
Przemysław Szczeciński
Zakład Chemii Organicznej
Wydział Chemiczny PW
LABORATORIUM Z CHEMII ORGANICZNEJ
Materiały pomocnicze
Spis treści
BROMEK BUTYLU (1-Bromobutan)
m-NITROANILINA
JODOBENZEN
p-JODOTOLUEN
p–JODONITROBENZEN
ACETANILID
p–BROMOACETANILID i p–BROMOANILINA
p–NITROACETANILID i p–NITROANILINA
KWAS CYNAMONOWY
ANILINA
p-METYLOACETOFENON (destylacja pod zmniejszonym ciśnieniem)
MRÓWCZAN ETYLU
BENZOESAN METYLU I ETYLU, SALICYLAN METYLU I ETYLU
OCTAN BUTYLU
Uwaga. Przepisy preparatywne zostały zaczerpnięte z Preparatyki Organicznej A. Vogla. Do niektórych z
nich wprowadzono pewne modyfikacje.
1
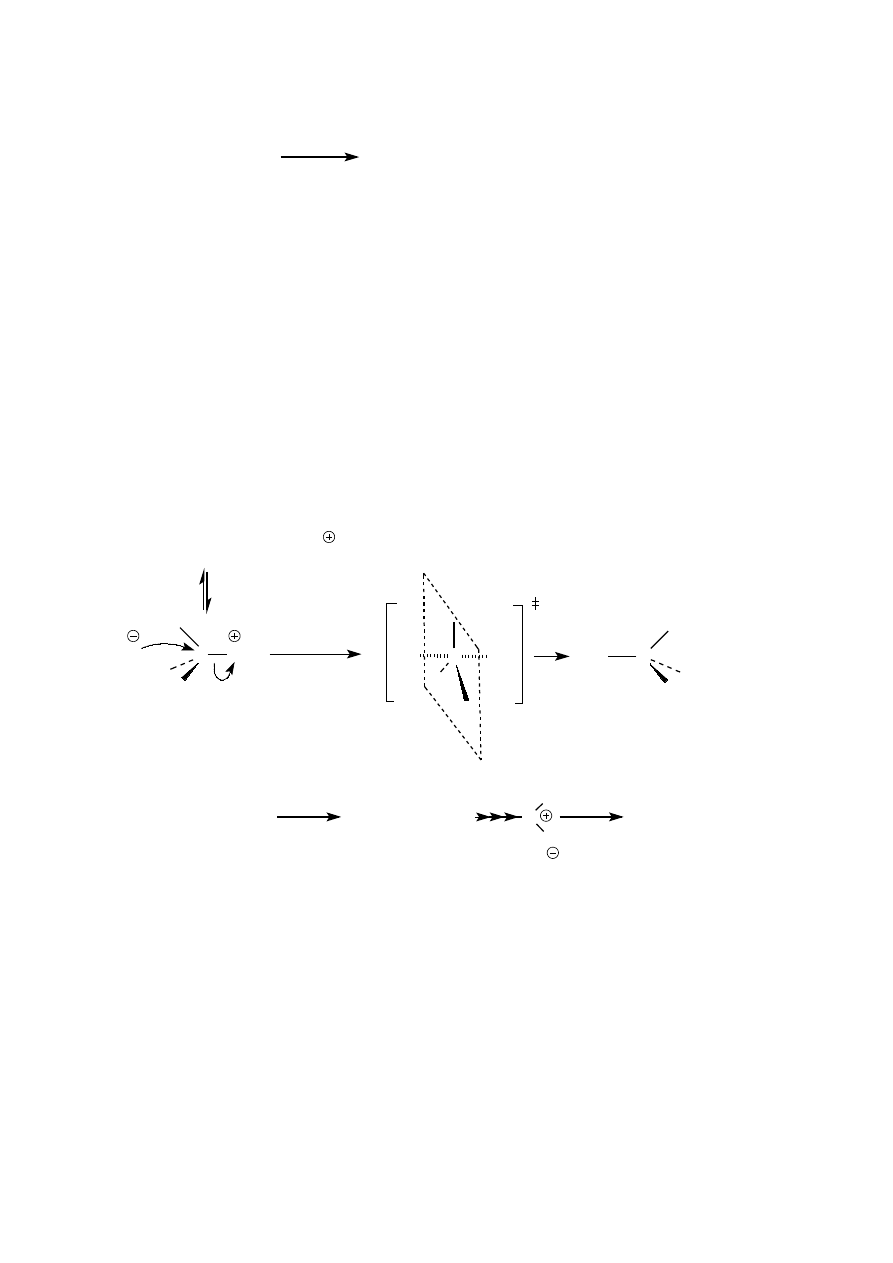
BROMEK BUTYLU (1-Bromobutan)
1-bromobutan
butan-1-ol
CH
3
CH
2
CH
2
CH
2
Br
H
2
SO
4
stęż.
48% HBr,
t
CH
3
CH
2
CH
2
CH
2
OH
W syntezie wykorzystuje się reakcję substytucji nukleofilowej. Grupa hydroksylowa jest grupą bardzo źle
odchodzącą i jej bezpośrednie podstawienie innym nukleofilem nie jest możliwe; należy ją najpierw
przekształcić w grupę łatwiej odchodzącą. Jeden ze sposobów modyfikowania grupy hydroksylowej, polegający
na jej protonowaniu, a więc przekształceniu w dobrze odchodzącą, trwałą, obojętną cząsteczkę wody,
wykorzystuje się w reakcjach alkoholi z halogenowodorami. Wszystkie alkohole I rz. reagują z kwasami
halogenowodorowymi w obecności katalizatorów. W przypadku kwasów bromo- i jodowodorowego są nimi
stężony kwas siarkowy lub fosforowy. Ich zadaniem jest ułatwienie powstania soli oksoniowej. Reakcje
przebiegają według mechanizmu S
N
2. Anion chlorkowy jest słabszym nukleofilem niż anion jodkowy czy
bromkowy, dlatego do zastąpienia grupy hydroksylowej atomem chloru podczas reakcji alkoholu
pierwszorzędowego z kwasem solnym konieczne jest efektywniejsze osłabienie wiązania C
O, niż to, jakie
następuje w wyniku protonowania atomu tlenu. Zadowalający skutek osiąga się kompleksując alkohol, który
dzięki posiadanym na atomie tlenu parom elektronów jest zasadą Lewisa, chlorkiem cynku (kwasem Lewisa).
CH
3
CH
2
CH
2
CH
2
OH + H
C
H
7
C
3
OH
2
H
H
C
C
3
H
7
Br
H
H
powoli
SN2
C
H
H
C
3
H
7
Br
OH
2
+
Br
+ H
2
O
+
CH
3
CH
2
CH
2
CH
2
O
H
ZnCl
2
t
CH
3
CH
2
CH
2
CH
2
Cl
ZnCl2
HClstęż.
CH
3
CH
2
CH
2
CH
2
OH
butan-1-ol
1-chlorobutan
Pierwszym etapem reakcji alkoholi II i III rz. z halogenowodorami jest także utworzenie soli okoniowej. Dalej
reakcja przebiega jednak według mechanizmu S
N
1. Skutkiem tego w niektórych przypadkach położenie atomu
fluorowca w produkcie jest inne w stosunku do położenia grupy hydroksylowej w substracie. Taka sytuacja ma
miejsce wtedy, gdy powstający karbokation może ulec przegrupowaniu do karbokationu trwalszego, wyżej
rzędowego.
2
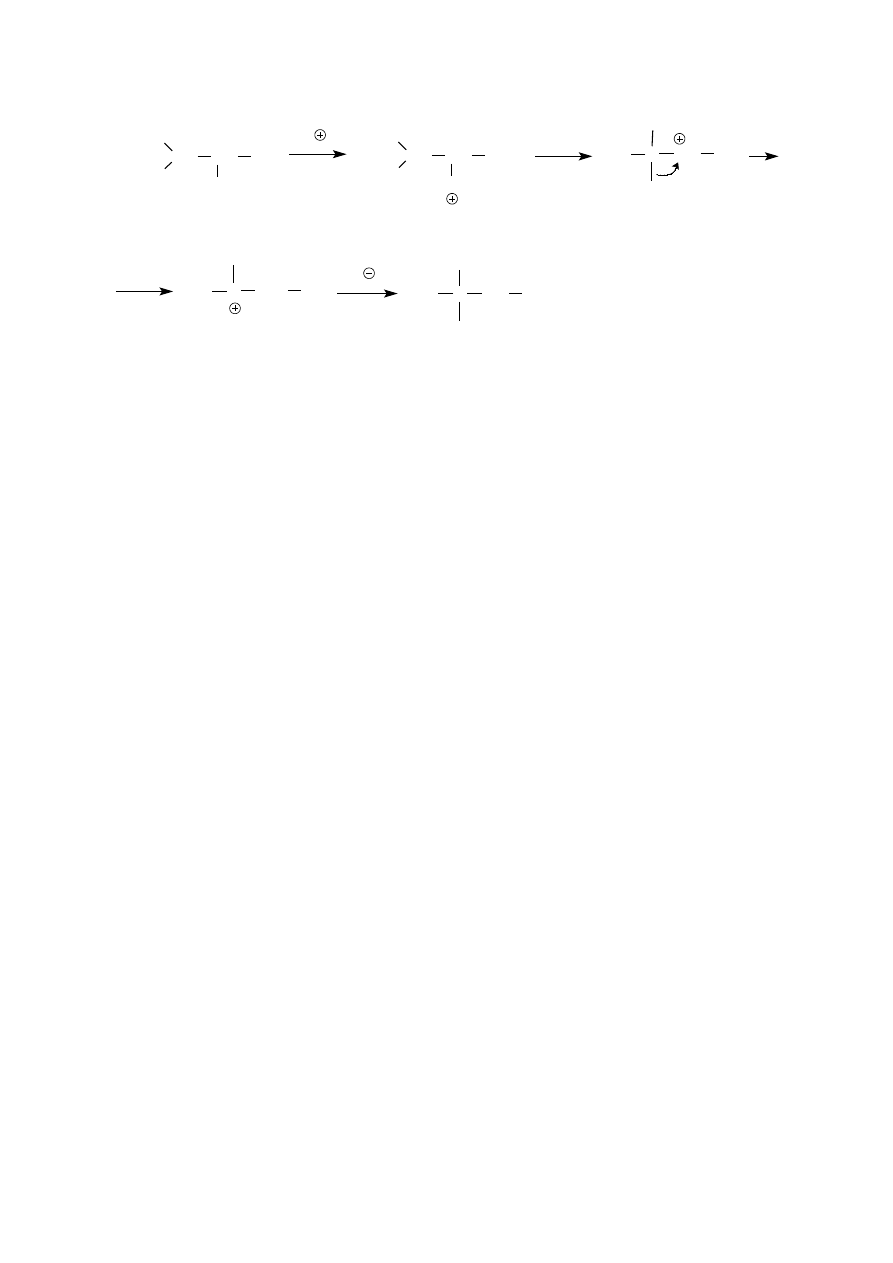
Br
H
3
C
CH
H
3
C
CH CH
3
OH
H
-H2O
H
3
C
C
H
3
C
CH CH
3
H
H
3
C
C
H
3
C
CH
2
CH
3
H
3
C
C
H
3
C
CH CH
3
Br
3-metylobutan-2-ol
2-bromo-2-metylobutan
H
3
C
CH
H
3
C
CH CH
3
OH
2
kation oksoniowy
karbokation II rz.
karbokation III rz.
Innymi reagentami służącymi do otrzymywania halogenków alkilowych z alkoholi są: chlorek tionylu (SOCl
2
),
chlorki i bromki fosforu (PX
3
i PX
5
), brom lub jod w obecności czerwonego fosforu. Do syntezy halogenków
alkilowych z innych substratów wykorzystuje się np. reakcje substytucji rodnikowej (alkany) a także addycji
elektrofilowej lub rodnikowej (alkeny, alkiny). Halogenki arylowe otrzymuje się głównie z soli diazoniowych
lub w reakcji substytucji elektrofilowej.
Preparat wykonuje się pod wyciągiem.
Do 250 g (1,48 mol) 48-proc. kwasu bromowodorowego znajdującego się w kolbie kulistej poj. 500 ml dodaje
się porcjami 75 g (41 ml) stęż. kwasu siarkowego. Po dodaniu każdej porcji należy wstrząsnąć zawartością kolby
– może się przy tym wydzielić pewna ilość bromowodoru. Do zawartości dodaje się 88,8 g (110 ml, 1,2 mol)
butan-1-olu a następnie, wstrząsając, w kilku porcjach, 60 g (32,5 ml) stęż. kwasu siarkowego. Do kolby wrzuca
się kilka kamyków wrzennych i instaluje chłodnicę zwrotną. Wylot chłodnicy łączy się wężem z nóżką
odwróconego lejka szklanego umocowanego łapą tuż nad powierzchnią wody znajdującej się w zlewce. Dzięki
temu wydzielające się podczas reakcji bromowodór i ditlenek siarki będą absorbowane w wodzie. Zawartość
kolby ogrzewa się do łagodnego wrzenia przez 2 – 3 godziny. Po tym czasie powstawanie bromku butylu jest
niemal zakończone; produkt stanowi odrębną, górną warstwę (1). Gdy zawartość kolby ostygnie, usuwa się
chłodnicę zwrotną a w jej miejsce montuje zestaw do destylacji. Mieszaninę destyluje się do momentu, aż
przestaną spływać oleiste krople bromku butylu (30 – 40 minut) (2). Destylat przenosi się do rozdzielacza i
oddziela halogenek, który tworzy tym razem dolną warstwę (dlaczego?). Produkt przemywa się kolejno wodą,
równą objętością stężonego kwasu solnego (3), wodą, 5-proc. roztworem wodorowęglanu lub węglanu sodu
(może wydzielać się CO
2
; trzeba często wyrównywać ciśnienie otwierając kran) i wodą. Po możliwie dokładnym
oddzieleniu wody produkt suszy się za pomocą 2 – 3 g bezw. chlorku wapnia lub siarczanu magnezu; środek
suszący powinien pozostawać w kontakcie z bromkiem przez co najmniej 30 minut a zawartością należy co
pewien czas wstrząsnąć. Wysuszony produkt sączy się przez niewielki lejek z sączkiem fałdowanym do 200 ml
kolby, dodaje się kilka kamyków wrzennych i destyluje ogrzewając kolbę czaszą grzejną. Zbiera się frakcję
wrzącą 100 – 103
°C. Wydajność 155 g (95%).
Uwagi. (1) Odpowiednie medium reakcyjne można przygotować inaczej. Rozpuszcza się 240 g bromku potasu
w 400 ml ciepłej wody i po ostudzeniu, ostrożnie dodaje mieszając, 200 ml stęż. kwasu siarkowego tak, aby
temperatura nie przekroczyła 40 °C. Mieszaninę chłodzi się do temp. 15
°C i odsącza wydzielony kwaśny
3

siarczan potasu. Do przesączu dodaje się butan-1-ol a następnie, ostrożnie kolejną porcję 120 ml stężonego
kwasu siarkowego. Mieszaninę ogrzewa się w temp. wrzenia, pod chłodnicą zwrotną przez 3 – 4 godziny.
