
Ćwiczenie 3
Aktywność enzymu
Charakterystyczną cechą wszystkich enzymów jest ich specyficzność działania i duża
aktywność katalityczna. Przyspieszają reakcje chemiczne co najmniej milionkrotnie, a przy
ich braku reakcje te zachodziłyby tak wolno, że praktycznie niezauważalnie. Enzymy
obniżają wymaganą ilość energii aktywacji, dzięki czemu zwiększa się szybkość reakcji.
Wpływają jedynie na takie procesy chemiczne, które są termodynamicznie możliwe,
przyspieszając osiągnięcie stanu końcowej równowagi chemicznej, do którego dąży dana
reakcja. Jednocześnie działają specyficznie zarówno pod względem rodzaju katalizowanej
reakcji, jak i substratów.
Ogólny schemat reakcji katalizowanej przez enzym jest następujący:
E+S ES E+P
1. Enzym łączy się z substratem w miejscu nazywanym centrum aktywnym enzymu,
tworząc przejściowy, nietrwały kompleks enzym-substrat. Następują wówczas
przesunięcia w układzie elektronów zgrupowanych dookoła określonych wiązań
chemicznych w substracie, sprzyjające rozluźnieniu tych wiązań, dlatego substrat staje się
bardziej aktywny.
2. Kompleks enzym-substrat rozpada się, czemu towarzyszy powstawanie produktów reakcji
i zregenerowanie enzymu.
Dowody na tworzenie kompleksu enzym-substrat są następujące:
•
badania kinetyki reakcji enzymatycznych,
•
zmiany właściwości fizykochemicznych enzymu w tym m.in. rozpuszczalności
czy widma pochłaniania promieniowania elektromagnetycznego po związaniu
substratu,
•
obserwacje metodami rentgenograficznymi innych form krystalicznych dla
samego enzymu niż dla jego kompleksu z substratem lub analogiem substratu,
•
w przypadku dużych struktur można niekiedy oglądać połączenie enzymu
z substratem w mikroskopie elektronowym.
Zdolność katalityczna enzymu, czyli jego aktywność, wyraża wzrost szybkości rekcji
w ściśle określonych warunkach. Zazwyczaj szybkość reakcji jest wyrażana jako zmiana
stężenia substratu lub produktu w jednostce czasu [mol/( l
x
s)].
Jednostką aktywności enzymatycznej stosowaną w układzie SI jest katal. Odpowiada
