ENZYMY
1. Klasyfikacja i nazewnictwo enzymów:
- enzymy - biologiczne makromolekuły o dużej masie cząsteczkowej, katalizujące reakcje chemiczne, niezbędne w procesach biologicznych; katalizują przekształcanie substratów w produkty zwiększając szybkość reakcji co najmniej 106 razy; nie zużywają się ani trwale nie zmieniają w wyniku udziału w reakcji; wysoce efektywne i selektywne (specyficzne w stosunku do typu katalizowanej reakcji i substratu lub zestawu podobnych substratów); stereospecyficzne (zwykle katalizują reakcje tylko dla jednego stereoizomeru danego związku; wysoka specyficzność enzymów nadaje komórkom zdolność równoczesnego prowadzenia i niezależnej kontroli procesów chemicznych
- powszechnie używane nazwy większości enzymów zakończone końcówką -aza opisują typ katalizowanej reakcji (według której klasyfikowane są enzymy)
- nazewnictwo enzymów:
+ zwykle nazwy enzymów pochodzą od substratów przetwarzanych w reakcji lub od samej reakcji przez nie obsługiwanych, do których dodaje się przyrostek -aza; często nazwa jest dwuczłonowa, gdzie pierwszy człon określa reakcję, a drugi jej substrat; czasami nazwa enzymu nie pochodzi od reakcji obsługiwanej w warunkach fizjologicznych, ale od jego aktywności in vitro (np. izomeraza glukozowa, wykazująca in vitro aktywność izomeracji glukozy we fruktozę, ale in vivo będąca izomerazą ksylozową); enzymy są niekiedy nazywane od ogólnego procesu, jaki przeprowadzają (np. gyraza DNA, której nazwa pochodzi od greckiego słowa γύρος, czyli obracać); nazwy niektórych enzymów powstały poprzez dodanie przyrostka -zym do ogólnej funkcji cząsteczki lub opisu (np. lizozym)
+ duża część enzymów jest nazywana zwyczajowo, historycznie (np. pepsyna, papaina)
+ enzymy restrykcyjne są nazywane według własnej nomenklatury - ich nazwa składa się z litery nazwy rodzajowej i dwóch pierwszych liter nazwy gatunkowej mikroorganizmu-źródła enzymu, pisanymi kursywą, po których występują cyfry arabskie lub litery oznaczających szczep mikroorganizmu; nazwa zakończona jest cyfrą rzymską wskazującą, jako który w kolejności chronologicznej został wyizolowany z danego organizmu dany enzym restrykcyjny
+ część enzymów jest natomiast bardziej znana pod skrótami pochodzącymi od ich pełnych nazw (np. RuBisCO) lub od skrótu nazwy opisowej (np. BACE2)
+ w celu uregulowania i ujednoznacznienia nazewnictwa enzymów, Komitet Nazewnictwa Międzynarodowej Unii Biochemii i Biologii Molekularnej w latach 1956-1972 opracował dla nich nomenklaturę naukową (numer EC) - według niej, każdy enzym jest opisany przez ciąg czterech segmentów cyfr, oddzielonych od siebie kropką, poprzedzonych literami "EC" (EC x.xx.xx.xx):
- pierwsza cyfra dzieli enzymy, według mechanizmu reakcji przez nie katalizowanych, na sześć głównych klas
- następne liczby numeru EC oddzielone kropkami klasyfikują dany enzym odpowiednio do podklasy i podpodklasy, natomiast ostatnia liczba określa miejsce enzymu w podpodklasie
+ w nazewnictwie enzymów zaleca się stosowanie numeru EC, jednak dopuszcza się stosowanie nazw zwyczajowych z przyrostkiem -aza (gdzie pierwszy człon określa reakcję, a drugi jej substrat), a także innych ogólnie przyjętych nazw roboczych i zwyczajowych, co także jest regulowane stosownymi zaleceniami
- klasyfikacja enzymów:
1) oksydoreduktazy - katalizują reakcje utleniania i redukcji:
- działające na grupę CH-OH donora z NAD lub NADP jako akceptorem (np. oksydoreduktaza alkoholowa: NAD-dehydrogenaza alkoholowa)
alkohol ![]()
aldehyd/keton
- działające na grupę aldehydową lub ketonową donora z NAD lub NADP jako akceptorem (np. oksydoreduktaza mrówczanowa: NAD-dehydrogenaza mrówczanowa)
HCOOH ![]()
CO2
