
dni
ć
równie
ż
st
ęż
enie agresywnego CO
2
. Tlen w takim
przypadku sprzyja korozji. Je
ż
eli w wodzie agresywny CO
2
nie wyst
ę
puje, obecny w niej tlen
nie nadaje wodzie wła
ś
ciwo
ś
ci koroduj
ą
cych.
Do oznaczania tlenu rozpuszczonego w wodzie stosuje si
ę
metod
ę
Winklera lub
przeprowadza si
ę
to oznaczenie przy u
ż
yciu sondy tlenowej.

39
Rys. 4. Galwaniczna sonda DurOx 325.
Chrom metaliczny i nierozpuszczalne zwi
ą
zki chromu(III) uwa
ż
a si
ę
za nieszkodliwe,
natomiast wła
ś
ciwo
ś
ci kancerogenne, mutagenne i teratogenne wykazuj
ą
zwi
ą
zki chromu(VI).
Najwy
ż
sze dopuszczalne st
ęż
enie chromu(VI) w wodzie przeznaczonych do spo
ż
ycia wynosi
0,05 mg/dm
3
.
Ź
ródłem chromu w wodach powierzchniowych s
ą
ś
cieki przemysłowe
pochodz
ą
ce z garbarni, galwanizerni, z zakładów lotniczych i przemysłu samochodowego.
Natomiast w wodach podziemnych zwi
ą
zku chromu wyst
ę
puj
ą
rzadko.
Chrom mo
ż
e wyst
ę
powa
ć
w wodzie w postaci sze
ś
ciu – i trójwarto
ś
ciowego. Cr(III)
jest trwały i w zwykłych warunkach nie zachodzi jego utlenianie do Cr(VI). W
ś
rodowisku
oboj
ę
tnym sole chromu cz
ęś
ciowo zhydrolizowane, w zale
ż
no
ś
ci od rodzaju anionu. W
ś
rodowisku alkalicznym Cr(III) ulega wytraceniu w postaci wodorotlenku. Cr(VI) mo
ż
e
wyst
ę
powa
ć
w roztworach alkalicznych w postaci jonów chromianowych i w kwa
ś
nych – jako
jony dwuchromianowe. W tej postaci chrom jest trwały w wodach nie zawieraj
ą
cych substancji
redukuj
ą
cych. Gdy
ś
rodowisko b
ę
dzie miało charakter redukuj
ą
cy nast
ą
pi redukcja
chromu(VI) do chromu(III).
W wodach wodoci
ą
gowych zwi
ą
zki chromu s
ą
rzadko spotykane. Ich obecno
ść
mo
ż
e
by
ć
tłumaczona niedostatecznym oczyszczaniem wód powierzchniowych, zanieczyszczeniem
ś
ciekami przemysłowymi lub zanieczyszczeniem sieci wodoci
ą
gowej wodami chłodniczymi,
do których dodaje si
ę
zwi
ą
zki chromu w celu ochrony rur przed korozj
ą
. Ze wzgl
ę
du na
toksyczne i rakotwórcze wła
ś
ciwo
ś
ci chromu(VI) dopuszczalne st
ęż
enie w wodzie
przeznaczonej do picia nie powinno przekracza
ć
0,05 mg/dm
3
.
Do oznaczania chromu stosowane s
ą
nast
ę
puj
ą
ce metody:
- miareczkowa jodometryczna
- kolorymetryczna z dwufenylokarbazydem.

40
Literatura
1. Buczkowski R., Wybrane zagadnienia proekologiczne w chemii, Wydawnictwo
Uniwersytetu M. Kopernika w Toruniu, Toru
ń
2002.
2.
Cyga
ń
ski A., Chemiczne metody analizy ilo
ś
ciowej, WNT Warszawa 1999.
3.
Hermanowicz W., Dojlido J., Do
ż
a
ń
ska W., Kosiorowski B., Zerze J., Fizyczno-
chemiczne badanie wody i
ś
cieków, Arkady, Warszawa 1999.
4. Kiedry
ń
ska L., Papciak D., Granops M., Chemia sanitarna, Wydawnictwo SGGW,
Warszawa 2006.
5. Krzechowska M., Podstawy chemii ogólnej i
ś
rodowiska przyrodniczego.
Ć
wiczenia
laboratoryjne. Oficyna Wydawnicza Politechniki Warszawskiej, Warszawa 2007.
6. Pokojska U., Przewodnik metodyczny do analizy wód, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
1999.
7. Szłyk E., Kurzawa M., Szydłowska-Czerniak A., Jastrz
ę
bska A., Ilo
ś
ciowa analiza
chemiczna. Metody wagowe i miareczkowe, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
2005.
8. Szmal S., Lipiec T., Chemia analityczna z elementami analizy instrumentalnej
PZWL, 1997.

