
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 1
I.
Patogeneza chorób genetycznych
Patogeneza schorzeń genetycznych wiąże się ze zmianami jakie zachodzą w DNA, a więc z mutacjami.
Czynnikami istotnymi dla zrozumienia patogenezy poszczególnych chorób są:
• rodzaj mutacji na poziomie DNA np. delecja, mutacja z przesunięciem ramki odczytu, mutacja zmiany sensu
• znajomość uszkodzonego produktu genowego – białka lub RNA
• znaczenie funkcjonalne mutacji np. utrata lub zmiana funkcji białka albo ekspresja na innym poziomie
• znajomość efektu fenotypowego dla danego allela – dominujący lub recesywny
• rodzaj produktu genowego – enzym, receptor, białka transportujące, białko strukturalne, czynnik
transkrypcji itp. oraz sposób jego funkcjonowania w systemie biologicznym
Postęp w dziedzinie biologii molekularnej zachodzący w sposób dynamiczny od lat 80 przyczynił się znacząco do
postępu w sferze diagnostyki schorzeń genetycznych. Umożliwił on przede wszystkim mapowanie i identyfikacje
genów odpowiedzialnych za wrodzone zaburzenia monogenowe i nowotwory komórek somatycznych.
1.
Mutacja
- stała dziedziczna zmiana w DNA, a więc zmiana pierwotnej sekwencji nukleotydów lub struktury i
liczby chromosomów. Zmiany te mogą być bardzo duże np. delecja, duplikacja lub przegrupowanie całego
chromosomu lub jego części (tzw. aberracje chromosomowe), mogą być to również zmiany mniejsze dotyczące
tylko drobnych fragmentów DNA (delecje, duplikacje), często dotyczą także jednego nukleotydu (mutacje
punktowe). Wszystkie wymienione wyżej zmiany mogą być przyczyną chorób. W związku z powyższymi
wyróżniamy:
• mutacje genowe (nukleotydowe)
• mutacje chromosomowe (aberracje chromosomowe)
a) Przyczyny powstawania mutacji dzielimy na:
• spontaniczne – samoistne, najczęściej jest do błąd polimerazy DNA
• indukowane – powodowane przez czynniki mutagenne
b) Mutacje w komórkach rozrodczych są przekazywane potomstwu, a mutacje w komórkach somatycznych
prowadzą czasami do nowotworów, lub do starzenia się komórek.
2. Najczęstsze mechanizmy mutacji w chorobach genetycznych:
1) Duże mutacje
Ø
delecja – utrata określonej liczby nukleotydów
• całego genu – np. większość mutacji w α-talasemii
• części genu - delecja trójki nukleotydów powoduje brak jednego aminokwasu w łańcuchu, delecja liczby
nukleotydów nie będącej wielokrotnością liczby 3 prowadzi do przesunięcia ramki odczytu i zmiany wszystkich
kodonów od miejsca delecji począwszy. Np. 60% mutacji w dystrofii mięśniowej typu Duchenne’a oraz
70% mutacji w dystrofii mięśniowej typu Beckera
Ø
insercja – wstawienie krótkiej sekwencji nukleotydów w obrębie genu, insercja trójki nukleotydów
powoduje powstanie dodatkowego aminokwasu w łańcuchu. Insercja liczby nukleotydów nie będącej
wielokrotnością liczby 3 prowadzi do przesunięcia ramki odczytu i zmiany wszystkich kodonów od
miejsca insercji począwszy. Np. mutacja będąca przyczyną hemofilii A
Ø
translokacja - przeniesienie odcinków między niehomologicznymi chromosomami np. dystrofia
mięśniowa typu Duchenne’a z translokacją chromosom X/autosom
Ø
inwersja - odwrócenie fragmentu chromosomu o 180 stopni np. w patogenezie hemofilii A (inwersja
genu F8)
Ø
duplikacja - powielenie odcinka chromosomu np. w patogenezie dystrofii mięśniowej typu Duchenne’a
Ø
efekt dawki genu – np. choroba Charcot-Marie-Tooth (neurogenny zanik mięśni)
Ø
mutacje dynamiczne (niestabilne sekwencje tripletów) – mimo tego iż w zasadzie tradycyjne określenie
mutacji jako stałej zmiany w DNA jest słuszne, bywają też mutacje zmienne, czego przykładem są mutacje
dynamiczne, charakteryzujące się obecnością niestabilnych, powtarzających się sekwencji
trinukleotydowych mogących występować w regionach niepodlegających translacji 5’, kodujących
(egzonach) lub nie podlegających translacji 3’. Najczęściej im większe jest zwielokrotnienie tripletów, tym
wcześniejszy jest początek i cięższy przebieg choroby (antycypacja). Przykładami chorób, których
patomechanizm polega na mutacji dynamicznej w obrębie określonych genów są: choroba Huntingtona,
dystrofia miotoniczna, zespół łamliwego chromosomu X, niezborność opuszkowo – rdzeniowa typu I

Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 2
2) Mutacje punktowe – polegające na zmianie jednej pary zasad na inną, mogą one występować:
a) Na poziomie DNA:
Ø
tranzycja
Ø
transwersja
Ø
delecja
Ø
insercja
b) Na poziomie powstającego polipeptydu
Ø
mutacja przesunięcia ramki odczytu - na skutek delecji lub insercji jednej pary nukleotydów,
powodujących zmianę ramki odczytu podczas translacji. Prowadzi to do zasadniczych zmian sekwencji
aminokwasów w tworzonym białku, a często wiąże się z całkowitym przerwaniem jego syntezy. W
niektórych przypadkach w związku z lokalizacją mutacji dochodzi do zaburzeń transkrypcji lub
składania mRNA
Ø
mutacja milcząca – aminokwas nie ulega zmianie, ponieważ zamiana zasady doprowadziła do
powstania innego kodonu synonimowego, wyznaczającego ten sam aminokwas
Ø
mutacja nonsensowna – zmiana zasady prowadzi do powstania kodu stop np. w patogenezie
mukowiscydozy
Ø
mutacja zmiany sensu – dochodzi do zamiany na kodon, który wyznacza inny aminokwas, nie zmienia
się jednak cała sekwencja aminokwasowa tworzonego białka np. anemia sierpowata
* w niektórych chorobach występuje wiele mutacji punktowych np. w przebiegu β-talasemii występuje
mutacja nonsensowna, składania mRNA, mutacje promotora i delecje.
3. Mutacje chromosomowe
Każdy gatunek ma określoną liczbę i skład chromosomów określanych mianem kariotypu. Komórki
somatyczne posiadają diploidalną liczbę chromosomów – 46, gamety natomiast są haploidalne i posiadają ich o
połowę mniej – 23 chromosomy. Chromosomy dzielimy na autosomy oznaczone numerami 1-22 oraz na
chromosomy płciowe – dwa chromosomy X u kobiet (kariotyp – 46, XX), oraz chromosomy X i Y u mężczyzn
(kariotyp 46, XY).
a) Geny dziedziczone od ojca i od matki znajdują swoją ekspresję u potomstwa. Wyjątek stanowi
zjawisko znane jako rodzicielskie piętnowanie genomu.
b) Rodzicielskie piętnowanie genomu – w przypadkach tych zachodzi ekspresja tylko jednej kopii genu,
wyłącznie od matki lub ojca. Mechanizm tego zjawisko ma związek z metylacją powodującą
inaktywację DNA. Piętnowanie odgrywa ważną rolę w ujawnianiu się niektórych chorób genetycznych,
najczęściej dochodzi do niego w obrębie chromosomów 6, 7, 14 i 15. Jest to sposób zapobiegania
homozygotyczności, poprzez regulację ekspresji genów zaangażowanych w rozwój i różnicowanie zarodka.
Klasyczny przykład stanowi zespół Pradera – Willego i zespół Angelmana. Obie choroby powoduje
delecja fragmentu chromosomu 15
• w zespole Pradera – Willego stwierdza się brak fragmentu chromosomu 15 pochodzenia ojcowskiego
• w zespole Angelmana stwierdza się brak fragmentu chromosomu 15 pochodzenia matczynego
Oba zespoły mogą być również spowodowane disomią chromosomową (jednorodzicielska) – w zespole
Pradera-Willego obie kopie chromosomu 15 pochodzą od
matki a w zespole Angelmana od ojca.
