
Maciej Duda 168516
Wojciech Ślęczek 168560
SPRAWOZDANIE
Inżynieria genetyczna – Laboratorium
Ćwiczenie nr 4 i 5:
• Przygotowanie insertu EcRDBD do klonowania (generowanie miejsc restrykcyjnych BamHI) –
reakcja PCR
• Przygotowanie plazmidu pGEX – 2T do klonowania – elektroforetyczna analiza plazmidowego
DNA oczyszczonego po trawieniu i defosforylacji oraz szacowanie jego stężenia
• Elektroforeza analityczna produktów PCR
• Izolacja produktu PCR (protokół Clean – up) (A&A Biotechnology)
• Ligacja wektora pGEX – 2T zlinearyzowanego enzymem BamHI i defosforylowanego
(pGEX - 2T/BamHI) z fragmentem DNA kodującym domenę wiążącą DNA receptora
ekdysteroidowego (EcRDBD/BamHI) przy użyciu ligazy T4
• Transformacja komórek kompetentnych (Maniatis i wsp. 1989)
1. Procedura i metodyka
• W celu przeprowadzenia reakcji PCR przygotowano następujące roztwory w probówkach
Eppendorfa:
a) Roztwór 1 – mieszanina reakcyjna:
3 μl pBS – EcR/EcoRI (około 15ng) – matryca DNA
4,1 μl WR5 (50 pmoli) – starter przedni („forward”)
4,2 μl WR6 (50 pmoli) – starter tylni (”reverse”)
8 μl dNTP (mieszanina nukleotydów, C = 1,25mM każdego)
5 μl bufor dla polimerazy [10x]
1 μl polimerazy – dodawanej po tzw. „gorącym starcie”
24,7 μl H
2
O
= 50 μl razem
b) Roztwór 2 – kontrola (K):
4,1 μl WR5 (50 pmoli)
4,2 μl WR6 (50 pmoli)
8 μl dNTP (mieszanina nukleotydów, C = 1,25mM każdego)
5 μl bufor dla polimerazy [10x]
1 μl polimerazy – dodawanej po tzw. „gorącym starcie”
27,7 μl H
2
O
= 50 μl razem
• Na powierzchnię roztworów nałożono 50 μl oleju parafinowego.
• Tak przygotowane próbki umieszczono w termocyklerze i ustawiono program reakcji PCR
zgodnie z instrukcją.

• Po „gorącym starcie” dodano 1 μl termostabilnej polimerazy, po czym „puszczono” reakcję
dalej według nastawionych wcześniej cyklów.
• W następnym kroku zlinearyzowany, oczyszczony i defosforylowany wektora pGEX – 2T
poddano analizie densytometrycznej; w tym celu przeprowadzono horyzontalną
elektroforezę analityczną w żelu agarozowym o stężeniu 0,8% przygotowanym w 1xTBE
i zawierającym bromek etydyny w stężeniu 0,4 μg/ml. Elektroforezę prowadzono w buforze
1xTBE przy stałym napięciu 75 V przez około 50min.
• W celu przeprowadzenia powyżej opisanej elektroforezy przygotowano następujący roztwór:
a) Roztwór do elektroforezy analitycznej:
3 μl roztworu plazmidowego DNA po oczyszczeniu
7 μl TE
2 μl 5x buforu do próbek
= 12 μl razem
• Oprócz analizowanych próbek, na żel naniesiono również 5 μl markera wagowego FastRuler™
DNA Ladder, Middle Range, (MBI Fermentas).
• Kolejnym krokiem było wykonanie elektroforezy analitycznej produktów przeprowadzonej
reakcji PCR. W tym celu przygotowano następujące roztwory:
a) Roztwór 1 – próbka:
5 μl próbki po reakcji PCR (pobranej spod oleju)
5 μl TE
2 μl 6xSB
= 12 μl razem
b) Roztwór 2 – kontrola (K):
5 μl próbki po reakcji PCR (spod oleju z roztworu kontroli)
3 μl TE
2 μl 6xSB
= 12 μl razem
• Oprócz analizowanych próbek na żel naniesiono też 5 μl markera wagowego FastRuler™ DNA
Ladder, Middle Range, (MBI Fermentas).
