
CUKRY PROSTE. BUDOWA I METABOLIZM.
Węglowodany - inaczej: cukry lub sacharydy są najobficiej występującą w przyrodzie grupą
związków organicznych.
Większość z nich składa się z trzech pierwiastków: węgla, wodoru i tlenu, a stosunek liczby
atomów H do O wynosi (podobnie jak w wodzie) 2: 1.
Stąd wywodzi się termin: węglowodany.
Najwyżej utlenionym jest węgiel w grupie aldehydowej lub ketonowej.
Pozostałe atomy węgla (poza nielicznymi wyjątkami) są nośnikami atomów wodoru i grup
hydroksylowych.
Ze względu na te cechy struktury; część cukrów zalicza się do aldehydoalkoholi, a pozostałe
do ketoalkoholi wielowodorotlenowych.
Cukry posiadające od 3 do 7 atomów węgla w cząsteczce, jedną grupę aldehydową lub keto-
nową oraz co najmniej dwie grupy hydroksylowe przy różnych atomach węgla, noszą nazwę
cukrów prostych, czyli monosacharydów.
Pełnią one wiele funkcji biologicznych.
Są substratami energetycznymi, uczestniczą w budowie związków wielkocząsteczkowych,
tworzących ściany bakterii, pancerze skorupiaków i włókien roślinnych.
Są elementami składowymi glikoprotein i glikolipidów.
Klasyfikacja i nazewnictwo cukrów prostych
Funkcjonują różne podziały cukrów prostych:
- w zależności od liczby atomów węgla w cząsteczce cukru prostego,
- od charakteru i lokalizacji grup funkcyjnych,
- kierunku skręcalności światła spolaryzowanego, typu pierścienia etc.
Monosacharydy
Nazwa monosacharyd, czyli cukier prosty oznacza, iż jest to najprostszy związek zachowujący
cechy cukru.
W odróżnieniu od oligosacharydów czy polisacharydów nie podlega hydrolizie do prostszych
związków zachowujących właściwości cukrowe.
Monosacharydy mogą być klasyfikowane według różnych kryteriów, jak:
-liczba atomów węgla w cząsteczce,
- charakter grup czynnych,
- lokalizacja grup czynnych,
- kierunek skręcalności światła spolaryzowanego, - charakter pierścienia etc.
W organizmie człowieka występują przede wszystkim cukry proste, zawierające od 3 do 7
atomów węgla.
Najważniejsze z nich są przedstawione na ryc. 5. 1, 5.2 i 5.3.
Rys. 5.1. Cukry trójwęglowe (triozy) i czterowęglowe (tetrozy).
Rys. 5.2. Cukry pięciowęglowe (pentozy).
Rys. 5.3. Cukry sześciowęglowe (heksozy).
Cukry trójwęglowe to triozy: aldehyd glicerynowy i jego izomer dihydroksyaceton.
Występują w postaci estrów fosforanowych, jako aldehyd 3-fosfoglicerynowy i fosfodihydrok-
syaceton, które są ważnymi metabolitami pośrednimi zarówno w procesach rozpadu, jak i
biosyntezy glukozy i fruktozy - glikoliza i glukoneogeneza.
Cukry czterowęglowe to tetrozy.
Należy do nich erytroza, występująca w postaci estru fosforanowego.
Jest ona ważnym pośrednikiem w przekształcaniu glukozy drogą szlaku pentozofosforanowe-
go.
Cukry pięciowęglowe noszą nazwę pentoz.
Najważniejsze z nich to ryboza, deoksyryboza, rybuloza, ksyloza i ksyluloza.
Najobficiej występują cukry sześciowęglowe, zwane heksozami.
Wśród nich glukoza, galaktoza, mannoza, fruktoza i fukoza.
Ta ostatnia jest deoksyheksozą (stosunek H : O jest w niej wyższy niż 1 : 2).
Cukry siedmiowęglowe (czyli heptozy), występują rzadko, w organizmie człowieka są reprezen-
towane jedynie przez sedoheptulozę.
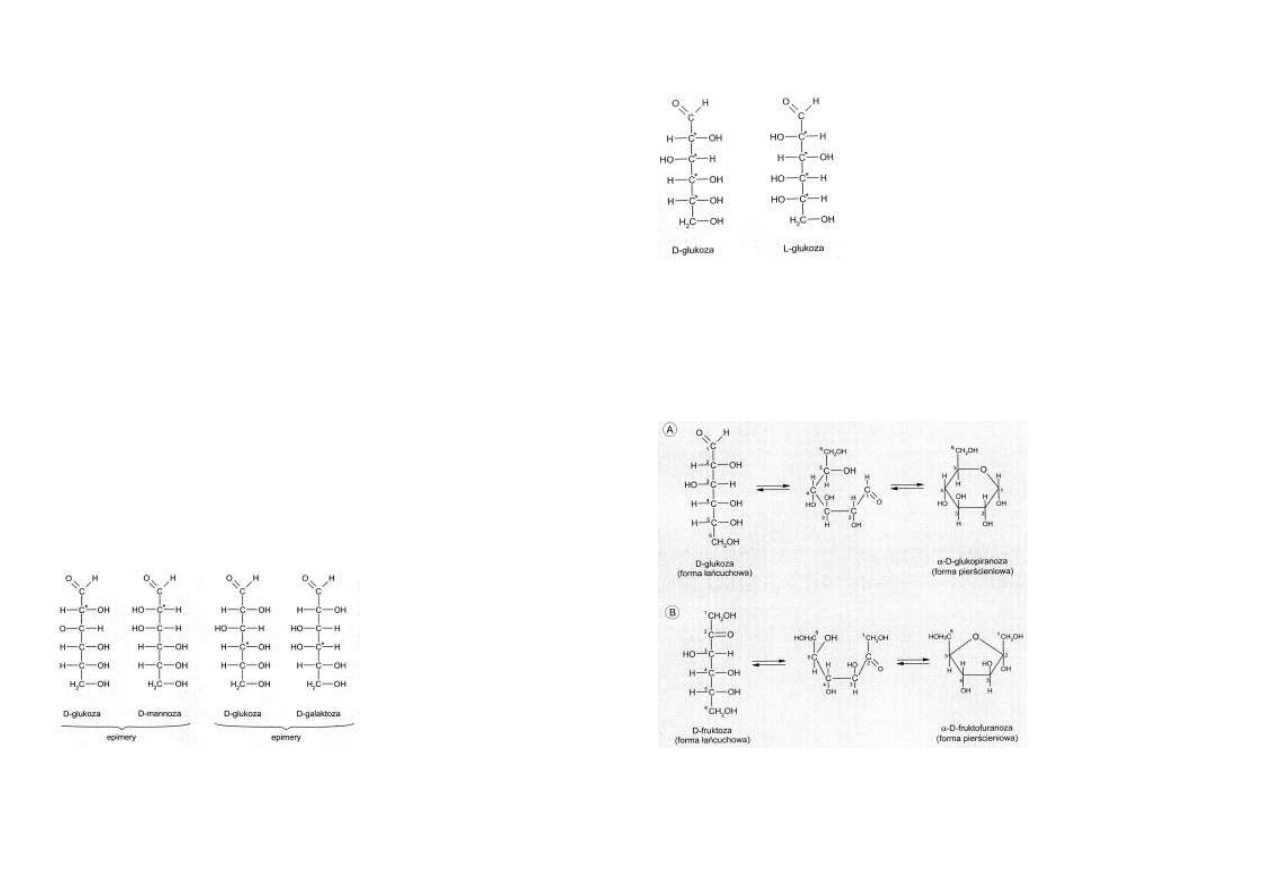
Cukry, których najwyżej utlenioną grupą funkcyjną jest grupa aldehydowa, noszą nazwę
aldoz,
Cukry, których najwyżej utlenioną grupą funkcyjną jest grupa ketonowa, noszą nazwę ketoz.
Na przykład, aldehyd glicerynowy jest aldozą, podczas gdy jego izomer: dihydroksyaceton jest
ketozą (ryc. 5. 1).
Cukry z grupy aldoz noszą nazwy z końcówką „oza”, np. glukoza, mannoza, ryboza.
Cukry z grupy ketoz noszą nazwy z końcówką „uloza”, np. heptuloza, rybuloza, ksyluloza.
Wyjątek stanowi fruktoza, która chociaż jest ketozą, nosi nazwę z końcówką „oza”.
Izomeria monosacharydów
Wiele cukrów, a szczególnie heksozy i pentozy występują w formie wielu izomerów.
Na przykład: glukoza, fruktoza, mannoza i galaktoza mają identyczny wzór sumaryczny –
C6H12O6.
Są względem siebie izomerami.
Charakteryzują się występowaniem szczególnych, niżej przedstawionych form izomerii.
Epimeria. Jeżeli dwa monosacharydy różnią się położeniem podstawników (-H i -OH) przy
jednym atomie węgla, z wyjątkiem węgla grupy karbonylowej, (aldehydowej lub ketonowej) są
określane epimerami, a ta forma izomerii nosi nazwę epimerii.
Na przykład, glukoza i galaktoza są C4 epimerami, ponieważ ich struktura różni się jedynie
położeniem grupy -OH w pozycji C4.
Glukoza i mannoza są C2 epimerami.
Różnią się bowiem położeniem grupy -OH przy C2 (ryc. 5.4).
Natomiast galaktoza i mannoza nie są względem siebie epimerami, gdyż różnią się położeniem
grup -OH przy dwu atomach węgla: C2 i C4.
Rys. 5.4. Przykłady epimerii. Glukoza i
mannoza różnią się położeniem grupy –
OH w pozycji C2 a glukoza i galaktoza
różnią się położeniem grupy –OH w
pozycji C4.
Enancjomeria jest formą izomerii,
polegającą na występowaniu pewnych
związków w dwóch postaciach, z któ-
rych jedna jest odbiciem zwierciadla-
nym drugiej.
Izomery takie noszą nazwy enancjomerów.
Enancjomerycznym formom cukrów przypisano symbole D i L.
W organizmie człowieka zdecydowanie dominują cukry szeregu D.
Cukry szeregu L występują sporadycznie, np. w glikoproteinach występuje L-fukoza, a w
glikozoaminoglikanach występuje kwas L-iduronowy będący
pochodną L-idozy (ryc. 5.5).
Rys. 5.5. Przykład enancjomerii. D – glukoza i L- glukoza
różnią się położeniem podstawników przy węglach asyme-
trycznych, oznakowanych gwiazdkami. Jeden z enancjome-
rów jest „lustrzanym” odbiciem drugiego.
Izomeria łańcuchowo-pierścieniowa. Cukry proste mogą
występować w postaci łańcuchowej lub pierścieniowej,
jednak ta pierwsza występuje w zdecydowanej mniejszości.
Co najwyżej 1 % monosacharydów, zawierających 5 lub
więcej atomów węgla, występuje w postaci łańcuchowej.
Większość występuje w formach pierścieniowych.
Grupa aldehydowa lub ketonowa, reagując z grupą alkoholową (-OH) tej samej cząsteczki
cukru, tworzy pierścień hemiacetalowy lub hemiketalowy.
Jeżeli pierścień zawiera sześć członów (5 C i 1 O), to powstały cukier jest piranozą, jeżeli
zawiera pięć członów (4 C i 1 O), jest furanozą.
Większość heksoz tworzy pierścienie piranozowe, jedynie fruktoza tworzy pierścień furanozo-
wy (ryc. 5.6).
Rys. 5.6. Izomeryzacja cu-
krów
poprzez
wzajemne
przechodzenie z formy łań-
cuchowej w formę pierście-
niową.
A – glukoza poprzez wiąza-
nie hemiacetalowe wytwa-
rza pierścień sześcioczłono-
wy (piranozowy), B – frukto-
za poprzez wiązanie hemike-
talowe wytwarza pierścień
pięcioczłonowy
(furanozo-
wy).
Sposób, w jaki przedstawio-
no wzory cukrów na ryc. 5.6
po stronie lewej, jest nazy-
wany projekcją Fischera.
Łańcuch węglowy jest pisa-
ny pionowo, z węglem C1 na
szczycie oraz podstawnikami, wodorem i grupami hydroksylowymi, na prawo i na lewo.
Drugi sposób, jak na ryc. 5.6 po stronie prawej, nazywany jest projekcją Hawortha.
Hemiacetalowy lub hemiketalowy atom węgla jest wysunięty najbardziej na prawo.
W przypadku aldoz jest to C1, a w przypadku ketoz C2.
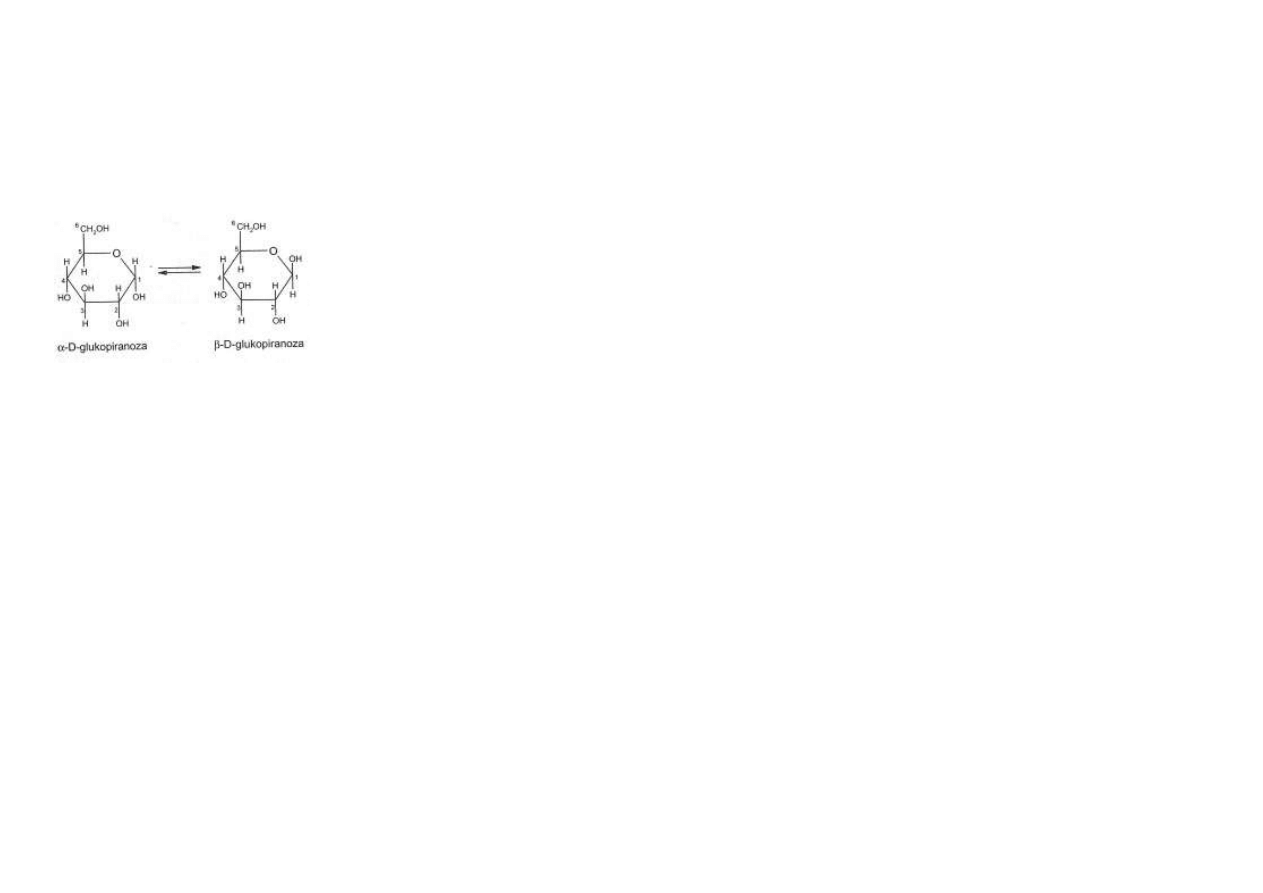
Płaszczyzna pierścienia jest płaska, natomiast podstawniki -H, -OH i –CH2OH są skierowane
powyżej lub poniżej płaszczyzny pierścienia.