Znaczenie piętnowania genomu widać na przykładzie
zaśniadu groniastego, w którym stwierdza się prawidłowy
kariotyp 46, XY jednak cały zestaw chromosomów jest
pochodzenia ojcowskiego wskutek podwojenia ojcowskiego
haploidalnego zestawu chromosomów.
c) Lyonizacja – jest przykładem innego zjawiska związanego z
ekspresją u potomstwa genów dziedziczonych od obojga
rodziców. Polega on na inaktywacji chromosomu X w
komórkach somatycznych kobiet. Kobieta ma dwa
chromosomy X, ale tylko jedna kopia podlega ekspresji w
komórkach somatycznych. Jeden z chromosomów X ulega
inaktywacji i jest to zjawisko losowe. Nieaktywny
chromosom X nie podlega transkrypcji i dzięki temu
dochodzi do syntezy produktów białkowych kodowanych
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 3
przez tylko jeden aktywny chromosom X. Nieaktywny chromosom X może być widoczny w jądrze
komórkowym w postaci ciałka Barra (chromatyny płciowej)
Ø
liczenie ciałek Barra jest podstawowym testem chromatyny płciowej do określania aneuplodii
chromosomów płciowych
Ø
zachodzi we wczesnej embriogenezie
Ø
ma na celu zrównoważenie ilości aktywnych genów występujących w komórkach męskich.
d) Aberracje chromosomowe – mutacje dotyczące całych chromosomów lub ich części, występują w 8%
wszystkich poczęć i większość z nich ulega eliminacji przez samoistne poronienia, u noworodków
występują dużo rzadziej (ok. 0,6%). Aberracje dzielimy na strukturalne i liczbowa (aneuploidia)
e) Konwersja genów – zmiana sekwencji nukleotydów w zrekombinowanych cząsteczkach, wynikająca z
niedopasowania, które mogło powstać podczas rekombinacji.
f) Aberracje strukturalne – powstają w wyniku pęknięć chromosomu, wyróżniamy wśród nich:
Ø
translokacja – polega na wymianie materiału między dwoma chromosomami, rzadziej między trzema.
Zazwyczaj translokacja ma charakter zrównoważony tj. bez utraty lub nadmiaru DNA. Nosiciel takiej
aberracji jest klinicznie zdrowy, problemy mogą pojawić się u potomstwa, u którego translokacja może
wystąpić w postaci niezrównoważonej. Wśród translokacji wyróżniamy:
• translokacja wymienna – dotyczy jakiejkolwiek
pary chromosomów homologicznych lub
niehomologicznych
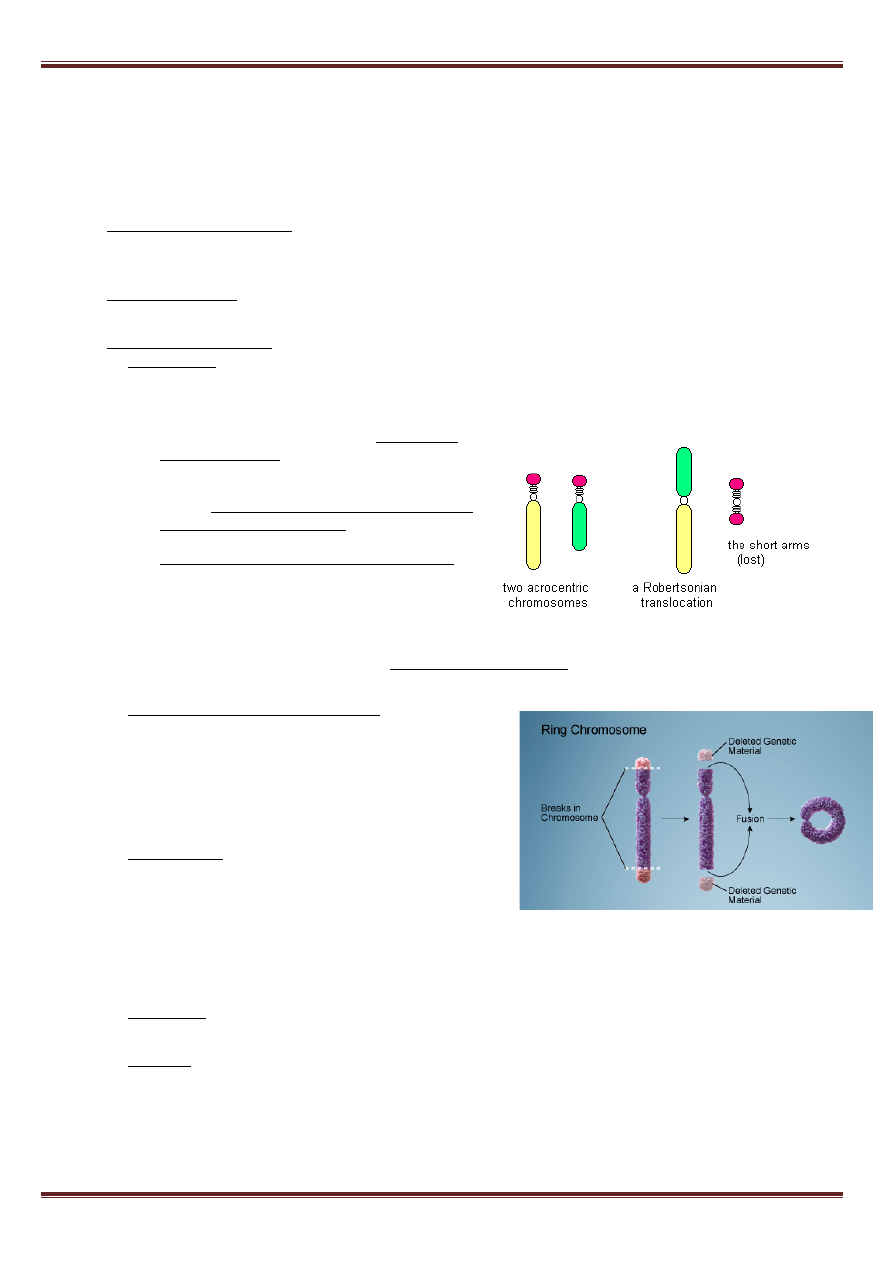
• translokacja robertsonowska (fuzja centryczna) –
dotyczy chromosomów akrocentrycznych, wynika z
pęknięć w pobliżu centromeru. W większości
przypadków powstaje wówczas pojedynczy
chromosom dicentryczny i fragment acentryczny.
Fuzja centryczna wynika często z przypadkowej
wymiany między homologicznymi sekwencjami na
różnych chromosomach podczas mejozy (crossing-
over). Najczęściej dochodzi do fuzji chromosomów
13 i 14, ponieważ fragment centryczny gubi się, powstaje 45 chromosomów.
• translokacja insercyjna – występują trzy pęknięcia chromosomów; następuje delecja interstycjalna w jednym
chromosomie i włączenie go (insercja) w miejsce pęknięcia w drugim chromosomie
Ø
Delecja i chromosom pierścieniowy – delecja oznacza
utratę części chromosomu, chromosom pierścieniowy
natomiast powstaje po pęknięciu obu ramion
chromosomu, jego lepkie końce proksymalne łączą
się, tworząc pierścień a terminalnie ulegając delecji.
Delecjom w obrębie autosomów mogą towarzyszyć
liczne wady wrodzone i upośledzenia umysłowe.
Ø
Mikrodelecja – są to utraty bardzo małych odcinków
chromosomów, prowadzące jednak do powstania
chorób takich jak:
• zespół Hirschhorna
• zespół miauczenia kota (cri du chat)
• zespół Williams
• zespół Pradera-Willego
• zespół Angelmana
Ø
Duplikacja – jest to obecność dwóch końców kopii odcinka chromosomu, zaburzenia chorobowe,
które może powodować, są zwykle nasilone w słabszym stopniu niż w przypadku delecji
Ø
Inwersja – jest wynikiem dwóch pęknięć chromosomu i odwrócenia odcinka między nimi o 180°.
Inwersja może być:
• paracentryczna (pęknięcia znajdują się na jednym ramieniu)
• pericentryczna (inwersją objęty jest również centromer).
Inwersja zwykle nie powoduje objawów klinicznych, ale wiąże się z ryzykiem niezrównoważenia
chromosomowego u potomstwa. Najczęściej obserwowaną inwersją jest inwersja para centryczna
chromosomu 9, która nie wywołuje zaburzeń chromosomowych u potomstwa.