• Elektroforeza prowadzona była w żelu agarozowym o stężeniu agarozy równym 2%,
przygotowanym w 1xTBE i zawierającym bromek etydyny w stężeniu 0,4 μg/ml. Elektroforezę
prowadzono w buforze 1xTBE przy stałym napięciu 75 V przez około 50min.
• Następnie przeprowadzono izolację produktu PCR z pozostałej części mieszaniny nie
poddanej elektroforezie. W tym celu opisano nową probówkę Eppendorfa i przeniesiono do
niej pozostałą część mieszniany po PCR spod oleju.
• Do takiego roztworu dodano pięciokrotny nadmiar roztworu G (223 μl).
• Mieszaninę krótko odwirowano w celu usunięcia resztek roztworu z wieczka i ścianek
probówki.
• Mieszaninę naniesiono na kolumnę do oczyszczania DNA i wirowano przez 30 s przy około
10 000 x g.
• Następnie wyciągnięto kolumnę z probówki, przesącz wylano i kolumnę ponownie włożono
do probówki.

• Na złoże w kolumnie naniesiono 600 μl roztworu A1 do płukania.
• Wirowano przez 30 s przu 10 000 x g.
• Wyciągnięto kolumnę z probówki, odrzucono przesącz i kolumnę ponownie włożono do
probówki.
• Na złoże w kolumnie naniesiono 300 μl roztworu A1 do płukania.
• Wirowano przez 2 min przy 10 000 x g.
• Wyjęto kolumnę z probówki, przesącz ponownie odrzucono i kolumnę ponownie włożono do
probówki.
• Kolumnę z probówką inkubowano przez 3 min w temperaturze pokojowej w celu osuszenia
złoża.
• Osuszoną kolumnę umieszczono w nowej probówce Eppendorfa i naniesiono na złożę 30 μl
buforu TE tak, by pokrył on całkowicie złoże.
• Ponownie inkubowano w temperaturze pokojowej przez 3 min.
• Następnie odwirowano przez 1 min przy 10 000 x g.
• Przesącz na dnie probówki, zawierający docelowe DNA, przeniesiono do czystej opisanej
probówki Eppendorfa i zanotowano jego objętość.
• Tak oczyszczony produkt PCR znajdujący się w buforze TE przechowywano w temperaturze
-20ᴼ C.
• Kolejnym krokiem doświadczenia było przeprowadzenie ligacji wektora pGEX – 2T
zlinearyzowanego BamHI i defosforylowanego (pGEX – 2T/BamHI) z fragmentem DNA
kodującym domenę wiążącą DNA receptora ekdysteroidewego (EcRDBD/BamHI) przy użyciu
ligazy T4. W tym celu przygotowano następujące roztwory:
a) Roztwór 1 – reakcja na ligację, próbka:
1,5 μl pGEX–2T/BamHI (C = 15 ng/μl) (wektor)
1,65 μl EcRDBD/BamHI (C = 3 ng/μl) (insert)
1 μl ligazy T4 (1U) (MBI Fermentas) (enzym)
2 μl buforu do ligazy T4 [10x] (MBI Fermentas)
13,85 μl H
2
O
= 20 μl razem
b) Roztwór 2 – kontrola (K):
1,5 μl pGEX–2T/BamHI (C = 15 ng/μl) (wektor)
1 μl ligazy T4 (1U) (MBI Fermentas) (enzym)
2 μl buforu do ligazy T4 [10x] (MBI Fermentas)
15,5 μl H
2
O
= 20 μl razem
• Ligację prowadzono w stosunku molowym 1:4, n
insertu
= 4*n
wektora
, w sterylnej i opisanej
probówce Eppendorfa. Reakcja była prowadzona w temperaturze pokojowej ~20ᴼ C przez 1h
i 20 min.
