ĆWICZENIE 3
Kolumnowa i planarna chromatografia adsorpcyjna w skali analitycznej i
preparatywnej w układach A) faz odwróconych (RP-HPLC), B) faz normalnych
(NP-HPLC i NP-TLC)
Chromatografia jest techniką rozdzielania mieszanin w układzie dwufazowym (faza
stacjonarna i faza ruchoma). W chromatografii cieczowej (LC- liquid chromatography) fazą
ruchomą jest ciecz, a fazą stacjonarną ciało stałe, rzadziej ciecz osadzona na nośniku.
Proces chromatograficzny to wielokrotna sorpcja i desorpcja substancji z fazy stacjonarnej do
fazy ruchomej i odwrotnie, z fazy ruchomej do stacjonarnej. W zależności od “siły”
oddziaływań z każdą z faz, składniki mieszaniny szybciej lub wolniej przemieszczają się
wzdłuż warstwy wypełnienia i opuszczają kolumnę w różnym czasie. Rozdzielenie analitów
możliwe jest tylko wówczas, gdy dobrane zostaną odpowiednie warunki chromatografowania,
takie jak rodzaj fazy ruchomej i stacjonarnej, prędkość przepływu eluentu oraz temperatura, w
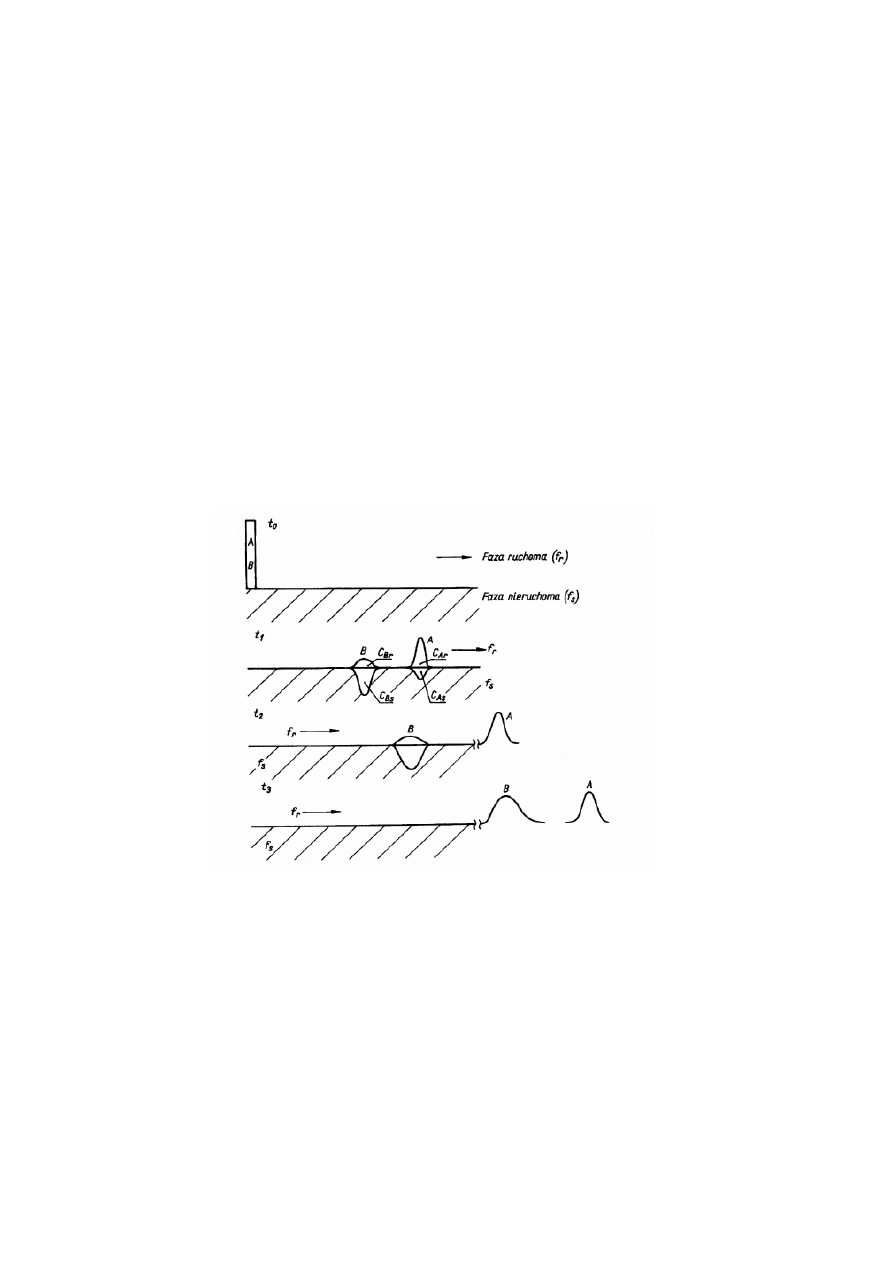
której prowadzony jest proces. Na Rysunku 1 przedstawiono przebieg rozdzielenia
chromatograficznego mieszaniny składającej się z dwóch substancji A i B.