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 4
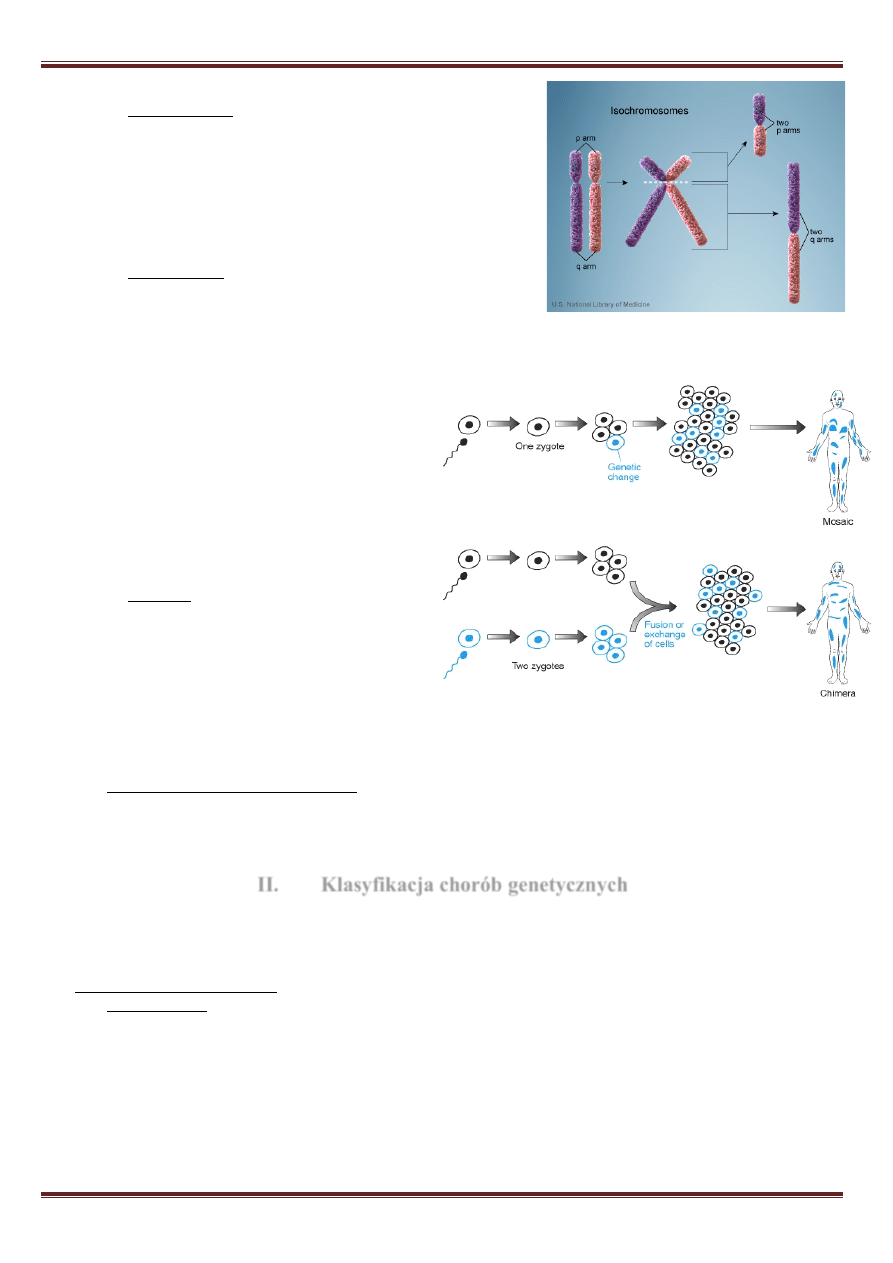
Ø
Izochromosom – nieprawidłowy chromosom z delecją
jednego ramienia i duplikacją drugiego. Może powstać w
wyniku poprzecznego pęknięcia centromeru. Najczęściej
spotyka się izochromosom długich ramion chromosomu X,
powoduje to wystąpienia zespołu Turnera ze względu na
delecję ramienia p. Izochromosomy autosomów obejmują
zwykle krótkie ramiona chromosomu 9 i 12. Delecji ulegają
ramiona krótkie chromosomów.
Ø
Mozaikowość - występowanie u jednego osobnika dwóch
lub więcej linii komórkowych pochodzących od jednej
zygoty. Różnorodność linii komórkowych może być
przyczyną powstawania charakterystycznego układu pigmentacji skóry, najczęściej na plecach.
Prawdopodobnie mozaikowość może też wiązać się z zaburzeniami pigmentacji tęczówki. Najczęściej
występującą tego typu mutacją jest mozaikowatość u osób z zespołem Downa, a więc trisomią 21
chromosomu, dotyczy ona 1-2% chorych. W
ich linii trisomicznej występuje linia
komórek prawidłowych, co powoduje, że u
osób tych choroba daje mniejsze objawy.
Znane są ponadto przypadki występowania
linii nieprawidłowych komórek wyłącznie w
gonadach (mozaikowość gonad), wówczas
ryzyko posiadania chorego potomstwa jest
duże, pomimo prawidłowego kariotypu w
limfocytach krwi obwodowej obu rodziców.
Ø
Chimera – mutacja, w której u jednej osoby
występują dwie linie komórkowe,
zazwyczaj prawidłowe, pochodzące z
dwóch oddzielnych zygot. Wśród zwierząt,
chimery mogą powstać np. wskutek
podwójnego zapłodnienia komórki jajowej,
połączenia zarodków lub częściowej
wymiany komórek pomiędzy zarodkami. Powstały osobnik ma niektóre narządy zbudowane z komórek o
innym składzie chromosomów niż reszta jego organizmu.
g)
Aberracje liczbowe (aneuplodia)
– jest to występowanie dodatkowych chromosomów w kariotypie
człowieka, lub całkowity brak, niektórych z nich. Wyróżniamy wśród nich: monosomię, disomię
(prawidłowa w komórkach somatycznych), trisomię. W zależności od lokalizacji dodatkowego
chromosomu, bądź jego braku, wyróżnia się różne aberracje liczbowe.
II.
Klasyfikacja chorób genetycznych
Częstość występowania chorób genetycznych jest stosunkowo wysoka, mniej więcej około 5% osób poniżej 25 r.ż.
może mieć objawy chorobowe związane z komponentem genetycznym. W późniejszym wieku odsetek ten
gwałtownie wzrasta nawet do 60% uwzględniając, że nadciśnienie tętnicze, choroba wieńcowa czy cukrzyca są
zaburzeniami wieloczynnikowymi. Wyróżniamy następujące klasy chorób genetycznych:
1.
Aberracje chromosomowe
, opisane powyżej, zaliczamy do nich takie schorzenia jak:
a) Zespół Downa (trisomia 21 chromosomu) - spowodowana nondysjunkcją chromosomów w
podziale mejotycznym, taki sposób powstawania trisomii dotyczy 90% zaistnienia choroby. Innymi
możliwościami są:
• mozaikowatość (por. wyżej)
• translokacja robertsonowka (długie ramię chromosomu 21 jest przyłączone do innego chromosomu, często jest
to chromosom 14)
• duplikacja fragmentu chromosomu 21
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 5
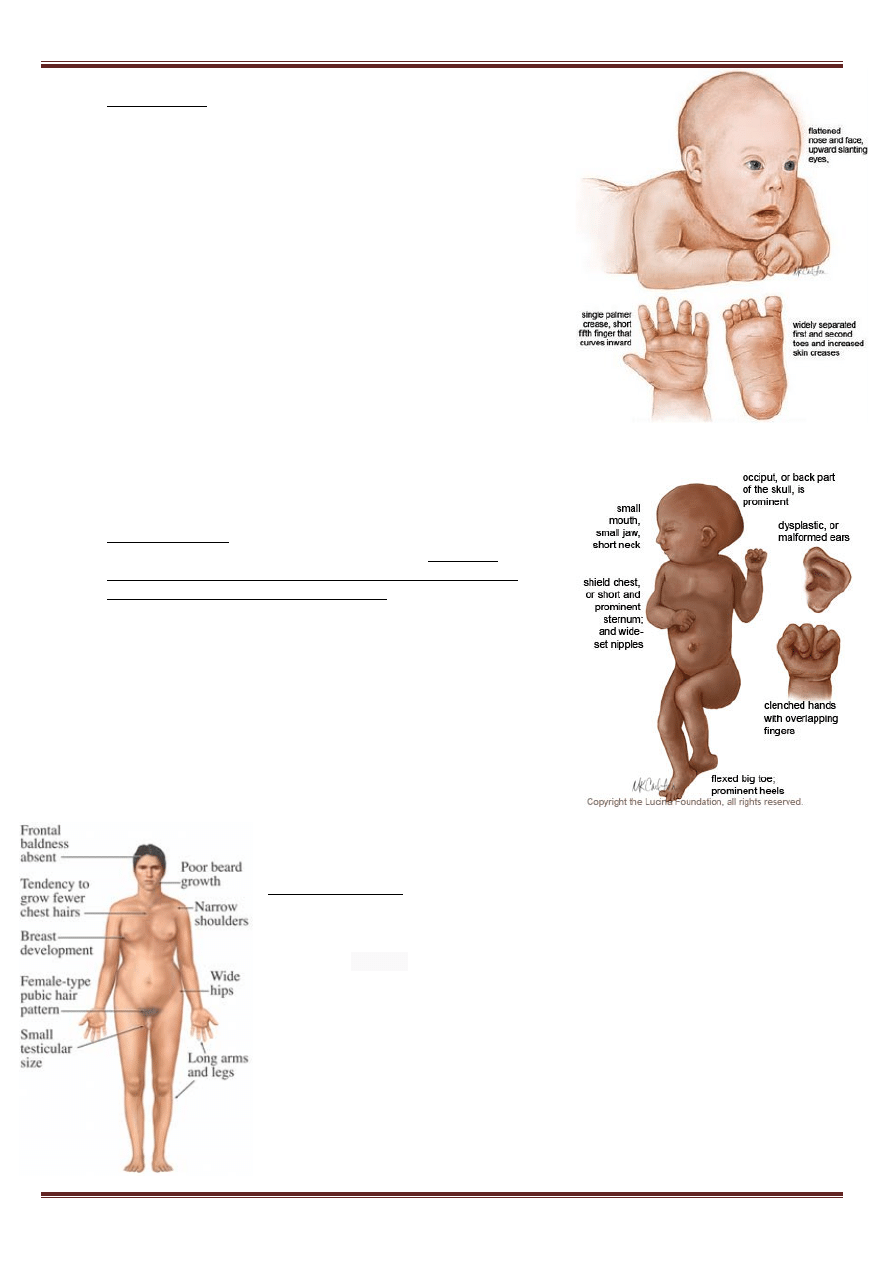
Zespół Downa występuje i 1/700 żywo urodzonych dzieci. Osoby z
zespołem Downa charakteryzują się:
• krótkogłowie, umiarkowane małogłowie, spłaszczona potylica
• płaski profil twarzy
• mongoloidalne ustawienie szpar powiekowych
• zmarszczka nakątna
• niewielki hiperteloryzm oczny
• jasne plamki na tęczówce (zwane plamkami Brushfielda)
• zaburzenia refrakcji, najczęściej krótkowzroczność (70%)
• oczopląs (35%)
• zez (45%)
• mały nos z płaską nasadą i szerokim grzbietem
• nisko osadzone, małe małżowiny uszne, często mały lub nieobecny płatek
• zaburzenia słuchu (66%)
• pojedyncza bruzda dłoni, nazwana też bruzdą małpią (45%)
• zwiększony odstęp między paluchem a drugim palcem stopy
• zwiększona ilość pętli linii papilarnych po łokciowej stronie (35%)
• krótka szyja, rzadziej płetwistość szyi
• fałd skóry na karku w okresie niemowlęcym
• miękkie, delikatne i rzadkie włosy skóry głowy
• wrodzone wady serca
• nieprawidłowy przebieg tętnicy podobojczykowej
• wady kośćca
• wady układu
• moczowo-płciowego
b) Zespół Edwardsa (trisomia 18 chromosomu) - około 95% płodów
z trisomią 18 ulega spontanicznemu poronieniu, 30% żywo
urodzonych dzieci z zespołem Edwardsa umiera w pierwszym
miesiącu życia, tylko 10% przeżywa 1 rok. Częstość zespołu
Edwardsa wzrasta z wiekiem matki podobnie jak w zespole Downa.
Objawy: niska masa urodzeniowa
noworodka, dysplastyczne małżowiny uszne, nadmiar skóry na
szyi, anomalie szkieletu, wady serca, atrezja przełyku, wady
narządów płciowych, niepełnosprawność intelektualna,
charakterystyczna pojedyncza bruzda zgięciowa dłoni,
nakładające się na siebie palce.
Ø
przyczyną choroby jest brak rozdziału chromatyd podczas
pierwszego podziału mejotycznego u matki
Ø
występuje u 1/3000 żywych urodzeń
Zespół Klinefeltera (trisomia XXY) - grupa chorób spowodowanych aberracją
chromosomalną polegającą na obecności przynajmniej jednego
dodatkowego chromosomu X w części lub we wszystkich komórkach organizmu
mężczyzny. Klasyczny zespół Klinefeltera występuje u mężczyzn
o kariotypie 47,XXY i jest najczęstszą aneuploidią człowieka, której częstość ocenia
się na 1:500 noworodków płci męskiej. Często bywa rozpoznawany dopiero u
dorosłych mężczyzn w związku z występującą u nich bezpłodnością. Obserwuje
się również takie nieprawidłowości jak: skrzywienie kręgosłupa, osteoporoza i
rozedma płuc. Wtórne cechy płciowe często są słabo wykształcone, obserwuje
się natomiast powiększenie sutków (ginekomastia). Poziom intelektu może być
lekko obniżony, zazwyczaj jednak mieści się w normie.
Ø
obecne jest w komórkach ciałko Barra
Ø
przyczyną choroby jest nondysjunkcja w pierwszym lub drugim podziale
mejotycznym
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 6
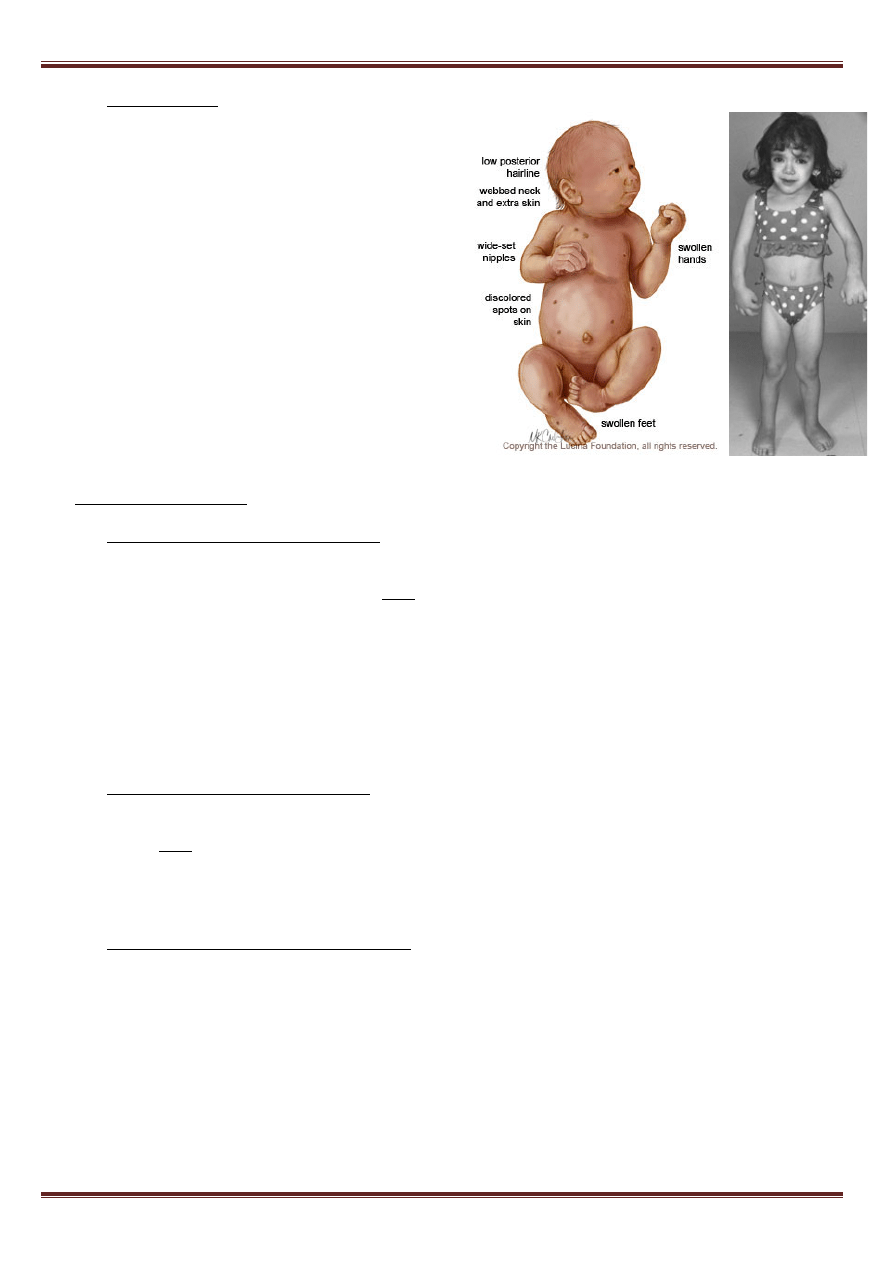
c) Zespół Turnera (monosomia chromosomu X) - spowodowany całkowitym lub częściowym brakiem
jednego z chromosomów X we wszystkich
komórkach organizmu lub w pewnej ich części.
Występuje u 1 na 2000 – 2500 urodzonych
dziewczynek. U chorych obserwuje się: nadmiar
skóry na szyi noworodka („szyja płetwiasta”),
obrzęki limfatyczna na skutek niedrożności
naczyń chłonnych. Wzrost od wczesnego
dzieciństwa jest niski, ale proporcjonalny, klatka
piersiowa szeroka. Występuje brak rozwoju
wtórnych cech płciowych. W 20% przypadków
obecna wrodzona wada serca, poziom intelektu
nie odbiega od normy.