(2) Destylacja jest właściwie destylacją z parą wodną. Skraplające się w chłodnicy pary tworzą dwie
niemieszające się ciecze. Stąd mowa o oleistych kroplach związku organicznego spływających z chłodnicy
razem z wodą. Po wydestylowaniu bromku butylu zaczyna spływać jednorodna, klarowna ciecz - woda.
(3) Surowy bromek zawiera niewielką ilość nieprzereagowanego alkoholu i prawdopodobnie nieco eteru
dibutylowego (tw. 141
°C) (pomyśl jak dochodzi do powstania tego związku?). W większości przypadków do
usunięcia nieprzereagowanego alkoholu wystarczy przemywanie stężonym kwasem solnym. Zarówno alkohol
jak i eter można usunąć przemywając 11- 12 ml stęż. kwasu siarkowego, który nie reaguje z bromkiem butylu.
m-NITROANILINA
Równania zachodzących kolejno reakcji:
Na
2
S + Sx
NaSSx-1SNa
H
2
O
t
+
tiosiarczan sodu
m-nitroanilina
wielosiarczek sodu
m-dinitrobenzen
+ Na
2
S
2
O
3
+ Sn
NaSSnSNa
NH
2
NO
2
NO
2
NO
2
Wszystkie aminy mają wspólną cechę wynikającą z obecności tzw. wolnej pary elektorów na atomie azotu. Jest
nią zasadowość. Jej konsekwencją jest tworzenie z kwasami Broensteda soli amoniowych. Zdolność atomu
azotu do wiązania protonu, a więc jego zasadowość, zależy od rodzaju i liczby podstawników z nim związanych.
Aminy alifatyczne są zasadami silniejszymi niż amoniak i aminy aromatyczne. Można to wytłumaczyć dodatnim
efektem indukcyjnym grup alkilowych, który zwiększając gęstość elektronową na atomie azotu ułatwia
przyłączenie protonu. Alifatyczna amina drugorzędowa jest silniejszą zasadą niż amina pierwszorzędowa.
Zasadowość odpowiedniej aminy trzeciorzędowej jest jednak mniejsza niż drugorzędowej. Należy pamiętać, że
zasadowość amin odnosi się do roztworów wodnych, w których kationy amoniowe są w różny, zależny od
rzędowości aminy, sposób solwatowane. Wpływa to na ich stabilność, a więc także na położenie równowagi
kwasowo-zasadowej. Oczekiwany monotoniczny wzrost zasadowości ze wzrostem rzędowości został
stwierdzony w fazie gazowej (brak solwatacji).
Aminy aromatyczne są znacznie słabszymi zasadami niż aminy alifatyczne i amoniak. Jest to związane z
występującym w tych związkach sprzężeniem orbitalu p atomu azotu z układem
elektronowym pierścienia
aromatycznego. Dzięki wynikającej stąd delokalizacji elektronów cząsteczka aminy uzyskuje pewną stabilizację.
Dlatego przyłączenie protonu, które likwiduje tę stabilizację, wymaga dostarczenia wyższej energii, niż w
przypadku amin alifatycznych. Zasadowość amin aromatycznych zależy od podstawników znajdujących się przy
4
pierścieniu aromatycznym. Podstawniki elektronoakceptorowe w pozycjach orto i para zmniejszają a
elektronodonorowe zwiększają zasadowość.
Preparat wykonuje się pod wyciągiem.
W zlewce rozpuszcza się 40 g (0,167 mol) krystalicznego siarczku sodu, Na
2
S
9H
2
O, w 150 ml wody, dodaje 10
g (0,31 mol) sproszkowanej siarki i ogrzewa np. palnikiem przez płytkę metalową umieszczoną na trójnogu, do
otrzymania klarownego, pomarańczowego roztworu.
W kolbie kulistej z trzema szyjami poj. 1 l zaopatrzonej w mieszadło mechaniczne, chłodnicę zwrotną i
wkraplacz umieszcza się 25,2 g (0,15 mol) m-dinitrobenzenu i 200 ml wody. Kolbę umieszcza się w czaszy
grzejnej. (Czaszę należy zamontować tak, aby w każdej chwili można ją było łatwo usunąć.) Mieszając
doprowadza się mieszaninę do wrzenia. m-Dinitrobenzen ulega stopieniu; stanowi odrębną, niemieszającą się z
wodą fazę. Do wkraplacza wlewa się roztwór wielosiarczku sodu i wkrapla go do kolby (30-45 min) utrzymując
jej zawartość w temperaturze wrzenia i energicznie mieszając dla zwiększenia powierzchni styku między fazami.
Ogrzewanie kontynuuje się przez dalsze 20 min. Kolbę chłodzi się do temperatury pokojowej cały czas
energicznie mieszając. Mieszanie zapobiega zakrzepnięciu mieszaniny organicznej w bryłę trudną do wyjęcia z
kolby. (Jeśli to jednak nastąpi, to mieszaninę ponownie ogrzewa się do stopienia warstwy organicznej operację
schładzania powtarza się.) Wytrącony osad zawierający produkt, nieprzereagowany substrat i siarkę odsącza się
pod zmniejszonym ciśnieniem, przemywa wodą i przenosi do zlewki poj. 600 ml zawierającej 150 ml H
2
O i 35
ml stęż. kwasu solnego. Zawartość zlewki ogrzewa się w temp. wrzenia przez 15 min. Powstała sól amoniowa
produktu rozpuszcza się w roztworze kwasu. Pozostały osad odsącza się pod zmniejszonym ciśnieniem. Do
ostudzonego przesączu dodaje się nadmiar stęż. wodnego roztworu amoniaku do uzyskania odczynu silnie
zasadowego (wg papierka uniwersalnego). Następuje wytrącenie m-nitroaniliny (wyjaśnij dlaczego), którą po
odsączeniu krystalizuje się z wody (ok. 700 ml) używając węgiel aktywny. Otrzymuje się zwykle 12 g (58%)
produktu o tt. 114
C.
NH
2
NO
2
NH
3
Cl
NO
2
chlorek m-nitrofenyloamoniowy
lub chlorek m-nitroaniliniowy
HCl
+NH
4
OH
-NH
4
Cl
NH
2
NO
2
chlorek amonu
JODOBENZEN
Kwas azotawy [azotowy(III)] jest związkiem nietrwałym. Wytwarza się go bezpośrednio w środowisku reakcji
działając na azotyn sodu silnym kwasem mineralnym, np. solnym lub siarkowym.
NaNO
2
+ HCl
HO N O + NaCl
HO N O + H
3
O
H
2
O N O + H
2
O
2H
2
O + N O
kation
nitrozoniowy
5

W warunkach reakcji kwas azotawy jest źródłem jonów nitrozoniowych, które natychmiast reagują z aminami.
Produkt reakcji zależy od budowy aminy. Aromatyczne aminy pierwszorzędowe reagują z kwasem azotawym
tworząc sole diazoniowe, które są trwałe w temperaturze 0
5 C.
PhNH
2
+ N=O
Ph N N=O
H
H
H
2
O
-H
3
O
Ph N N=O
H
Ph N N OH
-H
2
O
+H
3
O
Ph N N OH
2
Ph N N
Ph N N + H
2
O
C
6
H
5
NH
2
+ NaNO
2
+ 2HCl
C
6
H
5
N N Cl + 2H
2
O + NaCl
anilina
chlorek benzenodiazoniowy
0-5 °C
Ilość użytego do diazowania kwasu musi być większa (zwykle stosuje się 3 mole kwasu mineralnego na 1 mol
diazowanej aminy) niż to wynika z równania stechiometrycznego (2 mole kwasu na 1 mol aminy). W silnie
kwaśnym środowisku amina w trakcie całego procesu diazowania występuje w postaci soli aniliniowej. Przy
zbyt małym stężeniu kwasu następuje cofnięcie równowagi protonowania aminy, co prowadzi do sprzężenia
powstałej już soli diazoniowej z substratem.
PhN
2
Cl + PhNH
2
PhN=N NHPh + HCl
chlorek
benzenodiazoniowy
diazoaminobenzen
Dodanie kwasu, np. solnego cofa tę reakcję.
Podobnie reagują aminy pierwszorzędowe alifatyczne, ale powstające z nich sole diazoniowe są nietrwałe i
nawet w niskiej temperaturze samorzutnie rozkładają się z wydzieleniem cząsteczki azotu i utworzeniem
karbokationu alkilowego, który następnie ulega rozmaitym możliwym przegrupowaniom oraz reakcjom z
różnymi, obecnymi w środowisku reakcji nukleofilami.
Aminy drugorzędowe alifatyczne i aromatyczne reagują z kwasem azotawym podobnie. W obu przypadkach
powstają żółte, oleiste pochodne N-nitrozowe.
Ph N CH
3
NO
[HONO]
Ph NH CH
3
N-nitrozodimetyloamina
dimetyloamina
(CH
3
)
2
N N O
[HONO]
(CH
3
)
2
NH
N-metylo-N-nitrozoanilinaa
Aminy alifatyczne trzeciorzędowe reagują z kwasem azotawym dając złożoną mieszaninę produktów i dlatego
reakcja ta nie ma znaczenia preparatywnego. Trzeciorzędowe aminy aromatyczne ulegają reakcji nitrozowania w
pierścieniu (reakcja substytucji elektrofilowej) dając p-nitrozo podstawione pochodne.
6
N(CH
3
)
2
[HONO]
0-5 °C
N(CH
3
)
2
NO
N,N-dimetyloanilina
N,N-dimetylo-4-nitrozoanilina
Grupę diazoniową można zastąpić innym podstawnikiem, np.:
NaBF
4
KI
Ar I
t
Ar CN
t
CuCN, NaCN
Ar Br
t
CuBr, HBr
Ar Cl
t
CuCl, HCl
Ar
N
2
Cl
Ar OH
Ar
N
2
BF
4
piroliza
Ar F
NaBF
4
Ar
N
2
BF
4
NaNO
2
aq
Cu
Ar NO
2
Ar
N
2
OSO
3
H
H
2
SO
4
,aq/
t
H
3
PO
2
Ar H
reakcje Sandmayera
lub Cu
2
O, Cu ,H
2
O
Kation diazoniowy może brać udział w reakcji aromatycznej substytucji elektrofilowej. Jednak, ze względu na
jego słabo elektrofilowy charakter, do zajścia reakcji niezbędne jest, aby pierścień aromatyczny reagującego z
nim arenu miał znacznie zwiększoną gęstość elektronową. Dlatego reakcje S
E
z udziałem kationu diazoniowego
zachodzą tylko z pochodnymi aniliny i fenolanów. Reakcję tego typu nazywa się sprzęganiem. W jej wyniku
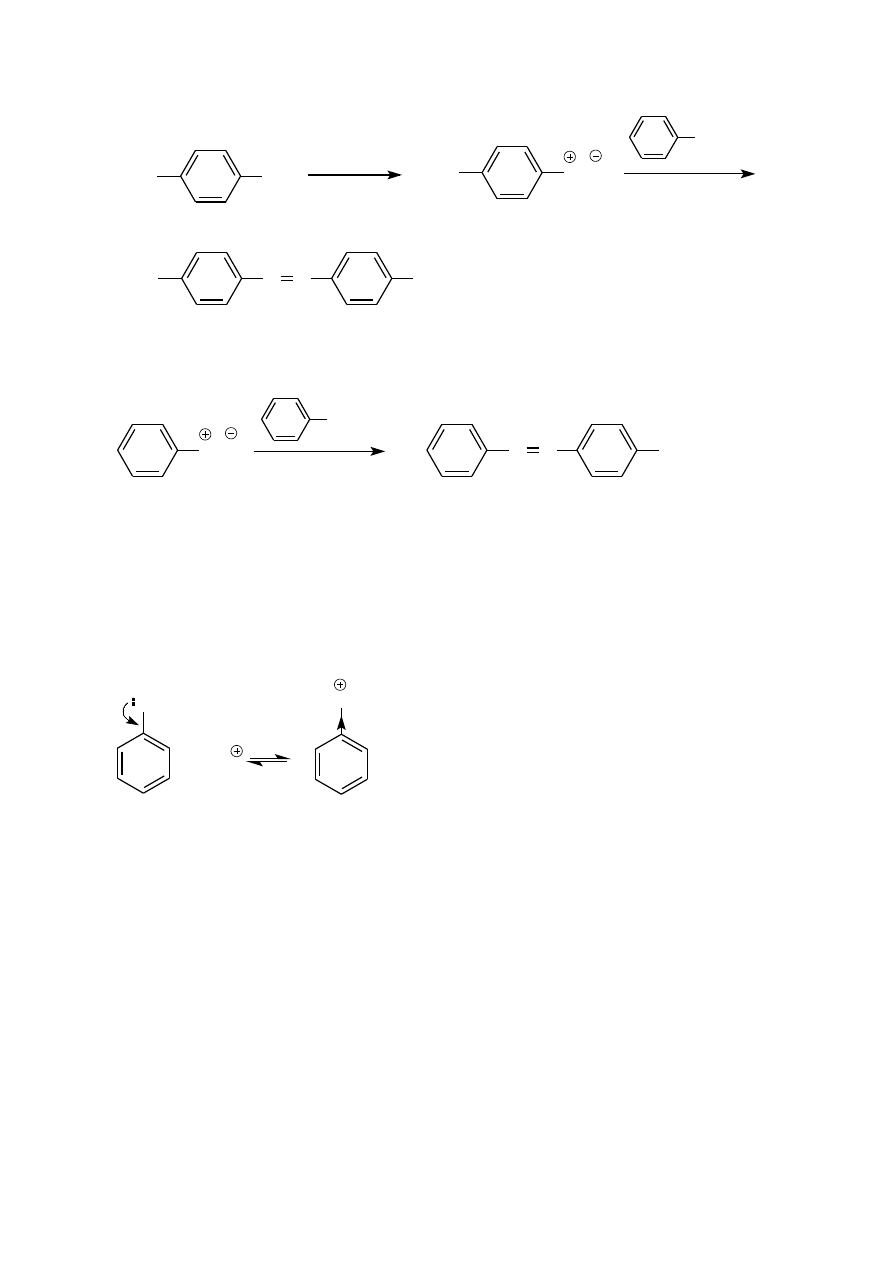
powstają związki azowe, które w zależności od budowy posiadają różne barwy. Związki azowe są szeroko
stosowane jako barwniki w przemyśle farbiarskim i spożywczym a także jako wskaźniki kwasowo-zasadowe.
7
HCl, 0-5 °C
HO
3
S
N
2
Cl
NaNO
2
,
HO
3
S
NH
2
N(CH
3
)
2
pH ~5
HO
3
S
N N
N(CH
3
)
2
kwas sulfanilowy
N,N-dimetyloanilina
oranż metylowy
kwas 4-(4-dimetyloaminofenyloazo)benzenosulfonowy
N
2
Cl
pH ~5
N,N-dimetyloanilina
N N
N(CH
3
)
2
żółcień masłowa; 4-dimetyloaminoazobenzen
N(CH
3
)
2
Z podanych przykładów można wywnioskować, że sole diazoniowe sprzęgają się z aminami w środowisku słabo
kwaśnym. Przy zbyt niskim pH cała ilość aminy występuje w postaci soli aniliniowej, w której pierścień
aromatyczny jest zubożony w elektrony (grupa
NR
3
+
jest podstawnikiem silnie elektronoakceptorowym), co
uniemożliwia zajście reakcji ze słabym elektrofilem jakim jest kation arenodiazoniowy.
