1
CHOROBY
DZIEDZICZONE
AUTOSOMALNIE
RECESYWNIE
Na przyk
ładzie mukowiscydozy i
fenyloketonurii.
Zak
ład Genetyki i Patomorfologii PAM
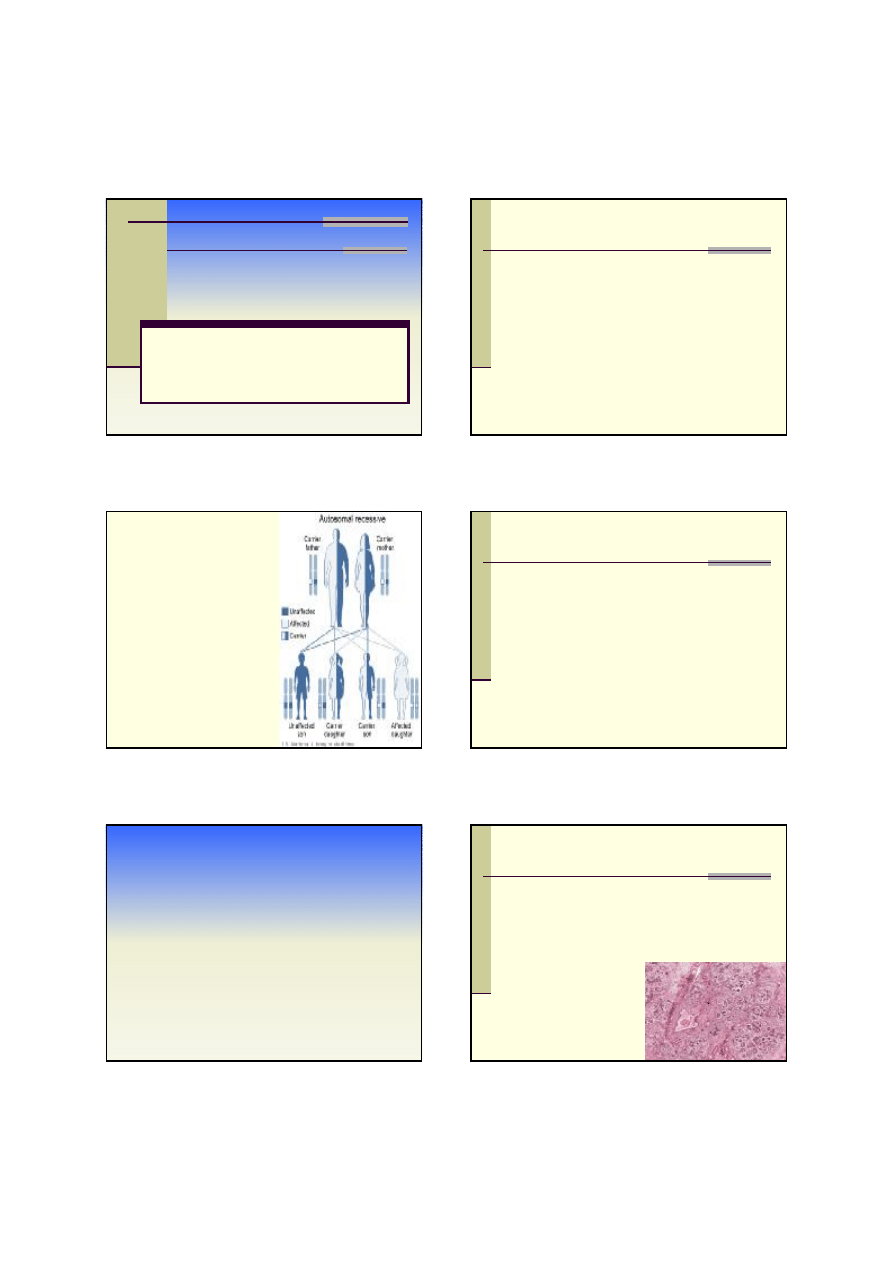
Cechy dziedziczenia AR
1. Choroba wyst
ępuje u homozygot (lub
heterozygot z
łożonych)
2. Ma
łe ryzyko dla potomstwa
3. Jednakowa cz
ęstość i ciężkość objawów
u obu p
łci
4. Sta
ła ekspresja w rodzinie
5. Poziomy wzór rodowodu
6. Wa
żne pokrewieństwo rodziców
n
25% ryzyko posiadania chorych
dzieci przez zdrowych rodziców nosicieli
(heterozygot)
n
50% ryzyko posiadania chorych
dzieci przez rodziców:
chory + nosiciel
n
100% ryzyko posiadania chorych
dzieci przez chorych rodziców
n
66% ryzyko rodze
ństwa osoby chorej
że jest heterozygotycznym nosicielem.
n
ryzyko posiadania chorego
potomstwa przez osob
ę chorą wynosi
zazwyczaj < 1%. Zale
ży od częstości
nosicielstwa choroby w populacji ogólnej.
n
cz
ęstość 2/1000
Przykłady chorób dziedziczonych
AR
n
CF
n
PKU
n
W PN
n
Hemochromatoza
n
Choroba W ilsona
n
Albinizm
n
Anemia sierpowata
n
Rdzeniowy zanik mi
ęśni
n
Galaktozemia
n
Tyrozynemia
n
G
łuchota wrodzona
n
Ślepota recesywna
n
Upo
śledzenie umysłowe recesywne
n
Zespó
ł nagich limfocytów
n
Zespó
ł niebieskich pieluszek
MUKOWISCYDOZA
CZ
ĘSTOŚĆ NOSICIELSTWA:
1:25
CZ
ĘSTOŚĆ CHOROBY:
1:2500
Mukowiscydoza. Synonimy.
