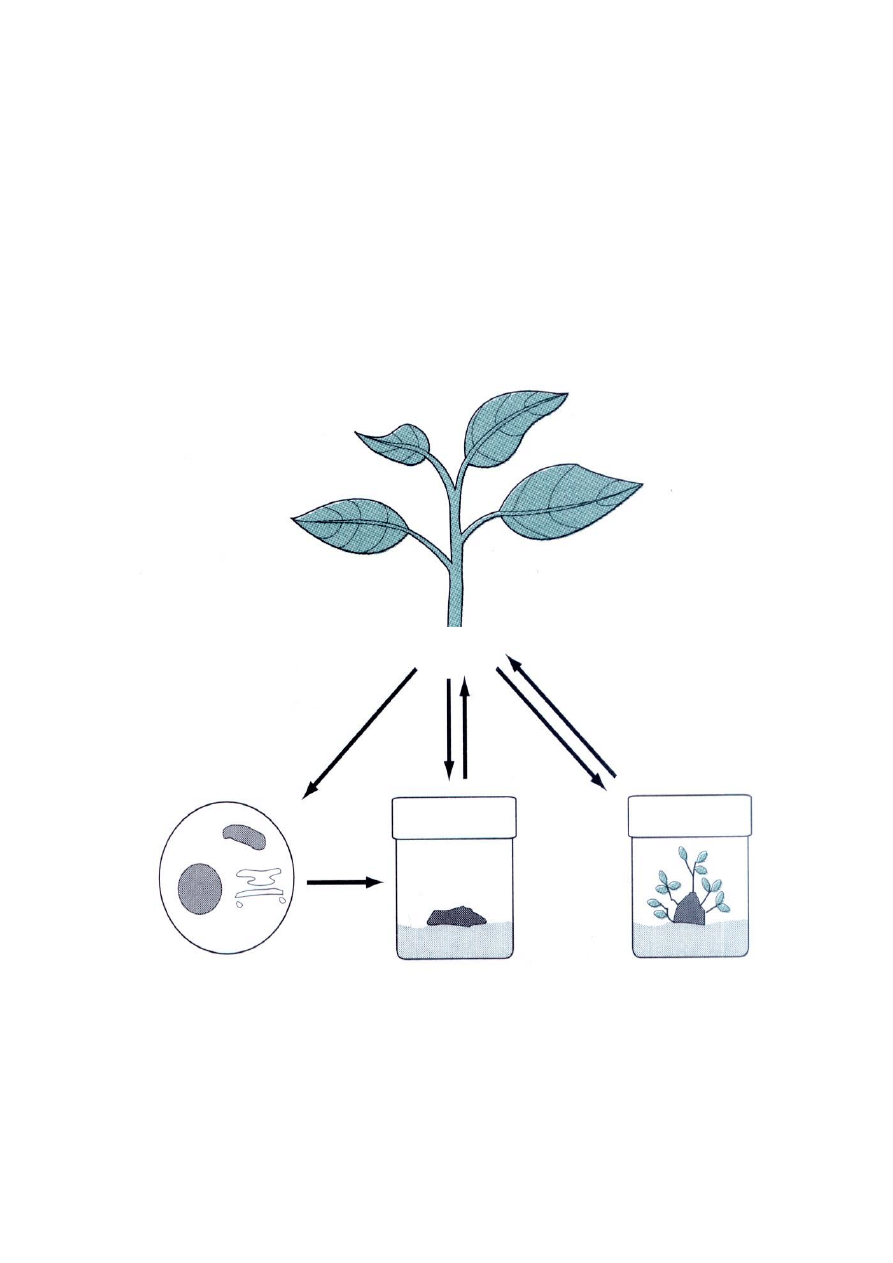
roślina
Protoplast Kultura kalusowa Kultura tkankowa
Fizjologia roślin M-2
Skrypt do dwiczeo
Instytut Biologii Eksperymentalnej Roślin
Warszawa 2010

1
Zarówno biolog molekularny jak i biotechnolog często prowadzi eksperymenty na roślinach.
Badania takie wymagają przygotowania odpowiedniego materiału roślinnego. W zależności od potrzeb i
celu doświadczeo używane są całe rośliny, tkanki, zawiesiny komórkowe bądź izolowane protoplasty.
Eksperymentator nie tylko powinien umied przygotowad materiał badawczy tzn. wyhodowad w
kontrolowanych warunkach rośliny, tkanki lub komórki, ale również zdawad sobie sprawę z ograniczeo
oraz interpretacji wyników w zależności od użytego materiału roślinnego.
Kontrolowane warunki hodowli są bardzo istotną sprawą przy powtórzeniu doświadczeo. Warunki
takie zapewniają komory fitotronowe, w których rośliny mogą rosnąd na różnorodnych podłożach.
Umiejętnośd dobrania odpowiednich warunków (długośd dnia, temperatura, podłoże, skład pożywki) to
bardzo istotne czynniki wpływające na wzrost roślin. Często w badaniach stosuje się tzw. kultury in vitro
(tkankowe bądź komórkowe). Materiał taki jest nieco trudniejszy do otrzymania, ale za to daje większe
możliwości wprowadzania do wnętrza komórki różnych związków oraz, co bardzo istotne dla biologa
molekularnego lub biotechnologa, manipulacji na poziomie genu. Kultury tkankowe in vitro dały np.
możliwośd szybkiego uzyskiwania jednorodnych genetycznie roślin (mikrorozmnażanie). Izolowane
protoplasty – komórki pozbawione ścian komórkowych używane są do badao metabolizmu, ale przede
wszystkim umożliwiają transformacje genetyczne tj. wprowadzenie określonych genów do komórki
roślinnej.
Dwiczenia będą uczyły podstawowych technik hodowli materiału roślinnego. Zaznajomią z
funkcjonowaniem komór fitotronowych, warunków szklarniowych, przygotowaniem podłoża oraz metodą
kultur hydroponicznych. Pokażą jak założyd i prowadzid kultury in vitro oraz nauczą podstawowych technik
manipulacji. Podczas zajęd postaramy się również zaznajomid Paostwa z podstawami izolacji organelli
roślinnych oraz oznaczeniami biochemicznymi, którymi możemy charakteryzowad materiał roślinny.
Dwiczenia nie są ilustracją wykładu, natomiast mamy nadzieję, ze zachęcą Paostwa do udziału w innych
zajęciach fakultatywnych IBER rozszerzających wiedzę na temat funkcjonowania roślin.
Dwiczenia będą prowadzone przez Zespół Pracowników Instytutu Biologii Eksperymentalnej
Roślin z Zakładów: Bioenergetyki Roślin, Anatomii i Cytologii Roślin, przy udziale Pracowni Fitotronowej.