NH
2
+ H
NH
3
Z drugiej strony w środowisku zbyt alkalicznym sól diazoniowa przekształca się w kwas benzenodiazowy, Ph-
N=N-OH, lub jego sól, Ph-N=N-O¯ Na
+
; żaden z tych związków nie ma właściwości elektrofilowych.
Stwierdzono doświadczalnie, że sprzęganie soli diazoniowych z aminami osiąga maksymalną szybkość przy pH
5-9.
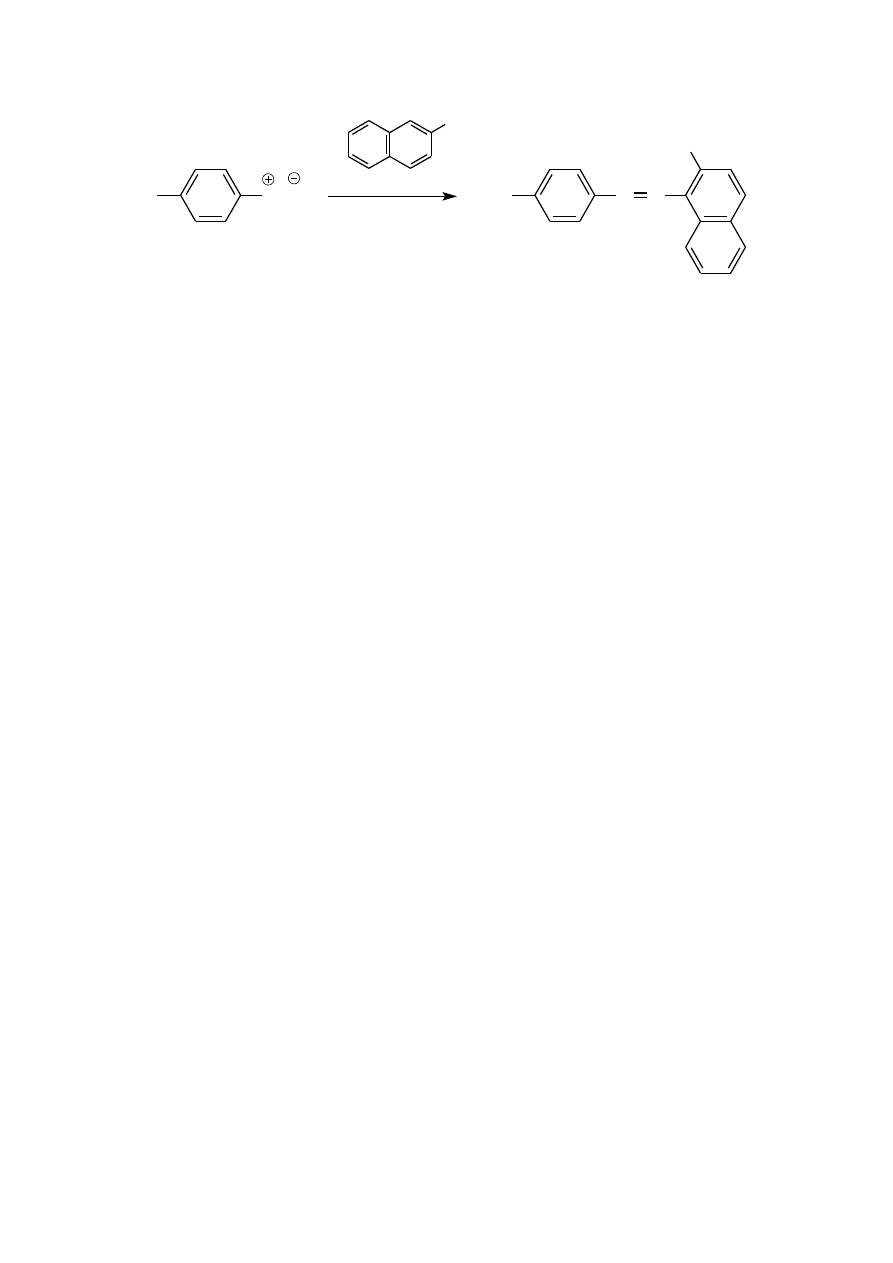
W przypadku sprzęgania z fenolami korzystniejsze jest środowisko słabo zasadowe (pH ~10), w którym fenole
występują w postaci anionów fenolanowych; pierścień aromatyczny anionu jest silniej wzbogacony w elektrony
niż sam fenol, dlatego łatwiej ulega reakcji S
E
z kationem diazoniowym.
8
HO
3
S
N
2
Cl
OH
naft-2-ol
pH ~10
HO
3
S
N N
HO
oranż II; kwas
4-(2-hydroksynaft-1-yloazo)-
benzenosulfonowy
Reakcję wykonuje się pod wyciągiem.
W kolbie kulistej poj. 500 ml z trzema szyjami zaopatrzonej w mieszadło mechaniczne, termometr sięgający
poniżej górnego poziomu mieszaniny reakcyjnej i wkraplacz z wyrównywaczem ciśnienia rozpuszcza się 20,0 g
(19,6 ml, 0,215 mol) aniliny w mieszaninie 55 ml stęż. kwasu solnego (1) i 55 ml wody. Kolbę zanurza się w
łaźni z pokruszonym, obficie posolonym lodem i niewielką ilością wody; roztwór chłodzi się mieszając do temp.
poniżej 5 ºC. Sporządza się roztwór 16,0 g (0,23 mol) azotynu sodu w 75 ml wody i chłodzi go w łaźni lodowej i
umieszcza we wkraplaczu. Wkraplacza nie należy zamykać korkiem, aby ciśnienie w kolbie mogło się bez
przeszkód wyrównywać z atmosferycznym. (Zamiast wkraplacza z wyrównywaczem ciśnienia można też użyć
rozdzielacza. Należy go tak zamocować, aby między nóżką a ścianką szyi kolby była szczelina umożliwiająca
wyrównywanie ciśnienia). Roztwór azotynu sodu wkrapla się następnie (2) do chłodzonego i energicznie
mieszanego roztworu chlorowodorku aniliny. W trakcie reakcji wydziela się ciepło. Nie należy dopuścić do
tego, aby temperatura mieszaniny reakcyjnej wzrosła powyżej 10 ºC (jeśli zajdzie potrzeba, do kolby można
dodać kilka kawałków lodu); w przeciwnym razie związek diazoniowy i kwas azotawy rozłożą się w znacznym
stopniu. Ostatnie ok. 5% roztworu azotynu sodu dodaje się małymi porcjami. Po 3–4 min mieszania od dodania
porcji, kroplę mieszaniny reakcyjnej (rozcieńczoną 3–4 kroplami wody) bada się za pomocą papierka
jodowoskrobiowego (3 ); jeśli nie zaobserwuje się natychmiastowego niebieskiego zabarwienia papierka w
miejscu zetknięcia z roztworem, do mieszaniny reakcyjnej należy dodać nową porcję roztworu azotynu. Po 3–4
min ponownie wykonuje się test z papierkiem jodowoskrobiowym. W ten sposób postępuje się do momentu, aż
mieszana reakcyjna będzie zawierać niewielki, utrzymujący się w czasie nadmiar kwasu azotawego. Kwas
azotawy ma właściwości utleniające; utlenia np. jony jodkowe do jodu: 2NaI + 2NaNO
2
+
4HCl à I
2
+ N
2
O
2
+
4NaCl + 2H
2
O. (Ta reakcja jest podstawą działania papierków jodowoskrobiowych. Są one nasączone jodkiem
sodu i skrobią. W obecności utleniacza powstaje jod tworzący ze skrobią fioletowy kompleks.) Obecność zbyt
dużego nadmiaru kwasu azotawego byłaby więc niepożądana w następnym etapie syntezy. Dlatego do
mieszaniny reakcyjnej dodaje się mocznik w takiej ilości, aby test z papierkiem jodoskrobiowym był negatywny.
Mocznik reaguje z kwasem azotawym zgodnie z równaniem: H
2
NCONH
2
+ 2HNO
2
à 2N
2
+ CO
2
+ 3H
2
O.
Do mieszanego roztworu chlorku benzenodiazoniowego dodaje się powoli roztwór 36,0 g (0,216 mol)
jodku potasu w 40 ml wody. Obserwuje się wydzielanie azotu. Mieszaninę pozostawia się na kilka godzin.
Kolbę zaopatruje się w chłodnicę powietrzną i ostrożnie ogrzewa na wrzącej łaźni wodnej do chwili, gdy
przestanie wydzielać się gaz. (Jeśli mieszanina jest pozostawiona na dłuższy okres czasu ogrzewanie jest
zbędne.) Po ochłodzeniu górną warstwę wodną dekantuje się możliwie dokładnie, a pozostałość w kolbie
wstrząsając alkalizuje, ostrożnie dodając taką ilość 10–proc. roztworu wodorotlenku sodu, aby kropla
mieszaniny (pobrana za pomocą szklanej bagietki) zabarwiła papierek wskaźnikowy na niebiesko. Fenol obecny
9

w mieszaninie jest za pomocą wodorotlenku przekształcany w fenolan sodu: C
6
H
5
OH + NaOH à C
6
H
5
ONa +
H
2
O, który (w przeciwieństwie do fenolu) nie jest lotny z parą wodną. Kolbę umieszcza się w zestawie do
destylacji z parą wodną (boczne szyje kolby należy zatkać korkami). Destylację prowadzi się do czasu, gdy z
chłodnicy zacznie spływać jednorodna ciecz – woda. Destylat przenosi się do rozdzielacza, dolną warstwę
jodobenzenu zlewa się do małej kolbki stożkowej. Surowy jodobenzen powinien mieć barwę jasnożółtą. Jeżeli
jest ciemny (zabarwienie pochodzi zwykle od rozpuszczonego jodu), to zawraca się go do rozdzielacza i
wytrząsa z niewielką ilością roztworu pirosiarczynu sodu ( 2I
2
+ Na
2
S
2
O
5
+ 3H
2
O à 4HI + 2NaHSO
4
) , aż do
zmiany barwy na jasnożółtą. Po oddzieleniu jodobenzenu od warstwy wodnej do czystej erlenmajerki suszy się
go bezwodnym chlorkiem wapnia lub siarczanem magnezu (1 g) i sączy przez sączek karbowany do małej
kolbki destylacyjnej zaopatrzonej w krótką chłodnicę powietrzną. Destyluje się (można dodać małą ilość pyłu
miedzi) z czaszy grzejnej zbierając frakcję wrzącą w temp. 185–190 ºC (4). Wydajność jodobenzenu (w postaci
prawie bezbarwnej cieczy) wynosi 33 g (74 %); na słońcu (świetle) związek stopniowo żółknie.
Uwagi. (1) Podczas obliczania ilości kwasu potrzebnego do reakcji diazowania warto pamiętać, że 100 ml
stężonego kwasu solnego (d 1,18) zawiera 42,4 g HCl, a 100 ml stężonego kwasu siarkowego (d 1,84) – 176 g
H
2
SO
4
.
(2) Szczególnie przy produkcji na dużą skalę, wskazane jest dodawanie roztworu azotynu sodu z wkraplacza,
umieszczonego w taki sposób, aby koniec nóżki był zanurzony głęboko pod powierzchnię cieczy. Wówczas
zapobiega się stratom kwasu azotawego, na skutek rozkładu do tlenków azotu zachodzącego na powierzchni
cieczy.
(3) Jakość papierków jodowoskrobiowych należy sprawdzić za pomocą zakwaszonego roztworu azotynu sodu.
Zdarza się, że dostępne w handlu papierki jodowoskrobiowe stają się bezużyteczne, jeśli są zbyt długo
przechowywane. Roztwór musi przez cały czas zawierać nadmiar kwasu mineralnego, tzn. barwić na czerwono
papierek uniwersalny.
(4) Jodobenzen lepiej jest destylować pod zmniejszonym ciśnieniem i zbierać frakcję wrzącą w temp. 77–80
ºC/20 mmHg lub 63–64 ºC/8 mmHg.
p-JODOTOLUEN
Syntezę przeprowadza się analogicznie do syntezy jodobenzenu wprowadzając niewielkie zmiany przedstawione
poniżej. Dlatego przed przystąpieniem do wykonania preparatu należy zapoznać się z wiadomościami
wstępnymi, przepisem i uwagami dotyczącymi otrzymywania jodobenzenu. Używa się 26,8 g (0,25 mol) p–
toluidyny, 63 ml stężonego kwasu solnego i 63 ml wody. W razie potrzeby, np. gdy toluidyna jest w postaci
większych bryłek, mieszaninę podgrzewa się do czasu rozpuszczenia aminy. Roztwór chłodzi się (energicznie
mieszając) do temp. 0–5 ºC przez zanurzenie kolby w mieszaninie lodu z solą i dodanie do niego niewielkiej
ilości pokruszonego lodu. Aminę diazuje się, dodając roztwór 18,5 g (0,27 mol) azotynu sodu w 40 ml wody,
jednocześnie mieszając zawartość kolby i w miarę możliwości utrzymując jej temperaturę w przedziale 0–5 ºC;
nie należy pozwolić aby temp. wzrosła powyżej 10 ºC. Następnie stopniowo dodaje się roztwór 44 g (0,265 mol)
jodku potasu w równoważnej (wagowo) ilości wody, przez cały czas mieszając zawartość kolby. Mieszaninę
pozostawia się na 1 godz. w temp. pokojowej, a następnie ostrożnie ogrzewa na łaźni wodnej do czasu
zakończenia wydzielania azotu. Po ochłodzeniu, na dnie kolby wydziela się ciemno zabarwiony olej, który
10

wkrótce się zestala. Z mieszaniny oddziela się jak największą część warstwy wodnej, a do ciemnej pozostałości
dodaje 1–2 g pirosiarczynu sodu, w celu usunięcia ciemnego zabarwienia (wskazane jest podgrzanie
mieszaniny). Mieszaninę alkalizuje się 10–proc. roztworem wodorotlenku sodu (aby związać powstały
ewentualnie krezol) i destyluje z parą wodną. p–Jodotoluen zestala się w chłodnicy; należy wówczas na pewien
czas zakręcić wodę w chłodnicy i zaczekać aż ciało stałe stopi się pod wpływem gorącej pary wodnej i spłynie z
chłodnicy do odbieralnika. Ciało stałe zebrane w odbieralniku odsącza się i suszy. Surowy produkt można
przekrystalizować z etanolu. Wydajność p–jodotoluenu (w postaci bezbarwnych płatków) wynosi 50 g (92%); tt.
35 ºC, tw. 211–212 ºC.
p–JODONITROBENZEN.
Niektóre czynności wykonuje się podobnie jak przy syntezie jodobenzenu. Dlatego przed przystąpieniem do
wykonania preparatu należy zapoznać się z wiadomościami wstępnymi, przepisem i uwagami dotyczącymi
otrzymywania jodobenzenu. Mieszaninę 49,7 g (0,36 mol) p–nitroaniliny, 75 g (41 ml) stężonego kwasu
siarkowego i 300 ml wody miesza się przez 1 godz., a następnie chłodzi do temp. 0–5 ºC i diazuje roztworem 25
g (0,36 mol) azotynu sodu w 75 ml wody. Zimny roztwór sączy się przez lejek Büchnera. Przesącz umieszcza
się w zlewce i mieszając dodaje do niego roztwór 99,6 g (0,6 mol) jodku potasu w 300 ml wody. Wydzielone
ciało stałe odsącza się pod zmniejszonym ciśnieniem i krystalizuje z etanolu. Wydajność p–jodonitrobenzenu
wynosi 73 g (83%), tt. 171 ºC.
