
Pierwotne
niedobory
odporności
Dr med.Małgorzata Bartkowiak-Emeryk
Katedra i Zakład Immunologii Klinicznej Uniwersytetu Medycznego w Lublinie

Pierwotne niedobory
odporności
• Grupa genetycznie uwarunkowanych zaburzeń
budowy, dojrzewania i różnicowania się oraz funkcji
narządów i komórek układu odpornościowego
Upośledzenie humoralnej lub komórkowej
odpowiedzi immunologicznej
• Zdefiniowanych – ponad 50 pierwotnych
niedoborów odporności u ludzi
• Częstość występowania: 1:10 000 urodzeń
wyższa niż dla mukowiscydozy i niedoczynności
tarczycy, porównywalna z fenyloketonurią

Pierwotne niedobory
odporności:
OBRAZ KLINICZNY
1. Zwiększona predyspozycja na zakażenia:
•
Wirusowe
•
Bakteryjne
•
Grzybicze
•
Pasożytnicze
•
Często bardzo ciężkie, zagrażające życiu
Przebieg i częstość infekcji zależna od:
–
Jednostki chorobowej
–
Rodzaju czynnika etiologicznego
–
Stopnia dysfunkcji układu odpornościowego
–
Poważne infekcje- głównie w zaburzeniach odporności
komórkowej
2. Wysoka częstość występowania rozrostów
nowotworowych i procesów autoimmunizacyjnych
zaburzenia związane z limfocytami T
zaburzenia odpowiedzi humoralnej
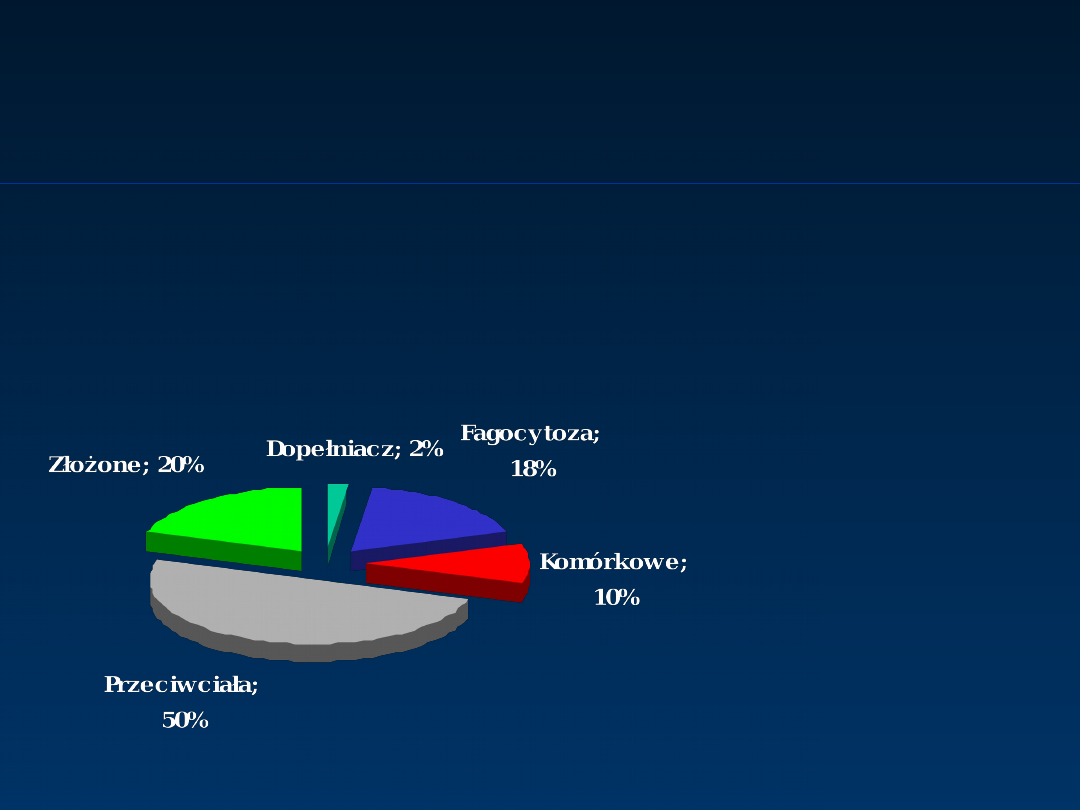
Pierwotne niedobory
odporności
• Włochy 1:77 000
• Japonia 1:200 000
• Szwajcaria 1:54
000
• Szwecja 1:55 000
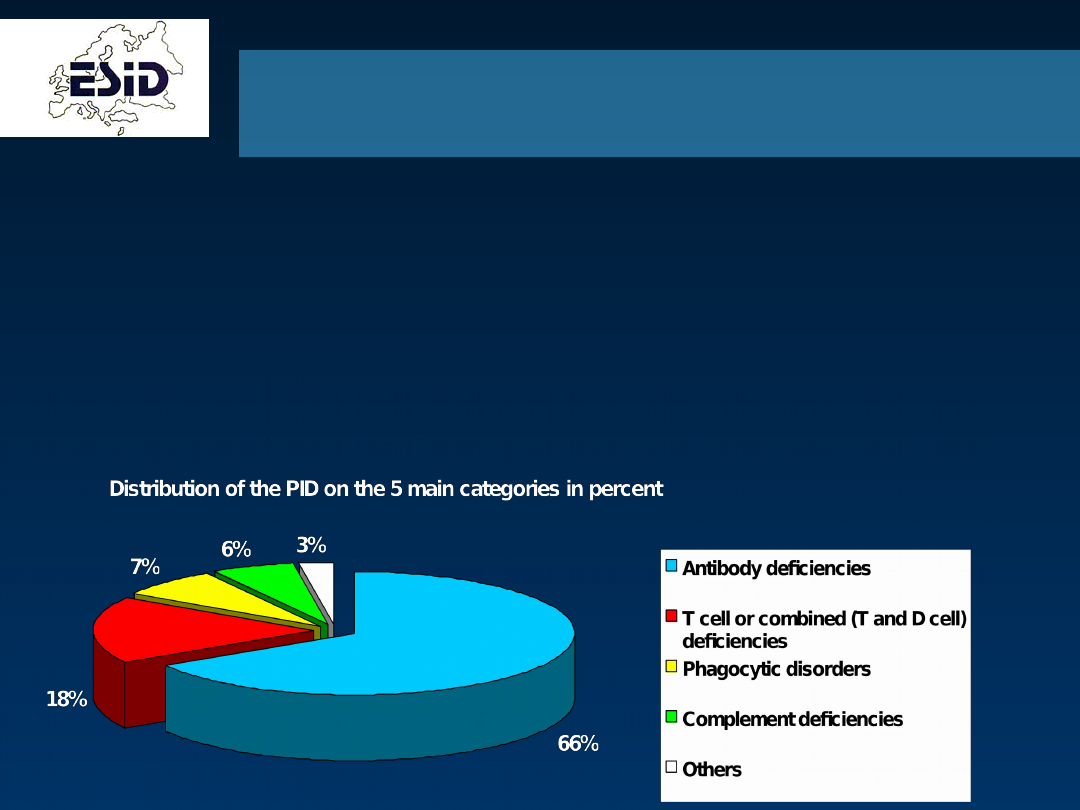
•participation of
27 European countries
27 European countries (incl. Israel
and Turkey)
•registration of
~10.700 cases
~10.700 cases (one-time
documentation)
•
from Jan 1994 until Dec 2003
from Jan 1994 until Dec 2003 (>450 Mill. inhabitants
in Europe)
All ESID
Countries
Austria
Belgium
Croatia
Czech-
Republic
Denmark
Estonia
Finland
France
Germany
Greece
Hungary
Iceland
Ireland
Israel
Italy
Netherland
s
Norway
Poland
Portugal
Russia
Serbia-
Montenegr
o Spain
Sweden
Switzerland
Turkey
United-
Kingdom
„
„
old“ ESID registry on FileMaker Pro
old“ ESID registry on FileMaker Pro
5.5
5.5
ESID database network:
background

ESID database network:
background
• prevalence of patients with PID
requiring
medical attention estimated to be
500 per 1
mln inhabitants !
• ~10.700 registered cases
and a European
population of >450 mln:
<5% registered
• how many may have been
diagnosed ?
• to know this, we would need to encourage
• all physicians taking care of patients
• with PID to register
100% of their patients !