Dwiczenia będą trwały przez połowę semestru, w każdym tygodniu w dwóch kolejnych dniach. Obecnośd
na wszystkich zajęciach jest obowiązkowa, zarówno w pierwszym jak i drugim dniu zajęd. Kolokwium
koocowe będzie dotyczyło materiału omawianego na zajęciach.
Anna Rychter
2
Spis dwiczeo:
Zadanie 1. Kultury roślinne (mgr Monika Ostaszewska, Zakład Bioenergetyki Roślin)
Zadanie 2. Kultury tkanek roślinnych in vitro, organogeneza (dr Izabela Juszczuk, Zakład
Bioenergetyki Roślin)
Zadanie 3. Otrzymywanie kultury zawiesinowej (mgr Katarzyna Tracz, Zakład Anatomii i
Cytologii Roślin)
Zadanie 4. Izolacja protoplastów (dr Bożena Szal, Zakład Bioenergetyki Roślin)
Zadanie 5. Oznaczanie stężenia chlorofilu i ATP w tkankach roślinnych (dr Bożena Szal,
Zakład Bioenergetyki Roślin)
Zadanie 6. Podstawy izolacji i charakterystyki organelli (dr Izabela Juszczuk, Zakład
Bioenergetyki Roślin
Plan dwiczeo:
Data
Grupa I A
11.00 - 13.00
Grupa I B
11.00 - 13.00
Grupa II A
13.45 - 15.45
Grupa II B
13.45 - 15.45
5. 10. 2010
Zadanie 2
Zadanie 4
Zadanie 2
Zadanie 4
6. 10. 2010
Zadanie 2
Zadanie 4
Zadanie 2
Zadanie 4
12. 10. 2010
Zadanie 1
Zadanie 2
Zadanie 1
Zadanie 2
13. 10. 2010
Zadanie 1
Zadanie 2
Zadanie 1
Zadanie 2
19. 10. 2010
Zadanie 3
Zadanie 1
Zadanie 3
Zadanie 1
20. 10. 2010
Zadanie 3
Zadanie 1
Zadanie 3
Zadanie 1
26. 10. 2010
Zadanie 4
Zadanie 3
Zadanie 4
Zadanie 3
27. 10. 2010
Zadanie 4
Zadanie 3
Zadanie 4
Zadanie 3
2. 11. 2010
Zad. 2/Zad. 3
Zadanie 5
Zad. 2/Zad. 3
Zadanie 5
3. 11. 2010
Zadanie 1
Zadanie 6
Zadanie 1
Zadanie 6
9. 11. 2010
Zadanie 5
Zad. 2/Zad. 3
Zadanie 5
Zad. 2/Zad. 3
10. 11. 2010
Zadanie 6
Zadanie 1
Zadanie 6
Zadanie 1
16. 11. 2010
Konsultacje
Konsultacje
Konsultacje
Konsultacje
17. 11. 2010
Egzamin
Egzamin
Egzamin
Egzamin

3
Dwiczenie 1. Kultury roślinne
Celem dwiczenia jest poznanie podstawowych sposobów prowadzenia kultur roślin
stanowiących materiał eksperymentalny lub będących źródłem tkanek do innych doświadczeo.
Doświadczenie umożliwia także obserwację skutków niewłaściwego prowadzenia kultur -
deficytu fosforu (P) oraz symbiozę bakterii rodzaju Rhizobium z korzeniami roślin motylkowych
na przykładzie fasoli.
Materiałem na dwiczeniach są rośliny fasoli (Phaseolus vulgaris) i rzodkiewnika (Arabidopsis
thaliana) hodowane w kulturze hydroponicznej, na naturalnym i sztucznym podłożu stałym oraz
w kulturze in vitro na pożywce zestalonej agarem. Hodowle prowadzone są w komorach
fitotronowych oraz w szklarni.
Kultury roślinne zostają założone w drugim tygodniu dwiczeo, zaś obserwacje i pielęgnacja kultur
prowadzona jest przez trzy następne tygodnie.
1 dzieo doświadczenia:
Kultura hydroponiczna:
Przygotowad 3 pojemniki do hodowli hydroponicznej fasoli oraz 2 do hodowli
rzodkiewnika.
Przygotowad pożywki Knopa (pożywkę pełną lub bez fosforu) wg wskazao osoby
prowadzącej.
Kultura torfowa i perlitowa
Przygotowad 12 doniczek z substratem torfowym, podlad wodą dejonizowaną.
Przygotowad 4 doniczki z perlitem, podlad wodą dejonizowaną.
Hodowla w pojemnikach typu „rhizobox”
Przygotowad pojemnik do hodowli napełniając go substratem torfowym, podlad wodą
dejonizowaną.
Kultura in vitro
Przygotowad słoiki do kultur in vitro (3 szt.) i szalki szklane 10 cm (9 szt.).
Przygotowad pożywkę Murashige-Skooga (MS).
Do erlenmayerki na 200 ml odważyd 600 mg agaru i dodad 90 ml pożywki MS. Rozpuścid
agar podgrzewając zawartośd erlenmayerki w kuchence mikrofalowej. Do każdego
słoiczka wylad po ok. 30 ml pożywki z agarem.
4
Do dwóch erlenmayerek na 200 ml odważyd po 600 mg agaru i dodad po 90 ml pożywki
MS. Rozpuścid agar podgrzewając zawartośd erlenmayerek w kuchence mikrofalowej,
erlenmayerki zamknąd folią aluminiową.
Przygotowad do sterylizacji koocówki do pipet automatycznych (żółte).
Szklane pipety Pasteura (15 szt.) zawinąd w folię aluminiową.
2 butelki (o poj. 100 ml) napełnid wodą destylowaną do 1/2 objętości, 1 butelkę napełnid
pożywką MS (bez agaru).
Sterylizowad wszystko w autoklawie w temp. 121°C.
2 dzieo doświadczenia
Kultura hydroponiczna
1 pojemnik do kultur hydroponicznych (do hodowli fasoli) napełnid pożywką pełną Knopa
1 pojemnik (do hodowli fasoli) napełnid pożywką Knopa -P.
1 pojemnik (do hodowli fasoli) napełnid wodą dejonizowaną.
Skiełkowane nasiona fasoli umieścid w pokrywkach, przykryd wilgotną gazą.
Koszyczki do hodowli rzodkiewnika (umieszczone w odpowiednich pojemnikach) napełnid
1% agarem w pożywce MS.
Nasiona rzodkiewnika zawieszone w 0,2% pożywce MS wysiad do koszyczków (przykryd
pokrywami).
Pojemniki ustawid w komorze fitotronowej.
Kultura torfowa
Skiełkowane nasiona fasoli umieścid w 8 doniczkach z substratem torfowym (5-6 na
doniczkę), podlad wodą dejonizowaną.