• Kolejnym etapem była transformacja komórek kompetentnych. W tym celu rozmrożono na
lodzie 100 μl komórek kompetentych (XL1 – Blue).
• Natychmiast przeniesiono je do schłodzonej sterylnej probówki Eppendorfa, która zawierała
mieszaninę po ligacji.
• Całość delikatnie wymieszano i inkubowano przez 20 min na lodzie.
• Następnie próbki poddano szokowi cieplnemu w 37ᴼC przez 5 min w termomikserze.

• Dodano 90 μl pożywki płynnej LB i inkubowano w termomikserze w temperaturze 37ᴼC przez
20 min.
• W ostatnim kroku przeniesiono 200 μl zawiesiny transformowanych komórek na szalkę
Petriego z podłożem LB–agar, zawierającym karbenicylinę w stężeniu końcowym 100 μg/ml.
• Wykonano posiew dywanowy za pomocą sterylnej głaszczki.
• Następnie prowadzono inkubację płytek przez 12h w 37ᴼC, po czym policzono kolonie
transformantów wyrosłe na płytce po ligacji i na płytce kontrolnej.
2. Wyniki i obserwacje
• Objętości starterów użytych w reakcji PCR w próbce i kontroli otrzymano w wyniku poniższych
obliczeń:
a) Starter WR5 (forward)
MW = 29 nukleotydów x 320 g/mol nul. = 9280 g/mol
9280 g
-
1 mol
x
1
-
50 x 10
-12
mola
x
1
=
4,64 x 10
-7
g masa startera WR5 [g]
C
1
= 1,14 x 10
-7
g/μl stąd:
1,14 x 10
-7
g -
1 μl
4,64 x 10
-7
-
V
1
V
1
=
4,1 μl objętość startera WR5 [μl]
b) Starter WR6 (reverse)
MW = 30 nukleotydów x 320 g/mol nul. = 9600 g/mol
9600 g
-
1 mol
x
2
-
50 x 10
-12
mola
x
2
=
4,80 x 10
-7
g masa startera WR5 [g]
C
2
= 1,14 x 10
-7
g/μl stąd:
1,14 x 10
-7
g -
1 μl
4,80 x 10
-7
-
V
2
V
2
=
4,2 μl objętość startera WR6 [μl]
• Przeprowadzony program PCR:
Temp 1
94ᴼC
Czas 1
5 min
„gorący start”
Cykl 1
-----
Dodanie 1 μl termostabilnej polimerazy DNA
Temp 2
94ᴼC
Czas 2
1 min
denaturacja DNA
Cykl 2
-----
Temp 3
58ᴼC
Czas 3
0,5 min
5x
stapianie
MATRYCA
Cykl 3
-----
PIERWOTNA
Temp 4
72ᴼC
Czas 4
0,5 min
polimeryzacja
Cykl 4
do 2-giego
Temp 5
94ᴼC
Czas 5
1 min
denaturacja
Cykl 5
-----
15x
Temp 6
72ᴼC
Czas 6
1 min
stapianie i polimeryzacja
Cykl 6
do 5-ego
MATRYCA
WTÓRNA
Temp 7
72ᴼC
Czas 7
2 min
dokończenie polimeryzacji
Cykl 7
-----
Temp 8
4ᴼC
Czas 8
PAUZA
zakończenie (schłodzenie mieszaniny reakcyjnej)
Cykl 8
-----
• Do przygotowana próbki do elektroforezy plazmidu pGEX-2T zastosowano końcową objętość 12
μl, tak aby roztwor 2 μl 6xSB rozcieńczony był w niej 6 razy (do stężenia 1x) i to z niej wynikają
ilości pozostałych składników. W ten sposób użyto 3 ěl plazmidowego DNA i 7 μl roztworu TE.