41
Celem
ć
wiczenia jest zapoznanie si
ę
z miareczkowaniem redoksometrycznym na
przykładzie oznacze
ń
jodometrycznych.
Zakres
ć
wiczenia obejmuje wykonanie oznaczenia zawarto
ś
ci tlenu rozpuszczonego
metod
ą
Winklera, obliczenie nasycenia wody tlenem oraz wykonanie oznaczenia zawarto
ś
ci
chromu(VI) w badanej wodzie.
Uwaga: roztworów mianowanych pobranych z butelki do biurety lub zlewki a nie
wykorzystanych do analizy, NIE WOLNO wlewa
ć
z powrotem do butelki, w której s
ą
przechowywane.
A. Oznaczanie zawarto
ś
ci tlenu rozpuszczonego metod
ą
Winklera.
Zasada metody
Oznaczanie tlenu rozpuszczonego przebiega w dwóch etapach. W pierwszym etapie
wprowadzony do zalkalizowanej roztworem jodku potasu (KJ + KOH) próbki badanej wody
mangan w formie MnSO
4
wytr
ą
ca si
ę
w postaci białego, kłaczkowatego osadu Mn(OH)
2
.
Zawarty w wodzie tlen rozpuszczony utlenia mangan(II) do manganu(IV) – powstaje osad
wodorotlenku manganu(IV) o barwie jasnobr
ą
zowej. W drugim etapie, po zakwaszeniu
roztworu próbki kwasem siarkowym(VI), nast
ę
puje rozpuszczenie osadu i redukcja
manganu(IV) do manganu(II). W roztworze z dodanego KJ wydziela si
ę
wolny jod w ilo
ś
ci
równowa
ż
nej zawarto
ś
ci tlenu rozpuszczonego, który nast
ę
pnie odmiareczkowuje si
ę
tiosiarczanem sodu w obecno
ś
ci skrobi jako wska
ź
nika. Na podstawie ilo
ś
ci zu
ż
ytego
tiosiarczanu oblicza si
ę
zawarto
ść
tlenu w badanej wodzie. Przebieg procesów ilustruj
ą
poni
ż
sze reakcje:
Mn
2+
+ 2OH
-
→
Mn(OH)
2
2Mn(OH)
2
+ O
2
→
2MnO(OH)
2
2MnO(OH)
2
+ 4H
+
→
Mn
4+
+
3H
2
O
Mn
4+
+
2J
-
→
Mn
2+
+ J
2
J
2
+ 2S
2
O
3
2-
→
S
4
O
6
2-
+ 2J
-
Uwaga: pojawienie si
ę
białego osadu w pierwszej fazie oznaczania
ś
wiadczy o braku tlenu
w próbce.
Odczynniki: alkaliczny roztwór KJ, MnSO
4
, H
2
SO
4
, 0,0125 M Na
2
S
2
O
4
, skrobia
Szkło: butelki inkubacyjne z ciemnego szkła z doszlifowanym, uko
ś
nie
ś
ci
ę
tym korkiem,
kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Sprz
ę
t:
termometr z podziałk
ą
o dokładno
ś
ci 0,1
0
C
Materiał: woda wodoci
ą
gowa
Wykonanie oznaczenia:
- wod
ę
do oznaczenia pobra
ć
z kranu – wylot kranu dokładnie umy
ć
- nast
ę
pnie wypuszcza
ć
wod
ę
przez całkowicie otwarty w ci
ą
gu 10 minut
Ć
wiczenie nr 6
REDOKSOMETRIA. CHEMIA SANITARNA.
OZNACZANIE TLENU ROZPUSZCZONEGO i CHROMU(VI) - instrukcja.
42
- pobra
ć
próbk
ę
wody wprost do przepłukanego ni
ą
naczynia
- po napełnieniu naczynia wyla
ć
nieco wody, tak by pod korkiem zostawi
ć
warstw
ę
powietrza
o grubo
ś
ci 4 cm, naczynie zamkn
ąć
,
- do 3 butelek inkubacyjnych o pojemno
ś
ci 250 ml nala
ć
do pełna otrzymanej do analizy
próbki, nadmiar cieczy usun
ąć
zamykaj
ą
c butelk
ę
korkiem tak, by pod korkiem nie pozostał
p
ę
cherzyk powietrza,
- do ka
ż
dej butelki doda
ć
po 2 ml roztworu MnSO
4
i po 2 ml alkalicznego roztworu jodku
potasu KJ + NaOH (wprowadzaj
ą
koniec pipety pod powierzchni
ę
cieczy),
- zamkn
ąć
butelk
ę
korkiem, tak aby nie powstał p
ę
cherzyk powierza pod korkiem,
- wymiesza
ć
(przekr
ę
caj
ą
c butelk
ę
15 razy) i odstawi
ć
na 20 minut w ciemne miejsce,
wydzielaj
ą
cy si
ę
osad to Mn(OH)
4
- nast
ę
pnie doda
ć
po 2 ml st
ęż
onego H
2
SO
4
wprowadzaj
ą
c koniec pipety pod powierzchni
ę
cieczy,
- zamkn
ąć
butelk
ę
korkiem i dokładnie wymiesza
ć
, a
ż
osad si
ę
rozpu
ś
ci,
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml cieczy,
- miareczkowa
ć
0,0125 N roztworem Na
2
S
2
O
3
do uzyskania jasno
ż
ółtego zabarwienia,
nast
ę
pnie doda
ć
1 ml roztworu skrobi i szybko miareczkowa
ć
tym samym roztworem
Na
2
S
2
O
3
do odbarwienia roztworu
jasno
ż
ółty po dodaniu skrobi po dodaniu Na
2
S
2
O
3
Uwaga: powtórnego zabarwienia nie bierze si
ę
pod uwag
ę
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu
Na
2
S
2
O
3
i obliczy
ć
zawarto
ść
rozpuszczonego tlenu
wg wzoru:
Z
O
2
=
)
(
b
V
V
a
−
∗
[mg O
2
/dm
3
]
gdzie: V – obj
ę
to
ść
butelki inkubacyjnej, 250 ml
a – obj
ę
to
ść
roztworu Na
2
S
2
O
3
zu
ż
yta na zmiareczkowanie jodu wydzielonego w
100 ml próbki, ml
b – obj
ę
to
ść
dodanych roztworów MnSO
4
, KJ + NaOH
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
43
B. Oznaczanie zawarto
ś
ci chromu (VI).
Odczynniki: 5% ZnSO
4
, H
2
SO
4
, fenoloftaleina, 20%
NaOH, 10% KJ, 0,0125 M Na
2
S
2
O
4
,
skrobia
Szkło: kolby sto
ż
kowe o poj. 200 – 300 ml, pipety wielomiarowe, biureta, cylinder miarowy
100 ml, podci
ą
garka do pipetowania
Materiał: badana woda
Wykonanie oznaczenia:
- do 3 zlewek o pojemno
ś
ci 250 ml przela
ć
po ok. 170 ml otrzymanej do analizy próbki,
- doda
ć
po 1 ml 5% roztworu ZnSO
4
, dokładnie wymiesza
ć
,
- doda
ć
2 – 3 krople fenoloftaleiny,
- do ka
ż
dej zlewki doda
ć
(ci
ą
gle mieszaj
ą
c) kroplami roztwór NaOH do pojawienia si
ę
trwałego czerwonego zabarwienia, odstawi
ć
do sklarowania,
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml klarownego roztworu znad osadu,
- doda
ć
po 10 ml 10% roztworu KJ i po 1 ml roztworu H
2
SO
4
(1+1),
- dobrze wymiesza
ć
i odstawi
ć
w ciemne miejsce na 10 minut,
- miareczkowa
ć
0,0125 N roztworem Na
2
S
2
O
3
do uzyskania jasno
ż
ółtego zabarwienia,
nast
ę
pnie doda
ć
1 ml roztworu skrobi i szybko miareczkowa
ć
tym samym roztworem
Na
2
S
2
O
3
do odbarwienia roztworu,
jasno
ż
ółty po dodaniu skrobi po dodaniu Na
2
S
2
O
3
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu Na
2
S
2
O
3
i obliczy
ć
zawarto
ść
Cr (VI) wg wzoru:
[Cr
VI
]
=
pr
V
V
a
1
35
,
17
1000
∗
∗
∗
[mg/dm
3
]
gdzie: V
1
– obj
ę
to
ść
roztworu Na
2
S
2
O
3
zu
ż
yta na zmiareczkowanie próbki, ml
a – normalno
ść
roztworu Na
2
S
2
O
3
, 0,0125 N
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
Wyniki otrzymane z oznacze
ń
zawarto
ś
ci tlenu rozpuszczonego i chromu(VI) wpisa
ć
do
tabeli.
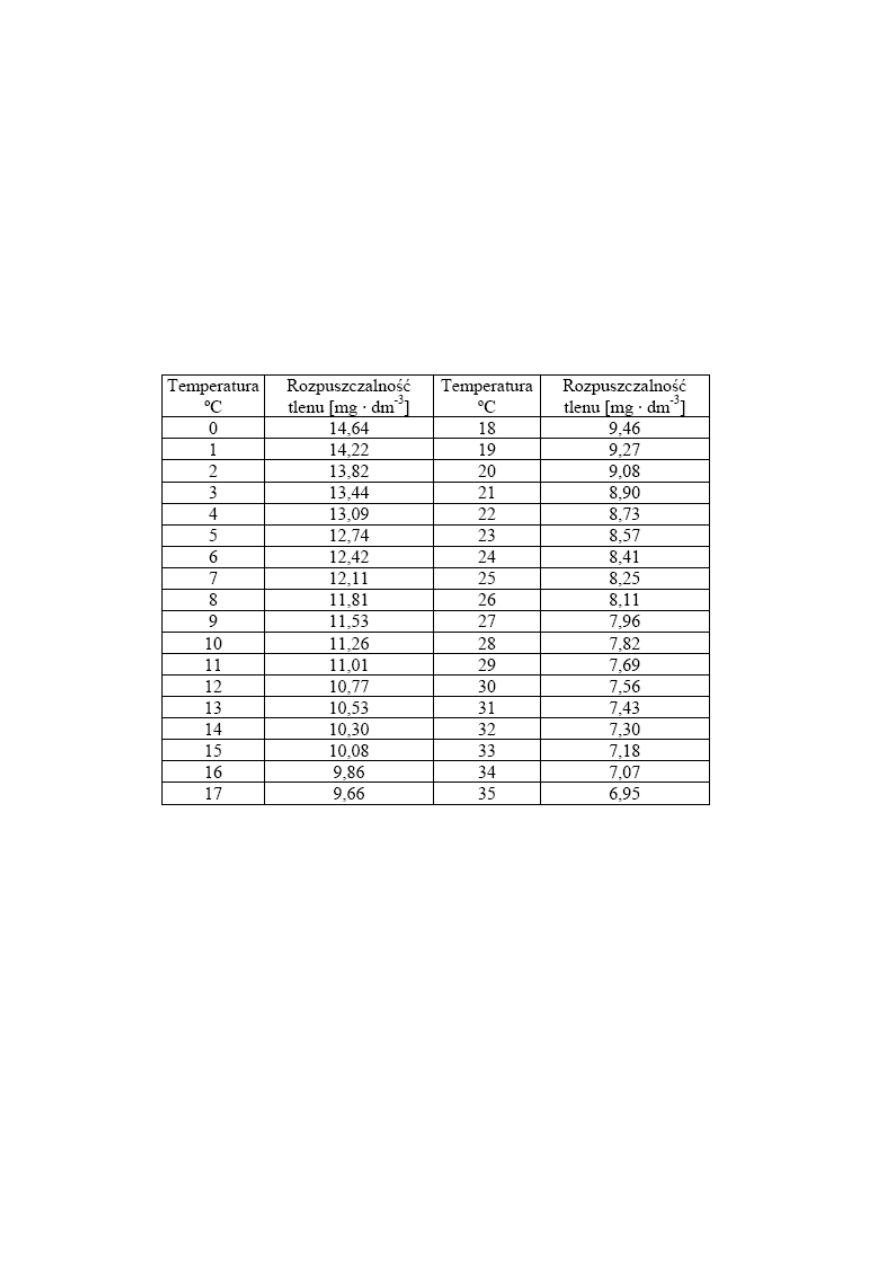
44
Sprawozdanie z
ć
wiczenia powinno zawiera
ć
:
- stron
ę
tytułow
ą
- cel i zakres
ć
wiczenia
- opis wykonania
ć
wiczenia (zasada oznaczenia, odczynniki, szkło, sprz
ę
t, materiały,
wykonanie
ć
wiczenia)
- otrzymane wyniki (tabela podpisana przez prowadz
ą
cego
ć
wiczenia)
- wnioski – odnie
ść
si
ę
do aktualnych przepisów prawnych dotycz
ą
cych jako
ś
ci wody
przeznaczonej do spo
ż
ycia przez ludzi.
Przy opracowywaniu wniosków skorzysta
ć
z poni
ż
szej Tabeli 12.
Tabela 12 . Rozpuszczalno
ść
tlenu w wodzie destylowanej stykaj
ą
cej si
ę
z powietrzem
o zawarto
ś
ci 20,9% tlenu pod ci
ś
nieniem 1013, 25hPa.

45
POLITECHNIKA LUBELSKA
Wydział In
ż
ynierii
Ś
rodowiska
In
ż
ynieria
Ś
rodowiska
SPRAWOZDANIE
Z
Ć
WICZE
Ń
LABORATORYJNYCH Z CHEMII
Nr
ć
wiczenia
6
Temat
ć
wiczenia
Redoksymetria. chemia sanitarna.
Oznaczanie tlenu rozpuszczonego metod
ą
Winklera
i chromu(VI).
Imi
ę
i nazwisko studenta
……………………………………………
……………………………………………
……………………………………………
Rok studiów
Semestr
Data
Imi
ę
i nazwisko prowadz
ą
cego
ć
wiczenia
Uwagi prowadz
ą
cego:

46
Oznaczanie tlenu rozpuszczonego i chromu (VI)
...................................................
…………………………………
………………………………….
Imię i Nazwisko
Grupa BDi .............
Data.......................
Oznaczanie tlenu rozpuszczonego
Nr próbki
a [ml]
Z
O
2
[mg O
2
/dm
3
]
1
2
3
Z
O
2
= ......................
[mg O
2
/dm
3
]
Obliczanie stopnia nasycenia wody tlenem N [%]
Nr próbki
Z
O
2
[mg O
2
/dm
3
]
temp. wody [
0
C]
n [mg/ dm
3
O
2
]
N [%]
1
2
3
N = ………… [%]
Oznaczanie zawartości chromu
Nr próbki
V
1
Na
2
S
2
O
3
[ml]
[Cr
VI
] [mg/dm
3
]
1
2
3
[Cr
VI
]
=
.........................
[mg/dm
3
]
gdzie:
2
(
)
O
a V
Z
V
b
∗
=
−
[mg O
2
/dm
3
]
2
1 0 0
O
Z
N
n
∗
=
[%]
pr
V
V
c
VI
Cr
1
35
,
17
1000
)
(
∗
∗
∗
=
[mg/dm
3
]
V
1
– objętość roztworu Na
2
S
2
O
3
zużyta na zmiareczkowanie próbki (ml),
V
– pojemność butelki inkubacyjnej ( 250 ml),
a – objętość roztworu Na
2
S
2
O
3
zużyta na zmiareczkowanie jodu wydzielonego w 100 cm
3
próbki (ml),
b – objętość dodanych roztworów MnSO
4
, KJ + NaOH (ml),
n - liczba mg tlenu zawarta w 1 dm
3
wody destylowanej potrzebna do nasycenia wody tlenem w danej
temperaturze i p =1013,25 hPa,
N – stopień nasycenia wody tlenem (%),
c - normalność roztworu Na
2
S
2
O
3
– 0,0125 n w oznaczaniu chromu ,
V
pr
- objętość próbki wody użytej do analizy (ml)
Uwaga: jako wynik podać średnią arytmetyczną z trzech oznaczeń