n
(z)w
łóknienie torbielowate trzustki
n
choroba w
łóknisto-torbielowata trzustki
(morbus fibrosocysticus pancreatis)
n
cystic fibrosic (CF) lub fibrosis cystica
n
„choroba krwi,
łez i potu”
n
„choroba o wielu
maskach”

2
MUKOWISCYDOZA
n
Jest to wrodzona, nieuleczalna choroba
ogólnoustrojowa o różnorodnej ekspresji
klinicznej.
n
Jest najczęściej występująca chorobą jednogenową
n
Kliniczne objawy spowodowane są wydzielaniem
nieprawidłowej gęstej wydzieliny, co prowadzi do
upośledzenia drożności przewodów
wyprowadzających.
n
W CF zaburzenia dotyczą przede wszystkim
układów:
Oddechowego
Pokarmowego
Rozrodczego
MUKOWISCYDOZA
n
Chorob
ą tą dotknięte są zarówno noworodki,
niemowl
ęta, dzieci starsze, jak i dorośli.
n
D
ługość przeżycia, jak i jakość życia
pacjentów w du
żej mierze zależą od
wczesnego rozpoznania i prawid
łowego
leczenia.
n
Mimo znacznego post
ępu konwencjonalnych
metod terapii,
średnia długość życia chorych
nie przekracza 30 r.
ż.
Mukowiscydoza
n
Przy częstości urodzeń 400 000, w Polsce rocznie
rodzi się około 200 dzieci z CF
n
Około 2 milionów Polaków, w równym stopniu
M i K, jest nosicielami zmutowanego genu CFTR.
n
Polski Rejestr Mukowiscydozy, prowadzony w
Instytucie Gruźlicy i Chorób Płuc (Zespół
Pediatryczny im Jana i Ireny Rudników w Rabce), w
2000r. zawierał informacje o 977 żyjących chorych z
CF (wiek do 43 lat)
Mutacje w CFTR:
n
Mutacje dziel
ą się na poszczególne rodzaje w
zale
żności od mechanizmu ich powstawania
w nast
ępujących proporcjach:
- mutacje missense – 48%
- mutacje frameshift – 19%
- mutacje splicingowe – 15%
- mutacje nonsens – 12%
- pozosta
łe – 6%
CF
ü
Przyczyn
ą choroby jest mutacja genu
odpowiedzialnego za syntez
ę błonowego kanału
chlorkowego CFTR (cystic fibrosis transmembrane
conductance regulator).
ü
W ok. 55% dominuj
ącą mutacją jest mutacja ∆F508.
Najcz
ęstszym genotypem jest układ ∆F508/∆F508
(35%)
ü
Ok. 1500 mutacji !
ü
W przypadku niewydolno
ści zewnątrzwydzielniczej
trzustki wykazano zale
żność od genotypu mutacji
silna/silna. Pacjenci z mutacj
ą słabą nie wymagają
suplementacji enzymów trzustkowych.
Z czego wynika różnorodność
fenotypowa w CF?
n
Rodzaj mutacji: silna (nonsense, frameshift, splice,du
że
delecje i insercje), s
łaba (missense, małe delecje i insercje).
Mutacja s
łaba redukuje funkcję białka CFTR o ok. 10-30%.
n
Miejsce mutacji w genie (s
ą bardziej i mniej ważne regiony
dla funkcji bia
łka).
n
Wzajemne relacje pomi
ędzy zmutowanymi allelami:
silna/silna, s
łaba/silna, słaba/słaba
n
% funkcjonalnego bia
łka CFTR: homozygoty dla deltaF508
<2% funkcji bia
łka CFTR (zawsze mają niewydolność
trzustki); >5% to zazwyczaj sprawna trzustka; >10% g
łównie
bezp
łodność męska bez chorób płuc, niewydolność trzustki
czy nieprawid
łowej czynność gruczołów potowych.
n
Aktywno
ść genów modyfikujących z poza locus CFTR np.
PTGS1 i 2
n
Obecno
ść złożonych alleli

3
Genotype and phenotype in cystic fibrosis. Respiration. 2000;67(2):117-33.
Źródło:www.cfgenetherapy.org.uk/cysticfibrosismore.htm
Objawy ze strony ukł. oddechowego
(występują u > 90% chorych)
n
g
ęsty i lepki śluz, który zalega w oskrzelach (→
atelectasis) i jest pod
łożem dla rozwoju bakterii
(Pseudomonas aeruginosa, Burkholderia cepacia, St.
aureus)
n
wyst
ępuje uciążliwy kaszel, niekiedy duszność (pierwsze
objawy mog
ą wystąpić już w wieku niemowlęcym)
n
nawracaj
ące zapalenia oskrzeli i płuc, trudno
poddaj
ące się typowemu leczeniu, prowadzą do rozstrzeni
oskrzeli (bronchectases) oraz ropni p
łuc (abscessus
pulmonum)
n
Cor pulmonum, niewydolno
ść oddechowa i
w
łóknienie miąższu.
n
MIKRO: hiperplazja i hipertrofia komórek wydzielaj
ących
śluz oraz bronchitis et bronchiolitis chronica.
n
G
łówną przyczyną śmierci (ponad 90%) jest niewydolność
oddechowa.
Cd.