Pierwotne niedobory
odporności:
obraz kliniczny
• Pierwsze objawy
– w okresie
noworodkowym lub niemowlęcym
– pod postacią nawracających zakażeń
– Upośledzenie rozwoju fizycznego dziecka
• Diagnostyka różnicowa
– Alergia
– Udział czynników środowiskowych
– Niedobory enzymatyczne (np. niedobór a1-
antytrypsyny
– Choroby metaboliczne (mukowiscydoza)
– Wady anatomiczne

Wskazania do diagnostyki
układu odpornościowego
1.
Osiem lub więcej zakażeń dróg oddechowych lub
uszu w ciągu roku
2. Dwa lub więcej zapalenia zatok w ciągu roku
3.
Trwająca dwa miesiące lub dłużej
antybiotykoterapia bez wyraźnej poprawy
4.
Dwa lub więcej zapalenia płuc w ciągu roku
5. Brak przyrostu masy ciała lub zahamowania
prawidłowego wzrostu u dziecka z nawracającymi
infekcjami
Wg The Jeffrey Modell Foundation Medical Advisory Board 1999
Wg Bernatowska E i wsp. Stand Med 1999

Wskazania do diagnostyki
układu odpornościowego
6.
Powtarzające się głębokie ropnie skórne lub
narządowe
7.
Przewlekające się grzybice jamy ustnej lub skóry
u dzieci >1 roku życia
8.
Konieczność długotrwałego stosowania
antybiotyków dożylnych dla opanowania
zakażenia
9.
Dwa lub więcej ciężkie zakażenia, takie jak:
mózgu, kości, skóry, posocznica
10. Wywiad rodzinny wskazujący na występowanie
pierwotnych niedoborów odporności
Wg The Jeffrey Modell Foundation Medical Advisory Board 1999
Wg Bernatowska E i wsp. Stand Med 1999

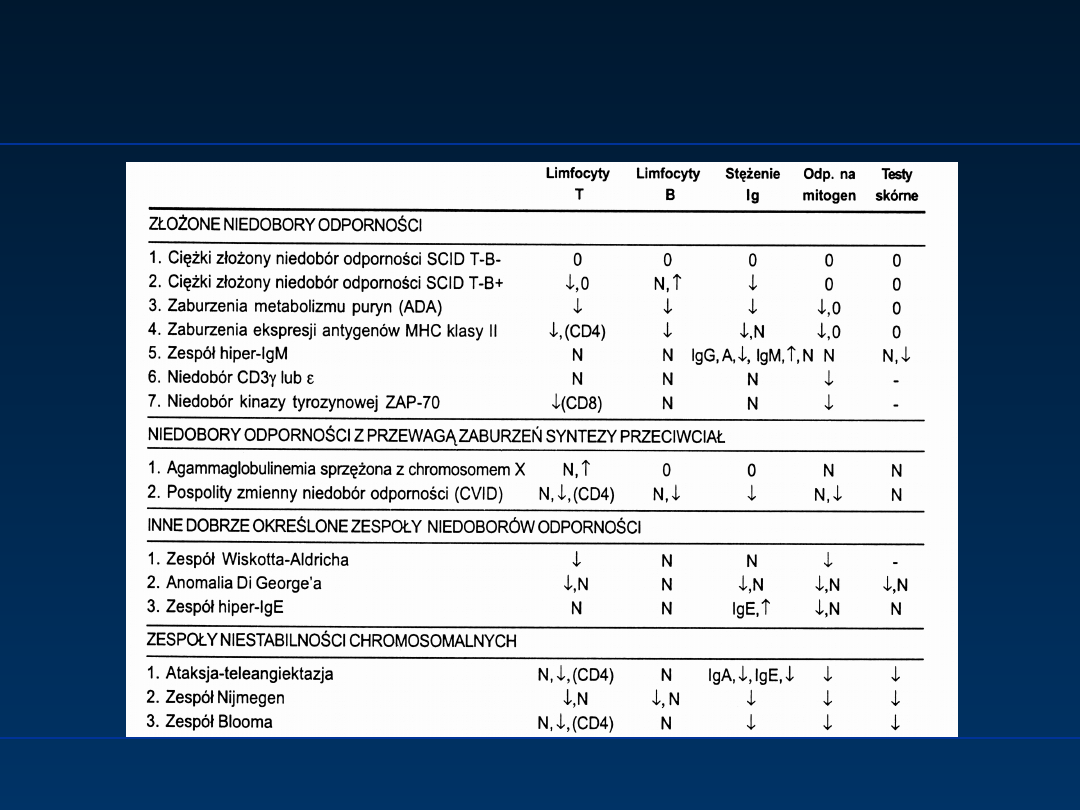
Pierwotne niedobory
odporności
podział wg WHO 1999
ZŁOŹONE NIEDOBORY ODPORNOŚCI

Złożone niedobory
odporności
Combined
immunodeficiencies
Grupa ciężko przebiegających zaburzeń
Wynik defektów ilościowych i/lub czynnościowych limfocytów T
i skojarzonych zaburzeń limfocytów B
Obraz kliniczny
– charakterystyczny
Pierwotny niedobór związany z limfocytami T
:
zaburzenia odpowiedzi
komórkowej
• Bardzo wczesny początek objawów:
zakażenie wewnątrzmaciczne, poważna infekcja, źle reagująca na
antybiotykoterapię, najczęściej dolnych dróg oddechowych
• Tendencja do nawracania, przewlekania się i uogólniania zakażeń
• Zakażenia oportunistyczne: Candida, Pneumocystis, wirus
cytomegalii
• Powikłania miejscowe i ogólne po szczepieniach żywymi
drobnoustrojami (BCG, polio, odra, świnka, różyczka)

Złożone niedobory
odporności
Combined immunodeficiencies
OBRAZ KLINICZNY
• Tendencja do przewlekłego utrzymywania się na błonach
śluzowych aft lub pleśniawek
• Ciężki przebieg z tendencją do uogólniania się zakażeń wirusami
opryszczki lub ospy wietrznej
• Większa podatność na zakażenia powodujące nie poddające się
leczeniu biegunki (rotawirus, Gardia lamblia, cryptosporidioza)
• Upośledzenie rozwoju somatycznego dzieci
• Cechy fenotypowe:
• Nieprawidłowości chrząstek w obrębie żeber i łopatki
niedobór deaminazy adenozyny
• Zmiany skórne np. złuszczająca erytrodermia Zespół
Omena