on przekształceniu 1 mola substratu lub wytworzenia 1 mola produktu w ciągu 1s.
Standardowo stosowaną jednostką jest jednak międzynarodowa jednostka aktywności
enzymu (IU). Jest to taka ilość enzymu, która katalizuje przemianę jednego µM substratu
w ciągu jednej minuty w temperaturze 30
o
C w standardowych warunkach (1 IU = 16,7 nkat).
Na szybkość przebiegu reakcji enzymatycznej wpływają:
• temperatura
• pH
• siła jonowa
• stężenie substratu
• stężenie enzymu
• obecność aktywatorów
• obecność inhibitorów
Wpływ temperatury na aktywność enzymów
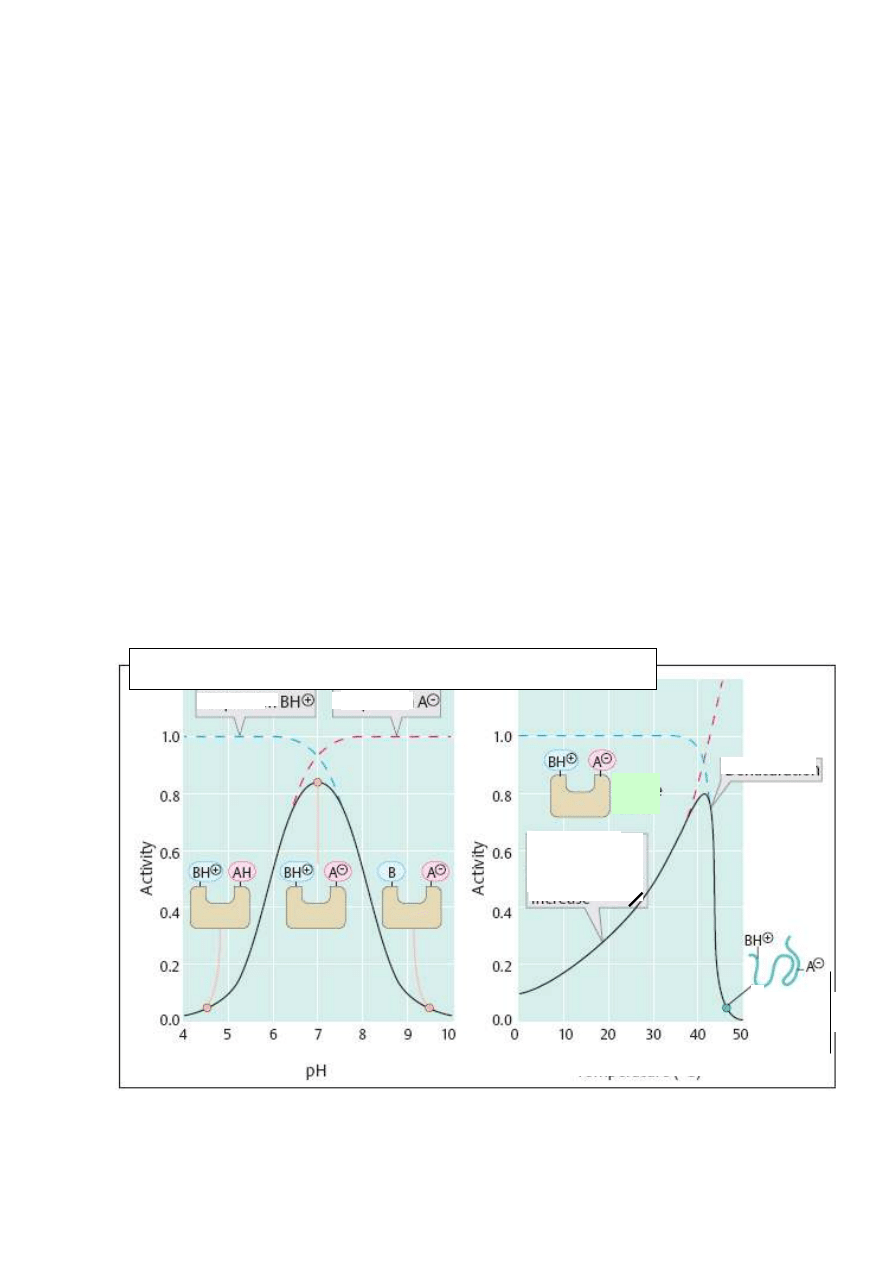
Wzrost temperatury zwiększa szybkość reakcji enzymatycznych, ponieważ wywołuje
wzrost energii kinetycznej reagujących cząsteczek i generuje większą częstość ich zderzeń.
Z reguły van’t Hoffa wiadmo, że podwyższenie temperatury o 10
o
C powoduje dwu- -
trzykrotny wzrost szybkości reakcji chemicznych. Ta sama reguła może być stosowana do
reakcji enzymatycznych, ale tylko w pewnym zakresie. Ponieważ enzym jest substancją
białkową, wzrost temperatury powyżej temperatury optymalnej dla jego działania powoduje
stopniową denaturację i zanik zdolności katalitycznych. Generalnie uznaje się, że w granicach
temperatury 0-30
o
C następuje wyłącznie wzrost szybkości reakcji zgodny z powyższą zasadą,
jednak przy dalszym wzroście temperatury szybkość reakcji rośnie nadal, jednak
równocześnie postępuje denaturacja enzymu i przyrosty szybkości są coraz mniejsze, aż
wreszcie denaturacja postępuje tak bardzo, że szybkość gwałtownie maleje.
Należy przy tym pamiętać, że optymalna temperatura dla działania enzymów i temperatura
przy której ulegają denaturacji zależy od ich pochodzenia.
Wpływ odczynu środowiska na aktywność enzymów.
Do utrzymania aktywności katalitycznej enzymy wymagają odpowiedniego odczynu pH
środowiska. Dla większości enzymów optymalne jest pH 5,5 – 7,4. Znane są jednak enzymy,
które działają najlepiej w środowisku kwaśnym (np. pepsyna w pH 1,5 – 2,7) lub zasadowym
( trypsyna, chymotrypsyna – pH 8 – 9 ).
