
1
Lipidy i lipoproteiny
1. Trawienie i wchłanianie lipidów
Lipidy są nierozpuszczalne w wodzie, dobrze w rozpuszczalnikach organicznych. W organizmie tworza
błony komórkowe, są źródłem energii, witaminami lub koenzymami a także hormonami. Najczęściej
występują pod postacią kwasów tłuszczowych, fosfolipidów, sfingolipidów i cholesterolu.
Kwasy tluszczowe
Składają się z łańcucha węglowodorowego zakończonego grupą –COOH (COO
-
). Łańcuch zawiera
zazwyczaj parzystą ilość atomów węgla. Część jest nasycona (najczęściej 16 palmitynowy i 18
stearynowy) a część nienasycona z konfiguracją cis. Wiązania podwójne są zawsze rozdzielone
przyjamniej jedną grupą CH2.
Są amfipatyczne, posiadają część hydrofobową i hydrofilową. Numeracja od węgla COOH, ostatni
węgiel to węgiel omega i od niego liczy się odległość do wiązania podwójnego.
Acyloglicerole
Estry glicerolu i kwasów tłuszczowych. Wyróżnia się mono/di/tri-acyloglicerole. W skład często
wchodzą różne kwasy tłuszczowe (niejednakowej długości). Grupa OH środkowego atomu węgla
glicerolu najczęściej wiąże kwas nienasycony.
Triacyloglicerole są rezerwą energetyczną organizmu. Są zredukowane, stanowią 60-90% masy tkanki
tłuszczowej, a stosunek wodoru do tlenu jest wyższy niż w cukrach.
Trawienie tłuszczów
Trawienie tłuszczów, czyli lipoliza zaczyna się w żołądku w niewielkim stopniu. Głównie przebiega w
jelicie cienkim. Wymaga enzymów – lipaz.
Lipaza kwasostabilna – powstaje w gruczołach tylnej części języka, nastepnie spływa przełykiem i
trafia do żołądka, gdzie wykazuje niewielką aktywność. Enzym działa w układzie dwuwarstwowym na
styku fazy wodnej (ENZYM) i fazy lipidowej(SUBSTRAT)
Lipaza żołądkowa – aktywna tylko w pH obojętnym (noworodki), wiec u dorosłego człowieka nie
wykazuje wysokiej aktywności.
W dwunastnicy zachodzi proces emulsyfikacji tłuszczów dzięki solom kwasów żółciowych, które
działają jak detergenty. Zmniejszają napięcie powierzchniowe kulek tłuszczu i przy ruchach
perystaltycznych powoduje emulsyfikację co zwiększa powierzchnię styku lipidów z enzymem.
Podobnie działają fosfolipidy obecne w żółci.
Triacyloglicerole, estry cholesterolu i fosfolipidy są hydrolizowane przez enzymy zawarte w soku
trzustkowym.
Cholecystokinina – hormon produkowany w odpowiedzi na obecność lipidów i częściowo
strawionych białek powoduje skurcz pęcherzyka żółciowego, powodując wypływ żółci do
dwunastnicy i pobudza komóeki egzokrynne trzustki do produkcji enzymów trawiennych.
Sekretyna – w odpowiedzi na niskie pH papki pokarmowej z żołądka. Pobudza trzustkę do
wydzielania soku bogatego w wodorowęglany co powoduje podniesienie wartości pH do optymalnej
wartości.
Lipaza trzustkowa – odłącza kwasy tłuszczowe przy C1 i C3 glicerolu. Produktem są wolne kwasy
tłuszczowe, 2-monoacyloglicerole.
Kolipaza - zakotwicza i stabilizuje lipazy na granicy fazy wodnej i lipidowej.
Fosfolipaza A2 – rozkłada fosfolipidy
Esteraza cholesterolowa - hydrolizuje estry cholesterolu cholesterol + wolne kwasy tłuszczowe
Tworzą się mieszane micele gdzie grupy hydrofobowe schowane są wewnątrz struktury a na
zewnątrz wystają hydrofilowe elementy. Micele dzięki temu są rozpuszczalne w wodzie i wchłaniają

2
się przez rąbek szczoteczkowy nabłonka jelitowego. Kwasy tłuszczowe o krótkich i średnich
łańcuchach mogą wchłaniać się bezpośrednio bez tworzenia micel.
Resynteza triacylogliceroli –kwasy tłuszczowe przechodza w postacie aktywne przez wiązanie CoA-
SH przy udziale syntetazy acylo~S-CoA cytosolowej (tiokinaza) a następnie przez acylotransferazy
przenoszone na 2monoacyloglicerol.
R-CH2-CH2-COOH + HS-CoA ----- ATP – AMP+PPi ---> R-CH2-CH2-CO-S-CoA
2 R-CH2-CH2-CO-S-CoA + 2-monoacyloglicerol triacyloglicerol + 2 CoA-SH
Krótkie kwasy tłuszczowe – krótsze niż 12C przechodzą do krążenia wrotnego, wiążą się z albuminą i
przechodzą do wątroby.
Cząsteczki triacylogliceroli i estry cholesterolu agregują w środowisku wodnym, otoczone zostają
hydrofilna powłoką (białka, fosfolipidy, cholesterol) i przechodzą do limfy nadając mętny, mleczny
wygląd. Chylomikrony są wydzielane droga egzocytozy z błony śluzowej j. cienkiego do naczyń
limfatycznych, wędrują przez przewój piersiowy do lewej żyły podobojczykowej i mieszają się z krwią.
Triacyloglicerole zawarte w chylomikronach są wyłapywane przez mięśnie szkieletowe, komórki tk.
tłuszczowej, mięsień serca, płuca, nerki i wątrobę.
Lipaza lipoproteinowa – degraduje triacyloglicerole do wolnych kwasów tłuszczowych i glicerolu.
Produkowany przez lipocyty i komórki mięśniowe, wędruje wraz z krwią i przylega do śródbłonka
naczyń krwionośnych. Wolne kwasy tłuszczowe wnikają bezpośrednio do lipocytów i mieśni, lub łączą
się z albuminami i krążą we krwi. Większośc komórek utlenia kwasy tłuszczowe, a lipocyty
magazynują je. Glicerol jest uwalniany z triacylogliceroli i jest zużywany przez wątrobę do produkcji
glicerofosforanu a następnie utlenianiu do fosfodihydroksyacetonu i wchodzi w glikolizę lub
glukoneogenezę.
2. Transport lipidów w chłonce i osoczu
Rodzaje i budowa lipoprotein
Hydrofobowy charakter lipidów powoduje potrzebę powstawania kompleksów białkowo-lipidowych.
Lipoproteiny mają wnętrze hydrofobowe gdzie znajdują się triacyloglicerole i estry cholesterolu.
Powłoka jest hydrofilna i zbudowana jest z lipidów majacych grupy polarne oraz białek apoprotein
(apolipoproteiny).
Apoproteiny zapewniają rozpuszczalność i rozpoznawanie lipoprotein przez receptory. Aktywują
enzymy lipolityczne, które rozkładają lipoproteiny. Dzielą się na podklasy.
Chylomikrony są lipoproteinami o najniższej gęstości i największej średnicy – mają duzo lipidów i
niewiele apoprotein
VLDL i LDL – mają zwiększajacą się gęstość, mniej lipidów i więcej apoprotein, ich srednica się
zmniejsza
HDL – najwyższa gęstość, mają najwięcej apoprotein i najmniej lipidów. Ich średnica jest najmniejsza
Przy elektroforezie najbardziej wędrują do [+] HDL, potem VLDL, LDL i Chylomikrony (które pozostają
w miejscu)
Chylomikrony – powstają w ścianie jelita cienkiego, syntezowane w RE szorstkim a następnie w RE
gładkim. Przemiana apolipoprotein i lipidów w chylomikrony zachodzi w czasie ich przechodzenia z
RE gładkiego do AG. Przez limfę trafiają do krwi, przenoszą triacyloglicerole TAG, cholesterol i inne
lipidy. Nowo powstałe chylomikrony zawierają apoB-48 i podlegają szybkiej modyfikacji przed
wydzieleniem do osocza przez wbudowanie apoE. ApoE i apoB-48 rozpoznawane SA przez receptory
w wątrobie. Wbudowywane są również apoC i apoC-II (pochodzenia HDL) – potrzebne do aktywacji
lipazy lipoproteinowej. Na powierzchni komórek śródbłonka naczyń wątroby lipaza lipoproteinowa
rozkłada lipoproteiny do wolnych kwasów tłuszczowych i monoacylogliceroli. Część jest wchłaniana
przez komórki a część wiąże się z albuminami i przechodzi do odległych tkanek.

3
Lipaza lipoproteinowa po podaniu heparyny egzogennej aktywuje się. TAG w kompleksach
lipoproteinowych ulegają hydrolizie a osocze staje się słomkowożółtym płynem.
Chylomikrony których rdzeń został zdegradowany zwiększając gęstość stają się chylomikronami
resztkowymi. Następnie wymieniają apoproteiny z innymi lipoproteinami i dzięki apoE i apoB-48
zostają zatrzymane na komórkach wątrobowych i wchłonięte na drodze endocytozy. Pęcherzyk
endocytarny ulega fuzji z lizosomem uwalniając aminokwasy, cholesterol, kwasy tłuszczowe i zasady
azotowe.
VLDL – wytwarzane przez wątrobę, zawierają apoB-100 i apoA-I i TAG endogenne. Przenoszą lipidy z
wątroby do tkanek. We krwi pobierają apoC-II i apoE z HDL. Lipaza lipoproteinowa zlokalizowana na
śródbłonku zaktywowana przez apoC-II rozkłada TAG zawarte w VLDL, powodując zmniejszenie ich
średnicy i zwiększenie gęstości. Następuje wymiana apoE i apoC-II z HDL. Estry cholesterolu
przenoszone są z HDL do VLDL, a TAG i fosfolipidy z VLDL do HDL – dzięki białku przenoszącemu estry
cholesterolu. VLDL przemienia się w LDL pośrednio tworząc IDL.
LDL – transporter cholesterolu z wątroby do innych tkanek, głównie mięśnie, nerki i kora nadnerczy.
Zawierają apoB-100, mniej TAG niż VLDL, wiecej cholesterolu i jego estrów niż VLDL. Receptory LDL są
ujemnie naładowanymi glikoproteinami skupionymi w dołkach opłaszczonych. Powierzchnia dołka
jest pokryta latryną, która stabilizuje strukturę dołka. Przy związaniu LDL następuje wchłanianie na
drodze endocytozy. Klatryna po uformowaniu endosomu jest ponownie używana. ATPaza
endosomalna ma właściwości pompy protonowej i przy działaniu powoduje spadek pH, który
powoduje oddzielenie LDL od receptora. Receptory powracają na błone komórkową. Rózna ilośc
receptorów pozwala na regulację ilości wchłanianego cholesterolu.
- Cholesterol pochodzenia HDL i LDL hamuje biosynteze cholesterolu endogennego
- Nadmiar cholesterolu zostaje estryfikowany przez acetylotransferazę
acylo-S-CoA:cholesterol, aktywność wzrasta ze wzrostem zawartości cholesterolu w komórce.
Estry cholesterolu mogą być przechoywane w komórce.
- Zwiekszona ilość cholesterolu w komórce wpływa na obniżenie transkrypcji genu
odpowiedzialnego za receptor LDL.
LDL jest wychwytywany też przez makrofagi, nadmierny pobór zmodyfikowanych LDL powoduje
transformację makrofagów w komórki piankowate, które uczestniczą w powstawaniu blaszek
miażdżycowych.
HDL – syntezowane w wątrobie i scianie jelita. Oczyszcza osocze z cholesterolu, bo wolny cholesterol
jest przyłączany do HDL. HDL stanowi rezerwę apoprotein apoC-II (aktywator lipazy lipoproteinowej).
Pod działaniem acylotransferazy lecytyna:cholesterol powstają estry cholesterolu przenoszone do
VLDL i LDL i wymieniane na TAG. HDL jest degradowany w wątrobie a cholesterol uwalniany. HDL
wychwytują apoproteiny z chylomikronów resztkowych i LDL, zabezpiecza to apoproteiny przed
przedwczesną degradacją. Świeżo wydzielone HDL SA bezkształtne, zawierają cholesterol, fosfolipidy
i apoproteiny apoE, apoA, apoC. W miare akumulacji cholesterolu są przekształcane w kuliste
struktury. Cholesterol jest estryfikowany przez enzym osoczowy acylotransferazę
lecytyna:cholesterol syntetyzowaną w wątrobie a aktywowaną przez apoA-I zawartą w HDL. Reszta
kwasu tłuszczowego związana ze środkowym atomem węgla glicerolu jest przenoszona na
cholesterol pozostawiając lizolecytynę.
Ester cholesterolu jest tak hydrofobowy że nie może być wykorzystany do budowy błon
biologicznych. Usunięcie estru cholesterolu z HDL jest możliwe tylko przy udziale białka
przenoszącego estry cholesterolu. Kuliste HDL są pobierane przez komórki wątrobowe, estry
cholesterolu ulegają hydrolizie i cholesterol jest używany do budowania innych lipoprotein, syntezy
kwasów żółciowych lub wydzielonyu do żółci.

4
Zaburzenia przemiany lipidów
Prawidłowe stężenie lipoprotein 10-190mg/dl a cholesterolu 150-200mg/ml Jeżeli te wartości
zostaną przekroczone mówi się o hiperlipoproteinemii. Najłatwiej zbadać to elektroforezą.
Utworzono wiele obrazów hiperlipoproteinemii, która może być wrodzona lub nabyta. Pojawiają się
choroby wieńcowe i zmiany skórne w postaci kępek żółtych.
Jeżeli nastąpi spadek stężenia lipoprotein mówi się o hipolipoproteinemii. Pojawia się spadek masy
ciała, zaburzenia widzenia, zmieny neurologiczne.
Rodzinny niedobór lipazy lipoproteinowej – brak lipazy lipoproteinowej lub aktywatora apoC-II.
Uniemożliwia to rozkładu TAG, a ich stężenie jest większe niż 2000mg/dl. Zawartość cholesterolu jest
prawidłowa. Próbka osocza rozwarstwia się w temp=4*C, na górze pojawia się śmietankowa warstwa
lipidów a poniżej warstwa przejrzysta, wolna od chylomikronów. Pojawia się zapalenie trzustki, bo
chylomikrony są rozkładane przez lipazę trzustkową w naczyniach włosowatych trzustki. Produkty
hydrolizy wnikają do komórek trzustki powodując ich uszkodzenie i rozpad, co zwiększa uwalnianie
lipazy trzustkowej do krwi i dalszy rozpad lipidów.