47
Zagadnienia do kartkówki:
1. Barwa – definicja,
ź
ródła, rodzaje barwy, jednostka, metody oznaczania.
2. M
ę
tno
ść
–
ź
ródła, jednostki, metody oznaczania.
3. Podstawy kolorymetrii, nefelometria, turbidymetria.
4.
Ź
ródła i rodzaje zapachu, metody oznaczania, sposoby wyra
ż
ania intensywno
ś
ci
i rodzaju zapachu.
5. Przewodnictwo - definicja, jednostka, metody pomiaru przewodnictwa, cel oznaczania
przewodnictwa.
6. BZT
n
– definicja, jednostka, metody oznaczania, cel oznaczania.
7. Fazy biochemicznego utleniania zwi
ą
zków organicznych, krzywa przebiegu BZT
n
.
8. ChZT – definicja, metody oznaczania ChZT, oznaczanie ChZT metod
ą
dwuchromianow
ą
.
9. Utlenialno
ść
– definicja,
ź
ródła, jednostka, zale
ż
no
ść
mi
ę
dzy warto
ś
ci
ą
utlenialno
ś
ci
wody a jej czysto
ś
ci
ą
, metody oznaczania utlenialno
ś
ci.
Przy ocenie jako
ś
ci wody wa
ż
n
ą
rol
ę
odgrywaj
ą
oznaczenia zarówno wska
ź
ników
fizycznych (barwa, m
ę
tno
ść
, zapach, temperatura, przewodnictwo wła
ś
ciwe) jak równie
ż
wska
ź
ników
charakteryzuj
ą
cych
zawarto
ść
zwi
ą
zków
organicznych
(BZT,
ChZT,
utlenialno
ść
).
Barwa wody
Woda chemicznie czysta jest bezbarwna w cienkiej warstwie, natomiast w warstwie
grubszej ma odcie
ń
niebieskawy. Wody naturalne całkowicie bezbarwne nale
żą
do rzadko
ś
ci
(wody potoków górskich). Wody podziemne i powierzchniowe wykazuj
ą
barw
ę
zielonkawo
ż
ółt
ą
o ró
ż
nej intensywno
ś
ci. Zabarwienie to jest spowodowane obecno
ś
ci
ą
zwi
ą
zków chemicznych wyługowanych z gleby (np. zwi
ą
zków
ż
elaza) lub barwnych zwi
ą
zków
humusowych. Zabarwienie wody powierzchniowej mo
ż
e by
ć
te
ż
wywołane rozwijaj
ą
cymi si
ę
w niej organizmami. Taka sytuacja ma miejsce podczas zakwitów zbiorników wodnych – przy
masowym rozwoju zielenic woda przybiera zabarwienie jasnozielone, w czasie rozwoju
okrzemek zielonoszare a przy rozwoju sinic bł
ę
kitnozielone. W przypadku wód
powierzchniowych barw
ę
mog
ą
wywoływa
ć
odprowadzone do nich nieczyszczone
ś
cieki
przemysłowe.
Wyró
ż
nia si
ę
nast
ę
puj
ą
ce rodzaje barwy:
- barw
ę
pozorn
ą
– wywołan
ą
obecno
ś
ci
ą
drobnej zawiesiny bogatej w
ż
elazo, cz
ą
steczki
gliny; barwa ta zanika po ods
ą
czeniu zawiesiny,
- barw
ę
rzeczywist
ą
– wywołan
ą
przez rozpuszczone w wodzie substancje.
Ponadto ze wzgl
ę
du na rodzaj zanieczyszczenia mo
ż
na wyró
ż
ni
ć
barw
ę
:
- naturaln
ą
– spowodowan
ą
naturalnymi zanieczyszczeniami lub domieszkami
- specyficzn
ą
– wywołan
ą
obecno
ś
ci
ą
ś
cieków przemysłowych.
PRACOWNIA nr 9
WSKA
Ź
NIKI ZANIECZYSZCZENIA WODY. CHEMIA SANITARNA.
OZNACZANIE UTLENIALNO
Ś
CI, BARWY, ZAPACHU, PRZEWODNICTWA i pH.

48
Barw
ę
mo
ż
na oznacza
ć
:
- wg skali platynowo-kobaltowej
- wg skali dwuchromianowo-kobaltowej
- metod
ą
opisow
ą
w przypadku barwy specyficznej
- metod
ą
oznaczania liczby progowej
- metod
ą
spektrofotometryczn
ą
.
Jednostk
ą
barwy jest zabarwienie, jakie nadaje 1 mg platyny zawartej w
chloroplatynianie potasu K
2
PtCl
6
rozpuszczonym w 1 dm
3
wody destylowanej z dodatkiem 0,5
mg CoCl
2
*6H
2
O. Zasada oznaczania barwy z wykorzystaniem skali platynowo-kobaltowej i
dwuchromianowo-kobaltowej polega na wizualnym porównaniu badanej próbki wody ze skal
ą
wzorców. Je
ż
eli woda jest m
ę
tna nale
ż
y usun
ąć
zawiesin
ę
poprzez odwirowanie lub s
ą
czenie
na s
ą
czku ilo
ś
ciowym o
ś
redniej grubo
ś
ci. Je
ż
eli woda ma barw
ę
odbiegaj
ą
c
ą
od skali
platynowo-kobaltowej nale
ż
y próbk
ę
rozcie
ń
czy
ć
do momentu a
ż
jej intensywno
ść
b
ę
dzie
mo
ż
liwa do oznaczenie wg dost
ę
pnej skali wzorców. Je
ż
eli barwa wody jest bardzo
intensywna i odbiega od skali wzorców nale
ż
y scharakteryzowa
ć
j
ą
w sposób opisowy.
Oznaczenie barwy wg liczby progowej (L
p
) polega na ustaleniu obj
ę
to
ś
ci badanej wody
rozcie
ń
czonej do 100 ml wod
ą
destylowan
ą
, przy której po raz pierwszy zaobserwowano
pojawienie si
ę
trwałego zabarwienia. Jako próbk
ę
odniesienia u
ż
ywa si
ę
100 ml wody
destylowanej. Liczb
ę
progow
ą
oblicza si
ę
ze wzoru:
100
p
L
V
=
gdzie:
V – obj
ę
to
ść
wody u
ż
yta do oznaczenia, przy której pojawił si
ę
zabarwienie
Najcz
ęś
ciej wody naturalne charakteryzuj
ą
si
ę
barw
ą
w zakresie 5 – 25 mg Pt/dm
3
.
Spotykane s
ą
równie
ż
wody o wi
ę
kszej barwie – pochodz
ą
one z terenów le
ś
nych, bagnistych
lub torfowiskowych. Dopuszczalna barwa wody wodoci
ą
gowej zgodnie z obowi
ą
zuj
ą
cymi
przepisami prawnymi wynosi 15 mg Pt/dm
3
. Wszelkie odcienie wody poza zielonkawo
ż
ółtym
dyskwalifikuj
ą
j
ą
jako wod
ę
pitn
ą
.
M
ę
tno
ść
wody
Jest to optyczna wła
ś
ciwo
ść
wody, któr
ą
nadaj
ą
jej drobne zawiesiny (cz
ą
stki gliny, iły,
zwi
ą
zki
ż
elaza, manganu, glinu, rozdrobnione substancje organiczne, plankton) powoduj
ą
ce
zjawisko rozproszenia
ś
wiatła. Woda do picia powinna by
ć
klarowna i wykazywa
ć
m
ę
tno
ść
poni
ż
ej 1 NTU.
M
ę
tno
ść
wody oznacza si
ę
nast
ę
puj
ą
cymi metodami:
- wizualn
ą
, turbidymetryczn
ą
w zakresie 0 - 5 mg/dm
3
i 5 - 50 mg/dm
3
- nefelometryczn
ą
.
Oznaczenie m
ę
tno
ś
ci metod
ą
wizualn
ą
polega na porównaniu intensywno
ś
ci
przepuszczania
ś
wiatła przez próbk
ę
wody w odniesieniu do skali wzorców.
Wody naturalne wykazuj
ą
m
ę
tno
ść
powy
ż
ej 5 NTU.
Nefelometryczna metoda oznaczania m
ę
tno
ś
ci wody polega na porównaniu nat
ęż
enia
ś
wiatła rozproszonego na koloidalnych cz
ą
stkach i drobnych zawiesinach z odpowiednio
przygotowanymi wzorcami. Nat
ęż
enie rozproszonego
ś
wiatła jest proporcjonalne do m
ę
tno
ś
ci
badanej wody. Przyrz
ą
dy wykorzystywane w tej metodzie nosz
ą
nazw
ę
nefelometrów.
Za jednostk
ę
m
ę
tno
ś
ci przyjmuje si
ę
m
ę
tno
ść
jak
ą
posiada wzorzec sporz
ą
dzony z 1
mg zawiesiny krzemionki rozpuszczonej w 1 dm
3
wody destylowanej (1 mg SiO
2
/dm
3
). Jako
wzorzec mo
ż
na równie
ż
zastosowa
ć
formazyn
ę
. Jako jednostka m
ę
tno
ś
ci stosowana jest
tak
ż
e jednostka NTU (nephelometric turbidity unit), która jest równowa
ż
na stosowanej w
Polsce jednostce mg SiO
2
/dm
3
.