W 4 doniczkach z substratem torfowym umieścid skiełkowane nasiona fasoli (5-6 na
doniczkę), uprzednio umoczone w zawiesinie Rhizobium.
Doniczki umieścid w szklarni.
Kultura na perlicie
Skiełkowane nasiona fasoli wysadzid do doniczek z perlitem (5-6 na doniczkę) po
uprzednim umoczeniu w zawiesinie Rhizobium.
Doniczki umieścid w szklarni
Hodowla w pojemnikach typu „rhizobox”
Skiełkowane nasiona fasoli wysadzid do pojemnika (ewentualnie po uprzednim
umoczeniu w roztworze Rhizobium wg wskazania osoby prowadzącej).
5
Pojemniki umieścid w szklarni pod odpowiednim kątem (wg wskazao osoby
prowadzącej).
Kultura in vitro
Nasiona rzodkiewnika umieścid w probówce Eppendorfa, dodad 1 ml 70% etanolu,
wstrząsad przez 2 min.
Usunąd etanol, dodad 5% wodny roztwór wody Javelle'a + TWEEN 20 (w proporcji 1
kropla na 50 ml). Sterylizowad nasiona przez 15 min. wytrząsając.
NASTĘPNE CZYNNOŚCI PRZEPROWADZAMY W KOMORZE LAMINARNEJ
Usunąd roztwór sterylizujący, płukad nasiona w 5-ciu zmianach sterylnej wody po 5 min.
Usunąd wodę, dodad sterylną pożywkę.
Zawiesinę nasion wysiad sterylnie pipetą do słoików i na szalki wg wskazao
prowadzącego.
Słoiki zamknąd pokrywkami z filtrem, szalki zakleid parafilmem.
Słoiczki i szalki wyjąd z laminaru, podpisad i umieścid w komorze do hodowli w temp.
21°C (fotoperiod 16 godz).
W następnym tygodniu:
Kultura hydroponiczna
Oznaczyd pH zużytej pożywki.
Wymienid pożywkę w pojemnikach na świeżą.
Usunąd liścienie z roślin.
Hodowle na podłożu stałym
Podlad pożywką kontrolną 4 doniczki z substratem torfowym i doniczki z perlitem.
Podlad pożywką 4x stężoną 4 doniczki z substratem torfowym.
Podlad pożywką (bez azotu) pojemniki z nasionami moczonymi w roztworze Rhizobium.
Kultura in vitro
Porównad morfologię korzeni siewek rosnących w słoikach i w pionowo umieszczonych
szalkach.
Kolejny tydzieo:
Kultura hydroponiczna
Porównad wzrost roślin w poszczególnych pojemnikach (pożywka pełna, pożywka -P,
woda dejonizowana).

6
Oznaczyd pH zużytej pożywki.
Wymienid pożywki na świeże.
Hodowle na podłożu stałym
Porównad wzrost roślin podlewanych pożywką kontrolną i 4x stężoną.
Porównad wzrost roślin hodowanych na podłożu torfowym i perlicie.
Zaobserwowad wzrost korzeni w rhizobox’ach.
Kolejny tydzieo:
Podsumowanie hodowli wg wskazao osoby prowadzącej.
Przygotowanie pożywek
POŻYWKA KNOPA (do kultur hydroponicznych)
1/ Przygotowujemy po 1 l wyjściowych roztworów soli w następujących stężeniach:
Ca(NO
3
)
2
-
144 g/l, KNO
3
- 30 g/l, MgSO
4
- 68 g/l , KH
2
PO
4
- 30 g/l, KCl - 30 g/l
Z każdego z w/w roztworów wyjściowych bierzemy po 5 ml na 1 l pożywki, poza tym:
Fe EDTA 28 g/l (1 ml na 1 l pożywki)
2/ Roztwór wyjściowy mikroelementów
W 1 l wody dejonizowanej rozpuszczamy:
50 mg CuSO
4
x 5 H
2
O, 100 mg ZnSO
4
x 7 H
2
O, 50 mg Co(NO
3
)
2
x 6 H
2
O, 50 mg Al
2
(SO
4
)
3
, 350 mg MnCl
2
x 4 H
2
O, 50 mg
NiSO
4
x 4 H
2
O, 5 mg KJ, 25 mg KBr, 50 mg Na
2
MoO
4
x 2 H
2
O, 2 krople nasyconego roztworu LiCl,
Roztwór mikroelementów dokładnie mieszamy - z tego roztworu bierzemy 1 ml na 1 l pożywki
Po dokładnym wymieszaniu składników pożywki doprowadzamy pH do 5,5 za pomocą 1M NaOH lub 1M HCl.
POŻYWKA HOAGLANDA-ARNOLDA (do podlewania kultur torfowych i perlitowych )
1/ Przygotowujemy po 1 l roztworu soli w następujących stężeniach:
Ca(NO
3
)
2
x 4 H
2
O - 1180 g/l, KNO
3
- 505 g/l, KH
2
PO
4
-
136 g/l, MgSO
4
x 7 H
2
O - 241 g/l,, Fe-cytrynian 5% - 50 g/l
Z każdego z tych roztworów bierzemy po 3,3 ml na 1 l pożywki
2/ Roztwór wyjściowy mikroelementów:
W 900 ml wody destylowanej rozpuszczamy:
50 mg CuSO
4
x 5 H
2
O, 100 mg ZnSO
4
x 7 H
2
O, 50 mg Co(NO
3
)
2
x 6 H
2
O, 50 mg Al
2
(SO
4
)
3
,
350 mg MnCl
2
x 4 H
2
O, 50 mg
NiSO
4
x 4 H
2
O, 25 mg KJ, 25 mg KBr, 50 mg Na
2
MoO
4
x 2 H
2
O, 550 mg H
3
BO
3
2 krople nasyconego roztworu LiCl
Roztwór mikroelementów dokładnie mieszamy - z tego roztworu bierzemy 1 ml na 1 l pożywki
Po dokładnym wymieszaniu składników pożywki doprowadzamy pH do 5,5 za pomocą 1M HCl lub 1M NaOH.

7
Dwiczenie 2. Kultury tkanek roślinnych in vitro, organogeneza
Rozwój roślin jest wynikiem wzrostu i różnicowania się komórek. Procesy te zachodzą w
trakcie kolejnych etapów rozwoju osobniczego (ontogenezy). Jednym z przejawów procesu
różnicowania jest powstawanie organów roślinnych czyli organogeneza.