•
przewlek
łe zapalenie zatok bocznych nosa z polipami
(g
łównie u starszych dzieci i dorosłych)
•
deformacje twarzy spowodowane sta
łym uciskiem dużych
ilo
ści gęstego śluzu na komórki sitowia (poszerzenie
nasady nosa, zmniejszenie
światła nozdrzy)
•
niedorozwój zatok czo
łowych z powodu ich słabego
upowietrznienia
Objawy ze strony przewodu pokarmowego
(75% chorych)
n
G
ęsta wydzielina zamyka światło kanalików
żółciowych cirrhosis biliaris hepatis
secundaria (5%)
n
Przed
łużająca się żółtaczka okresu
noworodkowego
n
Kamica
żółciowa
n
powi
ększenie objętości brzucha, wypadanie
odbytnicy
n
niedro
żność smółkowa (meconium ileus) jelit w
okresie noworodkowym (cz
ęsto 1 objaw CF)
n
Zmiany mikroskopowe w
śliniankach podobne do
tych w trzustce

4
Objawy ze strony przewodu pokarmowego
Niedro
żność smółkowa, występująca u ok.10-20%
noworodków z CF, jest wskazaniem do przeprowadzenia
diagnostyki w kierunku CF.
Nale
ży pamiętać, że do epizodów
niedro
żności może też dochodzić w
pó
źniejszych okresach życia pod
postaci
ą tzw. zespołu zaburzeń
dro
żności dystalnego odcinka
jelita kr
ętego, objawiającego się
przede wszystkim ostrymi,
kolkowymi bólami brzucha,
zlokalizowanymi w prawym dolnym
kwadrancie; u
żywana jest również
nazwa ekwiwalenty niedro
żności
smó
łkowej (MIE - Meconium Ileus
Equivalent).
Objawy ze strony przewodu
pokarmowego
n
u dzieci chorych na CF ryzyko zachorowania na
chorob
ę trzewną (celiakię) jest większe niż w
ogólnej populacji. Wspó
łwystępowanie obydwu
chorób dotyczy 0,45% pacjentów z CF.
Poniewa
ż objawy kliniczne celiakii wynikają z
zaburze
ń wchłaniania, które występują także w
CF, postawienie trafnej diagnozy wymaga du
żej
wnikliwo
ści.
n
wspó
łistnienie CF i nieswoistych zapaleń jelita
dotyczy 0,2-1,0% chorych na CF, a najcz
ęściej
stwierdza si
ę chorobę Crohna (17x częściej niż
w grupie kontrolnej).
Zmiany w trzustce (>90%)
• Nagromadzenie g
ęstego śluzu w świetle małych
przewodów
• Rozszerzenie przewodów i zanik gruczo
łów
zewn
ątrzwydzielniczych trzustki oraz włóknienie.
• Niewydolno
ść zewnątrzwydzielnicza trzustki (niedobór
lipazy)
• Utrudnienie trawienia i wch
łaniania lipidów
• Steatorrhoea (najcz
ęściej od wczesnego dzieciństwa),
niedobór ADEK, zahamowanie wzrostu i
↓ masy ciała,
malnutritio.
Objawy ze strony układu
rozrodczego
n
Prawie 95% m
ężczyzn z CF jest niepłodnych
w wyniku azoospermii spowodowanej
w
łóknieniem, atrofią lub całkowitym brakiem
nasieniowodów, naj
ądrza, pęcherzyków
nasiennych.
n
Mo
że to być jedyny objaw kliniczny
nietypowej postaci CF.
n
U kobiet zap
łodnienie może być utrudnione z
powodu zbyt g
ęstego śluzu w szyjce macicy.
Mutacje genu CFTR a niepłodność
n
>80% m
ężczyzn z CBAVD (congenital
bilateral absence of the vas deferens) to
nosiciele mutacji w CFTR
n
~30% m
ężczyzn z CUAVD to nosiciele
mutacji CFTR
n
5T allele s
ą częstsze u chorych z CBAVD niż
z CF
n
Wszystkich m
ężczyzn z CBAVD i CUAVD
nale
ży skierować na poradę genetyczną
Niektóre inne objawy:
n
upo
śledzenie rozwoju mięśni (np. brak
po
śladków)
n
nadmierna m
ęczliwość
n
obecno
ść palców pałeczkowatych
n
s
łony smak potu
wrodzony
obustronny
brak
nasieniowodów
(CBAVD)
Azoospermia
Niep
łodność

5
Cd.
s
odwodnienie hiponatremiczne i zasadowica
hipochloremiczna
s
hipoprotrombinemia po okresie
noworodkowym
s
hipoproteinemia z uogólnionymi obrz
ękami
s
nawracaj
ące obrzęki ślinianek przyusznych
W CF zdj
ęcie przeglądowe klp u dziecka
mo
że uwidocznić następujące objawy:
n
linijne cienie pogrubia
łych ścian oskrzeli, częściej występujące
w p
łatach górnych
n
obszary niedodmy segmentarnej lub subsegmentarnej,
rzadziej p
łatowej, zwykle dotyczącej płatów górnych
(zw
łaszcza prawego); niedodma ta może być odwracalna (gdy
jest wynikiem zatkania oskrzela przez g
ęstą śluzową
wydzielin
ę) lub nieodwracalna (gdy jest wynikiem
przewlek
łych, bliznowatych zmian w płucach)
n
nawracaj
ące zagęszczenia w płucach w przebiegu
odoskrzelowego zapalenia
n
rozstrzenie oskrzeli (dotyczy to ok. 90% starszych dzieci)
n
ropie
ń płuca
n
zast
ępcze rozdęcie (dotyczy głównie płatów dolnych)
n
rozedma p
ęcherzowa
n
inne powik
łania: odma i płyn w jamie opłucnej
Diagnostyka CF:
n
Wstępne rozpoznanie należy potwierdzić jednym z
badań wykrywających dysfunkcję genu CFTR:
- testem potowym, wykazującym znamiennie
wysokie wartości chlorków w pocie (Cl>60 mmol/l),
w co najmniej 2 odrębnie wykonanych badaniach,
- wykryciem mutacji w genie CFTR w obu allelach
- wysokimi wartościami przezbłonowej różnicy
potencjałów.