49
Kolorymetria - technika analityczna polegaj
ą
ca na wykorzystywaniu zdolno
ś
ci badanych
zwi
ą
zków chemicznych do pochłaniania
ś
wiatła. St
ęż
enie badanego roztworu badane jest
przy pomocy sporz
ą
dzonej krzywej wzorcowej. Krzywa ta jest funkcj
ą
absorpcji
ś
wiatła w
zale
ż
no
ś
ci od st
ęż
enia roztworu. Zale
ż
no
ść
ta jest cz
ę
sto liniowa.
Nefelometria - metoda analizy instrumentalnej polegaj
ą
ca na pomiarze st
ęż
enia roztworu na
podstawie pomiaru nat
ęż
enia
ś
wiatła rozproszonego przez zawiesin
ę
, wykorzystuj
ą
ca efekt
Tyndalla. Wi
ą
zka
ś
wiatła przechodz
ą
c przez roztwór koloidalny pod okre
ś
lonym k
ą
tem
wzgl
ę
dem wi
ą
zki padaj
ą
cej, staje si
ę
widoczna w postaci tzw. sto
ż
ka Tyndalla. Na tej
podstawie oznacza si
ę
st
ęż
enie tej zawiesiny lub rozmiary tworz
ą
cych j
ą
cz
ą
stek.
Turbidymetria - jedna z metod spektrofotometrycznych w chemii analitycznej; słu
ż
y do
pomiaru m
ę
tno
ś
ci zawiesin. Istota metody jest analogiczna, jak w przypadku innych metod
spektrofotometrycznych i opiera si
ę
na pomiarze relacji pomi
ę
dzy ilo
ś
ci
ą
ś
wiatła emitowanego
przez
ź
ródło, a ilo
ś
ci
ą
ś
wiatła docieraj
ą
c
ą
do detektora spektrofotometru, po przej
ś
ciu przez
komórk
ę
(kuwet
ę
) z badan
ą
próbk
ą
.
Zapach wody
Zapach wody jest spowodowany lotnymi substancjami, które mog
ą
mie
ć
pochodzenie
naturalne lub antropogeniczne (
ś
cieki). Zapach w wodach powierzchniowych pochodzi
najcz
ęś
ciej od substancji organicznych i gazów. Charakterystyczny zapach wodom
powierzchniowym mog
ą
nadawa
ć
wprowadzone do nich nieczyszczone
ś
cieki zarówno
komunalne jak i przemysłowe (zapach gnilny spowodowany siarkowodorem, indolem i
skatolem) W przypadku wprowadzania
ś
cieków rodzaj zapachu uzale
ż
niony jest od produkcji
zakładu. Wody podziemne w wi
ę
kszo
ś
ci przypadków s
ą
bezwonne.
Do naturalnych zapachów w wodzie powierzchniowej nale
żą
zapachy olejków
eterycznych wydzielanych przez ro
ś
liny wodne, ple
ś
nie, grzyby wodne i pierwotniaki. W
wodach podziemnych najcz
ęś
ciej jego
ź
ródłem mo
ż
e by
ć
siarkowodór. Ogólnie zapachy dzieli
si
ę
na pochodzenia naturalnego i nienaturalnego. Pierwszy rodzaj zapachów jest
spowodowany w wodzie obecno
ś
ci
ą
naturalnych substancji organicznych. Do tego rodzaju
zapachów mo
ż
na zaliczy
ć
zapach wilgotnej ziemi, korzy drzewnej, torfu, siana, olejków
eterycznych wydzielanych przez plankton. Zapachy te pochodz
ą
z gleby, osadów dennych
zbiornika wodnego. Okre
ś
la si
ę
je mianem zapachów ro
ś
linnych (R). Zapachy te nie
dyskwalifikuj
ą
wody przeznaczonej do picia. Zapachy naturalne spowodowane obecno
ś
ci
ą
substancji organicznych znajduj
ą
cych si
ę
w stanie rozkładu i nadaj
ą
ce wodzie przykry,
czasem odra
ż
aj
ą
cy zapach zgniłych jaj, st
ę
chły zapach gnij
ą
cych ryb, zbutwiały, zapach
ple
ś
ni, zapach fekalny nale
żą
do zapachów gnilnych (G). Zapach gnilny
ś
wiadczy o
zanieczyszczeniu wody (z wyj
ą
tkiem zapachu siarkowodoru dla wód podziemnych) i nawet
bardzo slaby zapach gnilny dyskwalifikuje przydatno
ść
wody do spo
ż
ycia.
Do nienaturalnych zapachów nale
żą
wszystkie zapachy nie wyst
ę
puj
ą
ce w wodach
naturalnych (zapach fenolu, benzyny, smoły). Okre
ś
la si
ę
je jako zapachy specyficzne (S),
przy czym podaje si
ę
bli
ż
sze okre
ś
lenie zapachu, np. S (nafta).
Tabela 13. Grupy i rodzaje zapachów.
Grupa zapachu
Oznaczenie
Rodzaj zapachu
roślinny
R
ziemisty, trawiasty, kwiatowy, rybny, aromatyczny itp.
gnilny
G
stęchły, pleśni, feralny, siarkowodoru
specyficzny
S
fenolu, benzyny, chloru, nafty, smoły, lakieru
Do zapachów pochodzenia naturalnego zalicza si
ę
dwie grupy zapachów – ro
ś
linny (R) i
gnilny(G). Do zapachów nienaturalnych zalicza si
ę
grup
ę
zapachów specyficznych (S).
W celu pełnego opisu zapachu badanej wody oprócz podania grupy zapachu, nale
ż
y
wskaza
ć
jego intensywno
ść
w skali 0 – 5 (Tabela 14). Poniewa
ż
zapach okre
ś
la si
ę
metod
ą
organoleptyczn
ą
w celu unikni
ę
cia subiektywnych pomyłek oznaczenia tego powinno
dokonywa
ć
równolegle kilka osób. Zapach wody do picia powinien by
ć
akceptowalny.

50
Tabela 14. Skala intensywno
ś
ci zapachów.
Intensywność
zapachu
Wyczuwalność zapachu i jego znaczenie
0
brak zapachu
1
zapach bardzo słaby
2
zapach słaby
3
zapach wyraźny
4
zapach silny – dyskwalifikuje wodę do celów bytowo-gospodarczych
5
zapach bardzo silny – dyskwalifikuje wodę do celów bytowo-gospodarczych
Przewodnictwo wła
ś
ciwe wody
Parametr ten pozwala na szybki pomiar zawarto
ś
ci substancji rozpuszczonych w
wodzie. Przewodnictwo wła
ś
ciwe wody
κ
(inne okre
ś
lenia: konduktywno
ść
, przewodno
ść
wła
ś
ciwa)jest to zdolno
ść
roztworu wodnego do przewodzenia pr
ą
du elektrycznego. Definiuje
si
ę
j
ą
jako odwrotno
ść
oporu wła
ś
ciwego słupa cieczy zawartego pomi
ę
dzy elektrodami o
powierzchni 1 cm
2
i odległo
ś
ci 1 cm. Jednostk
ą
tego parametru jest simens/cm (S/cm).
Poniewa
ż
roztwory cechuje bardzo małe przewodnictwo wła
ś
ciwe zazwyczaj wyra
ż
a si
ę
je w
µ
S/cm.
Ś
wie
ż
a destylowana woda wykazuje przewodnictwo wła
ś
ciwe w zakresie 0,5 - 2,0
µ
S/cm. Przewodnictwo wła
ś
ciwe wód naturalnych waha si
ę
w granicach 50 – 500
µ
S/cm, a
dla wód mineralnych mo
ż
e osi
ą
ga
ć
warto
ść
1000
µ
S/cm. Wody naturalne zanieczyszczone
przez
ś
cieki przemysłowe wykazuj
ą
przewodnictwo wła
ś
ciwe nawet 10 000
µ
S/cm.
Oznaczenie przewodnictwa wła
ś
ciwego wody wykonuje si
ę
w celu stwierdzenia czysto
ś
ci
wody destylowanej, zdemineralizowanej, kondensatów a tak
ż
e wody wodoci
ą
gowej. Nagły
wzrost przewodnictwa wła
ś
ciwego wody mo
ż
e
ś
wiadczy
ć
o jej zanieczyszczeniu zwi
ą
zkami
mineralnymi, gdy
ż
przewodnictwo wody wynikaj
ą
ce z obecno
ś
ci w niej zwi
ą
zków
organicznych jest niewielkie.
Na podstawie pomiaru przewodnictwa wła
ś
ciwego mo
ż
na okre
ś
li
ć
przybli
ż
on
ą
zawarto
ść
kationów i anionów (w mval/dm
3
) mno
żą
c wynik przewodnictwa w
µ
S/cm przez
0,01.
Biochemiczne zapotrzebowanie tlenu (BZT) jest terminem umownym. Oznacza si
ę
go zarówno dla wody i
ś
cieków. Okre
ś
la ilo
ść
tlenu wyra
ż
on
ą
w mgO
2
/dm
3
wody lub
ś
cieków,
potrzebn
ą
do utlenienia zwi
ą
zków organicznych zawartych w wodzie lub w
ś
ciekach metod
ą
biochemiczn
ą
, przy udziale mikroorganizmów, w warunkach tlenowych w temperaturze 20
0
C.
Zwi
ą
zki organiczne w wodach naturalnych mog
ą
by
ć
pochodzenia autogenicznego
(zanieczyszczenia naturalne – wymywane z gleb, produkty autotrofii, produkty przemiany
materii organizmów zwierz
ę
cych) oraz pochodzenia antropogenicznego (nieoczyszczone
ś
cieki). W przypadku silnego zanieczyszczenia wody du
żą
ilo
ś
ci
ą
zwi
ą
zków organicznych i
brakiem tlenu rozwijaj
ą
si
ę
mikroorganizmy beztlenowe powoduj
ą
ce anaerobowy rozkład
zwi
ą
zków organicznych (gnicie). Produktami rozkładu s
ą
siarkowodór, metan, amoniak i indol.
W warunkach tlenowych rozwijaj
ą
si
ę
natomiast organizmy aerobowe zdolne do rozkładu
zwi
ą
zków organicznych do dwutlenku w
ę
gla, wody, zwi
ą
zków azotowych, siarczanów i innych
produktów bezwonnych. Na pocz
ą
tku ulegaj
ą
biodegradacji zwi
ą
zki łatwo degradowane
(cukry proste, aminokwasy), pó
ź
niej za
ś
zwi
ą
zki zło
ż
one (cukry, lipidy, zwi
ą
zki humusowe).
Całkowity aerobowy rozkład zwi
ą
zków organicznych mo
ż
e trwa
ć
nawet 100 dni. Przebiega w
dwóch etapach. W pierwszym etapie rozkładowi ulegaj
ą
w
ę
glowodany (do 20 dni). Etap drugi
dotyczy rozkładu organicznych zwi
ą
zków azotu. Procesy biochemicznego utleniania
zwi
ą
zków organicznych najintensywniej w ci
ą
gu pierwszych 5 dni. W wodach
powierzchniowych w tym okresie uzyskuje si
ę
BZT na poziomie 68%. St
ą
d te
ż
przyj
ę
to w
praktyce okres 5 dni za wystarczaj
ą
cy do charakterystyki procesu biochemicznego rozkładu
zwi
ą
zków organicznych. Jest on opisany symbolem BZT
5
. Je
ż
eli BZT jest oznaczany w
okresie innym ni
ż
5 dni, to uwidacznia si
ę
to poprzez dodanie odpowiedniego indeksu
liczbowego, np. je
ż
eli oznaczano BZT dla okresu 20 dni to zapis wygl
ą
da nast
ę
puj
ą
co -
BZT
20
. Do prawidłowego przeprowadzenia procesu BZT oprócz tlenu mikroorganizmy
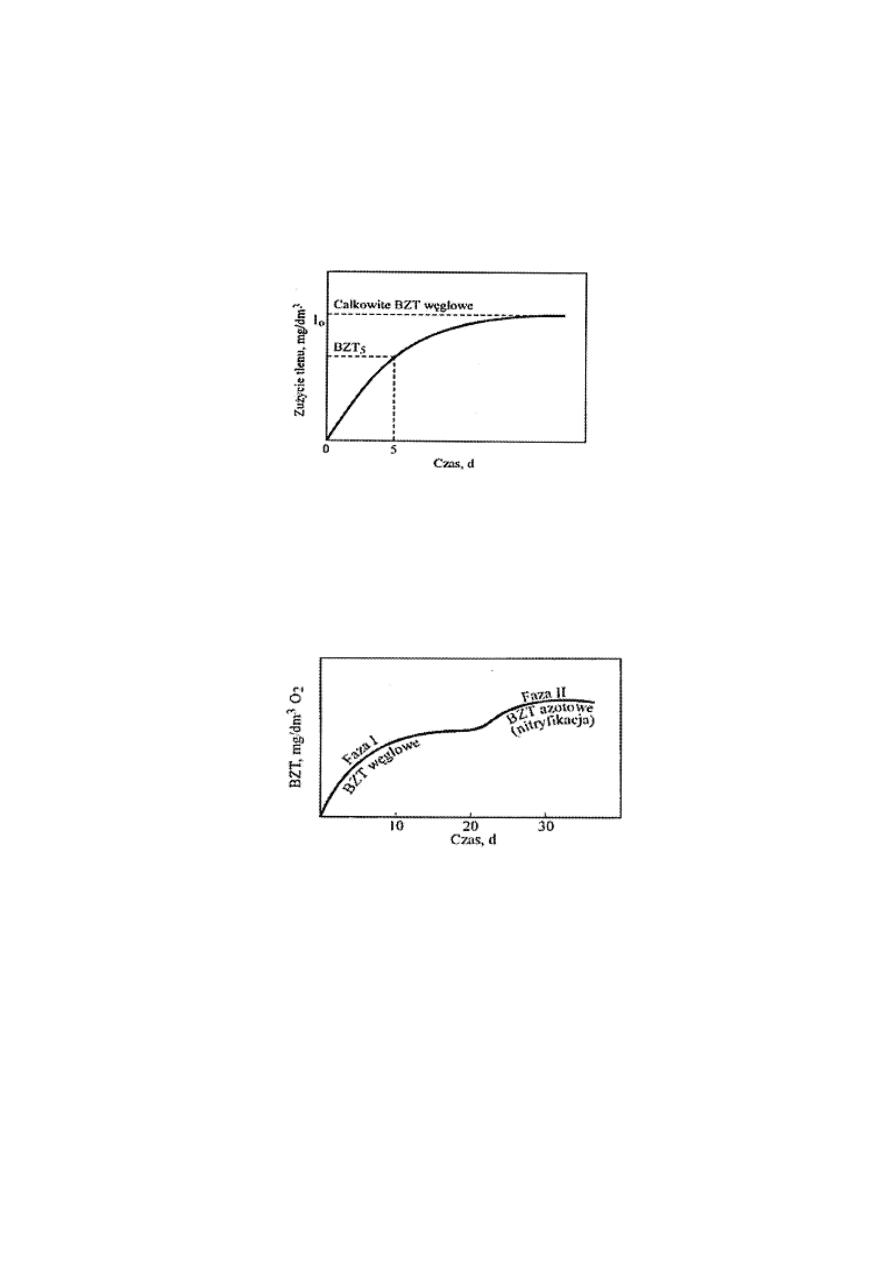
51
potrzebuj
ą
wielu substancji po
ż
ywkowych (głównie N i P), mikroelementów oraz
odpowiedniego pH (optymalne 7 – 8). Wiele z obecnych w wodzie lub
ś
ciekach zwi
ą
zków
mo
ż
e działa
ć
toksycznie na mikroby, skutkuj
ą
c opó
ź
nieniem w rozkładzie zwi
ą
zków
organicznych lub zahamowaniem procesu. Nale
żą
do nich metale ci
ęż
kie, pestycydy,
detergenty.
Krzywa przebiegu BZT
Rys. 5. Krzywa przebiegu BZT w
ę
glowego.
Podczas utleniania azotu anionowego nast
ę
puje zu
ż
ywanie tlenu. Przy badaniu procesu
BZT w warunkach laboratoryjnych obserwuje si
ę
na ogół dwie fazy :
- faz
ę
I - tzw. BZT w
ę
glowe
- faz
ę
II - tzw. BZT azotowe
.
Rys. 6. Krzywa BZT w
ę
glowego i azotowego.
W fazie I zachodzi zu
ż
ycie tlenu na rozkład zwi
ą
zków organicznych i powstanie amoniaku. W
fazie II zachodzi zu
ż
ycie tlenu na utlenienie amoniaku do azotanów (nitryfikacja). Je
ś
li woda
zawiera du
ż
e ilo
ś
ci amoniaku, procesy te mog
ą
si
ę
na siebie nakłada
ć
. Zu
ż
ycie tlenu na nitryfikacj
ę
w wodach mo
ż
na okre
ś
li
ć
, wykonuj
ą
c pomiary BZT wody z inhibitorem i bez inhibitora nitryfikacji.