Ø
brak rozwoju wtórnych cech płciowych,
brak ciałek Barra
Ø
przyczyną choroby jest brak rozdziału
chromatyd lub brak chromosomu
ojcowskiego, ponadto do choroby może
prowadzić tworzenie się izochromosomu.
2.
Choroby monogenowe
– zaburzenia spowodowane przez mutację pojedynczych genów, wykazujące główne
wzorce dziedziczenia zgodne z prawami Mendla:
a) Dziedziczenie autosomowe dominujące – wystarczy obecność jednego genu, aby dana cecha pojawiła
się w fenotypie. Choroba warunkowana jest obecnością genu A ujawnia się zarówno u homozygoty AA,
jak i u heterozygot Aa. Prawdopodobieństwo otrzymania od dziecka zmutowanego allela od chorego ojca
lub matki jest takie samo, ryzyko wynosi 50%. Obowiązuje pionowy wzór dziedziczenia. Niekiedy osoby
ze zmutowanym genem mają prawidłowy fenotyp, mówi się wówczas o „niepełnej penetracji genu”. Osoby
takie jednak przenoszą gen chorobowy na swoje potomstwo, co jest zjawiskiem przeskakiwania pokoleń.
Cechy dominujące dotyczą często zmienionych białek strukturalnych, transportujących i receptorowych,
rzadziej enzymów. Przykłady chorób:
• hipercholesterolemia rodzinna
• zwyrodnienie torbielowate nerek
• choroba Huntingtona
• zespół Marfana
• achondroplazja
b) Dziedziczenie autosomowe recesywne – tak uwarunkowana cecha ujawnia się jedynie u heterozygot aa,
czyli u osób mających gen chorobowy w podwójnej dawce. Rodzice dziecka są zwykle zdrowymi
nosicielami Aa. Proporcja osób chorych do zdrowych u potomstwa wynosi 1:3, a więc ryzyko zachorowania
wynosi 25%.Wzór dziedziczenia jest poziomy, wszystkie dzieci osoby chorej będą nosicielami mutacji
genowej. Przykłady chorób:
• mukowiscydoza
• fenyloketonuria
• choroba Taya i Sachsa
• anemia sierpowata
• galaktozemia
• wrodzona łamliwość kości
c) Dziedziczenie sprzężone z chromosomem X – geny odpowiedzialne za choroby sprzężone z
chromosomem X są zlokalizowane na tym chromosomie. Ryzyko genetyczne i ciężkość przebiegu
choroby zależy więc od płci pacjenta. Ponieważ kobieta ma dwa chromosomy X, może być heterozygotą
lub homozygotą dla mutacji określonego genu. Ekspresja takiego genu u kobiet jest zmienna.
Dużą rolę
odgrywa zjawisko losowej inaktywacji chromosomu X (lyonizacja
). Mężczyźni mają tylko jeden
chromosom X, a więc należy oczekiwać występowania choroby z wszystkimi jej objawami, jeżeli otrzymają
gen chorobowy od matki, bez względu na to, czy jest to u niej cecha recesywna czy dominująca. Przykłady
chorób:
• hemofilia A i B
• dystrofia mięśniowa typu Duchenne’a/Beckera
(DMD/BMD)
• albinizm
• zespół feminizujących jąder
• zespół kruchego chromosomu X
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 7
Skutki mutacji jednego genu, a więc odcinka DNA kodującego jedno białko, mogą być następujące:
• całkowity brak białka
• białko jest wytwarzane, ale nie wykazuje aktywności
• wytworzone białko ma zmienione właściwości funkcjonalne np. mutacja w locus kodującym enzym
lizosomalny może być powodem braku enzymu, wytwarzania enzymu który nie dociera do lizosomy, lub
wytwarzania enzymu, który dociera do lizosomy, lecz jest nietrwały bądź ma zmienione właściwości
katalityczne
Częstość zaburzeń monogenowych
wynosi 10:1000 urodzonych żywo noworodków, z czego 7:1000
stanowią zaburzenia dominujące, 2,5:1000 recesywne i około 0,4:1000 zaburzenia sprzężone z chromosomem X.
W wielu chorobach monogenowych zmieniony w skutek mutacji produkt białkowy jest jeszcze nieznany. Defekty
wywoływane przez choroby monogenowe mogą dotyczyć praktycznie wszystkich związków bedących białkami
np. enzymów, receptorów, białek nośnikowych, hormonów peptydowych, immunoglobulin, kolagenu, czynników
krzepnięcia, czynników transkrypcyjnych.
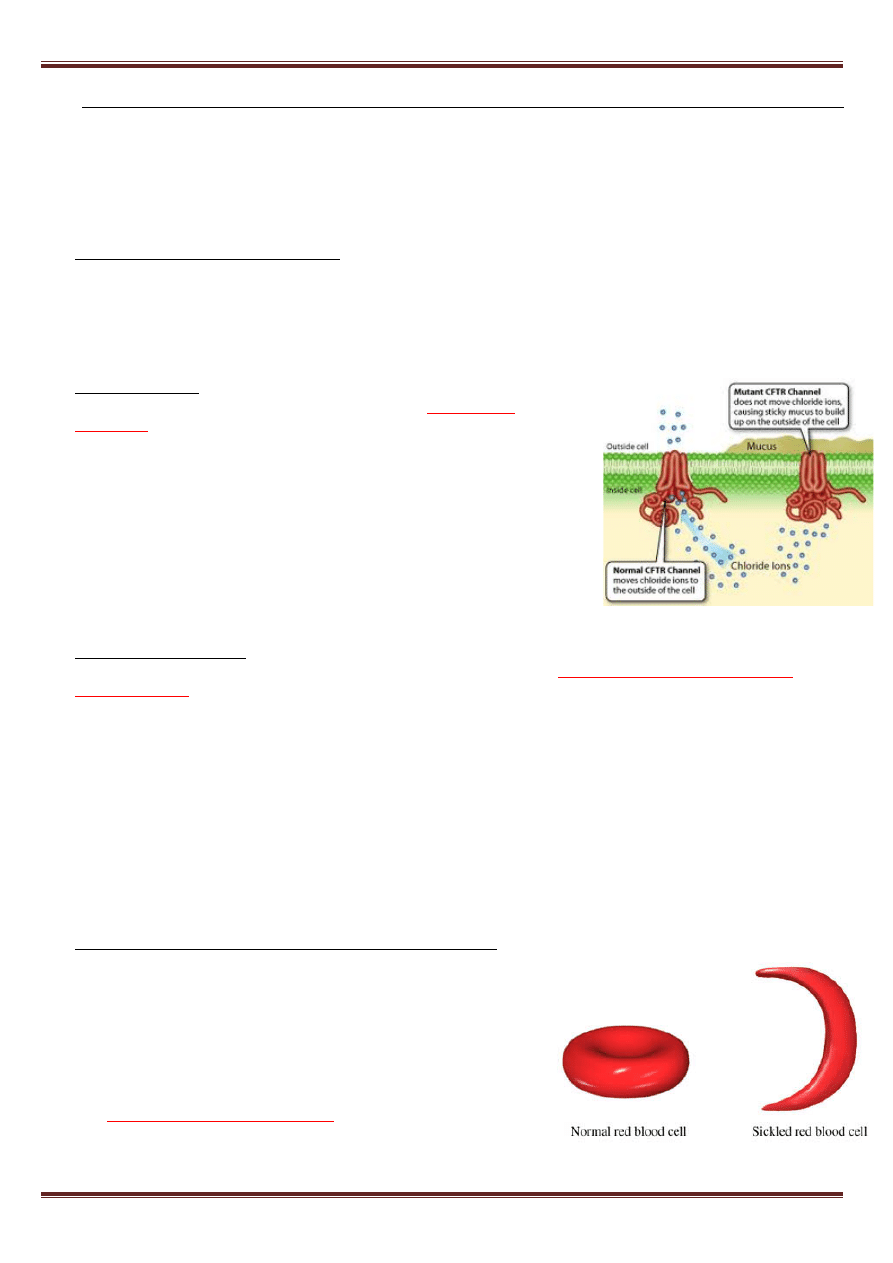
Mukowiscydoza
(zwłóknienie torbielowate) – ciężka i nieuleczalna
choroba wieku dziecięcego, przekazywana w sposób
autosomowy
recesywny
. Nosicielem genu chorobowego jest 1:20 Europejczyków. Gen
mukowiscydozy zlokalizowany jest w chromosomie 7q13, koduje on białko
odpowiedzialne za czynność kanału chlorkowego błon komórkowych
(CFTR). Wykryto liczne mutacje odpowiedzialne za tę chorobę – około
1000. Najczęśniej wystepującą jest delecja kodonu 508 (CTT) w egzonie
10, kodującego fenyloalaninę (mutacja delta-F508). Schorzenie to najczęściej
powoduje zmiany w:
• układzie oddechowym - nawracające zakażenia, które prowadzą do
uszkodzenia płuc i niewydolności oddechowej
• przewodzie pokarmowym - przewlekły stan zapalny trzustki, prowadzi
do uszkodzenia tego narządu i jego niewydolności, a niekiedy także wtórnej cukrzycy.