Anomeria. Przejście cukru z postaci łańcuchowej w postać pierścieniową wiąże się z powsta-
niem nowego węgla asymetrycznego, zwanego węglem anomerycznym, w pozycji C1 aldozy lub
C2 ketozy.
W zależności od położenia grupy -OH względem płaszczyzny pierścienia, powstająca struktura
może być dwojaka: α lub β.
Powstają izomery, zwane anomerami, np. α-D-glukoza lub β-D-glukoza (ryc. 5.7).
Rys. 5.7. Przykład anomerii. α-D-glukoza i β-D-
glukoza różnią się położeniem grupy –OH w
pozycji C1.
Przejście anomeru α w anomer β lub odwrotnie
zachodzi przez łańcuchową postać tego cukru i
wiąże się ze zmianą skręcalności optycznej.
Zjawisko to nosi nazwę mutarotacji.
W roztworze wodnym anomery α i β pozostają w
stanie równowagi.
Na przykład, w roztworze glukozy anomer α stanowi 36%, a anomer β 36% całkowitej ilości
rozpuszczonego cukru (ryc. 5.7).
Grupy -OH przy węglach anomerycznych, uczestniczą w tworzeniu wiązań (odpowiednio) α i β
–glikozydowych.
Utlenianie i redukcja cukrów
Obecność grup aldehydowych lub ketonowych oraz grup hydroksylowych sprawia, iż cukry
wykazują reakcje charakterystyczne dla aldehydów/ketonów i alkoholi.
Na szczególną uwagę zasługują właściwości oksydoredukcyjne cukrów.
Mogą łatwo utleniać się do odpowiednich kwasów aldonowych, kosztem redukcji czynnika
utleniającego.
Inne elementy składowe cząsteczki nie zmieniają się.
Grupa aldehydowa utlenia się do grupy karboksylowej.
Wprawdzie ketony, w odróżnieniu od aldehydów, nie dają odczynów redukcyjnych, jednak w
środowisku alkalicznym ketozy izomeryzują do aldoz, np. fruktoza (ketoza) izomeryzuje do
glukozy (aldoza), dlatego fruktoza jest także cukrem redukującym.
Jeżeli tlen grupy karbonylowej przy węglu anomerycznym cukru nie jest związany z żadną
inną strukturą, cukier ten ma właściwości redukujące.
Na przykład mogą redukować kationy metali: Cu2+ do Cu+ lub Ag+ do Ag0.
Jednocześnie węgiel anomeryczny utlenia się do grupy karboksylowej.
Cukier przekształca się w odpowiedni kwas aldonowy, np. glukoza w kwas glukonowy galak-
toza w kwas galaktonowy.
Grupy hydroksylowe cukru nie uczestniczą w tych reakcjach.
Utlenianie grupy –CH2OH na przeciwstawnym końcu cząsteczki prowadzi do przekształcenia
cukru w odpowiedni kwas uronowy.
Glukoza przekształca się w kwas glukuronowy, a galaktoza w kwas galakturonowy.
Wolne monosacharydy nie mogą jednak pełnić funkcji substratu w tych reakcjach.
Proces ten zachodzi drogą enzymatyczną poprzez utlenianie nukleotydowch pochodnych
niektórych cukrów.
Redukcja grupy karbonylowej (aldehydowej lub ketonowej) zamienia ją w grupę alkoholową.
Cukier staje się alkoholem wielowodorotlenowym -poliolem.
Tą drogą glukoza i fruktoza zamieniają się w sorbitol, mannoza w mannitol, a galaktoza w
galaktytol.
Redukcja grupy hydroksylowej prowadzi do powstania deoksycukrów.
Tą drogą ryboza przekształca się w 2-deoksyrybozę, składnik nukleotydów budujących kwas
deoksyrybonukleinowy (DNA).
Wolna ryboza nie może być substratem w tej reakcji.
Redukcja zachodzi na etapie rybozy zawartej w odpowiednich nukleotydach.
Glikoliza tlenowa
Wyjaśnienie mechanizmu rozkładu glukozy dla uzyskania energii zawdzięcza się takim bada-
czom, jak: Embden, Meyerhof, Robisson, Neuberg, Cori, Parnas, Warburg.
Znany ciąg reakcji glikolizy często nosi nazwę szlaku Embdena-Meyerhofa-Parnasa (EMP).
Jakub Parnas
Jakub Karol Parnas (ur. 16 stycznia 1884 w Mokrzanach, zm. 29 stycznia 1949 w Moskwie) –
polski chemik, pionier polskiej biochemii, twórca lwowskiej szkoły biochemicznej. Był synem
właściciela ziemskiego. Absolwent Lwowskiego gimnazjum (1902) i wyższych studiów w Tech-
nische Hochschule w Berlinie (1904). Studiował także w Strasburgu i Monachium, gdzie w
1907 roku obronił doktorat. Pracował naukowo na uniwersytetach w Strasburgu, Cambridge,
Neapolu oraz w Warszawie. Od 1920 do 1941 roku profesor Uniwersytetu Jana Kazimierza
we Lwowie, kierownik Zakładu Chemii Lekarskiej Wydziału Lekarskiego. W latach 1930-
1931 wykładał na Uniwersytecie w Zurychu. W 1934 roku został doktorem honoris causa
uniwersytetu w Atenach. Był członkiem niemieckiej Akademii Nauk Leopoldina w Halle, wy-
kładowcą na Uniwersytecie w Gandawie.Członek czynny Towarzystwa Naukowego we Lwowie.
Podczas pierwszej okupacji sowieckiej Lwowa (1939 – 1941) współpracował aktywnie z sowie-
tami. Po wybuchu wojny niemiecko-sowieckiej ewakuowany do Kijowa, a potem do Ufy. W
1942 roku laureat nagrody Stalina za całokształt badań nad chemizmem mięśni. W latach
1943 – 1949 dyrektor Wydziału Chemii i Filozofii Akademii Nauk Medycznych ZSRR w Mo-

skwie. Był członkiem Akademii Nauk ZSRR (od 1942) i Akademii Nauk Medycznych ZSRR (od
1944). W 1945 roku wybrany członkiem Francuskiej Akademii Medycyny w Paryżu, otrzymał
doktorat honorowy Sorbony oraz został członkiem towarzystw chemicznych w Londynie,
Paryżu i Moskwie. W 1946 roku przyjechał do Krakowa i Wrocławia z wykładami. 29 stycznia
1949 roku aresztowany w Moskwie pod fałszywymi oskarżeniami, zmarł tego samego dnia na
Łubiance. Po śmierci Stalina zrehabilitowany. Twórca pierwszej w Polsce tzw. lwowskiej szko-
ły biochemicznej, autor wielu odkrywczych prac naukowych tłumaczonych na wiele języków
obcych. Opublikował m.in. podręcznik Chemia Fizjologiczna (1922). Wraz ze swoimi uczniami
zajmował się przede wszystkim metabolizmem mięśni, odkrył m.in. fosforolizę glikogenu.
Fosforoliza (gr.), enzymatyczny, odwracalny proces rozkładu wiązania glikozydowego polisa-
charydów; umożliwia stopniowy rozkład glikogenu u zwierząt i skrobi u roślin; zachodzi przy
udziale nieorganicznego kwasu fosforowego.
Jako jeden z pierwszych na świecie zastosował metody izotopowe w biochemii (przy współpra-
cy z Instytutem Nielsa Bohra w Kopenhadze), za pomocą których prześledził etapy przemian
związków fosforowych w mięśniach.
Nazwisko Parnasa zapisano w nauce w nazwie schematu EMP (Schemat Embdena-
Meyerhofa-Parnasa), opisującego ciąg przemian związków fosforowych w mięśniach od fosfo-
rolizy glikogenu do wytworzenia kwasu mlekowego.
Glikoliza tlenowa jest szlakiem metabolicznym, przekształcającym glukozę do pirogronianu, w
celu dostarczenia komórce energii (w postaci ATP) oraz substratów do innych szlaków meta-
bolicznych.
Glikoliza jest centralnym punktem przemiany cukrów, ponieważ prawie wszystkie z nich
przekształcają się poprzez glukozę.
Glukoza jest głównym a dla niektórych komórek jedynym (erytrocyty, płytki krwi), lub prawie
jedynym (komórki mózgu), substratem energetycznym.
Przemiana glukozy do CO2 i H2O jest procesem wieloetapowym.
Pierwszym z nich jest glikoliza.
Produktem glikolizy w warunkach tlenowych jest pirogronian.
Ten ulega oksydacyjnej dekarboksylacji do acetylo~S-CoA.
Reszty acetylowe utleniają się w cyklu kwasów trikarboksylowych do CO2 i H2O.
Każdy z tych etapów generuje energię magazynowaną w postaci ATP.
Przemiana glukozy do pirogronianu przebiega poprzez 10 kolejno po sobie następujących
reakcji.
Pierwszy etap glikolizy obejmujący 5 reakcji, to faza zużywająca energię.
Zachodzi kosztem energii zainwestowanej w ten proces, poprzez zużycie 2 cząsteczek ATP.
Drugi etap to faza generująca energię, prowadząca do syntezy 4 cząsteczek ATP.
Zysk netto wynosi 2 cząsteczki ATP w przeliczeniu na 1 cząsteczkę glukozy.
Jednocześnie powstają 2 cząsteczki NADH+2H+.
Utlenianie NADH przez łańcuch oddechowy dostarcza dodatkowo 6 (2 x 3) cząsteczek ATP.
Faza zużywająca energię
Fosforylacja glukozy
W pierwszym etapie następuje fosforylacja glukozy w pozycji C6, z wytworzeniem glukozo-6-
fosforanu.
Dawcą reszty fosforanowej jest ATP, przekształcający się w ADP.
Reakcja jest nieodwracalna, prowadzi do zakotwiczenia glukozy w komórce.
Powstający
glukozo-6-fosforan
nie
przenika przez błonę plazmatyczną na
zewnątrz komórki.
Estry fosforanowe cukrów nie mogą
przenikać przez błonę komórkową,
ponieważ ta nie posiada odpowiednich
przenośników.
Muszą być przetworzone w komórce.
Fosforylacja glukozy jest katalizowana
przez heksokinazę lub glukokinazę
(wzór 5.1).
Izomeryzacja glukozo-6-fosforanu.
Izomeryzacja glukozo-6-fosforanu (aldo-
zy) do fruktozo-6-fosforanu (ketozy) jest
katalizowana przez fosfoheksoizomera-
zę.
Reakcja jest odwracalna.
Proces izomeryzacji zachodzi poprzez
łańcuchowe formy wspomnianych
cu-
krów (wzór 5.2).
Fosforylacja fruktozo-6-fosforanu.
Fosforylacja produktu poprzedniej
reakcji prowadzi do powstania frukto-
zo-1,6-bis-fosforanu jest katalizowana
przez fosfofruktokinazę (
wzór 5.3
).
W tej nieodwracalnej reakcji zużywa
się druga cząsteczka ATP.
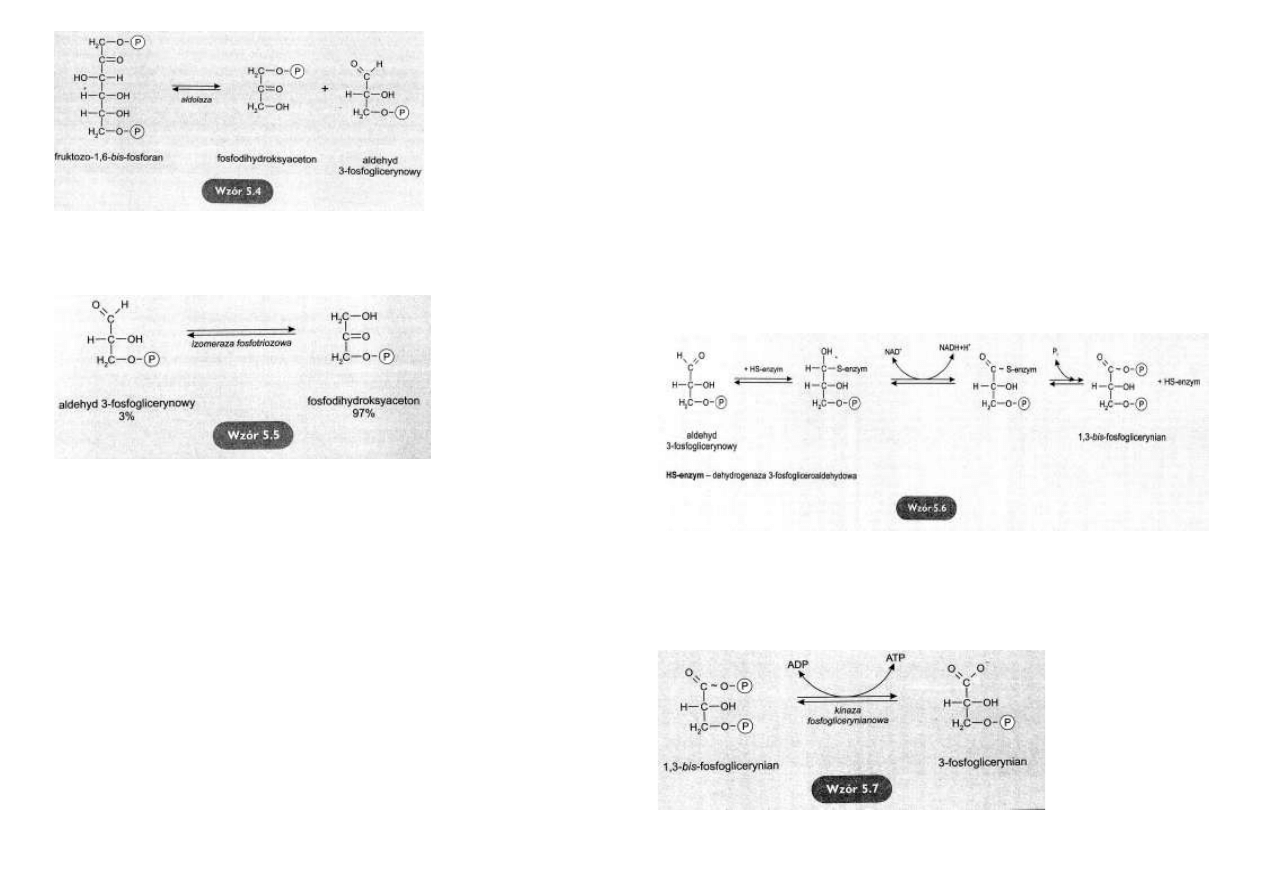
Rozpad fruktozo-1, 6-bis-
fosforanu
Pod działaniem aldolazy
fruktozo-
1,6-bis-fosforan
rozpada się odwracalnie na
dwa ufosforylowane cukry
trójwęglowe – fosfotriozy.