Rysunek 1. Schemat rozdzielenia chromatograficznego mieszaniny składającej się z dwóch składników A i B,
t=0- moment wprowadzania mieszaniny do układu chromatograficznego, t
1
, t
2
- wybrane czasy z ogólnego czasu
przebiegu rozdzielenia chromatograficznego, t
3
- czas zakończenia rozdzielenie składników mieszaniny, C-
stężenie składnika (A lub B) w fazie ruchomej (r) lub nieruchomej (s
)
Mieszaninę tę wprowadzono do fazy ruchomej w czasie, który przyjęto za zerowy i od
którego rozpoczął się proces rozdzielania składników w wyniku różnego sposobu ich
oddziaływania z fazą ruchomą i stacjonarną. Załóżmy, że składnik A oddziałuje z fazą
stacjonarną znacznie słabiej niż składnik B. Cząsteczki obu substancji dzielą się między obie
fazy w różnych stosunkach, charakterystycznych dla tych składników i opisanych przez stałe
podziału K
c
.

Dla składnika A
Am
As
cA
C
C
K
; dla składnika B
Bm
Bs
cB
C
C
K
W obu przypadkach c oznacza stężenie składnika w fazie stacjonarnej (s) i w fazie ruchomej
(m). Między liczbą cząsteczek związków chromatografowanych, obecnych w fazie ruchomej i
stacjonarnej, ustala się równowaga dynamiczna z wielokrotnym przechodzeniem tych
cząsteczek z jednej fazy do drugiej. Ich przenoszenie wzdłuż układu chromatograficznego jest
możliwe tylko wtedy, gdy znajdują się w fazie ruchomej. W czasie t
1
(Rys. 1) widoczny jest
różny podział składników między obie fazy układu chromatograficznego i rozdzielenie tych
składników. To rozdzielenie jest możliwe tylko wtedy, gdy stałe podziału tych składników
różnią się (K
A
K
B
). Z Rys. 1 wynika, że w czasie t
2
jeden ze składników (A) został już
wyniesiony z układu chromatograficznego i znajduje się w fazie ruchomej, a w czasie t
3
oba
składniki (A i B) są już w fazie ruchomej poza zasięgiem oddziaływania fazy stacjonarnej.
Pasma stężeniowe składników po przejściu układu chromatograficznego różnią się od pasma
początkowego mieszaniny. Różnica polega na poszerzeniu tych pasm i na przyjęciu kształtu
krzywej Gaussa. Pasma te noszą nazwę pików chromatograficznych. Na Rys. 1 widać, że pik
składnika B jest szerszy niż pik składnika A. Jest on wynikiem większego rozmycia
dyfuzyjnego składnika B, który dłużej przebywał w układzie chromatograficznym.
Wynik procesu rozdzielenia zapisywany jest w postaci pików chromatograficznych, których
kształt odpowiada pasmu stężeniowemu substancji opuszczającej kolumnę. Powstały w ten
sposób chromatogram jest źródłem informacji jakościowej, mówiącej o liczbie i rodzaju
składników mieszaniny, a także informacji ilościowej, określającej stężenie lub masę
poszczególnych substancji w próbce.
PARAMETRY I WIELKOŚCI CHROMATOGRAFICZNE OPISUJĄCE
PROCES CHROMATOGRAFICZNY
Czas retencji t
R
- czas od momentu zadozowania (wprowadzenia) substancji do pojawienia
się na chromatogramie maksimum piku (pasma stężeniowego) rozpatrywanej substancji.
Czas martwy t
M
- czas od momentu zadozowania substancji nie ulegającej efektom
chromatograficznym do pojawienia się na chromatogramie maksimum piku tej substancji.
Substancja nie oddziałuje z fazą stacjonarną w kolumnie i przemieszcza się z tą samą
szybkością, co faza ruchoma. Jest to równoważne z czasem, jaki związek spędza w fazie
ruchomej.