Złożone niedobory
odporności
Combined immunodeficiencies
BADANIA LABORATORYJNE
• Limfopenia < 10
3
/ml
• Brak lub znaczny niedobór odsetka limfocytów T
i/lub B, subpopulacji oraz zaburzenia
ekspresji powierzchniowych receptorów
• Proliferacja limfocytów pod wpływem specyficznych
antygenów i mitogenów – znacznie upośledzona
• Stężenia immunoglobulin w surowicy – niskie
• Odsetek i aktywność komórek NK - prawidłowa
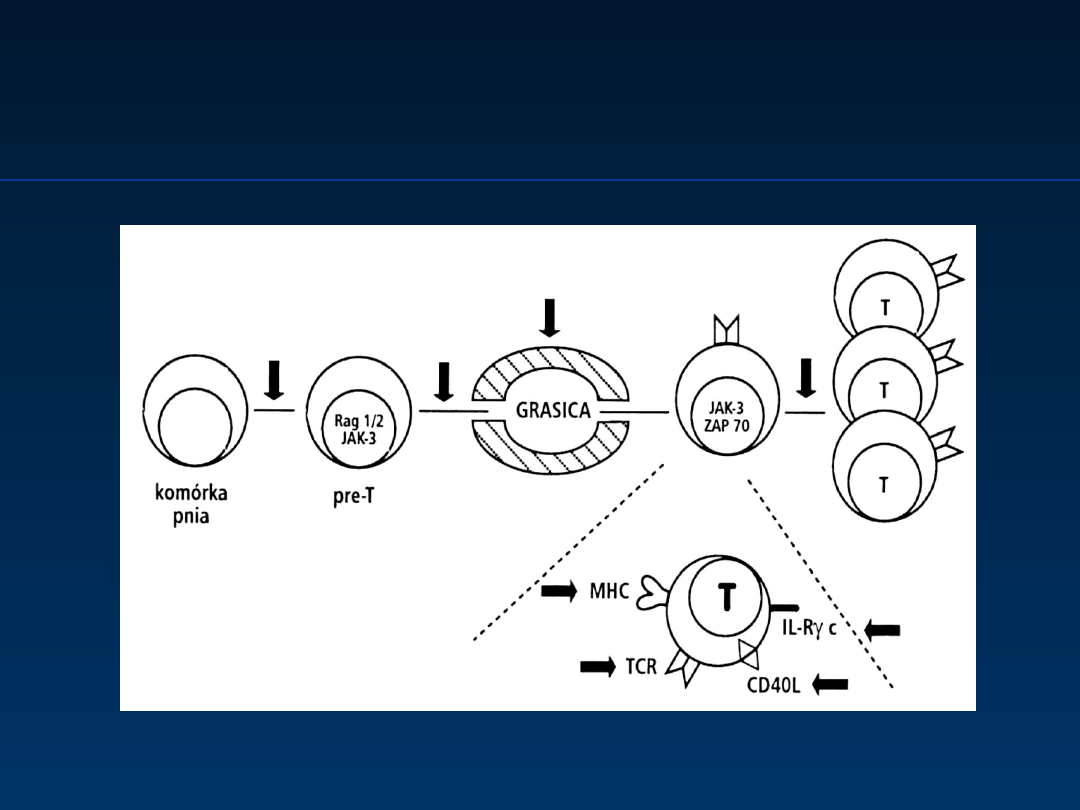
Złożone niedobory
odporności:
miejsca
potencjalnych defektów rozwoju limfocytów T
Wg Zeman K. Immunologia Kliniczna
2000
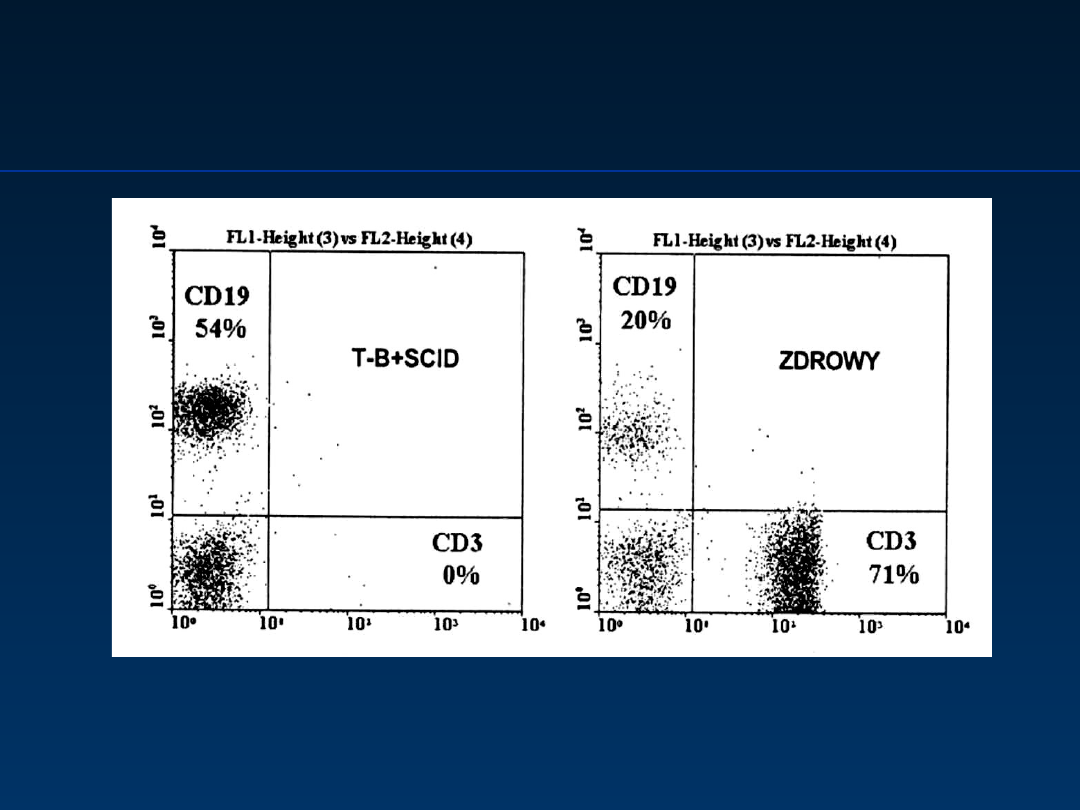
Ciężki złożony niedobór
odporności
Analiza odsetka limfocytów CD3 i CD19 krwi pępowinowej dziecka
z niedoborem IL-12
c
(T-B+SCID) zdiagnozowanego w okresie
prenatalnym
Wg Zeman K. Immunologia Kliniczna 2000

Ciężki złożony niedobór
odporności
Severe combined
immunodeficiency, SCID
T-B+SCID
• Duży niedobór lub brak limfocytów T przy prawidłowej lub
podwyższonej liczbie limfocytów B
• Dziedziczenie:
– W sprzężeniu z chromosomem X:
niedobór łańcucha dla
receptora IL-2
• eliminacja tymocytów w grasicy (u chorych zawiera
wyłącznie komórki nabłonkowe)
• W krwi obwodowej limfocyty B, ale upośledzone
wytwarzanie immunoglobulin i swoistych przeciwciał
(np. poszczepiennych)
– Autosomalnie recesywnie:
niedobór JAK
• Mutacje w obrębie genu kodującego kinazę tyrozynową
JAK-3 – niezbędna w procesie przekazywania sygnału z
powierzchni komórki do jej wnętrza

Ciężki złożony niedobór
odporności
Severe combined
immunodeficiency, SCID
T-B-SCID
• Brak limfocytów T i B
• Dysgenezja siateczki:
Komórka macierzysta limfocytów T i B w szpiku:
zahamowanie syntezy czynników wzrostowych lub
brak ekspresji receptorów obierających sygnał
• Zaburzenia dojrzewania i różnicowania
komórek macierzystych
• Zaburzenia rekombinacji genów
immunoglobulinowych i dla TCR
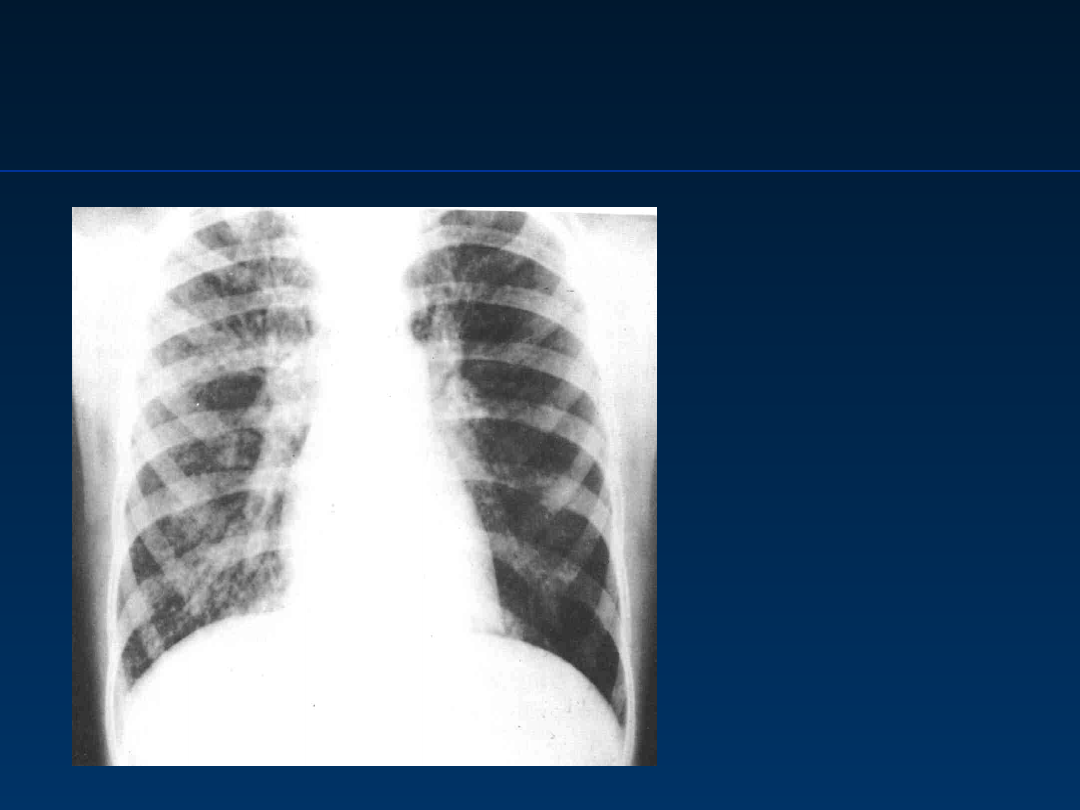
Ciężki złożony niedobór
odporności
Severe combined
immunodeficiency, SCID
Obraz RTG klatki
piersiowej
:
Cytoplazmatyczne
zapalenie płuc u
chorego z SCID
Wg Roitt I.Essential Immunology1994
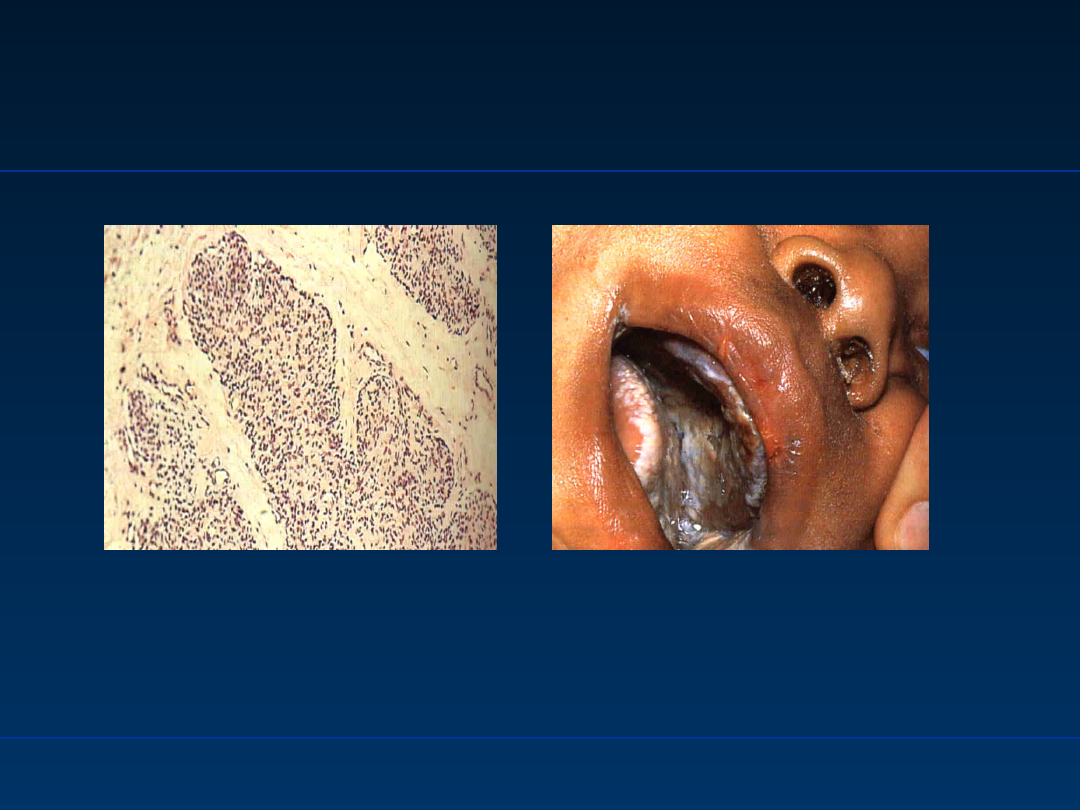
Ciężki złożony niedobór
odporności
Severe combined
immunodeficiency, SCID
Grasica w SCID –
brak ciałek Hassala i
prekursorów limfocytów,
grasica przypomina
gruczoł płodowy
Candida albicans w jamie ustnej
u chorego z SCID
Wg Rosen F. Immunologia 1996