Są
to:
aldehyd
3-
fosfoglicerynowy
i
fos-
fodihydroksyaceton
(wzór 5.4).
Izomeryzacja fosfotrioz
Obydwa produkty reakcji
aldolazowej, aldehyd 3-
fosfoglicerolowy
i
fos-
fodihydroksyaceton
są
izomerami, nawzajem w
siebie przechodzącymi.
Reakcję izomeryzacji kata-
lizuje izomeraza fosfotrio-
zowa
(wzór 5.5).
Gdyby doszło do ustalenia stanu równowagi, 97% fosfotrioz stanowiłby fosfodihydroksyacetn,
a jedynie 3% stanowiłby aldehyd 3-fosfoglicerynowy.
Jednakże aldehyd 3-fosfoglicerynowy jest natychmiast usuwany poprzez włączanie go do
kolejnego etapu glikolizy.
Ubytek ten jest rekompensowany poprzez izomeryzację fosfodihydroksyacetonu do aldehydu
3-fosfoglicerynowego.
Oznacza to w praktyce, że jedna cząsteczka fruktozo- 1,6-bis-fosforanu przekształca się w
dwie cząsteczki aldehydu 3-fosfoglicerynowego.
Inaczej bowiem fosfodihydroksyaceton nie mógłby być dalej przekształcany drogą glikolizy.
Faza generująca energię
Utlenianie aldehydu 3-fosfoglicerynowego
W kolejnym etapie aldehyd 3-fosfoglicerynowy jest utleniany do 1, 3-bis-fosfoglicerynianu.
Proces ten jest odwracalny i obejmuje dwie sprzężone ze sobą reakcje.
W pierwszej grupa aldehydowa jest utleniana do grupy karboksylowej.
Reakcję tę katalizuje dehydrogenaza 3-fosfogliceroaldehydowa.
Uczestniczy grupa -SH reszty cysteinylowej tego enzymu.
Powstaje przejściowo hemitioacetal, który przekazuje jon wodorkowy (H-, czyli H+ + 2e-) na
NAD+.
Powstaje NADH+H+.
Przedrostek bis oznacza, iż cząsteczka zawiera dwie odrębne reszty fosforanowe (przy różnych
atomach węgla), w odróżnieniu od przedrostka di, który oznacza, iż dwie reszty fosforanowe
są połączone ze sobą wiązaniem bezwodnikowym, a powstały pirofosforan wiąże się z jednym
atomem węgla, np. w ADP pirofosforan jest związany z weglem 5’rybozy.
Jest to jedyna reakcja oksydoredukcji zachodząca w przebiegu glikolizy.
Powstaje bogate w energię wiązanie tioestrowe między siarką enzymu a grupą karboksylową.
W drugim etapie fosforan nieorganiczny rozkłada wiązanie tioestrowe, uwalnia enzym z grupą
-SH i wytwarza wiązanie bezwodnikowe między grupą karboksylową 3-fosfoglicerynianu a
resztą fosforanową.
Powstaje 1,3-bis-fosfoglicerynian zawierający wiązanie bezwodnikowe (acylofosforanowe),
bogate w energię
(wzór 5.6).
Powstawanie ATP z 1,3-bis-fosfoglicerynianu i ADP
Reszta fosforanowa z 1,3-bis-fosfoglicerynianu zostaje przeniesiona na ADP z wytworzeniem
ATP natomiast 1,3-bis-fosfoglicerynian przekształca się w 3-fosfoglicerynian.
Reakcję tę katalizuje kinaza fosfoglicerynianowa
(wzór 5.7).
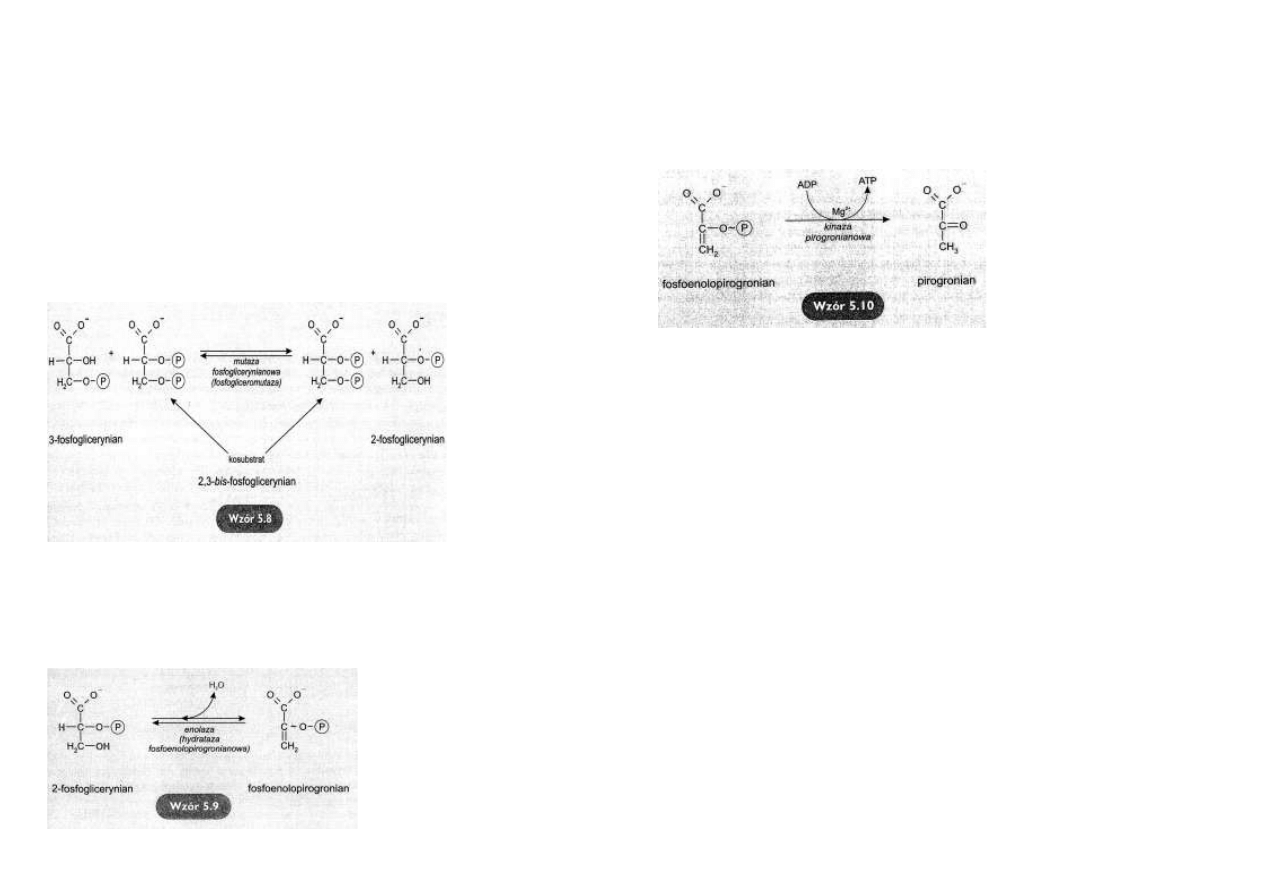
Reakcja ta jest przykładem fosforylacji substratowej, w której powstawanie wiązania bezwod-
nikowego bogatego w energię nie jest bezpośrednio związane z utlenianym substratem i za-
chodzi bez udziału łańcucha oddechowego.
Energia zmagazynowana w wiązaniu bezwodnikowym między grupą karboksylową 3-
fosfoglicerynianu i fosforanem zostaje przekazana bezpośrednio z tego substratu na nowe
wiązanie bezwodnikowe między ADP i Pi.
W przeciwieństwie do większości reakcji katalizowanych przez kinazy, ta jest odwracalna.
Biorąc pod uwagę fakt, iż z jednej cząsteczki glukozy powstają dwie fosfotriozy, łączny zysk
energetyczny, w przeliczeniu na 1 cząsteczkę glukozy, wyniesie 2 cząsteczki ATP.
Rekompensuje to utratę 2 cząsteczek ATP, zużytych do fosforylacji glukozy i fruktozo-6-
fosforanu.
Przemieszczanie fosfo-
ranu z C3 do C2
Powstały
3-
fosfoglicerynian
ulega
odwracalnej izomeryza-
cji
do
2-
fosfoglicerynianu.
Reakcja jest katalizo-
wana
przez
mutazę
fosfoglicerynianową
(fosfogliceromutazę)
i
polega na wewnątrzczą-
steczkowym przemiesz-
czeniu reszty fosfora-
nowej
(wzór 5.8).
Jest odwracalna.
Enzym wymaga katalitycznych ilości 2,3-bis-fosfoglicerynianu.
Dehydratacja 2-fosfoglicerynianu
Dehydratacja (odłączenie H2O) od 2-
fosfoglicerynianu przez enolazę powo-
duje redystrybucję energii w obrębie
przekształcanego substratu.
To prowadzi do wytworzenia wiązania
bogatego w energię między C2 a fosfo-
ranem.
Powstaje fosfoenolopirogronian, zawie-
rający bogate w energię wiązanie enolo-
fosforanowe
(wzór 5.9).
Energia wiązania fosforanu w fosfoenolopirogronianie jest prawie pięciokrotnie wyższa niż w
wiązaniu estrowym, występującym w 2-fosfoglicerynianie.
Reakcja jest odwracalna.
Powstawanie pirogronianu
Kinaza pirogronianowa przekształ-
ca fosfoenolopirogronian w piro-
gronian
(wzór 5.10).
Jest to trzecia z kolei reakcja nie-
odwracalna.
Następuje przekazanie reszty fosfo-
ranowej z fosfoenolopirogronianu
na ADP.
Powstaje pirogronian i cząsteczka ATP.
Po raz drugi zachodzi fosforylacja substratowa.
Ponieważ z jednej cząsteczki glukozy powstają 2 cząsteczki pirogronianu,
Łączny zysk energetyczny na tym etapie glikolizy wynosi 2 cząsteczki ATP.
Bilans energetyczny glikolizy tlenowej
W bilansie energetycznym glikolizy tlenowej należy uwzględnić fakt, iż powstające w jej prze-
biegu 2 cząsteczki NADH są utleniane przez mitochondrialny łańcuch oddechowy.
Wewnętrzna błona mitochondrialna nie jest przepuszczalna dla NADH, który jest nośnikiem
pary elektronów, zwanych równoważnikami redukcyjnymi.
Z tego powodu para elektronów i para protonów zawarte w cytosolowym NADH+H+, powstałe
w trakcie przemiany jednej cząsteczki glukozy, przemieszczają się do mitochondrium poprzez
specjalne mechanizmy zwane „mostkami” lub „czółenkami”.
Mechanizmy te przemieszczają równoważniki redukcyjne z NADH cytosolowego na NAD+
mitochondrialny.
W mitochondriach powstają 2 cząsteczki NADH+2H+.
Ich utlenianie przez łańcuch oddechowy dostarcza 6 (2 x 3) cząsteczek ATP.
Przebieg glikolizy tlenowej można przedstawić sumarycznie w postaci dwóch równań:
Glukoza + 2 Pi + 2 NAD+ + 2 ADP → 2 pirogronian + 2 ATP + 2 NADH + 2H+
2 NADH + 2H+ + O2 + 6 ADP + 6 Pi → 2 NAD+ + 2 H2O + 6 ATP
Tak więc przemiana 1 cząsteczki glukozy do 2 cząsteczek pirogronianu dostarcza (netto)
dwóch cząsteczek ATP na drodze fosforylacji substratowej.

Uwzględniając utlenianie powstających w tym procesie 2 cząsteczek NADH, bilans energe-
tyczny glikolizy tlenowej wzbogaca się dodatkowo o 6 (2 x 3) cząsteczek ATP, powstających na
drodze fosforylacji oksydacyjnej.
Reasumując, należy stwierdzić, iż jedna cząsteczka (lub jeden mol) glukozy, utleniająca się do
dwóch cząsteczek (lub dwóch moli) pirogronianu dostarcza 8 cząsteczek (lub 8 moli) ATP.
Pirogronian, końcowy produkt glikolizy tlenowej, zachowuje większość energii zawartej w
glukozie.
Energia ta uwalnia się w kolejnych etapach rozpadu pirogronianu.
Glikoliza beztlenowa
W niektórych warunkach glikoliza tlenowa nie może funkcjonować.
Dotyczy to komórek nieposiadających mitochondriów, jak erytrocyty lub płytki krwi, lub
komórek niedostatecznie zaopatrywanych w tlen, np. komórki mięśni szkieletowych podczas
wysiłku.
Glikoliza tlenowa jest zahamowana także w sytuacji, gdy ilość powstającego NADH przekracza
możliwości jego utleniania przez łańcuch oddechowy.
W tej sytuacji zostaje uruchomiona glikoliza beztlenowa.
Przebieg glikolizy beztlenowej
Większość reakcji glikolizy beztlenowej pokrywa się z przebiegiem glikolizy tlenowej.
Jedyna różnica polega na tym, że NADH + H+ będący wytworem reakcji katalizowanej przez
dehydrogenazę 3-fosfogliceroaldehydową nie jest utleniany przez mitochondrialny łańcuch
oddechowy, lecz przez końcowy produkt glikolizy, jakim jest pirogronian.