Badania diagnostyczne CF
n
Oznaczenie immunoreaktywnej trypsyny lub trypsynogenu (metod
ą
ELISA) w kropli krwi wysuszonej na bibule. Ich aktywno
ść jest 5-10x
wi
ększa u noworodków z CF niż u dzieci zdrowych. Czułość 98%. Niski
koszt.
n
TEST POTOWY: Do skóry przedramienia wprowadza si
ę pilokarpinę -
przyk
łada się elektrodę stymulując miejscowo gruczoły potowe do
wydzielania potu. Nast
ępnie pot zbierany jest na bibule, lub gazie czy
naczyniu szklanym jest wa
żony. Aby oznaczyć zawartość Cl w próbce
u
żywa się chlorometru a aby wynik był wiarygodny należy poddać badaniu
przynajmniej 50 mg potu.
http://www.youtube.com/watch?v=8UCWoz6gUp8
[Link dla
Pani z gr A która pyta
ła jak dokładnie taki test wygląda]
n
POMIAR PRZEZB
ŁONOWEJ RÓŻNICY POTENCJAŁÓWU W NOSIE: U
chorych na CF stwierdza si
ę zwiększoną, bardziej ujemną, różnicę
potencja
łów nosa niż u zdrowych, związaną z zaburzeniami funkcji kanału
Cl. Pomiar wykonywany jest przez wprowadzenie elektrody (cewnik
Foley'a) do nosa poni
żej małżowiny dolnej. Elektroda referencyjna
umieszczona jest na uprzednio poddanej punktowej abrazji skórze
przedramienia. Wynik jest
średnią z 3 pomiarów wykonanych w każdym
przewodzie nosowym. Badanie ma zastosowanie u chorych z ujemnym
testem potowym pomimo charakterystycznych dla CF objawów
klinicznych. Test ten nie jest diagnostyczny w przypadku
przeprowadzonych jakichkolwiek zabiegów na
śluzówce nosa (np.
polipektomia).
Ygy
Źródło: Guidelines for Diagnosis of Cystic Fibrosis in Newborns through Older Adults: Cystic Fibrosis Foundation
Consensus Report. J Pediatr. 2008 Aug;153(2):S4-S14.

6
Leczenie
n
dieta wysokoenergetyczna, wysokobia
łkowa i
wysokot
łuszczowa (zapotrzebowanie kaloryczne dzieci z CF jest
zazwyczaj wi
ększe o ok. 30–50% w stosunku do zdrowych
rówie
śników)
n
fizjoterapia oddechowa
n
suplementacja enzymów trzustkowych
n
mukolityki (np. ludzka rekombinowana dezoksyrybonukleaza -
dornaza alfa)
n
bronchodilatatory
n
GKS (systemowe- w zaostrzeniach, wziewne – przewlekle)
n
antybiotyki (w zaostrzeniach)
n
tlenoterapia (w niewydolno
ści oddechowej)
n
leczenie chirurgiczne: leczenie odmy op
łucnowej, częściowa
resekcja p
łuca w krwotokach zagrażających życiu, zabiegi
paliatywne resekcji tkanki p
łucnej w przypadku ograniczonych
rozstrzeni oskrzeli i marsko
ści tkanki płucnej, szczególnie w
przypadku uporczywego ropienia.
FENYLOKETONURIA
(oligofrenia fenylopirogronianowa)
Częstość występowania PHU na
świecie.
Kraj
Cz
ęstość
w yst
ępowania
Turcja
1:2600
Irlandia
1:3000
Izrael
1:5000
Polska
1:7000
W
łochy
1:7200
Estonia
1:8100
Litw a
1:8700
Wielka Brytania
1:9100
Niemcy
1:10000
USA
1:10000
Australia
1:11200
Kanada
1:15000
Szw ajcaria
1:16000
Chiny
1:16500
Francja
1:17000
Szw ecja
1:30850
Japonia
1:70000
Finlandia
1:100000
Dolny
Śląsk 1:6216
Wielkopolska 1:10000
FENYLOKETONURIA
n
Przyczyna: 1. niedobór hydroksylazy
fenyloalaninowej przekszta
łcającej
fenyloalanin
ę w tyrozynę (98%) lub 2.
niedobór enzymów kofaktora tej reakcji (2%).
n
Cech
ą wspólną jest podwyższenie stężenia
fenyloalaniny.