Objętość retencji V
R
- objętość retencji substancji od momentu jej zadozowania do
pojawienia się na chromatogramie maksimum piku tej substancji
Objętość martwa V
M
- objętość martwa kolumny powszechnie definiowana jako objętość
cieczy znajdująca się w przestrzeniach międzyziarnowych i porach fazy stacjonarnej, oraz
objętość dozownika, detektora i łączników.
Współczynnik retencji k- współczynnik retencji ocenia czas przebywania substancji w fazie
stacjonarnej w stosunku do czasu przebywania w fazie ruchomej, opisany wzorem:
m
m
s
s
m
s
V
C
V
C
V
V
K
k
gdzie:
K – stała podziału z równania Nernsta
V
s
– objętość fazy stacjonarnej
V
m
– objętość fazy ruchomej
C
s
– stężenie substancji w fazie stacjonarnej
C
m
– stężenie substancji w fazie ruchomej

W praktyce jednak parametr k można wyznaczyć z chromatogramu, wykorzystując do tego
celu zależność zwaną równaniem elucji, przedstawioną za pomocą równania:
k
V
V
M
R
1
lub
k
t
t
M
R
1
gdzie:
V
R
– objętość retencji substancji
t
R
– czas retencji substancji, liczony od momentu wprowadzenia substancji do kolumny do uzyskania maksimum
piku tej substancji na chromatogramie
Współczynnik rozdzielenia α- zwany również współczynnikiem selektywności, czy też
retencją względną; jest on miarą selektywności układu chromatograficznego, przedstawiony
za pomocą równania:
1
2
k
k
gdzie:
k
1
– współczynnik retencji substancji 1
k
2
– współczynnik retencji substancji 2
Selektywność układu chromatograficznego charakteryzuje wzajemne oddziaływania
substancji rozdzielanych z fazą ruchomą i fazą stacjonarną. Parametr ten zależy głównie od
chemicznej budowy analitów, a także rodzaju i właściwości obu faz. Przy założeniu, że
mieszanina jest całkowicie rozdzielona, selektywność układu można opisać jako odległość
pomiędzy maksimum sąsiednich pików chromatograficznych.
Sprawność układu chromatograficznego
Miarą sprawności układu chromatograficznego (stopnia poszerzenia piku) jest liczba półek
teoretycznych kolumny (N) lub też wysokość równoważna półce teoretycznej (H).
Zakładając, że pik danej substancji ma kształt gaussowski, liczbę półek kolumny można
wyznaczyć ze wzoru:
2
16
b
R
w
l
N
gdzie:
l
R
– odległość maksimum piku od punktu zadozowania
w
b
– szerokość piku wyznaczona u jego podstawy