Środowisko silnie kwaśne i silnie zasadowe z reguły działa na enzymy denaturująco,
niszcząc nieodwracalnie ich aktywność. Niewielkie zmiany pH nie dezaktywują enzymu, ale
obniżają szybkość reakcji, ponieważ wpływając na stopień jonizacji enzymu i substratu,
zmieniają warunki tworzenia się kompleksu enzym-substrat.
Dla większości enzymów optymalne jest środowisko obojętne lub słabo kwaśne.
Rys. 2. Wpływ pH środowiska i temperatury na aktywność enzymu.
temperatura (
o
C)
enzym
zdenaturowany
wzrost
aktywności
denaturacja
aktywny
enzym
udział
Wpływ pH środowiska i temperatury na aktywność enzymu
udział
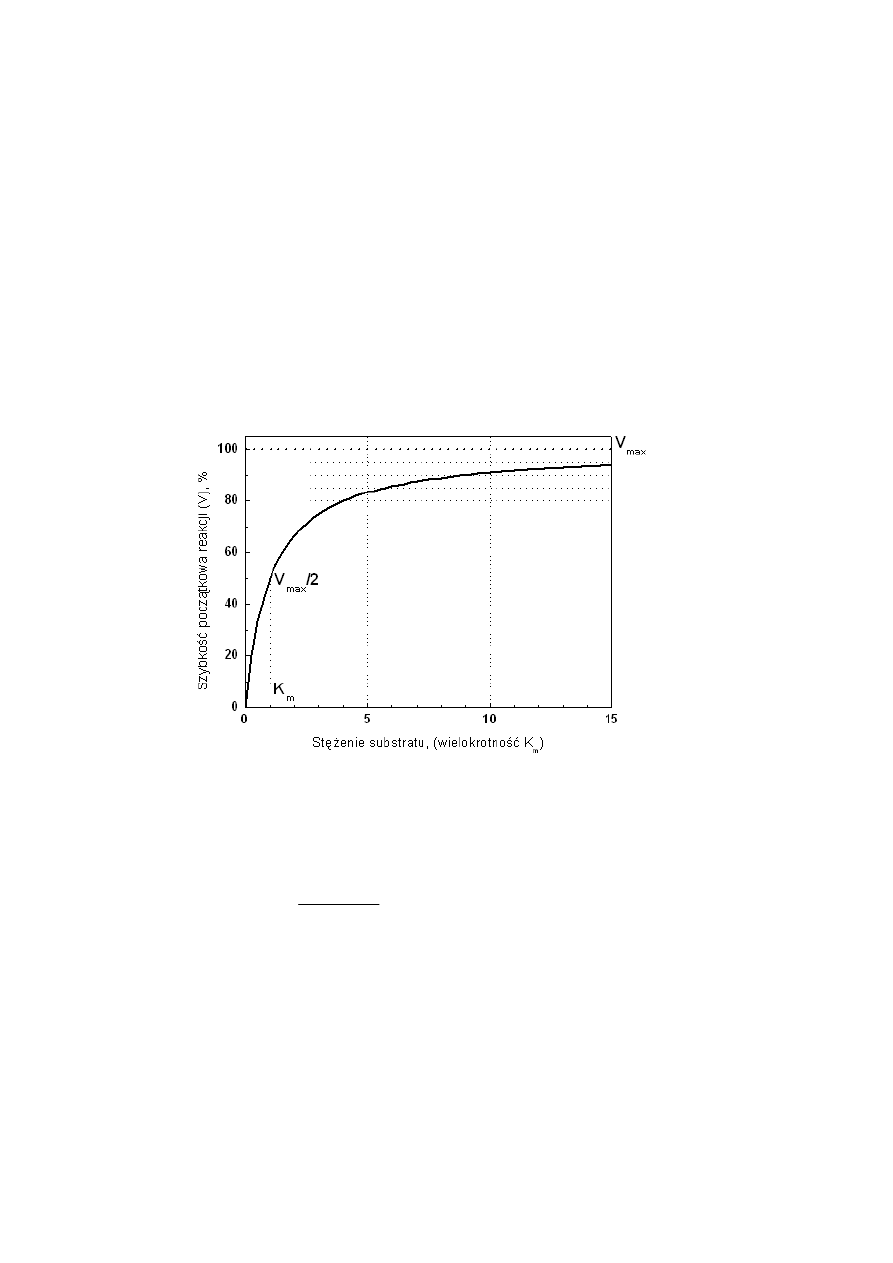
Wpływ stężenia substratu i stężenia enzymu na szybkość reakcji enzymatycznej
Dla wielu enzymów szybkość katalizy zmienia się ze stężeniem substratu. Zależność ta
została opisana przez Michaelisa i Menten. Model przez nich opracowany zakłada, że:
•
koniecznym etapem prowadzenia katalizy jest utworzenie kompleksu pośredniego
enzym-substrat
•
istnieją dwie możliwości rozpadu komplesku pośredniego: na enzym i produkt lub
enzym i substrat reakcji
•
nie jest możliwa reakcja odwrotna, tj. przekształcenie produktu w substrat.