Zaburzenia metabolizmu
puryn
NIEDOBÓR DEAMINAZY ADENOZYNY (ADA)
• Punktowa mutacja i delecja w obrębie genu
kodującego deaminazę adenozyny w obrębie
chromosomu 20 (20% wszystkich
przypadków złożonych niedoborów odporności)
• Deaminaza adenozyny – katalizuje przemianę
adenozyny do inozyny
• Brak lub zmniejszenie aktywności enzymu <10% :
• gromadzenie się wewnątrz komórek toksycznych
metabolitów: deoksyadenozyny i deoksy ATP,
• hamowanie syntezy DNA
• zaburzenia proliferacji limfocytów
• indukcja apoptozy limfocytów (?)
• postępująca limfopenia: limfocyty T, B, komórki
NK

Zaburzenia metabolizmu
puryn
NIEDOBÓR FOSFORYLAZY NUKLEOZYDÓW
PURYNOWYCH (PNP)
• Defekt genu na chromosomie 14
• Obserwowany u chorych z zespołem Nezelofa
• Zaburzenia przemiany: guanozyny do guaniny oraz
inozyny do hipoksantyny
• Gromadzenie w komórkach toksycznych metabolitów:
deoksyguanozyna i deoksy GTP
– upośledzenie proliferacji komórek
– niedobory limfocytów T i komórek NK
– brak zaburzeń odpowiedzi humoralnej
• Test: niskie stężenie moczanów w surowicy -
brak
substratów dla oksydazy moczanowej

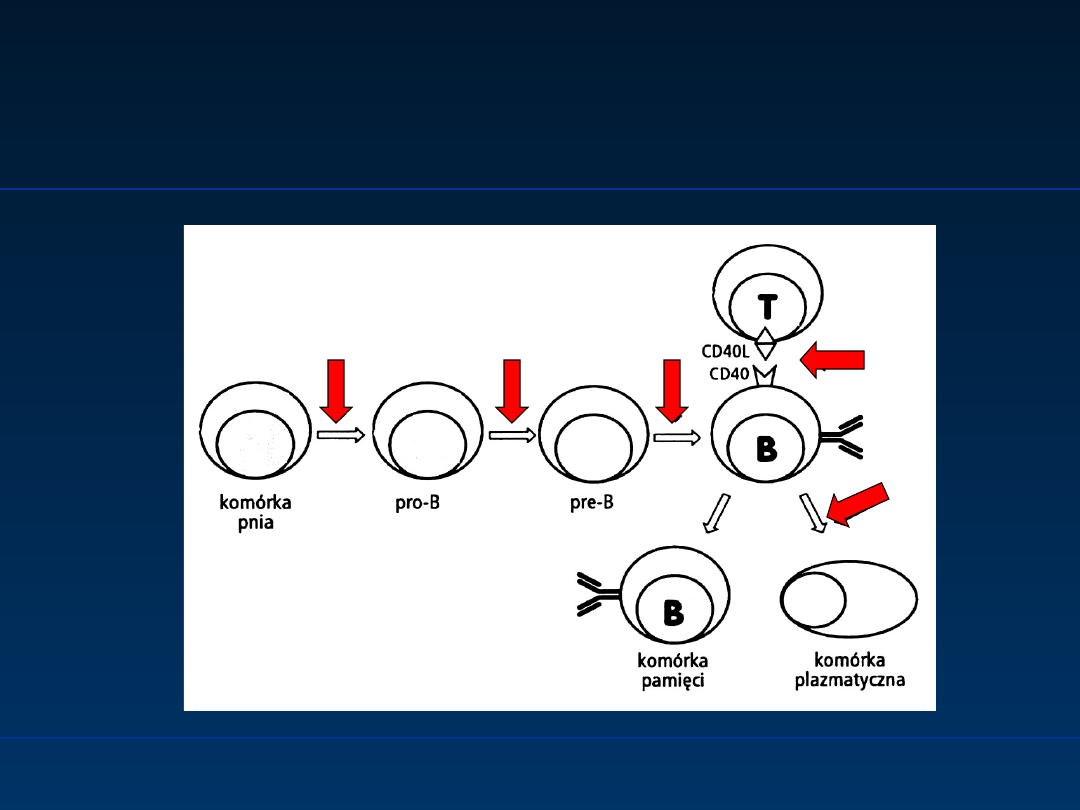
Zespół hiper-IgM
X-linked hyper IgM syndrome, HIGMI
•
70% - dziedziczenie w sprzężeniu z chromosomem X
•
Mutacja genu (punktowa lub delecja) dla ligandu
CD40 na ramieniu długim chromosomu X
– Interakcja CD40L (ligandu dla cząsteczki CD40)
na powierzchni aktywowanych limfocytów T z
CD40 (na prawidłowych limfocytach B) –
indukcja limfocytów B do produkcji IgG, IgA lub
IgE
– Mutacja genu dla CD40L
• brak interakcji limfocytów T z B
• brak interakcji limfocytów T z makrofagami