Ponieważ bardziej dokładne okazuje się wyznaczenie szerokości piku w połowie jego
wysokości, do obliczenia liczby półek teoretycznych można wykorzystać zależność (5) [2]:
2
2
/
1
545
,
5
w
l
N
R
gdzie:
w
1/2
– szerokość piku w połowie wysokości
Na Rysunku 2 zobrazowano sposób wyznaczania sprawności kolumny w oparciu o wyżej
wymienione wzory. Szerokości piku gaussowskiego na różnych wysokościach oraz czas
retencji są wykorzystywane do obliczania liczby półek teoretycznych.
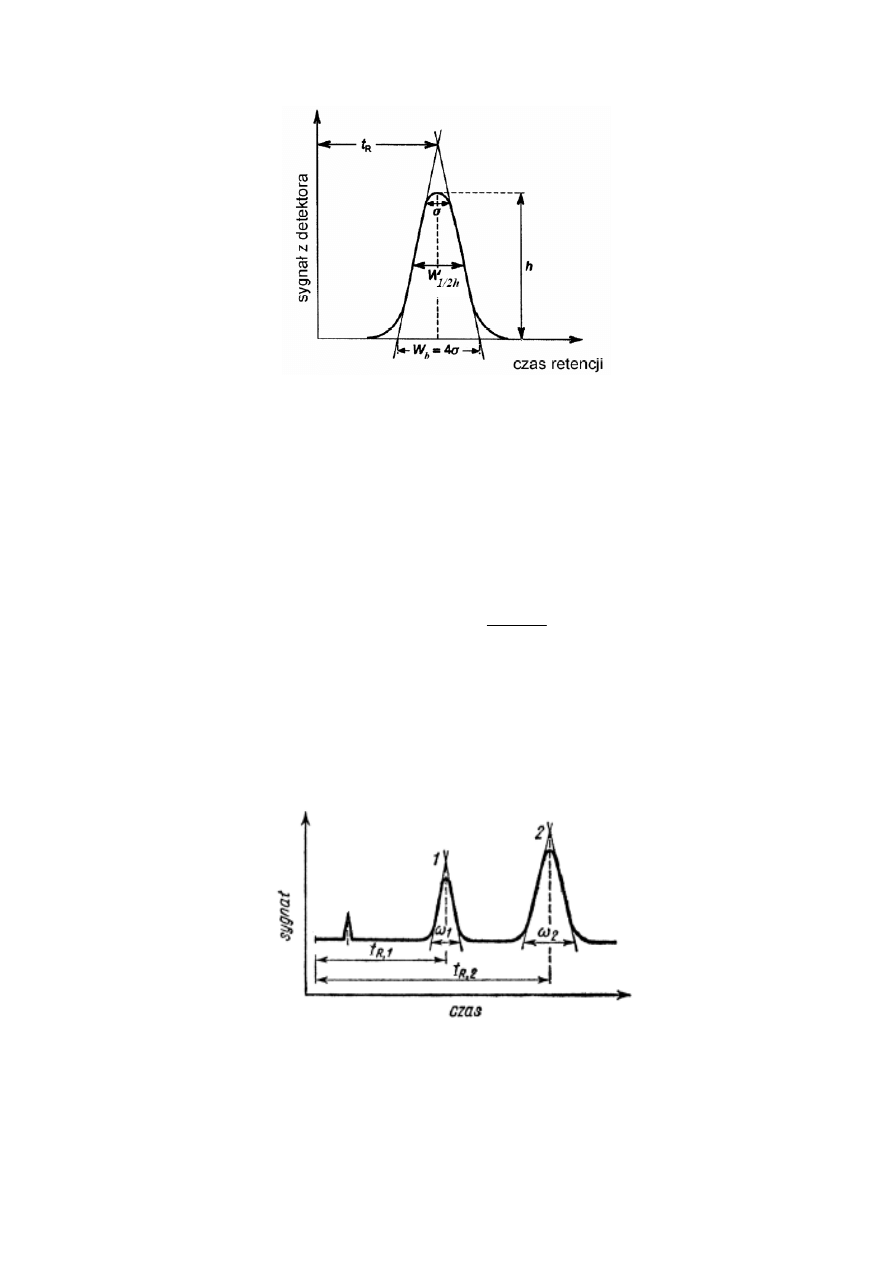
Rysunek 2. Obliczanie liczby półek teoretycznych kolumny; t
R
- czas retencji, h- wysokość piku, w
b
-
szerokość piku przy podstawie, w
1/2h
- szerokość piku w połowie jego wysokości
Stopień rozdzielenia Rs
Oceny efektu rozdzielenia substancji można dokonać wyznaczając współczynnik rozdzielenia
R
s
, zwany także stopniem rozdzielenia lub rozdzielczością pików. Dla dwóch sąsiadujących
pików chromatograficznych wartość współczynnika R
s
można obliczyć z równania:
2
1
1
2
2
w
w
t
t
R
R
R
s
gdzie:
t
R2
– czas retencji substancji B
t
R1
– czas retencji substancji A
w
2
– szerokość podstawy piku substancji B
w
1
– szerokość podstawy piku substancji A
Na Rysunku 3 zobrazowano sposób wyznaczenia stopnia rozdzielenia dwóch sąsiednich
pików w oparciu o wyżej wymieniony wzór.
Rysunek 3. Schemat chromatogramu mieszaniny dwuskładnikowej obrazujący sposób wyznaczenia
stopnia rozdzielenia dwóch sąsiadujących pików
.
Najbardziej znaczącym równaniem w chromatografii jest jednak równanie, które uwzględnia
retencję, sprawność i selektywność układu.

2
2
1
1
4
k
k
N
Rs
gdzie:
N – liczba półek teoretycznych, charakteryzująca sprawność kolumny
α – współczynnik selektywności
k
2
– współczynnik retencji substancji później eluowanej
Z powyższej zależności wynika, iż wzrost sprawności kolumny ułatwia rozdzielenie
substancji o zbliżonych własnościach. Uzyskanie rozdzielenia jednak możliwe jest wówczas,
gdy α > 1.
Im większa jest wartość współczynnika R
s
tym piki są lepiej rozdzielone.
Rozdzielenie pików do linii podstawowej uzyskuje się wówczas, gdy wartość Rs wynosi 1,5,
natomiast, gdy współczynnik ten przyjmuje wartość od 0,9 do 1, to piki o podobnej podstawie
są rozdzielone w około 96%. Przekroczenie optymalnej wartości współczynnika rozdzielenia
znacznie wydłuża czas analizy.