ACETANILID
Pierwszorzędowe aminy aromatyczne ogrzewane z bezwodnikiem octowym łatwo reagują dając monoacetylowe
pochodne.
Ar
NH
2
+ (CH
3
CO)
2
O à Ar
NHC(O)CH
3
+ CH
3
CO
2
H
Jest to reakcja przebiegająca według mechanizmu addycji-eliminacji charakterystycznego dla przekształcania
kwasów karbosylowych w ich pochodne (np. estryfikacja: kwas à ester) oraz jednych pochodnych kwasów
karboksylowych w inne (np. omawiany przykład: bezwodnik à amid).
11
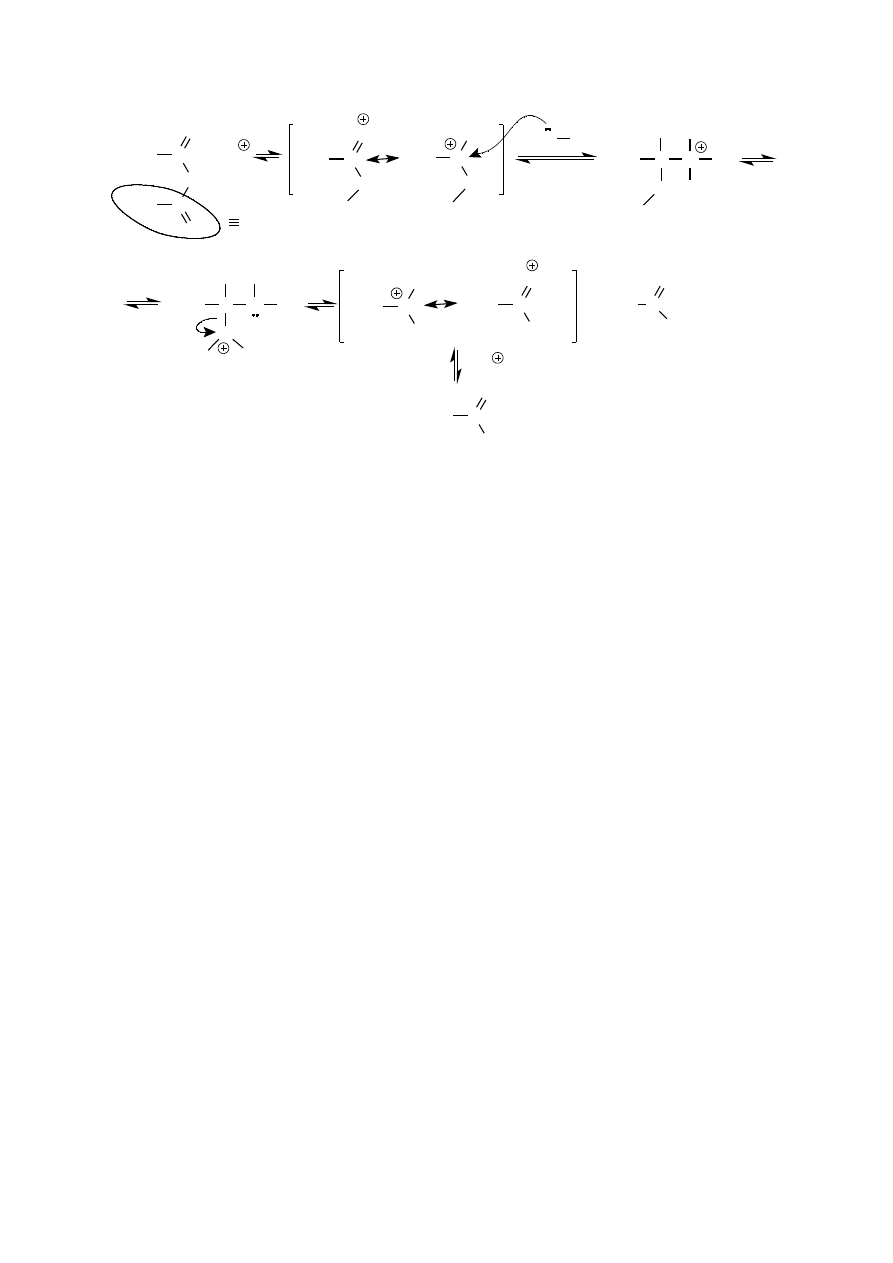
H
3
C C
O
C
H
3
C
O
O
Ac
+ H
H
3
C C
O
OH
Ac
H
3
C C
O
OH
Ac
H
2
N Ph
H
3
C C
O
OH
N
H
H
Ph
Ac
H
3
C C
O
OH
N
H
Ph
H
Ac
H
3
C C
NHPh
OH
H
3
C C
NHPh
OH
+ CH
3
C
O
OH
- H
H
3
C C
NHPh
O
acetanilid
(N-fenyloacetamid)
addycja
eliminacja
Stosując nadmiar bezwodnika octowego można też otrzymać pochodne diacetylowe.
Ar
NHC(O)CH
3
+ (CH
3
CO)
2
O à Ar
N[C(O)CH
3
]
2
+ CH
3
CO
2
H
Jednak są one na tyle nietrwałe, że krystalizowane z wody ulegają hydrolizie do pochodnych monoacetylowych.
Dzięki temu acetanilidy otrzymuje się łatwo i z wysokimi wydajnościami.
W kolbie kulistej poj. 500 ml, zaopatrzonej w chłodnicę zwrotną, umieszcza się 20,5 g (20 ml, 0,22 mol) aniliny,
21,5 g (20 ml, 0,21 mol) bezwodnika octowego, 21,0 g (20 ml, 0,35 mol) lodowatego (czyli 100%) kwasu
octowego i 0,1 g pyłu cynkowego (dzięki redukującym właściwościom pył cynkowy zapobiega utlenianiu się
aniliny podczas ogrzewania). Mieszaninę ogrzewa się łagodnie w temperaturze wrzenia przez 30 min a następnie
gorącą ciecz wylewa się cienkim strumieniem do zlewki poj. 1 l zawierającej 500 ml zimnej wody. Po
oziębieniu, najlepiej w lodzie, surowy produkt odsącza się pod zmniejszonym ciśnieniem, przemywa niewielką
ilością zimnej wody, dokładnie odsysa, rozkłada na bibule i suszy na powietrzu. Otrzymuje się ok. 26 g
acetanilidu o tt. 112-113
C. Surowy produkt krystalizuje się z ok. 500 ml wody z dodatkiem 10 ml etanolu.
Wydajność czystego związku o tt 113-114
C– 21 g (70%).
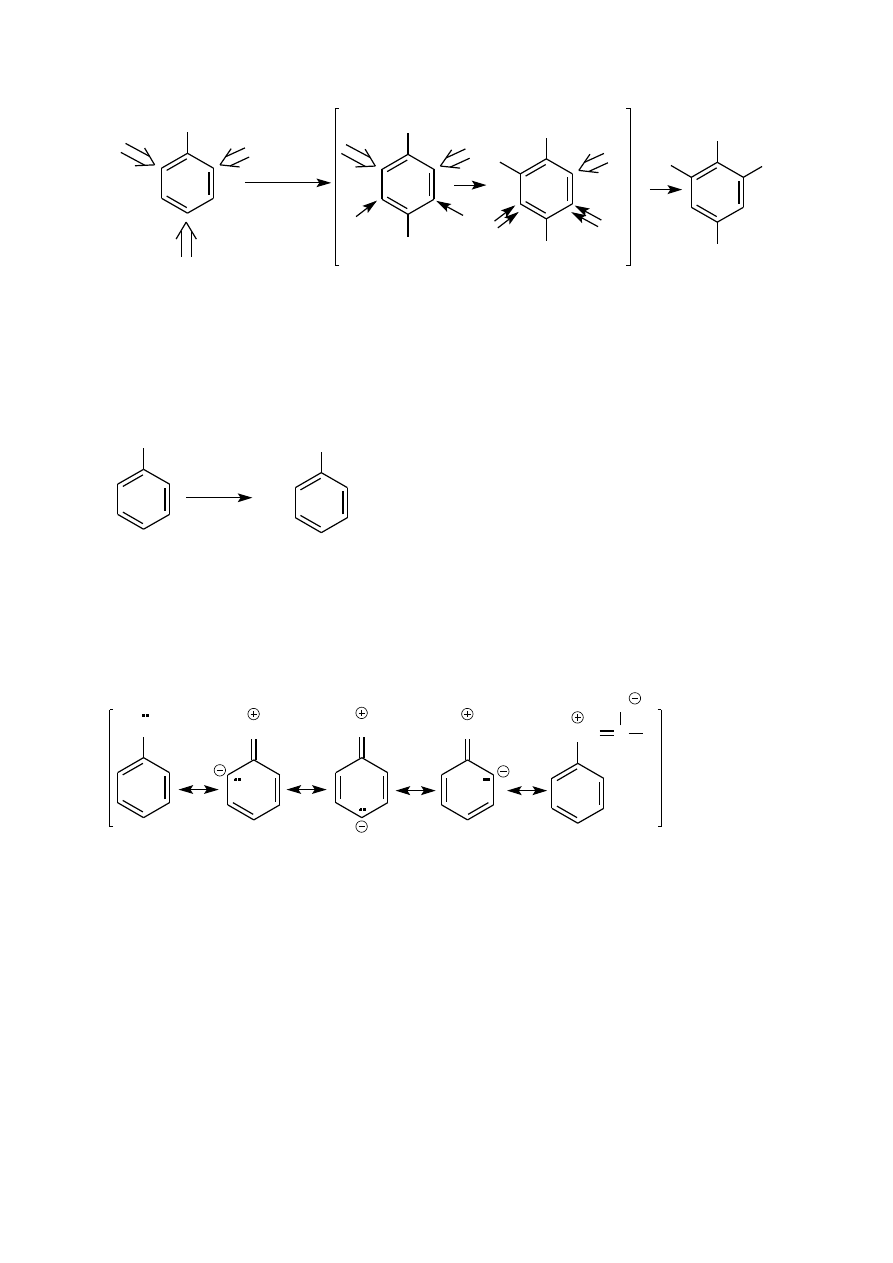
p–BROMOACETANILID i p–BROMOANILINA
Z uwagi na silny aktywujący wpływ grupy NH
2
bromowanie aniliny przebiega natychmiastowo w temperaturze
pokojowej i nie wymaga katalizatora. Reakcja wprowadzenia pierwszego podstawnika przebiega równie łatwo w
pozycji orto jak i para. Ponadto monobromo i dibromopochodne aniliny, podobnie jak sama anilina, łatwo
ulegają dalszemu bromowaniu, przy czym w każdym przypadku kierujący wpływ grupy aminowej (
)
dominuje nad wpływem bromu (à). Z tego powodu jedynym produktem jaki w omawianej reakcji można
otrzymać z dobrą wydajnością jest 2,4,6-tribromoanilina (zob. Preparatyka).
12
NH
2
3Br
2
bez kat.
temp. pok.
NH
2
Br
NH
2
Br
Br
NH
2
Br
Br
Br
anilina
2,4,6-tribromoanilina
Osłabienie elektrodonorowości grupy NH
2
osiąga się przekształcając ją w grupę amidową NHC(O)R w reakcji
acylowania. Jako odczynnika acylującego używa się chlorku lub bezwodnika kwasowego. W praktyce
najczęściej stosuje się bezwodnik octowy; reakcja acetylowania. (Zwróć uwagę na różnicę między terminami
acylowanie i acetylowanie).
NH
2
RCOCl
lub (RCO)
2
O
NHC(O)R
Para elektronowa atomu azotu jest w tych związkach sprzężona z elektronami
grupy karbonylowej, co osłabia
jej sprzężenie z układem
elektronowym pierścienia aromatycznego a tym samym zmniejsza jego podatność na
substytucję elektrofilową.
NHC(O)R
NHC(O)R
NHC(O)R
NHC(O)R
NH C
O
R
Dzięki temu acetanilid można bromować, zachowując łagodne warunki (nadal niepotrzebny jest katalizator, np.
FeBr
3
), otrzymując z dobrą wydajnością p-monopodstawioną pochodną, która po etanolizie w środowisku
kwaśnym (zaproponuj mechanizm tej reakcji) a następnie zalkalizowaniu mieszaniny poreakcyjnej daje p-
bromoanilinę.
13
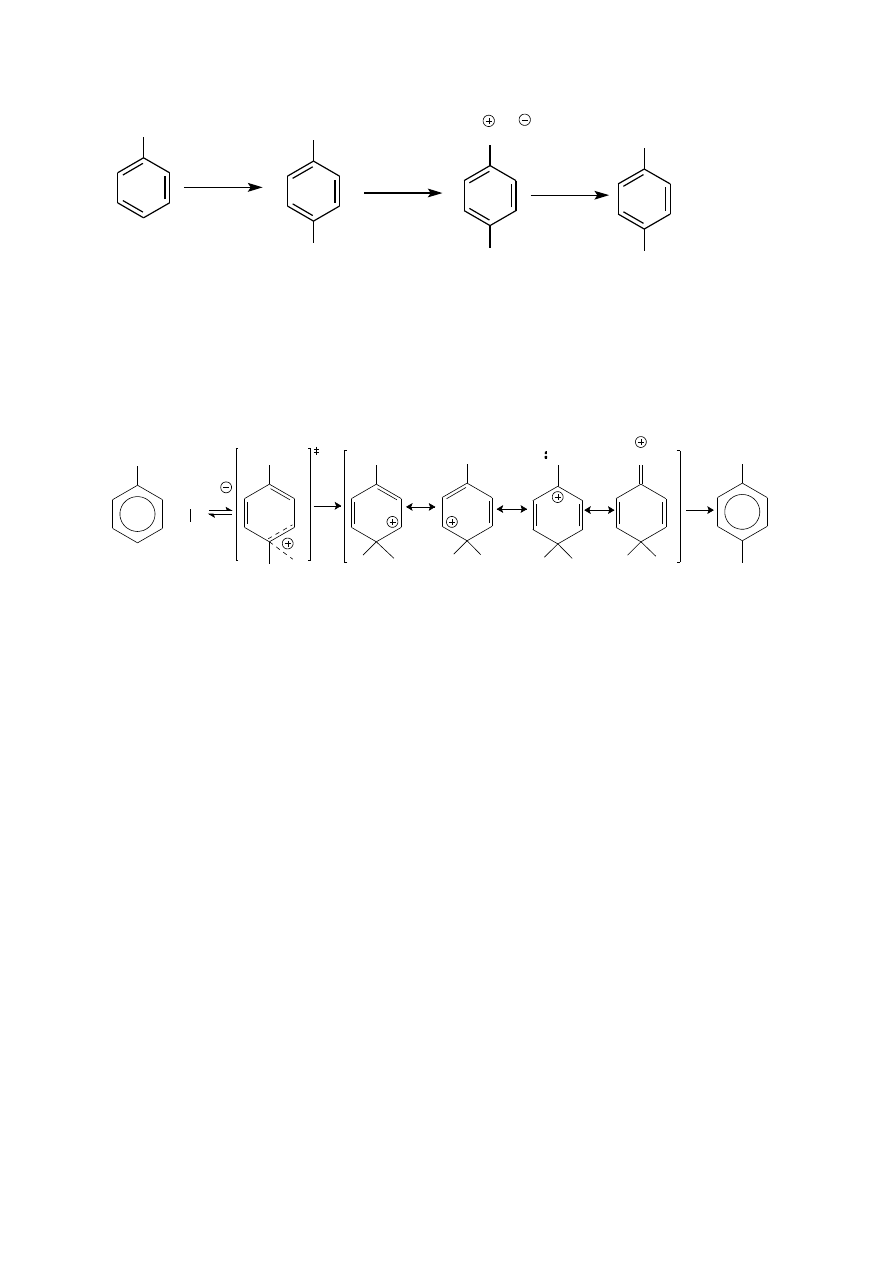
p-bromoanilina
p-bromoacetanilid
acetanilid
NH
3
Cl
Br
NaOHaq
C
2
H
5
OH, HCl,
t
NHC(O)CH
3
Br
Br
2
CH
3
CO
2
H
t. pok.