Zespół hiper-IgM
X-linked hyper IgM syndrome, HIGMI
• Odchylenia laboratoryjne:
– Prawidłowe lub podwyższone wartości surowiczej
IgM
– Obniżone lub niewykrywalne stężenia IgA, IgG i IgE
– Liczba krążących limfocytów B – prawidłowa,
posiadają jedynie powierzchniowe IgM/IgD
+
– Brak ośrodków rozmnażania w węzłach chłonnych
• Obraz kliniczny:
– Nawracające ropne infekcje, często
oportunistyczne
(Cryptosporidium, Pneumocystis)
– Biegunki
– Splenomegalia, limfadenopatia
– Wysoka skłonność do chorób limfoproliferacyjnych
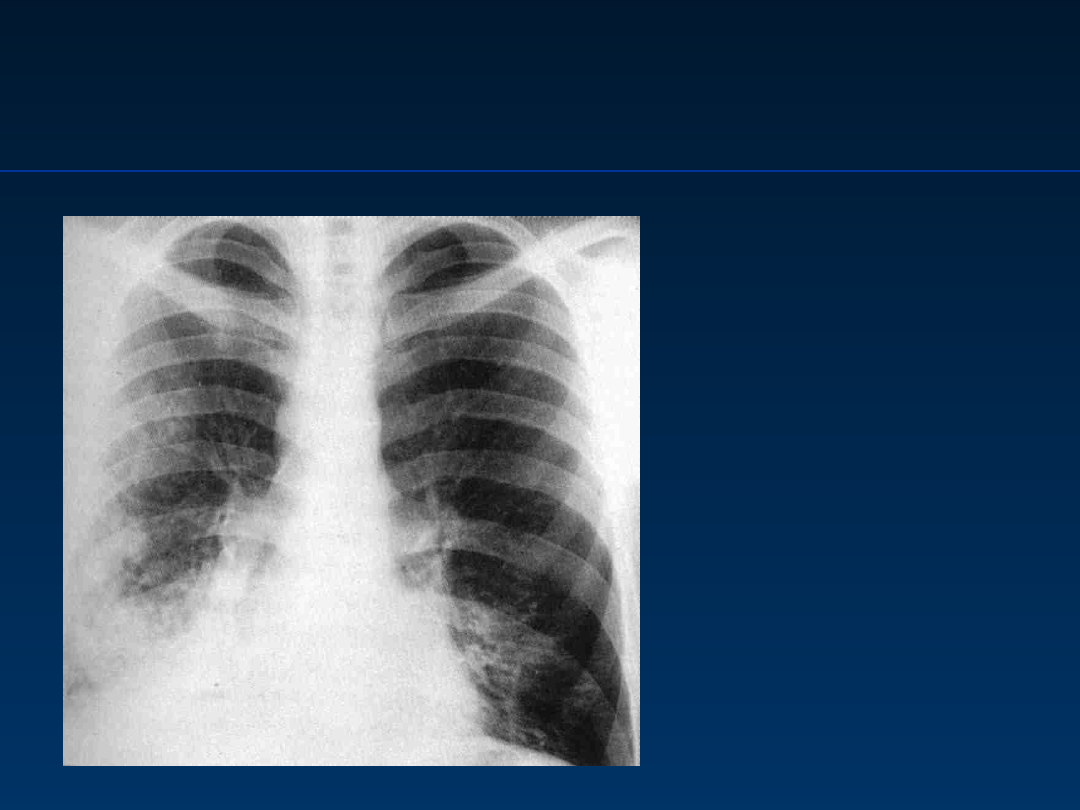
Zespół hiper-IgM
X-linked hyper IgM syndrome, HIGMI
Obraz RTG klatki
piersiowej
:
Pneumokokowe zapalenie
płuc u mężczyzny 26-
letniego
z hipogammaglobulinemią
IgM
Hayward AR. Clinical Immunology 1994

Leczenie
złożonych niedoborów odporności
• Przeszczepienie szpiku
(1968) – leczenie z wyboru
– Skuteczność: 62%, przy identycznych HLA dawcach: 79%
• Przeszczep komórek CD34+ in utero u płodów z
defektem odporności potwierdzonym badaniem
prenatalnym
• PEG-ADA, Adagen: bydlęca deaminaza adenozyny
modyfikowana glikolem polietylenowym
– W zespole ADA
– Terapia efektywna i bezpieczna
– Roczny koszt: > 100 000 USD
• Preparaty dożylnych immunoglubulin (zespół hiper-
IgM)
• Terapia genowa

Pierwotne niedobory
odporności
podział wg WHO 1999
NIEDOBORY ODPORNOŚCI Z PRZEWAGĄ ZABURZEŃ SYNTEZY
PRZECIWCIAŁ
Pierwotne niedobory odporności z
przewagą zaburzeń syntezy
przeciwciał
miejsca potencjalnych defektów limfocytów B
Wg Zeman K. Immunologia Kliniczna
2000

Pierwotne niedobory odporności z
przewagą zaburzeń syntezy
przeciwciał
OBRAZ KLINICZNY
• Objawy chorobowe > 6 miesiąca życia
• Nawracające zakażenia
:
Heamophilus influenza i Streptococcus
pneumoniae
• Zapalenia:
– Ucha środkowego
– Płuc
– Oskrzeli
– Zatok
Upośledzenie rozwoju somatycznego
Przewlekle zmiany w płucach
Rozstrzenie oskrzeli
Wg Zeman K. Immunologia Kliniczna
2000

Agammaglobulinemia sprzężona z
chromosomem X
choroba Brutona; X-linked agammaglobulinemia, XLA
Obraz kliniczny
• Pierwsze objawy
: 6-12 miesiąc życia, rozpoznanie: około 3
roku życia
• Nawracające, o średnio-ciężkim przebiegu
infekcje górnych i
dolnych dróg oddechowych
– rozstrzenia oskrzeli
– Czynniki patogenne zakażeń
: Heamophilus influenzea,
Streptococcus pneumoniae, Staphylococcus aureus
• Zmiany zapalne w stawach
(Chlamydia)
• Zapalenia skóry o typie pyodermii
• Skłonność do odczynów alergicznych
• Zakażenia wirusowe: neurotropowe (ECHO 19)
• Powikłania poszczepienne (Polio)
• Zaburzenia rozwoju somatycznego, nieprawidłowości w
budowie klatki piersiowej, pałeczkowate palce
• Pierwszy opisany niedobór odporności (1952- Ogden
Bruton)
• U chłopców
• Częstość występowania- 1:100 000

Agammaglobulinemia sprzężona z
chromosomem X
choroba Brutona; X-linked agammaglobulinemia, XLA
• Tkanka limfatyczna: hipoplastyczna
– Brak lub małe migdałki podniebienne
– Niewyczuwalne węzły chłonne szyjne,
podżuchwowe i inne obwodowe
• Odchylenia laboratoryjne
– Stężenia IgG, IgM i IgA w surowicy – śladowe
– Brak przeciwciał poszczepiennych
– Brak limfocytów B i komórek plazmatycznych
– W szpiku: limfocyty pre-B prawidłowa liczba
– Liczba i funkcja limfocytów T i neutrofilów -
prawidłowa

Agammaglobulinemia sprzężona z
chromosomem X
choroba Brutona; X-linked agammaglobulinemia, XLA
PATOGENEZA:
– Defekt genetyczny na długim ramieniu
chromosomu X (XQ21.3-22)
– Mutacja genu dla cytoplazmatycznej kinazy
tyrozynowej btk niezbędnej dla przekazania sygnału
transdukcji w procesie różnicowania limfocytów B
Zaburzenie przejścia limfocytów pre-B
w B
Brak klonów zdolnych do syntezy
przeciwciał
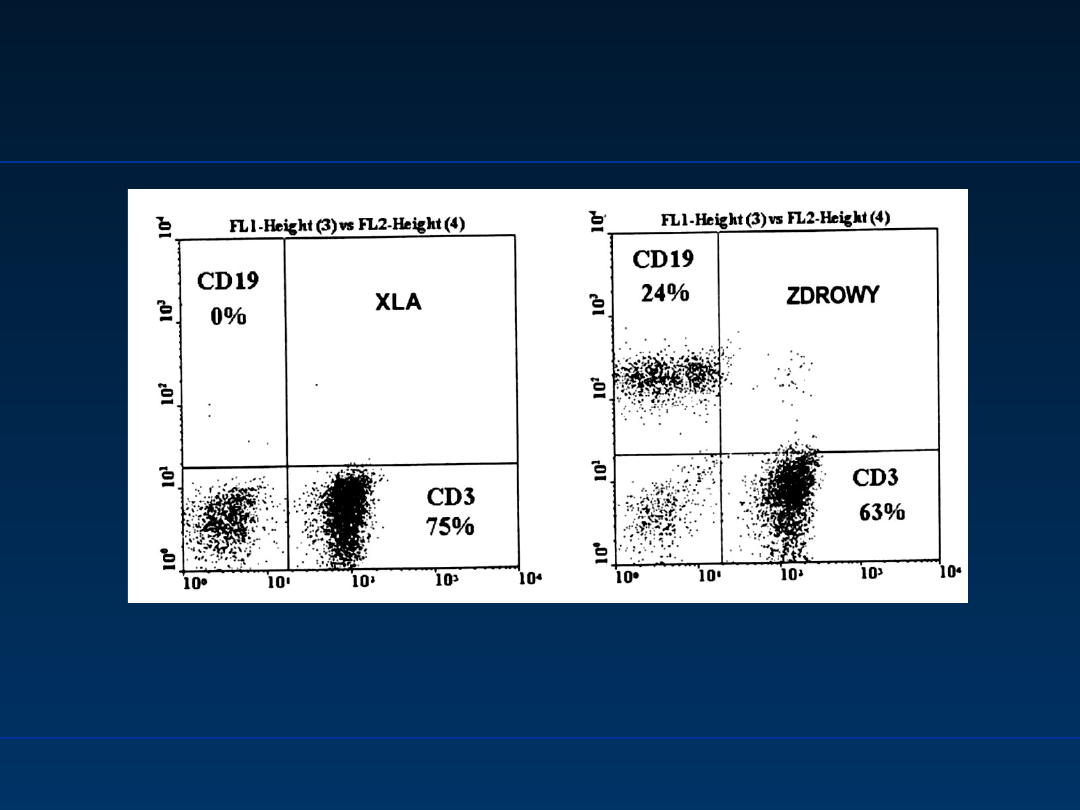
Agammaglobulinemia sprzężona z
chromosomem X
choroba Brutona; X-linked agammaglobulinemia, XLA
Cytometryczna analiza odsetka limfocytów T (CD3+) i limfocytów B (CD19+)
w krwi obwodowej dziecka z agammaglobulinemią dziedziczącą się w
skojarzeniu z chromosomem X
Wg Zeman K. Immunologia Kliniczna 2000
Pospolity zmienny niedobór
odporności
Common
variable immunodeficiency, CVID
Występowanie:
• U obojga płci
• Częstość: 1:10 000 – 1:50 000
Obraz kliniczny:
• Pierwsze objawy: od okresu niemowlęctwa do późnego
dzieciństwa, często w 3 dekadzie życia
• Nawracające ropne infekcje:
• zespół zatokowo-oskrzelowy
• rozstrzenie oskrzeli
• Nawracające infekcje Herpes zoster i Pneumocystis
• Objawy ze strony przewodu pokarmowego
przypominające chorobę trzewną, infestacje Giardia
lamblia
• 30% - splemomegalia i limfadenopatia
• Rozwój chłoniaków, nowotworów przewodu
pokarmowego lub indukcji procesów
autoimmunizacyjnych (niedokrwistość hemolityczna,
małopłytkowość, neutropenia)