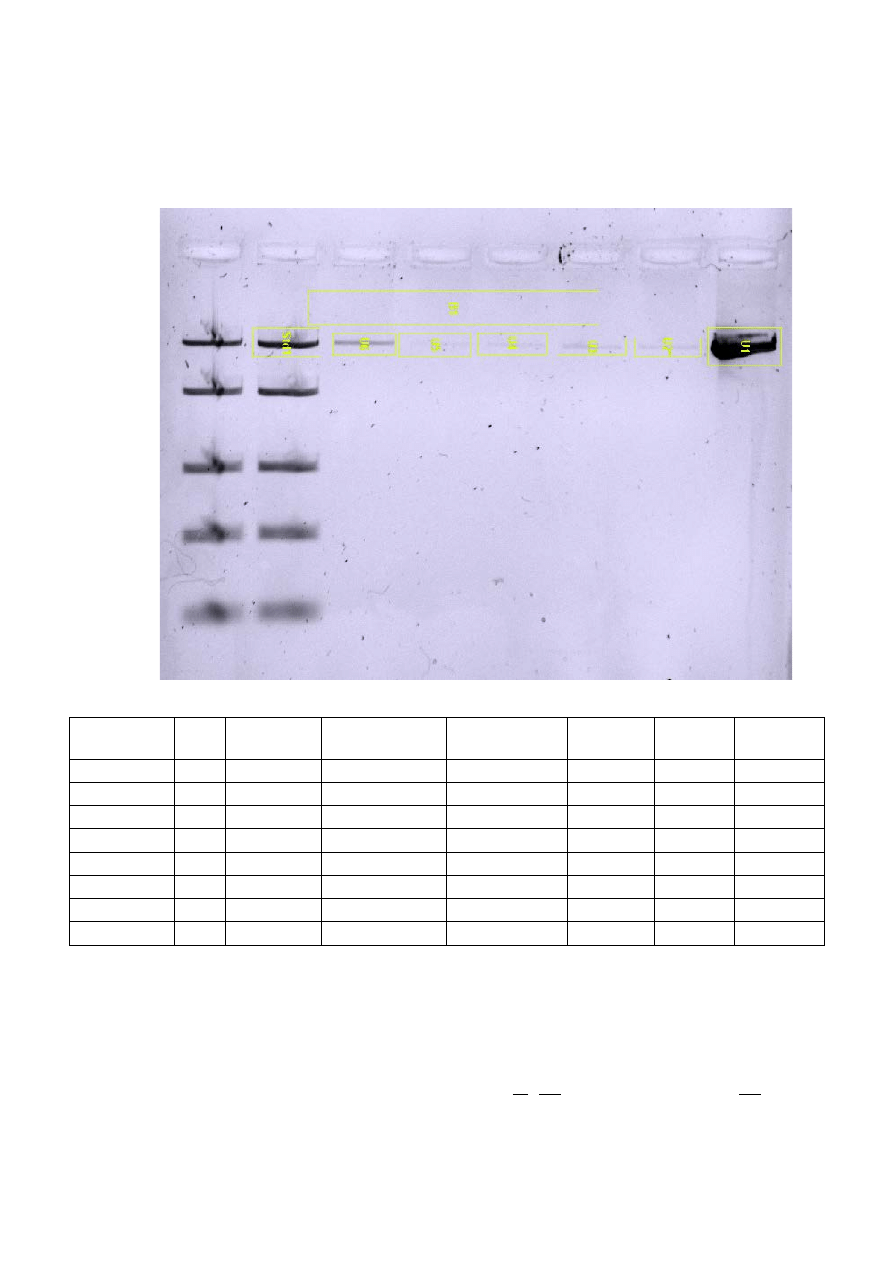
• Wyniki elektroforetycznej analizy plazmidowego DNA, oczyszczonego po trawieniu i
defosforylacji przedstawione są poniżej. Żel analizowano w świetle UV przy krótszej długości fali
254 nm.
No.
Label
Type
Mean Bkgd.
(Int)
Abs. Quant.
(ng)
Rel.
Quant.