52
Do oznaczania BZT w wodzie i
ś
ciekach wykorzystuje si
ę
nast
ę
puj
ą
ce metody:
- metod
ę
rozcie
ń
cze
ń
- metody manometryczne Sierpa i Warburga
- metody automatyczne (np. przy u
ż
yciu systemu OxiTop Control).
OZNACZENIE BZT METOD
Ą
ROZCIE
Ń
CZE
Ń
W przypadku próbek wody lub
ś
cieków bardziej zanieczyszczonych ilo
ść
tlenu w nich
zawarta jest niewystarczaj
ą
ca, dlatego stosuje si
ę
rozcie
ń
czanie wod
ą
, która jest specjalnie
przygotowana i stanowi
ź
ródło tlenu. Skład wody do rozcie
ń
cze
ń
: woda destylowana z
dodatkiem: buforu fosforanowego, siarczanu magnezu, chlorku wapnia i chlorku
ż
elaza. (pH
= 7,2). W celu maksymalnego wysycenia przygotowanej wody tlenem nale
ż
y przepuszcza
ć
przez ni
ą
powietrze minimum kilkana
ś
cie godzin. Odstawi
ć
w 20
0
C na 5-7 dni w celu
stabilizacji. Zawarto
ść
tlenu w tak przygotowanej wodzie powinna waha
ć
si
ę
w przedziale 8-9
mg0
2
/dm
3
. Skal
ę
rozcie
ń
czenia przygotowa
ć
zgodnie z tabel
ą
1. W celu oznaczenia BZT
napełni
ć
dwie butelki inkubacyjne badan
ą
rozcie
ń
czon
ą
próbk
ą
, zamkn
ąć
je korkami
szklanymi w taki sposób by nie pozostał p
ę
cherzyk powietrza. W jednej butelce oznaczy
ć
zawarto
ść
tlenu niezwłocznie po napełnieniu (metod
ą
Winklera), drug
ą
inkubowa
ć
w
temperaturze 20
0
C przez okres 5 dni, po czym oznaczy
ć
zawarto
ść
tlenu t
ą
sam
ą
metod
ą
.
Równolegle oznaczy
ć
zawarto
ść
tlenu w samej wodzie do rozcie
ń
cze
ń
(przed i po inkubacji).
Oznaczanie BZT w wodach zanieczyszczonych substancjami refrakcyjnymi oraz
zawieraj
ą
cych substancje toksyczne dla organizmów
ż
ywych (bior
ą
cych udział w
biodegradacji zanieczyszcze
ń
organicznych) jest mało przydatne w ocenie jako
ś
ci takiej
wody. W wodzie u
ż
ywanej do celów komunalnych warto
ść
BZT
5
nie powinna by
ć
wi
ę
ksza
ni
ż
4,0 gO
2
/m
3
.
SYSTEM OxiTop
Pomiary OxiTop
®
polegaj
ą
na pomiarach ci
ś
nienia w
zamkni
ę
tym systemie: mikroorganizmy znajduj
ą
ce si
ę
w
próbie zu
ż
ywaj
ą
tlen i produkuj
ą
przy tym CO
2
,absorbowany
przez NaOH. Powstaje podci
ś
nienie, które jako warto
ść
pomiarowa mo
ż
e by
ć
bezpo
ś
rednio odczytana w mg/l.
Obj
ę
to
ść
próby mo
ż
e by
ć
regulowana w zale
ż
no
ś
ci od ilo
ś
ci
tlenu do dyspozycji, aby móc przeprowadzi
ć
zupełny pomiar
BZT.
Dzi
ę
ki zró
ż
nicowanym obj
ę
to
ś
ciom mo
ż
na dokonywa
ć
pomiaru w zakresie pomiarowym do 4000 mg/l. Główki
OxiTop
®
(zielone i
ż
ółte) posiadaj
ą
funkcj
ę
AutoTemp - kiedy
temperatura próby jest za zimna, automatycznie nast
ę
puje
opó
ź
nienie pomiaru, dopóki temperatura nie ustabilizuje si
ę
, najcz
ęś
ciej trwa to 1 godzin
ę
.
Obok automatycznej pami
ę
ci 5 warto
ś
ci pomiarowych (1 warto
ść
na dzie
ń
) mo
ż
na
odczytywa
ć
warto
ś
ci pomiarowe r
ę
cznie zarówno w trakcie, jak równie
ż
po upływie 5 dni, co
usprawnia kontrol
ę
(www.wtw.pl).