Objawy: nawracające bóle brzucha (zapalenie trzustki), kepki żółte (makrofagi wypełnione lipidami),
powiększenie wątroby i śledziony. Zmniejszenie podazy tłuszczów w diecie łagodzi objawy
Rodzinna hipercholesterolemia – defekt receptora tkankowego lipoprotein LDL. Częściowa lub
całkowita niemożność wiązania LDL lub transportowania lipoprotein do wnętrza komórki. Wzrasta
stężenie cholesterolu w osoczu. Czasem pojawia się podwyższone stężenie TAG, LDL i VLDL. Osocze
nie rozwarstwia się w T=4*C.
Objawy: wczesny rozwój miażdżycy, komórki piankowe w ścianach tętnic wieńcowych. Zawały
pojawiają się u M w wieku 30 lat a u K w wieku 40. Kępki żółte w ścięgnach. Leczenie polega na
żywieniu dietą ubogą w cholesterol i podawaniu nienasyconych kwasów tłuszczowych.
Rodzinna Dys-β-lipoproteinemia – brak apoE-III odpowiedzialnego za wychwytywanie resztek VLDL
przez wątrobę. Zwiększa się stężenie lipidów w osoczu, a reszty VLDL wędrują w elektroforezie
między LDL i pre-VLDL.
Objawy: kepki żółte na dłoniach i w okolicach stawów, rozwój miażdżycy w okolicy t. głównej,
niedoczynność tarczycy i cukrzycy sprzyjają pojawianiu się objawów.
Rodzinna hipertriacyloglicerolemia – zwiększenie zawartości VLDL i wzrost TAG 200-500mg/dl.
Objawy: nie ukazują się. Ujawniają się w przypadku zaburzeń hormonalnych – cukrzycy,
niedoczynności tarczycy. Zwiększenie stężenia chylomikronów wywołuje objawy chorobowe
obserwowane w rodzinnym niedoborze lipazy lipoproteinowej.
Hiperlipidemia spowodowana różnymi defektami lipoprotein – zwiekszenie stężenia VLDL lub LDL.
VLDL SA przenośnikami TAG, a LDL cholesterolu. Nadmiar VLDL z powodu braku możliwości rozkładu
wywołuje hipertriacyloglicerolemię. Część VLDL jest zamieniana na LDL, które nie mogą być
metabolizowane prawidłowo i zwiększa się stężenie cholesterolu.
Objawy: przedwczesna miażdżyca, otyłość i nietolerancja glukozy.
A-β-lipoproteinemia – brak apoproteiny B-48. W osoczu nie występują chylomikrony, LDL i VLDL. W
elektroforegramie występuje pasmo HDL. Objawy złego wchłaniania, zburzenia neurologiczne,
wzrokowe i objawy hematologiczne. W cytoplazmie komórek nabłonka jelitowego krople tłuszczu.
Treść jelitowa jest wypełniona niestrawionym tłuszczem.
Objawy: w dzieciństwie zaburzenia równowagi, osłabienie siły, skurcze spastyczne uniemożliwiające
poruszanie się. Ubytki w polu widzenia. Erytrocyty zmieniają kształt i pojawiaja się kolce –
akantocyty. Z tego powodu rozwija się niedokrwistość akantocytoza. W akantocytach zwiększa się
ilość cholesterolu do fosfolipidów. Upośledzone wchłanianie witamin ADEK. Leczenie przez dietę z
tłuszczami krótkołańcuchowymi.

5
Rodzinna hipo-β-lipoproteinemia – zmniejszona zawartość apoB i frakcji LDL i VLDL. Stężenie
cholesterolu jest niskie, fosfolipidów zmniejszone, a TAG mieści się w normie. Nie daje objawów
klinicznych.
Choroba tangerska - pierwotną przyczyną – wrodzony niedobór apoA-I i apoA-II. Zmniejsza się
stężenie HDL i budowa jest nieprawidłowa. Ponieważ HDL jest rezerwuarem różnych apolipoprotein,
więc równocześnie VLDL i chylomikrony mają nieprawidłową budowę. Takie nieprawidłowe
lipoproteiny są wyłapywane przez komórki fagocytujące, przez co w wielu narządach kumulują się
estry cholesterolu. Objawy są spowodowane odkładaniem się estrów cholesterolu w narzadach.
Migdałki są powiększone i mają pomarańczową barwę. Powiększenie śledziony, wątroby i węzłów
chłonnych. Zaburzenie czucia i osłabienie mięśni.
Niedobór acetylotransferazy lecytyna : cholesterol – brak przenoszenia reszt kwasów tłuszczowych z
lecytyny na cholesterol. Zaburzony proces estryfikacji powoduje zmniejszenie stężenia estrów
cholesterolu w osoczu i zwiększenie stężenia lecytyny. Zmiany składu frakcji LDL i VLDL również w
wielkości. Zamiast HDL występują dwie inne frakcje.
Objawy: zmętnienie rogówki, niedokrwistość hemolityczna, białkomocz, niewydolność nerek.
3. Spalanie kwasów tłuszczowych
W komórkach proces jest regulowany hormonalnie. Hormon (głównie adrenalina) działa na receptor
błonowy. Następuje aktywacja cyklazy adenylanowej, która przekształca ATP w cykliczny 3’5’-AMP.
Cykliczny 3’5’-AMP aktywuje kinazę białkową, która z kolei fosforyluje lipazę hormonowrażliwą.
Karboksylaza acetylo-S-CoA sterująca biosyntezą kwasów tłuszczowych jest hamowana przez
zwiększone stężenie cyklicznego 3’5’-AMP. Związanie hormonu i produkcja cyklicznego 3’5’-AMP
powoduje lipolizę z zahamowaniem lipogenezy.
Lipaza hormonowrażliwa jest defosforylowana przy wysokim stężeniu glukozy i insuliny we krwi.
WKT przenikają przez błonę komórkową do krwi, gdzie wiążą się a albuminą i są transportowane ddo
innych tkanek. Erytrocyty, tkanka nerwowa i rdzeń nadnerczy nie może zużywać kwasów
tłuszczowych do celów energetycznych.
Kwasy tłuszczowe o krótkich łańcuchac do 10C przenikają bezpośrednio do wnętrza mitochondium.
Są aktywowane przez COA-SH kosztem energii ATP AMP + PP. Powstaje acylo-S-CoA. Reakcja jest
katalizowana przez mitochondrialną syntetazę acylo-S-CoA (tiokinaza). PP 2Pi dzięki pirofosfatazie,
reakcja jest nieodwracalna.
Kwasy tłuszczowe o długich łańcuchach powiżej 12C nie wnikają bezpośrednio do mitochonrium,
przeszkodę stanowi wewnętrzna błona. Aktywowane są w cytosolu przez przyłączenie cytosolowego
CoA-SH kosztem energii ATP AMP + PP. Reakcja katalizowana przez cytosolową syntetazę acylo-S-
CoA (tiokinazę). PP 2Pi dzięki pirofosfatazie, reakcja nieodwracalna. Następnie transport do
wnętrza mitochondrium zachodzi dzieki (mostkowi) czółenku karnitynowemu.
Czółenko karnitynowe – wytworzenie acylokarnityny z acylo-S-CoA i karnityny w cytosolu dzięki
działaniu acylotransferazy karnitynowej I, która związana jest z zewnętrzną powierzchnią
wewnętrznej błony mitochondrialnej. Acylokarnityna przenika do mitochondrium do macierzy i
zostaje rozłożona na karnitynę i acylo-S-CoA przez acylotransferazę karnitynową II związaną z
wewnętrzną powierzchnią wewnętrznej błony mitochondium.
Malonylo-S-CoA jest inhibitorem acylotransferazy karnitynowej I. Hamuje to transport kwasów
tłuszczowych do macierzy mitochondrialnej. Zwiększona ilość malonylo-S-CoA powstaje przy syntezie
kwasów tłuszczowych w cytosolu, dzięki czemu nowopowstałe kwasy tłuszczowe nie SA
degradowane w mitochonrium.
6
Brak acylotransferazy karnitynowej lub niskie stężenie karnityny w mięśniach powodują niemozność
wykorzystania energii z kwasów tłuszczowych i pojawiaja się osłabienia, bóle mięśni i przenikanie
mioglobiny do krwi z uszkodzonych mięśni.
Utlenianie kwasów tłuszczowych w mitochondrium
Składa się z 2 etapów:
- β-okydacja kwasów tłuszczowych – wielokrotne odwodornienie przy węglu β skraca łańcuch
kwasu tłuszczowego o 2C. Produktem tej części są cząstki acetylo-S-CoA.
- utlenianie reszt acetylowych w cyklu kwasów trój karboksylowych do CO2 i H2O.
β-oksydacja kwasów tłuszczowych o parzystej liczbie atomów węgla
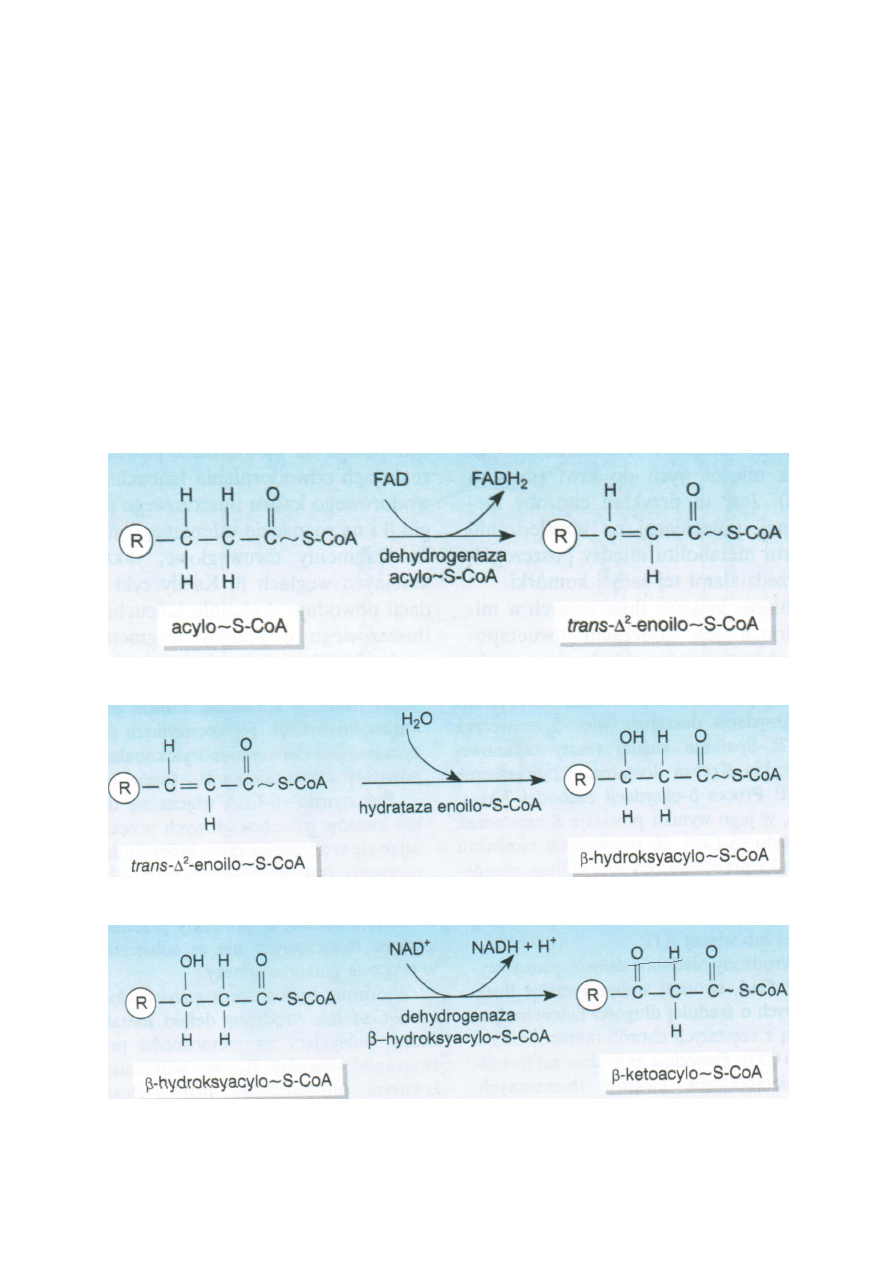
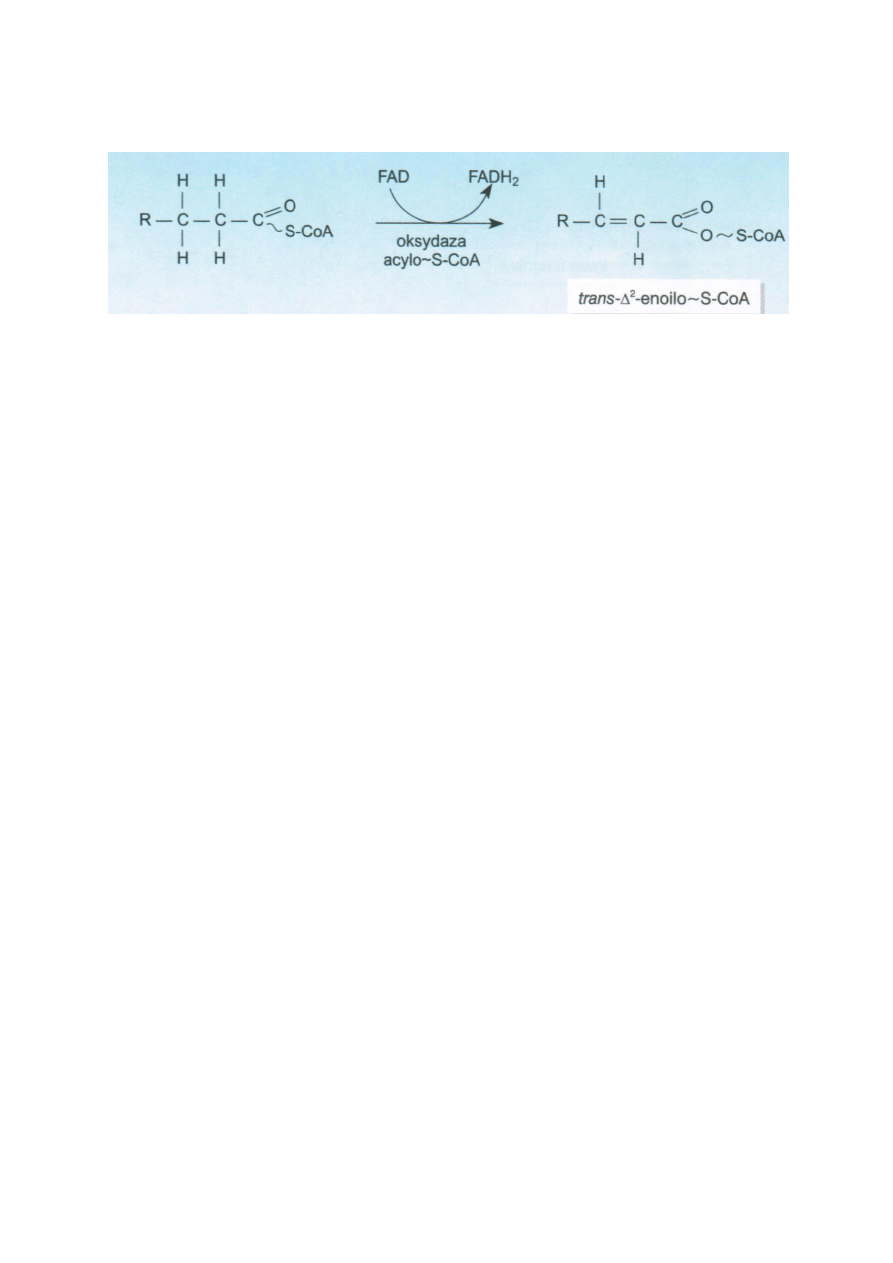
Acylo-S-CoA ulega odwodornieniu dzięki dehydrogenazie acylo-S-CoA. W mitochondrium występują 3
typu tego enzymu do długich, średnich i krótkich łancuchów. Akceptorem wodoru jest FAD. Powstaje
trans-Δ2-enoilo-S-CoA.