# of
Pixels
Area
(mm2)
1
U1
Unknown
489,61
103,23
263,87
5885,00
24,60
2
U2
Unknown
338,98
0,39
1,00
2592,00
10,84
3
U3
Unknown
332,32
0,93
2,38
2574,00
10,76
4
U4
Unknown
330,42
0,66
1,70
3162,00
13,22
5
U5
Unknown
325,43
0,46
1,18
3605,00
15,07
6
U6
Unknown
330,27
3,12
7,99
3003,00
12,55
7
B1
Background
344,04
N/A
N/A
20496,00
85,68
8
Std1
Standard
364,50
20,00
51,12
4158,00
17,38
• Wymagana była minimalna ilość 5 ng wektora, dlatego też do dalszych badań użyto próbki grupy I
(U1), gdzie wektor znajdował się w najwyższym stężeniu. Elektroforezę przeprowadzono przy 3 μl
roztworu plazmidowego. Stężenie to policzono wg. poniższego schematu:
23
,
103
=
m
[ng]
3
=
V
[μl]
=
l
ng
V
m
C
µ
=
l
ng
C
wekt
µ
41
,
34
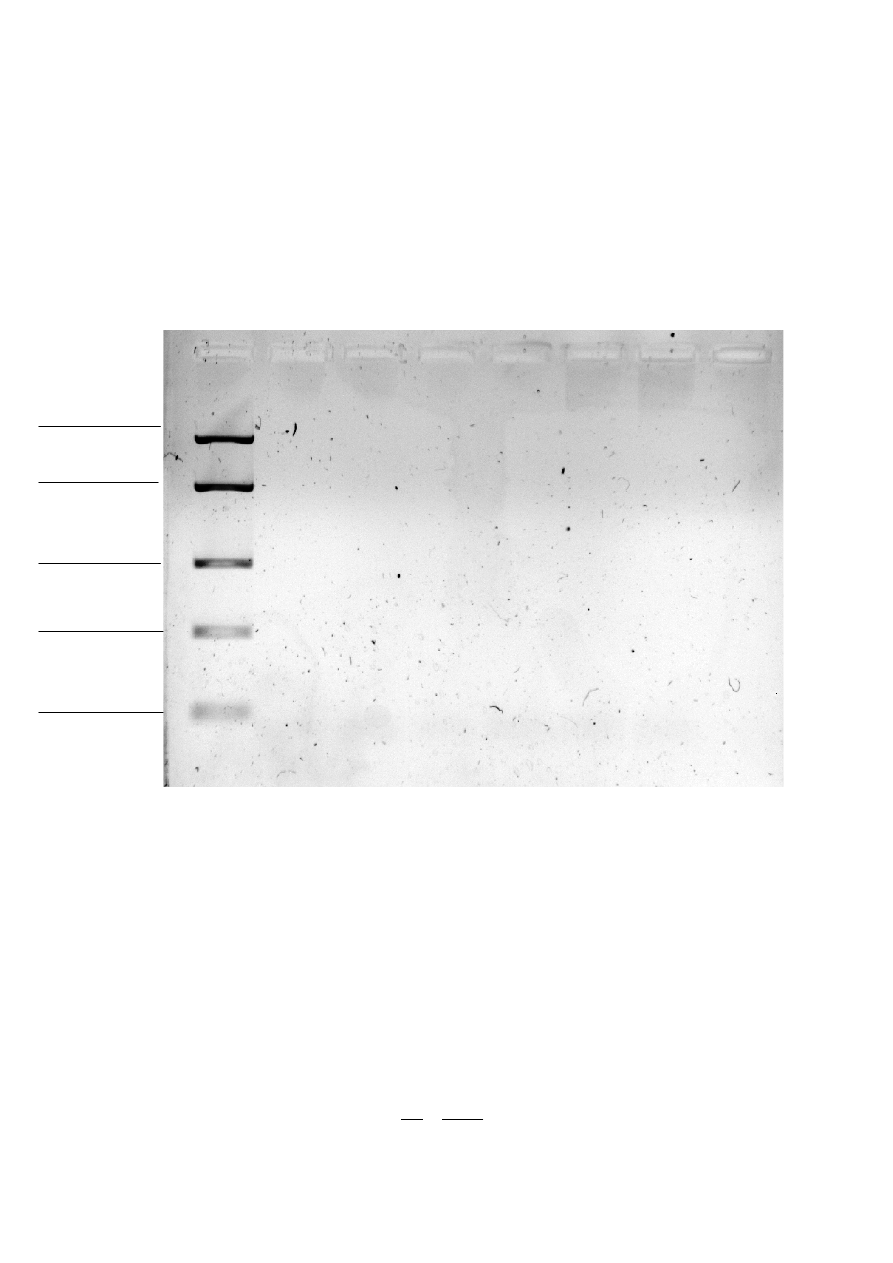
400 bp
100 bp
• Podobnie, w elektroforezie analitycznej produktów PCR zastosowano końcową objętość 12 μl.