Pospolity zmienny niedobór
odporności
Common
variable immunodeficiency, CVID
Heterogenna grupa chorób ze
zmniejszonym stężeniem
immunoglobulin w surowicy
Odchylenia laboratoryjne:
– Stężenia IgG i IgA: niskie
– Stężenie IgM: niewykrywalne lub obniżone
– Brak wytwarzania przeciwciał poszczepiennych
– Liczba limfocytów B we krwi obwodowej:
prawidłowa lub niska
– Ilość lub czynność limfocytów T: zaburzona
( u części chorych)

Pospolity zmienny niedobór
odporności
Common variable
immunodeficiency, CVID
PATOGENEZA:
•
Genetyczne podstawy: nieznane
•
Defekt przekształcenia limfocytów B w komórki plazmatyczne
•
Defekt wydzielania zsyntetyzowanej Ig poza komórkę
•
Nadmierna aktywność limfocytów supresorowych
•
Aktywność autoprzeciwciał skierowanych przeciwko limfocytom B
•
Zaburzenia aktywacji limfocytów T - zaburzenia transdukcji
sygnału
•
Obniżony stosunek CD4:CD8
•
Obniżona ilość limfocytów CD4/CD45RA
•
Obniżona odpowiedź limfocytów T na stymulację receptora TCR
•
Obniżona ekspresja ligandu CD40

Niedobór IgA
IgA deficiency,
IgAD
Najczęściej występujący pierwotny niedobór odporności:
1:500 – 1:700 (Europa)
1: 18 500 (Japonia)
OBRAZ KLINICZNY
• Często przebieg bezobjawowy
• 25% - nawracające infekcje zatokowo-oskrzelowe
bakteryjne i wirusowe
• Choroby przewodu pokarmowego
• Skłonność do alergii (20-30%)
• Skłonność do chorób autoimmunizacyjnych
• Reakcje potransfuzyjne- przeciwciała anty-IgA

Niedobór IgA
IgA deficiency,
IgAD
ODCHYLENIA W BADANIACH LABORATORYJNYCH
• Stężenie IgA w surowicy:
– niskie (33%)
– Brak IgA (<5mg/dl, 67%)
• Stężenia wydzielniczej IgA: w normie
• Stężenia IgG, IgM: w normie
– 20-30%: niedobór podklas IgG2 i IgG4
PATOGENEZA
• Zatrzymanie różnicowania limfocytów B IgM do
komórek plazmatycznych
• Defekt genetyczny (?):
autosomalny recesywny lub dominujący

Niedobory podklas IgG
Selective IgG subclass deficiency
Najczęściej niedobór:
– IgG2 i IgG3
– IgG2 i IgG4
Obraz kliniczny:
– 25% - nawracające infekcje górnych i
dolnych dróg oddechowych wywołane
przez bakterie ropotwórcze
– Współistnienie z astmą oskrzelową
– Współistnienie niedoboru IgA

Przemijająca hipogammaglobulinemia
niemowląt
Transient hypogammaglobulinaemia of
infancy
• Fizjologicznie: 3-5 miesiąc życia
• Głównie u niemowląt urodzonych przedwcześnie,
synteza IgG i IgA opóźniona:
w 18-36 miesiącu
życia
• U części dzieci brak objawów chorobowych
• Patogeneza:
– Zaburzenie funkcji limfocytów T CD4:
zaburzenie aktywacji limfocytów T i
przekazywania sygnału limfocytom B
• Stężenia Ig w surowicy: niskie
• Liczba limfocytów B w krwi obwodowej: prawidłowa

Pierwotne niedobory odporności z
przewagą zaburzeń syntezy
przeciwciał
Agammaglobulinemia
• Monomeryczne preparaty IgG (co 4 tyg., w
dawkach 100-800mg/kg m.c., dożylnie lub
podskórnie)
• Terapia genowa (?)
Niedobory selektywnych IgG, niedobór IgA
• Antybiotykoterapia
• Preparaty immunoglobulin dożylnie (?)
Wg Zeman K. Immunologia Kliniczna 2000

Pierwotne niedobory
odporności
podział wg WHO 1999
INNE DOBRZE OKRESLONE ZESPOŁY NIEDOBORÓW
ODPORNOŚCI

Zespół Wiskott-Aldricha
Wiskott-Aldrich syndrome, WAS
DZIEDZICZENIE:
w sprzężeniu z chromosomem X
OBRAZ KLINICZNY:
• Trombocytopenia: już w okresie noworodkowym, małe
płytki krwi, nasilenie po szczepionkach
streptokokowych
• Nawracające zakażenia:
– ucha środkowego, płuc i opon mózgowo-
rdzeniowych,
– bakteryjne, wirusowe, grzybicze
• Zmiany skórne o charakterze wyprysku atopowego
• Hepatosplenomegalia
• Wzmożona skłonność do chorób autoimmunizacyjnych
i rozrostów nowotworowych (chłoniaki)
Wg Zeman K. Immunologia Kliniczna
2000

Zespół Wiskott-Aldricha
Wiskott-Aldrich syndrome, WAS
ODCHYLENIA W BADANIACH LABORATORYJNYCH
• Brak mikrowypustek limfocytów T- „łysy” wygląd
w mikroskopie skaningowym
• W błonie komórkowej limfocytów T- niestabilna
sialoglikoproteina CD43
• Odpowiedź limfoproliferacyjna limfocytów T –
obniżona (anergia w testach
skórnych)
• Limfopenia
• Stężenia IgM w surowicy: niskie
• Stężenia IgE: podwyższone
• Brak naturalnych przeciwciał w układzie ABO

Zespół Wiskott-Aldricha
Wiskott-Aldrich syndrome, WAS
PATOGENEZA:
• Defekt genu na krótkim ramieniu chromosomu
Xp11.22
• Produkt genu: białko WASP, niezbędne dla
prawidłowego sygnału transdukcji dla
reorganizacji cytoskeletonu komórki
LECZENIE:
• Przeszczepienie szpiku
• Leczenie przeciwbakteryjne,
przeciwkrwotoczne (przetoczenia
napromienionych świeżych płytek krwi lub
osocza), przeciwalergiczne
• Splenektomia (?)
• Preparaty immunoglobulin