Do trans-Δ2-enoilo-S-CoA przyłączana jest woda dzięki hydratazie enoilo-S-CoA. Wodór wiąże się z
węglem α a OH z β. Powstaje β-hydroksyacylo-S-CoA.
β-hydroksyacylo-S-CoA jest substratem dehydrogenazy β-hydroksyacylo-S-CoA, która przekształca go
przy udziale NAD do β-ketoacylo-S-CoA.
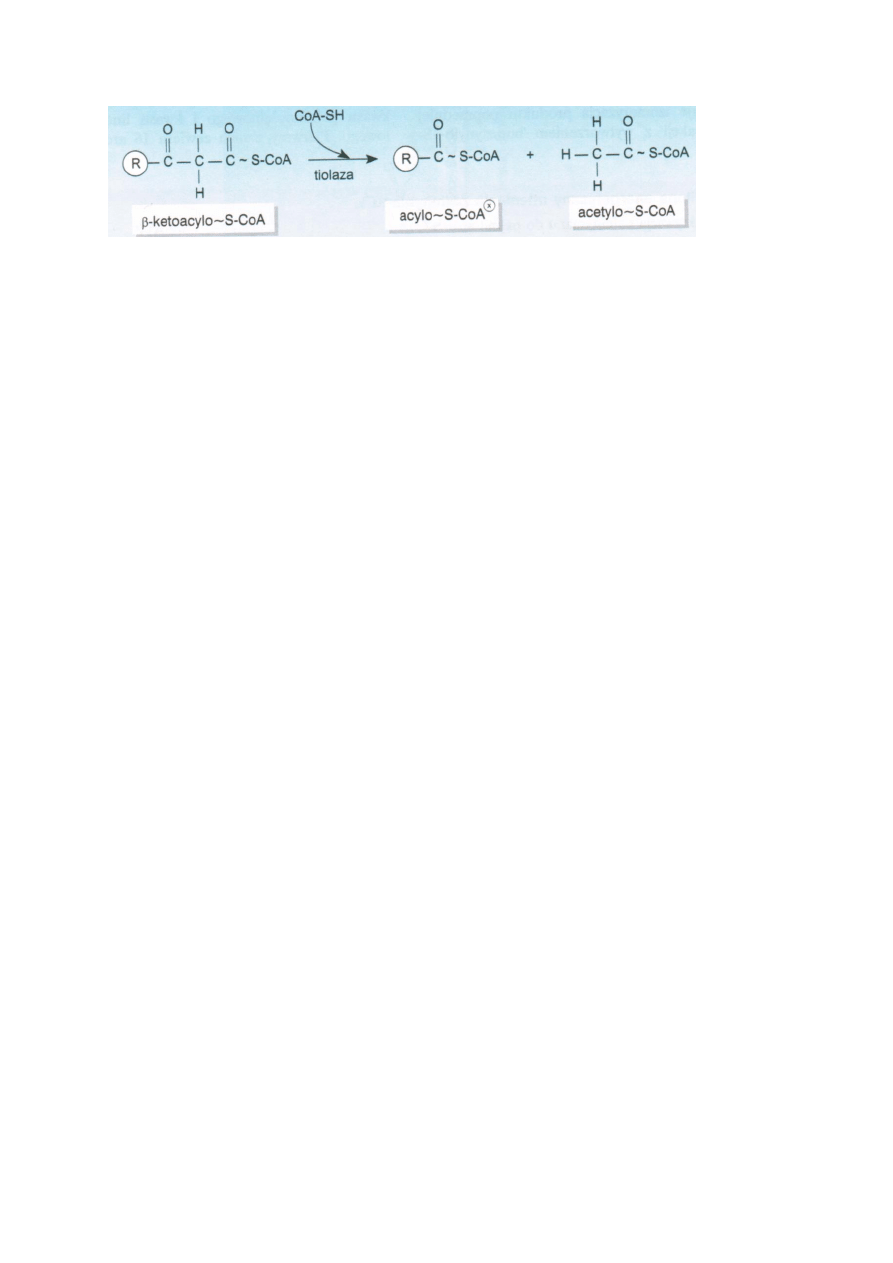
β-ketoacylo-S-CoA przy udziale tiolazy rozpada się na acetylo-S-CoA i acylo-S-CoA.
7
Powyższe reakcje się powtarzają i kwas tłuszczowy rozpada się na n-cząstek acetylo-S-CoA. Liczba
zachodzących β-oksydacji jest równa n-1, bo ostatnia daje 2x acetylo-S-CoA.
Bilans reakcji zależy od długości łańcucha kwasu tłuszczowego. Aby kwas tłuszczowy został włączony
do β-oksydacji musza zostać zuzyte 2xATP. Na każdą β-oksydację dostajemy czastkę FADH2 (2ATP) i
cząstkę NADH+H+ (3ATP). Z kazdego octanu w cyklu Krebsa powstaje 12 ATP.
Spalenie 1 cząstki palmitynianu (16C) daje: -2 ATP + 7x5 ATP + 8x12 ATP = 129 ATP
β-oksydacja kwasów tłuszczowych o nieparzystej liczbie atomów węgla
W wyniku ostatniej β-oksydacji powstaje fragment trój węglowy propionylo-S-CoA, który nie ulega
dalszej β-oksydacji. Propionylo-S-CoA ulega karboksylacji tworząc metylomalonylo-S-CoA dzięki
karboksylazie propionylo-S-CoA. Mutaza metylomalonylo-S-CoA wraz z koenzymem
deoksyadenozylokobalamina (powstaje z Wit B12) przekształca metlylomalonylo-S-CoA do
bursztynylo-S-CoA, który jest przekształcany do szczawiooctanu w cyklu Krebsa.
Wady:
Wrodzony niedobór dehydrogenazy acylo-S-CoA powoduje zakłócenia w metabolizmie kwasów
tłuszczowych, które są przczyną zespołu nagłej śmierci niemowląt. SIDS sudden infant death
syndrome.
Wrodzony brak mutazy metlomalonylo-S-CoA lub wrodzony defekt metaboliczny polegający na
niemożności przetwarzania Wit B12 uniemozliwia przekształcanie metlomalonylo-S-CoA w
bursztynylo-S-CoA. Propionian i metlomalonian przenikają do krwi i wydalane są z moczem. Prowadzi
to do kwasicy i opóźnien w rozwoju.
β-oksydacja kwasów tłuszczowych nienasyconych
Wymaga obecności izomerazy i reduktazy.
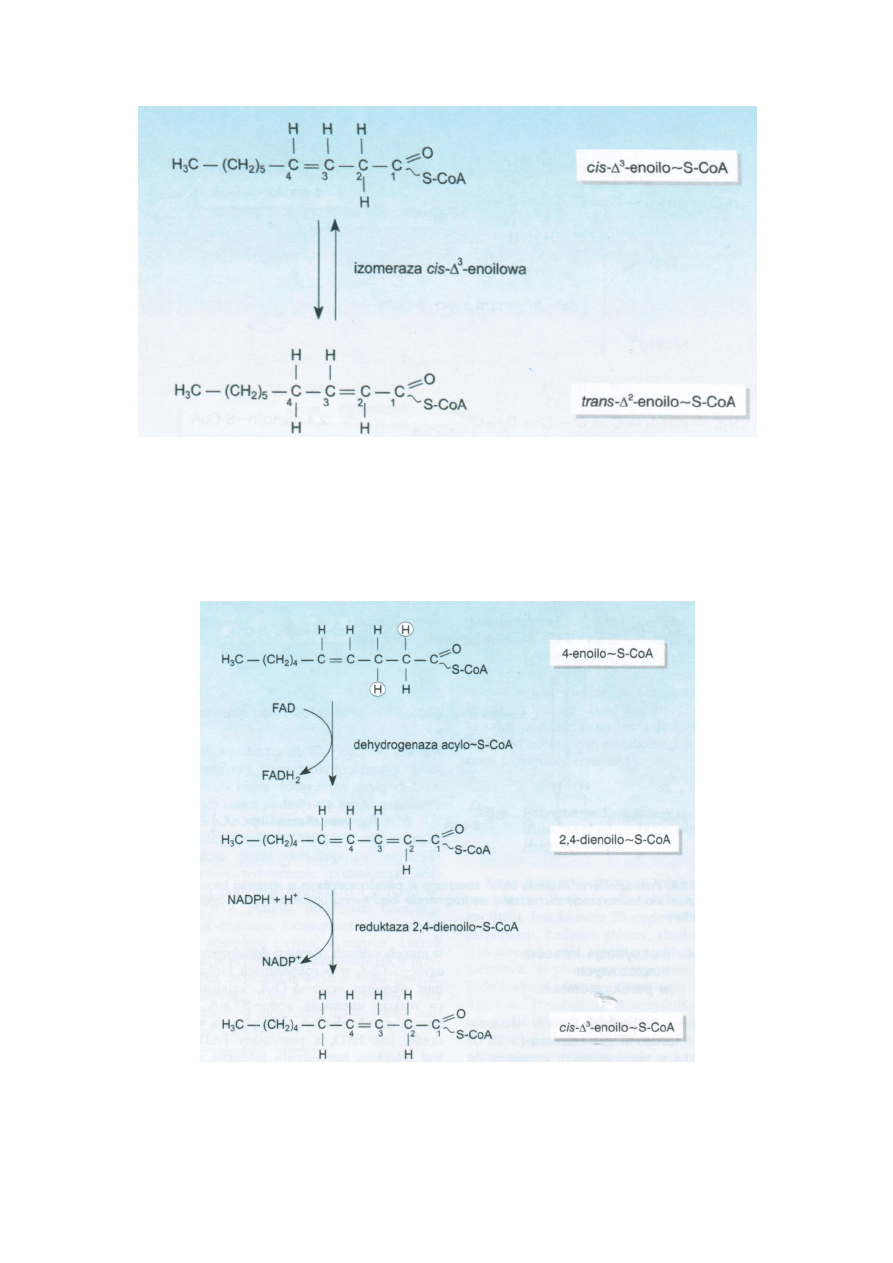
Palmitooleinian – 16C, który przy β-oksydacji dochodzi do długości 10C i w pozycji Δ3 znajduje się
wiązanie podwójne o kionfiguracji cis między węglami β i γ. Uniemożliwia to odwodornienie przez
dehydrogenazę acylo-S-CoA i wytworzenie podwójnego wiązania między węglami α i β. Izomeraza
cis-Δ3-enolinowa przekształca podwójne wiązanie cis-Δ3 na trans-Δ2. Dalsze reakcje przebiegają tak
jak w przypadku nasyconych kwasów tłuszczowych.
8
Linolan – 18C, który przy β-oksydacji do długości 12C posiada wiązanie cisΔ3. Za pomocą reakcji
opisanych w palmitooleinianie przekształcone zostaje ono w trans-Δ2. Orzy długości 10C pojawia się
wiązanie cis-Δ4 i odwodornienie tego acylo-S-CoA powoduje powstanie 2,4-dienoilo-S-CoA. Dwa
wiązania w bezpośrednim sąsiedztwie uniemożliwiają dalszą β-oksydację. Reduktaza 2,4-dienoilo-S-
CoA z udziałem NADPH+ H+ redukuje jedno z podwójnych wiązań i przekształca 2,4-dienoilo-S-CoA w
cis-Δ3-enoilo-S-CoA, a ten przez izomerazę cis-Δ3-enoilową przekształca się w trans-Δ2-enoilo-S-CoA,
który ulega β-oksydacji.
β-oksydacja kwasów tłuszczowych rozgałęzionych
Kwas fitanowy – C20 z czego C16 łańcuch i 4C podstawniki metylowe.
Hydroksylacja kwasu fitanowego przy udziale hydroksylazy i powstaje kwas α-hydroksyfitanowy.