WYSOKOSPRAWNA CHROMATOGRAFIA CIECZOWA (HIGH
PERFORMANCE LIQUID CHROMATOGRAPHY- HPLC)
Wysokosprawna chromatografia cieczowa to rodzaj chromatografii kolumnowej. Oznacza to,
iż oznaczana substancja jest rozpuszczona w fazie ruchomej tzw. eluencie i w tej formie
kierowana jest do kolumny wypełnionej fazą stacjonarną- specjalnym złożem.
Aparaty do HPLC (Rys 4.) składają się zwykle z:
1) zbiorników na eluent ( fazę ruchomą)
2) pompy
3) dozownika
4) prekolumny
5) kolumny chromatograficznej (może być termostatowana)
6) termostatu
7) detektora
8) zbiornika na ścieki
9) systemu zbierania danych (np.komputer)
Rysunek 4. Schemat blokowy chromatografu cieczowego

Pompa ze zbiornika (lub zbiorników) zasysa fazę ruchomą i przez dozownik tłoczy do
kolumny chromatograficzne- serca układu chromatograficznego. Kolumny są zbudowane z
wysokiej jakości stali kwasoodpornej, albo szkła oraz teflonu. Wypełnienie: porowaty
sorbent, albo faza stacjonarna o możliwie kulistych i jak najmniejszych ziarnach. Często
kolumna jest umieszczana w termostacie. Analizowaną próbkę wstrzykuje się za pomocą
dozownika na szczyt kolumny chromatograficznej, a następnie składniki mieszaniny
rozdzielają się w kolumnie i na wyjściu z niej są wykrywane przez detektor. Sygnał
elektryczny z detektora po wzmocnieniu jest rejestrowany za pomocą komputera w postaci
piku chromatograficznego.
Rodzaje detektorów stosowanych w chromatografii cieczowej:
- spektrofotometryczny w zakresie UV-VIS
- UV-VIS typu DAD
- refraktometryczny RI
- fluorescencyjny FL
- elektrochemiczny EC
- radiometryczny
- spektrometr mas
CHROMATOGRAFIA W UKŁADZIE FAZ NORMALNYCH (NP- ang. normal phase)
Układem faz normalnych (NP) nazywa się układ chromatograficzny, w którym faza
stacjonarna jest bardziej polarna niż faza ruchoma, a podstawowym zjawiskiem decydującym
o rozdzielaniu jest adsorpcja na powierzchni fazy stacjonarnej.
Fazy stacjonarne:
1. adsorbenty nieorganiczne
-żel krzemionkowy
-tlenek glinu
-tlenek cyrkonu
-siarczan magnezu itp.
2. fazy wiązne- otrzymuje się je w wyniku modyfikacji polarnych sorbentów
nieorganicznych np. żelu krzemionkowego za pomocą:
a) związków chemicznych z grupami
- cyjanową
- aminową
-
nitrową
- diolową
- fenolową
b) cyklodekstrynami
Faza ruchoma- eluent
Zazwyczaj dobiera się dwuskładnikowa fazę ruchomą stosując rozpuszczalnik o dużej sile
elucyjnej (polarny) i rozpuszczalnik niepolarny, o małej sile elucyjnej (“rozcieńczalnik”
eluentu). Eluent w układzie faz normalnych składa się z niepolarnego rozpuszczalnika o małej
sile elucyjnej. W wielu przypadkach do fazy ruchomej- niepolarnego rozpuszczalnika dodaje
się niewielkiej ilości rozpuszczalnika polarnego (ok. 0,1- 2%).

W tabeli 1 zestawiono rozpuszczalniki najczęściej stosowane w układzie faz normalnych
uszeregowane według rosnącej siły elucyjnej (ε
0
) dla żelu krzemionkowego.
Tabela 1
ROZPUSZCZALNIK
0
pentan, heksan,oktan 0,00
chloroform
0,26
chlorek etylenu 0,30
eter di-izo-propylowy
0,32
1,3 dichloroetan
0,34
eter di-etylowy
0,38
eter metylo-terta butylowy 0,48
octan etylu
0,48
dioksan 0,51
acetonitryl 0,52
1-lub 2- propanol
0,60
metanol
0,70
kwas octowy
duża
woda Bardzo
duża
Kolejność elucji:
Związki niepolarne są najsłabiej oddziałują z fazą stacjonarną i dlatego są wymywane z
kolumny jako pierwsze. Natomiast związki polarne silnie oddziałują z wypełnieniem
kolumny- są później wymywane z kolumny.