Cząsteczka pirogronianu przejmuje parę atomów wodoru (2H+ + 2e-) z NADH + H+, redukując
się do mleczanu i utleniając NADH + H+ do NAD+.
Ten ostatni może wziąć udział w kolejnej
reakcji
utleniania
aldehydu
3-
fosfoglicerynowego
do
1,3-bis-
fosfoglicerynianu.
Gdyby nie funkcjonował sprawny me-
chanizm regeneracji NAD+, doszłoby do
szybkiego wyczerpania jego zapasów w
cytosolu i do zatrzymania glikolizy na
etapie aldehydu 3-fosfoglicerynowego.
Redukcja pirogronianu do mleczanu przy
równoczesnym utlenianiu NADH + H+ do NAD+ zachodzi w cytosolu pod katalitycznym dzia-
łaniem dehydrogenazy rnleczanowej
(wzór 5.11).
Wprawdzie mięśnie szkieletowe posiadają mitochondria, to jednak ilość NADH + H+ wytwa-
rzanego przez dehydrogenazę 3-fosfogliceroaldehydową w pracującym mięśniu przewyższa
możliwości jego utleniania przez mitochondrialny łańcuch oddechowy.
Powoduje to wzrost stosunku NADH/NAD+, co sprzyja redukcji pirogronianu do mleczanu.
Z tego powodu wysiłek fizyczny prowadzi do akumulacji mleczanu w mięśniach, powodując
ich zakwaszenie.
Następuje obniżenie pH wewnątrz komórki.
Większość mleczanu przenika do krwi i przemieszcza się do wątroby, gdzie następuje jego
utlenienie do pirogronianu.
Reakcja katalizowana przez dehydrogenazę mleczanową jest odwracalna.
Kierunek jej przebiegu zależy od wartości stosunku pirogronian/mleczan i NADH/NAD+.
W mięśniu szkieletowym (pracującym) stosunek NADH/NAD+ jest duży, reakcja przebiega w
kierunku mleczanu.
Duże stężenie mleczanu we krwi wywołuje stan, określany mianem kwasicy mleczanowej.
Powstaniu jej przeciwdziała wątroba i mięsień sercowy, w których stosunek NADH/NAD+ jest
niski.
Reakcja przebiega w kierunku pirogronianu.
Reakcja ta zachodzi pod katalitycznym działaniem innego izoenzymu dehydrogenazy mlecza-
nowej.
Bilans energetyczny glikolizy beztlenowej
Glikoliza beztlenowa jest mniej wydajna pod względem energetycznym niż glikoliza tlenowa.
Jej sumaryczny efekt przedstawia poniższe równanie:
Glukoza + 2 Pi + 2 ADP → 2 mleczan + 2 ATP + 2 H2O
Tak więc przemiana 1 cząsteczki glukozy do 2 cząsteczek mleczanu dostarcza jedynie 2 czą-
steczki ATP.
Obydwie powstają drogą fosforylacji substratowej.
W warunkach beztlenowych nie funkcjonuje bowiem łańcuch oddechowy, a tym samym fosfo-
rylacja oksydacyjna.
Mleczan zachowuje więcej energii zawartej w glukozie niż pirogronian.
Energia ta może być uwolniona poprzez utlenianie mleczanu w innych tkankach.
Jakkolwiek glikoliza beztlenowa uwalnia tylko niewielką ilość energii zawartej w glukozie, jest
ona ważnym źródłem energii w sytuacjach, gdy przetwarzanie glukozy szlakiem tlenowym jest
niemożliwe lub ograniczone.
Metaboliczne losy pirogronianu
Pirogronian ulega wielokierunkowym przemianom.

Może być zużyty w procesach katabolicznych - jako substrat energetyczny.
Jednocześnie może być wykorzystany w procesach anabolicznych - jako substrat do biosynte-
zy innych metabolitów.
Fermentacja alkoholowa
Drożdże i niektóre bakterie przekształcają pirogronian w alkohol etylowy.
Proces ten obejmuje dwie reakcje:
dekarboksylację, która polega na odłączeniu grupy karboksylowej pirogronianu w postaci
CO2 z wytworzeniem aldehydu octowego,
redukcję powstałego aldehydu octowego przez NADH + H+ do etanolu.
Aldehyd octowy pełni więc funkcję taką, jak pirogronian w glikolizie beztlenowej.
Staje się akceptorem 2H++2e- z NADH + H+, powstałego w reakcji utleniania aldehydu 3-
fosfoglicerynowego.
Uczestniczy w regeneracji (zapobiega wyczerpaniu zapasów) NAD+, który jest konieczny do
kolejnych reakcji utleniania aldehydu 3-fosfoglicerynowego.
Proces przemiany glukozy poprzez pirogronian do etanolu i CO2 nosi nazwę fermentacji alko-
holowej.
Jest to proces beztlenowy, a jego bilans energetyczny jest identyczny jak w przypadku glikoli-
zy beztlenowej, zachodzącej u organizmów wyższych.
Z jednej cząsteczki glukozy powstają dwie cząsteczki etanolu i dwie cząsteczki CO2.
Towarzyszy temu powstanie dwóch cząsteczek ATP na drodze fosforylacji substratowej.
Fermentacja alkoholowa jest jedynym źródła energii dla drożdży i niektórych bakterii.
Transaminacja
Pirogronian może pełnić funkcję akceptora grup aminowych z aminokwasów, przechodząc w
alaninę.
Proces ten nosi nazwę transaminacji.
Glukoneogeneza
W wątrobie dominującym szlakiem przetwarzania pirogronianu jest jego karboksylacja, pro-
wadząca do powstania szczawiooctanu i włączenia produktu tej reakcji do procesu glukoneo-
genezy, czyli syntezy glukozy ze składników niecukrowych.
Oksydacyjna dekarboksylacja
Jest zasadniczym kierunkiem przemiany pirogronianu.
Proces ten zachodzi w macierzy mitochondrialnej.
Grupa karboksylowa pirogronianu odłącza się w postaci CO2, a pozostający fragment dwu-
węglowy utlenia się równocześnie do reszty kwasu octowego (reszty acetylowej), która wchodzi
w reakcję z CoA-SH, tworząc acetylo~S-CoA.
Proces ten jest katalizowany przez dehydrogenazę pirogronianową - będącą kompleksem
trzech enzymów: dekarboksylazy pirogronianowej, transacetylazy dihydroliponianowej i dehy-
drogenazy dihydroliponianowej.
Istnienie tego kompleksu sprawia, iż wiąże on substrat (pirogronian) i uwalnia produkt koń-
cowy (acetylo~S-CoA).
Nie uwalnia natomiast metabolitów pośrednich, będących produktami reakcji katalizowanych
przez poszczególne enzymy składowe tego kompleksu.
Dzięki temu omawiany proces przebiega sprawniej niż w sytuacji, gdyby każdy z enzymów
składowych działał niezależnie od siebie.
Dehydrogenaza pirogronianowa współdziała z 5 koenzymami, które pełnią funkcję przenośni-
ków lub utleniaczy dla pośredników powstających w procesie oksydacyjnej dekarboksylacji
pirogronianu.
Są to: pirofosforan tiaminy (TPP), NAD+, FAD, CoA-SH oraz kwas liponowy w postaci utlenio-
nej (kwas dehydroliponowy).
W pierwszym etapie dekarboksylaza pirogronianowa wiąże pirogronian z TPP.
Połączenie to sprawia, iż grupa karboksylowa pirogronianu staje się bardziej „mobilna”.
Odłącza się w postaci CO2, a pozostały fragment dwuwęglowy - odpowiadający strukturą
aldehydowi octowemu - wchodzi w reakcję z kwasem dehydroliponowym.
Reakcję katalizuje transacetylaza dihydroliponianowa.
Następuje utlenienie aldehydu octowego do reszty acetylowej kosztem redukcji mostka dwu-
siarczkowego zawartego w kwasie dehydroliponowym.
Jeden z atomów siarki wiąże atom wodoru - odłączony z utlenianego aldehydu (powstaje
grupa -SH), a drugi atom siarki wiąże wspomnianą resztę acetylową.
Powstaje kwas acetyloliponowy.
Grupa acetylowa z kwasu acetyloliponowego zostaje przeniesiona na atom siarki CoA-SH, a
jej miejsce zajmuje wodór pochodzący z grupy -SH tego koenzymu.
Powstaje kwas liponowy z dwiema grupami -SH.
Odtwarzanie kwasu dehydroliponowego zachodzi pod działaniem dehydrogenazy dihydrolipo-
nianowej, współdziałającej z FAD i NAD+.
Powstaje NADH + H+, a jego utlenienie przez łańcuch oddechowy dostarcza trzech cząsteczek
ATP w przeliczeniu na jedną cząsteczkę pirogronianu.
Sumaryczny efekt procesu oksydacyjnej dekarboksylacji pirogronianu przedstawia ryc. 5.8.
Biorąc pod uwagę fakt, iż jedna cząsteczka glukozy rozpada się do dwu cząsteczek pirogro-
nianu,sumaryczny zysk energetyczny oksydacyjnej dekarboksylacji pirogronianu, w przeli-
czeniu na jedną cząsteczkę glukozy wynosi (2 x 3) 6 cząsteczek ATP.
Rys. 5.8. Uproszczony schemat
przebiegu oksydacyjnej dekar-
boksylacji pirogronianu.
Grupy acetylowe związane w
postaci acetylo~S-CoA są sub-
stratem energetycznym włą-
czanym do cyklu kwasów trikarboksylowych, w którym utleniają się do CO2 i H2O, dostar-
czając energii w postaci ATP.
Mogą być użyte także do procesów anabolicznych, jak: synteza kwasów tłuszczowych, ciał
ketonowych, cholesterolu (a pośrednio innych steroidów) lub do reakcji acetylacji.
Dehydrogenaza pirogronianowa, a w konsekwencji proces oksydacyjnej dekarboksylacji piro-
gronianu, są podatne na działanie czynników regulacyjnych, które mogą zarówno pobudzać,
jak i hamować wspomniany proces.
Kompleks dehydrogenazy pirogronianowej jest hamowany przez produkt reakcji, acetylo~S-
CoA, który akumuluje się w sytuacji, gdy jest produkowany szybciej, niż może być utleniany
przez cykl kwasów trikarboksylowych.
Enzym jest także hamowany przez wysokie stężenia NADH, co występuje w sytuacji, gdy
funkcjonowanie łańcucha oddechowego jest spowolnione przez niedobór tlenu, końcowego
akceptora elektronów i protonów.
Kompleks dehydrogenazy pirogronianowej występuje w dwóch postaciach: aktywnej (niefosfo-
rylowanej) i nieaktywnej (fosforylowanej).
Wzajemne przechodzenie w siebie formy fosforylowanej i niefosforylowanej pozostaje pod
kontrolą dwóch enzymów: kinazy i fosfatazy.
Aktywacja kinazy sprzyja fosforylacji, a w konsekwencji inaktywacji dehydrogenazy pirogro-
nianowej i zahamowaniu oksydacyjnej dekarboksylacji pirogronianu.
Natomiast aktywacja fosfatazy sprzyja defosforylacji, a w konsekwencji aktywacji dehydroge-
nazy pirogronianowej i nasilaniu omawianego procesu.
Cykl kwasów trikarboksylowych
Cykl kwasów trikarboksylowych, zwany także cyklem Krebsa lub cyklem kwasu cytrynowego,
odgrywa podstawową rolę w metabolizmie.
Jego zasadnicza funkcja polega na utlenianiu grup acetylowych związanych z CoA (acetylo~S-
CoA) do CO2 i H2O.
Acetylo~S-CoA pochodzi z metabolizmu substratów energetycznych.
Jego głównym źródłem jest oksydacyjna dekarboksylacja pirogronianu - pochodzącego głów-
nie z glikolizy tlenowej.
Z tego powodu cykl Krebsa został włączony do wykładu poświęconego metabolizmowi cukrów
prostych.
Cykl kwasów trikarboksylowych jest także miejscem utleniania reszt acetylowych zawartych
w acetylo~S-CoA, powstałym w procesie β-oksydacji kwasów tłuszczowych, podczas rozpadu
ciał ketonowych i w trakcie przemiany szkieletów węglowodorowych niektórych aminokwa-
sów.
Reszta acetylowa włącza się do cyklu Krebsa poprzez wiązanie się ze szczawiooctanem i wy-
tworzenie cytrynianu.
W wyniku kilku kolejno po sobie następujących reakcji utleniania reszta acetylowa prze-
kształca się w CO2 i H2O, natomiast szczawiooctan odtwarza się.
Utlenianie grup acetylowych w cyklu Krebsa zużywa 2/3 całkowitej ilości tlenu pobieranego
przez organizm człowieka i dostarcza 2/3 ATP powstającego w organizmie.
Ponadto cykl ten dostarcza substratów do różnych biosyntez np. przetwarza szkielety węglo-
wodorowe niektórych aminokwasów do szczawiooctanu, zużywanego w glukoneogenezie,
dostarcza α-ketokwasów do reakcji transaminacji i syntezy aminokwasów oraz bursztynianu
do syntezy hemu.
Cykl Krebsa funkcjonuje wyłącznie w mitochondriach.
Jest sprzężony z mitochondrialnym łańcuchem oddechowym i z reakcjami fosforylacji oksy-
dacyjnej.
Reakcje cyklu kwasów trikarboksylowych
Wiązanie reszt acetylowych ze szczawiooctanem
Kondensacja reszty acetylowej, pochodzącej z acetylo~S-CoA, ze szczawiooctanem jest katali-
zowana
przez
syntazę
cytrynianową.
Powstaje cytrynian.
Równowaga reakcji kon-
densacji aldolowej jest
silnie przesunięta w kie-
runku syntezy cytrynianu
(wzór 5.12),
ze względu na
silnie
ujemną
wartość
ΔG°, wynoszącą około - 38
kJ/mol.
Izomeryzacja cytrynianu
Cytrynian izomeryzuje do izocytrynianu pod działaniem akonitazy.Pośrednikiem w tej reakcji
jest cis-akonitan.
Reakcja polega na dehydra-
tacji cytrynianu do cis-
akonitanu i hydratacji cis-
akonitanu z wytworzeniem
izocytrynianu.Produkt reak-
cji różni się od substratu
położeniem grupy -OH
(wzór
5. 13).