Kwas ten pod wpływem α-oksydazy fitynianowej ulega dekarboksylacji z utlenieniem węgla α do
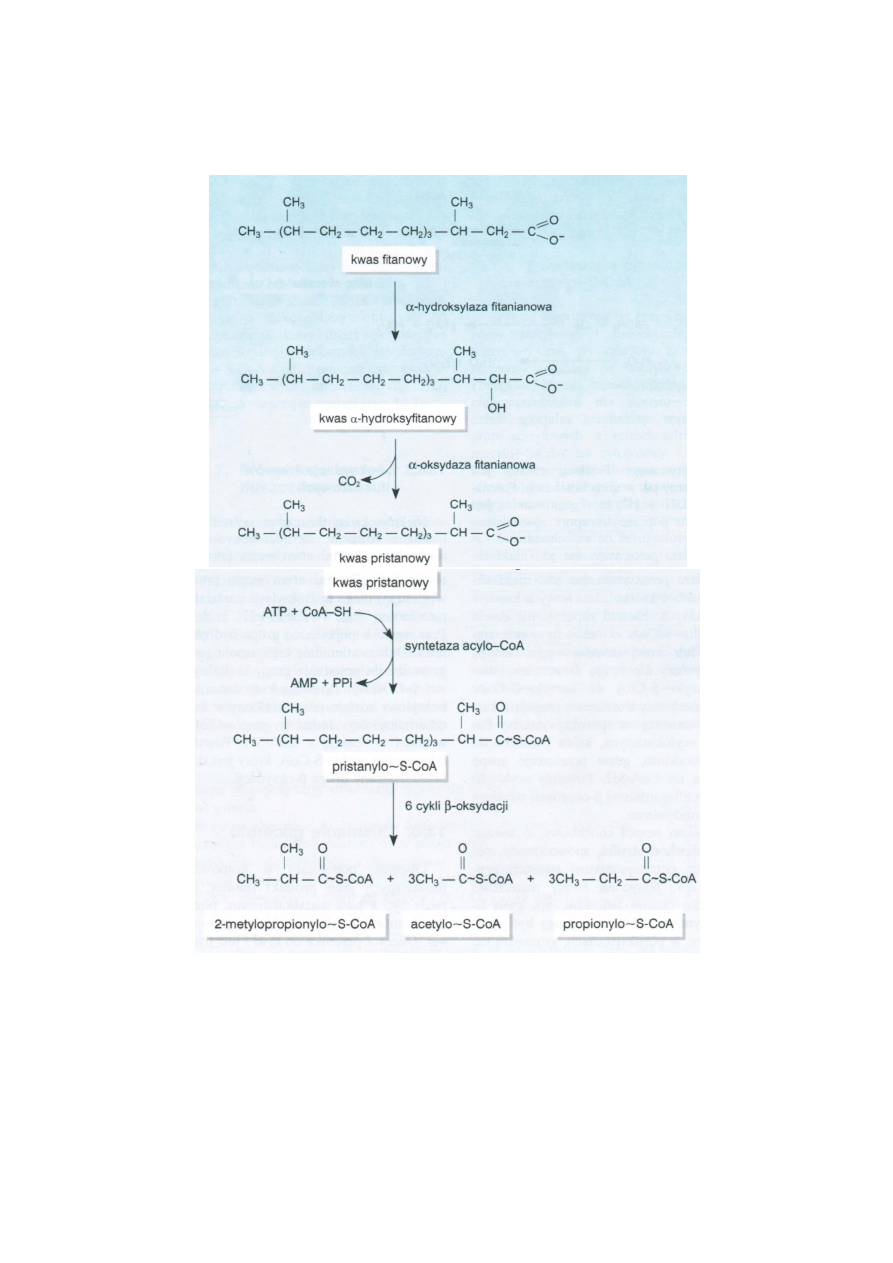
9
karboksylowej. Powstaje 19C kwas pristanowy który przez syntetazę acylo-S-CoA jest aktywowany.
Pristanylo-S-CoA zostaje poddany reakcjom β-oksydacji. Powstają 3 acetylo-S-CoA, 3 propionylo-S-
CoA. β-oksydacja kończy się przy 4-węglowym fragmencie 2-metylopropionylo-S-CoA.
β-oksydacja kwasów tłuszczowych w peroksysomach
β- oksydacja w peroksysomach dotyczy kwasów tłuszczowych o długości powyżej 22C. Transport do
peroksysomu nie wymaga karnityny, choć w peroksysomie istnieją odpowiednie enzymy i karnityna.
Peroksysomalna syntetaza acylo-S-CoA aktywuje kwas tłuszczowy. Acylo-S-CoA ulega β-oksydacji.
Skrócony kwas tłuszczowy wiąże się z karnityna i zostaje przekazany do mitochondrium, gdzie ulega
dalszej β-oksydacji.
β-oksydacja w peroksysomach zachodzi troche inaczej. Zamiast dehydrogenazy acylo-S-CoA
wystepuje tu oksydaza acylo-S-CoA, która katalizuje przejście acylo-S-CoA do trans-Δ2-enoilo-S-CoA.
Akceptorem wodoru jest FAD, a powstający FADH2 jest utleniany bez łańcucha oddechowego, z kolei
10
2H i 2e SA przekazywane bezpośrednio na tlen cząsteczkowy. Powstaje nadtlenek wodoru H2O2,
który rozkładany jest przez katalazę na H2O i O2.
Nie wystepuje tu fosforylacja substratowa ani inna forma wytwarzania energii, więc utlenianie
kwasów tłuszczowych w peroksysomach jest niekorzystne energetycznie, zysk jednego cyklu β-
oksydacji jest o 2ATP mniejszy niż cykl w mitochondrium.
Hydrataza enoilo-S-CoA i dehydrogenaza β-hydroksyacylo-S-CoA są aktywnościami jednego białka i
powstaje NADH+H+, który jest utleniany dzięki transportowi równoważników redukcyjnych do
mitochondriow. Tiolaza peroksysomalna jest mało aktywna dla krótkich kwasów tłuszczowych, a
wykazuje brak aktywności w stosunku do kwasów o C8 i mniej. Kwasy łącza się z karnityna i wedrują
do mitochondrium.
ω-oksydacja kwasów tłuszczowych – o średnim i długim łańcuchu. Za pomocą cytochromu P450,
NADPH+H+ i tlenu utleniony zostaje ostatni atom kwasu tłuszczowego do grupy COOH, która
następnie łączy się z CoA-SH i kwas tłuszczowy jest poddawany beta-osydacji.
α-oksydacja kwasów tłuszczowych – w mózgu – nie wymaga istnienia pośrednich metabolitów w
postaci CoA i nie przyczynia się do powstawania wysokoenergetycznych wiązań fosforanowych.
Ciała ketonowe – powstawanie
β-oksydacja i oksydacyjna dekarboksylacja pirogronianu są źródłem acetylo-S-CoA w wątrobie. Część
acetylowa może wejść do cyklu Krebsa (1), do syntezy kwasów tłuszczowych (2), do syntezy
steroidów (3). Jeżeli ilość acetylo-S-CoA w wątrobie przekroczy możliwości jego dalszego
przetwarzania zostaje uruchomiony proces ketogenezy (4). Acetylo-S-CoA przekształcany jest w ciała
ketonowe – acetooctan, β-hydroksymaślan i aceton.
Ciała ketonowe przechodzą do krwi i są zuzywane przez inne tkanki – mięśnie szkieletowe, mięsień
sercowy, korę nerki. Dziękli temu wątroba zaopatruje w energetyczne substraty inne tkanki. Ciała
ketonowe rozpuszczalne są w wodzie i nie potrzebują przenośników w postaci białek czy lipoprotein.
W warunkach normalnych istnieje równowaga w ilości wytwarzanych siał ketonowych przez wątrobę
i zuzywanych przez inne narządy.
W cukrzycy i choroby głodowej nastepuje nasilenie powstawania ciał ketonowych. Zużywanie ciał
ketonowych nie nadąża i wzrasta ich stężenie we krwi, przez co pojawiają się w moczu. Powodem są
zaburzenia w przemianie octanów w wątrobie, które towarzyszą w chorobach.
Metabolizm wątrobowy cukrów jest sprzężony z metabolizmem kwasów tłuszczowych. Przemiana
glukozy droga glikolizy dostarcza pirogronian, który przechodzi w szczawiooctan, który z kolei jest
akceptorem octanu i włącza go do cyklu Krebsa.
W cukrzycy i chorobie głodowej spada ilość szczawiooctanu i utrudnia włączanie octanu do cyklu
Krebsa (1).
W cukrzycy i chorobie głodowej upośledzony jest szlak pentozo fosforanowy i obniża to wytwarzanie
NADPH + H+, co uniemożliwia włączenie acetylo-S-CoA do syntez kwasów tłuszczowych (2) i
steroidów (3).
Z powodu zahamowania szlaku 1, 2 i 3, octan podąża szlakiem 4 – tworzenie ciał ketonowych.
11
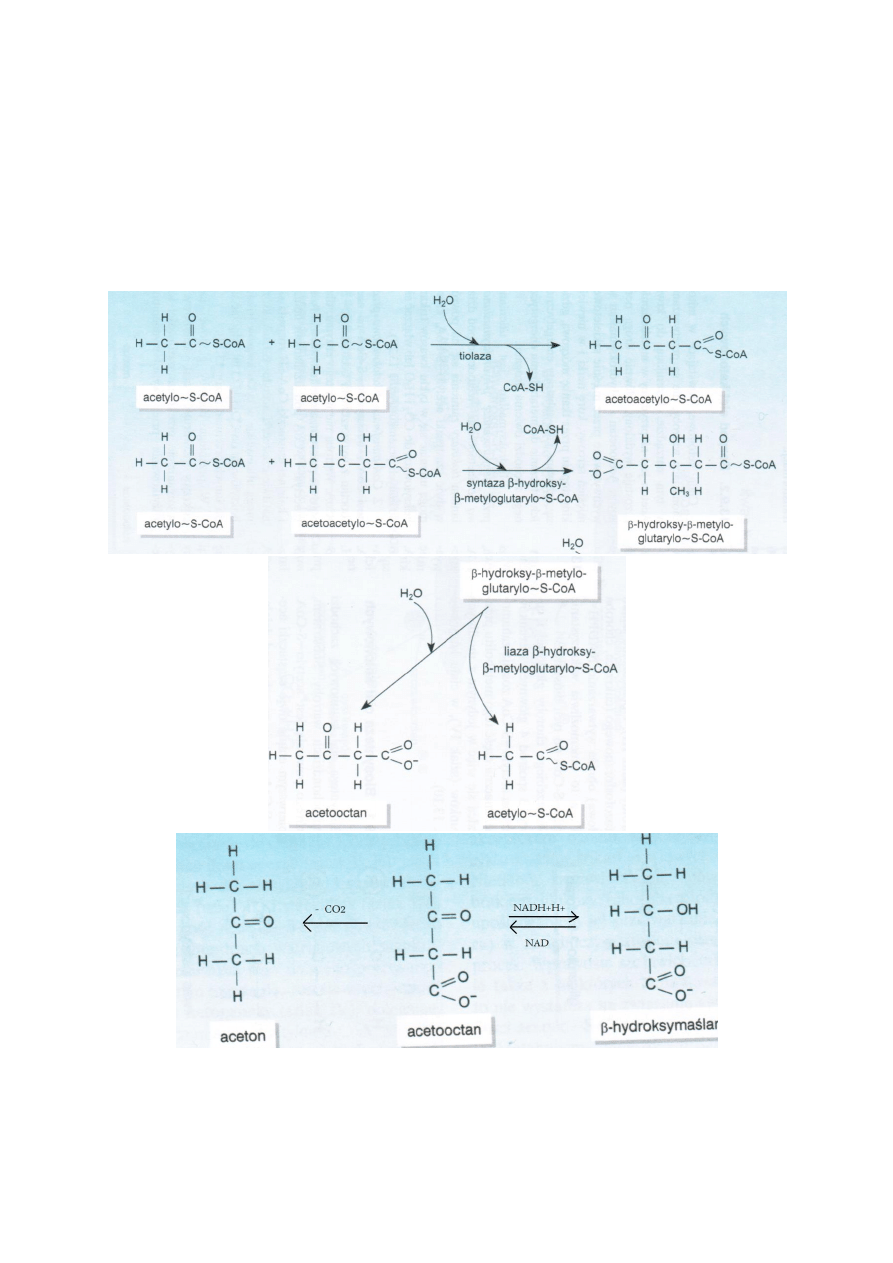
Biosynteza ciał ketonowych
Zachodzi w mitochondriach wątroby. Substratem jest acetylo-S-CoA. 2 czasteczki reagują ze sobą pod
działaniem tiolazy, od jednego dołącza się CoA-SH i powstaje acetoacetylo-S-CoA. Odłacza się kolejna
czastka CoA-SH pod działaniem syntazy β-hydroksy- β-metyloglutarylo-S-CoA i liazy β-hydroksy- β-
metyloglutarylo-S-CoA, która powoduje rozpad β-hydroksy- β-meytloglutarylo-S-CoA na acetooctan i
acetylo-S-CoA. Acetooctan może przekształcać się w dwóch kierunkach z udziałem dehydrogenazy β-
hydroksymaślanowej do β –hydroksymaślanu lub ulega samodzielnej dekarboksylacji z
wytworzeniem acetonu.
Rozpad ciał ketonowych
Ciała ketonowe przechodzą do krwi. Aceton jest wydalany, a acetooctan i β-hydroksymaślan są
wychwytywane przez mięśnie szkieletowe, sercowy i korę nerki. Ich włączenie do metabolizmu
energetycznego zachodzi na 2 sposoby:

12
1. β-hydroksymaślan jest utleniany przez dehydrogenazę β-hydroksymaślanową do
acetooctanu. Acetooctan pod działaniem tiokinazy zamienia się w dwie cząstki acetylo-S-CoA,
które wchodzą do cyklu Krebsa.
2. CoA-transferaza β-ketokwasowa przenosi CoA z bursztynylo-S-CoA na acetooctan z
wytworzeniem acetoacetylo-S-CoA. Wątroba pozbawiona tego enzymu nie przekształca w
ten sposób ciał ketonowych. Acetoacetylo-S-CoA od działaniem tiolazy i kolejnej cząsteczki
CoA-SH rozpada się na dwie czasteczki acetylo-S-CoA, które wchodzą w cykl Krebsa.
Zwiększone stężenie we krwi ciał ketonowych to ketonemia, a obecność w moczu ketonuria. Aceton
jest obecny w powietrzu wydechowym.
Kwas acetooctowy i β-hydroksymasłowy powodują kwasice metaboliczną i są wydalane przez nerki
wraz z jonami sodowymi. Utrata jonów Na wiąze się ze zwiększonym wydalaniem wody i nasileniem
kwasicy.