Choroba Huntingtona
– postępująca nieuleczalna choroba zwyrodnieniowa ośrodkowego układu
nerwowego, szczególnie jąder podstawy mózgu, dziedziczona w sposób
autosomowy dominujący o pełnej
penetracji genu.
W obrazie histologicznym charakterystyczne jest przedwczesne obumieranie neuronów,
częstość występowania waha się od 1:10 000 do 1:20 000. Gen chorobowy IT15 zlokalizowany jest na
chromosomie 4p16.3 kodując huntingtynę, białko o nieznanej funkcji, przypuszcza się jednak że jest ona jednym
z inhibitorów apaptozy komórek nerwowych.
• mutacja związana jest z niestabilnością sekwencji CAG, na końcu 5’ genu IT15, liczba powtórzeń tej
sekwencji u chorych wzrasta nawet do 120, podczas gdy u zdrowej osoby wynosi do 30 powtórzeń. Istnieje
korelacja pomiędzy wiekiem zachorowań a stopniem zwielokrotnienia tripletu CAG w obrębie genu.
• pierwsze objawy pojawiają się między 35-40 r.ż. i są to objawy psychotyczne, depresja, otępienie, objawy
neurologiczne – głównie mimowolne ruchy pląsawicze
• w 10% przypadków w tzw. postaci młodzieńczej, objawy chorobowe występują przed 20 r.ż., przebieg
jest wówczas cięższy
• chorzy umierają zazwyczaj w ciągu 15-20 lat od wystąpienia choroby
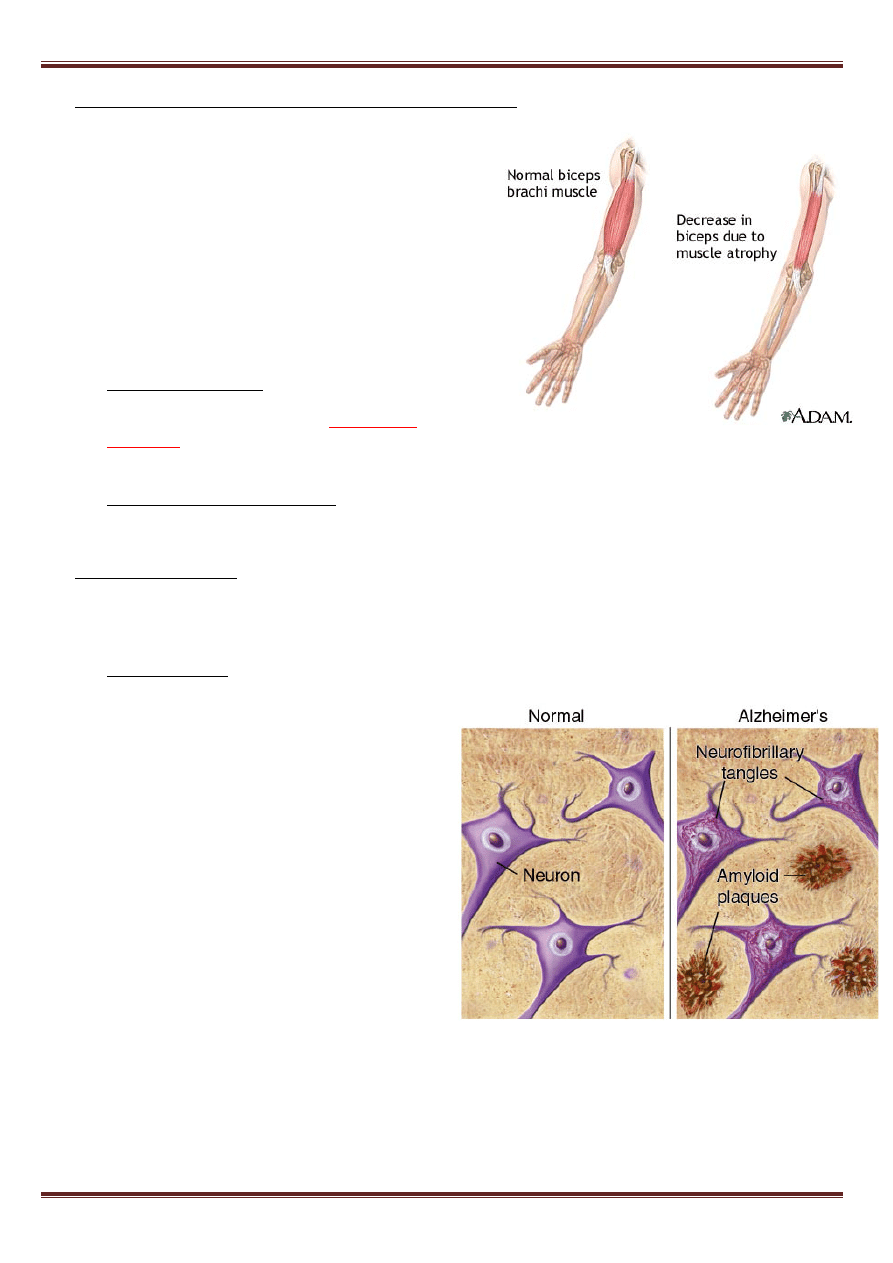
Niedokrwistość sierpowokrinkowa (anemia sierpowata)
-
mutacja punktowa w genie łańcucha β (HBB) hemoglobiny
powoduje zmianę pojedynczego aminokwasu w
sekwencji białka (z k. glutaminowego na walinę). Hemoglobinę z
tak zmienioną, nieprawidłową strukturą I-rzędową określa się jako
hemoglobinę S (HbS) w przeciwieństwie do normalnej,
występującej u dorosłych hemoglobiny A (HbA). Hemoglobina S
charakteryzuje się zmienionymi w porównaniu z hemoglobiną A
własnościami fizykochemicznymi.
•
choroba autosomowa recesywna
• występuje znacznie częściej w krajach, gdzie panuje malaria,
ponieważ nosiciele genu (heterozygoty) są odporni na malarię
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 8
Rdzeniowy zanik mięśni (SMA – spinal muscular atrophy)
– polega na postępującym osłabieniu mięśni,
które jest wynikiem zaniku komórek rogów przednich rdzenia kręgowego. Głębokie odruchy ścięgniste są
zniesione lub osłabione, występują drgania pęczkowe
mięśni, drżenie palców. Osłabienie jest bardziej zaznaczone
w mięśniach kończyn dolnych.
• gen zlokalizowano w 5q11-13, z regionu tego
izolowano dwa leżące obok siebie geny:
ü
gen odpowiedzialny za przeżywanie neuronów
ruchowych (SMN – survival motor neuron)
ü
gen kodujący białko inhibitora apoptozy neuronów
(NAIP – neuronal apoptosis inhibitor protein)
• oba geny ulegają delecji u pacjentów z rdzeniowym
zanikiem mięśni
• produktem genu jest białko SMN, gen NAIP ma
znaczenie modyfikujące obraz kliniczny choroby
• Dziecięca postać SMA (choroba Werdniga-
Hoffmana) jest drugą po mukowiscydozie chorobą
dziecięcą dziedziczoną w sposób
autosomowy
recesywny
, charakterystyczna jest wiotkość dziecka, badania wykazują odnerwienie i zanik neurogenny w
obrębie mięśni. Przebieg choroby jest bardzo ciężki, kończy się śmiercią w ciągu pierwszego lub drugiego
roku życia
• Pośrednia i łagodna postać SMA (typu Kugelberg-Welander) postęp choroby jest wolniejszy i skrócenie
długości życia może być nieduże.