Pobrano 5 μl próbki po PCR spod oleju, 5 μl roztworu TE i 2 μl 6xSB (aby jego końcowe stężenie
w 12 μl wynosiło 1x). Jednak po nabraniu całej zawartości do pipety okazało się, że dokładna ilość
mieszaniny reakcyjnej wynosi 11,7 μl.
• Kontrola przygotowana została tak samo, jak próbka, jednak 5 μl próbki po PCR pobierano
z kontroli z reakcji PCR. Końcowa objętość wynosiła 12 μl.
• Elektroforeza analityczna produktów PCR niestety nie przyniosła pożądanych rezultatów.
Widoczna jest jedynie smuga po użytych starterach przy każdej studzience. Żel analizowano
w świetle UV przy krótszej długości fali 254 nm.
• Podczas izolacji produktu PCR zastosowano 5-krotny nadmiar roztworu G (223 μl). Dokładną
objętość wyliczono na podstawie posiadanych ilości produktu PCR: 39,3 μl i 5,3 μl – razem 44,6 μl.
223
5
6
,
44
=
⋅
μl roztworu G
• Do ligacji wektora pGEX-2T, zlinearyzowanego enzymem BamHI i defosforylowanego
(pGEX-2T/BamHI) fragmentem DNA kodującym domenę wiążącą DNA receptora
ekdysteroidowego (EcRDBD/BamHI) przy użyciu ligazy T4 użyto następujących ilości
poszczególnych składników:
Wyjściowe stężenie wektora, obliczone na podstawie wyników elektroforezy, wynosiło 34,41
ng/μl. Wymagane było rozcieńczenie do stężenia 15 ng/μl, zatem policzono rozcieńczenie:
29
,
2
15
41
,
34
≅
=
=
p
k
V
V
R
5000 bp
850 bp
2000 bp
Zatem mamy objętość 7,5 μl wektora
p
k
V
V
R
=
p
k
V
R
V
⋅
=
175
,
17
5
,
7
29
,
2
=
⋅
=
k
V
μl
Objętość roztworu TE:
7
,
9
675
,
9
5
,
7
175
,
17
≅
=
−
=
TE
V
μl
(wtedy właśnie stężenie wektora wynosi 15 ng/μl)
Wektor pGEX-2T:
15
=
wektora
C
ng/μl
20
=
wektora
m
ng (20 x 10
-9
g)
V
m
C
wektora
=
C
m
V
wektora
=
l
V
wektora
µ
5
,
1
33
,
1
15
20
≅
=
=
W przypadku insertu DNA stosowany był 4-krotny nadmiar molowy (n
insertu
= 4n
wektora
):
3
=
insertu
C
ng/μl
4948
=
wektora
L
pz
306
=
insertu
L
pz
640
=
bp
M
g/mol pz
3166720
4948
640
=
⋅
=
wektora
M
g/mol
195840
306
640
=
⋅
=
insertu
M
g/mol
15
9
10
316
,
6
3166720
10
20
−
−
⋅
=
⋅
=
=
wektora
wektora
wektora
M
m
n
mol
14
15
10
526
,
2
10
316
,
6
4
4
−
−
⋅
=
⋅
⋅
=
⋅
=
wektora
insertu
n
n
mol
95
,
4
10
95
,
4
195840
10
526
,
2
9
14
≅
⋅
=
⋅
⋅
=
⋅
=
−
−
g
M
n
m
insertu
insertu
insertu
ng
insertu
insertu
insertu
C
m
V
=
l
C
m
V
insertu
insertu
insertu
µ
65
,
1
3
95
,
4
=
=
=
• Po inkubacji bakterii w 37ᴼC policzono kolonie transformantów wyrosłe na płytce po ligacji i
płytce kontrolnej. Stopień religacji wektora policzono, dzieląc liczbę kolonii wyrosłych na płytce