- działające na grupę CH-CH donora z tlenem jako akceptorem (np. oksydoreduktaza L-4,5-dihydroortoranowa: tlen-dehydrogenaza dwuhydroortoranowa)
- z innymi akceptorami (np. oksydoreduktaza bursztynianowa; akceptor-dehydrogenaza bursztynianowa)
COOH-CH2-CH2-COOH ![]()
COOH-CH=CH-COOH
bursztynian kwas fumarowy
- działające na nadtlenek wodoru jako akceptor:
+ katalaza H2O2 + H2O2 = O2 + 2H2O
+ peroksydaza donor + H2O2 = utleniony donor + 2H2O
- działające na wodór jako donor
- działające na jeden donor z wyłączeniem tlenu - oksygenazy (np. oksygenaza tryptofanowa)
L-tryptofan ![]()
L-formylokimureina
2) transferazy - katalizują przenoszenie takich jednostek, jak reszty glikozylowe, grupy metylowe czy fosforanowe):
- przenoszące grupy jednowęglowe - metylotransferazy (np. N-metylotransferaza S-adenozynometioninowa: guanidynooctan-metylotransferaza guanidynooctanowa)
- przenoszące ugrupowania aldehydowe lub ketonowe:
+ transketolaza
+ transaldolaza
- przenoszące grupy zawierające azot - aminotransferazy (transaminazy)
3) hydrolazy - katalizują hydrolityczne rozszczepienie wiązań C-C, C-O, C-N i innych:
- działające na wiązania estrowe C-O - hydrolazy estrów karboksylowych (np. lipaza)
- działające na związki glikozydowi - hydrolazy glikozydów (np. α i β-amylaza)
- hydrolazy peptydylopeptydów:
+ pepsyna - hydroliza wiązań peptydowych między resztami L-aminokwasów aromatycznych lub dwukarboksylowych a innymi aminokwasami
+ podpuszczka - hydroliza peptydów
+ trypsyna - hydroliza peptydów, amidów, estrów w miejscach wiązań grup karboksylowych L-argininy lub L-lizyny
+ chymotrypsyna A
+ enteropeptydaza
+ katepsyna C
+ papaina
+ trombina
+ renina
+ peptydaza A
+ keratynaza
4) liazy - katalizują rozszczepienie wiązań C-O, C-C, C-N i innych przez eliminację atomu i pozostawienie wiązania podwójnego:
- liazy wiązania C-C - karboksylazy (np. dekarboksylaza pirogronianowa)
CH3-CO-CH3 → H-CO-CH3 + CO2
kwas pirogronowy aldehyd octowy
- liazy wiązania C-O:
+ hydroliazy - np. hydrataza fumaranowa, fosfopirogronianowa, L-serynowa, synteza cysternowa
+ inne liazy wiązania C-O (np. liaza poliglukuronidowa)
- liazy wiązania C-N:
+ amoniakoliazy (np. amoniakoliaza asparaginianowa)
+ amidynoliazy (np. liaza argininobursztynianowa)
- liazy wiązania C-S - np. desulfohydrataza cysternowa
5) izomerazy - katalizują geometryczne lub strukturalne zmiany w obrębie cząsteczki:
- racemazy i epimerazy:
+ działające na aminokwasy i ich pochodne (np. racemaza glutaminianowi)
+ działające na hydroksykwasy i ich pochodne (np. racemaza mleczanowa)
+ działające na węglowodany i ich pochodne
- wewnątrzcząsteczkowy izomerazy - np. izomeraza rybozofosforanowa, glukozofosforanowa, glukuronianowa)
glukozo-6-fosforan ↔ fruktozo-6-fosforan
- przemieszczające wiązania C=L - np. izomeraza sterydowa, izopentenylopirofosforanowa)
6) ligazy (syntetazy) - katalizują wiązanie się dwóch cząsteczek połączone z hydrolizą ATP (łączą cząsteczki wiązaniami kowalencyjnymi):
- wytwarzające wiązanie C-O - np. ligazy wytwarzające aminoacylo-N-tRNA i związki pokrewne (syntetazy aminoacylo-tRNA)
- wytwarzające wiązanie C-N:
+ ligazy kwas-amoniak (syntetazy amidów)
+ ligazy kwas-aminokwas (syntetazy peptydów)
+ C-N syntetazy z glutaminą jako donorem azotu amidowego (syntetaza GMP)
- wytwarzające wiązania C-C - np. karboksylaza pirogronianowa
2a. Budowa enzymów:
- apoenzym = holoenzym (część białkowa) + grupa prostetyczna
- grupy prostetyczne, kofaktory, koenzymy - małe, niebiałkowe cząsteczki i jony metali, które bezpośrednio uczestniczą w przyłączaniu substratu lub katalizie; rozszerzają możliwości katalityczne enzymu (wynikające z właściwości reszt aminokwasowych występujących w jego łańcuchach peptydowych)