Szybkość reakcji enzymatycznej katalizowanej przez enzymy podlegające kinetyce
Michaelisa-Menten jest zależna od stężenia substratu. Przy niskich stężeniach substratu
wzrasta proporcjonalnie do wzrostu jego stężenia. Reakcja osiąga szybkość maksymalną przy
takim stężeniu substratu, gdy wszystkie centra aktywne enzymu są wysycone substratem.
Dalsze zwiększanie stężenia substratu nie powoduje zmian szybkości katalizy.
Rys. 3. Zależność hiperboliczna opisująca równanie Michaelisa-Menten .
Szybkość reakcji enzymatycznych opisuje równanie Michaelisa – Menten:
]
[
]
[
max
S
Km
S
V
V
+
×
=
gdzie: K
m
– stała Michaelisa, równa takiemu stężeniu substratu [S], przy którym szybkość
reakcji V jest połową szybkości maksymalnej V
max
i opisywana równaniem:
K
m
=(k
2
+k
3
)/k
1
gdzie: k
1
– stała szybkości tworzenia kompleksu enzym-substrat
k
2
– stała szybkości rozpadu kompleksu enzym substrat w kierunku uwolnienia
substratu
k
3
– stała szybkości rozpadu kompleksu enzym-substrat z wytworzeniem produktu
Stała Michaelisa-Menten stanowi wartość charakterystyczną dla danego enzymu
w odpowiednich warunkach pH i temperatury. Określa powinowactwo enzymu do substratu.

Jeżeli jest niskie, K
m
jest wysoka, jeżeli enzym ma wysokie powinowactwo do substratu –
niska.
Znając wartość Km można tak dobierać stężenie substratu, aby osiągnąć stan
nasycenia enzymu. Odpowiada ona takiemu stężeniu substratu, dla którego szybkość reakcji
enzymatycznej osiąga połowę swojej maksymalnej wartości.
Wpływ aktywatorów i inhibitorów na aktywność enzymów
Enzymy mogą podlegać zarówno inhibicji, jak i aktywacji przez różne specyficzne
cząsteczki lub jony. Ma to szczególnie istotne znaczenie dla kontroli fizjologicznej ich
działania w układach biologicznych. W ten sam sposób działa również wiele leków
i czynników toksycznych.
Jak już wspomniano, aktywność enzymu w organizmie podlega kontroli
fizjologicznej. Niektóre z enzymów są syntezowane w postaci nieczynnych prekursorów
i ulegają aktywacji w pożądanym czasie oraz miejscu, np. enzym trawienny – trypsyna
wydzielana jest w trzustce jako trypsynogen, a jako czynny enzym pojawia się w jelicie
cienkim dopiero po hydrolizie wiązania peptydowego, która powoduje odczepienie krótkiego
peptydu i przejście proenzymy w formę aktywną.
Enzymy mogą również podlegać modyfikacji kowalencyjnej. Aktywność jest
wówczas regulowana przez przyłączenie małej grupy chemicznej do cząsteczki enzymu, np.
w wyniku fosforylacji, adenylacji czy rybozylacji.