Regulacja ketogenezy
Zwiększone stężenie wolnych kwasów tłuszczowych w tkance tłuszczowej.
Przez regulacje aktywności acylotransferazy karnitynowej I
+ POBUDZANIE
- HAMOWANIE
Głodzenie
Sytość
Obniżenie stosunku Insulina/Glukagon
Malonylo-S-CoA
Ograniczenie wydatku energetycznego przez wątrobę – powstawanie ciał ketonowych umożliwia
powstanie 30% energii co w b-oksydacji.
4. Synteza kwasów tłuszczowych
Zachodzi przede wszystkim w wątrobie i tkance tłuszczowej, nerce i gruczole mlekowym.
Substancjami zużywanymi na to są acetylo-S-CoA i NADPH+H+, źródełem energii ATP, a substratem
jest acetylo-S-CoA pochodzący z oksydacyjnej dekarboksylacji pirogronianu lub β-oksydacja
tłuszczów, ciała ketonowe itp. NADPH+H+ pochodzi z cyklu pentozo fosforanowego i z
dekarboksylacji jabłczanu.
Cytosolowy acetylo-S-CoA. – Przeniesienie grupy acetylowej z mitochondrialnego acetylo-S-CoA do
cytosolu z wytworzeniem acetylo-S-CoA. Jako że błona mitochondrium jest nieprzepuszczalna dla
acetylo-S-CoA, dlatego wymagany jest mechanizm mostka cytrynianowego. Grupa acetylowa w
mitochondrium przenoszona jest z acetylo-S-CoA na szczawiooctan dając cytrynian reakcje katalizuje
Syntaza cytrynianowa. Cytrynian przenika do cytosolu gdzie rozpada się pod działaniem ATP-liazy
13
cytrynianowej. Reszta acetylowa przeniesiona zostaje na cytosolowy CoA-SH i powstaje ponownie
acetylo-S-CoA i szczawiooctan.
Szczawiooctan + acetylo-S-CoA ----syntaza cytrynianowa---> cytrynian cytrynian ---ATP-liaza
cytrynianowa ---> szczawioooctan + acetylo-S-CoA
Przenoszenie cytrynianu przez błonę mitochondrium tylko wtedy gdy jego stężenie w mitochondrium
jest wysokie, oraz wystepuje dostatek ATP. Wysokie stężenie ATP hamuje dehydrogenazę
izocytrynianową przez co kumuluje się cytrynian i izocytrynian. Wysokie stężenie ATP jest potrzebne
do syntezy kwasów tłuszczowych. W przypadku niskiego stężenia cytrynian przechodzi do cyklu
Krebsa i wytwarza się odpowiednia ilość ATP.
Szczawiooctan nie może wrócic bezpośrednio przez błonę do mitochondrium, musi zostać
zredukowany przez NADH+H+ do jabłczanu, lub związać grupe aminową i przejść w asparaginian. Oba
związki mogą przenikać do mitochondrium.
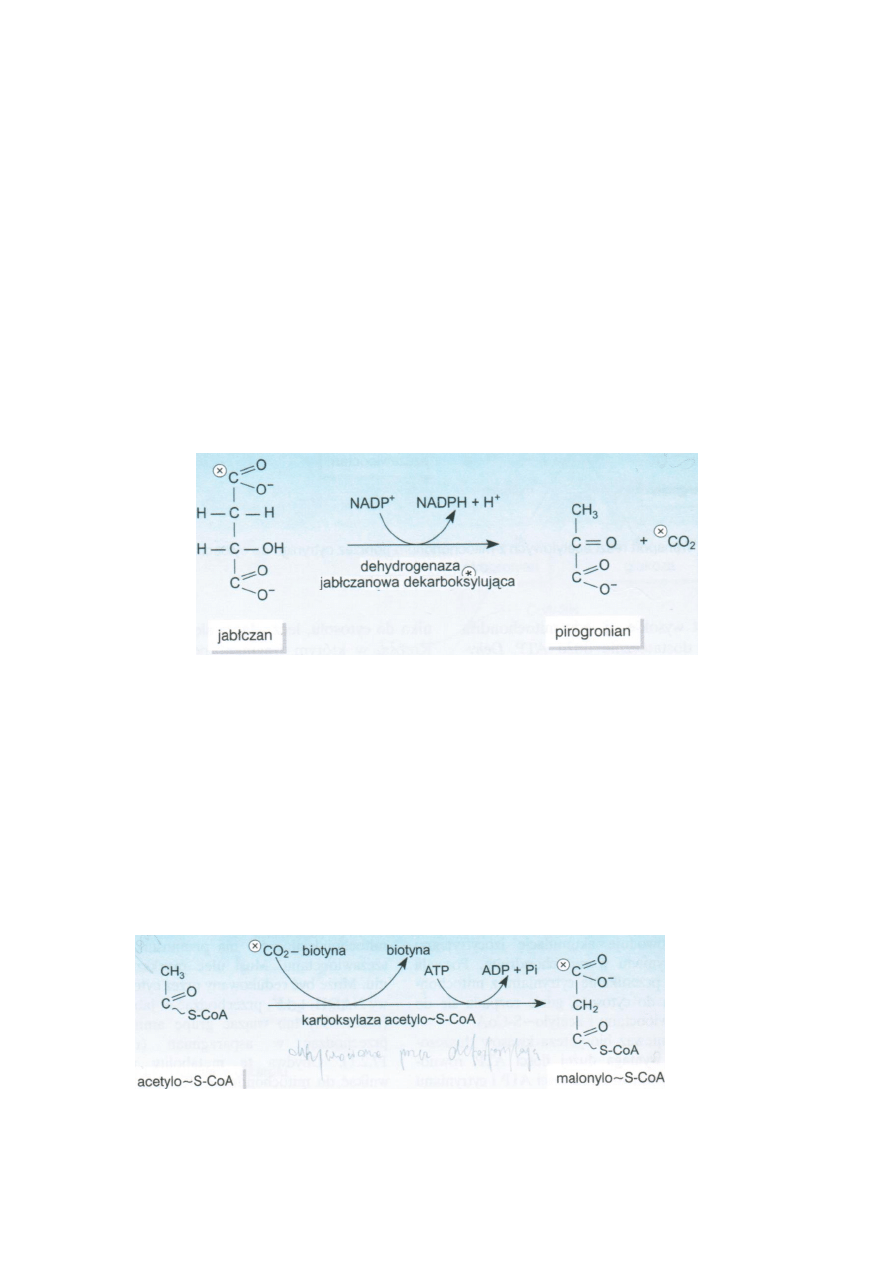
Jabłczan w cytosolu może być przekształcany przez dehydrogenazę jabłczanową dekarboksylującą w
pirogronian. Powstaje przy tym NADPH+H+.
Karboksylacja acetylo-S-CoA
Acetylo-S-CoA jest karboksylowany przez karboksylazę acetylo-S-CoA. Koenzymem jest
karboksybiotyna. Reakcja jest energiochłonna ATP ADP + Pi. Powstaje Malonylo-S-CoA.
Regulacja zachodzi na drodze:
1. Odwracalnej asocjacji i dysocjacji podjednostek karboksylazy acetylo-S-CoA. W postaci
monomeru jest nieaktywny, a aktywuje go cytrynian i podjednostki asocjuja i tworza
kompleks złozony z kilkunastu monomerów. Malonylo-S-CoA i palmitoilo-S-CoA powoduja
dysocjację polimeru na podjednostki.
2. Fosforylacja i de fosforylacja enzymu. Adresnalina pobudza fosforylację białka, co powoduje
jego inaktywację. Insulina pobudza de fosforylację karboksylazy acetylo-S-CoA. Dieta bogata
w tłuszcze powoduje obniżenie poziomu tego enzymu. Dieta uboga w tłuszcze powoduje
podwyższenie poziomu tego enzymu.
+ insulina; cytrynian
- acylo-CoAdługołańcuchowe; glukagon; adrenalina
Dołączanie kolejnych fragmentów dwuweglowych

14
Pod działaniem syntazy kwasów tłuszczowych – syntaza KT – dimer, w którym każdy monomer ma 7
aktywności enzymatycznych i jedną domenę wiążącą fosfopanteteiny (Pan-SH). Domena wiążąca
fosfopanteteinę jest zwana białkiem przenoszącym grupy acylowe ACP. ACP jest nośnikiem grup
acylowych wiążących się z siarką grup –SH fosfopanteteiny.
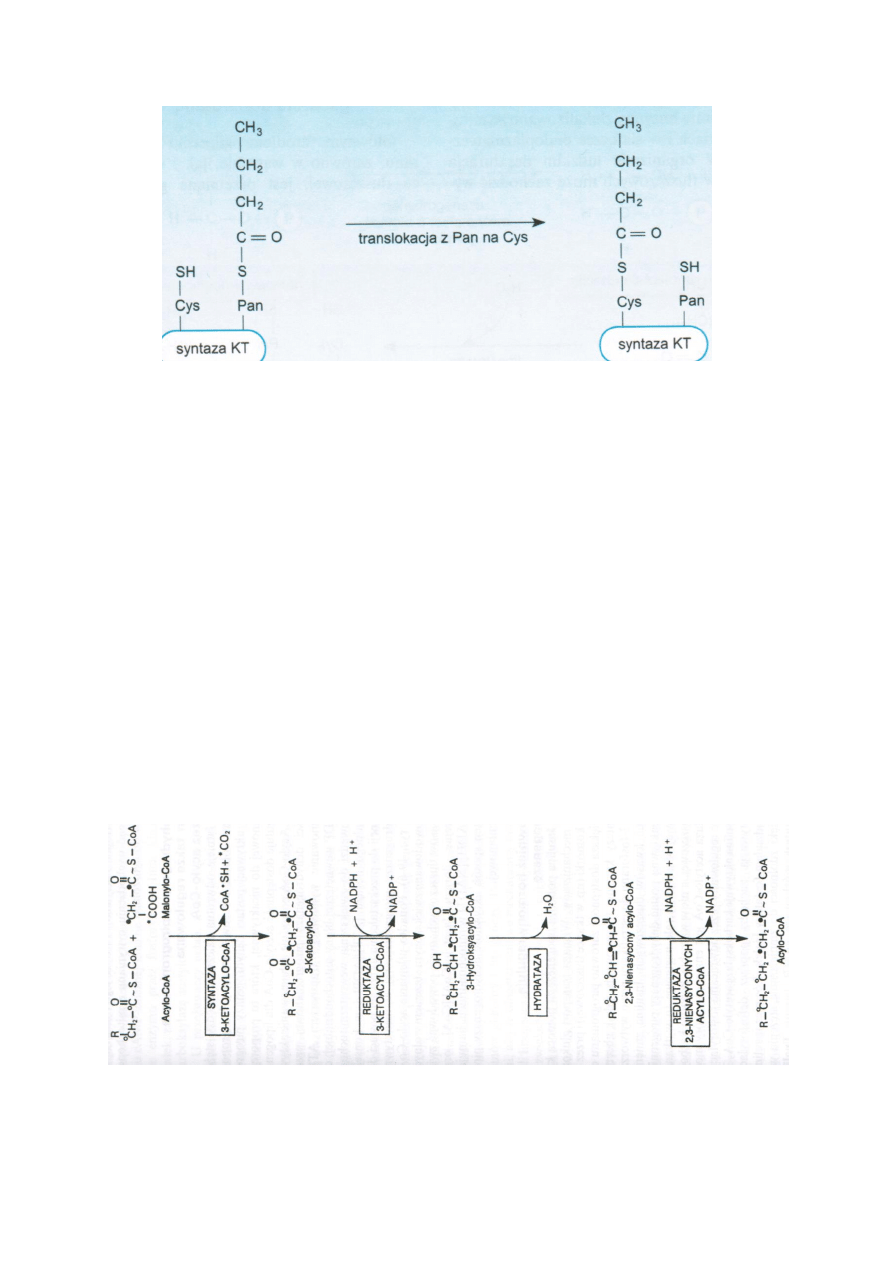
1. Grupa acetylowa jest przenoszona z acetylo-S-CoA na siarkę należącą do grupy –SH reszty
cysteinowej jednej z domen syntazy KT. Reakcję katalizuje acetylotransferaza, CoA-SH
odłącza się. Równocześnie malonylo-S-CoA przez malonylotransferazę przenoszona jest na
grupę –SH fosfopanteteiny, odłącza się CoA-SH.
2. Grupa acylowa reaguje z grupa malonylową, odłącza się CO2 od reszty malonylowej i jego
miejsce zajmuje reszta acetylowa. Reakcję katalizuje syntaza β-ketoacylowa. Powstaje
acetoacetylo-S-ACP. Grupa –SH cysteiny odtwarza się.
3. Grupa ketonowa reszty acetoacetylowej jest redukowana do grupy alkoholowej. Dawcą 2H i
2e jest NADPH+H+, a reakcję katalizuje reduktaza β-ketoacylowa. Powstaje β-
hydroksybutyrylo-S-ACP

15
4. Odłączona zostaje cząsteczka wody i wprowadzone zostaje wiązanie podwójne. Reakcję
katalizuje dehydrataza β-hydroksyacylo-ACP. Powstaje krotonoilo-S-ACP.
5. Kolejna reakcja redukcji zachodzi dzieki reduktazie enoilowej. Grupa krotonoilowa zostaje
przekształcona w grupę butyrylową (maślanową) – butyrylo-S-ACP.
6. Następuje translokacja 4-weglowego kwasu masłowego z siarki ACP na siarkę Cys-SH. Grupa
Pan-SH zostaje uwolniona i przyłącza kolejną cząstkę malonylo-S-CoA.
16
Cykl ten powtarza się 7-krotnie, za każdym razem wydłużając kwas połączony z cysteiną. Obie
podjednostki współpracują ze sobą, bo mają blisko siebie położone grupy Cys-SH i Pan-SH. Gdy kwas
tłuszczowy ma 16C proces syntezy zostaje zatrzymany. Tioesteraza uwalnia powstały kwas
tłuszczowy.