Żywe komórki roślinne (niezależnie z jakiej tkanki pochodzą), uwolnione spod wpływu
rośliny macierzystej, dośd łatwo podlegają odróżnicowaniu. Możliwośd zmiany rozwoju
komórek w innym (niż dotychczasowy) kierunku świadczy o tym, że proces różnicowania nie
wiąże się ze zmianą jej podstawowej informacji genetycznej. Obserwowane w trakcie
różnicowania zmiany w rozwoju są wynikiem zróżnicowanej ekspresji genów. Komórki roślinne
są zatem totipotencjalne tzn. każda żywa komórka zawiera pełną informację genetyczną i w
może podjąd rozwój w każdym kierunku.
W regulacji różnicowania komórek uczestniczą hormony produkowane przez rośliny.
Świadczy o tym m.in. fakt, że wpływ rośliny macierzystej (produkującej hormony) na
różnicowanie się organów można zastąpid i przyśpieszyd działaniem podanego z zewnątrz
(egzogennego) hormonu.
Najbardziej przekonujące dowody na udział hormonów w różnicowaniu pochodzą z
doświadczeo z zastosowaniem hodowli izolowanych części roślin w warunkach sterylnych in
vitro. Izolacja fragmentów roślin eliminuje korelacyjne oddziaływania rośliny macierzystej, które
mogą byd zastąpione przez odpowiednio dobrane warunki hodowli. Typ wzrostu wyizolowanego
fragmentu rośliny (eksplantatu) i różnicowanie się organów zależą od ilościowej równowagi
pomiędzy dwoma hormonami roślinnymi auksyną i cytokininą. Obecnośd obu tych hormonów
jest niezbędna do przekształcenia zróżnicowanych tkanek eksplantatu w tkankę kalusową
(odróżnicowanie). Zwiększenie w pożywce hodowlanej stosunku cytokininy do auksyny indukuje
powstawanie pędów, a nadmiar auksyny względem cytokininy – tworzenie się korzeni.
Hodowla in vitro jest nie tylko prostym modelem doświadczalnym do badania procesów
związanych z różnicowaniem się tkanek i organów, lecz jest także intensywnie wykorzystywana
do celów praktycznych jak np. mikrorozmnażanie roślin.
Celem dwiczenia jest zapoznanie się z metodą hodowli roślin w warunkach in vitro oraz
wykazanie na przykładzie stymulowanego hormonami powstawania organów (organogenezy), że
komórki roślinne są zdolne do zmiany swojego programu różnicowania i rozwoju.
8
Indukcja powstawania pędów i korzeni u lnu (Linnum usitatissimumm L.) w kulturach in vitro
Materiał do doświadczeo stanowią fragmenty hipokotyli lnu izolowane z 9 dniowych siewek
wyhodowanych na stałym podłożu agarowym w sterylnych warunkach, przy 16 godzinnym
fotoperiodzie.
1 dzieo doświadczenia
Przygotowad 12 słoików przeznaczonych do hodowli in vitro (po 2 na każdy wariant
doświadczenia). Skalpele i pęsety sterylizowad w autoklawie przez 25 min. w
temperaturze 121
°
C.
Przygotowanie pożywek do indukcji organogenezy w izolowanych hipokotylach siewek lnu.
W zlewce na 500 ml przygotowad 300 ml pożywki Murashige i Skooga (4,43g/l).
Odpowiednią naważkę pożywki rozpuścid w wodzie, a następnie dodad sacharozy – tak
aby koocowe stężenie cukru w pożywce wynosiło 3%. Pożywkę rozlad po 50 ml do 6
odpowiednio oznakowanych zlewek (w zależności od wariantu doświadczenia). Do
pożywki podstawowej dodad odpowiednie ilości hormonów- auksyny i/lub cytokininy, w
celu uzyskania wariantów A-F (patrz poniżej). Obliczyd jaką objętośd każdego z
hormonów należy dodad do 50 ml pożywki aby uzyskad odpowiednie stężenie koocowe.
Doprowadzid pH każdej pożywki do wartości 5,8 (przy pomocy 0,1 M KOH) i przelad
pożywki do 12 opisanych słoików (po 25 ml na jedno powtórzenie każdego wariantu
doświadczenia). Następnie do każdego słoika dodad agar w takiej ilości, aby koocowe
jego stężenie wynosiło 8g/l. Tak przygotowane słoiki z pożywkami umieścid w
autoklawie i sterylizowad przez ok. 25 min. w temperaturze 121
°
C. Po wyjęciu z
autoklawu pozostawid pożywkę pod laminarem do zestalenia do następnego dnia.
Uwaga ! – wyjściowe stężenie BA i 2,4 D wynosi 1 mg/ml
A - kontrola (pożywka bez hormonów)
B - 2, 4 D (1mg/l)
C - BA (1mg/l)
D - 2, 4 D (1mg/l) + BA (1mg/l)
E - 2,4 D (1mg/l) + BA (0,1mg/l)
F - 2,4 D (0,1mg/l) + BA (1mg/l)
2,4 D- kwas 2,4-dwuchlorofenoksyoctowy
BA - benzyloaminopuryna
9
2 dzieo doświadczenia
Praca pod wyciągiem laminarnym. Z otrzymanych sterylnych hodowli delikatnie wyjąd
siewki lnu. Umieścid na sterylnych szalkach Petri’ego. Uważad aby nie uszkodzid
młodych siewek! Następnie sterylnym skalpelem wyciąd 2 cm fragmenty hipokotyli.
Umieścid po 3 szt. hipokotyli (lub więcej, w zależności od ilości dostępnego materiału) w
przygotowanych wcześniej słoikach zawierających pożywkę z różnymi kombinacjami
hormonów roślinnych (A,B,C,D,E,F). Sterylną kulturę hipokotyli umieścid w komorze
hodowlanej na 4 tygodnie, w temperaturze 24
°
C w dzieo i 22
°
C w nocy, przy 16
godzinnym fotoperiodzie (tzn. 16 godz. światło-dzieo, 8 godz. ciemnośd-noc).
W trakcie 4-tygodniowej hodowli w w/w warunkach prowadzid obserwacje, co tydzieo,
rozwoju fragmentów hipokotyli w poszczególnych wariantach doświadczenia. Wyniki
zestawid w tabeli, zaznaczając pojawianie się tkanki kalusowej, pędów i korzeni.
Tabela 1. Wpływ hormonów na powstawanie tkanki kalusowej i odtwarzanie organów
roślinnych w kulturach izolowanych fragmentów hipokotyli lnu.
stężenie hormonów w pożywce efekt morfogenetyczny
mg/l kalus pęd korzeo
2,4 D BA
0
0
1
0
0
1
1
1
0,1
1
1
0,1
Zwrócid uwagę na rolę auksyn i cytokinin w regulacji różnicowania się organów in vitro, oraz na
wpływ stężenia hormonów i wzajemnych ich proporcji na pojawianie się pędów i korzeni.