• łączna częstość zachorowań wszystkich wariantów SMA wynosi 1:10 000 urodzeń żywych
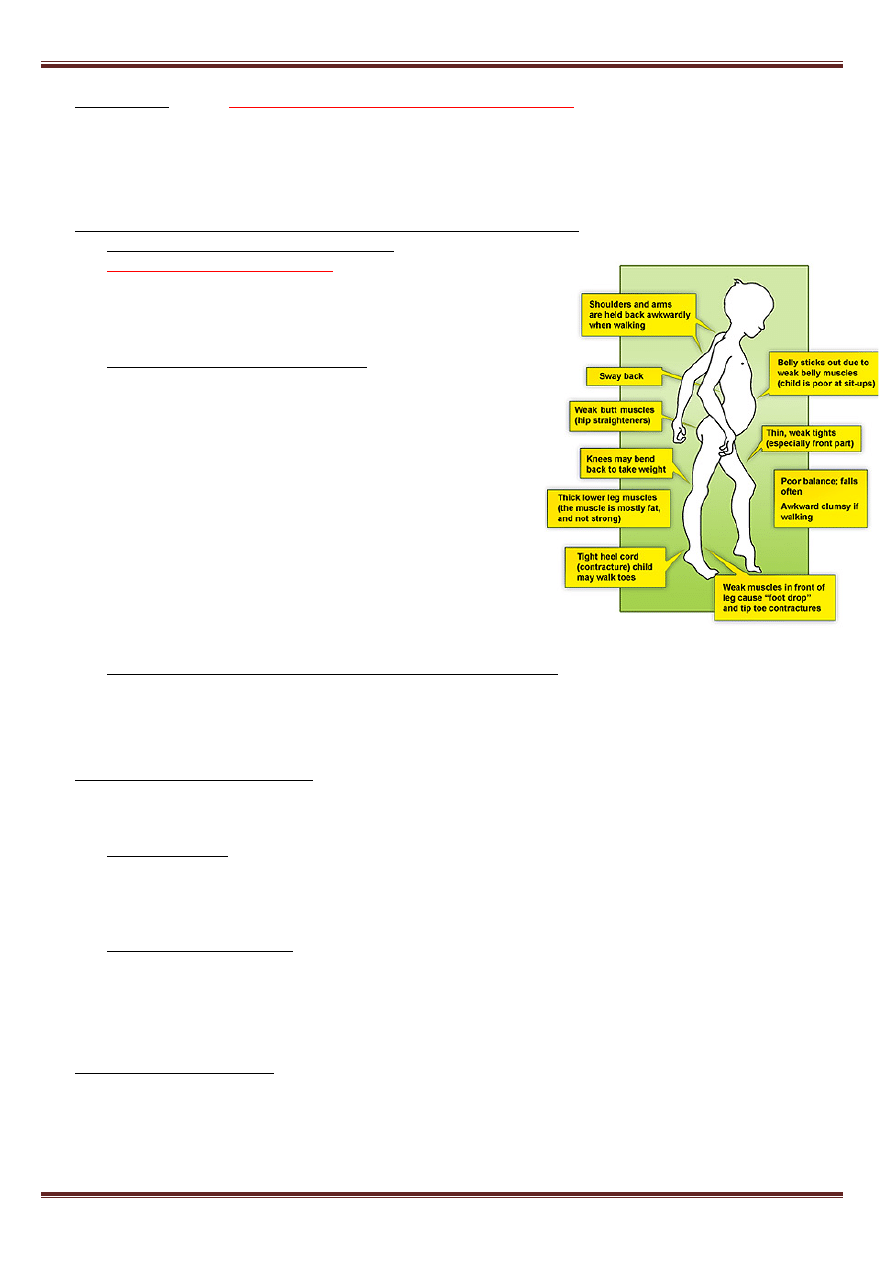
Choroba Alzheimera
– w szarej substancji mózgu obecne są tzw. płytki starcze i zwyrodnienie włókienkowe
neuronów, głównym składnikiem płytek starczych jest peptyd β-amyloidowy. Odgrywa on zasadniczą rolę w
patogenezie choroby Alzheimera typu I, a także w otępieniu obserwowanym w zespole Downa.
• Gen APP, kodujący prekursor β-amyloidowy został zlokalizowany na chromosomie 21q21 i jego mutacje
wykryto w rodzinach z chorobą Alzheimera
• Złogi β-amyloidu w zespole Downa są efektem dawki genu – w trisomii 21 występują trzy kopie genu APP
• W patogenezie choroby Alzheimera odgrywają rolę trzy inne geny:
ü
gen apolipoproteiny E (Apo E) w 19q13.2 –
białko kodowane przez ten gen ma różne
właściwości: tworzy kompleksy z β-amyloidem.
Wariant genu E – E4 wykazuje wyraźny
związek z chorobą Alzheimera typu 2.
ü
gen S182 w 14q24.3 – wykazano związek w
niektórych rodzinach pomiędzy mutacjami w
tym genie a chorobą Alzheimera z wczesnym
początkiem zachorowania (typu 3)
ü
gen STM2 w 1q31-42
• W przebiegu choroby dochodzi do wystąpienia
następujących objawów:
ü
zaburzenia pamięci
ü
zmiany nastroju
ü
zaburzenia funkcji poznawczych
ü
zaburzenia osobowości i zachowania
• Charakterystyczne dla demencji (i w tym choroby
Alzheimera) objawy to:
ü
agnozja – nieumiejętność rozpoznawania
przedmiotów, szczególnie jeżeli chory ma podać nazwę przedmiotu, w odpowiedzi na pytanie zadane z
zaskoczenia.
ü
afazja – zaburzenia mowy, jej spowolnienie
ü
apraksja – zaburzenia czynności ruchowych, od prostych do złożonych, np. ubieranie, kąpanie
• Zmianom otępiennym mogą towarzyszyć objawy neurologiczne, spośród których najczęstszym jest tzw.
zespół parkinsonowski – spowolnienie psychoruchowe, zaburzenia mimiki twarzy i sztywność mięśni.
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 9
Hemofilia A
– jest to
choroba recesywna sprzężona z chromosomem X
, gen zlokalizowany w regionie q28,
koduje tzw. czynnik VIII krzepnięcia krwi. U chorych brak jest tego czynnika, lub następuje jego
niedostateczną aktywność poniżej 30%.
• występują nawracające krwotoki
• długość życia przy leczeniu czynnikiem VIII nie odbiega od normy
• mutacja następuje przez insercje oraz duże inwersje (50% przypadków)
Choroba Duchenne’a/Beckera (dystrofia mięśniowa; DMD/BMD)
• Dystrofia mięśniowa Duchenne’a (DMD) - jedna z najczęściej występujących chorób recesywnych
sprzężonych z chromosomem X,
częstość występowania
wynosi 1:3000 żywo urodzonych chłopców.
ü
jest ciężką, postępującą chorobą zwyrodnieniową,
powodującą znaczne uszkodzenia mięśni, zwłaszcza
kończyn dolnych, prowadząca do śmierci około 20 r.ż.
• Dystrofia mięśniowa Beckera (BMD) – występuje rzadziej i
ma lżejszy przebieg, czas przeżycia może być normalny lub
nieznacznie skrócony w porównaniu z populacja ogólną
ü
w tej odmianie choroby obserwuje się dużą zmienność w
nasileniu objawów chorobowych
• Gen zlokalizowano w Xp21, jest to największy z dotąd
poznanych genów, koduje białko dystrofinę. Wchodzi ona w
skład kompleksu białkowego uszczelniającego błonę
komórkową, który zawiera m.in. aktynę, dystroglikan,
lamininę. Defekty tych białek są odpowiedzialne za inne
postacie dystrofii mięśniowych
• Mutacje powodujące DMD/BMD to delecje, duplikacje oraz
mutacje punktowe, z których delecje i duplikacje mogą
zmieniać ramkę odczytu, co uważa się za charakterystyczne
dla DMD i wiąże z cięższym przebiegiem tej odmiany dystrofii
mięśniowej – w DMD brak jest w mięśniach dystrofiny
• W BMD dystrofina występuje w mniejszej ilości, lub jej brakuje
• Czasem chorują kobiety – tzw. nosicielki objawowe, ale przebieg choroby jest u nich lżejszy niż u
mężczyzn. Wyjątek stanowią kobiety z nosicielkami translokacji zrównoważonych obejmujących
chromosom X i jeden z autosomów – choroba jest u nich wynikiem wybiórczej inaktywacji prawidłowego
chromosomu X w komórkach somatycznych, aktywny bowiem pozostaje chromosom X objęty translokacją
3.
Dziedziczenie wieloczynnikowe
– w przeciwieństwie do dziedziczenia monogenowego cechy
wieloczynnikowe są uwarunkowane oddziaływaniem wielu genów w różnych loci, każdy z małym,
addytywnym efektem, a także przez czynniki środowiskowe. Przykładem cech wieloczynnikowych są: wzrost,
masa ciała, inteligencja, ciśnienie krwi itp.
a) Wady wrodzone:
•
rozszczep wargi i podniebienia
•
wrodzona wada serca
•
wady cewy nerwowej
•
wrodzone zwężenie odźwiernika
b) Choroby wieku dorosłego:
•
padaczka
•
wrzód trawienny
•
nadciśnienie tętnicze
•
cukrzyca
•
gościec stawowy, pierwotnie przewlekły
4.