NHC(O)CH
3
-CH
3
CO
2
C
2
H
5
NH
2
Br
chlorek p-bromoaniliniowy
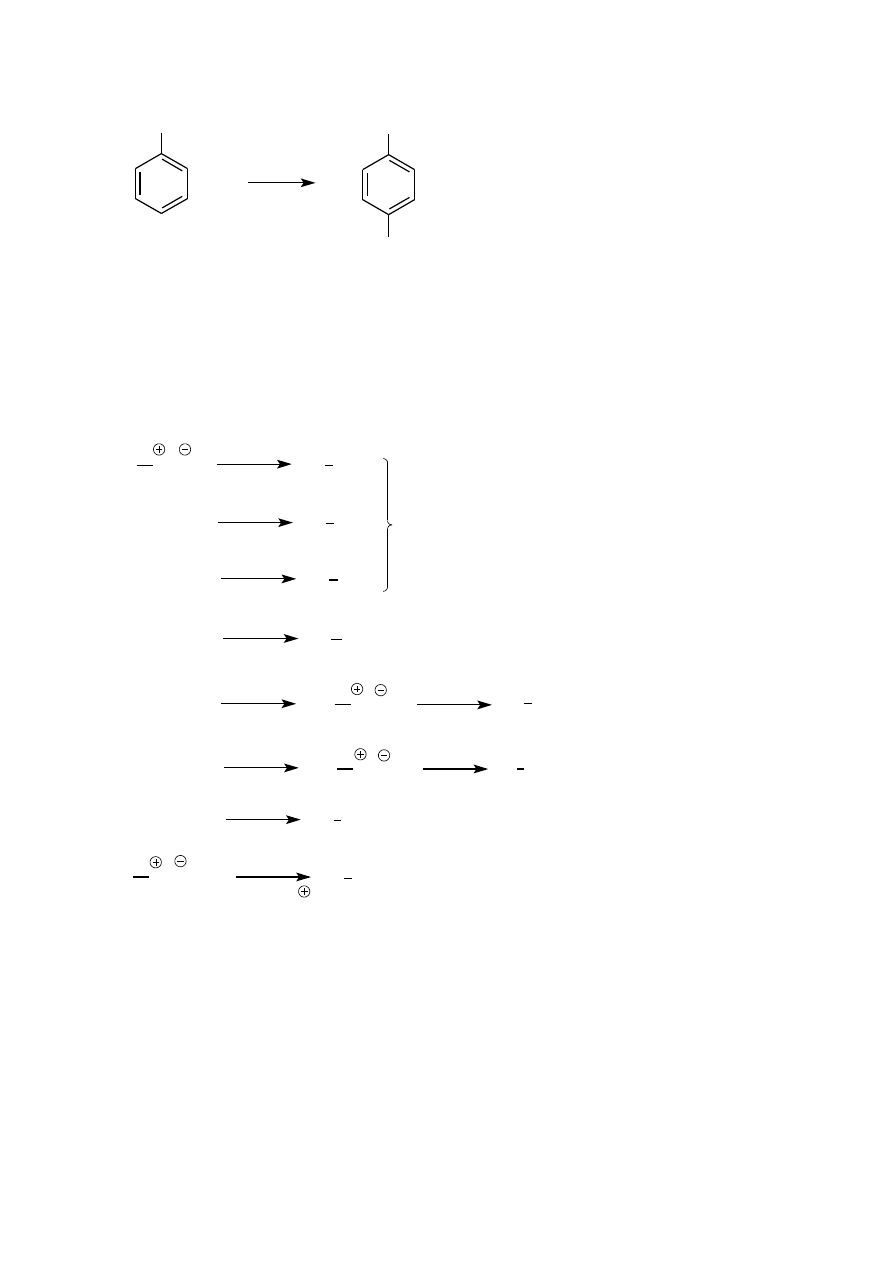
Reakcja bromowania przebiega według niżej przedstawionego mechanizmu. (Wyjaśnij regioselektywność tej
reakcji).
acetanilid
NHAc
Br
H
NHAc
NHAc
Br
H
NHAc
Br
H
NHAc
Br
H
NHAc
Br
H
NHAc
Br
-H
+
kompleks
p
stan przejściowy
Br
Br
+
-Br
Bromowanie acetanilidu. OSTRZEŻENIE: Reakcję należy wykonywać pod sprawnie działającym wyciągiem.
Brom jest substancją w najwyższym stopniu żrącą. Jako ciecz wywołuje bolesne poparzenia. Podczas
operowania bromem należy używać rękawiczek (np. jednorazowych z lateksu). Miejsca oparzone należy
natychmiast zmyć dużą ilością gliceryny. Pary bromu działają drażniąco na drogi oddechowe. Podrażnienia
wywołane wdychaniem par bromu można złagodzić oddychając przez chusteczkę zwilżoną etanolem. Wszystkie
operacje związane z bromem muszą być wykonywane wyłącznie pod wyciągiem. Naczynia zanieczyszczone
bromem należy pod wyciągiem przemyć roztworem pirosiarczynu sodu.
W kolbie stożkowej poj. 350 ml rozpuszcza się 13,5 g (0,1 mol) drobno sproszkowanego acetanilidu w
45 ml lodowatego kwasu octowego. W innej erlenmajerce rozpuszcza się 17 g (5,5 ml, 0,106 mol) bromu w 25
ml lodowatego kwasu octowego; roztwór przenosi się do wkraplacza umieszczonego nad kolbą reakcyjną.
Roztwór bromu dodaje się powoli do stale wstrząsanej (można użyć mieszadła magnetycznego) i wstawionej do
zimnej wody kolby zawierającej roztwór acetanilidu. Po dodaniu całej ilości bromu, zabarwienie mieszaniny
reakcyjnej jest pomarańczowe ze względu na obecność w niej niewielkiego nadmiaru bromu. Część produktu
rekcji może wykrystalizować. W celu zakończenia reakcji mieszaninę reakcyjną pozostawia się w temp.
pokojowej na 30 min, wstrząsając nią od czasu do czasu. Potem mieszaninę reakcyjną wylewa się do 400 ml
wody, a kolbę reakcyjną przemywa się ok. 100 ml wody. Zawiesinę miesza się dokładnie; jeśli jest
pomarańczowo zabarwiona, dodaje się do niej pewną ilość roztworu pirosiarczynu sodu redukującego brom do
rozpuszczalnego w wodzie anionu bromkowego (2Br
2
+ Na
2
S
2
O
5
+ 3H
2
O à 4HBr + 2NaHSO
4
). Wytrącony
osad odsącza się pod zmniejszonym ciśnieniem na lejku Büchnera, dokładnie przemywa zimną wodą, starannie
14

odciska szerokim szklanym korkiem, aby usunąć jak najwięcej wody i krystalizuje z rozcieńczonego metanolu
lub etanolu. Wydajność p–bromoacetanilidu, w postaci bezbarwnych kryształów, wynosi 18 g (84%), tt. 167 ºC.
Etanoliza p–bromoacetanilidu. W kolbie kulistej o poj. 500 ml, zaopatrzonej w chłodnicę zwrotną, rozpuszcza
się 18 g (0,084 mol) p–bromoacetanilidu w 35 ml wrzącego etanolu. Następnie do wrzącego roztworu dodaje się
z wkraplacza z wyrównywaczem ciśnienia umieszczonego na szczycie chłodnicy zwrotnej (górny otwór
wkraplacza powinien być otwarty!) 22 ml stężonego kwasu solnego. Mieszaninę ogrzewa się przez 30–40 min
lub do czasu, gdy pobrana próbka rozcieńczona wodą będzie klarownym roztworem (na czym polega ten test?).
Wtedy mieszaninę rozcieńcza się 150 ml wody, a kolbę zaopatruje się w układ do destylacji. Mieszaninę
destyluje się z czaszy grzejnej zbierając ok. 100 ml destylatu zawierającego octan etylu (w jaki sposób dochodzi
do powstania tego związku?), etanol i wodę. Pozostały w kolbie roztwór chlorku p–bromoaniliniowego wylewa
się do 100 ml lodowatej wody i energicznie mieszając, dodaje taką ilość 5–proc. roztworu wodorotlenku sodu,
aby odczyn mieszaniny był słabo alkaliczny. p–Bromoanilina wydziela się w postaci krzepnącego oleju. Osad
odsącza się pod zmniejszonym ciśnieniem, przemywa zimną wodą, rozkłada na bibule filtracyjnej i suszy na
powietrzu. Wydajność produktu wynosi 14 g (97%), tt. 66 ºC. Zwykle krystalizacja z rozcieńczonego alkoholu
(związana z dużymi stratami) nie jest konieczna.
p–NITROACETANILID i p–NITROANILINA
Aniliny nie poddaje się bezpośredniemu nitrowaniu. Po pierwsze, kwas azotowy ma właściwości utleniające a
wzbogacony w elektrony pierścień aromatyczny jest podatny na utlenianie; prowadzi to do jego destrukcji. Po
drugie, w silnie kwaśnym środowisku mieszaniny nitrującej grupa aminowa przekształca się w grupę amoniową,
która jest podstawnikiem o silnych właściwościach elektronoakceptorowych, kierującym podstawienie
elektrofilowe w pozycję meta. W rezultacie otrzymuje się z niezbyt dobrą wydajnością mieszaninę
regioizomerów nitroanilin.
Osłabienie elektrodonorowości grupy NH
2
osiąga się przekształcając ją w grupę amidową NHC(O)R w reakcji
acylowania. Jako odczynnika acylującego używa się chlorku lub bezwodnika kwasowego. W praktyce
najczęściej stosuje się bezwodnik octowy; reakcja acetylowania. (Zwróć uwagę na różnicę między acylowaniem
a acetylowaniem).
NH
2
RCOCl
lub (RCO)
2
O
NHC(O)R
Para elektronowa atomu azotu jest w tych związkach sprzężona z elektronami
grupy karbonylowej, co osłabia
jej sprzężenie z układem
elektronowym pierścienia aromatycznego a tym samym zmniejsza jego podatność na
substytucję elektrofilową.
15
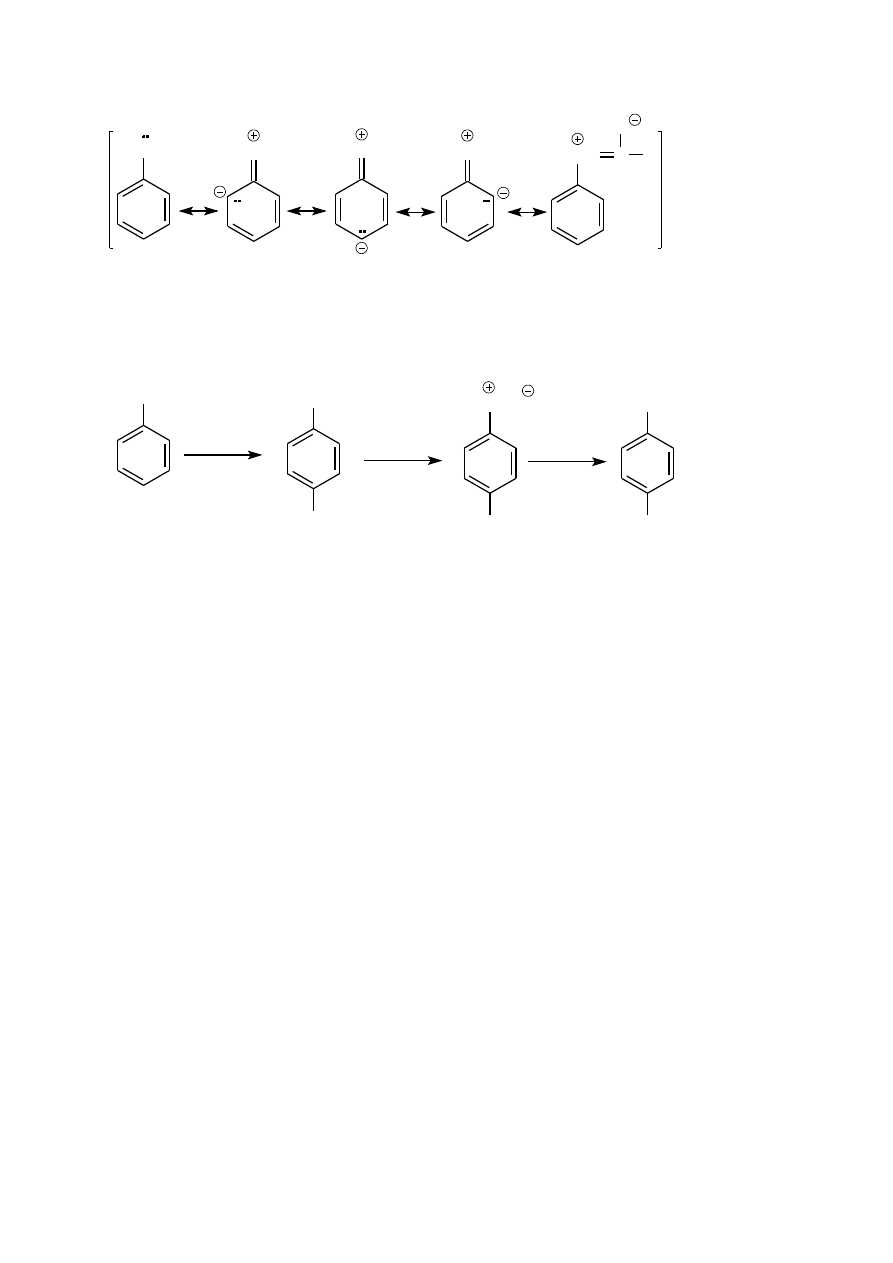
NHC(O)R
NHC(O)R
NHC(O)R
NHC(O)R
NH C
O
R
Dzięki zmniejszonej reaktywności acetanilid można nitrować, zachowując łagodne warunki, otrzymując z dobrą
wydajnością p-monopodstawioną pochodną, która po hydrolizie w środowisku kwaśnym i zalkalizowaniu
mieszaniny poreakcyjnej daje p-nitroanilinę.
wodorosiarczan
p-nitroaniliniowy
p-nitroacetanilid
acetanilid
NH
3
OSO
3
H
NO
2
NaOHaq
H
2
O, H
2
SO
4
,
t
NHC(O)CH
3
NO
2
HNO
3
, H
2
SO
4
CH
3
CO
2
H
t. < 10 °C
NHC(O)CH
3
NH
2
NO
2
p-nitroanilina
Nitrowanie acetanilidu.