10
Dwiczenie 3. Otrzymywanie kultury zawiesinowej
Kultura zawiesinowa składa się ze zbioru pojedynczych komórek oraz różnej wielkości
agregatów komórkowych zwieszonych w pożywce płynnej. Utrzymanie takiej kultury wymaga
stałego dostarczania tlenu, co osiąga się dzięki zastosowaniu wytrząsarek lub użyciu
bioreaktorów (fermentatorów) wyposażonych w specjalne systemy napowietrzania. Dodatkowo
ciągły ruch, w jakim znajduje się kultura, zapewnia lepsze rozdrobnienie agregatów
komórkowych oraz utrzymuje komórki w zawieszeniu.
Zawiesiny komórkowe można uzyskad na dwa sposoby:
- przez kulturę określonych fragmentów tkanki roślinnej (tzw. eksplantatów) w pożywkach
płynnych,
- przez mechaniczne rozdrobnienie materiału roślinnego, filtrowanie go na sitach o określonej
wielkości oczek i zawieszenie w pożywce płynnej.
Fragmentami tkanki stosowanymi do zakładania kultury zawiesinowej najczęściej są: młody,
luźny kalus, zarodki oraz różne części siewek (np. hipokotyle). Tkanka roślinna musi byd
całkowicie sterylna i dlatego materiał używany do zakładania kultury zawiesinowej najczęściej
pochodzi z innej kultury in vitro. Przy inicjowaniu kultury zawiesinowej należy pamiętad, że do
tego celu nadają się tylko te fragmenty tkanki, które cechuje duża żywotnośd (ładnie i zdrowo
wyglądające). Stosunek masy wyjściowej eksplantatów do objętości pożywki powinien wynosid
od 0,3 do 1,5 g na 100 cm
3
pożywki. Stosowana w kulturze zawiesinowej pożywka płynna ma z
reguły taki sam skład, jaki pożywki przeznaczone do innych typów kultur in vitro dla danego
gatunku rośliny. Najczęściej spotykaną modyfikacją jest obniżenie o połowę lub więcej stężenia
soli mineralnych (z reguły makroelementów).
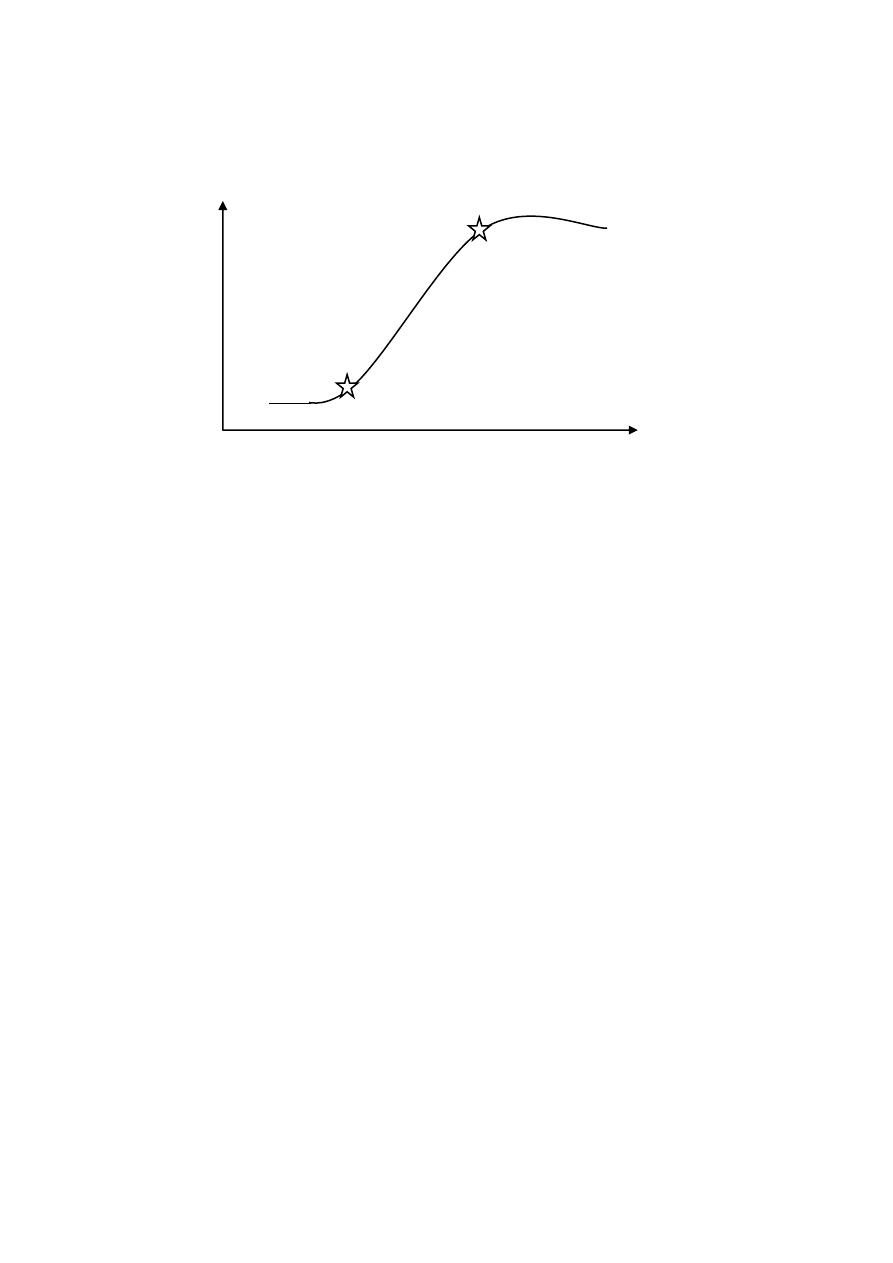
Przebieg wzrostu każdej kultury zawiesinowej ma swoisty charakter, jest mniej lub
bardziej zbliżony do krzywej wykładniczej. W każdej kulturze należy ustalid przynajmniej dwa
charakterystyczne punkty tej krzywej: rozpoczęcie fazy wzrostu logarytmicznego i zakooczenie
tej fazy wzrostu. Z reguły dokonuje się tego przez okresowe pomiary świeżej masy kultury. Faza
wzrostu logarytmicznego kooczy się najczęściej po 12 – 24 dniach od zainicjowania kultury. W
momencie zbliżania się tej fazy do kooca można zawiesinę przenieśd do nowej pożywki (pasaż)
lub, po prostu, zlikwidowad.
11
Krzywa wzrostu kultury zawiesinowej.