Większość enzymów wymaga dla zachowania swojej aktywności aktywatorów.
Aktywatorami nazywamy związki niskocząsteczkowe, których obecność w miejscu katalizy
enzymatycznej przyspiesza przebieg reakcji. Aktywatorami enzymów mogą być jony metali
(np. Mn
2+
, Co
2+
, Zn
2+
), aniony współdziałające z białkami (np. Cl
-
), a także związki
regulujące potencjał oksydoredukcyjny środowiska, od których zależy budowa centrów
aktywnych.
Inhibitory są substancjami hamującymi działanie enzymów. Inhibicja cząsteczki
enzymu może zachodzić pod wpływem małych cząsteczek lub jonów, zarówno
nieodwracalnie jak i odwracalnie.
W inhibicji nieodwracalnej inhibitor wiąże się kowalencyjnie z enzymem tak silnie, że
jego dysocjacja jest bardzo powolna (np. działanie gazów paraliżujących na układ nerwowy).
Te inhibitory nazywane są często „truciznami enzymów”, ponieważ powodują chemiczną
modyfikację aminokwasów enzymu. Najczęściej brak jest podobieństwa pomiędzy
inhibitorem i substratem, ale jeżeli takie podobieństwo występuje, wówczas związanie
substratu przez enzym działa na enzym ochronnie.
W inhibicji odwracalnej szybko osiągany jest układ enzym-inhibitor. Podstawowe
typy odwracalnej inhibicji, to:
♦ Inhibicja kompetycyjna, kiedy inhibitor jest podobny do substratu i wiąże się
w miejscu aktywnym enzymu, blokując wiązanie substratu. Wówczas inhibitor
współzawodniczy z enzymem o centrum aktywne. Często taki typ inhibicji
występuje, gdy produkt reakcji jest podobny do substratu. Wówczas reakcja
enzymatyczna hamowana jest przez produkt (zjawisko sprzężenia zwrotnego).
♦ Inhibicja niekompetycyjna, kiedy inhibitor wiąże się z cząsteczką enzymu
w innym miejscu niż centrum aktywne enzymu. Wtedy następuje zmiana
konformacji enzymu, która pociąga za sobą zmianę konformacji centrum
aktywnego.
Aktywatory i inhibitory mają szczególne znaczenie dla podniesienia szybkości
katalizy prowadzonej przez enzymy allosteryczne. W cząsteczce takich enzymów poza