Dla hipotetycznej mieszaniny substancji, w której znajdują się wszystkie znane klasy
związków chemicznych, siłę oddziaływań wyrażoną opisowym pojęciem “słaba” “średnia” i
“silna” adsorpcja, można przedstawić w następującej kolejności:
Tabela 2
Nie adsorbowane i słabo adsorbowane
Węglowodory alifatyczne i cykliczne (parafiny i nafteny)
Słabo adsorbowane
Alkeny (olefiny), merkaptany, sulfidy, jednopierścieniowe węglowodory
aromatyczne (tzw. monoaromaty) i chloroaromatyczne ( z jednym lub
więcej pierścieniami aromatycznymi)
Średnio adsorbowane
WWA- wielopierścieniowe węglowodory aromatyczne, etery, nitryle,
związki nitrowe i większość związków karbonylowych
Silne
adsorbowalne
Alkohole, fenole, aminy, amidy, imidy, sulfotlenki i kwasy
karboksylowe
Zastosowanie układu NP
Generalnie można powiedzieć, że chromatografię w układzie faz normalnych (NP), można
stosować do rozdzielania związków niejonowych i raczej nisko oraz średnio polarnych, jako
alternatywę dla układów faz odwrócony. Bardzo ważne znaczenie praktyczne ma szczególna
“zdolność” układów faz normalnych do wysoce selektywnego rozdzielania izomerów
strukturalnych, a w przypadku stosowania chiralnych faz stacjonarnych, do rozdzielanie
izomerów optycznych. Na podkreślenie zasługuje zdolność adsorbentów do rozróżniania
substancji pod względem polarności grup funkcyjnych. To powoduje, że praktycznie tylko
układy faz normalnych znajdują zastosowanie do rozdzielenia mieszanin substancji na klasy
związków chemicznych. Rozdzielanie grupowe, np. składników ropy naftowej i produktów
ropopochodnych, nasyconych i nienasyconych kwasów tłuszczowych, mono-, di-, i tri-
glicerydów itp., może być z powodzeniem wykonywane, tylko, z zastosowaniem układów faz
normalnych.
CHROMATOGRAFIA W UKŁADZIE FAZ ODWRÓCONYCH (RP- ang. reversed
phase)
Układ faz odwróconych (RP), to taki układ, w którym faza stacjonarna jest mniej polarna
niż faza ruchoma.
Fazy stacjonarne
fazy związne- otrzymywanie niepolarnych faz związanych opiera się na reakcji
powierzchniowych grup OH z odpowiednimi silanami.
Fazy stacjonarne stosowane w układzie faz odwróconych, zamiast grup OH, o charakterze
kwasowym, mają na powierzchni niepolarne łańcuchy węglowodorowe, ewentualnie
alkilonitrylowe, albo podobne. Powierzchnia jest tym bardziej hydrofobowa, im większy jest
stopień pokrycia niepolarną fazą stacjonarną oraz im więcej atomów węgla zawiera łańcuch
węglowodorowy.
Tabela 3
Rodzaj modyfikacji
Grupa funkcyjna
Najczęściej stosowany skrót nazwy
-CH2CH3 C2
-CH2(CH2)2CH3 C4
-CH2(CH2)6CH3 C8
n-alkanami
-CH2(CH2)28CH3
C18
grupą fenylową -CH2(CH2)xC6H5
fenylowa
grupą propylocyjanową -CH2(CH2)2CN
cyjanową
grupą perfluorową
-CH2(CF2)xCF3
grupą polarną, np
amidową,
karbaminianową, eterową
-CH2(CH2)2NHCO(CH2)nCH3
Faza ruchoma- eluent
W chromatografii w układach faz odwróconych fazami ruchomymi są mieszaniny wody i
rozpuszczalników organicznych (tzw. modyfikatorów) mieszających się z wodą. Najczęściej
stosowane rozpuszczalniki organiczne to acetonitryl, metanol i tetrahydrofuran..

Tabela 4 Podział rozpuszczalników ze względu na zbliżone wartości indeksu polarności P'
Grupa Rozpuszczalnik
I eter
metylowo-t-butylowy (etery alifatyczne)
II
metanol (alkohole alifatyczne)
III tetrahydrofuran
IV
kwas octowy, formamid
V
chlorek metylenu, chlorek etylenu
VI dioksan,
acetonitryl
VII toluen
VIII woda,
chloroform
Kolejność elucji
Substancje eluują w kolejności od najbardziej polarnych (ściśle najbardziej hydrofilowych) do
niepolarnych (najmniej hydrofilowych), a w szeregach homologicznych od nisko- do
wysokocząsteczkowych.