Choroby mitochondrialne
– DNA mitochondrialne są głównie odpowiedzialne za kodowanie enzymów cyklu
oddechowego
Ø
mitochondrialne DNA jest dziedziczone wyłącznie w linii matczynej
Ø
objawy chorobowe występują, gdy mutacja obecna jest w wystarczającej liczbie mitochondriów (efekt
progowy)
Ø
przykłady chorób:
mitochondrialna miopatia, dziedziczny zanik nerwu wzrokowego Lebera, padaczka
miokloniczna
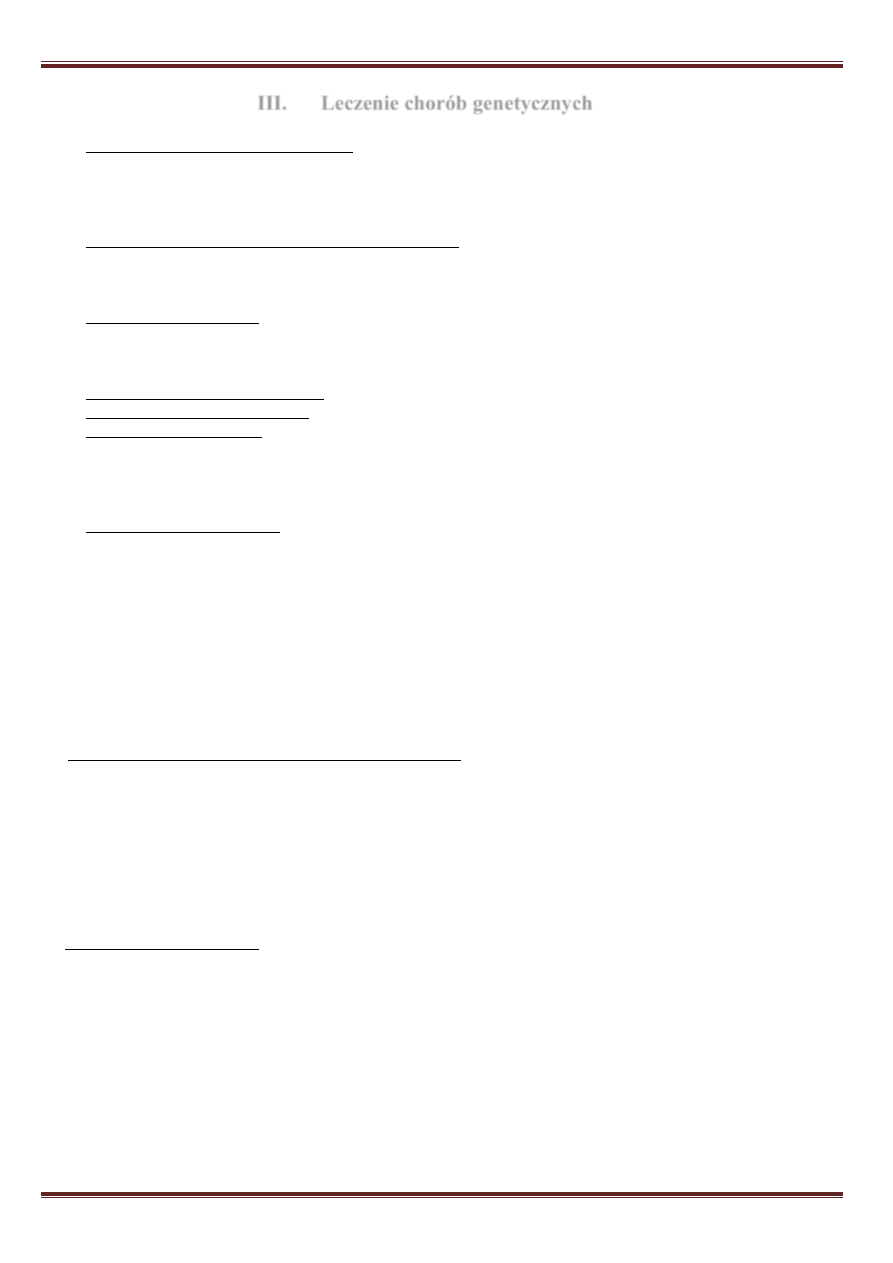
Czynniki genetyczne w etiopatogenezie chorób
m.piktel@hotmail.com
Strona 10
III.
Leczenie chorób genetycznych
Leczenie zaburzeń genetycznych wymaga właściwej diagnozy, wczesnego rozpoczęcia i znajomości patogenezy.
a)
leczenie farmakologiczne i operacyjne – bez próby zmiany podstawowego defektu genetycznego np. leczenie
operacyjne w polipowatości jelit
•
podawanie środków przeciwpadaczkowych w wielu chorobach układu nerwowego
•
podawanie blokerów β-andrenergicznych w celu opóźnienia postępującego poszerzania aorty w zespole
Marfana
b)
pouczanie pacjenta aby unikał niektórych czynników – niekiedy dobre wyniki osiąga się przez
poinformowanie pacjenta, że musi on unikać czynników wywołujących chorobę
•
niektórych leków w chorobach farmakogenetycznych
•
promieni słonecznych w albinizmie oraz xeroderma pigmentosum)
c)
ograniczenia dietetyczne
•
fenyloketonuria
•
choroba syropu klonowego
•
galaktozemia
d)
usuwanie produktów toksycznych np. przez stosowanie penicylaminy w chorobie Wilsona
e)
aktywacja uszkodzonego białka przez stosowanie pirydoksyny w homocystynurii
f)
transplantacja narządów – dzięki przeszczepianu narządowi jest dostarczane nie tylko prawidłowe białko, ale
także nowa informacje genetyczna, chociaż nie włączana w genom biorcy
•
przeszczep szpiku – lizosomalne choroby spichrzeniowe, β- talasemia
•
przeszczep wątroby - hipercholesterolemia rodzinna, choroba glikogenowa typu I
•
przeszczep nerki – cystynoza
g)
terapia genowa somatyczna – polega na korygowaniu defektu genetycznego poprzez zmiane genotypu.
Dotyczy to tylko niektórych komórek somatycznych, nie obejmuje natomiast linii komórek rozrodczych –
zmiany nie są przekazywane potomstwu. Istnieje kilka sposobów takiej terapii:
•
dodanie prawidłowo działającego genu w chorobach, które charakteryzują się brakiem odpowiednich
enzymów np. fenyloketonuria
•
zastąpienie prawidłowego genu genem nieprawidłowym, metoda ta mogałby być stosowana w chorobach,
w których zmutowany gen wytwarza nieprawidłowe białko, mogące naruszać integralność strukturalną
tkanek, jak ma to miejsce w ciężkich postaciach wrodzonej łamliwości kości
•
naprawienie nieprawidłowego genu – byłaby to właściwa metoda leczenia chorób, w których
nieprawidłowy gen ma działanie patogenne
•
wybiórcze zablokowanie wybranego genu przez siRNA
Wskazania do przeprowadzenia analizy chromosomów:
•
cechy dysmorficzen sugerujące zespół chromosomowy
•
uspośledzenie umysłowe o niewyjaśnionej przyczynie
•
badanie rodzinne w przypadkach strukturalnie nieprawidłowych ccchromosomów
•
obecność mnogich wrodzonych nieprawidłowości
•
obojnaczy rozwój płciowy
•
niektóre typy nowotworów
•
pierwotna bezpłodność
•
nawracające poronienia samoistne
Wskazania do analizy DNA
•
rozpoznanie lub podejrzenie choroby monogenowej u pacjenta
•
rozpoznanie lub podejrzenie choroby monogenowej u członków rodziny pacjenta
•
podejrzenie choroby mitochondrialnej
•
zgon dziecka w okresie okołoporodowym o podejrzeniu choroby etiologicznej
Wyszukiwarka
Podobne podstrony:
Biomedyczne podstawy rozwoju i wychowania Czynniki genetyczne i środowiskowe wpływające na rozwój
CZYNNIKI GENETYCZNE A TERAPIA CHOROBY LEŚNIOWSKIEGO, bilogia molekularna
Czynniki genetyczne i środowiskowe wpływające na rozwój człowieka, biomedyczne podstawy rozwoju i wy
Czynniki genetyczne w patogenez Nieznany
Modul 2 Czynniki genetyczne i srodowiskowe a rozwoj czlowieka
Czynniki genetyczne i środowiskowe wpływające na rozwój człowieka
Czynniki genetyczne w powstawaniu chorób
Wpływ czynników genetycznych na skutecznośc i bezpieczeństwo farmakoterapii informacje wstępne
Wpływ czynników genetycznych na skutecznośc i bezpieczeństwo farmakoterapii podsumowanie
10446-mutacje genomu ludzkiego i czynniki je wywołujące, semestr IV, genetyka, Genetyka
biologiczne podstawy, czynniki endogenne, Czynniki endogenne genetycznie
Zaburzenia afektywne i depresje – epidemiologia, etiopatogeneza, uwarunkowania genetyczne
Czynniki endogenne genetycznie
Czynniki mutagenne, Ogrodnictwo UP Wrocław, Semestr IV, Genetyka
Biologiczne skutki działania czynnikow mutagennych, AM, rozne, genetyka, genetyka, GENETYKA, Genetyk
więcej podobnych podstron