centrum aktywnym występuje również centrum regulatorowe, ulokowane najczęściej na innej
podjednostce białka. Przyłączenie do tego centrum efektora (tj. aktywatora lub inhibitora)
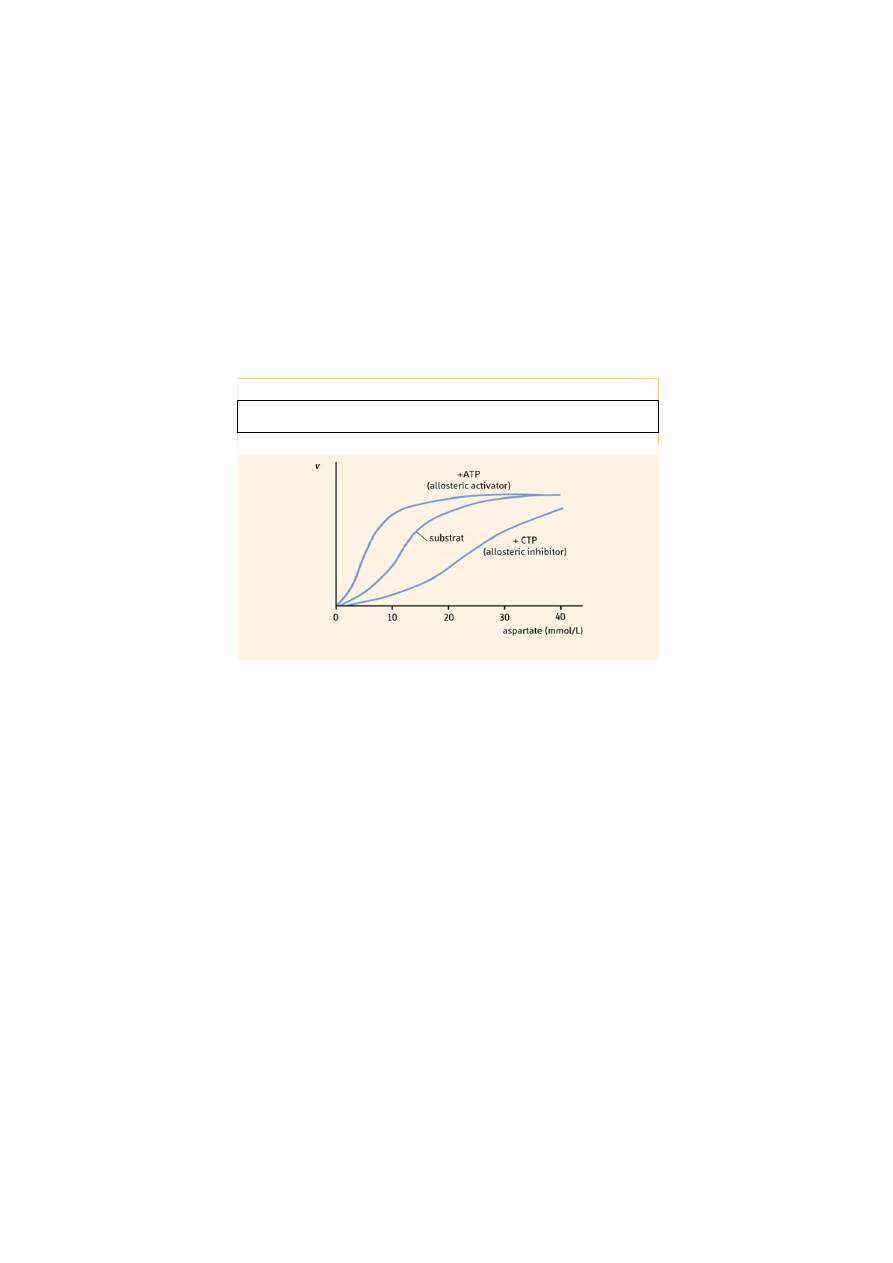
powoduje gwałtowną zmianę szybkości katalizy. Proces aktywacji bądź inhibicji takiego
enzymu jest procesem odwracalnym. Enzymy takie katalizują kluczowe reakcje szlaków
metabolicznych i nie podlegają kinetyce Michaelisa-Menten.
Kinetykę allosterycznych enzymów opisuje model sigmoidalny. Szybkość reakcji
katalizowana przez enzym allosteryczny przed związaniem efektora jest bardzo niska
i zmienia się nieznacznie w zależności od stężenia substratu. Związanie aktywatora powoduje
gwałtowny wzrost szybkości reakcji, aż do poziomu pełnego wysycenia enzymu substratem.
Natomiast związanie inhibitora powoduje jeszcze większe opóźnienie uwalniania produktu
z kompleksu przejściowego.
Charakterystyczne dla tych enzymów jest występowanie zjawiska pozytywnej
kooperacji, czyli ułatwienie wiązania cząsteczek substratu do kolejnych centrów aktywnych
wskutek zmian konformacyjnych, jakie wywołane zostały w białku poprzez przyłączenie
pierwszej cząsteczki substratu.
Rys. 4. Model sigmoidalny dla regulacji ATC-azy. ATP – allosteryczny aktywator,
CTP – allosteryczny inhibitor enzymu.
Wśród czynników, które wpływają na szybkość reakcji enzymatycznej należy
dodatkowo wyróżnić grupę inaktywatorów, czyli czynniki, które doprowadzają do
niespecyficznego i nieodwracalnego hamowania enzymu poprzez jego denaturację. Należą
do nich zarówno czynniki chemiczne (np. niektóre rozpuszczalniki organiczne czy stężone
roztwory soli metali ciężkich), jak i czynniki fizyczne (np. promieniowanie jonizujące lub
ultradźwięki).
W ćwiczeniu ostatnim z cyklu analizowana będzie aktywność wyizolowanych
i oczyszczonych amylaz oraz wpływ różnorodnych, wyżej wymienionych czynników na ich
aktywność.