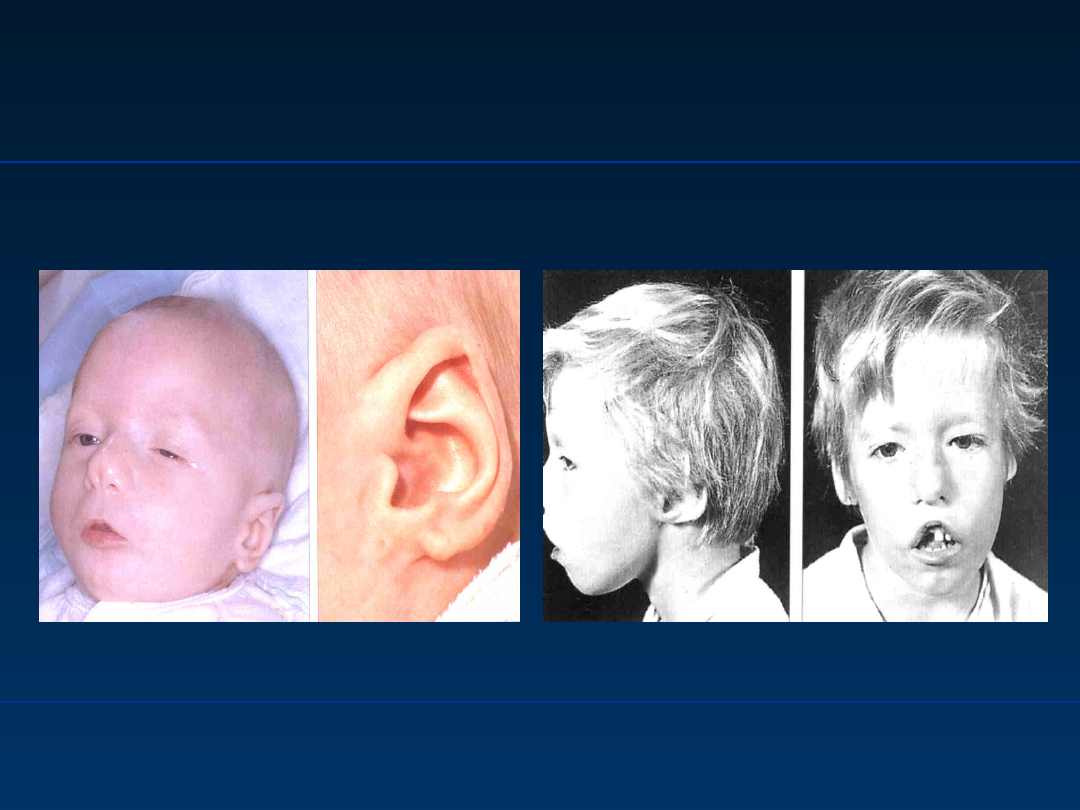
Anomalia Di George’a
Di George
syndrome
OBRAZ KLINICZNY
• Aplazja lub hipolazja grasicy
• Tężyczka związana z niedorozwojem przytarczyc
• Wada serca
– Prawostronny łuk aorty
– Zwężenie stożka prawej komory
– VSD
• Charakterystyczny fenotyp: mikrogracja, hiperteloryzm,
rozszerzenie podniebienia, niskie osadzenie uszu, wady
małżowiny
• zakażenia wirusowe (wirus paragrypy, adenowirusy)
• Zakażenia grzybicze dróg oddechowych i przewodu
pokarmowego
• Infekcje Pneumocystis
• Skłonność do procesów autoimmunizacyjnych
(młodzieńcze przewlekle zapalenie stawów)
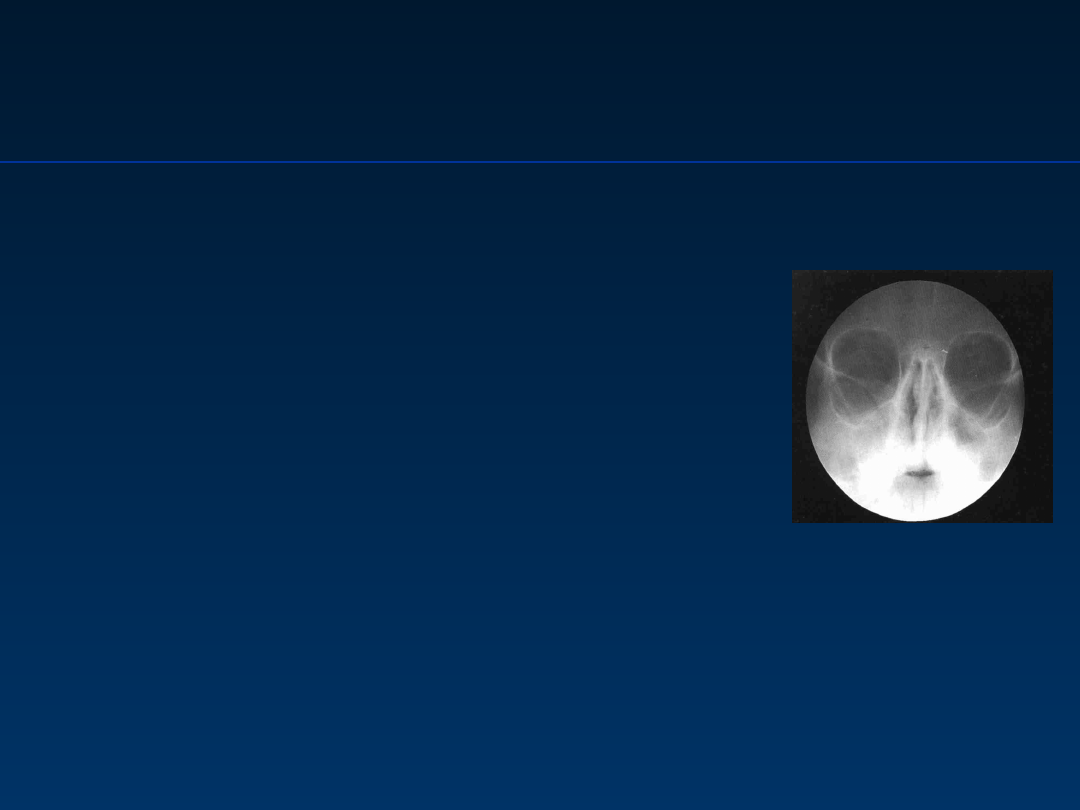
Anomalia Di George’a
Di George
syndrome
Wg Hayward AR. Clinical Immunology 1994

Anomalia Di George’a
Di George
syndrome
I. Forma kompletna
z poważnymi zaburzeniami
odpowiedzi komórkowej i humoralnej (20%
przypadków)
II. Forma częściowa
– nieznacznie upośledzona odpowiedź
immunologiczna
ODCHYLENIA W BADANIACH LABORATORYJNYCH
• Stężenie Ca w surowicy: niskie
• Stężenie P w surowicy: podwyższone
• Liczba limfocytów T, CD8+: obniżona
• Odpowiedź limfocytów na stymulację mitogenami:
obniżona
• Stężenie Ig w surowicy: prawidłowe
• Brak cienia grasicy na zdjęciu RTG

Pierwotne niedobory
odporności
podział wg WHO 1999
ZESPOŁY NIESTABILNOŚCI CHROMOSOMALNYCH

Ataksja-teleangiektazja
Ataxia
teleangiectasia, AT
• Dziedziczenie: autosomalnie recesywnie
• Częstość występowania: 1: 40 000 – 1: 100 000 (w
Polsce 100 chorych)
OBRAZ KLINICZNY:
• Pierwsze objawy: 12-18 miesiąc życia
• Postępująca ataksja móżdżkowa
• Rozszerzenia naczyń krwionośnych spojówek i skóry
• Nawracające zakażenia (zatokowo-oskrzelowe)
• Hipoplastyczna grasica
• Upośledzenie wzrostu
• Brak rozwoju wtórnych cech płciowych
• Rozwój choroby nowotworowej lub procesów
autoimmunizacyjnych

Pierwotne niedobory
odporności
podział wg WHO 1999
DEFEKTY KOMÓREK ŻERNYCH

Wrodzone zaburzenia
liczby i funkcji komórek
żernych
• Przewlekła choroba ziarniniakowa
• Zespół Chediak-Higashi
• Zespół Griscelli
• Defekt adhezji krwinek białych
• Wrodzone neutropenie

Przewlekła choroba
ziarniniakowa
Chronic granulomatous disease, CGD
• Dziedziczenie: recesywne sprzężone z chromosomem X
(chłopcy) lub autosomalne recesywne
• Częstość występowania:
1: 160 000 (Dania) – 1: 300 000 (Japonia)
W Polsce: 20 chorych
OBRAZ KLINICZNY:
• Pierwsze objawy: w okresie niemowlęctwa (75%)
• Nawracające zapalenia skóry, błon śluzowych i węzłów chłonnych
• Zapalenia oskrzeli i płuc
• Ropnie: okołoodbytnicze, wątroby, mózgu
• Zapalenia szpiku i kości
• Ziarniniaki w przewodzie pokarmowych i drogach moczowo-
płciowych
• Drobnoustroje chorobotwórcze: E.coli, Serratia marcescens,
Bulholderia cepacia, Aspergillus sp., Candida albicans
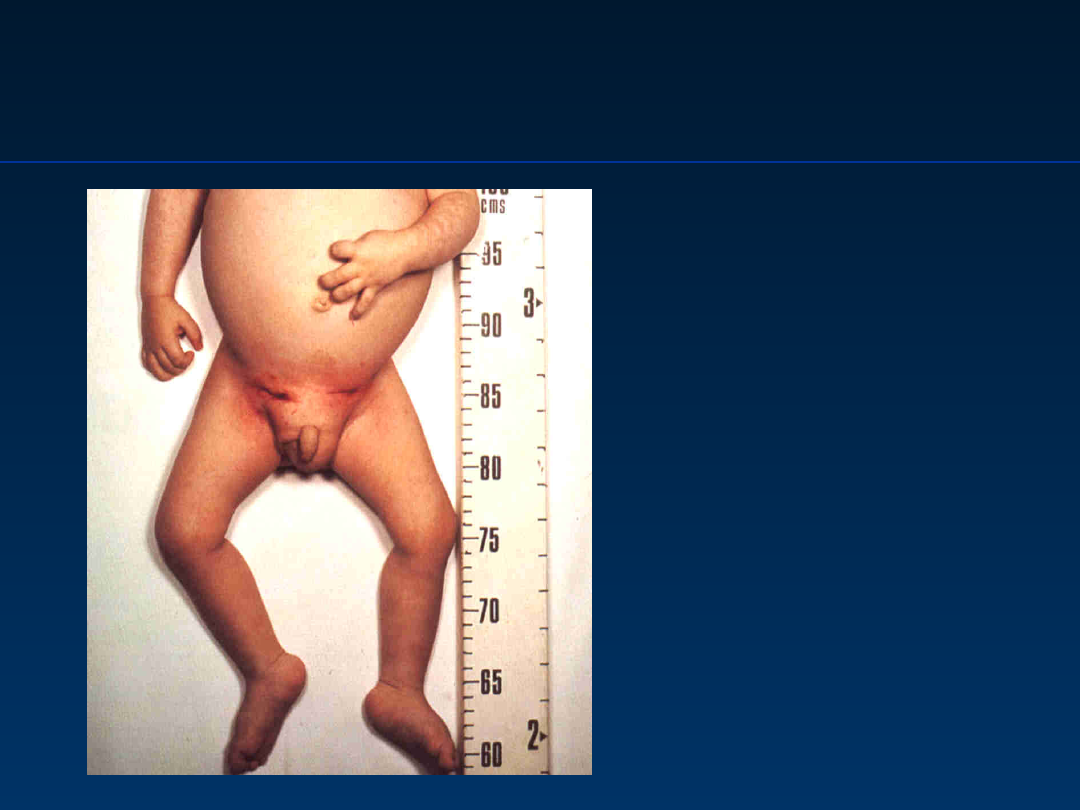
Przewlekła choroba
ziarniniakowa
Chronic granulomatous disease, CGD
Chłopiec 18-miesięczny z
CGD:
obustronne ropnie węzłów
chłonnych
pachwinowych – po drenażu
chirurgicznym
Wg Hayward AR. Clinical Immunology 1994