8acetylo-S-CoA + 14NADPH + 14H+ + 7ADPkw. Palmitynowy + 8CoA-SH + 14NADP+ + 7ADP + 7Pi + 7H2O
Palmitynian jest końcowym produktem reakcji katalizowanej przez syntazę KT. Dalsze wydłużanie i
desaturacja jest prowadzona przez inne enzymy, które zlokalizowane są w mitochondriach i w RE. W
organizmie ludzkim destaturacja zachodzi głównie między węglem 9 i 10. Wielonienasycone kwasy
tłuszczowe muszą zostać wprowadzone z zewnątrz. Desaturacja zachodzi przy działaniu układu Δ9-
desaturazy, która znajduje się w RE. Do reakcji potrzebny jest tlen i NADH+ H+. Układ ten zawiera:
reduktazę NADH-cytochrom b5, cytochrom b5 i desaturazę z żelazem niehemowym.
Regulacja: Przy nadmiarze glukozy i związków przemiany jak pirogronian, mleczan i acetylo-CoA
pobudzona jest synteza kwasów tłuszczowych.
Insulina wzmaga transport glukozy do komórek. Aktywuje karboksylazę acetylo-CoA. Obniża stężenie
cAMP równocześnie hamując lipolizę.
Glukagon i adrenalina działają odwrotnie – obniżają aktywność karboksylazy acetylo-CoA. Zwiększają
stężenie cAMP i pobudzają lipolizę.
Elongacja łańcucha kwasów tłuszczowych w RE (mikrosomy). Wydłużany jest łańcuch o 2 atomy
węgla.
Elongacja stearylo-CoA w mózgu ulega przyspieszeniu w okresie mielinizacji, aby dostarczyć kwasów
C22 i C24.

17
5. Synteza triglicerydów
Proces zachodzi w wątrobie i tkance tłuszczowej. Jest podzielony na 2 etapy:
1. Powstawanie fosfoglicerolu
2. Estryfikacja fosfoglicerolu kwasami tłuszczowymi
Glicerolo-3-fosforan i fosfodihydroksyaceton pochodzą ze szlaku przemian glukozy.
Fosfodihydroksyaceton dzięki dehydrogenazie glicerolo-3-fosforanowej przechodzi w glicerolo-3-
fosforan. Dzieje się to przy obecności insuliny. Przy braku insuliny lipocyty mają ograniczoną
biosyntezę glicerolo-3-fosforanu wraz ze zużyciem NADH+H+.
Glicerolo-3-fosforan powstaje też w wątrobie w specyficznym szlaku. Wolny glicerol dzięki kinazie
glicerolowej przechodzi w glicerolo-3-fosforan wraz ze zużyciem energii.
Aby kwasy tłuszczowe mogły być użyte do estryfikacji muszą zostać aktywowane do acylo-S-CoA
dzięki syntetazie acylo-S-CoA (tiokinaza).
Wytwarzanie fosfatydanu czyli 1,2-diacyloglicerolo-3-fosforanu zachodzi w dwóch etapach. Najpierw
działa acylotransferaza glicerolo-3-fosforanowa i powstaje lizofosfatydan. Następnie działa
acylotransferaza-1-acyloglicerolo-3-fosforanowa i powstaje fosfatydan. Fosfatydan pod wpływem
fosfohydrolazy fosfatydanowej przekształcany jest do 1,2-diacyloglicerolu. Acylotransferaza
diacyloglicerolowa katalizuje przeniesienie trzeciej reszty acylowej z acylo-S-CoA na diacyloglicerol.
Powstaje tak triacyloglicerol.
Regulacja polega na dostępności wolnych kwasów tłuszczowych, które zazwyczaj przekształcane są
do fosfolipidów, a kiedy zapotrzebowanie na fosfolipidy jest zaspokojone pojawiają się triglicerydy.
6. Metabolizm cholesterolu
Cholesterol występuje w tkankach i lipoproteinach osocza jako wolny cholesterol lub estry
cholesterolu z długimi łańcuchami kwasów tłuszczowych. Syntezowany jest z acetylo-CoA i wydalany
jako wolny cholesterol w żółci lub jako sole kwasów żółciowych.
Służy do syntezy soli kwasów żółciowych, witaminy D, hormonów steroidowych.
Transportowany jest przez lipoproteiny LDL do tkanek. Z tkanek pobierany i transportowany do
wątroby przez HDL (odwrócony transport cholesterolu). Najważniejszą patologią związaną z
cholesterolem jest odkładanie się płytek miażdżycowych w tętnicach, które są niezwykle ważne do
życia. Nasilenie miażdżycy obserwuje się w wysokim stosunku stężenia LDL/HDL.
Synteza cholesterolu
Zachodzi w RE gładkim i cytozolu. Większość cholesterolu pochodzi z syntezy, pozostała część
dostarczana jest wraz z dietą. Źródłem wszystkich atomów węgla w cholesterolu jest acetylo-CoA.
Cząstki acetylo-CoA łączą się tworząc mewalonian. Mewalonian traci CO2 i powstają jednostki
izoprenoidowe. Sześć jednostek izoprenoidowych kondensuje tworząc skwalen. Skwalen cyklizuje i
powstaje steroid – lanosterol. Lanosterol przekształca się do cholesterolu.
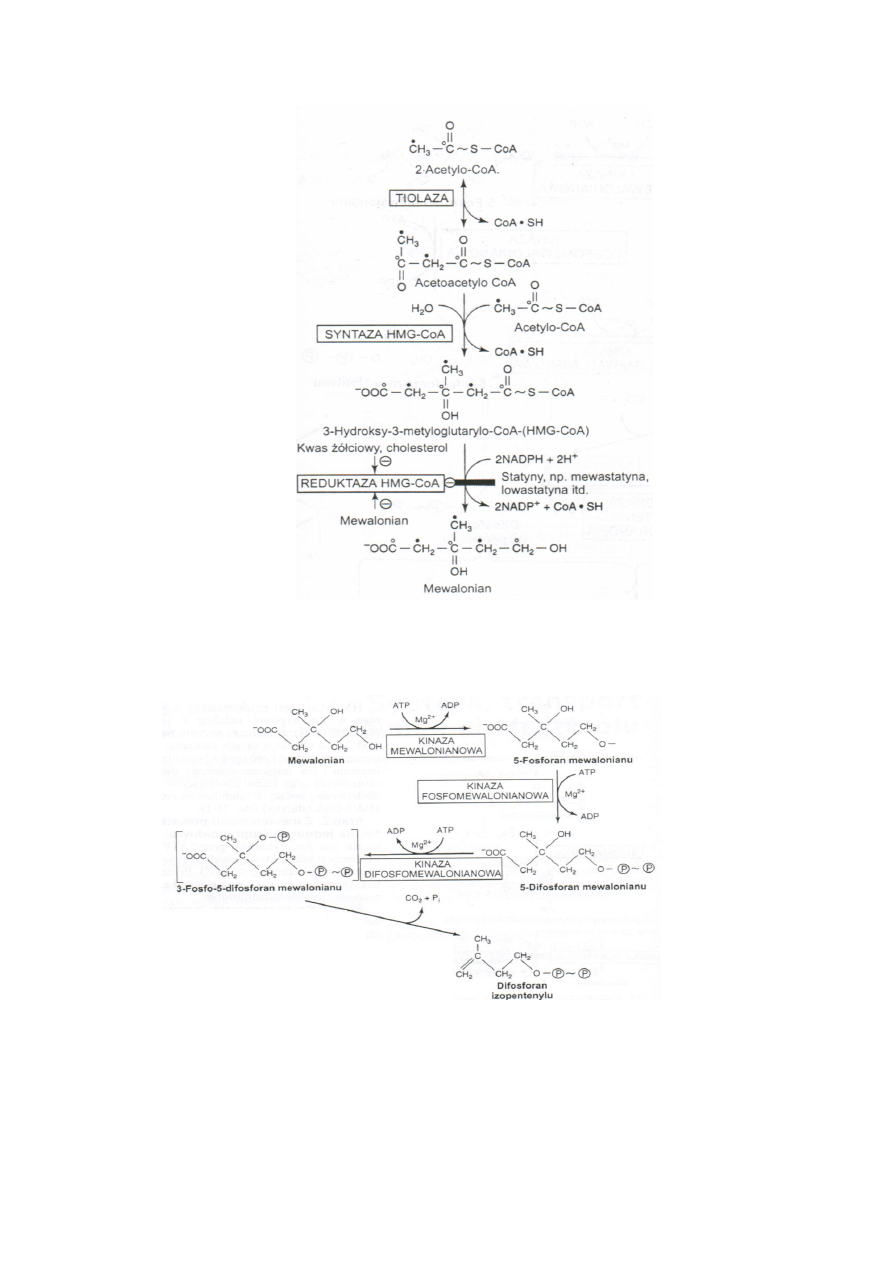
1. Powstawanie HMG-CoA i mewalonianu z acetylo-CoA.
2 acetylo-CoA reagują ze sobą przy działaniu tiolazy i powstaje acetoacetylo-CoA.
Acetoacetylo-CoA kondensuje z acetylo-CoA dzięki syntezie HMG-CoA dając HMG-CoA czyli 3-
hydroksy-3metyloglutarylo-CoA.
HMG-CoA dzięki NADPH i mikrosomalnego enzymu reduktazy HMG-CoA przechodzi w
mewalonian. Na reduktazę HMG-CoA działa wiele leków i przez hamowanie obniża produkcje
cholesterolu.
18
2. Powstawanie jednostek izoprenoidowych z mewalonianu.
Mewalonian jest fosforyzowany przez ATP z wytworzeniem ufosforylowanych związków
pośrednich. Następuje dekarboksylacja i odłaczenie H2O. Powstaje jednostka izoprenoidowa
– izopentenylopirofosforan.
3. Sześć jednostek izoprenoidowych tworzy skwalen
Trzy cząstki izopentenylodifosforanu kondensuje i powstaje difosforan farnezylu.
Izopentenylopirofosforan izomeryzuje do pirofosforanu dimetyloallilu
pirofosforan dimetyloallilu kondensuje z izopentenylopirofosforanem i wytwarza się pośredni
związek – pirofosforan geranylu.
pirofosforan geranylu kondensując z izopentenylopirofosforanem tworzy pirofosforan
farnezylu
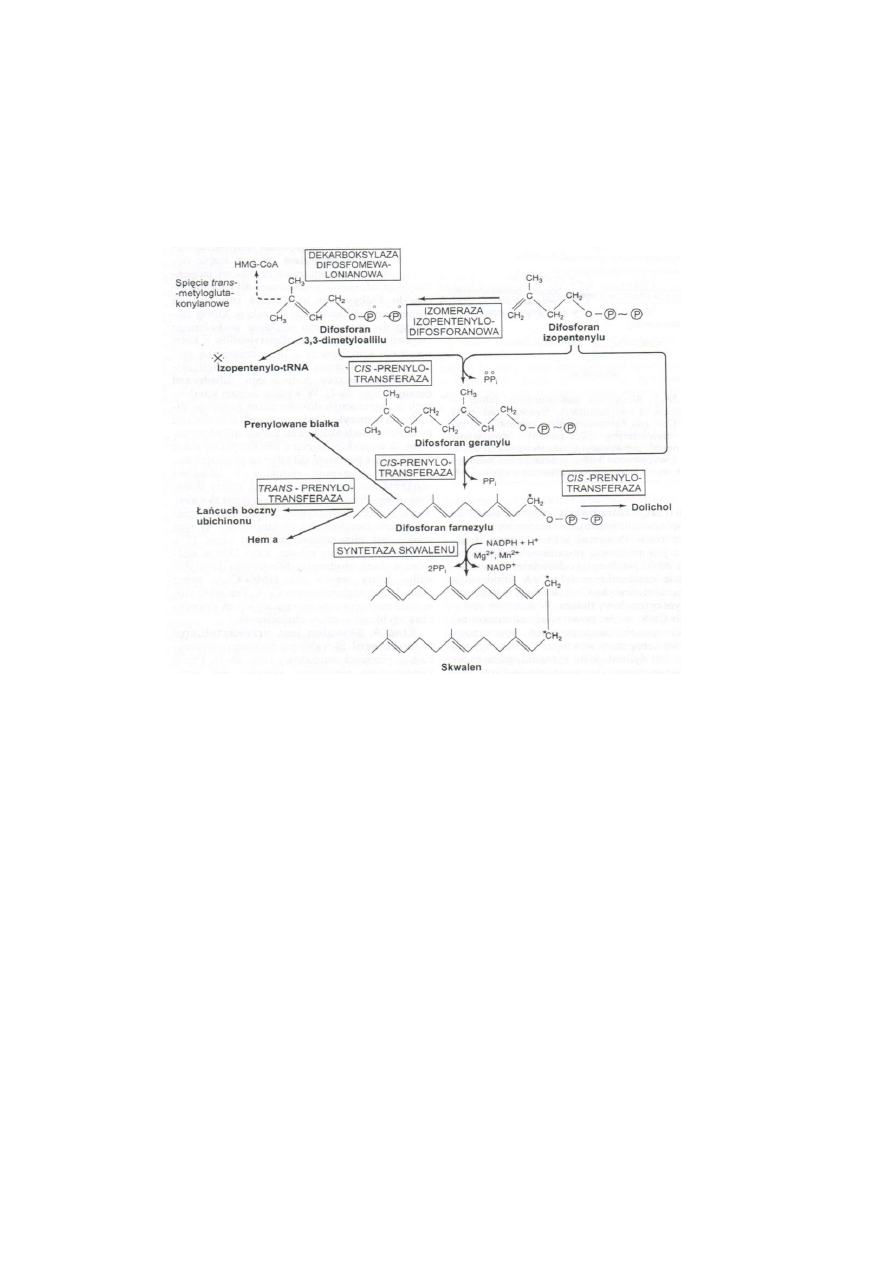
19
Dwie cząstki pirofosforanu farnezylu kondensują od końca pirofosforanowego odłączając
jedną grupę PP wytwarzając preskwalen.
Druga grupa PP odlączona zostaje w reakcji redukcji z udziałem NADPH. Powstaje skwalen.
Szlak „spięcia trans-metyloglutakonylanowego” umożliwia eliminowanie pirofosforanu
dimetyloallilu, który wraca do HMG-CoA przez trans-3-metyloglutakonylo-CoA regulując
szybkość syntezy cholesterolu
4. Skwalen jest przekształcany w lanosterol.
W RE gładkim zachodzi reakcja katalizowana przez epoksydazę skwalenową i skwalen
przekształcany jest do 2,3-epoksydu (tlenek skwalenu)
Lanosterolocyklaza 2,3-oksydoskwalenowa umożliwia cyklizację i przeniesienie grupy
metylowej przy C14 na C13 i C8 na C14.