Metody oznaczania aktywności amylaz opierają się najczęściej na wyznaczaniu ilości
cukrów redukujących, powstających podczas hydrolizy skrobi. Wykorzystuje się w tym celu
reakcję z kwasem 3,5-dinitrosalicylowym (DNS), który w obecności cukrów redukujących
przekształcany jest do kwasu 3-amino-5-nitrosalicylowego, tworząc barwne kompleksy
z fenolem. Pomiar absorbancji następuje przy długości fali λ= 530 nm. Stężenie cukrów
redukujących odczytuje się z krzywej wzorcowej sporządzonej dla glukozy.
Allosteryczna regulacja ATC-azy

Badanie wpływu temperatury, pH, inhibitorów, aktywatorów, czasu
przechowywania na aktywność enzymu.
1\ Odczynniki
(1)
bufor uniwersalny:
dla amylazy trzustkowej o pH 7,0
dla amylazy zbożowej o
pH 5,5
(2)
1% roztwór skrobi (odczynnik do samodzielnego przygotowania)
(3)
DNS
(4)
bufor uniwersalny z 1 M NaCl
(5)
bufor uniwersalny z 10 mM FeSO
4
(6)
0,2 M Na
2
HPO
4
(7)
0,1 M kwas cytrynowy
(8)
roztwór I w KI (0,3% I
2
w 3% KI)
(9)
roztwór 1% NaCl
2\ Przygotowanie kleiku skrobiowego
Odmierzyć 50 ml odpowiedniego buforu (1). Odważyć 0,5 g skrobi i przygotować
zawiesinę w ok. 5 ml buforu (1), 40 ml buforu (1) zagotować. Do wrzącego buforu wlać
zawiesinę skrobiową, przepłukać zlewkę od zawiesiny pozostałą częścią buforu, zagotować
i szybko schłodzić otrzymany 1 % kleik skrobiowy (2).
3\ Bilans aktywności dla wykonanego procesu oczyszczania
Rozmrozić próbkę A, B, C i D. Próbki A, B i D zawierające amylazy izolowane ze
słodu rozcieńczyć 10 razy, dodając do 1 ml roztworu enzymu 9 ml buforu uniwersalnego o
pH 5,5 (1). Próbki A, B i D zawierające amylazy trzustkowe rozcieńczyć 50 razy, dodając do
100 µl roztworu enzymu 4900 µl buforu uniwersalnego o pH 7,0 (1). Próbki C nie
rozcieńczać.
Z otrzymanych rozcieńczeń i próbki C pobrać po 100 µl i dodać do 1000 µl 1% skrobi
(2). Zamieszać i inkubować przez 5 min w temp. 35
o
C. W ten sam sposób przygotować próbę
zerową, zamiast enzymu dodając bufor (1). Próby wykonać w dwóch powtórzeniach.
Reakcję zatrzymać przez dodanie 1000 µl DNS (12) i ogrzewanie we wrzącej łaźni
wodnej przez 5 min. Gwałtownie schłodzić próbki i dodać po 10 ml H
2
O
dest
. Odczytać
absorbancję przy 530 nm.
4\ Działanie inhibitorów i aktywatorów na aktywność enzymu.
Równolegle do próbek przygotowywanych w celu oznaczenia bilansu aktywności α-
amylazy przygotować próbki do wyznaczenia aktywności enzymu w obecności inhibitora
(mocznika) i aktywatora (jonów chlorkowych).
W tym celu rozcieńczyć odpowiednio (enzymy słodowe – 10 razy, trzustkowe 50
razy) próbkę D dodając:
A\ bufor uniwersalny z 1 M NaCl (4)
B\ bufor uniwersalny z 10 mM FeSO
4
(5)
Pobrać z przygotowanych próbek po 100 µl, dodać probówek zawierających po 1000 µl
roztworu skrobi (2). Dalszy tok postępowania jest analogiczny do metody w punkcie 3\.