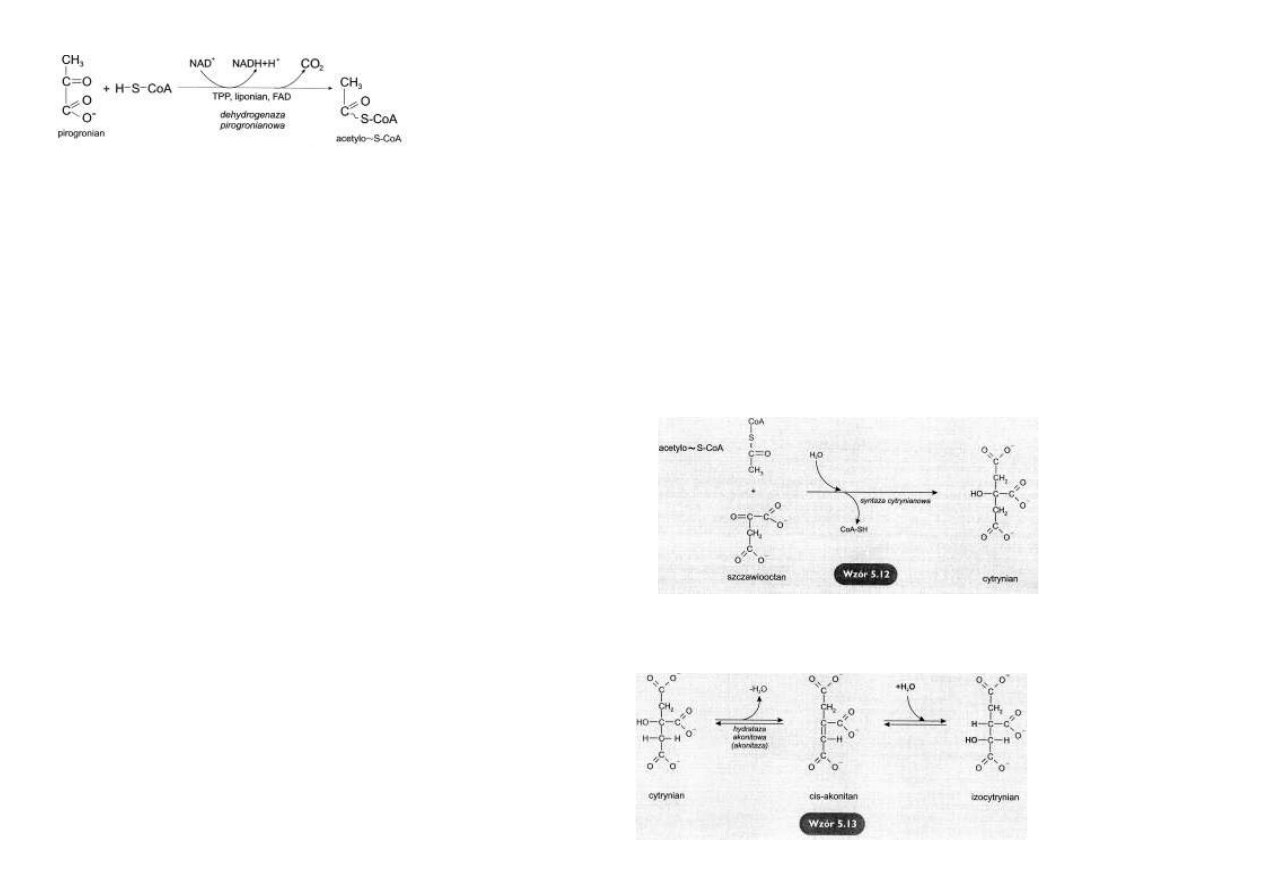
Utlenianie i dekarboksylacja izocytrynianu
Pod działaniem dehydrogenazy izocytrynianowej zachodzi oksydacyjna dekarboksylacja izocy-
trynianu do α-ketoglutaranu.
Przebieg reakcji jest dwuetapowy.
W pierwszym następuje utlenienie izocytrynianu przy udziale NAD+ do bardzo nietrwałego
szczawiobursztynianu.
Powstaje pierwsza cząsteczka NADH+H+.
Szczawiobursztynian ulega samoistnej dekarboksylacji do α-ketoglutaranu.
Atom węgla odłącza się w postaci CO2
(wzór 5. 14).
Oksydacyjna dekarboksylacja α-ketoglutaranu
Konwersja α-ketoglutaranu do bursztynylo~S-CoA (inaczej: sukcynylo~S-CoA jest katalizowa-
na przez dehydrogenazę α-ketoglutaranową, która jest kompleksem trójenzymatycznym,
podobnym do wcześniej omówionej dehydrogenazy pirogronianowej.
Kompleks ten współdziała z TPP, NAD+, FAD, kwasem liponowym i CoA-SH.
Funkcje każdego z enzymów składowych tego kompleksu oraz wymienionych koenzymów są
analogiczne do wyżej opisanych.
W przebiegu tej reakcji uwalnia się druga cząsteczka CO2.
W ten sposób obydwa atomy węgla
wniesione do cyklu przez grupę acety-
lową zostały utlenione do CO2.
Powstaje druga cząsteczka NADH +
H+.
Produktem reakcji jest bursztyny-
lo~S-CoA (sukcynylo~S-CoA) - tioes-
ter bogaty w energię, podobnie jak
acetylo~S-CoA
(wzór 5.15).
Rozpad bursztynylo~S-CoA
Tiokinaza bursztyniano-
wa (syntetaza bursztyny-
lo~S-CoA) rozkłada wią-
zanie tioestrowe w bursz-
tynylo~S-CoA.
Reakcja ta jest sprzężona
z fosforylacją ADP do ATP
lub GDP do GTP
(wzór
5.16).
W wątrobie funkcjonuje
syntetaza bursztynylo~S-
CoA, fosforylująca GDP,
natomiast w mózgu i sercu dominuje enzym fosforylujący ADP.
Energia zawarta w GTP jest równoważna energii zawartej w ATP.
Te dwa nukleotydy mogą przechodzić w siebie nawzajem pod działaniem kinazy difosfonukle-
ozydowej.
GTP + ADP ↔GDP +ATP
Powstawanie GTP lub ATP z bursztynylo~S-CoA jest kolejnym przykładem fosforylacji sub-
stratowej, w której produkcja (GTP lub ATP) jest skojarzona z konwersją bogatego w energię
substratu (w tym przypadku bursztynylo~S-CoA) w ubogi w energię produkt (w tym przypad-
ku bursztynian).
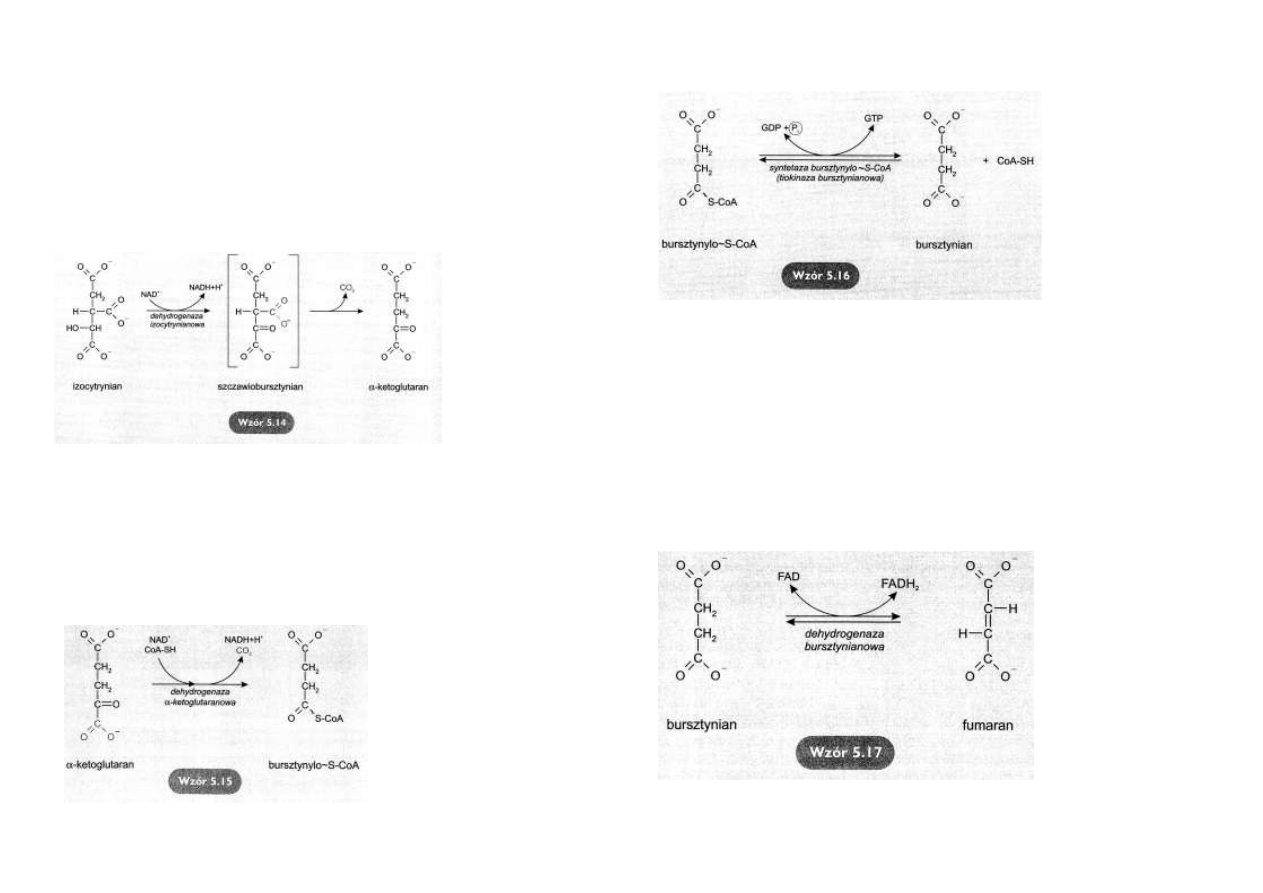
Utlenianie bursztynianu
Bursztynian jest utleniany
przez
dehydrogenazę
bursztynianową do fuma-
ranu.
Akceptorem dwóch atomów
wodoru (2H+ +2e-) jest
FAD,
przechodzący
w
FADH2
(wzór 5.17).
Utlenianie zredukowanego
FADH2
przez
łańcuch
oddechowy pomija miejsce
pierwszej fosforylacji oksy-
dacyjnej.
Para atomów wodoru jest przekazywana bezpośrednio na koenzym Q.
Efekt energetyczny tego procesu to jedynie 2 cząsteczki ATP w przeliczeniu na parę atomów
wodoru.
Hydratacja fumaranu
Fumaran ulega hydratacji do jabłczanu w odwracalnej reakcji katalizowanej przez fumarazę,
zwaną też hydratazą fumaranową.
Enzym ten wiąże cząsteczkę wody z fumaranem, przekształcając go w L-jabłczan.
Zanika podwójne wiązanie między atomami węgla
(wzór 5. 18).
Utlenianie jabłczanu
Jabłczan jest utleniany przez dehydrogenazę jabłczanową do szczawiooctanu.
Reakcja ta jest źródłem trzeciej cząsteczki NADH + H+ powstającej w cyklu Krebsa
(wzór
5.19).
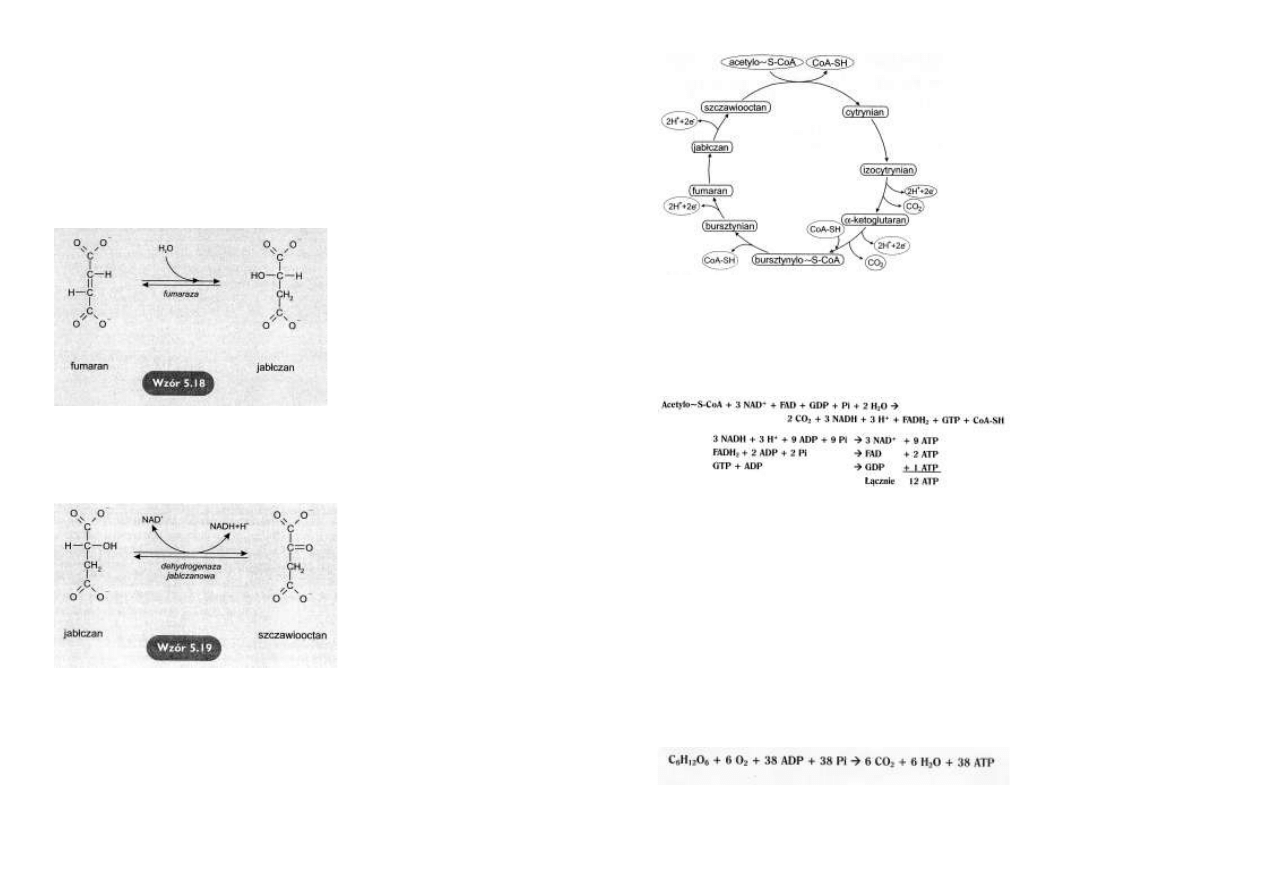
Bilans cyklu kwasów trikarboksylowych
Do cyklu Krebsa wchodzą dwa atomy węgla w postaci reszty acetylowej (octanowej), a opusz-
czają ten cykl w postaci dwóch cząsteczek CO2.