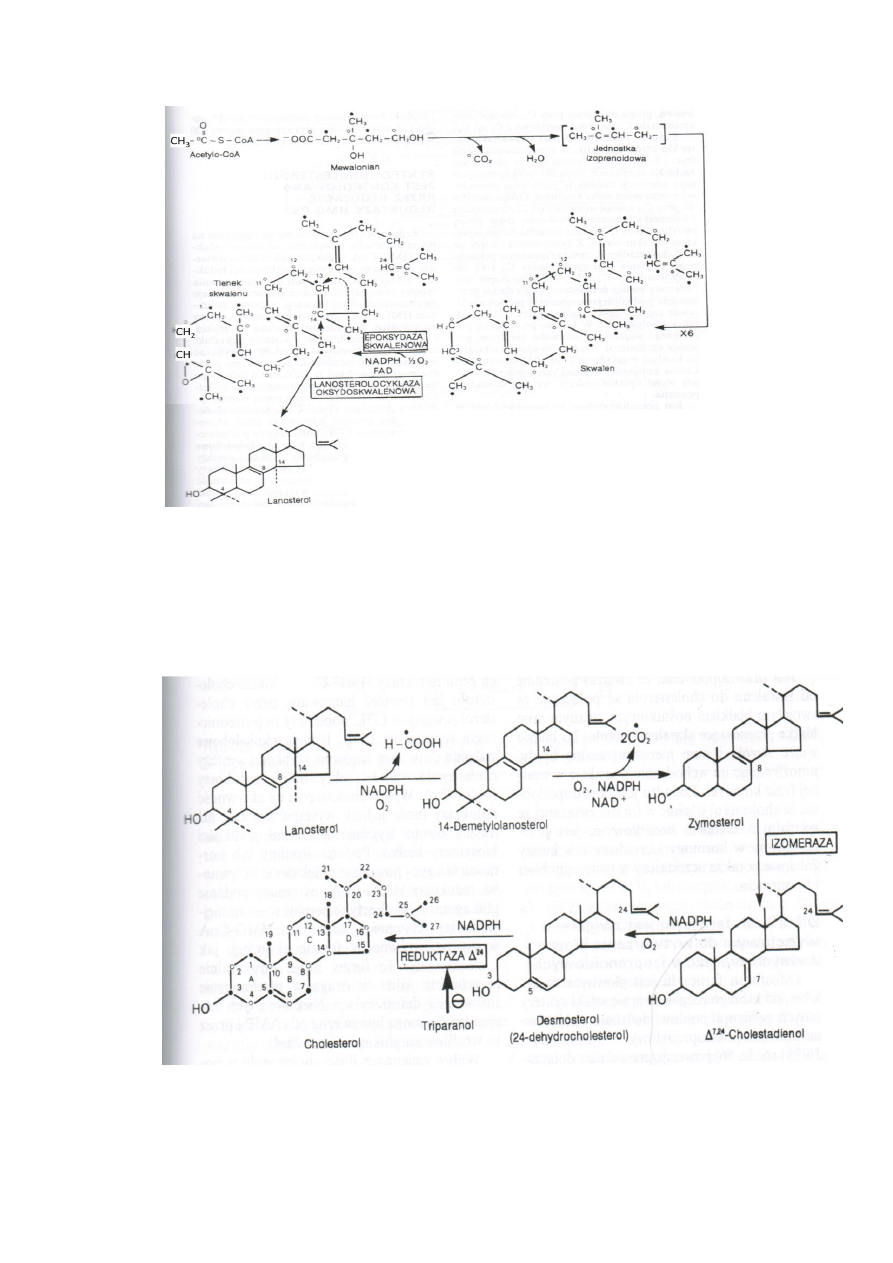
20
5. Lanosterol przekształcany jest do cholesterolu
W RE grupa metylowa C14 zostaje utleniona do CO2 i powstaje 14-demetylolanosterol.
Grupy metylowe przy C4 zostają usunięte i powstaje zymosterol.
Z zymosterolu powstaje Δ7,24-cholestadienol przez przesunięcie podwójnego wiązania z
pomiędzy C8 i C9 na C8 i C7.
Przez dalsze przesunięcia podwójnego wiązania między C5 i C6 powstaje desmosterol.
Redukcja podwójnego wiązania w bocznym łańcuchu i powstanie cholesterolu.
Możliwe że związki pośrednie od skwalenu do cholesterolu SA połączone z białkiem nośnikowym –
białkiem przenoszącym skwalen i sterole.
Difosforan farnezylu jest związkiem wyjściowym do syntezy dolicholu (dołączenie do 16 jednostek
izoprenoidowych) i ubichinonu (dołączenie 3-7 jednostek izoprenoidowych). Niektóre białka wiążące

21
GTP w błonie komórkowej mogą ulegać poliprenylacji, co umożliwia łatwiejsze zakotwiczanie się
prenylowanych białek w lipidowych błonach.
Regulacja syntezy cholesterolu przez regulację reduktazy HMG-CoA:
+ POBUDZANIE
- HAMOWANIE
Dobre żywienie
Głodzenie
Insulina
Mewalonian
Hormony tarczycy
Cholesterol
Cholesterol zawarty w LDL
Glukagon
glikokortykosteroidy
defosforylacja
fosforylacja
Regulacja stężenia cholesterolu w tkankach:
+ ZWIĘKSZANIE
- ZMNIEJSZANIE
Pobieranie lipoprotein zawartych w LDL przez
receptory LDL
Wypływ cholesterolu z błon do lipoprotein HDL
pobudzane przez LCAT acylotransferazę
lecytyna:cholesterol
Pobieranie cholesterolu z lipoprotein bogatych w
cholesterol
Zużywanie cholesterolu do innych syntez
Hydroliza estrów cholesterolu – hydrolaza
estrów cholesterolowych
Estryfikacja cholesterolu przez ACAT –
acetylotransferazę acylo-CoA:cholesterol
Synteza cholesterolu
Regulacja przez receptor LDL:
Reaguje na apoB-100, po przyłączeniu cząstka LDL całkowicie zostaje pochłonięta przez endocytozę.
Nastepuje rozkład w lizosomach na białka, cholesterol i inne lipidy. Receptory wracają na
powierzchnię komórki. Napływ cholesterolu hamuje syntazę HMG-CoA i reduktazę HMG-CoA
hamując syntezę cholesterolu. Pobudza natomiast ACAT i przez to zmniejsza syntezę receptora LDL.
Cholesterol jest głównie przenoszony przez LDL i VLDL. Istnieje białko, które ułatwia przenoszenie
estrów cholesterolu z HDL do innych lipoprotein w zamian pobierając z nich triacyloglicerole.
Wydalanie cholesterolu
Jako wolny cholesterol albo jako kwasy żółciowe (sole). Około 1g cholesterolu wydalamy wraz z
kałem jako kwasy żółciowe. Koprostanol jest wytwarzany w j. grubym z cholesterolu przez bakterie.
Duża część soli kwasów żółciowych jest reabsorbowanych do krążenia wrotnego i wychwytywana
przez wątrobę i ponownie wydalana z żółcią – krążenie jelitowo-wątrobowe.
Kwasy żółciowe pierwotne – kwas cholowy i chenodeoksycholowy. Powstają one ze wspólnego
prekursora, który powstaje z cholesterolu.
Cholesterol jest przekształcany przez 7α-hydroksylazę do 7 α-hydroksycholesterolu w
mikrosomach, wymaga to udziału NADPH i cytochromu P-450. Niedobór witaminy C zaburza
tworzenie kwasów żółciowych i kumulacje cholesterolu.
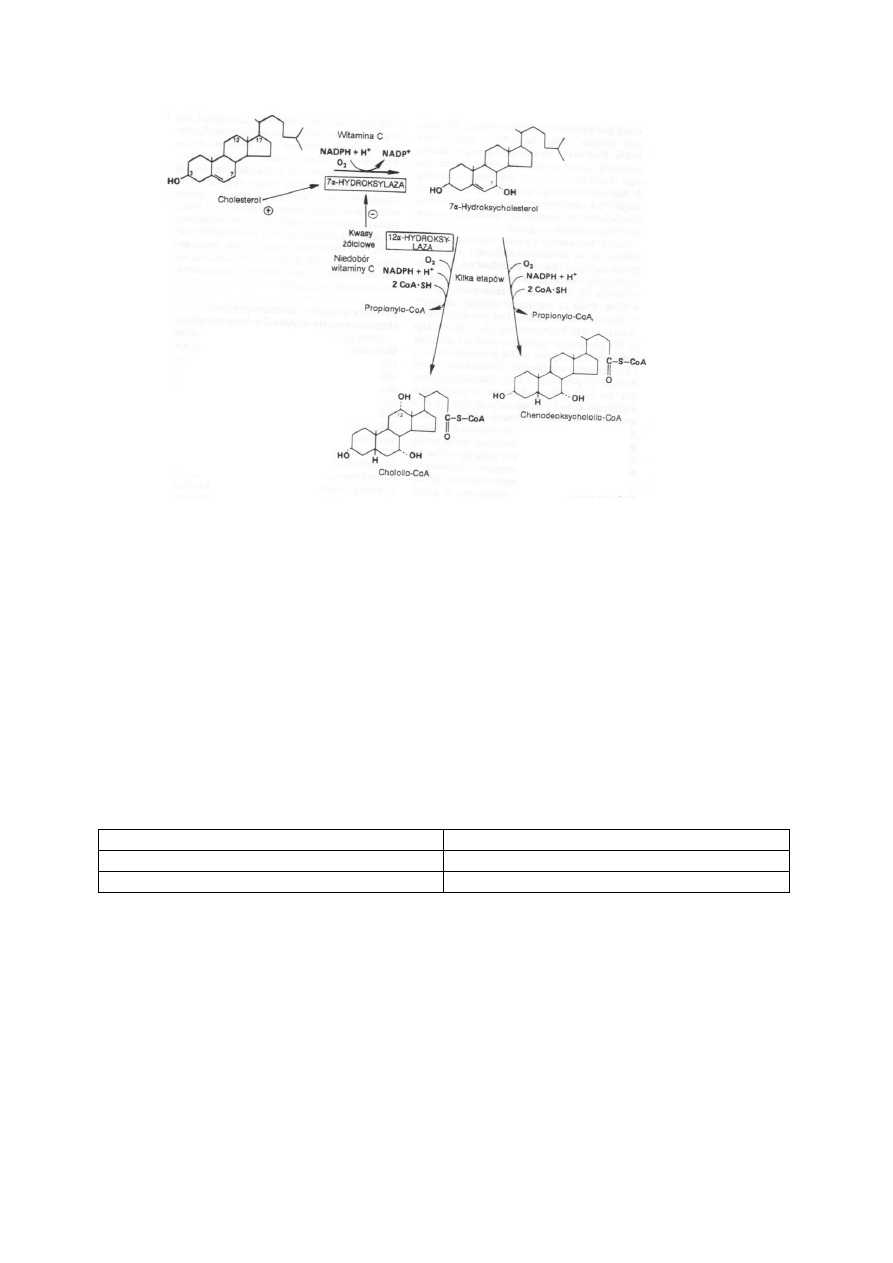
22
Szlak tworzenia kwasów żółciowych szybko się rozgałęzia. Podobne reakcje zachodzą w
każdym ze szlaków i powstaje choloilo-CoA z grupą α-OH w pozycji 12 i chenodeoksycholoilo-
CoA bez grupy α-OH. Łańcuch boczny ulega skróceniu.
Pierwotne kwasy żółciowe przechodzą do żółci jako połączenia z glicyną lub tauryną.
Część kwasów żółciowych podlega dalszym przemianom w jelicie, dzięki czemu powstają
wtórne kwasy żółciowe – kwas deoksycholowy z kwasu cholowego i kwas litocholowy z
kwasu chenodeoksycholowego.
Pierwotne kwasy żółciowe SA wchłaniane w jelicie krętym, powraca około 98% kwasów żółciowych
wydzielonych do jelita. Kwas lito cholowy nie wchłania się zwrotnie w dużych ilościach, bo jest
nierozpuszczalny.
Synteza kwasów żółciowych jest regulowana na etapie 7 α-hydroksylazy.
+ pobudzanie
- hamowanie
Spadek powracających kwasów żółciowych
Reduktaza HMG-CoA
fosforylacja
Defosforylacja
Hormony kory nadnerczy
Można je zakwalifikować do 3 grup: glikokortykoidów, mineralokortykoidów i androgenów. Hormony
steroidowe wiążą się do receptora śródkomórkowego i jako kompleks hormon-receptor wiąże się z
fragmentami DNA i indukuje ekspresję genów.
Mineralokortykoidy są ważne dla utrzymania równowagi sodowej i potasowej
Glukokortykoidy syntetyczne działają jako środki przeciwbólowe.
Warstwa kłębkowa ta – mineralokortykosteroidy – aldosteron 21C
Warstwa pasmowata – glukokortykoidy – kortyzol i kortykosteron 21C
Warstwa siatkowata – glukokortykoidy i androgeny – dehydroepiandrosteron i androstendion.
Hormony steroidowe zawierają pierścień cyklopentanoperhydrofenantrenowy i w pozycjach 10, 13 i
17 mogą występować dodatkowe atomy węgla.