5\ Wpływ temperatury na aktywność enzymu
Do 5-u probówek dodać po 500 µl roztworu skrobi (2), 500 µl odpowiedniego dla
danego enzymu buforu (1) oraz 10 kropli 1% NaCl (9). Zamieszać i inkubować przez 5 min

w temp. 4, 25, 35, 60 i 100
o
C. Następnie do każdej probówki dodać po 100 µl enzymu D
(nierozcieńczonego) zmieszać i inkubować dalszych 10 min. Do wszystkich probówek dodać
po 1-2 kropli roztworu I w KI (8), zmieszać. Określić temperaturę, w której amylaza
wykazuje największą aktywność.
6\ Wyznaczanie pH optymalnego dla działania α
α
α
α
-amylazy
Do dziesięciu probówek wlać 0,2 M roztwór Na
2
HPO
4
(6) oraz 0,1 M roztwór kwasu
cytrynowego (7) w ilościach przedstawionych w tabeli 1. Otrzymuje się roztwory buforowe o
pH od 4,4 do 8,0. Do każdej probówki dodać 10 kropli 1% NaCl (9) oraz 10 kropli roztworu
skrobi (2). Po zmieszaniu wlać do każdej probówki po 100 µl próbki D (nierozcieńczonej).
Roztwory zmieszać i umieścić na 10 min w łaźni o temp. 35
o
C. Do wszystkich probówek
dodać po 1-2 kropli roztworu I w KI (8), zmieszać.
Określić pH, przy którym amylaza wykazuje największą aktywność.
Lp. 0,2 M Na
2
HPO
4
[ml]
0,1 M roztwór kwasu cytrynowego
[ml]
pH mieszaniny
1
2
3
4
5
6
7
8
9
10
0,44
0,49
0,56
0,59
0,63
0,69
0,77
0,87
0,94
0,97
0,56
0,51
0,44
0,41
0,37
0,31
0,23
0,13
0,06
0,03
4,4
4,8
5,2
5,6
6,0
6,4
6,8
7,2
7,5
8,0
Tab.1. Przygotowanie buforu Mc Ilvaine’a w zakresie pH 4,4-8,0.
7\ Obliczenia.
Wyznaczyć aktywność α- amylazy, przyjmując jako jednostkę aktywności (JAA) ilość
maltozy (µM) uwolnionej podczas 1-minutowej hydrolizy 1 %-owej skrobi prowadzonej
w temp. 35
o
C przy odpowiednim dla danego enzymu pH.
JAA=
5
1
558
,
0
1
100
1000
1
1
×
×
×
=
×
×
×
E
t
Em
Vpr
V
E
gdzie:
V – objętość, w której wyrażamy aktywność enzymu (1000 µl)
V pr – objętość próbki enzymu (100 µl)
t – czas inkubacji (5 min)
E – odczytana wartość ekstynkcji
Em – wartość ekstynkcji dla 1 µM maltozy
Wyznaczyć bilans aktywności dla procesu oczyszczania α- amylazy oraz wpływ aktywatorów
i inhibitorów na aktywność enzymu.
Podsumowanie ćwiczeń
Porównać wyniki z ćwiczeń 1-3 dla amylazy trzustkowej i zbożowej, zestawić
w tabelach.
Wyszukiwarka
Podobne podstrony:
Czynniki wpływające na aktywność enzymów
czynniki wpływające na zmeczenie psychiczne w pracy
(2,3) Działania nieporządane, toksytczne leków Metabolizm, czynniki wpływające na działanie substanc
CZYNNIKI WPŁYWAJĄCE NA KSZTAŁTOWANIE SIĘ POSTAW
Podstawowe czynniki wpływające na wartość opcji na akcje
85 Omow czynniki wplywajace na lepkosc krwi
Uczenie się - czynniki wpływające na nabieranie wprawy, Prace z socjologii, pedagogiki, psychologii,
Czynniki wplywajace na rentownosc bankow w polskim sektorze bankowym
czynniki wpływające na starość+ rozwój w późnej dorosłości, tradycje opieki i pomocy społecznej, Kon
Czynniki wpływające na wzrost roślin, Akwarium
gegra-powietrze, Czynniki wpływające na temperaturę powietrza:
czynniki wpływające na Wielkość PPM
Czynniki wpływające na zachowanie konsumenta
(), biochemia L, Wpływ temperatury na aktywność enzymów (ćw E)
Czynniki wpływające na szybkość biodeodoryzacji
więcej podobnych podstron