Retencja substancji rośnie ze wzrostem:
- stopnia pokrycia powierzchni związaną fazą organiczną
- długości łańcucha fazy związanej
- hydrofobowości grupy funkcyjnej decydującej o charakterze powierzchni sorpcyjnej
- hydrofobowości substancji rozdzielanych
- zawartości wody w fazie ruchomej
Zastosowanie układu RP
Nazwa „układ faz odwróconych” jest nazwą zwyczajową wynikającą z historii rozwoju
chromatografii cieczowej. W początkowym okresie stosowania technik chromatograficznych
fazami stacjonarnymi były polarne sorbenty, a fazami ruchomymi mniej polarne
rozpuszczalniki (układ faz normalnych). Obecnie chromatografia w układzie faz
odwróconych ma ogromny zakres zastosowań, natomiast chromatografie w układzie faz
odwróconych stosuje się wyłącznie w omówionych wcześniej przypadkach. RP-HPLC służy
ona do rozdzielania mieszanin substancji istotnie różniących się właściwościami, zarówno
polarnych, średniopolarnych, jak i niepolarnych.
CHROMATOGRAFIA CIENKOWARSTWOWA/PLANARNA (TLC- ang. Thin layer
chromatography)
Chromatografia planarna lub cienkowarstwowa (Thin Layer Chromatography - TLC) jest
jednym z przykładów chromatografii cieczowej gdzie rozdzielenie mieszaniny zachodzi na
fazie stacjonarnej w postaci cienkiej warstwy. Gdy fazą jest warstwa bibuły jest to
chromatografia bibułowa a kiedy mamy do czynienia z warstwą rozprowadzoną na płytce
szklanej, aluminiowej lub z tworzywa sztucznego jest to chromatografia cienkowarstwowa.
Mechanizm rozdzielenia polega na migracji składników mieszaniny wraz z fazą ruchomą
wzdłuż warstwy sorbentu. W zależności od energii oddziaływań składników z fazami
wykazują one różny stopień retencji tzn. mają różną drogę migracji i znajdują się w różnych
miejscach warstwy. Głównym mechanizmem oddziaływań międzycząsteczkowych jest
adsorpcja a najczęściej stosowanymi adsorbentami są żel krzemionkowy, tlenek glinu,
krzemian magnezu i krzemian wapnia. Warstwy tych substancji mogą być modyfikowane
różnymi związkami chemicznymi, w zależności od użytego czynnika impregnującego
otrzymuje się warstwy hydrofilowe lub hydrofobowe. Warstwy hydrofilowe uzyskuje się
stosując dimetyloformamid, dimetylosulfoamid, dimetylosulfotlenek a warstwy hydrofobowe