W kolbie kulistej z trzema szyjami poj. 500 ml zaopatrzonej w mieszadło mechaniczne, termometr i umieszcza
się 25,0 g (0,185 mol) drobno sproszkowanego i suchego acetanilidu oraz 25 ml lodowatego kwasu octowego.
Do energicznie mieszanej zawartości dodaje się 92 g (50 ml) stężonego kwasu siarkowego. Mieszanina
rozgrzewa się i powstaje klarowny roztwór. W bocznych szyjach kolby umieszcza się termometr i wkraplacz z
wyrównywaczem ciśnienia (lub nóżkę umocowanego do statywu rozdzielacza) zawierający przygotowaną
wcześniej i ochłodzoną mieszaniną 15,5 g (11 ml) stężonego kwasu azotowego i 12,5 g (7 ml) stężonego kwasu
siarkowego. Kolbę umieszcza się w mieszaninie chłodzącej składającej się z lodu i soli, i jej zawartość miesza
się mechanicznie. Kiedy temperatura mieszaniny spadnie do 0–2 ºC, powoli wkrapla się do niej mieszaninę
kwasów, utrzymując temperaturę poniżej 10 ºC. Po zakończeniu dodawania kwasów, usuwa się łaźnię chłodzącą
i pozostawia kolbę na 1 godz. w temp. pokojowej. Potem mieszaninę reakcyjną wylewa się na 250 g
pokruszonego lodu (lub do 500 ml zimnej wody); surowy nitroacetanilid natychmiast się wytrąca. Zawiesinę
pozostawia się na 15 min, a następnie sączy pod zmniejszonym ciśnieniem na lejku Büchnera. Osad przemywa
się kilkakrotnie zimną wodą (do czasu gdy odczyn przesączu będzie obojętny) i dokładnie odciska (1).
Jasnożółty produkt krystalizuje się z etanolu, sączy pod zmniejszonym ciśnieniem, przemywa niewielką ilością
zimnego etanolu i suszy na powietrzu na bibule filtracyjnej. (Żółty o–nitroacetanilid pozostaje w przesączu.)
Wydajność p–nitroacetanilidu (w postaci bezbarwnego, krystalicznego ciała stałego) wynosi 20 g (60%), tt. 214
ºC.
Hydroliza p–nitroacetanilidu. Mieszaninę 15,0 g (0,083 mol) p–nitroacetanilidu i 75 ml 70–proc. roztworu
kwasu siarkowego (2) ogrzewa się pod chłodnicą zwrotną przez 20–30 min lub do czasu, gdy próbka mieszaniny
16
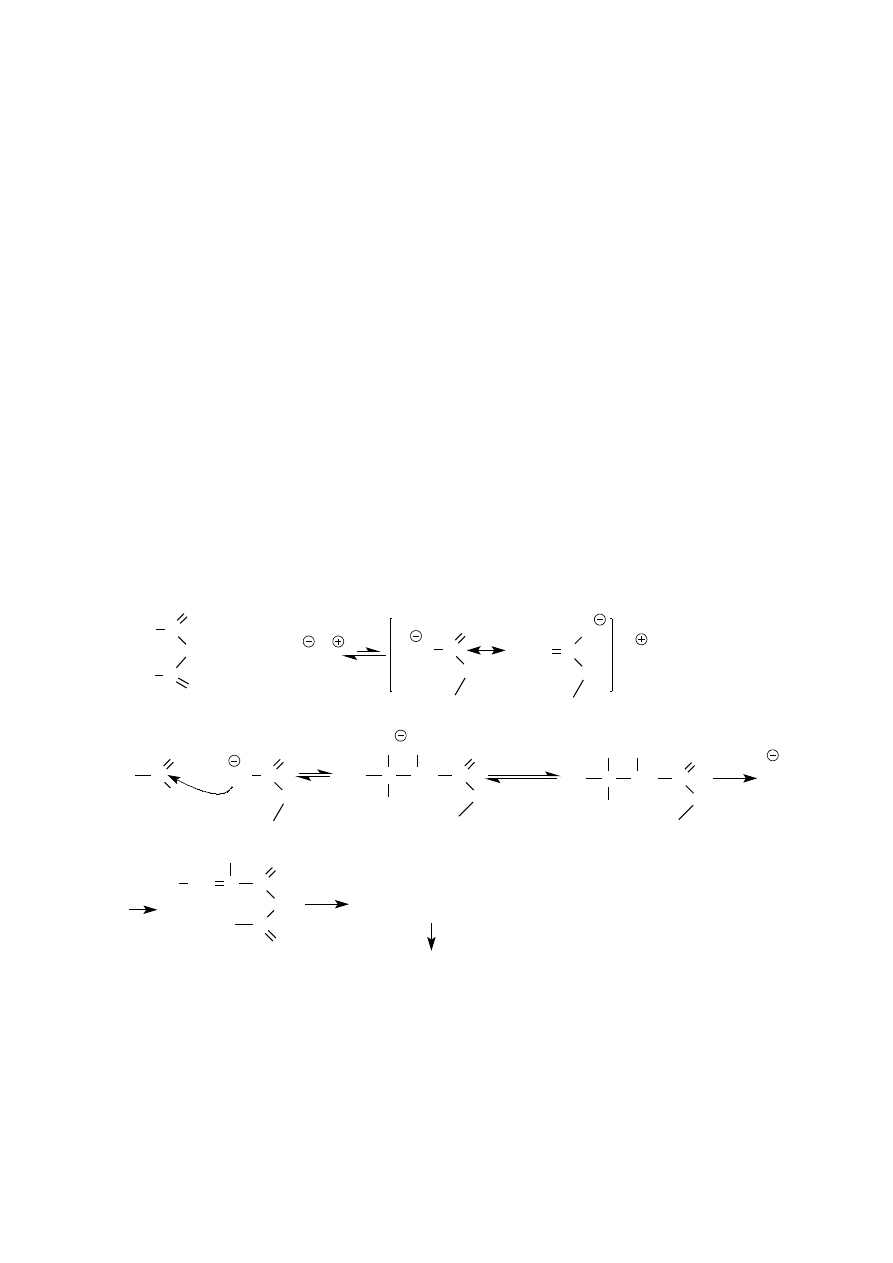
reakcyjnej rozcieńczona 2–3 częściami wody będzie stanowić klarowny roztwór (wyjaśnij zasadę tego testu). p–
Nitroanilina jest w roztworze, w postaci wodorosiarczanu amoniowego. Klarowny, gorący roztwór wylewa się
do 500 ml zimnej wody, i wytrąca p–nitroanilinę dodając nadmiar 10–proc. roztworu wodorotlenku sodu lub
stężonego roztworu amoniaku. Z zimnej zawiesiny (jeśli to konieczne, należy ją schłodzić) odsącza się żółty
osad pod zmniejszonym ciśnieniem, przemywa starannie wodą, dokładnie odciska, a następnie krystalizuje z
mieszaniny rektyfikat–woda (1 : 1) lub gorącej wody. Po odsączeniu, przemyciu i wysuszeniu osadu, otrzymuje
się 11 g (96%) p–nitroaniliny, tt. 148 ºC.
Uwagi. (1) Przemywanie osadu jest znacznie efektywniejsze, jeśli przenosi się go zlewki, dokładnie rozciera z
wodą, a następnie sączy.
(2) 70–Proc. roztwór kwasu siarkowego przygotowuje się przez dodanie 60 ml stężonego kwasu siarkowego
(ostrożnie i wolno, cienkim strumieniem) do 45 ml wody.
KWAS CYNAMONOWY
W syntezie wykorzystuje się reakcję Perkina będącą przykładem reakcji addycji nukleofilowej anionu
enolanowego do grupy karbonylowej. Jej przebieg jest podobny do przebiegu kondensacji aldolowej.
Ponieważ anion enolanowy jest tu wytwarzany z bezwodnika w reakcji z solą odpowiedniego kwasu
karboksylowego proces musi przebiegać w warunkach bezwodnych.
PhCH=C(R)CO
2
Na + RCH
2
CO
2
Na
Na
2
CO
3
Ph CH C C
O
O
C
O
R
RCH
2
+RCH
2
CO
2
Ph C
OH
H
CH C
O
O
R
RCH
2
CO
2
H
Ph C
O
H
CH C
O
O
R
+ RCH C
O
O
Ph C
O
H
Na + RCH
2
CO
2
H
RCH C
O
O
RCH C
O
O
+ RCH
2
CO
2
Na
RCH
2
C
O
O
C
O
RCH
2
t
-H
2
O
HCl
PhCH=C(R)CO
2
H + RCH
2
CO
2
H
W suchej kolbie kulistej o poj. 250 ml, zaopatrzonej w chłodnicę powietrzną zabezpieczoną rurką z chlorkiem
wapnia, umieszcza się 21 g (20 ml, 0,2 mol) czystego aldehydu benzoesowego (1), 30 g (28 ml, 0,29 mol)
bezwodnika octowego i 12 g (0,122 mol) świeżo stopionego i dokładnie sproszkowanego octanu potasu (2). Po
starannym wymieszaniu substratów kolbę ogrzewa się w czaszy grzejnej w temperaturze łagodnego wrzenia
przez 4 godz. Jeszcze gorącą (80—100 °C) mieszaninę reakcyjną przelewa do kolby kulistej o poj. 1 l, w
której znajduje się 100 ml wody. Kolbę reakcyjną przepłukuje się jeszcze niewielką ilością gorącej wody.
17

Następnie dodaje się, energicznie wstrząsając, nasyconego wodnego roztworu węglanu sodu (3) dotąd, aż
mieszanina przybierze odczyn zasadowy (papierek wskaźnikowy). Wówczas kolbę łączy się z aparaturą do
destylacji z parą wodną i prowadzi destylację dotąd, aż oddestyluje nieprzereagowany aldehyd benzoesowy;
zacznie destylować klarowna ciecz – woda. Do pozostałego w kolbie destylacyjnej roztworu dodaje się dwie
łopatki węgla aktywnego i ogrzewa go w temp. wrzenia przez kilka minut. Nieco tylko ochłodzony roztwór
sączy się pod zmniejszonym ciśnieniem, aby oddzielić węgiel wraz z zaadsorbowanymi na nim smolistymi
produktami ubocznymi. Przesącz, energicznie mieszając, zakwasza się stęż. kwasem solnym, przy czym kwas
dodaje się powoli, tak długo, aż przestanie wydzielać się CO
2
(odczyn kwaśny wg papierka wskaźnikowego). Po
ochłodzeniu odsącza się wydzielony kwas cynamonowy pod zmniejszonym ciśnieniem, przemywa go zimną wodą
i dobrze odciska. Produkt krystalizuje się z gorącej wody albo z mieszaniny 3 obj. wody i 1 obj. rektyfikatu.
Wydajność suchego kwasu cynamonowego w postaci bezbarwnych kryształów o tt. 133 °C wynosi 18 g (62%).
Uwagi. (1) Aldehyd benzoesowy nie może zawierać kwasu benzoesowego. Oczyszczanie aldehydu opisano w
Preparatyce: II str. 535; III str. 991.
(2)
Stopiony octan potasu powinien być świeżo przygotowany metodą opisaną dla octanu sodu (zob.
Preparatyka). Można go jednak zastąpić równoważną molowo ilością świeżo stopionego octanu sodu, ale
wówczas reakcja przebiega wolniej i ogrzewanie mieszaniny trzeba przedłużyć o 3 godz.
(3)
W tym etapie syntezy do alkalizowania nie można używać roztworu wodorotlenku sodu, ponieważ
mogłoby to spowodować reakcję Cannizzaro (Prep.: II str. 676; III str. 990) prowadzącą do powstania kwasu
benzoesowego z nieprzereagowanego aldehydu benzoesowego.
ANILINA
Redukcja grupy nitrowej zwłaszcza w nitroarenach jest ważną metodą otrzymywania aniliny i jej pochodnych.
Najczęściej stosowanymi oczynnikami używanymi do redukcji są np. wodór wobec katalizatora (np. platyny),
metal (cyna, cynk, żelazo) w środowisku kwaśnym lub zasadowym a także siarczki i wielosiarczki metali
alkalicznych lub amonu. Anilinę można otrzymać w reakcji nitrobenzenu z cyną w stężonym kwasie solnym
zgodnie z równaniem:
2C
6
H
5
NO
2
+ 12HCl + 3Sn à 2C
6
H
5
NH
2
+ 3SnCl
4
+ 4H
2
O
Powstająca podczas reakcji anilina jest wiązana przez kwas chlorocynowy dając odpowiednią sól aniliniową -
[C
6
H
5
NH
3
+
]
2
[SnCl
6
]
2
. Dzięki temu z mieszaniny poreakcyjnej można łatwo usunąć nieprzereagowany
nitrobenzen przez destylację z parą wodną. Sól aniliniową rozkłada się wodorotlenkiem sodu:
[C
6
H
5
NH
3
+
]
2
[SnCl
6
]
2
+ 8NaOH à C
6
H
5
NH
2
+ Na
2
SnO
3
+ 6NaCl
i oddziela od wodnej zawiesiny zasadowych soli cyny za pomocą destylacji z parą wodną.
Produktem redukcji nitrobenzenu w środowisku kwaśnym, w trakcie której niewyodrębnialnymi związkami
pośrednimi są ntrozobenzen (C
6
H
5
N=O) i fenylohydroksyloamina (C
6
H
5
NHOH), jest anilina. Jeśli redukcję
prowadzi się w środowisku zasadowym lub/i stosuje łagodniejsze środki redukujące możliwe jest otrzymanie np.
18

fenylohydroksyloaminy oraz związków powstających w wyniku reakcji między produktami pośrednimi np.
azoksybenzenu (C
6
H
5
N
+
(O
)=NC
6
H
5
). Synteza tych i pokrewnych związków jest opisana w Preparatyce: II str.
617-619; III str. 920-922.
Preparat wykonuje się pod wyciągiem.
Zalecane jest, aby do reakcji użyć cyny możliwie dobrze rozdrobnionej. Można ją otrzymać w następujący
sposób. Palnik Meckera umocowuje się poziomo do statywu na wysokości ok. 50 cm od podstawy i umieszcza
się pod nim większą parownicę porcelanową. Granulkę cyny trzymaną szczypcami metalowymi stapia się w
płomieniu palnika. Spadająca kropla zastyga na powierzchni porcelany w postaci cienkiej warstwy. Otrzymane
blaszki cynowe kroi się nożyczkami na możliwie wąskie paski.