kontrolnej przez liczbę kolonii na płytce „po ligacji”. Wynik podano w procentach:
Liczba kolonii na płytce kontrolnej:
2
Liczba kolonii na płytce „po ligacji”:
14
Stopień religacji:
%
28
,
14
%
100
14
2
%
=
⋅
=
religacji

3. Wnioski
Zastosowane zostały startery WR5 i WR6 o długości odpowiednio 29 i 30 nukleotydów o
temperaturze stapiania równej 58ᴼC, wyliczonej na podstawie obecności poszczególnych par zasad
we fragmentach. Im starter jest dłuższy, tym specyficzność reakcji PCR jest wyższa.
Jony Mg
2+
obecne w mieszaninie reakcyjnej PCR w buforze tworzą kompleksy z DTP, starterami i
matrycą DNA, stąd też wymagane jest ich odpowiednie stężenie. Jeśli jest ono zbyt niskie, produktów
PCR jest mało i wydajność reakcji spada. Z kolei jeśli występuje zbyt wysokie stężenie, może powstać
wiele niespecyficznych produktów.
Ważne są równe ilości dNTP (dATP, dCTP, dGTP i dTTP) w mieszaninie reakcyjnej, gdyż różnica w
stężeniu nawet jednego z deoksynukleotydów drastycznie zwiększa poziom źle włączonych zasad.
50 μl oleju parafinowego dodano na powierzchnię mieszaniny reakcyjnej w celu zapobieżenia jej
parowaniu. Tzw. „gorący start” zastosowany został w celu stworzenia optymalnych warunków dla
działania polimerazy DNA. Nastąpiła denaturacja ewentualnych zanieczyszczeń białkowych i
zwiększona została wydajność enzymu. Dopiero po przeprowadzeniu „gorącego startu” dodano 1 μl
enzymu.
Z racji wprowadzania miejsca restrykcyjnego, przeprowadzone zostały 2 cykle podczas reakcji PCR.
Wynika to z faktu, że użyta matryca nie zawierała wcześniej miejsca restrykcyjnego.
W wyniku elektroforezy analitycznej nie otrzymano produktów PCR. Możliwe jest, że do mieszaniny
reakcyjnej nie dodano matrycy DNA, co objawia się właśnie brakiem tej matrycy na żelu. Widoczna
jest jedynie smuga po użytych starterach.
Wyszukiwarka
Podobne podstrony:
El sprawko 5 id 157337 Nieznany
plastiki sprawko id 362078 Nieznany
przetwarzanie sprawko 3 id 4066 Nieznany
automatyka sprawko 2 id 73363 Nieznany
3 Sprawko id 34095 Nieznany
Elektronika cw6 sprawko id 1589 Nieznany
DC impulsowo sprawko id 132337 Nieznany
Metrologia Sprawko id 297285 Nieznany
jeszcze raz sprawko5 (1) id 227 Nieznany
El sprawko 5 id 157337 Nieznany
plastiki sprawko id 362078 Nieznany
przetwarzanie sprawko 3 id 4066 Nieznany
IG Konferencja ID Formatka DK i Nieznany
Fortuna sprawko Figo id 179936 Nieznany
IG Konferencja ID Formatka DK i Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
więcej podobnych podstron