Podczas jednego obrotu cyklu cztery pary elektronów są przenoszone z substratów na akcep-
tory:
trzy pary na NAD+, który redukuje się do NADH,
jedna para na FAD, który redukuje się do FADH2 (ryc. 5.9).
Rys. 5.9. Schemat cyklu kwasów trikar-
boksylowych.
Utlenianie jednej cząsteczki NADH przez
łańcuch oddechowy prowadzi do powsta-
nia trzech cząsteczek ATP.
Podczas jednego obrotu cyklu Krebsa
powstają 3 cząsteczki NADH.
Ich utlenianie dostarcza więc 9 (3 x 3)
cząsteczek ATP.
Utlenianie jednej cząsteczki FADH2 wy-
twarza 2 cząsteczki ATP.
Łącznie, w wyniku procesów oksydore-
dukcyjnych, powstaje jedenaście cząsteczek ATP.
Dodatkowo powstaje jedna cząsteczka GTP lub ATP na drodze fosforylacji substratowej.
Utlenienie jednej reszty acetylowej w cyklu kwasów trikarboksylowych dostarcza więc 12
cząsteczek ATP.
Bilans cyklu Krebsa można przedstawić za pomocą następujących równań:
Ponieważ z cząsteczki glukozy powstają dwie cząsteczki acetylo~S-CoA, łączny zysk energe-
tyczny, w przeliczeniu na cząsteczkę glukozy, wynosi (2 x 12) 24 cząsteczki ATP.
BILANS ENERGETYCZNY UTLENIANIA GLUKOZY do CO2 i H2O
Przemiana glukozy drogą glikolizy, wraz z oksydacyjną dekarboksylacją powstającego z niej
pirogronianu, jest głównym źródłem metabolicznym acetylo~S-CoA.
Reszty acetylowe utleniają się w cyklu kwasów trikarboksylowych do CO2 i H2O.
Z podsumowania efektów energetycznych glikolizy tlenowej (8 ATP), oksydacyjnej dekarboksy-
lacji dwóch cząsteczek pirogronianu (2 x 3 ATP = 6 ATP) oraz utleniania dwóch reszt acetylo-
wych w cyklu Krebsa (2 x 12 ATP = 24 ATP) wynika, że: 1 cząsteczka glukozy utleniając się do
6 cząsteczek CO2 i 6 cząsteczek H2O, dostarcza 38 cząsteczek ATP.
Można to zapisać następującym równaniem sumarycznym:
Glukoneogeneza
Niektóre narządy, jak mózg, rdzeń nerki, soczewka, rogówka, jądro, pracujący mięsień, a
także krwinki czerwone wymagają stałego dopływu glukozy jako substratu energetycznego.
W sytuacji, gdy stężenie glukozy we krwi maleje, następuje uruchomienie glikogenolizy (roz-
pad glikogenu wątrobowego), która dostarcza glukozy do krwi, a poprzez krew do innych
tkanek.
Zapas glikogenu wątrobowego może zaspokoić potrzeby energetyczne wymienionych narzą-
dów oraz krwinek czerwonych przez 10-18 godzin.
Wyczerpanie tego zapasu uruchamia biosyntezę glukozy z substratów nie będących cukrami,
jak mleczan, pirogronian, glicerol oraz szkielety węglowodorowe niektórych aminokwasów.
Proces ten nosi nazwę glukoneogenezy.
Glukoneogeneza nie jest prostym odwróceniem glikolizy.
Ta bowiem charakteryzuje się nieodwracalnością trzech spośród dziesięciu reakcji.
Miejscem glukoneogenezy jest przede wszystkim wątroba, gdzie powstaje około 90% glukozy i
w mniejszym stopniu kora nerki, która syntetyzuje około 10% glukozy.
Tylko w tych narządach występują enzymy potrzebne w procesie glukoneogenezy.
Mięśnie są wielkim „konsumentem”, a nie producentem” glukozy.
Zachodzi w nich glikoliza, nie zachodzi glukoneogeneza.
Zawierają enzymy glikolizy, nie zawierają enzymów glukoneogenezy.
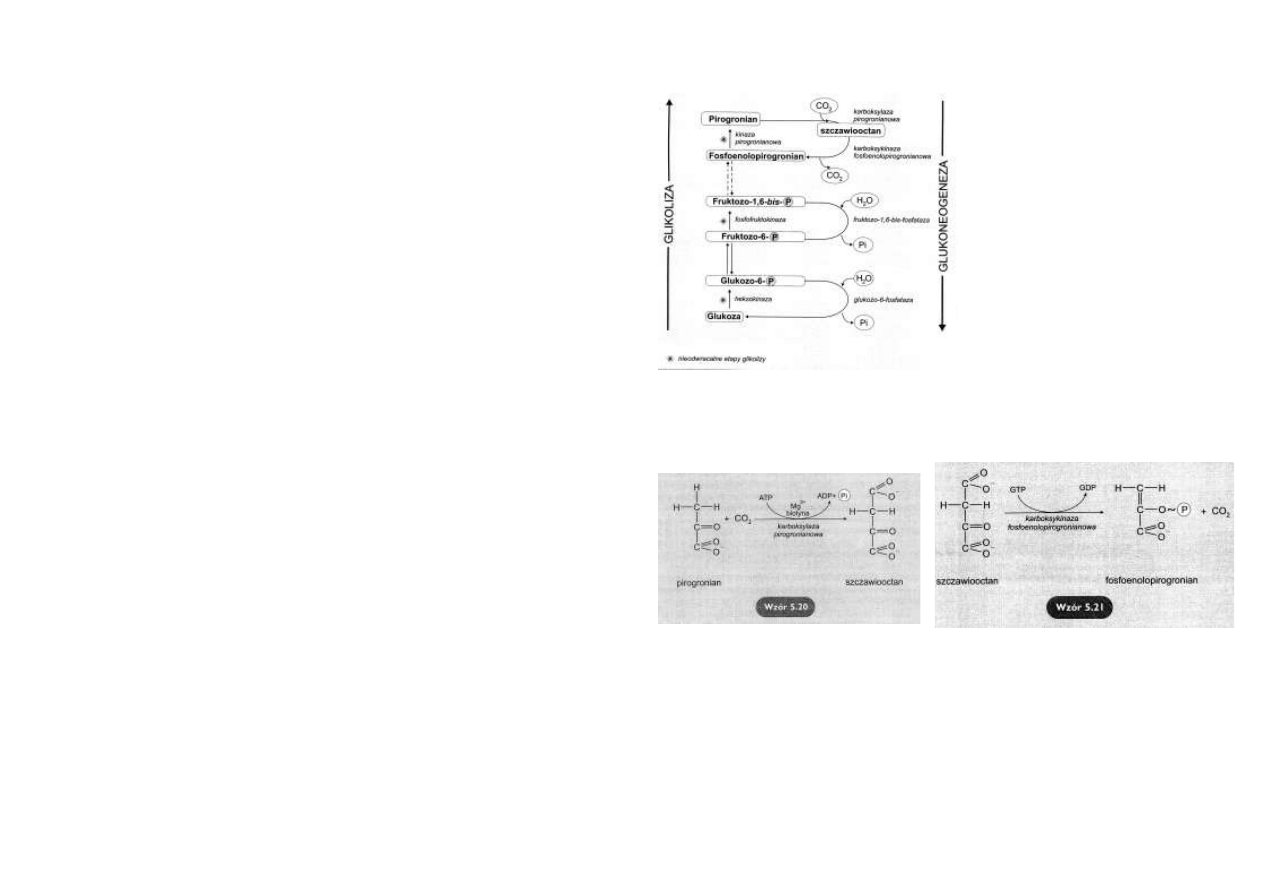
Reakcje glukoneogenezy
Siedem spośród dziesięciu etapów glikolizy to reakcje odwracalne.
W procesie glukoneogenezy przebiegają one w kierunku odwrotnym niż w glikolizie, przy
udziale tych samych enzymów.
Nieodwracalne są natomiast wszystkie reakcje katalizowane przez kinazy: heksokinazę lub
glukokinazę, fosfofruktokinazę i kinazę pirogronianową.
Reakcje katalizowane przez te enzymy w procesie glikolizy nie zachodzą w procesie glukoneo-
genezy.
Ich przebieg jest katalizowany przez inne enzymy.
Przemiana pirogronianu w fosfoenolopirogronian jest reakcją dwuetapową, zachodzącą przy
udziale dwóch enzymów: karboksylazy pirogronianowej i karboksykinazy fosfoenolopirogro-
nianowej oraz biotyny ATP i GTP.
Rozpad fruktozo-1,6-bis-fosforanu do fruktozo-6-fosforanu i Pi jest katalizowany przez fruk-
tozo-l,6-bis-fosfatazę, a hydroliza glukozo-6-fosforanu do glukozy i Pi jest katalizowana przez
glukozo-6-fosfatazę.
Ogólny, uproszczony schemat procesu glikolizy i glukoneogenezy przedstawia ryc. 5.10.
Zaznaczono gwiazdkami nieodwracalne etapy glikolizy oraz wymieniono enzymy, które funk-
cjonują wyłącznie w glikolizie lub wy-
łącznie w glukoneogenezie.
Rys. 5.10. Schemat przebiegu glikolizy i
glukoneogenezy.
Wskazano
enzymy
katalizujace nieodwracalne etapy glikoli-
zy oraz enzymy glukoneogenezy pozwa-
lające na ominięcie nieodwracalnych
reakcji.
Karboksylacja pirogronianu
Reakcja katalizowana w glikolizie przez
kinazę pirogronianową jest nieodwra-
calna, dlatego pirogronian nie może być
przekształcany w fosfoenolopirogronian
przez ten sam enzym.
Przemiana pirogronianu w fosfoenolopi-
rogronian zachodzi innym szlakiem, w
dwóch etapach.
Najpierw pirogronian (w mitochondriach) jest karboksylowany przez karboksylazę pirogronia-
nową do szczawiooctanu (
wzór 5.20),
a ten jest transportowany do cytosolu, gdzie ulega
przemianie do fosfoenolopirogronianu (
wzór 5.21).
Karboksylaza pirogronianowa zawiera biotynę, związaną kowalencyjnie poprzez grupę ε-
aminową reszty lizylowej białka enzymatycznego.
Biotyna jest przenośnikiem CO2.
Koenzym ten po związaniu CO2 staje się karboksybiotyną.
Powstanie kompleksu: apoenzym-biotyna-CO2 zachodzi kosztem energii pochodzącej z rozpa-
du cząsteczki ATP.
Energia zawarta w tym kompleksie zostaje wykorzystana w reakcji wiązania CO2 z pirogro-
nianem, prowadzącej do powstania szczawiooctanu.
Szczawiooctan, powstały w mitochondrium, musi wniknąć do cytosolu, gdzie są zlokalizowa-
ne pozostałe enzymy glukoneogenezy.
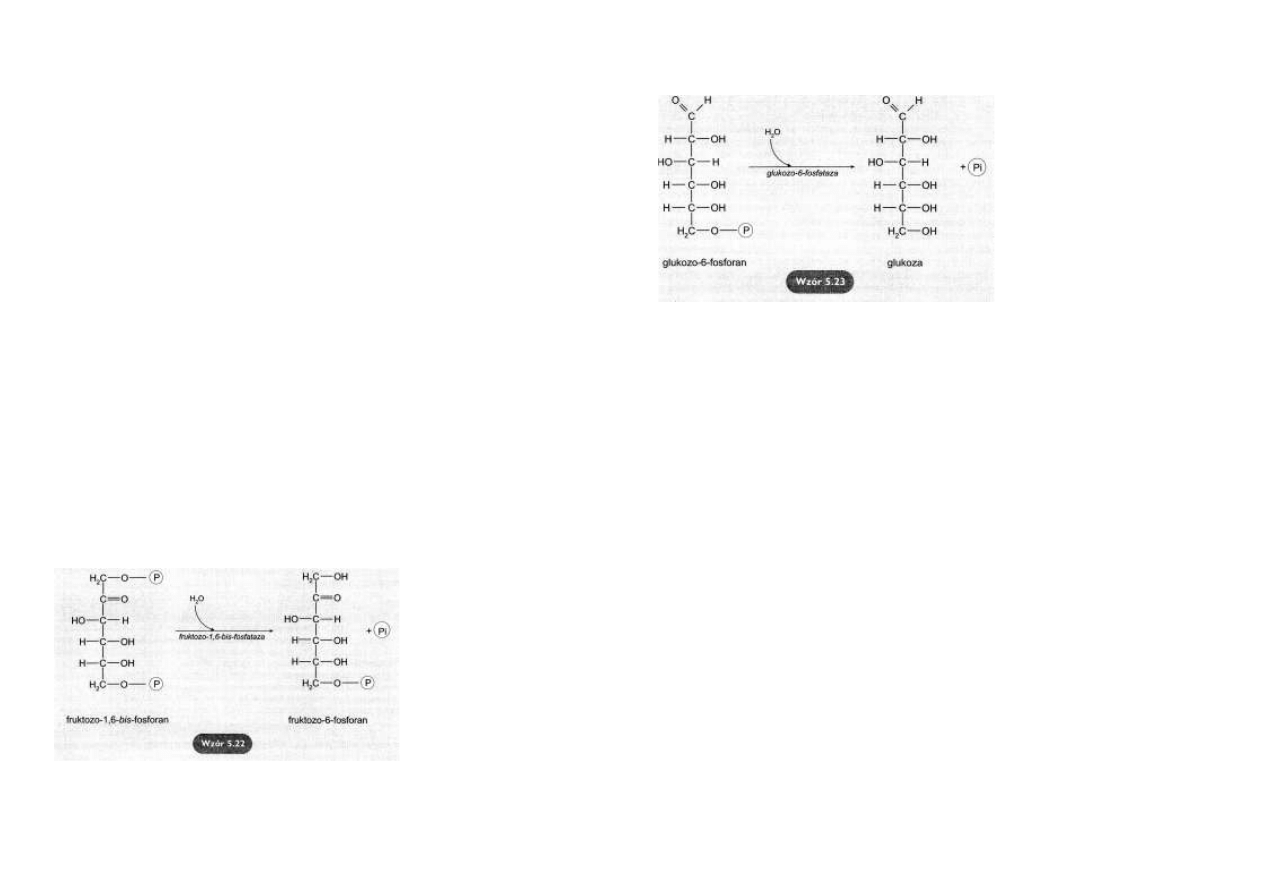
Jednakże wewnętrzna błona mitochondrialna jest nieprzepuszczalna dla szczawiooctanu.
Musi on przekształcić się w metabolit, który przenika przez wspomnianą błonę.