W kolbie kulistej o poj. 1000 ml, zaopatrzonej w chłodnicę zwrotną (przy wprawnym dalszym postępowaniu nie
jest konieczne podłączanie węży do chłodnicy; alternatywnie – chłodnicę napełnia się wodą i łączy oba jej
króćce kawałkiem węża), umieszcza się 24,6 g (21 ml, 0,20 mol) nitrobenzenu i 45 g (0,38 mol) cyny. Kolbę
umocowuje się łapą do statywu. Przygotowuje się czaszę grzejną lub łaźnię z wrzącą wodą i miskę z zimną
wodą. Odmierza się w cylindrze 100 ml stężonego kwasu solnego (1.2 mol), a następnie ok. 15 ml tego kwasu
wprowadza się do kolby przez chłodnicę zwrotną. Kolbę wraz z łapą odłącza się od statywu i trzymając jedną
ręką za łapę a drugą za chłodnicę energicznie wstrząsa się zawartością kolby. Mieszanina reakcyjna rozgrzewa
się, a po niedługim czasie reakcja powinna przebiegać dosyć energicznie. Jeśli to nie nastąpi kolbę ogrzewa się
aż do zapoczątkowania reakcji. Jeżeli mieszanina wrze bardzo energicznie, to należy ją schłodzić, na chwilę
zanurzając kolbę w łaźni z zimną wodą. Należy unikać zbytniego ochłodzenia. Wytrąca się wtedy chlorocynian
aniliniowy w postaci białej lub żółtej krystalicznej masy. Może być wtedy konieczne ogrzanie kolby, aby
przywrócić właściwe tempo przebiegu reakcji. Podczas całego procesu zawartość kolby powinna być klarowna i
łagodnie wrzeć. Kiedy początkowa szybkość reakcji zacznie samorzutnie maleć, przez chłodnicę zwrotną dodaje
się nową porcję 15 ml kwasu solnego i zawartość kolby silnie wytrząsa w celu dokładnego wymieszania
składników, a jeśli reakcja zachodzi zbyt gwałtownie, ponownie chłodzi. W ten sposób postępuje się dotąd, aż
cała ilość kwasu zostanie dodana. Następnie mieszaninę reakcyjną ogrzewa się na wrzącej łaźni wodnej (przez
30–60 min) do czasu gdy zapach nitrobenzenu przestanie być wyczuwalny, a próbka pobrana z kolby (kilka
kropli) i rozcieńczona wodą utworzy całkowicie klarowny roztwór (jaki jest cel i sens tej próby?). (Jeśli po 60
min ogrzewania nadal stwierdza się obecność nitrobenzenu kolbę reakcyjną umieszcza się w zestawie do
destylacji z parą wodną. Destylację prowadzi się tak długo, aż z chłodnicy zacznie spływać klarowna ciecz
woda.)
Mieszaninę reakcyjną chłodzi się do temperatury pokojowej i stopniowo dodaje do niej roztwór 75 g
wodorotlenku sodu w 125 ml wody. Jeśli w trakcie dodawania wodorotlenku mieszanina zacznie wrzeć, to
należy ją ochłodzić. Wytrącający się początkowo wodorotlenek cyny, powinien się całkowicie rozpuścić, a
odczyn mieszaniny powinien być silnie alkaliczny. Anilina wydziela się w postaci oleju. Kolbę umieszcza się w
układzie do destylacji z parą wodną. Destylację prowadzi się tak długo aż, po oddestylowaniu mętnej cieczy,
zostanie zebranych dodatkowo 120 ml klarownego destylatu. Anilina jest częściowo rozpuszczalna w wodzie
(ok. 3%), dlatego należy ją „wysolić”, czyli zmniejszyć jej rozpuszczalność nasycając destylat chlorkiem sodu.
Na każde 100 ml destylatu używa się ok. 20 g soli. Następnie nasycony solą destylat przenosi się do
rozdzielacza, dodaje do ok. 40 ml eteru (lub chlorku metylenu) i całość dokładnie wytrząsa w celu dokładnego
19

wyekstrahowania aniliny z warstwy wodnej; należy pamiętać o częstym wyrównywaniu ciśnienia w
rozdzielaczu, w zależności od położenia rozdzielacza, albo przez uniesienie na chwilę korka, albo otworzenie
kranu. (Podczas ekstrakcji wszystkie palniki znajdujące się w pobliżu muszą być zgaszone.) Następnie
mieszaninę pozostawia się do czasu rozdzielenia warstw. Zlewa się obie warstwy do osobnych naczyń. Warstwę
wodną (dolną; uwaga: w przypadku użycia CH
2
Cl
2
warstwa wodna jest na górze) zawraca się do rozdzielacza i
ekstrahuje nową porcją 40 ml eteru. Połączone roztwory eterowe umieszczone w erlenmajerce suszy się kilkoma
gramami bezwodnego węglanu potasu (2), wytrząsając zakorkowaną kolbą przez kila minut.
Roztwór eterowy sączy się przez sączek karbowany do wkraplacza. Eter oddestylowuje się metodą
destylacji równowagowej (zob. str. 27), stosując kolbę kulistą o poj. 50 ml, do której dodaje się kilka kamyków
wrzennych. Kolbę ogrzewa się łagodnie łaźnią elektryczną, pamiętając o zachowaniu środków ostrożności
związanych z wyjątkową lotnością i łatwopalnością eteru. Kiedy do kolby zostanie wprowadzony cały roztwór,
wkraplacz zastępuje się termometrem i oddestylowuje się resztę rozpuszczalnika (wskazówką jest temperatura
wrzenia). Wówczas z chłodnicy usuwa się wodę i zwiększa intensywność ogrzewania. Kiedy temperatura
wrzenia osiągnie 180 ºC zmienia się odbieralnik (sucha zważona erlenmajerka) i zbiera frakcję wrzącą w
temp.180–184 ºC. Wydajność aniliny wynosi 18 g (97%).
Czysta anilina wrze w temp. 184 ºC. Świeżo przedestylowana jest bezbarwna, jednak podczas
przechowywania, zwłaszcza na świetle, zabarwia się, na skutek utleniania tlenem z powietrza. Zabarwienie to
można usunąć, destylując anilinę znad niewielkiej ilości pyłu cynkowego.
Uwaga. (1) Do ekstrakcji aniliny zamiast eteru zaleca się użycie chlorku metylenu. W tym przypadku warstwa
wodna jest warstwą górną.
(2) Roztworu nie należy suszyć chlorkiem wapnia, ponieważ tworzy on z aniliną (oraz innymi aminami)
związki cząsteczkowe. Najlepszym czynnikiem suszącym jest wodorotlenek sodu lub potasu (w postaci
granulek).
p-METYLOACETOFENON
Reakcja acylowania metodą Friedla-Craftsa.
Jest to reakcja substytucji elektrofilowej. Reagentem acylującym jest w niej bezwodnik kwasu karboksylowego
lub chlorek acylu. Bezwodnik kwasowy (podobnie jak chlorek kwasowy) ma właściwości zasady Lewisa dzięki
obecności nie zaangażowanych w wiązania par elektronów na atomach tlenu (i chloru). Oddziaływanie kwasu
Lewisa, chlorku glinu, z którymkolwiek z tych atomów zwiększa polaryzację odpowiedniego wiązania a tym
samym zwiększa elektrofilowość karbonylowego atomu węgla. Należy zatem oczekiwać, że kompleksy tego
typu (RCOClàAlCl
3
), chociaż mniej reaktywne od będącego z nimi w równowadze wolnego jonu acyliowego,
mogą również pełnić rolę elektrofila w reakcji acylowania.
20
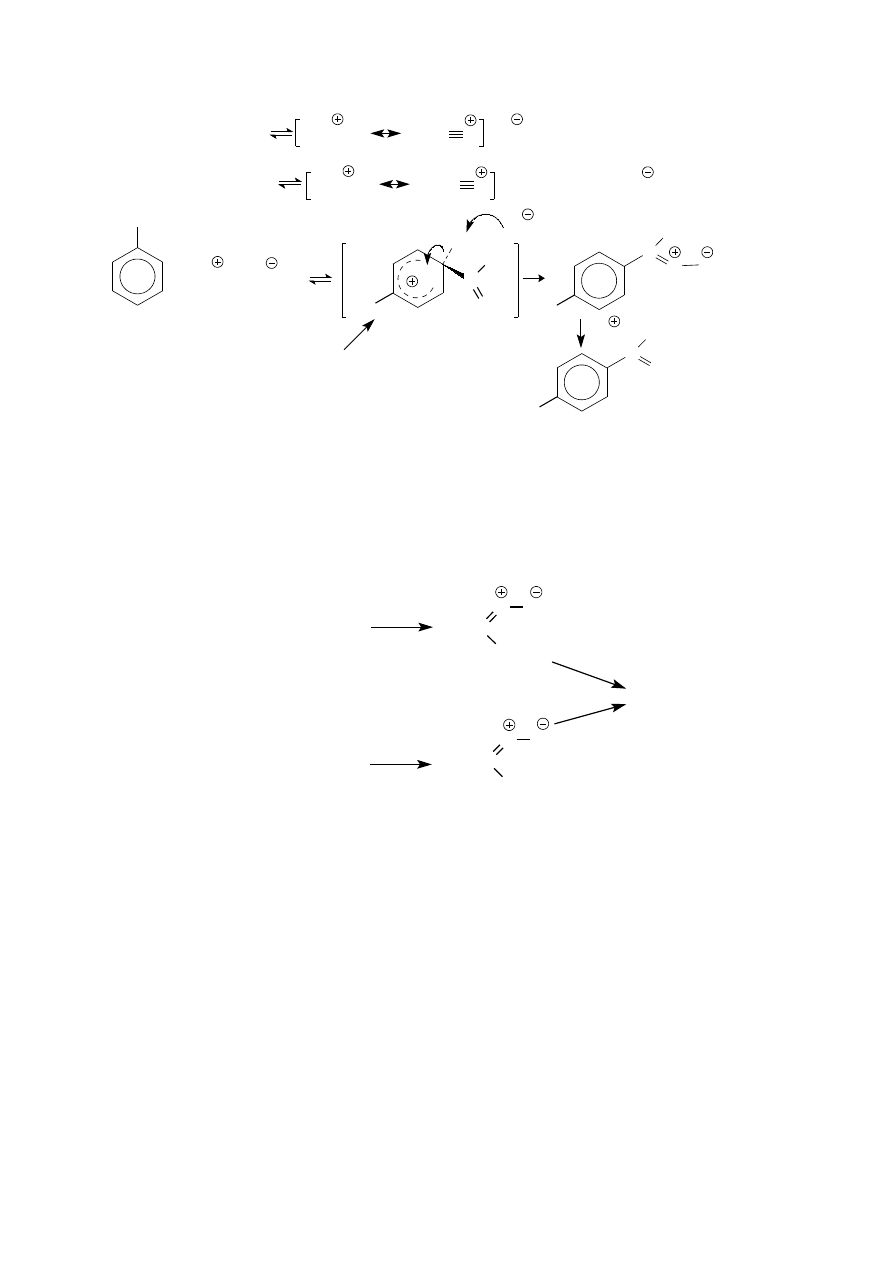
H
C
CH
3
O
H
3
C
ClAlCl
3
+ AlCl
4
CH
3
C O
CH
3
C=O
kompleks
CH
3
C O
CH
3
C=O
CH
3
COCl + AlCl
3
(CH
3
CO)
2
O + 2AlCl
3
+ CH
3
CO
2
AlCl
2
+ AlCl
4
CH
3
+ CH
3
C=O + AlCl
4
C
O
CH
3
AlCl
3
H
3
C
+HCl
H
3
O
C
O
CH
3
H
3
C
p-metyloacetofenon
kation acyliowy
przedstaw rozkład elektronów w kompleksie
za pomocą struktur mezomerycznych
i wyjaśnij regioselektywność reakcji
kompleks produktu
z chlorkiem glinu
Odmiennie niż w reakcji alkilowania, chlorek glinu jest nie tylko katalizatorem, ale także reagentem ponieważ
tworzy trwały kompleks z powstającymym produktem. Z tego powodu do reakcji acylowania używa się 1,1 mol
AlCl
3
na 1 mol chlorku kwasowego lub 2,1 mol AlCl
3
na 1 mol bezwodnika.
Sumaryczne równania reakcji:
AlCl
3
C
6
H
5
C
O
CH
3
AlCl
3
C
6
H
6
+ (CH
3
CO)
2
O + 2AlCl
3
AlCl
3
C
6
H
6
+ CH
3
COCl + AlCl
3
+ CH
3
COOAlCl
2
+ HCl
+ HCl
C
6
H
5
C
O
CH
3
AlCl
3
HClaq
C
6
H
5
C(O)CH
3
acetofenon
Dla uniknięcia hydrolizy bezwodnika octowego (do kwasu octowego) i chlorku glinu (do zasadowych soli glinu)
reakcję przeprowadza się w warunkach bezwodnych. Elementy aparatury używane do reakcji muszą być suche.
Wszystkie operacje (ważenie lub odmierzanie reagentów, napełnianie nimi reaktora itp.) powinny być wykonane
sprawnie, aby kontakt chlorku glinu i bezwodnika octowego z wilgocią zawartą w powietrzu był jak najkrótszy.
Toluen powinien być pozbawiony wody. Można to zrobić za pomocą destylacji. Na początku destylacji destyluje
azeotrop wody i toluenu. Jego skład jest taki, że po skropleniu par w chłodnicy następuje oddzielenie warstwy
wodnej od toluenowej; w odbieralniku zbiera się mętny destylat. Destylację przerywa się wtedy, gdy z chłodnicy
zacznie spływać klarowna ciecz – pozbawiony wody toluen. Pozostały w kolbie toluen po ochłodzeniu używa
się do reakcji. (Jeśli potrzebny jest toluen bardziej suchy, do wstępnie osuszonego azeotropowo związku dodaje
się metaliczny sód, najczęściej w postaci drutu otrzymanego za pomocą specjalnej prasy; prep.: II str. 224; III
str. 379).
21

Reakcję wykonuje się pod wyciągiem.
Kolbę kulistą z trzema szyjami, o poj. 500 ml, zaopatruje się w chłodnicę zwrotną, mieszadło z uszczelnieniem
(zob. rysunek niżej) oraz wkraplacz z wyrównywaczem ciśnienia (sprawdź szczelność kranu przed na
pełnieniem). Na szczycie chłodnicy umieszcza się rurkę z chlorkiem wapnia (najlepszy do tego celu jest
CaCl
2
granulowany; wypełnienie rurki nie powinno hamować przepływu HCl), którą łączy się z
urządzeniem do pochłaniania wydzielającego się w reakcji chlorowodoru (zob. prep.: II str. 50, rys. 1.50a;
III str. 88, rys. 2.61a). W kolbie umieszcza się 75 g (0,56 mol) bezwodnego, dobrze sproszkowanego chlorku
glinu i 120 g (140 ml, 1,30 mol) bezw. toluenu i zanurza się ją w łaźni z zimną wodą. (Łaźnię, metalową
miskę z wodą, należy zamontować w taki sposób, aby w każdej chwili można ją było usunąć spod kolby -
najlepiej na pierścieniu metalowym zamocowanym do statywu – i na takiej wysokości, aby później można było
ogrzać palnikiem zawartą w niej wodę.) Po uruchomieniu mieszadła powoli wkrapla się w ciągu pół godziny 25,5
g (24 ml, 0,25 mol) bezwodnika octowego (jeśli reagent jest „niewiadomego pochodzenia” albo „stary” należy go
przedestylować, aby usunąć kwas octowy mogący powstawać w wyniku powolnej hydrolizy bezwodnika pod
wpływem wilgoci). Reakcja jest egzotermiczna i mieszanina łagodnie wrze; jednocześnie wydziela się
chlorowodór. Niekiedy po wewnętrznej stronie kolby pod wkraplaczem osadza się osad. Przed zakończeniem
wkraplania bezwodnika należy go zsunąć do roztworu za pomocą suchej bagietki wyjmując na chwilę wkraplacz.