53
Chemiczne zapotrzebowanie tlenu (ChZT)
Jest to ilo
ść
tlenu równowa
ż
na ilo
ś
ci utleniacza zu
ż
ytego do utlenienia substancji
zawartych w jednostce obj
ę
to
ś
ci badanej wody. W zale
ż
no
ś
ci od u
ż
ytego
ś
rodka
utleniaj
ą
cego rozró
ż
nia si
ę
zapotrzebowanie tlenu nadmanganianowe ChZT-Mn
(utlenialno
ść
, indeks nadmanganianowy) oraz dwuchromianowe ChZT-Cr
ChZT-Cr polega na utlenianiu zawartych w wodzie zwi
ą
zków organicznych i niektórych
nieorganicznych za pomoc
ą
mieszaniny utleniaj
ą
cej składaj
ą
cej si
ę
z K
2
Cr
2
O
7
i st
ęż
onego
H
2
SO
4
a nast
ę
pnie odmiareczkowaniu K
2
Cr
2
O
7
, który nie przereagował, za pomoc
ą
siarczanu
ż
elaza(II)amonu wobec ferroiny jako wska
ź
nika. Utlenianie prowadzi si
ę
przy
u
ż
yciu katalizatora Ag
2
SO
4
w zestawie destylacyjnym z chłodnica zwrotn
ą
utrzymuj
ą
c próbk
ę
w stanie wrzenia przez 10 minut.
W oznaczaniu ChZT utlenianie s
ą
głownie substancje organiczne i w ilo
ś
ci 95-100% warto
ś
ci
teoretycznej. Utlenieniu nie ulega jedynie benzen, toluen i pirydyna.
Utlenialno
ść
wody (U) – to umowny wska
ź
nik okre
ś
laj
ą
cy zdolno
ść
wody do pobierania
tlenu z KMnO
4
, w roztworze kwa
ś
nym lub alkalicznym wskutek utleniania si
ę
obecnych w
wodzie zwi
ą
zków organicznych a tak
ż
e niektórych łatwo utleniaj
ą
cych si
ę
zwi
ą
zków
nieorganicznych (np. sole
ż
elaza(II), azotyny, siarczki, chlorki i inne). Utlenialno
ść
jest
jedynie
wska
ź
nikiem
orientacyjnym,
gdy
ż
daje
jedynie
ogóln
ą
charakterystyk
ę
zanieczyszczenia próbki zwi
ą
zkami organicznymi. Oznaczona t
ą
metod
ą
ilo
ść
zwi
ą
zków
organicznych, zwłaszcza azotowych, stanowi jedynie 60 – 70 % rzeczywistego st
ęż
enia tych
substancji. Pełniejsz
ą
informacj
ę
na temat zawarto
ś
ci zwi
ą
zków organicznych w badanej
próbce daje oznaczenie ChZT-Cr (chemiczne zapotrzebowanie tlenu), gdy
ż
przy
zastosowaniu silniejszego utleniacza ulega utlenieniu wi
ę
cej zwi
ą
zków organicznych.
Pomimo, ze utlenialno
ść
jest jedynie wska
ź
nikiem orientacyjnym ilo
ść
zwi
ą
zków
organicznych zawartych w wodzie, jej warto
ść
ma du
ż
e znaczenie przy ocenie stopnia
zanieczyszczenia wód naturalnych zwi
ą
zkami organicznymi.
Przy wysokiej utlenialno
ś
ci wywołanej obecno
ś
ci
ą
zwi
ą
zków organicznych
pochodzenia zwierz
ę
cego woda dodatkowo wykazuje podwy
ż
szone st
ęż
enie chlorków i
zwi
ą
zków azotowych, oraz opalizacj
ę
przy niekoniecznie zwi
ę
kszonej barwie. Gdy
utlenialno
ść
wywołana jest przez organiczne zwi
ą
zki pochodzenia ro
ś
linnego, obok
podwy
ż
szonej utlenialno
ś
ci woda wykazuj
ę
podwy
ż
szon
ą
barw
ę
.
Z higienicznego punktu widzenia utlenialno
ść
wody ma istotne znaczenie wówczas,
gdy wywołana jest zwi
ą
zkami organicznymi pochodzenia zwierz
ę
cego, co sugeruje
zanieczyszczenie fekaliami i potencjalne zagro
ż
enie mikrobiologiczne.
Wody naturalne niezanieczyszczone maj
ą
utlenialno
ść
od ułamka do 2-3 mgO
2
/dm
3
(dotyczy to wód gł
ę
binowych i niezanieczyszczonych wód powierzchniowych). W przypadku
zawarto
ś
ci du
ż
ej ilo
ś
ci substancji humusowych – utlenialno
ść
mo
ż
e osi
ą
gn
ąć
warto
ść
nawet
do kilkuset mgO
2
/dm
3
. Utlenialno
ść
wody do picia nie powinna przekracza
ć
3 mgO
2
/dm
3
(wg
rozporz
ą
dzenia 5 mgO
2
/dm
3
). W przypadku wód zabarwionych utlenialno
ść
mo
ż
e by
ć
wy
ż
sza, pod warunkiem
ż
e inne cechy wody nie wskazuj
ą
na jej zanieczyszczenie.
Do oznaczania utlenialno
ś
ci stosuje si
ę
dwie metody w zale
ż
no
ś
ci od st
ęż
enia
chlorków w próbce analizowanej wody. Gdy st
ęż
enie chlorków nie przekracza 300 mg/dm
3
to
utlenialno
ść
oznacza si
ę
w
ś
rodowisku kwa
ś
nym, a gdy chlorki wyst
ę
puj
ą
w koncentracji
powy
ż
ej 300 mg/dm
3
to stosuje si
ę
metod
ę
opart
ą
na utlenianiu w
ś
rodowisku alkalicznym.
Odczyn wody (pH)
Odczyn wody jest wa
ż
nym wska
ź
nikiem składu chemicznego wody i ma du
ż
e
znaczenie w procesach uzdatniania wody, kontroli korozyjno
ś
ci wody w wodoci
ą
gach, a
przede wszystkim dla organizmu człowieka. Zwykle wody naturalne powierzchniowe
posiadaj
ą
pH w zakresie 6,5 – 8,5 co odpowiada wymaganiom stawianym wodzie do picia.

54
Obni
ż
enie odczynu dla tych wód jest spowodowane wprowadzeniem kwa
ś
nych
ś
cieków.
Wysokie warto
ś
ci pH s
ą
typowe po wprowadzeniu do wód powierzchniowych
ś
cieków
alkalicznych a tak
ż
e jest charakterystyczne dla zbiorników zeutrofizowanych. Na warto
ść
pH
wód podziemnych wpływa rodzaj podło
ż
a – przy kontakcie z warstwami kwarcowymi wody
maj
ą
odczyn kwa
ś
ny a gdy jest to podło
ż
e wapienne – odczyn zasadowy.
Do oznaczania pH wody wykorzystuje si
ę
metod
ę
elektrometryczn
ą
, której zasada
polega na pomiarze siły elektromotorycznej SEM ogniwa zło
ż
onego z elektrody pomiarowej i
elektrody odniesienia zanurzonych w badanym roztworze. Zmierzony potencjał jest
proporcjonalny do warto
ś
ci pH.
Istnieje
wiele
ró
ż
nych
konstrukcji
elektrod
pH-metrów.
Najbardziej
rozpowszechnione s
ą
jednak pH-metry ze zintegrowanymi elektrodami (wzorcow
ą
i pomiarow
ą
) w jednej, szklanej sondzie, o kształcie rurki. Układ ten jest zwykle
oparty na wzorcowym roztworze chlorku srebra i układzie elektrod wykonanych ze
srebra:
1. kulka z porowatego szkła, przez który mog
ą
swobodnie przenika
ć
jony
hydroniowe odpowiadaj
ą
ce za pH analizowanego roztworu
2. czasami na dnie kulki zbiera si
ę
nieco stałego chlorku srebra, co jest
zjawiskiem normalnym, nie wpływaj
ą
cym na czuło
ść
pomiaru
3. wewn
ę
trzny roztwór wzorcowy - zwykle 0,1 M HCl
4. elektroda pomiarowa - wykonana ze srebra
5. szklana obudowa całego układu elektrod
6. elektroda wzorcowa - wykonana ze srebra i zanurzona we wzorcowym roztworze
chlorku srebra
7. membrana ł
ą
cz
ą
ca roztwór wzorcowy z roztworem, którego pH si
ę
mierzy -
membrana ta jest zwykle wykonana z g
ę
stego spieku ceramicznego, który zapobiega
mieszaniu si
ę
obu roztworów ale zapewnia ich elektryczne poł
ą
czenie.
Literatura
1. Buczkowski R., Wybrane zagadnienia proekologiczne w chemii, Wydawnictwo
Uniwersytetu M. Kopernika w Toruniu, Toru
ń
2002.
2.
Cyga
ń
ski A., Chemiczne metody analizy ilo
ś
ciowej, WNT Warszawa 1999.
3.
Hermanowicz W., Dojlido J., Do
ż
a
ń
ska W., Kosiorowski B., Zerze J., Fizyczno-
chemiczne badanie wody i
ś
cieków, Arkady, Warszawa 1999.
4. Kiedry
ń
ska L., Papciak D., Granops M., Chemia sanitarna, Wydawnictwo SGGW,
Warszawa 2006.
5. Krzechowska M., Podstawy chemii ogólnej i
ś
rodowiska przyrodniczego.
Ć
wiczenia
laboratoryjne. Oficyna Wydawnicza Politechniki Warszawskiej, Warszawa 2007.
6. Pokojska U., Przewodnik metodyczny do analizy wód, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
1999.
7. Szłyk E., Kurzawa M., Szydłowska-Czerniak A., Jastrz
ę
bska A., Ilo
ś
ciowa analiza
chemiczna. Metody wagowe i miareczkowe, Wydawnictwo Uniwersytetu
M. Kopernika w Toruniu, Toru
ń
2005.
8. Szmal S., Lipiec T., Chemia analityczna z elementami analizy instrumentalnej
PZWL, 1997.