Może być zredukowany do jabłczanu, może wiązać resztę acetylową z acetylo~S-CoA i prze-
kształcać się w cytrynian, albo drogą transaminacji przekształcać się w asparaginian.
Produkty tych reakcji przenikają do cytosolu, gdzie przekształcają się na powrót do szczawio-
octanu.
Dekarboksylacja szczawiooctanu
Szczawiooctan jest dekarboksylowany i fosforylowany w cytosolu przez karboksykinazę fos-
foenolopirogronianową.
Reakcja ta zachodzi kosztem energii uwalnianej przez hydrolizę GTP.
Produktem reakcji staje się fosfoenolopirogronian (wzór 5.21).
Reakcje odwracalne
Dalsza przemiana fosfoenolopirogronianu poprzez fosfotriozy aż do fruktozo-l,6-bis-fosforanu
zachodzi poprzez odwrócenie reakcji glikolizy, z wykorzystaniem enzymów glikolizy.
Należy zwrócić uwagę na fakt, iż przemiana 3-fosfoglicerynianu w 1,3-bis-fosfoglicerynian
zużywa kolejną cząsteczkę ATP, a redukcja 1,3-bis-fosfoglicerynianu do aldehydu 3-
fosfoglicerynowego zużywa cząsteczkę NADH + H+.
Ponieważ do powstania 1 cząsteczki glukozy potrzeba dwóch cząsteczek fosfotrioz, łączny
nakład energetyczny komórki na tym etapie wynosi 2 cząsteczki ATP oraz 2 cząsteczki NADH
+ 2H+.
Defosforylacja fruktozo-1,6-bis-fosforanu
Hydrolityczne odłączenie fosfo-
ranu w pozycji C1 fruktozo-1,6-
bis-fosforanu prowadzi do po-
wstania fruktozo-6-fosforanu.
Reakcja jest katalizowana przez
fruktozo-1,6-bis-fosfatazę
(wzór
5.22).
Enzym ten pozwala na ominię-
cie nieodwracalnego etapu gli-
kolizy, katalizowanego przez
fosfofruktokinazę.
Defosforylacja glukozo-6-fosforanu
Hydroliza glukozo-6-fosforanu przez glukozo-6-fosfatazę przekształca ten ester w wolną glu-
kozę.
Pozwala na ominięcie nieodwra-
calnego etapu glikolizy, katalizo-
wanego przez heksokinazę lub
glukokinazę
(wzór 5.23).
Wolna glukoza, w odróżnieniu od
jej estrów fosforanowych, może
opuszczać komórkę, przenikać do
krwi i tą drogą docierać do odle-
głych narządów.
Bilans glukoneogenezy
Powstanie cząsteczki glukozy z 2 cząsteczek pirogronianu wiąże się z rozpadem sześciu wią-
zań bogatych w energię, po 3 na każdą cząsteczkę pirogronianu.
Rozpadają się 4 cząsteczki ATP i 2 cząsteczki GTP.
Zużywają się 2 cząsteczki NADH + 2H+.
Przebieg procesu glukoneogenezy można zapisać za pomocą następującego równania:
2 pirogronian + 4 ATP + 2 GTP + 2 NADH + 2 H+
→ glukoza + 4 ADP + 2 GDP + 2NAD+
Szlak pentozofosforanowy
Szlak pentozofosforanowy jest cytosolowym mechanizmem przetwarzania glukozy, niezwiąza-
nym bezpośrednio z potrzebami energetycznymi komórki.
Jego głównym celem jest dostarczenie komórce zredukowanej postaci fosforanu dinukleotydu
nikotynoamidoadeninowego: NADPH+H+ oraz rybozo-5-fosforanu do niżej omówionych celów
metabolicznych.
Szlak pentozofosforanowy składa się z dwóch faz.
W pierwszej, zwanej fazą oksydacyjną, następuje dwukrotne utlenienie glukozo-6-fosforanu z
udziałem NADP+.
Produktami tej fazy jest rybulozo-5-fosforan, CO2 i dwie cząsteczki NADPH.
W drugim etapie, zwanym fazą nieoksydacyjną, rybulozo-5-fosforan przekształca się w rybo-
zo-5-fosforan (substrat do biosyntezy nukleotydów) lub ulega wieloetapowym przekształce-
niom w metabolity glikolizy i glukoneogenezy.
W odróżnieniu od glikolizy lub cyklu kwasów trikarboksylowych, w których kierunek prze-
mian jest ściśle określony w szlaku pentozofosforanowym przekształcanie cukrów może na-
stępować w różnych kierunkach.
Szybkość i kierunek reakcji w danym czasie są określone przez dostępność substratu lub
zapotrzebowanie na określone metabolity tego szlaku.
Szlak pentozofosforanowy funkcjonuje w cytosolu.
Nie zależy od łańcucha oddechowego.
Poza pierwszą reakcją, w której zachodzi fosforylacja glukozy w pozycji C6, nie zużywa ani nie
produkuje ATP.
Funkcjonuje niezależnie od mitochondriów, jest natomiast sprzężony z glikolizą i glukoneoge-
nezą.
Szlak ten dostarcza większości NADPH, który pełni funkcję reduktora w przebiegu różnych
biosyntez.
Jest to szczególnie istotne w wątrobie i gruczołach mlecznych, gdzie zachodzi intensywna
biosynteza kwasów tłuszczowych i cholesterolu.
Oraz w korze nadnerczy, która jest miejscem biosyntezy wielu hormonów steroidowych.
Ponadto szlak pentozofosforanowy dostarcza rybozo-5-fosforanu, niezbędnego do biosyntezy
nukleotydów wchodzących w skład kwasów nukleinowych, lub funkcjonujących jako przeno-
śniki energii, oraz koenzymów, jak: CoA-SH, NAD+, NADP+ i FAD.
Cząsteczka glukozy włączająca się do szlaku pentozofosforanowego podlega fosforylacji w
pozycji C6.
Powstaje glukozo 6-fosforan.
Przebieg tej reakcji jest identyczny jak w przypadku fosforylacji glukozy włączanej do glikoli-
zy.
Faza oksydacyjna
Oksydacyjna część szlaku pentozofosforanowego składa się z trzech reakcji, które prowadzą
do przekształcenia glukozo-6-fosforanu w rybulozo-5-fosforan.
Procesowi temu towarzyszy odłączenie CO2 i redukcja 2 cząsteczek NADP+ z powstaniem 2
cząsteczek NADPH+2H+.
Każda spośród sześciu cząsteczek glukozy włączonych do szlaku pentozofosforanowego w
fazie oksydacyjnej ulega jednako-
wym
przekształceniom
(czym
różni się od fazy nieoksydacyjnej).
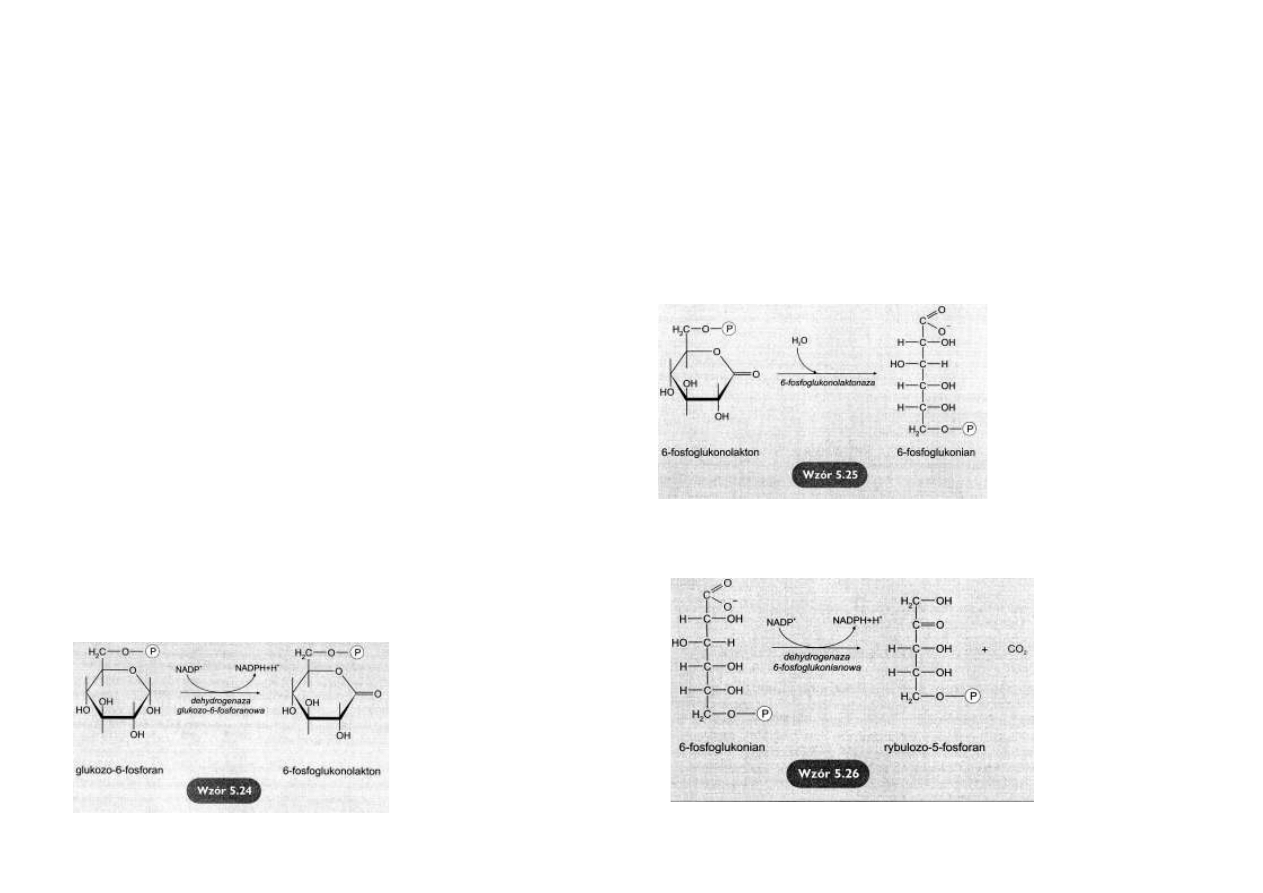
Utlenianie glukozo-6-fosforanu
Dehydrogenaza
glukozo-6-
fosforanowa katalizuje nieodwra-
calną reakcję utlenienia glukozo-
6-fosforanu
do
6-
fosfoglukonolaktonu (
wzór 5.24).
Akceptorem
elektronów
jest
NADP+ przechodzący w NADPH +
H+.
Etap ten jest regulowany przez stosunek NADPH/NADP+.
Jeżeli stosunek NADPH/NADP+ jest wysoki, aktywność enzymu jest silnie hamowana, a szlak
pentozofosforanowy zostaje spowolniony.
Jednak w miarę wzrostu zapotrzebowania na NADPH następuje jego zużycie.
Maleje ilość NADPH rośnie ilość NADP+.
Stosunek NADPH/NADP+ maleje, aktywność dehydrogenazy glukozo-6-fosforanowej rośnie, a
szlak pentozofosforanowy zostaje przyspieszony, aż do odtworzenia wyjściowego stosunku
NADPH/NADP+.
Hydroliza 6-fosfoglukonolaktonu
Produkt poprzedniej reakcji – 6-
fosfoglukonolakton - jest związ-
kiem nietrwałym.
Ulega hydrolizie nawet bez udziału
enzymu, jednak jego rozpad jest
katalizowany
przez
6-
fosfoglukonolaktonazę
(wzór 5.25).
Reakcja jest nieodwracalna.
Nie podlega regulacji.
Jej
produktem
jest
6-
fosfoglukonian.
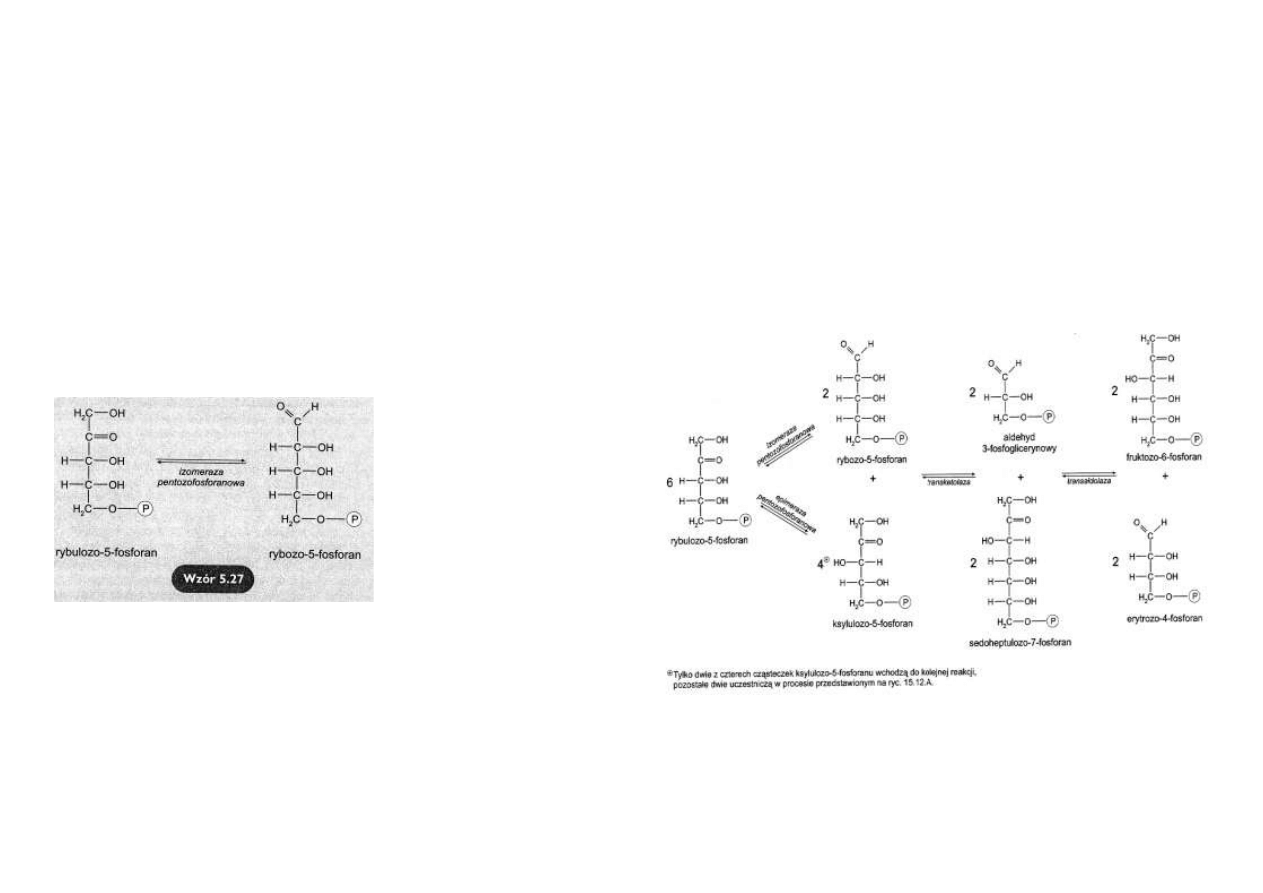
Utlenianie 6-fosfoglukonianu
Kolejna reakcja oksydacyjnej dekarboksylacji 6-fosfoglukonianu jest katalizowana przez de-
hydrogenazę
6-
fosfoglukonianową, enzym
współdziałający z jonem
Mg2+.