stosując substancje takie jak olej parafinowy, skwalan, undekan, olej silikonowy i inne.
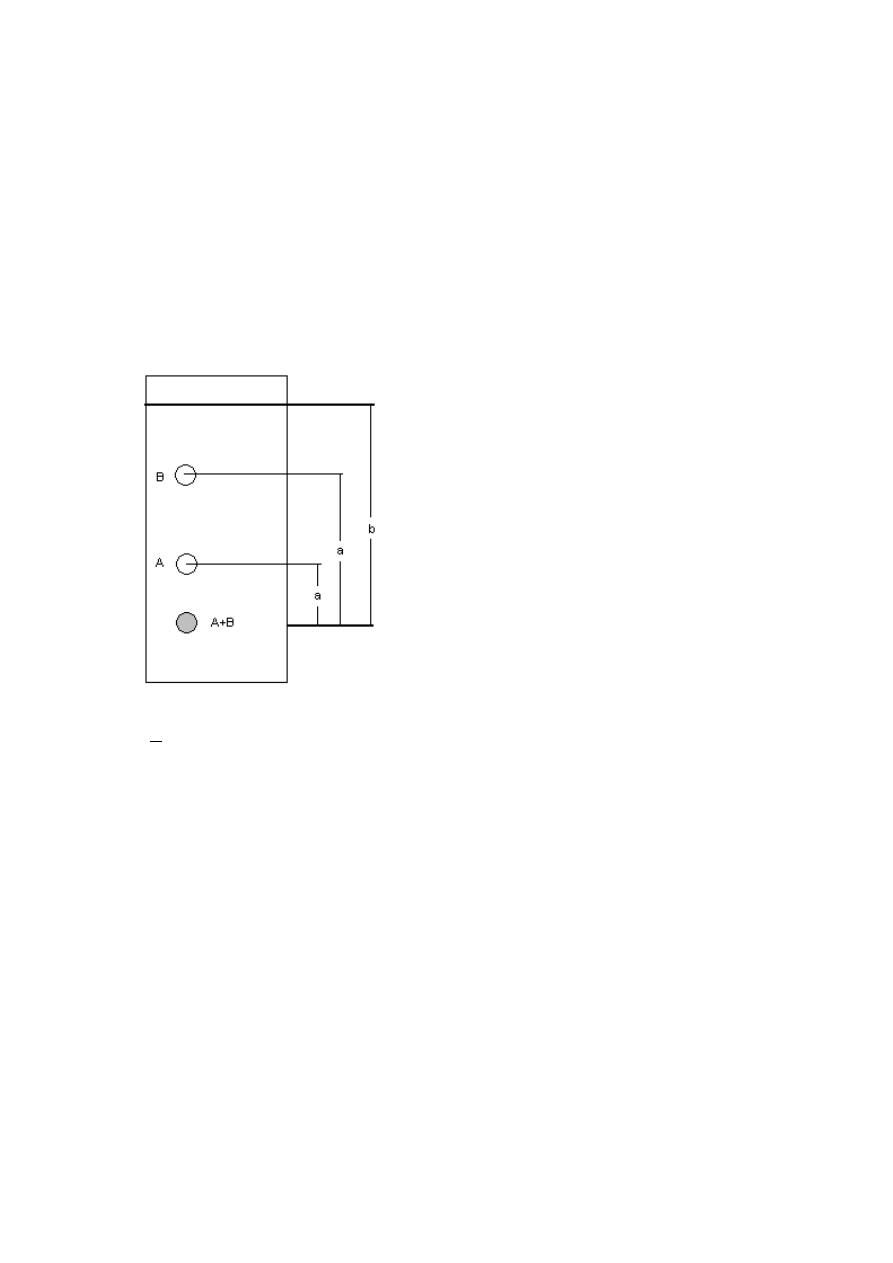
Parametrem, który stosuje się do opisu zjawisk zachodzących w warstwie chromatograficznej
jest współczynnik R
f
opisywany jako stosunek drogi przebytej przez środek plamy substancji
(a) do drogi czoła fazy ruchomej (b).
b
a
R
f
Rys. 1 Chromatogram cienkowarstwowy mieszaniny substancji; a-droga przebyta przez środek plamy
substancji A,B; b- droga przebyta przez czoło fazy ruchomej
Aby uzyskać informacje o rozdzielonych substancjach należy umiejscowić je na warstwie
chromatograficznej. Detekcję substancji przeprowadzić można za pomocą metod fizycznych,
chemicznych lub biologiczno-fizjologicznych. Do metod fizycznych zaliczyć możemy:
fotometrię absorpcyjną, fluorescencję, fosforescencję a w przypadku substancji znakowanych
izotopami promieniotwórczymi, metody radiometryczne. Najczęściej stosuje się lampę
emitującą promieniowanie UV, ponieważ większość związków organicznych wykazuje
absorpcję tego promieniowania. Dodatkową fluorescencje można wzbudzić impregnując
warstwy odpowiednim „wywoływaczem”. Na płytce obserwować można tzw. Świecenie
plamki.
Detekcja chemiczna polega na przeprowadzeniu badanych substancji w substancje barwne z
pomocą reagentów chemicznych, które reagują z wybranymi grupami funkcyjnymi.
Detekcja biologiczno-fizjologiczna wykorzystuje aktywność biologiczną rozdzielanych
substancji, gdy są one specyficzne. Metody te służą do oznaczania antybiotyków,
insektycydów, fungicydów i innych.

Document Outline
Wyszukiwarka
Podobne podstrony:
17 02 2011 2id 17062 Nieznany (2)
17 02 2011 2id 17062 Nieznany (2)
102 106 SUPLEMENT 53 2id 11668 Nieznany
2 PE 2012 2id 21154 Nieznany (2)
1 Wprowadzenie 2id 8727 Nieznany (2)
014 2id 3218 Nieznany (2)
IMG 17 id 210990 Nieznany
1informatyka 2id 19002 Nieznany (2)
2002 matura arkusz 2id 21667 Nieznany (2)
1 RNP 2id 9695 Nieznany (2)
08 2id 7222 Nieznany
1(1) 2id 10171 Nieznany
praca magisterska(17) jak napis Nieznany
2 Kurs Cubase Cz 2id 20482 Nieznany (2)
030 2id 4629 Nieznany (2)
1 teoria 1i 2 2id 9964 Nieznany
2009 02 17 test egzaminacyjny n Nieznany (2)
Matematyka 17 id 283105 Nieznany
więcej podobnych podstron