Celem dwiczenia jest założenie kultury zawiesinowej i zmierzenie jej żywotności. Kultury
zakładane będą poprzez rozdrobnienie kalusa i zawieszenie otrzymanego w ten sposób materiału
w pożywce płynnej. Cała procedura inicjacji kultury zawiesinowej przeprowadzona zostanie w
dwóch wariantach: w warunkach sterylnych (pod laminarem i przy wykorzystaniu wyjałowionych
narzędzi) i w warunkach niesterylnych (na stole laboratoryjnym). Materiałem do doświadczeo
jest kalus.
1 dzieo doświadczenia
Kalus należy delikatnie rozdrobnid, uważając przy tym, aby nie zmiażdżyd komórek.
Następnie tkankę zawiesid w pożywce płynnej Murashige Skooga (MS) i przesączyd przez sitko w
celu odrzucenia zbyt wielkich agregatów komórek. Przesącz pozostawid do odstania i
zdekantowad tak by pozostało około 0,5 cm starej pożywki, a następnie zawiesid w świeżej
pożywce. Zaszczepione kolby odstawid na wytrząsarkę.
Żywotnośd komórek i agregatów komórkowych oceniana jest metodą tetrazoliową, w
której określa się ilośd czerwonego barwnika, powstałego w komórkach w wyniku redukcji
chlorku tetrazoliowego (TTC). Ilośd powstałego barwnika jest proporcjonalna do stopnia
żywotności komórek. za miarę żywotności komórek przyjmuje się zmiany zawartości
wyekstrahowanego etanolem barwnika oznaczone w stosunku do czystego etanolu.
zakooczenie fazy wzrostu
rozpoczęcie fazy wzrostu
lo
g lic
zb
y
ko
m
ó
rek
czas kultury

12
Częśd prób nie wykorzystanych do zaszczepienia pożywki, należy podzielid na naważki o
masie 0,1 g; umieścid w probówce i zalad 3 ml odczynnika TTC o stężeniu 0,4 %. Odstawid na noc
w ciemne miejsce.
2 dzieo doświadczenia
Obejrzed i opisad wyniki testu TTC. Obejrzed pod mikroskopem i opisad komórki
zawiesiny. Wyniki przedstawid w tabeli.
W następnym tygodniu przeprowadzid dalsze obserwacje kultur zawiesinowych.

13
Dwiczenie 4. Izolacja protoplastów z liści jęczmienia (Hordeum vulgare L.)
Izolacja protoplastów mezofilu polega na uzyskaniu z tkanki miękiszu asymilacyjnego liści
pojedynczych „komórek” pozbawionych ściany komórkowej, a ograniczonych tylko błoną
komórkową (protoplasty), przy użyciu odpowiednich enzymów, do których należą: celulaza,
pektynaza, hemicelulaza. Enzymy te mają zdolnośd rozkładu poszczególnych składników
budujących ścianę komórkową. Komórki roślinne pozbawione ściany komórkowej, in vitro
przyjmują kształt kulisty i nie wykazują zdolności do podziału. W odpowiednich warunkach w
płynnej pożywce protoplasty odbudowują ścianę, a powstałe komórki wchodzą w fazę
podziałową. W ten sposób tworzą się agregaty komórkowe, a kultura protoplastów stopniowo
staje się kulturą zawiesinową.
Izolowane protoplasty stanowią doskonały obiekt do tzw. badao przyżyciowych komórek
roślinnych. Stanowią one użyteczne narzędzie do badao metabolizmu roślin. Uzyskiwanie
protoplastów z różnych typów tkanek znalazło również zastosowanie w biotechnologii roślin.
Protoplasty stanowią bowiem jednorodną populację komórek roślinnych pozbawionych ściany
komórkowej, do której każdy czynnik eksperymentalny dociera jednocześnie i w tym samym
stężeniu. Wprowadzanie do protoplastów obcego DNA umożliwia uzyskanie transgenicznych
roślin wykazujących cechy kodowane przez wprowadzony gen. Możliwa jest regeneracja in vitro
całych roślin z kultury protoplastów prowadzonej w odpowiednich warunkach laboratoryjnych.
Celem doświadczenia jest zapoznanie się z metodą izolacji nienaruszonych protoplastów.
Materiałem doświadczalnym będą liście 6-dniowego jęczmienia ozimego odm. Gregor
hodowanego w kulturach hydroponicznych
Izolacja protoplastów z liści jęczmienia zostanie przeprowadzona wg metody opisanej
przez Gardestrom i Wigge (1988, Plant Physiol. 88: 69-76). Fragmenty liści pozbawione epidermy
umieszczamy w szalkach Petri’ego w roztworze do płukania tkanek (TWM). Powierzchnia blaszki
liściowej pozbawiona epidermy powinna byd zanurzona w roztworze. Po kilku min. usuwamy
roztwór TWM, a następnie pipetujemy do szalki roztwór do trawienia. Fragmenty liści w szalce
nie powinny nakładad się na siebie lub byd pozwijane. Tak przygotowany materiał trawimy ok. 1
godz. w temperaturze 27-29 C w słabym świetle. Po tym czasie bardzo delikatnie usuwamy
roztwór do trawienia i do szalek delikatnie wlewamy niewielką ilośd buforu do płukania (WM)
(bufor powinien byd schłodzony do temp 4°C) i delikatnie wytrząsając uwalniamy protoplasty.
Zawiesinę protoplastów przesączamy napierw przez sitko a następnie przez filtr o średnicy oczek

14
100 µm. Pozostały materiał przemywamy dwukrotnie niewielką ilością roztworu do płukania.
Następnie zawiesinę protoplastów wirujemy 3 min. przy 200 x g, uzyskany osad zawiera zarówno
całe jak i uszkodzone protoplasty. Powstały osad zawieszamy ostrożnie (delikatnie potrząsając) w
schłodzonym do temp. 4°C buforze F1 i przenosimy do probówek wirówkowych, po czym na
powierzchnię tej warstwy delikatnie pipetujemy niewielkie ilości najpierw roztworu F2 a później
roztworu F3 (oba roztwory powinny mied temp. 4°C). Powstały gradient wirujemy 5 min przy
300 x g. Po wirowaniu protoplasty z nienaruszoną błoną powinny znaleźd się pomiędzy
warstwami F2 i F3.
Przeprowadzamy obserwację otrzymanych protoplastów w mikroskopie świetlnym.