55
Celem
ć
wiczenia jest zapoznanie ze wska
ź
nikami charakteryzuj
ą
cymi fizyczne wła
ś
ciwo
ś
ci
wody (barwa, zapach, m
ę
tno
ść
, przewodnictwo wła
ś
ciwe) oraz ze wska
ź
nikami
charakteryzuj
ą
cymi zawarto
ść
zwi
ą
zków organicznych w wodzie (utlenialno
ść
. BZT, ChZT).
Zakres
ć
wiczenia obejmuje wykonanie oznaczenia utlenialno
ś
ci, barwy, zapachu,
przewodnictwa i pH badanej wody.
Uwaga: roztworów mianowanych pobranych z butelki do biurety lub zlewki a nie
wykorzystanych do analizy, NIE WOLNO wlewa
ć
z powrotem do butelki, w której s
ą
przechowywane.
A. Oznaczanie utlenialno
ś
ci (chemicznego zapotrzebowania na tlen metod
ą
nadmanganianiow
ą
w
ś
rodowisku kwa
ś
nym).
Zasada metody
Metoda oznaczania utlenialno
ś
ci polega na okre
ś
leniu ilo
ś
ci KMnO
4
(w przeliczeniu na tlen)
potrzebnej do utlenienia zwi
ą
zków organicznych i niektórych łatwo utleniaj
ą
cych si
ę
zwi
ą
zków nieorganicznych obecnych w zakwaszonej kwasem siarkowym próbce wody.
Manganian(VII) potasu utlenia zwi
ą
zki organiczne do prostych zwi
ą
zków wg reakcji:
5C
6
H
12
O
6
+ 24MnO
4
-
+ 72H
+
→
24Mn
2+
+ 10CO
2
+ 66H
2
O
Nadmiar manganianu(VII) potasu redukuje si
ę
dodanym w ilo
ś
ci równowa
ż
nej kwasem
szczawianem sodu, którego nadmiar odmiareczkowuje si
ę
KMnO
4
według reakcji:
2MnO
4
-
+ 5C
2
O
4
2-
+ 16H
+
→
2Mn
2+
+ 10CO
2
+ 8H
2
O
Odczynniki: 0,0125N KMnO
4
, kwas siarkowy 1+3, 0,0125 M Na
2
C
2
O
4
Szkło: kolby sto
ż
kowe poj. 200-300 ml, pipeta jednomiarowa 100 ml, pipety wielomiarowe,
biureta.
Sprz
ę
t: ła
ź
nia wodna
Wykonanie oznaczenia:
- do 3 kolb sto
ż
kowych o pojemno
ś
ci 200 – 300 ml pobra
ć
pipet
ą
jednomiarow
ą
po
100 ml otrzymanej do analizy próbki,
- do ka
ż
dej kolby doda
ć
pipet
ą
wielomiarow
ą
10 ml roztworu kwasu siarkowego(VI) (1+3)
i 10 ml roztworu 0,0125N manganianu(VII) potasu KMnO
4
,
- wymiesza
ć
zawarto
ść
kolby i ogrzewa
ć
we wrz
ą
cej ła
ź
ni wodnej przez 30 minut,
- po wyj
ę
ciu z ła
ź
ni natychmiast doda
ć
10 ml roztworu 0,0125N szczawianu sodu Na
2
C
2
O
4
i wymiesza
ć
,
- po odbarwieniu miareczkowa
ć
na gor
ą
co roztworem 0,0125N manganianu(VII) potasu
KMnO
4
do wyst
ą
pienia lekko ró
ż
owego zabarwienia utrzymuj
ą
cego si
ę
przez kilka minut,
Ć
wiczenie nr 7
WSKA
Ź
NIKI ZANIECZYSZCZENIA WODY. CHEMIA SANITARNA.
OZNACZANIE UTLENIALNO
Ś
CI, BARWY, ZAPACHU, PRZEWODNICTWA i pH - instrukcja.
56
po miareczkowaniu KMnO
4
- odczyta
ć
obj
ę
to
ść
zu
ż
ytego roztworu KMnO
4
na zmiareczkowanie próbki i obliczy
ć
utlenialno
ść
wg poni
ż
szego wzoru:
U =
1
200*
pr
V
V
[mg O
2
/dm
3
]
gdzie: V
1
– obj
ę
to
ść
zu
ż
ytego roztworu KMnO
4
na miareczkowanie próbki, ml
V
pr
– obj
ę
to
ść
próbki u
ż
ytej do analizy, 100 ml
UWAGA: jako wynik poda
ć
ś
redni
ą
arytmetyczn
ą
z 3 oznacze
ń
B. Oznaczanie barwy.
Zasada metody polega na wizualnym porównaniu barwy badanej próbki wody ze skal
ą
wzorców.
Odczynniki: skala wzorców (platynowo-kobaltowa) o warto
ś
ciach barwy: 0, 5, 10, 20, 30,
40, 50, 60, 70, 80, 90, 100 mg Pt/dm
3
Szkło: cylindry Nesslera o poj. 100 ml
Sprz
ę
t: statyw (komparator)
Materiał: badana woda
Wykonanie oznaczenia:
- otrzyman
ą
do analizy próbk
ą
napełni
ć
cylinder Nesslera o pojemno
ś
ci 100 ml,
- cylinder z badan
ą
próbk
ą
umie
ś
ci
ć
w statywie,
- po obu stronach badanej próbki ustawi
ć
wzorce o barwie najbardziej zbli
ż
onej do barwy
próbki,
- barw
ę
okre
ś
li
ć
patrz
ą
c z góry na słup cieczy,
- wynik oznaczenia poda
ć
w mg Pt/dm
3
Uwaga: je
ż
eli intensywno
ść
barwy badanej próbki wody przewy
ż
sza 100 mg Pt/dm
3
nale
ż
y
próbk
ę
rozcie
ń
czy
ć
– do cylindra Nesslera pobra
ć
50 ml badanej wody i uzupełni
ć
wod
ą
destylowan
ą
do obj
ę
to
ś
ci 100 ml. Próbk
ę
nale
ż
y rozcie
ń
cza
ć
do momentu uzyskania

57
intensywno
ś
ci barwy mieszcz
ą
cej si
ę
w skali wzorców. Przy obliczaniu barwy uwzgl
ę
dni
ć
krotno
ść
rozcie
ń
cze
ń
.
B =
100
pr
a
V
∗
[mg Pt/dm
3
]
gdzie: a – odczytana ze skali wzorców barwa badanej wody, mgPt/dm
3
V
pr
– obj
ę
to
ść
badanej wody u
ż
yta do analizy, ml
np. je
ż
eli próbka analizowanej wody była rozcie
ń
czana 2-krotnie to V
pr
= 25 ml
lub
B = 2
n
*a
[mg Pt/dm
3
]
gdzie: a – odczytana ze skali wzorców barwa badanej wody, mg Pt/dm
3
n – ilo
ść
rozcie
ń
cze
ń
C. Oznaczanie zapachu metod
ą
organoleptyczn
ą
bezpo
ś
redni
ą
.
Zasada metody polega na okre
ś
leniu za pomoc
ą
powonienia grupy zapachu (ro
ś
linny – R,
gnilny – G, specyficzny – S) i jego intensywno
ś
ci według sze
ś
ciostopniowej skali (0 – 5).
Intensywno
ść
zapachu oznacza si
ę
na zimno w temperaturze 18 -20
0
C i na gor
ą
co w
temperaturze 60
0
C.
Szkło: kolby sto
ż
kowe z doszlifowanym korkiem o poj. 500 ml, butelka inkubacyjna, cylinder
miarowy 100 ml, szkiełko zegarkowe
Sprz
ę
t: ła
ź
nia wodna
Materiał: badana woda
Wykonanie oznaczenia intensywno
ś
ci zapachu na zimno:
- do butelki inkubacyjnej o poj. 200 - 300 ml odmierzy
ć
cylindrem miarowym 100 ml
otrzymanej do analizy próbki wody
- otworzy
ć
butelk
ę
i natychmiast w
ą
cha
ć
zawarto
ść
przy wylocie szyjki,
- wynik oznaczenia poda
ć
w postaci symbolu zawieraj
ą
cego informacj
ę
o rodzaju
i intensywno
ś
ci zapachu oraz o warunkach przeprowadzania próby (na zimno, gor
ą
co,
temperatura wykonania oznaczenia).
Wykonanie oznaczenia intensywno
ś
ci zapachu na gor
ą
co:
- do kolby sto
ż
kowej z doszlifowanym korkiem o poj. 500 ml odmierzy
ć
cylindrem miarowym
100 ml otrzymanej do analizy próbki wody,
- kolb
ę
przykry
ć
szkiełkiem zegarkowym i umie
ś
ci
ć
w ła
ź
ni wodnej,
- ogrzewa
ć
do temperatury 60
0
C,
- po wyj
ę
ciu z ła
ź
ni kolb
ę
zamkn
ąć
korkiem i kilkakrotnie wstrz
ą
sn
ąć
,
58
- otworzy
ć
kolb
ę
i natychmiast w
ą
cha
ć
zawarto
ść
przy wylocie szyjki,
- wynik oznaczenia poda
ć
w postaci symbolu zawieraj
ą
cego informacj
ę
o rodzaju
i intensywno
ś
ci zapachu oraz o warunkach przeprowadzania próby -na zimno, gor
ą
co,
np. z 4R oznacza, ze zapach okre
ś
lano na zimno, jest to zapach silny i nale
ż
y do grupy
zapachów ro
ś
linnych. Dla grupy zapachów specyficznych nale
ż
y dodatkowo poda
ć
rodzaj
zapachu, np. z 1S (benzyna).
Tabela 13. Grupy i rodzaje zapachów.
Grupa zapachu
Oznaczenie
Rodzaj zapachu
roślinny
R
ziemisty, trawiasty, kwiatowy, rybny, aromatyczny itp.
gnilny
G
stęchły, pleśni, feralny, siarkowodoru
specyficzny
S
fenolu, benzyny, chloru, nafty, smoły, lakieru
Tabela 14. Skala intensywno
ś
ci zapachów.
Intensywność
zapachu
Wyczuwalność zapachu i jego znaczenie
0
brak zapachu
1
zapach bardzo słaby
2
zapach słaby
3
zapach wyraźny
4
zapach silny – dyskwalifikuje wodę do celów bytowo-gospodarczych
5
zapach bardzo silny – dyskwalifikuje wodę do celów bytowo-
gospodarczych
D. Oznaczanie przewodno
ś
ci elektrolitycznej metod
ą
konduktometryczn
ą
.
Zasada metody
Przewodno
ść
elektrolityczna roztworu okre
ś
la jego zdolno
ść
do przewodzenia pr
ą
du
elektrycznego. Przeno
ś
nikiem pr
ą
du w roztworze s
ą
jony. Wielko
ść
przewodno
ś
ci jest
proporcjonalna do st
ęż
enia substancji mineralnych wyst
ę
puj
ą
cych w postaci jonowej.
Poniewa
ż
wielko
ść
przewodno
ś
ci elektrolitycznej zale
ż
y od sposobu pomiaru, przyj
ę
to
posługiwa
ć
si
ę
ś
ci
ś
le zdefiniowan
ą
przewodno
ś
ci
ą
elektrolityczn
ą
wła
ś
ciw
ą
, która odnosi si
ę
do słupa roztworu o przekroju 1 cm
2
i wysoko
ś
ci 1 cm. Jednostk
ą
przewodno
ś
ci jest simens
(odwrotno
ść
oma), symbol S. Przewodno
ść
elektrolityczn
ą
wła
ś
ciw
ą
wody podaje si
ę
zwykle
w mS/cm lub przy niskiej mineralizacji
µ
S/cm.
Szkło: zlewki poj. 25 ml
Sprz
ę
t: konduktometr, czujnik konduktometryczny, tryskawka
Materiał: badana woda
Wykonanie oznaczenia:
Badan
ą
próbk
ę
wody wla
ć
do zlewki. Elektrod
ę
konduktometru wyj
ąć
z kolby, opłuka
ć
wod
ą
destylowan
ą
z tryskawki, osuszy
ć
bibuł
ą
. Nast
ę
pnie według instrukcji obsługi konduktometru
zmierzy
ć
przewodno
ść
elektrolityczn
ą
wła
ś
ciw
ą
i odczyta
ć
warto
ść
temperatury. Po