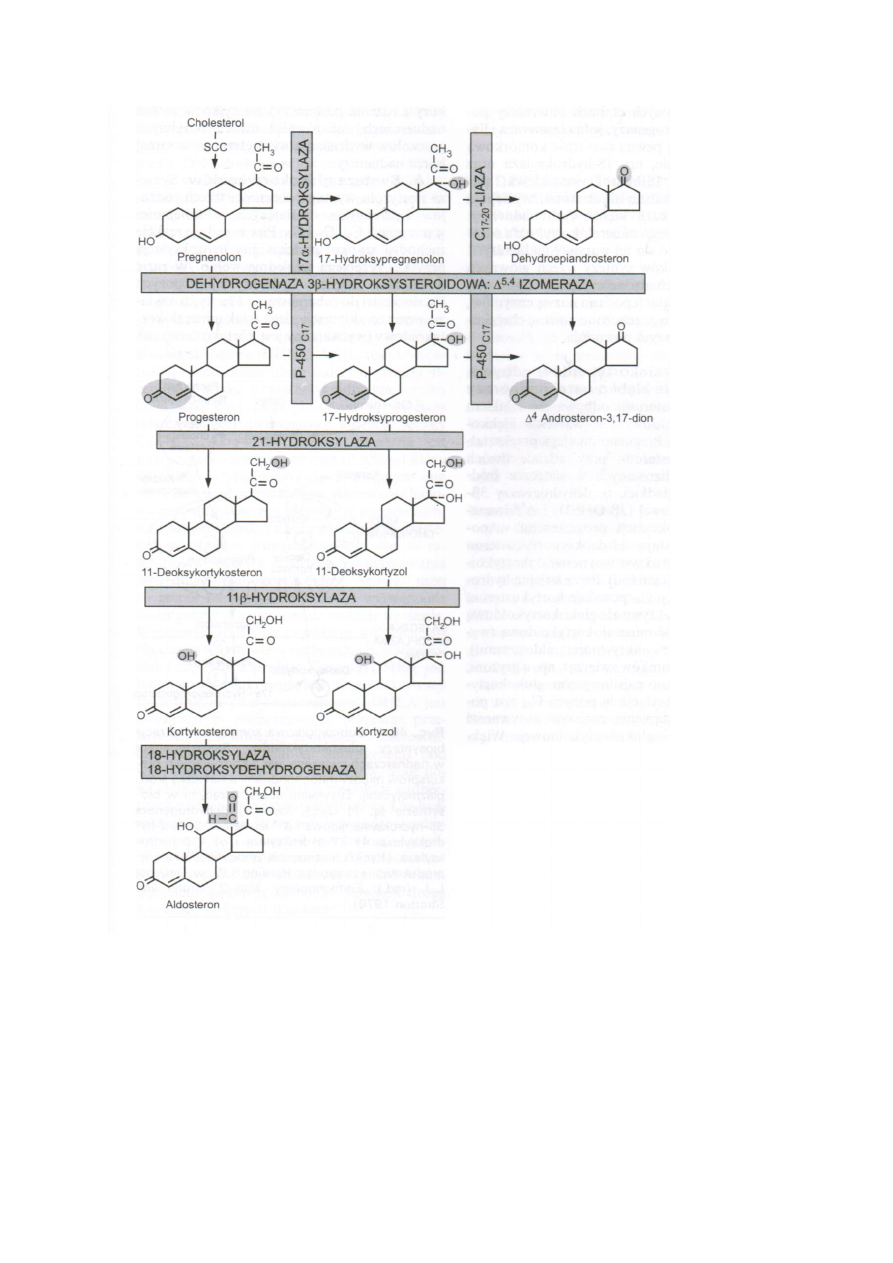
Powstawanie pregnenolonu

23
Cholesterol jest magazynowny w kroplach tłuszczowych w formie zestryfikowanej. W stymulacji
ACTH lub cAMP dochodzi do uruchomienia esterazy i uwolnienia wolnego cholesterolu. Wolny
cholesterol przechodzi do mitochondriów gdzie enzym rozszczepiający łańcuch boczny zawierający
cytochrom P-450 (P-450SCC) przekształca cholesterol w pregnenolon. Cholesterol ulega podwójnej
hydroksylacji najpierw przy węglu C22 a potem C20. Dochodzi do odszczepienia 6-węglowego
łańcucha izokaproaldehydu i powstaje 21C steroid. Aby cholesterol został przetransportowany do P-
450SCC potrzebne jest zależne od ACTH białko symulujące steroidogenezę STAR. Następnie
pregnenolon przechodzi do RE gładkiego.
Synteza mineralokortykoidów
Pregnenolon dzięki dehydrogenazie 3β-hydroksysteroidowej (3β-OHSD) i Δ5,4-izomerazie przechodzi
w progesteron. Progesteron przechodzi w 11-deoksykortykosteron pod działaniem 21-Hydroksylazy.
11-deoksykortykosteron dzięki 11β-hydroksylazie przechodzi w kortykosteron. Mitochondrialny
enzym 18-hydroksylaza i 18-hydroksydehydrogenaza przekształcają kortykosteron w aldosteron.
Synteza glukokortykoidów
Na pregnenolon działa dehydrogenaza 3β-hydroksysteroidowa i Δ5,4-izomeraza i przechodzi w
progesteron. Progesteron pod wpływem 17α-hydroksylazy przechodzi w 17α-hydroksyprogesteron.
17α-hydroksyprogesteron jest przekształcany przez 21-hydroksylazę ddo 11 deoksykortyzolu.
11-deoksykortyzol przechodzi do mitochondrium i jest przekształcany przez 11β-hydroksylazę do
kortyzolu.
Synteza androgenów
Pregnenolon przekształcany jest do dehydroepiandrosteronu przez 17α-hydroksylazę i C17-20 –Liazę,
które są częścią jednego enzymu P-450C17. Na dehydroepiandrosteron działa dehydrogenaza 3β-
hydroksysteroidowa i Δ5,4-izomeraza i przechodzi on w androstendion. Przez redukcję
androstendionu w pozycji C17 powstaje testosteron.
Regulacja wydzielania hormonów steroidowych nadnerczy
Glukokortykoidy – kortyzol jest wydzielany zależnie od ACTH. ACTH wydzielane jest przez przysadkę
gdy działa na nią kortykoliberdyna. Wszystko dzieje się na zasadzie sprzężenia zwrotnego.
Mineralokortykoidy – aldosteron wydzielany jest w odpowiedzi na zadziałanie czynników:
Układ reninowo-angiotensynowy – przy spadku ciśnienia tętniczego krwi, utracie płynów
ustrojowych, spadku stężenia NaCl w płynie kanalikowym komórki aparatu
przykłębuszkowego wydzielają reninę. Renina działa na angiotensyno gen uwalniając
deka peptyd – angiotensynę I – której synteze wzmagają glukokortykoidy i estrogeny.
Konwertaza przekształca angiotensynę I w angiotensynę II przez odcięcie 2C.
Angiotensyna podnosi ciśnienie tętnicze krwi. Łącyz się z receptorem komórek kory
nadnerczy i pobudza konwersję cholesterolu do aldosteronu.
Potas – wzrost stężenia o 0.1mmol/l pobudza sekrecję aldosteronu, spadek hamuje
sekrecję.
ACTH i sód
24
Brunatna tkanka tłuszczowa
Uczestniczy w metabolizmie gdy konieczne jest wytwarzanie ciepła. Występuje u noworodków i u
zdrowych ludzi u których jest odpowiedzialna za termo genezę indukowaną przez dietę – dzięki
czemu te osoby mogą jeść i nie być otyłe. U ludzi otyłych tkanki tłuszczowej brunatnej nie ma lub jest
znikoma. Termogeneza jest powodowana przez noradrenalinę, która przez cAMP pobudza Lipazę
wrażliwą na hormony. Lipaza rozkłada triacyloglicerole na wolne kwasy tłuszczowe, które
przekształcane są do acylo-CoA. Reakcjom towarzyszy utleniane i fosforyzowane. Wytwarza się dużo
ciepła przez gradient protonów, który w tkance tłuszczowej brunatnej przechodzi przez białko
termogeninę, którego mechanizm polega na tworzeniu drogi swobodnego przenikania protonów
przez błonę.

25
Wątroba spełnia funkcje
1. Ułatwia trawienie i wchłanianie lipidów – żółć z cholesterolem i sole kwasów żółciowych
2. Synteza i utlenianie kwasów tłuszczowych i synteza triacylogliceroli oraz fosfolipidów
3. Przekształcanie kwasów tłuszczowych w ciała ketonowe
4. Integrują syntezę i metabolizm liporptein osocza.
Stłuszczanie wątroby – brak równowagi między szybkością tworzenia triacyloglicerolu a jego
eksportem. Wyznacza się dwa główne rodzaje stłuszczenia wątroby.
1. Zwiększone stężenie wolnych kwasów tłuszczowych spowodowane przez mobilizację tłuszczu
z tk. tłuszczowej albo hydrolizą triacylogliceroli lipoprotein. Zwiększone ilości wolnych
kwasów tłuszczowych osocza są wychwytywane przez wątrobę i estryfikowane. Wytwarzanie
VLDL nie nadąża za napływem wolnych kwasów tłuszczowych i następuje akumulacja tłuszczu
w wątrobie – stłuszczenie. Ilość triacylogliceroli powiększa się podczas głodzenia i spożywania
produktów bogatych w tłuszcze. Spadek wytwarzania VLDL może być spowodowane niskim
stężeniem insuliny z powodu głodzenia.
2. Metaboliczne zablokowanie wytwarzania lipoprotein osocza.
Zablokowanie syntezy apolipoprotein
Zablokowanie tworzenia apolipoprotein z lipidów i apolipoproteiny
Niedobór dostarczania fosfolipidów w lipoproteinach
Niewydolność mechanizmu wydzielania
U szczura stłuszczenie wywołane było niedoborem choliny – czynnik lipotropowy. Niedobór
choliny jest spowodowany niedoborem grup metylowych np. pochodzenia metioniny (?)
Antybiotyk puromycyna – inhibitor syntezy białka i powoduje stłuszczenie wątroby i
zmniejszenie wydzielania VLDL. Podbne działanie: metionina, czterochlorek węgla, fosfor,
ołów, arsen.
Witamina E i Selen ma ochronne działanie na wątrobę i przeciwdziała peroksydacji lipidów.
Etanol powoduje stłuszczenie wątroby w alkoholizmie dochodzi do hiperlipidemii i marskości
wątroby. Dehydrogenaza alkoholowa powoduje wytworzenie NADH, który konkuruje o
łańcuch oddechowy z innymi równoważnikami hamując utlenianie innych substancji. Wzrost
stosunku NADH/NAD powoduje upośledzenie cyklu cytrynianowego przez przesunięcie
równowagi jabłczan szczawiooctan w lewo.
Metabolizm alkoholu w wątrobie powoduje wzrost syntezy triacylogliceroli i hamuje
utlenianie kwasów tłuszczowych.
Regulacja uwalniania kwasów tłuszczowych
W tkance tłuszczowej aktywność kinazy glicerolowej jest mała, glicerol nie może być wykorzystany w
większym stopniu do estryfikacji ecylo-CoA. Jeśli chodzi o dostępność glicerolo-3-fosforanu to tkanka
tłuszczowa jest zależna od glikolizy i dostępności glukozy dostarczanej przez GLUT 1 i 4.
Dostępność glicerolo-3-fosforanu reguluje estryfikację: lipoliza jest regulowana przez lipazę
wrażliwą na hormon. Produktami tej reakcji są wolne kwasy tłuszczowe i glicerol. Glicerol nie może
być wykorzystany przez tk. tłuszczową i przechodzi do osocza. Wolne kwasy tłuszczowe mogą być
przekształcany w tkance w acylo-CoA pod wpływem syntetazy acylo-CoA i mogą być re estryfikowane
z glicerolo-3-fosforanem tworząc triacyloglicerol. Jesto to ciągły cykl lipolizy i reestryfikacji. Jeżeli
szybkość reestryfikacji nie jest wystarczająco duża aby zrównoważyć lipolizę wolne kwasy tłuszczowe
przenikają do osocza i łączą się z albuminami.
Zwiększony metabolizm glukozy zmniejsza uwalnianie wolnych kwasów tłuszczowych – przez to, że
jest dużo glicerolo-3-fosforanu, który nasila estryfikację wolnych kwasów tłuszczowych po
przekształceniu ich w acylo-CoA. Gdy zużycie glukozy jest duże większa część pobranej glukozy jest

26
utleniana do CO2 i prztwarzana w kwasy tłuszczowe. Jeżeli większośc jest przekształcana w glicerolo-
3-fosforan, to wykorzystywany jest do estryfikacji acylo-CoA.
Lipaza lipoproteinowa wychwytuje wolne kwasy tłuszczowe – WKT pobierane z osocza w wyniku
działania lipazy na chylomikrony i VLDL wchodzą w skład WKT pula 2 i nie mieszają się z pulą 1, która
jest używana do syntezy triacylogliceroli.
Hormony mają wpływ na mobilizację tłuszczu.
1. Insulina zmniejsza wydzielanie WKT. Wzmaga lipogenezę i syntezę acyloglicerolu. Nasila
utlenianie glukozy do CO2. Wzmaga pobieranie glukozy przez komórki tk. tłuszczowej drogą
GLUT4. Wzmaga aktywność dehydrogenazy pirogronianowej, karboksylazy acetylo-CoA i
acetylotransferazy glicerolo-3-fosforanowej. Zasadniczym działaniem insuliny jest
hamowanie aktywności lipazy wrażliwej na hormon, przez co zmniejsza się uwalnianie WKT i
glicerolu.
2. Adrenalina, noradrenalina, glukagon, kortykotropina,α i β melanotrofina, tyreotropina,
hormon wzrostu. Aktywują one lipazę wrazliwą na hormon. Wymagana jest obecność
glikokortykoidów i hormonów tarczycy, które ułatwiają działanie lipolitycznych czynników.
Aminy katecholowe pobudzają cyklozę adenylanową, która przekształca ATP w cAMP.
Pobudza to cAMP kinazę białek i uaktywnia przez fosforylację lipazę triacyloglicerolową.
Lipoliza jest regulowana przez poziom cAMP. cAMP jest rozkładany do 5’-AMP przez
fosfodiesterazę cyklicznego 3’,5’-nukleotydu. Enzym ten jest hamowany przez kofeine i
teofilinę.
Wyszukiwarka
Podobne podstrony:
LIPIDY I LIPOPROTEINY OSOCZA(1)
LIPIDY I LIPOPROTEINY OSOCZA(1)
Lipoproteiny Lipidybł
Lipoproteina X-2, Lekarski WLK SUM, lekarski, biochemia, lipidy
lipidy2 2
Lipidy do wywieszenia
lipidy skrot
Biochemia 4 Lipidy
Lipidy Teoria 14
Lipidy w kosmetyc1
lipidy izoprenowe id 269491 Nieznany
ch zywnosci wyklad 4 lipidy low
Lipidy 6
2 Lipidy
lipidy - podział i metabolizm, Technologia żywności i żywienia człowieka, Biochemia
lipidy, Lekarski, FARMAKOLOGIA, 2. semestr, 2sem
biochemia lipidy
więcej podobnych podstron