ROZTWORY DO IZOLACJI PROTOPLASTÓW Z LIŚCI JĘCZMIENIA:
Roztwór do płukania tkanek (TWM): 0,5 M sorbitol; 1 mM CaCl
2
; 0,05% PVP; 5 mM Hepes pH 7,2
Roztwór do trawienia: 1% celulaza; 0,3% maceraza; 0,5 M sorbitol; 10 mM MES pH 5.5; 0,05% PVP; 1
mM MgCl
2
; 1mM CaCl
2
; 0,05% albumina, 20 mM askorbinian
Roztwór do płukania (WM): 0,5 M sorbitol; 1 mM CaCl
2
; 0,05% PVP; 5 mM Hepes pH 7,0; 0.05%
albumina (odtłuszczona)
Roztwór F1: 0,5 M sacharoza; 1 mM MgCl
2
; 5 mM Hepes pH 7,0
Roztwór F2: 0,4 M sacharoza; 0,1 M sorbitol; 1 mM MgCl
2
; 5 mM Hepes pH 7,0
Roztwór F3: 0,5 M sorbitol; 1 mM MgCl
2
; 5 mM Hepes pH 7,0

15
Dwiczenie 5. Oznaczenia stężenia chlorofilu i ATP w tkankach roślinnych
Badania metabolizmu roślin wymagają precyzyjnych metod oznaczania stężenia
poszczególnych związków (tzw. metabolitów). Do oznaczeo takich wykorzystujemy np. metody
spektrofotometryczne, luminometryczne, fluorymetryczne. Podstawową zasadą w oznaczaniu
metabolitów jest zastosowanie wydajnej ekstrakcji co wiąże się z doborem odpowiedniego
roztworu oraz warunków do ekstrakcji (np. temperatura). Procedura oznaczania danego
metabolitu również musi byd dobrze dobrana do osiągnięcia zamierzonego celu. W wielu
przypadkach metody oznaczeo opierające się na właściwościach fizykochemicznych danego
związku są wystarczające, metody te wykorzystują reakcje chemiczne oraz właściwości
spektralne badanego związku; w ten sposób oznaczyd możemy np. stężenie chlorofilu. Jednak do
oznaczenia sporej grupy metabolitów, szczególnie takich, które znajdują się które mogą
znajdowad się w komórkach w bardzo małych stężeniach (np. ATP), konieczne jest wykorzystanie
specyficzności enzymów.
Celem dwiczenia jest oznaczenie stężenia chlorofilu z wykorzystaniem znanych wartości
współczynników ekstynkcji otrzymanych dla chlorofilu a i chlorofilu b oraz oznaczenie statusu
energetycznego tkanek liści rzodkiewnika zbieranych po okresie światła lub ciemności z
wykorzystaniem specyficzności układu lucyferyna-lucyferaza.
Ekstrakcja oraz spektrofotometryczne oznaczanie stężenia chlorofilu w liściach rzodkiewnika
Oznaczanie stężenia chlorofilu a i b wykonujemy wg metody opisanej w Porra i wsp.
(1989) [Biochim Biophys Acta, 975: 384-394]. Zważyd i zanotowad masę krążka wyciętego z liścia
rzodkiewnika; tkankę umieścid w probówce typu eppendorff (procedurę ekstrakcji
przeprowadzamy w temp 4 °C). Następnie dodad 1 ml 80% schłodzonego acetonu oraz dwie
stalowe kulki do homogenizacji. Homogenizowad tkankę przez 3 min. w młynie kulkowym.
Ekstrakty zwirowad 10 min. przy 10 000 x g w temp. 4 °C (przed wirowaniem należy koniecznie
usunąd kulki z probówek!). Zebrad supernatant i zmierzyd absorbancję próbek przy 2 długościach
fali: λ = 663,3 nm; λ = 646,6nm.
Stężenia chlorofilu (µg ml
-1
) wyliczyd wg wzoru:
Chl a = 12,25 Abs
663,3
– 2,55 Abs
646,6
Chl b = 20,31 Abs
646,6
– 4,91 Abs
663,6
Chl a + b = 17,76 Abs
646,6
+ 7,34 Abs
663,6
Obliczyd zawartośd chlorofilu w 1 g świeżej masy tkanki oraz w 1 cm
2
powierzchni liścia.

16
Ekstrakcja i luminometryczne oznaczanie stężenia ATP w liściach rzodkiewnika
Ekstrakcja: Zważyd i zanotowad masę liścia rzodkiewnika; liśd umieścid w probówce typu
eppendorff (procedurę ekstrakcji przeprowadzamy w temp 4 °C). Następnie dodad 400 µl 3%
schłodzonego kwasu trójchlorooctowego oraz dwie stalowe kulki do homogenizacji.
Homogenizowad tkankę przez 3 min. w młynie kulkowym. Ekstrakty zwirowad 10 min. przy
10000g w temp. 4 °C (przed wirowaniem należy koniecznie usunąd kulki z probówek!).
Zebrad supernatant i oznaczyd w nim stężenie ATP.
Oznaczanie: Oznaczanie wykonujemy z wykorzystaniem zestawu do oznaczania ATP firmy
Biothema (Szwecja). Do probówki typu eppendorff odpipetowad: 60 µl buforu do oznaczeo
oraz 200 µl roztworu lucyferyny-lucyferazy i zmierzyd luminescencję (tło). Następnie dodad
3 ul ekstraktu tkankowego i ponownie zmierzyd luminescencję. Z różnicy luminescencji
wykorzystując krzywą wzorcową wyliczyd zawartośd ATP w próbce. Przeliczyd zawartośd ATP
w 1 g świeżej masy tkanki.
Wykonanie krzywej wzorcowej. Do probówki typu eppendorff odpipetowad: 60 µl buforu do
oznaczeo oraz 200 µl roztworu lucyferyny-lucyferazy i zmierzyd luminescencję (tło).
Następnie dodad 1-5 ul wzorcowego 10 µM roztworu ATP i ponownie zmierzyd
luminescencję. Wyrysowad krzywą wzorcową zależności ilości ATP od luminescencji roztworu
(wyrażanej w jednostkach CPS, counts per second)

17
Dwiczenie 6. Podstawy izolacji i charakterystyki organelli
Mitochondria to organella przetwarzające energię w komórce w procesie oddychania.