Po wkropleniu całej ilości bezwodnika kolbę ogrzewa się na wrzącej łaźni wodnej do czasu aż przestanie
wydzielać się chlorowodór (ok. 30 min). W tym czasie reakcja przebiega do końca. Następnie zawartość
kolby chłodzi się i wylewa do zlewki, w której znajduje się mieszanina 150 g pokruszonego lodu i 150 ml stęż.
kwasu solnego. Następuje rozkład kompleksu produktu z chlorkiem glinu. Reakcja jest silnie egzotermiczna
dlatego wskazane jest mieszanie zawartości zlewki bagietką. W razie potrzeby można do zlewki dodać kawałki
lodu. Zawartość zlewki składa się z dwóch warstw ciekłych; ciemnej górnej będącej roztworem p-
metyloacetofenonu w toluenie (toluen ma gęstość mniejszą niż woda) i jasnej dolnej zawierającej roztwór soli
glinu w rozcieńczonym kwasie solnym. Niekiedy z warstwy wodnej wytrącają się sole nieorganiczne. Należy
wtedy dodać wody w ilości wystarczającej do ich rozpuszczenia. Mieszaninę przelewa się do rozdzielacza i
oddziela górną warstwę. Warstwę wodną zawraca się do rozdzielacza i ekstrahuje 25-30 ml eteru. Ekstrakcję
powtarza się jeszcze raz. Ekstrakty eterowe łączy się z roztworem toluenowym i przemywa w rozdzielaczu 50
ml 10 proc. roztworu wodorotlenku sodu (dla uniknięcia tworzenia się emulsji można użyć roztwór Na
2
CO
3
;
warstwa wodna po przemyciu powinna wykazać odczyn alkaliczny), a następnie wodą. Oddzielone warstwy
organiczne suszy się bezw. siarczanem magnezu lub chlorkiem wapnia. Po odsączeniu środka suszącego na
sączku karbowanym eter i toluen oddestylowuje się pod normalnym ciśnieniem. Do tej operacji można
zastosować tzw. destylację równowagową. Montuje się zestaw do destylacji prostej, w którym kolbą
destylacyjną jest kolba Claisena; będzie ona później użyta w zestawie do destylacji próżniowej. W nasadce
destylacyjnej zamiast termometru umieszcza się wkraplacz. Boczną szyję zamyka się korkiem. Część
roztworu wlewa się do kolby Claisena. Zawartość kolby ogrzewa się do wrzenia; rozpoczyna się destylacja
eteru i toluenu. Z wkraplacza wkrapla się pozostałą część roztworu w takim tempie, aby poziom cieczy w
kolbie nie zmieniał się. Po wkropleniu całej ilości roztworu wkraplacz zamienia się na termometr a w bocznej
szyi umieszcza się kapilarę. (Alternatywnie – cały roztwór toluenowo-eterowy produktu umieszcza się w
kolbie kulistej i usuwa rozpuszczalniki za pomocą destylacji prostej. Pozostałość przenosi się do kolby
Claisena stanowiącej część zestawu do destylacji pod zmniejszonym ciśnieniem.) Kontynuuje się destylację
22
dotąd aż temperatura oparów osiągnie ok. 120
C. Po ostudzeniu i usunięciu kamyczków wrzennych kolbę z
surowym p-metyloacetofenonem przenosi się do zaakceptowanego przez asystenta zestawu do destylacji pod
zmniejszonym ciśnieniem. Po ustaleniu się i zmierzeniu ciśnienia w aparaturze wyznacza się za pomocą
diagramu spodziewaną temperaturę wrzenia wiedząc, że pod ciśnieniem atmosferycznym wynosi ona 225
C.
Wskazane jest owinięcie deflegmatora kartką papieru lub taśmą izolacyjną. Na początku destylują resztki
toluenu. Z powodu dużej prężności par tego rozpuszczalnika ciśnienie w aparaturze może nieco wzrosnąć.
Sprawdzamy to otwierając co jakiś czas kran manometru. (Kran ten powinien być zamknięty podczas destylacji.
Otwiera się go tylko na czas pomiaru ciśnienia.) Kiedy temperatura oparów zbliży się do temperatury wrzenia
oszacowanej dla danego ciśnienia (ze względu na dokładność wyznaczania rzeczywista temperatura wrzenia
może się różnić o kilka stopni od oszacowanej) i przestanie wzrastać przekręca się „krówkę” tak, aby produkt
spływał do starowanej uprzednio kolby. Destylację prowadzi się do momentu, kiedy temperatura oparów
zacznie wyraźnie wzrastać lub kiedy w kolbie destylacyjnej pozostanie ok. 1 ml cieczy (nie należy
wydestylowywać cieczy „do sucha”. Boczne ścianki kolby destylacyjnej niedostatecznie zwilżone cieczą
nagrzewają się do wysokiej temperatury. W zetknięciu z nimi spływające z deflegmatora ciecz może ulec
rozkładowi.) Usuwa się wtedy czaszę grzejną i pozostawia kolbę do ostygnięcia. Zapowietrza się aparaturę
ostrożnie zdejmując wąż z tubusa „krówki”. Należy zwrócić uwagę, aby w tym czasie kran manometru był
zamknięty oraz aby kolby z przedgonem i produktem nie zsunęły się ze szlifów „krówki”. Dopiero wtedy można
zakręcić kran pompki wodnej i ostrożnie zapowietrzyć manometr. Zwykle otrzymuje się ok. 29 g produktu, wyd.
86%.
korek gumowy
wąż gumowy
smarować olejem parafinowym
prowadnica
bagietka - mieszadło
23
MRÓWCZAN ETYLU
Kwasy karboksylowe alifatyczne i aromatyczne w rekcji z alkoholami pierwszo- i drugorzędowymi dają estry.
Reakcja estryfikacji jest katalizowana mocnymi kwasami, najczęściej kwasem siarkowym. Rola katalizatora
polega na protonowaniu atomu tlenu grupy karbonylowej, co zwiększa elektrofilowość karbonylowego atomu
węgla i ułatwia przyłączenie słabego nukleofila, alkoholu. Wszystkie etapy tej reakcji są odwracalne, a położenie
równowagi zależy od budowy reagentów. W celu zwiększenia wydajności estru stosuje się nadmiar jednego z
reagentów (zob. Benzoesan etylu i Octan butylu) lub usuwa produkty, wodę lub/i ester (zob. Mrówczan etylu), w
trakcie ich powstawania. Jeśli ester ma niższą niż substraty temperaturę wrzenia, to można go oddestylowywać.
Wodę można usuwać za pomocą środka suszącego albo oddestylowywać w postaci azeotropu, np. z benzenem.
Używany jako katalizator stężony kwas siarkowy protonując powstającą wodę, także przyczynia się do
zwiększenia wydajność estru.
+H
2
O
+ R
1
OH
+ H
3
O
R C
O
OR
1
R C
O
OR
1
H
R C
O
OR
1
H
- H
2
O
R C
OH
O
OR
1
H
H
R C
O
OH
H
R C
O
OH
H
R C
OH
OH
O
R
1
H
+ H
R C
O
OH
addycja
etap najwolniejszy
eliminacja
24

Według powyższego schematu nie można otrzymywać estrów alkoholi trzeciorzędowych. O szybkości
najwolniejszego etapu, a więc i o szybkości całej reakcji, w znacznym stopniu decydują czynniki steryczne. Jest
zatem oczywiste, że utworzenie wiązania między karbonylowym atomem węgla i atomem tlenu alkoholu
trzeciorzędowego, mającego dużą objętościowo grupę alkilową, będzie przebiegało powoli. Jednocześnie
obecność silnego kwasu mineralnego będzie sprzyjała reakcji dehydratacji alkoholu, której alkohole mogące
utworzyć trwałe karbokationy, np. alkohole trzeciorzędowe, ulegają szczególnie łatwo.
W przypadkach, w których z przyczyn technicznych lub ekonomicznych nie można zastosować nadmiaru
jednego z reagentów, do reakcji używa się chlorków kwasowych lub bezwodników (octany, ftalany). Rekcje
przebiegają wtedy szybko i dużymi wydajnościami.
W kolbie kulistej poj. 500 ml umieszcza się mieszaninę 135 ml (2,3 mol) alkoholu etylowego, 124 g (2,3 mol)
85-proc. kwasu mrówkowego i 20 g chlorku wapnia. Kolbę zaopatruje się w zestaw do destylacji zawierający
kolumnę Vigreux i ogrzewa powoli na łaźni wodnej. Wkrótce rozpoczyna się destylacja mrówczanu etylu.
Temperaturę łaźni wodnej reguluje się tak, aby zapewnić powolną i równomierną destylacją estru. Produkt (168
g) zbiera się w temp. 53-55
C. Surowy mrówczan etylu destyluje się ponownie znad 20 g bezw. węglanu potasu
(do czego służy K
2
CO
3
?) stosując ten sam (umyty i suchy!) zestaw destylacyjny. Zbiera się frakcję wrzącą w
temp. 53-54
C otrzymując 150 g (88%) estru.
BENZOESAN METYLU
Reakcja przebiega według mechanizmu addycji-eliminacji – patrz wstęp do Mrówczanu etylu.
W kolbie kulistej poj. 500 ml umieszcza się 30 g (0,246 mol) kwasu benzoesowego, 80 g(101 ml, 2,5 mol) bezw.
metanolu (odwadnianie metanolu – zob. Prep.: II str. 219; III str. 374) i 5 g (2,7 ml) stęż. kwasu siarkowego i
dodaje kilka kamyczków wrzennych. Kolbę zaopatruje się w chłodnicę zwrotną i ogrzewa w temp. wrzenia
przez 4 godz. Nadmiar alkoholu oddestylowuje się z łaźni wodnej (można użyć wyparki obrotowej; nie należy
natomiast raczej używać czaszy grzejnej – dlaczego?)) a pozostałość, po ochłodzeniu, wlewa się do rozdzielacza
zawierającego 250 ml wody. Kolbę przepłukuje się niewielką ilością wody, którą wlewa się do rozdzielacza.
Często oddzielenie estru od górnej warstwy wodnej jest trudne z powodu małej różnicy gęstości estru i wody. W
takim przypadku do rozdzielacza dodaje się 10-15 ml chlorku metylenu i energicznie wstrząsa; cięższy roztwór
estru w chlorku metylenu oddziela się wyraźnie i szybko. Dolną warstwę oddziela się dokładnie, usuwa z
rozdzielacza warstwę wodną, część organiczną zawraca do rozdzielacza i wytrząsa z nasyconym roztworem
wodorowęglanu sodu aż do zaprzestania wydzielania się ditlenku węgla, czyli do całkowitego usunięcia
kwasów. Roztwór estru przemywa się wodą i zlewa do małej kolby stożkowej zawierającej 5 g siarczanu
magnezu. Kolbę zamyka się korkiem i zostawia na ok. pół godz. wstrząsając od czasu do czasu. Roztwór sączy
się przez mały sączek karbowany do kolby kulistej, którą następnie zaopatruje się w zestaw do destylacji z
chłodnicą powietrzną i termometrem do 360
C. Po dodaniu kamyków wrzennych kolbę ogrzewa się w czaszy
grzejnej. Początkowo intensywność ogrzewania powinna być mała, aby chlorek metylenu nie destylował zbyt
gwałtownie. Po oddestylowaniu rozpuszczalnika zwiększa się intensywność ogrzewania. Benzoesan metylu
zbiera się w temp. 198-200
C. Wydajność 31 g (92%).
25

Analogicznie otrzymuje się:
BENZOESAN ETYLU
Z 30 g (0,246 mol) kwasu benzoesowego, 115 g (145 ml, 2,5 mol) bezw. etanolu i 5 g (2,7 ml) stęż. kwasu
siarkowego. Otrzymuje się 32 g (86%) estru o tw. 212-214
C.
SALICYLAN METYLU
Z 27,6 g (0,2 mol) kwasu salicylowego, 64 g (81 ml, 2 mol) bezw. metanolu i 5 g (2,7 ml) stęż. kwasu
siarkowego. Ogrzewanie 5 godz. Otrzymuje się 32 g (86%) estru o tw. 221-224
C. Ester można destylować pod
zmniejszonym ciśnieniem; tw. 115
C/20 mmHg.
SALICYLAN ETYLU
Z 27,6 g (0,2 mol) kwasu salicylowego, 92 g (116 ml, 2 mol) bezw. etanolu i 5 g (2,7 ml) stęż. kwasu
siarkowego. Ogrzewanie 5 godz. Tw. 234
C/760mmHg, 119 C/20 mmHg. Wydajność 75%.
OCTAN BUTYLU
W kolbie kulistej o poj. 250 ml (lub 500 ml) umieszcza się 37,0 g (46 ml, 0,5 mol) butan-1-olu i 60 g (60 ml, 1
mol) lod. kwasu octowego i dodaje ostrożnie (z małego cylinderka lub kalibrowanej pipety) 1 ml stęż. kwasu
siarkowego. Kolbę zaopatruje się w chłodnicę zwrotną i mieszaninę reakcyjną ogrzewa w temp. wrzenia przez 3-
6 godz. (1). Schłodzoną mieszaninę reakcyjną przenosi się do rozdzielacza, w którym znajduje się ok. 250 ml
wody, wytrząsa i oddziela górną warstwę surowego estru. Warstwę estrową przemywa się kolejno 100 ml wody,
25 ml nasyconego wodnego roztworu wodorowęglanu sodu i 50 ml wody, a następnie suszy 5-6 g bezw.
siarczanu sodu. Po odsączeniu środka suszącego na sączku karbowanym ester destyluje się ogrzewając kolbę
czaszą grzejną. Czysty octan butylu zbiera się w temp. 124-125
C; wydajność 40 g (69%).
Uwaga. (1) Aby uzyskać nieco wyższą wydajność należy wziąć do reakcji więcej kwasu octowego (90-120 g) i
przedłużyć ogrzewanie w temp. wrzenia pod chłodnicą zwrotną do 12-18 godz..
26
Document Outline
- Przemysław Szczeciński
- LABORATORIUM Z CHEMII ORGANICZNEJ
- Materiały pomocnicze
- Spis treści
- ACETANILID
- ANILINA
- JODOBENZEN
- KWAS CYNAMONOWY
- ANILINA
Wyszukiwarka
Podobne podstrony:
C021 cwiczenia laboratoryjne z chemii organicznej Wydz chemii UJ
LABORATORIUM CHEMII ORGANICZNEJ war zal TCh2013
metody syntezy związków organicznych, Laboratorium chemii organicznej
Warunki Zaliczenia LABORATORIUM CHEMII ORGANICZNEJ
Synteza octanu n-butylu, Biotechnologia PWR, Semestr 3, Podstawy chemii organicznej - Laboratorium (
Zajecia 4, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Zajecia 3, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Instrukcja NMR, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organic
CHEMIA ORGANICZNA I – laboratorium, podstawy chemii organicznej
PROGRAM CWICZEN Z CHEMII ORGANICZNEJ BIOLOGIA 2010 2011
I POPRAWKA EGZAMINU Z CHEMII ORGANICZNEJ, Technologia chemiczna, Chemia organiczna, 4 semestr, organ
więcej podobnych podstron