- enzymy mogą zawierać dodatkowe miejsca wiązania małych cząsteczek (np. pośrednich lub bezpośrednich produktów czy substratów szlaków metabolicznych obsługiwanych przez enzym), które związane mogą dodatkowo regulować aktywność enzymu na zasadzie sprzężenia zwrotnego
- grupy prostetyczne:
+ trwały element budowy enzymu; odznaczają się stabilnością wbudowania w strukturę białka za pośrednictwem wiązań kowalencyjnych lub niekowalencyjnych
+ występują zwykle w miejscu aktywnym i są bezpośrednio zaangażowane w katalizę oraz nie opuszczają miejsca aktywnego podczas reakcji
+ fosforan pirydoksalu, mononukleotyd flawinowy (FMN), dinukleotyd flawino-adeninowy (FAD), fosforan tiaminy (związki te są niekiedy zaliczane do koenzymów), biotyna, jony metali (Co, Cu, Mg, Mn, Zn); najbardziej powszechnymi grupami prostetycznymi są jony metali (ale często atom metalu pełni rolę aktywatora, a nie grupy prostetycznej)
+ metaloenzymy - enzymy zawierające silnie przyłączone jony metali; ok. 1/3 wszystkich enzymów; jony metalu uczestniczące w reakcjach redoks wchodzą w skład hemu lub centrum żelazo-siarkowego; metale mogą także ułatwiać przyłączanie i orientację substratów, tworzenie wiązań kowalencyjnych z produktami pośrednimi reakcji (np. Co2+ w koenzymie B12) lub współdziałać z substratami, aby uczynić je bardziej elektrofilowymi (ubogimi w elektrony) lub nukleofilowymi (bogatymi w elektrony)
- kofaktory:
+ asocjują odwracalnie z enzymami lub substratami (np. z ATP)
+ pełnią funkcje podobne do funkcji grup prostetycznych
+ muszą być obecne w środowisku otaczającym enzym, aby zaszła kataliza
+ najbardziej powszechnymi kofaktorami są jony metali; enzymy aktywowane przez metal - enzymy wymagające jonu metalu jako kofaktora
- koenzymy:
+ służą jako wahadłowce (transportery) substratów - są czynnikami przenoszącymi grupy funkcyjne; transportują wiele substratów z miejsca ich tworzenia do miejsca ich wykorzystania
+ asocjacja z koenzymem stabilizuje niektóre substraty (np. atomy wodoru, jony wodorkowe), niestabilne w wodnym środowisku komórki; inne grupy chemiczne transportowane przez koenzymy - grupy metylowe (folian), grupy acylowe (CoA), oligosacharydy (dolichol)
- witaminy B - stanowią ważne komponenty wielu koenzymów; niektóre koenzymy zawierają także reszty adeniny, rybozy i resztę fosforanową AMP lub ADP:
+ nikotynamid - komponent koenzymów reakcji utleniania i redukcji (NAD+ i NADP+)
+ ryboflawina - składnik koenzymów FMN i FAD
+ kwas pantotenowy - komponent CoA
+ tiamina (difosforan tiaminy) - udział w dekarboksylacji ketokwasów
+ kwas foliowy i Kobami - koenzymy w metabolizmie ugrupowań jednowęglowych
- centrum aktywne:
+ trójwymiarowe, najczęściej przyjmuje formę szczeliny lub kieszeni; szczelina zawiera głównie reszty niepolarne, ale zdarzają się czasem reszty polarne; aminokwasami występującymi szczególnie często w centrach aktywnych są His, Lys i Cys
+ nadaje enzymowi specyficzność substratową i wysoką sprawność katalityczną dzięki tworzeniu środowiska idealnie dostosowanego do pojedynczej reakcji
+ w enzymach multimetrycznych jest najczęściej zlokalizowane w regionie między podjednostkami i zawiera reszty z więcej niż jednego monomeru
+ chroni substraty przed rozpuszczalnikiem i ułatwia katalizę - substraty przyłączają się w regionie komplementarnym do tej części substratu, który nie będzie podległam zmianie chemicznej podczas przebiedu reakcji; jednocześnie ustawia to odpowiednio w stosunku do funkcjonalnych grup łańcuchów bocznych aminokwasów te części substratu, które będą podlegały zmianie
+ przyłącza i orientuje kofaktory lub grupy prostetyczne
+ dość duże ze względu na wiele reszt aminokwasowych pochodzących z różnych części łańcucha polipeptydowego