Zawartośd macierzy mitochondrialnej otoczona jest podwójną błoną białkowo-lipidową, w której
zlokalizowane są białka odpowiedzialne za transport elektronów i syntezę ATP. Izolacja
mitochondriów roślinnych opiera się na zastosowaniu serii wirowao różnicowych
zhomogenizowanych tkanek. Mieszanina do homogenizacji powinna: 1) byd schłodzona i
buforowana poprzez zapewnienie pH ok. 7,5 -8,0; 2) mied zbliżone do tego, jakie panuje w
cytosolu ciśnienie osmotyczne, które zapewnia najczęściej obecnośd cukru; 3) zawierad
substancje wiążące fenole, jony metali dwuwartościowych i wolne kwasy tłuszczowe, które po
wydostaniu się z wakuoli mogą powodowad uszkodzenia m.in. białek i/lub kwasów
nukleinowych; 4) zawierad związki o charakterze redukującym, które są odpowiedzialne za
utrzymanie struktury białek i mają właściwości przeciwutleniaczy. Homogenizacja tkanek
roślinnych ma na celu rozbicie ściany komórkowej i uwolnienie jak najmniej uszkodzonych
organelli i dlatego powinna trwad możliwie krótko. Najlepsze efekty przynosi zastosowanie
moździerza przy przeprowadzeniu całej procedury w chłodni. Wyizolowane mitochondria, po
oznaczeniu stopnia integralności błony zewnętrznej, wykorzystuje się do charakterystyki
oddychania komórkowego in vitro. Zastosowanie elektrody tlenowej Clarka pozwala na badanie
zużywania tlenu i wytwarzania ATP przez izolowane mitochondria po dodaniu różnych
substratów oddechowych oraz związków stymulujących lub hamujących oddychanie. Najprostsza
procedura preparatyki mitochondriów to otrzymanie aktywnych „mitochondriów płukanych”,
zanieczyszczonych przez inne organella lub ich fragmenty (np. błony chloroplastowe, jeśli
preparat został przygotowany z tkanek zielonych).
Celem dwiczenia jest wykonanie izolacji i przeprowadzenie krótkiej charakterystyki oddechowej
mitochondriów z liści rzodkiewnika (Arabidopsis thaliana L.).
Izolacja mitochondriów z liści rzodkiewnika
Izolacja mitochondriów zostanie przeprowadzona wg metody opisanej przez Keech i wsp. (2005,
Physiol Plant 124: 403-409). Około 5 g świeżych liści rzodkiewnika odciąd od rozet, rozdrobnid na
mniejsze fragmenty i umieścid bezpośrednio w schłodzonym moździerzu. Homogenizowad w
mieszaninie o pH 8,0 i składzie: 0,3 M sacharoza, 60 mM TES *(hydroksymethyl)-methyl-2-
aminoethanesulphonic acid], 10 mM EDTA [etylene diamine tetra-acetic acid], 10 mM KH
2
PO
4
,
25 mM pirofosforan czterosodowy, 1 mM glicyna, 1% (w/o) PVP-40 [poliwinylopirolidon], 1%

18
(w/o) odtłuszczona albumina (BSA), 50 mM askorbinian sodu i 20 mM cysteina. Homogenat
ostrożnie przesączyd przez nylonową siatkę o wielkości oczek 20 µm i odwirowad przez 5 min.
przy 2 500 x g w celu mechanicznego usunięcia większości całych chloroplastów i błon
tylakoidów. Nadsącz przelad do nowej probówki wirówkowej i wirowad 15 min. przy 15 000 x g.
Nadsącz odrzucid a osad zawierający nieoczyszczone „płukane” mitochondria zawiesid w 200 µl
buforu do płukania o pH 7,5 i składzie: 0,3 M sacharoza, 10 mM TES, 2 mM EDTA, 10 mM
KH
2
PO
4
.
Oznaczanie aktywności oddechowej „płukanych” mitochondriów
Pomiary zużywania tlenu przez izolowane mitochondria prowadzid w stałej temp. 25 °C,
wykorzystując połączoną z rejestratorem komputerowym elektrodę tlenową Clarka (Hansatech).
Objętośd koocowa mieszaniny reakcyjnej o pH 7,5 i składzie: 0,3 M sacharoza, 10 mM TES, 10
mM KCl, 2 mM MgSO
4
, 10 mM K H
2
PO
4
, 0,1% BSA, powinna wynosid 0,5 ml. Do oznaczania
aktywności oddechowej pobierad 10-100 µl zawiesiny mitochondriów. Jako substraty
oddechowe stosowad 10 mM jabłczan (w obecności 1 mM glutaminianu) lub 1 mM NADH.
Wytwarzanie ATP mierzyd w obecności 50 nmoli ADP. Inhibitory oddychania, KCN lub SHAM
stosowad w koocowym stężeniu 1 mM.

19
Regulamin pracy w laboratorium
1.
W pracowni nie wolno jeśd, pid oraz palid.
2.
Ubranie wierzchnie należy pozostawid w szatni, natomiast torby, siatki, itp. składad w miejscu do tego
przeznaczonym.
3.
Każda grupa ma wyznaczone miejsce pracy, za którego czystośd jest odpowiedzialna.
4.
Przed przystąpieniem do pracy należy zapoznad się z obsługą używanej aparatury.
5.
Alkohol używany w pracowni skażony jest w dawkach niebezpiecznych dla zdrowia.
6.
Mając do czynienia z silnie trującymi związkami zachowad szczególną czystośd rąk i miejsca pracy.
Prace wykonywad w rękawiczkach.
7.
Ze stężonymi kwasami, zasadami oraz innymi substancjami żrącymi pracowad można w wyznaczonych
miejscach. Szczególną ostrożnośd zachowad przy ich odmierzaniu. Skórę oparzoną kwasem lub
zasadą należy spłukad dokładnie wodą bieżącą, następnie przemyd 5% NaHCO
3
(kwas) lub 2%
CH
3
COOH (zasada). W przypadku stężonych kwasów skórę należy przed spłukaniem osuszyd.
8.
O każdym, nawet drobnym, wypadku należy powiadomid asystenta prowadzącego dwiczenie.
Wyszukiwarka
Podobne podstrony:
Suzuki Kizashi FR, od 2010
m2 c 2010
m2 w 2010
Wykład 4 Fotosynteza M2 2010
BIOCHEMIA skrypt 2010 id 86508 Nieznany
2010 KD mgr zaocz teczka, Skrypty, PK - materiały ze studiów, II stopień, pomoc, III semestr, KBI 2
prawo pracy skrypt 2009 2010 id Nieznany
poprawiony skrypt ze srodowiska 2010
Prasoznawstwo skrypt 2010, Prasoznawstwo
skrypt z finansow - borodo 2010, Prawo Finansów Publicznych(5)
Podstawy prawoznawstwa skrypt[1] 23.01.2010(2), Szkoła, Uczelnia
prawo materialy 2, Prawo administracyjne skrypt zaoczne 2010, DECYZJA
BMZ - pytania z zerówki 2009-2010, Egzamin + skrypt Trojana
Prawa CzB3owieka - skrypt2E, Politologia UMCS (2005 - 2010) specjalność samorząd i polityka lokalna,
TI-skrypt, Ekonomia UWr WPAIE 2010-2013, Semestr I, Technologie Informacyjne
Psychologia przemysłowa - skrypt 2010, Psychologia, Psychologia Przemysłowa
PROJEKT SKRYPT ROZMOWY tel. 2010, BAS, IG BAS, IG - Skrypty tele
2010 02 12 skrypt OTZ dzienne
więcej podobnych podstron