n
Gen PAH (ok. 500 mutacji, ok. 60% to
mutacje typu missense): najcz
ęstsza mutacja:
R408W (cz
ęstość w Europie ok. 80%)
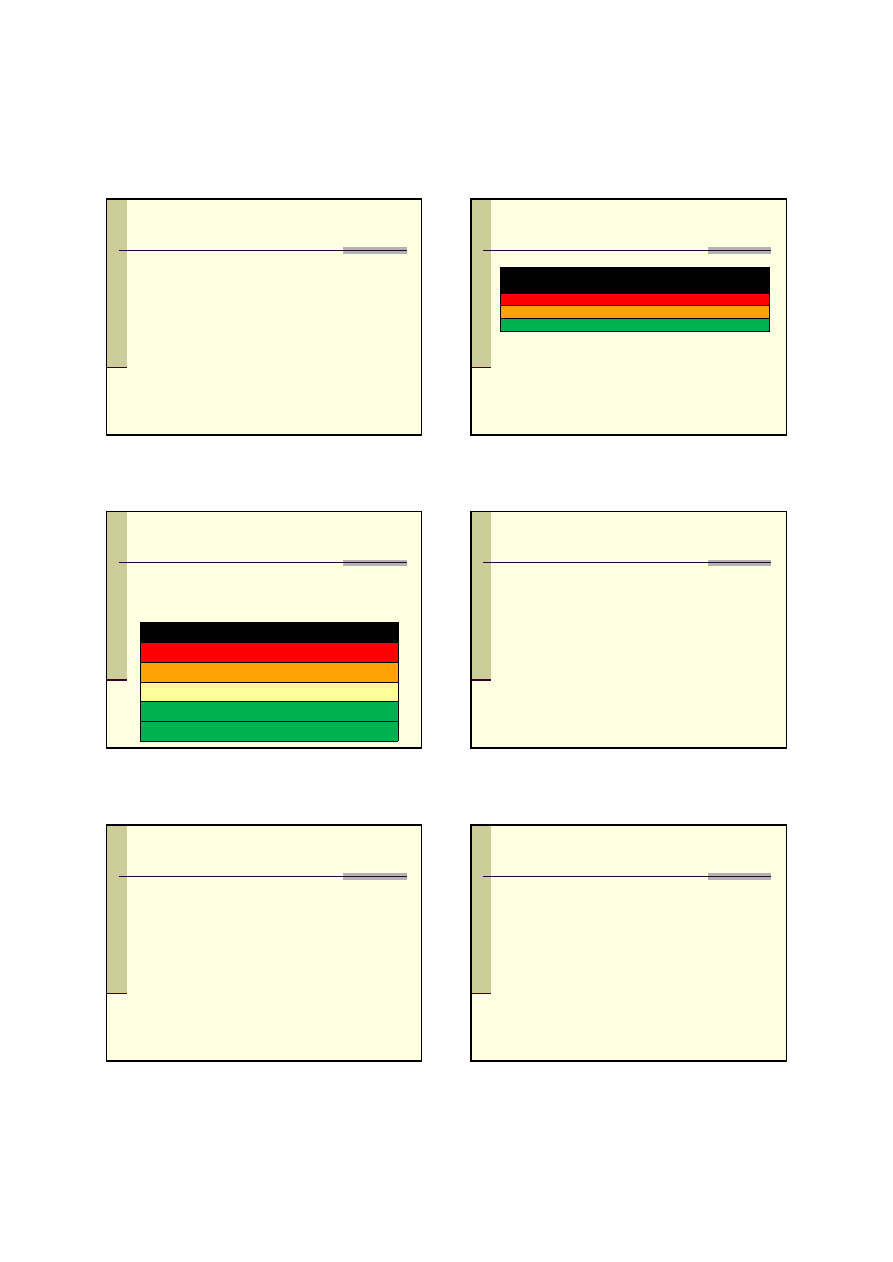
1 - cyklohydrolaza I GTP (GTP-CH)
2 - syntaza 6-pirogronylotetrahydrobiopterynowa (PTPS) – 57%
3 -
hydroksylaza fenyloalaninowa (PAH)
4 - dehydrataza pteryno-4-karbinoloaminowa (PCD)
5 - reduktaza dihydrobiopterynowa (DHPR) – 31%
↓ BH4
3
↑
MELANINA, SEROTONINA,
DOPAMINA, A, NA, TYROKSYNA,
3JTYRONINA
kw.fenylooctowy
(Pha)
7
PODZIAŁ
HIPERPENYLOALANINEMII
1.
FKU klasyczna o ostrym przebiegu.
2.
FKU o
łagodnym przebiegu.
3.
Łagodna hiperfenyloalaninemia (HPA)
4.
Atypowa posta
ć FKU: najczęściej tzw.
z
łośliwa hiperfenyloalaninemia
n
objawy nie ust
ępują pod wpływem leczenia dietą
n
burzliwe objawy neurologiczne (trudne do
opanowania napady drgawek)
n
wczesne, nag
łe zgony.
Uproszczona klasyfikacja hiperfenyloalaninemii
wynikającej z defektu genu PAH
Posta
ć choroby
Aktywno
ść PAH w
bioptatach w
ątroby (%
poziomu normalnego)
St
ężenie fenyloalaniny w
osoczu krwi (mg%)
Klasyczna PKU
< 1
> 20
Łagodna PKU
1-3
8-20
Łagodna HPA
3-6
2,5-7
N= ok. 0,6-2,5 mg%
Źródło: Diagnostyka molekularna w ybranych chorób uw arunkowanych genetycznie: rozprawa habilitacyjna. Med. Wieku Rozw . 2001: 5 (1) supl. 2 s.1-87 oraz
Program Bada
ń Przesiew ow ych Now orodków w Polsce 2006-2008.
Korelacja pomiędzy rodzajem mutacji w locus PAH
a postacią kliniczną HPA
n
Ró
żne mutacje w różnym stopniu ograniczają
aktywno
ść hydroksylazy fenyloalaninowej, można
wyró
żnić jednak w ich obrębie 3 grupy:
mutacje silne (S),
łagodne (Ł) i pośrednie (P).