Przewlekła choroba
ziarniniakowa
Chronic granulomatous disease, CGD
ODCHYLENIA LABORATORYJNE
• Fagocytoza: prawidłowa
• Zabijanie wewnątrzkomórkowe drobnoustrojów:
upośledzone (test NBT, chemiluninescencja,
wybuch tlenowy – obniżona produkcja metabolitów
tlenowych: O
2-
, H
2
O
2
)
• Zaburzenia immunoregulacji: obniżona synteza
IFN-g, niska proliferacja limfocytów T
PATOGENEZA
• Defekt produkcji tlenowych metabolitów
niezbędnych w niszczeniu pochłoniętych
drobnoustrojów
• Defekty genów (delecje, duplikacje, mutacje
punktowe) dla składowych oksydazy NADPH
(gp91
phox
, p47
phox)
– zaburzenia składników
cytochromu b
558
lub składników cytozolowych
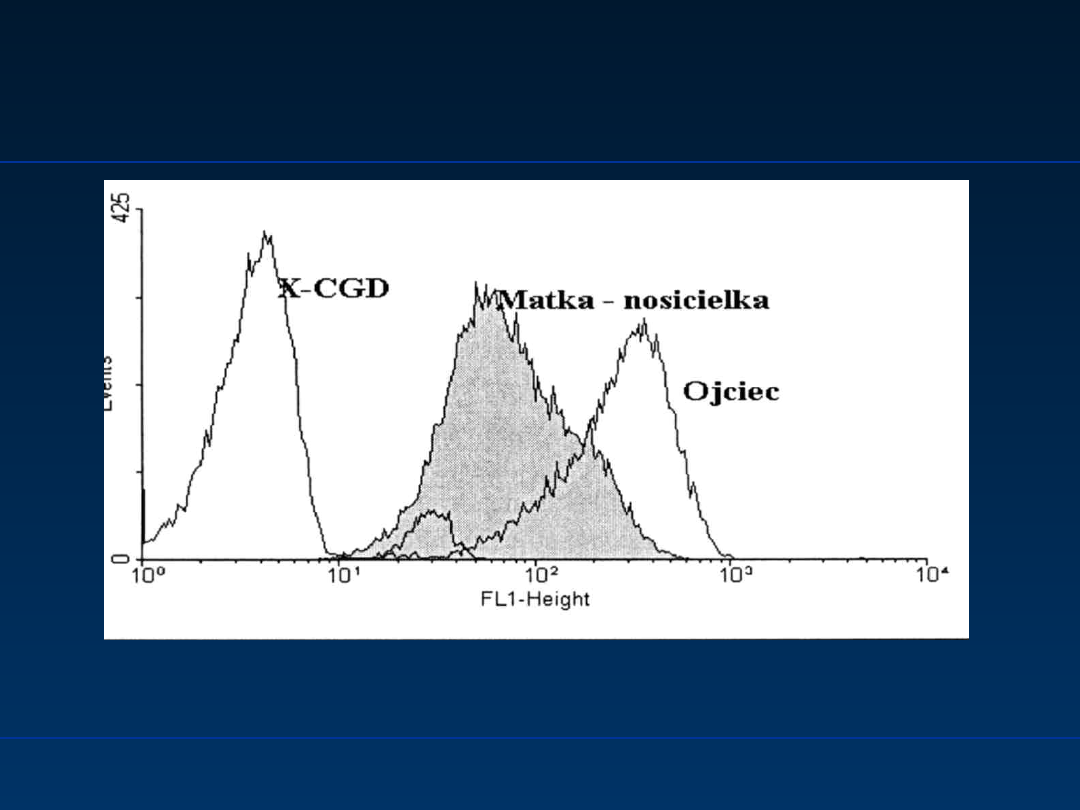
Przewlekła choroba
ziarniniakowa
Chronic granulomatous disease, CGD
Wg Zeman K. Immunologia Kliniczna 2000
Cytometryczna analiza wybuchu oddechowego neutrofilów u dziecka z X-CGD
(gp91
phox
), jego matki nosicielki i zdrowego ojca (Phagoburst)

Przewlekła choroba
ziarniniakowa
Chronic granulomatous disease, CGD
LECZENIE
• Profilaktyka zakażeń
• Antybiotykoterapia
• Interwencja chirurgiczna
• Profilaktycznie IFN-gamma
• Przeszczep szpiku
• Terapia genowa
(też in utero po
diagnostyce prenatalnej)

Zespół Chediaka-
Higashiego
Chediak-Higashi syndrome, CHS
Dziedziczenie:
autosomalne recesywne
OBRAZ KLINICZNY:
• Nawracające, ciężkie ropne zakażenia skóry i
zakażenia układowe (gronkowce,
paciorkowce)
• Hipopigmentacja lub albinizm
• Skłonność do krwawień i wybroczyn
• Nieprawidłowości neurologiczne
– Opóźnienie rozwoju, padaczka, postępująca
polineuropatia obwodowa
• Rozwój procesów limfoproliferacyjnych (85%)

Diagnostyka
pierwotnych niedoborów
odporności
• Wywiad rodzinny
• Badania laboratoryjne:
– ilość krwinek białych, limfocytów, neutrofilów,
eozynofilów, płytek krwi
– Ilość (odsetek) limfocytów T i B, subpopulacje
limfocytów T
– RTG klatki piersiowej, USG grasicy
– Poziom Ca i P w surowicy (Z. Di George’a)
– Poziom a-fetoproteina,antygen karcinoembrionalny
(AT)
• Diagnostyka molekularna
• Diagnostyka prenatalna
(krew płodu, komórki owodniowe, materiał z
biopsji trofoblastu)

Diagnostyka
pierwotnych niedoborów
odporności
BADANIA ODPORNOŚCI HUMORALNEJ
•
Ilość krążących limfocytów B (CD5+, CD19+,
CD20+)
•
Stężenie immunoglobulin w surowicy: IgG, IgA,
IgM – metoda nefelometryczna, ELISA)
•
Ocena zdolności do syntezy swoistych przeciwciał
po czynnej immunizacji szczepionkami (DTPa,
polio, wzwB, antygeny polisacharydowe)
•
Ocena syntezy przeciwciał in vitro:
–
Stymulacja limfocytów antygenem szkarłatki
–
Proliferacja limfocytów B w odpowiedzi na
CD40, IL-4, IL-10
Diagnostyka
pierwotnych niedoborów odporności
Wg Zeman K. Immunologia Kliniczna 2000
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
Wyszukiwarka
Podobne podstrony:
Niedobory odpornosci wersja dluzsza
Imm Cw 3 Wt rne niedobory odpornosci
00 Niedobory odpornoscioweid 1 Nieznany (2)
03 0000 014 02 Leczenie pierwotnych niedoborow odpornosci u dzieci immunoglobulinami
Niedobory odporności
09 pierwotne niedobory odporności
WT RNE NIEDOBORY ODPORNO CI
Niedobory odpornościowe(1), 1.Lekarski, II rok, Immunologia, Prelekcje
niedobory odporności
Imm Cw 4 NIEDOBORY ODPORNO UCI Nieznany
Diagnostyka niedoborów odporności
więcej podobnych podstron