Jej produktami są: rybulo-
zo-5-fosforan, CO2 (z C1
glukozy) i druga cząsteczka
NADPH
(wzór 5.26).
Rybulozo-5-fosforan staje
się substratem przekształ-
canym w fazie nieoksyda-
cyjnej.
Faza nieoksydacyjna
W odróżnieniu od fazy oksydacyjnej, kierunek przemian poszczególnych cząsteczek rybulozo-
5-fosforanu w fazie nieoksydacyjnej jest zróżnicowany.
W zależności od potrzeb metabolicznych komórki, rybulozo-5-fosforan zamienia się w rybozo-
5-fosforan (potrzebny do biosyntezy nukleotydów) albo przekształca się w metabolity pośred-
nie glikolizy lub glukoneogenezy.
Tak więc szlak pentozofosforanowy nie jest procesem wyodrębnionym, lecz zintegrowanym z
glikolizą i glukoneogenezą.
Enzymy uczestniczące w fazie nieoksydacyjnej nie podlegają kontroli regulacyjnej.
Przebieg reakcji jest regulowany głównie przez dostępność substratów.
Jedynym koenzymem potrzebnym do funkcjonowania tej fazy szlaku pentozofosforanowego
jest pirofosforan tiaminy pełniący funkcję koenzymu transketolazy.
Powstawanie rybozo-5-fosforanu
Zasadniczym kierunkiem przemiany rybulozo-5-fosforanu w fazie nieoksydacyjnej jest jego
izomeryzacja do rybozo-5-fosforanu katalizowana przez izomerazę pentozofosforanową
(wzór
5.27).
Tą drogą powstaje substrat do bio-
syntezy nukleotydów.
Ten kierunek przemiany rybulozo-5-
fosforanu dominuje w komórkach
dzielących się, potrzebujących sub-
stratów nukleotydowych do syntezy
DNA.
Powstawanie pośredników glikoli-
zy i glukoneogenezy
Wiele komórek, w których intensyw-
nie zachodzą procesy biosyntezy z udziałem reakcji redukcji, wykazuje większe zapotrzebo-
wanie na NADPH niż na rybozo-5-fosforan.
Brak zapotrzebowania na rybozo-5-fosforan sprawia, iż cząsteczki rybulozo-5-fosforanu prze-
kształcają się w innym kierunku: do aldehydu 3-fosfoglicerynowego i fruktozo-6-fosforanu,
które są metabolitami pośrednimi, zarówno glikolizy jak i glukoneogenezy.
Mogą przekształcać się w pirogronian lub odtwarzać glukozo-6 -fosforan.
Przebieg tego procesu najłatwiej jest przeanalizować na przykładzie 6 cząsteczek rybulozo-5-
fosforanu powstałych w fazie oksydacyjnej.
Dwie spośród sześciu cząsteczek rybulozo-5-fosforanu izomeryzują do dwóch cząsteczek
rybozo-5-fosforanu.
Reakcje katalizuje izomeraza pentozofosforanowa.
Cztery pozostałe izomeryzują do 4 cząsteczek ksylulozo-5-fosforanu.
Reakcje katalizuje epimeraza pentozofosforanowa.
Pod działaniem transketolazy (enzymu przenoszącego fragmenty dwuwęglowe) następuje
przeniesienie fragmentów dwuwęglowych z 2 cząsteczek ksylulozo-5-fosforanu na 2 cząsteczki
rybozo-5-fosforanu.
Powstają 2 cząsteczki aldehydu 3-fosfoglicerynowego i 2 cząsteczki sedoheptulozo-7-
fosforanu.
Pozostałe dwie cząsteczki ksylulozo-5-fosforanu nie uczestniczą w reakcji.
W kolejnym etapie nowo powstałe 2 cząsteczki aldehydu 3-fosfoglicerynowego i 2 cząsteczki
sedoheptulozo-7-fosforanu reagują ze sobą pod działaniem transaldolazy, która przenosi 2
fragmenty trójwęglowe z dwóch cząsteczek sedoheptulozo-7-fosforanu na 2 cząsteczki aldehy-
du 3-fosfoglicerynowego.
Powstają 2 cząsteczki fruktozo-6-fosforanu i odłączają się 2 cząsteczki erytrozo-4-fosforanu
(ryc. 5.11).
Rys. 5.11. Szlak pentozofosforanowy. Przekształcanie rybulozo-5-fosforanu w pośredniki gliko-
lizy i glukoneogenezy: aldehyd 3-fosfo-glicerynowy i fruktozo-6-fosforan.
Do kolejnego etapu włączają się 2 pozostałe cząsteczki ksylulozo-5-fosforanu, które w reakcji
z 2 nowo powstałymi cząsteczkami erytrozo-4-fosforanu, katalizowanej ponownie przez trans-
ketolazę, tworzą 2 cząsteczki fruktozo-6-fosforanu oraz 2 cząsteczki aldehydu 3-
fosfoglicerynowego (ryc. 5.12).
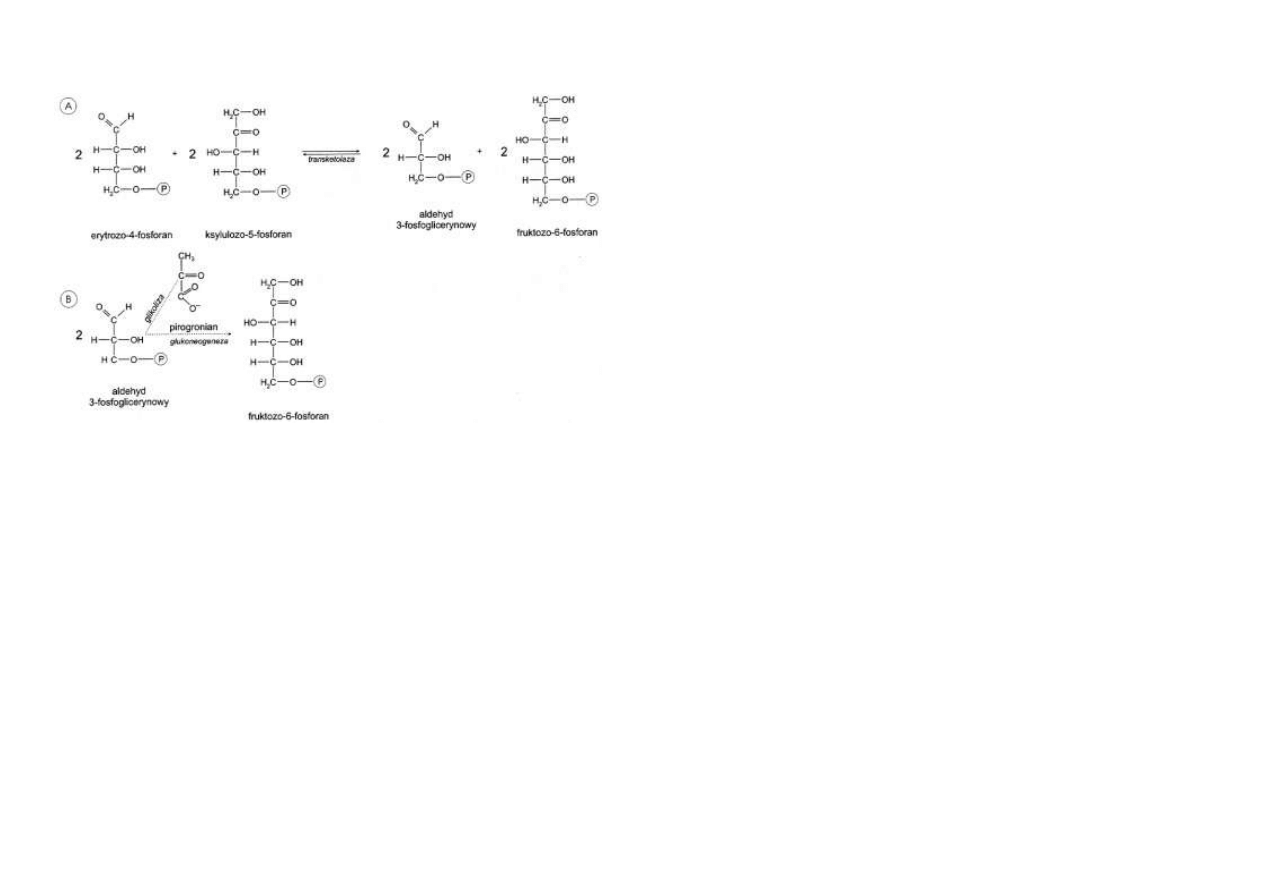
Produkty te mogą być dalej przetwarzane drogą glikolizy do pirogronianu lub drogą glukoneo-
genezy do glukozy.
Rys. 5.12. Szlak pentozofosforanowy – ciąg dalszy. Przekształcanie erytrozo-4-fosforanu i
ksylulozo-5-fosforanu w pośredniki glikolizy i glukoneogenezy: aldehyd 3-fosfoglicerynowy i
fruktozo-6-fosforan.
Jeżeli zostaną włączone do glikolizy podzielą losy pirogronianu, który poprzez oksydacyjną
dekarboksylację i cykl kwasów trikarboksylowych może utlenić się do CO2 i H2O.
Tak więc cząsteczka glukozy poprzez szlak pentozofosforanowy może być całkowicie degrado-
wana do CO2 i H2O.
Jeżeli zostaną włączone do glukoneogenezy, przekształcą się w glukozę.
W procesie tym 2 cząsteczki aldehydu 3-fosfoglicerynowego przetwarzają się w 1 cząsteczkę
glukozy, a 4 cząsteczki fruktozo-6-fosforanu w 4 cząsteczki glukozy.
Tą drogą odtwarza się 5 spośród 6 cząsteczek glukozo-6-fosforanu, które włączyły się do
cyklu pentozowego.
Bilans szlaku pentozofosforanowego
Oksydacyjna faza szlaku pentozofosforanowego powoduje „skrócenie” każdej heksozy o jeden
atom węgla, który odłącza się w postaci CO2.
Heksoza ulega przemianie w pentozę.
Bilans szlaku pentozofosforanowego najłatwiej przedstawić na przykładzie 6 cząsteczek glu-
kozy przekształcanych w tym szlaku.
Jeżeli do szlaku wejdzie 6 cząsteczek glukozy, odtworzy się 5 cząsteczek glukozy i uwolni się
6 cząsteczek CO2.
Efekt sumaryczny jest taki, jakby jedna spośród 6 cząsteczek glukozy utleniała się do CO2 i
H2O, a pozostałe uległy odtworzeniu.
Wielokrotny obrót szlaku pentozofosforanowego prowadzi do stopniowego zużywania glukozy.
Bilans tego procesu można przedstawić w postaci następującego równania:
6 glukozo-6-fosforan + 12 NADP+ + 7 H2O → 5 glukozo-6-fosforan + 6 CO2 + 12 NADPH + 12
H+ + Pi
Sześć spośród siedmiu cząsteczek H2O, wymienionych w powyższym równaniu, zużywa się w
reakcjach hydrolizy sześciu cząsteczek 6-fosfoglukonolaktonu do 6-fosfoglukonianu, a jedna
w hydrolitycznym odłączaniu fosforanu z produktu przekształcenia tej cząsteczki glukozo-6-
fosforanu, która ulega zużyciu.
Efekt rozpadu jednej spośród sześciu cząsteczek glukozo-6-fosforanu w szlaku pentozofosfo-
ranowym ilustruje następujące równanie:
glukozo-6-fosforan + 12 NADP+ + 7 H2O → 6CO2 + 12 NADPH + 12 H+ + Pi
Powyższe rozważania nad bilansem szlaku pentozofosforanowego dotyczą tylko jednej z moż-
liwych sytuacji, kiedy rybulozo-5-fosforan przekształca się wyłącznie torem generującym
pośredniki glukoneogenezy.
Wykluczają one z bilansu tę część rybulozo-5-fosforanu, która po izomeryzacji do rybozo-5-
fosforanu wbudowała się do nukleotydów i tę część aldehydu 3-fosfoglicerynowego, która
włączyła się do glikolizy.
Ponieważ szlak pentozofosforanowy jest głównym generatorem NADPH, rozdział ten jest naj-
właściwszym miejscem na omówienie jego roli metabolicznej.
Funkcje nadph
NADP+ różni się od NAD+ jedynie obecnością dodatkowej reszty fosforanowej w pozycji 2’
adenylanowej części tego dinukleotydu.
Ta niewielka różnica w budowie pozwala NADP+ na interakcje ze swoistymi enzymami oksy-
dacyjno-redukcyjnymi.
Udział NADPH w procesach biosyntezy
NADPH może być uważany za cząsteczkę o wysokiej energii, podobnie jak NADH.
Jednak elektrony z NADPH są przeznaczone raczej do użytku w redukcyjnych etapach bio-
syntezy niż do przenoszenia na tlen, jak to występuje w przypadku NADH.
Tak więc, w przebiegu szlaku pentozofosforanowego część energii zawartej w glukozo-6-
fosforanie jest przenoszona na NADP+ z wytworzeniem NADPH, który może być użyty w reak-
cjach wymagających wysokiego potencjału przenoszenia elektronów. ● NADPH jest źródłem
elektronów w reakcjach redukcji zachodzących przede wszystkim w przebiegu biosyntezy
kwasów tłuszczowych.
Wyszukiwarka
Podobne podstrony:
Cukry proste i z-o¬one, Studia II rok, Studia, PD materialy donauki, PD materialy donauki
Cukry proste i złożone, Chemia
cukry proste i zlozone budowa i wlasciwosci. (2), Prace pisemne
cw 9 cukry proste
cw.9-cukry proste
cw 8 cukry proste
cukry proste sciaga
Cukry proste i złożone
cukry cz 2 st
cukry
CUKRY
cukry
Cukry
andmp proste zagadki dla dzieci
biochemia cukry instrukcja id 8 Nieznany (2)
więcej podobnych podstron