Mutacje
Posta
ć
S+S
Klasyczna PKU
S+P
Łagodna PKU
P+P
Łagodna HPA, rzadziej łagodna PKU
S+
Ł
Łagodna HPA
P+
Ł, Ł+Ł
Łagodna HPA lub fenotyp prawidłowy
FENYLOKETONURIA
n
Fenotyp kliniczny chorych nios
ących jedną z mutacji
„silnych” zale
ży od mutacji w drugim allelu.
n
Korelacje pomi
ędzy genotypem i fenotypem są
istotne dla rutynowej diagnostyki molekularnej.
Znaj
ąc rodzaj mutacji można przewidywać fenotyp
metaboliczny oraz kliniczny chorego. Noworodek, u
którego zidentyfikowano dwie mutacje „silne”,
nawet je
śli wyjściowe poziomy Pha sugerowały
b
ędą łagodną postać hiperfenyloalaninemii,
b
ędzie w przyszłości prezentował klasyczną
posta
ć PKU. Z drugiej strony identyfikacja u
probanta mutacji
Ł stanowi podstawę do postawienia
rozpoznania
łagodnej HPA. Znalezienie mutacji
po
średniej sugeruje łagodną HPA lub łagodną PKU.
FENYLOKETONURIA
n
Łagodne postacie HPA – 5x rzadziej niż
klasyczna posta
ć
n
Mutacja R408W okre
ślana jest jako „silna”.
Odpowiada za ca
łkowity brak aktywności
hydroksylazy
n
wyst
ępuje z częstością ok. 55% w klasycznej
PKU, 25% w
łagodnej PKU i 32% a w
przypadku
łagodnej HPA.
FENYLOKETONURIA
n
W przypadku
łagodnej HPA nie występują zasadniczo
zaburzenia w sferze poznawczej czy emocjonalnej.
n
Wydaje si
ę, że w fenyloketonurii genotyp wpływa na
wyst
ępujące zawsze, niezależnie od stosowanej diety,
wahania poziomu Pha w krótszych odcinkach
czasu. Przypuszcza si
ę, że wahania te wpływają w
najwi
ększym stopniu na obraz neurologiczny chorych.
n
Pomi
ędzy ludźmi istnieją różnice w kinetyce
przyswajania, metabolizmu Pha i jej przenikania przez
barier
ę krew-mózg. W rezultacie wewnątrzmózgowe
st
ężenia Pha u chorych z PKU mogą być bardzo
ró
żne, mimo takich samych stężeń Pha we krwi i
takich samych genotypów w locus PAH.
n
Badania przesiewowe maj
ą znaczenie tylko u dzieci >
3-go dnia
życia.
8
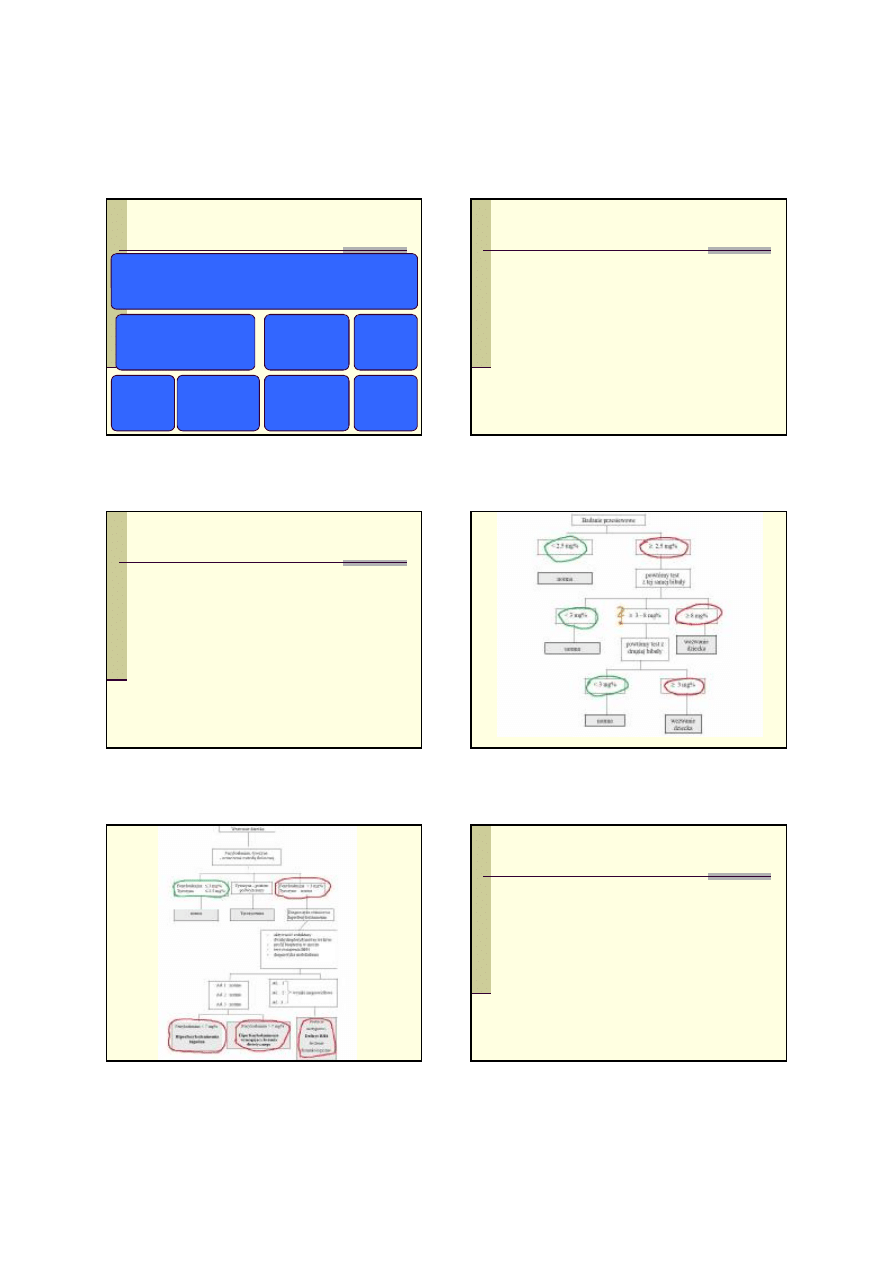
Patomechanizm zmian w OUN
Uszkodzenie OUN
↓Transportu aminokwasów przez
b
ł komórkowe i barierę krew-
mózg
Pha hamuje
kompetycyjne
transport Tyr,
Met, Leu i Wal
przez barier
ę
krew-mózg.