59
wykonanym pomiarze dokładnie opłuka
ć
elektrod
ę
konduktometru wod
ą
destylowan
ą
z
tryskawki i osuszy
ć
kawałkiem bibuły. Nast
ę
pnie elektrod
ę
odstawi
ć
do kolby. Badanie
wykona
ć
w trzech powtórzeniach.
Uwaga: wynik pomiaru zale
ż
y od temperatury, w zwi
ą
zku z czym nale
ż
y zaznaczy
ć
, jakiej
temperatury dotyczy (np. zapis EC
20
oznacza przewodno
ść
w temperaturze 20
0
C). Jako
standardow
ą
temperatur
ę
przyjmuje si
ę
25
0
C.
Co pewien czas nale
ż
y przeprowadzi
ć
kalibracj
ę
czujnika konduktometrycznego stosuj
ą
c w
tym celu roztwór o znanej przewodno
ś
ci elektrolitycznej (najcz
ęś
ciej jest to 0,01 M KCl i EC
25
= 1,412 mS/cm. Po dłu
ż
szej serii pomiarów zaleca si
ę
przepłuka
ć
elektrody konduktometru
2% HCl.
E. Oznaczanie odczynu (pH) roztworu metod
ą
potencjometryczn
ą
.
Zasada metody
Oznaczanie pH roztworu metod
ą
potencjometryczn
ą
polega na pomiarze siły
elektromotorycznej ogniwa zło
ż
onego z elektrody pomiarowej i elektrody odniesienia, które
zanurzone s
ą
w badanej próbce. Pomiar pH powinien by
ć
wykonany bezpo
ś
rednio po
pobraniu próbki wody.
Szkło: zlewki o poj. 25 ml
Sprz
ę
t: pehametr laboratoryjny, elektroda zespolona do pomiaru pH, tryskawka z wod
ą
destylowan
ą
, bibuła do osuszania elektrody.
Materiał: badana woda
Wykonanie oznaczenia:
Elektrod
ę
pehametru wyj
ąć
z kolby, dokładnie opłuka
ć
wod
ą
destylowan
ą
z tryskawki,
osuszy
ć
bibuł
ą
. Do zlewki przela
ć
odpowiedni
ą
ilo
ść
badanej wody, tak aby elektroda
zespolona był
ą
w nim zanurzona powy
ż
ej punktu kontaktu. Uruchomi
ć
pehametr zgodnie z
instrukcj
ą
obsługi. Odczyta
ć
wskazania pehametru - warto
ść
pH i temperatur
ę
. Po
wykonanym pomiarze dokładnie opłuka
ć
elektrod
ę
pehametru wod
ą
destylowan
ą
z tryskawki
i osuszy
ć
kawałkiem bibuły. Nast
ę
pnie elektrod
ę
odstawi
ć
do kolby. Pomiar pH wykona
ć
w
trzech powtórzeniach.
Wyniki otrzymane z oznacze
ń
zawarto
ś
ci utlenialno
ś
ci, barwy, zapachu, przewodnictwa i pH
wpisa
ć
do tabeli.
Sprawozdanie z
ć
wiczenia powinno zawiera
ć
:
- stron
ę
tytułow
ą
- cel i zakres
ć
wiczenia
- opis wykonania
ć
wiczenia (zasada oznaczenia, odczynniki, szkło, sprz
ę
t, materiały,
wykonanie
ć
wiczenia)
- otrzymane wyniki (tabela podpisana przez prowadz
ą
cego
ć
wiczenia)
- wnioski – odnie
ść
si
ę
do aktualnych przepisów prawnych dotycz
ą
cych jako
ś
ci wody
przeznaczonej do spo
ż
ycia przez ludzi.
60
POLITECHNIKA LUBELSKA
Wydział In
ż
ynierii
Ś
rodowiska
In
ż
ynieria
Ś
rodowiska
SPRAWOZDANIE
Z
Ć
WICZE
Ń
LABORATORYJNYCH Z CHEMII
Nr
ć
wiczenia
7
Temat
ć
wiczenia
Wska
ź
niki zanieczyszczenia wody.
Chemia sanitarna.
Oznaczanie barwy, zapachu, pH, przewodnictwa
wła
ś
ciwego i utlenialno
ś
ci wody.
Imi
ę
i nazwisko studenta
……………………………………………
……………………………………………
……………………………………………
Rok studiów
Semestr
Data
Imi
ę
i nazwisko prowadz
ą
cego
ć
wiczenia
Uwagi prowadz
ą
cego:
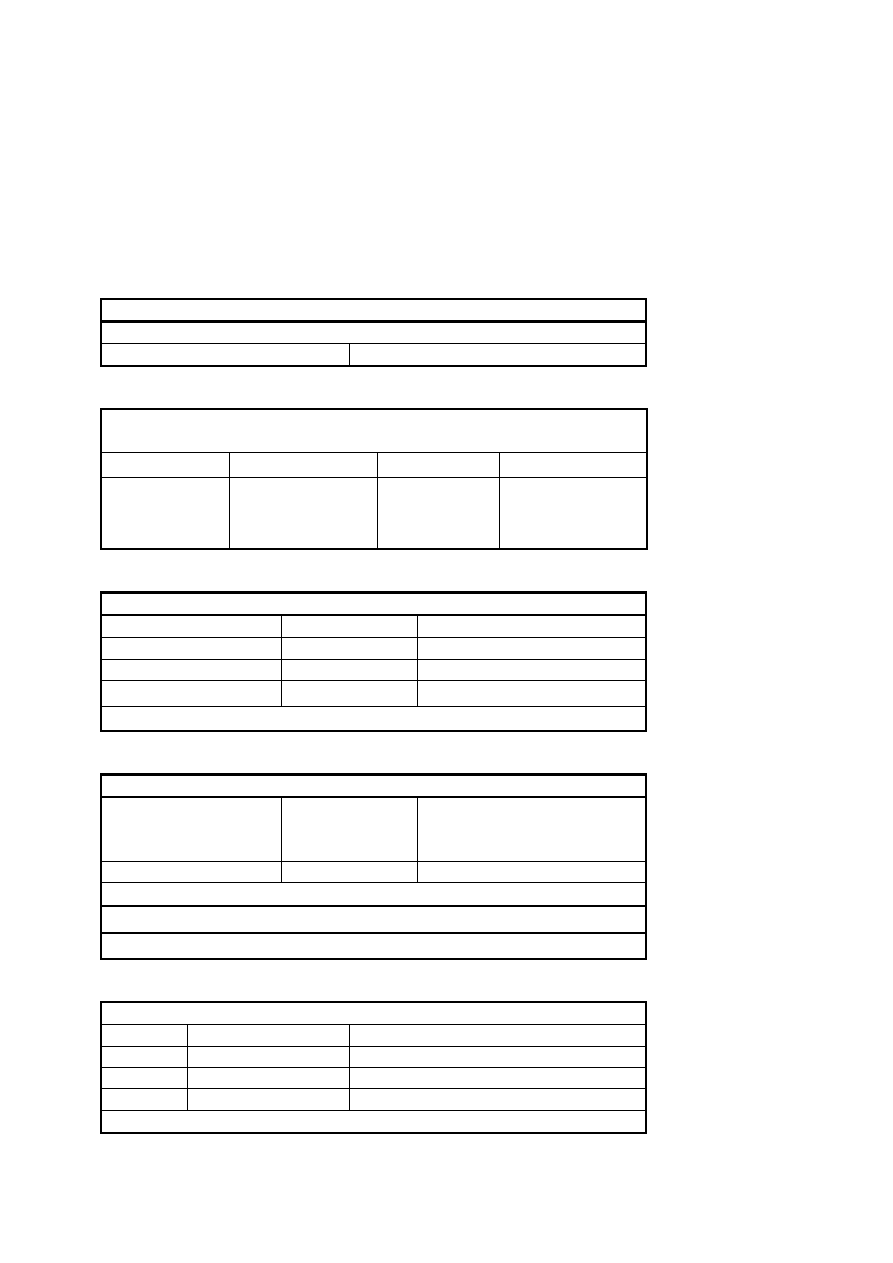
61
Wskaźniki zanieczyszczenia wody
...................................................
………………………………..
………………………………..
Imię i Nazwisko
Grupa BDi .............
Data.......................
Oznaczanie barwy
Barwa = ....................[mgPt/dm
3
]
Ilość rozcieńczeń
Oznaczanie zapachu
Na zimno/ gorąco
Grupa zapachu
Rodzaj zapachu
Intensywność
.......................
.............................
...........................
………………………
Oznaczanie pH
Nr pomiaru
Wartość pH
Temperatura pomiaru [
o
C]
1
2
3
pH =
Oznaczanie przewodnictwa
Nr pomiaru
Wartość
przewodnictwa
[S/cm]
Temperatura pomiaru [
o
C]
1
κκκκ
= ................. S/cm
Zawartość kationów i anionów
Z = ................. [mval/dm
3
]
Oznaczanie utlenialności
Nr próbki
V
1
[ml]
U [mg O
2
/dm
3
]
1
2
3
U =
......................
[mg O
2
/dm
3
]
Wyszukiwarka
Podobne podstrony:
W19 kompleksonometria, wska«niki i krzywe miareczkowania kompleks i
Analizowanie procesow technolog Nieznany (2)
ANALIZA MIARECZKOWA
analizy 2 id 62051 Nieznany
analiza 6 1 id 584986 Nieznany (2)
1d analiza interasariuszy, pro Nieznany
Lab 03 Analiza obwodu elektrycz Nieznany
Cw 5 10 Analiza tolerancji i od Nieznany
Analiza algorytmow ukrywania w Nieznany
analiza 3 id 59700 Nieznany (2)
,analiza matematyczna 2, elemen Nieznany (2)
1 Analiza kinematyczna manipula Nieznany (2)
Analiza cyklu koniunkturalnego Nieznany
Analizowanie dzialania ukladow Nieznany
06 Analizowanie ukladow elektry Nieznany (2)
więcej podobnych podstron