Pha zaburza
rozmieszczenie Tyr
w komórkach
co
hamuje transport
przez b
łony
p
ęcherzyków
synaptycznych.
Nieprawid
łowości
syntezy i
przemiany mieliny
Pha hamuje
sulfurylaz
ę
w
oligodendrocytach co
powoduje
↓syntezy
cerebrosulfatydów i
szybszy
rozpad
mieliny
↓Syntezy
neuro-
transmiterów
Niedobór
hydroksylazy
fenyloalaniny
OBJAWY FENYLOKETONURII
n
P
łód rozwija się prawidłowo. Wczesnym objawem u dzieci
nieleczonych mog
ą być chlustające wymioty nie powodujące
zahamowania przyrostu.
n
Ma
łogłowie (68-94%)
n
Spadek poziomu inteligencji i zahamowanie rozwoju psycho-
fizycznego, wi
ększość dzieci nie umie mówić a ok. 30% nie
chodzi. Nieleczone niemowl
ęta tracą 1-2 punkty IQ/tydzień. IQ
20-40.
n
Jasna karnacja skóry, blond w
łosy, niebieskie tęczówki.
n
Mysi zapach (kwas fenylooctowy) od 2 miesi
ąca
n
Objawy neurologiczne: dr
żenie mięśniowe, obniżenie napięcia
mi
ęśniowego, drgawki, zespoły spastyczne, nadpobudliwość
ruchowa.
n
Zaburzenia zachowania: nadpobudliwo
ść, napady złości, krótki
czas uwagi, zaburzenia snu, autoagresja
n
Zmiany skórne o typie
łojotokowym
n
Wystaj
ąca szczęka, duże odstępy pomiędzy zębami, hipoplazja
szkliwa.
Metody diagostycne
fenyloketonurii.
n
Test Guthrie’go (historia) – pó
łilościowy, dużo
wyników fa
łszywie+; wzrost bakterii jest
uwarunkowany obecno
ścią Pha i jest wprost
proporcjonalny do jego st
ężenia.
n
Analiza kolorymetryczna – test ilo
ściowy,
mniej wyników fa
łszywie+
n
Tandemowa spektrometria mas (badanie
ilo
ściowe kilku substancji jednoczasowo)
Źródło: Program Badań Przesiewowych Noworodków w Polsce 2006-2008.
1.
Źródło: Program Badań
Przesiewowych Noworodków
w Polsce 2006-2008.
2.
3.
Zespół fenyloketonurii matczynej
n
Podwy
ższony poziom Phe we krwi kobiety ciężarnej może
wywo
łać nieodwracalne zmiany rozwojowe płodu.
n
Kobiety chore, których krew by
ła badana dopiero od 8-go t.,
rodzi
ły dzieci ciężko upośledzone z wrodzoną wadą serca,
ma
łogłowiem i hipotrofią wewnątrzmaciczną.
Opisywano u tych dzieci tak
że inne patologie:
wady uk
ładu kostnego
wodog
łowie
centralne pora
żenie nerwu twarzowego
zro
śnięcie przełyku, przetoka tchawiczo-przełykowa
dysplazj
ę stawów biodrowych i inne.
n
Obraz uszkodzenia p
łodu dowodzi, że zachodzi ono w
I trymestrze ci
ąży.

9
Zespół fenyloketonurii matczynej
n
Kobiety chore na hiperfenyloalaninemi
ę
(
łącznie z łagodną HPA!) powinny
kontrolowa
ć poziom Phe już od okresu
przedkoncepcyjnego. St
ężenie Phe we
krwi nie powinno przekracza
ć 4 mg%.
n
W zale
żności od występującej u matki pary
alleli, dziecko wykazuje ró
żny poziom
wska
źnika IQ.
Kobiety o genotypie S/
Ł, rodziły dzieci,
których IQ = 96-99, natomiast w przypadku,
S/S lub S/P, IQ = 83-84.
Diagnostyka prenatalna:
n
CVS chorionic villus sampling – 10-12 t.
n
Amniocenteza – 15-18t.
n
Przed diagnostyk
ą prenatalną muszą być
zidentyfikowane oba zmutowane allele w
rodzinie.
Leczenie: jak najszybciej,
„diet for life”
n
1. Klasyczna dieta niskofenyloalaninowa
wzbogacona o tyrozyn
ę, v. z grupy B i
minera
ły (cynk, selen, żelazo). Optymalne
st
ężenie: 2-6 mg%. Pacjenci z łagodną HPA
nie wymagaj
ą ograniczenia podaży Pha w
diecie.
n
2. Atypowa: dieta niskofenyloalaninowa, BH4
(s
łaba penetracja przez barierę krew-mózg),
prekursory neurotransmiterów
Czy mukowiscydoza by
ła
przyczyn
ą śmierci
Chopina?
Rodzina Chopina
n
Matka: Tekla Justyna z Krzy
żanowskich – zdrowa
n
Ojciec: Miko
łaj – przez całe życie nawracające
dolegliwo
ści ze strony ukł. oddechowego, żył 73 lata.
n
Siostry: Izabela – zdrowa; Ludwika – nawracaj
ące
infekcje uk
ł. oddechowego, żyła 47 lat; Emilia – „była
delikatnym dzieckiem wyniszczonym przez
nawracaj
ące zapalenia płuc, z nieustającą
duszno
ścią, kaszlem, krwiopluciem oraz znacznym
niedoborem masy cia
ła”. Jej choroba miała początek
w dzieci
ństwie. Żyła 15 lat.
Izabela
Ludwika
Emilia
Jaka była przyczyna śmierci
Fryderyka Chopina?
n
Gru
źlica?
n
Mukowiscydoza?
n
Rozedma p
łuc?
n
Rozstrzenie oskrzeli?
n
Hemosyderoza p
łuc?
n
Hipogammaglobulinemia?
n
Alergiczna aspergiloza oskrzelowo-p
łucna?
n
Stenoza mitralna?
n
Przewlek
ły ropień płuc?
n
P
łucne malformacje tętniczo-żylne?
10
Ukł. oddechowy
n
Napady kaszlu oraz duszno
ść, krwioplucie,
sinica, trudno
ści z odkrztuszaniem wydzieliny
(„nadmiar
śluzu”), beczkowata klatka piersiowa
n
„Metody diagnostyczne”:
„Jeden w
ąchał, com pluł, drugi stukał skądem
plu
ł, trzeci macał i słuchał jakem pluł. Jeden
mówi
ł, żem zdechł, drugi – że zdycham, trzeci –
że zdechnę”
„Oni b
łądzą po omacku, a ulgi mi żadnej nie
przynosz
ą”
Przewód pokarmowy
n
Przewlek
łe biegunki, bóle brzucha,
nietolerancja t
łustego jedzenia, krwawe
wymioty („podczas nocnej podró
ży przez
morze krew usuwano ca
łymi miseczkami”)
n
„Strasznie schud
łem i zmizerniałem…nie
moja wina,
żem jak stary grzyb”
Ogólne
n
M
ęczliwość, niedobór masy ciała (44 kg/170
cm), blado
ść, przebarwienia skórne,
obwodowe obrz
ęki, zaniki mięśniowe, bóle
stawów, bóle g
łowy, objawy depresji.
n
We wrze
śniu 1849 Chopina odwiedza C.K.
Norwid: „zasta
łem go ubranego, ale leżącego
na
łóżku ze obrzękniętymi nogami, z głosem
zak
łócanym przez kaszel i duszącego się”
Kobiety...
n
Nigdy nie by
ł ojcem.
1.
Konstancja G
ładkowska (1829-31)
2.
Maria Wodzi
ńska (1835-36)
3.
George Sand (1838-47) („wiem,
że wiele
osób oskar
ża mnie – jedni, że
wyniszczy
łam go swoją gwałtowną
zmys
łowością, inni, że wybrykami”
4.
Jane Stirling (1848-49)
Metody lecznicze
n
Zażywał krople opium na cukrze i nacierał skronie wodą kolońską
n
Wezykatoria (kantardyna)
n
Serwatka z mleka koziego, oślego i krowiego
n
Płucnik czyli mech islandzki zawierający substancje bakteriostatyczne o
działaniu zbliżonym do streptamycyny.
n
Napary z ziół.
n
„flakony” do wdychania o działaniu wykrztuśnym, spazmolitycznym,
przeciwkrwotocznym, tonizującym.
n
Aeroterapia (pobyt na wsi, wyjazdy na południe, „wziąć mieszkanie od
południa”)
n
Wzmacniające kąpiele (Duszniki, Enghien, Marsylia)
n
Zabiegi: pijawki, masaże
n
Ciepłe okłady, kataplazmy
n
Dieta (owsianna, nabiałowa, buliony)
n
„Wczoraj Cruveillego się radziłem, który mi nic prawie nie każe brać,
tylko spokojnie siedzieć… Ale widzę, że mnie także za suchotnika ma”
Czy mukowiscydoza była
przyczyną śmierci Chopina?
n
Jedynie badania genetyczne mog
ą dzisiaj
ostatecznie odpowiedzie
ć na to pytanie.
n
Badania takie zostan
ą przeprowadzone z
tkanek serca Chopina znajduj
ącego się w
ko
ściele Świętego Krzyża w Warszawie.
Wyszukiwarka
Podobne podstrony:
Choroby AR2008 ppt [Tylko do odczytu] [tryb zgodności]
PCR [Tylko do odczytu] [tryb zgodności]
Funkcjonowanie UE prawo i instytucje Tylko do odczytu tryb zgodno ci
POiZ 1 do wyk 2 d [tryb zgodności]
finanse na ubezpieczenia do wysłania [tryb zgodności]
techniki i technologie ksztaltujace wyglad zewn [Tylko do odczytu]
2013 Wykład 1III r WPROWADZENIE [Tylko do odczytu]
OB diagramy 9x [Tylko do odczytu]
POiZ 1 do wyk 2 d [tryb zgodności]
kurs operatora suwnic tylko do odczytu
EtykaLekarskaWykład Cz 2 pptx (Tylko do odczytu)
(W7a Stale do kszta t na zimno cz I [tryb zgodno ci])
więcej podobnych podstron