+ wiązania wodorowe, jonowe i siły van der Vaalsa
+ model klucza i zamka Fishera - zakłada sztywność miejsca aktywnego enzymu, co uniemożliwia dynamiczne zmiany towarzyszące katalizie
+ model indukowanego dopasowania Koshlanda - zakłada, że zbliżenie substratów do enzymu indukuje zmianę konformacyjną, w efekcie której enzym indukuje wzajemnie zmiany w substratach, wykorzystując energię przyłączenia na ułatwienie transformacji substratów w produkty
+ model trzypunktowego dołączenia Ogstona - do specyficznego związania substratu przez enzym wystarczą tylko trzy miejsca wiązania; jeśli substrat może się przyłączyć do miejsca katalitycznego tylko z jednej strony i mogą ze sobą reagować tylko atomy i miejsca komplementarne, cząsteczka substratu może się przyłączyć tylko w jeden sposób - oznacza to, że symetryczna cząsteczka substratu staje się poniekąd asymetryczna dla wypadku katalizy enzymatycznej; model ten jest wyraźnie jakościowy, dostarczając tylko ograniczonego wyjaśnienia ilościowych i energicznych parametrów przebiegu stereospecyficznych procesów; w 2004 roku opisano rozszerzenie modelu dla większej ilości punktów wiązania, co zapewnia większą precyzję w doborze substratu
2b. Allosteria:
- enzymy allosteryczne - enzymy zmieniające swoją konformację w odpowiedzi na związanie efektora (inhibitora lub aktywatora); modulacja może być bezpośrednia (gdy efektor sam wiąże się z enzymem) lub pośrednia (gdy efektor wiąże się z innym białkiem lub podjednostką białka, która oddziałuje z enzymem, zmieniając jego aktywność katalityczną)
- hamowanie przez sprzężenie zwrotne - hamowanie aktywności enzymu szlaku biosyntezy przez końcowy produkt tego szlaku; związek końcowy łączy się z enzymem zwykle w miejscu allosterycznym, różnym od miejsca katalitycznego; inhibitory zwrotne - ujemne efektory allosteryczne (na ogół nie wykazujące podobieństwa strukturalnego do substratów hamowanego enzymu); kinetyka hamowania przez sprzężenie zwrotne może mieć charakter kompetycyjny, niekompetycyjny lub mieszany; hamujący wpływ dwóch lub kilku produktów końcowych może być sumą skutków wywołanych przez każdy z tych produktów z osobna (kumulatywne hamowanie przez sprzężenie zwrotne) lub też przewyższać efekt sumaryczny (kooperatywne hamowanie przez sprzężenie zwrotne)
- miejsce allosteryczne - odmienne fizycznie od miejsca katalitycznego; enzymy allosteryczne - enzymy, których aktywność miejsca katalitycznego może być regulowana przez efektory allosteryczne znajdujące się w miejscu allosterycznym; na ogół przyłączenie regulatora allosterycznego indukuje w enzymie zmiany konformacyjne obejmujące miejsce katalityczne (zmiany te mogą wpływać na sprawność katalityczną enzymu, powinowactwo enzymu do substratu lub na obie te właściwości)
- podział enzymów allosterycznych:
+ seria V - efektor allosteryczny zmniejsza wartość Vmax bez widocznego wpływu na wartość Km; skutkiem zmian konformacyjnych może być zmiana orientacji przestrzennej lub ładunku reszt katalitycznych, prowadząca do zmniejszenia wartości Vmax
+ seria K - kinetyka nasycenia substratem jest kompetycyjna w sensie zwiększania wartości Km i braku wpływu na wartość Vmax; skutkiem zmian konformacyjnych może być osłabienie wiązań między substratem i resztami aminokwasowymi w miejscu wiążącym substrat
+ w obu przypadkach zmiany konformacyjne mogą mieć wpływ zarówno na Vmax, jak i Km.
3a. Mechanizmy katalizy enzymatycznej:
- kataliza przez sąsiedztwo - aby cząsteczki przereagowały muszą znajdować się w odległości wystarczającej do utworzenia wiązania; jeśli ich stężenie będzie największe, będą się dużo częściej ze sobą spotykać; kiedy enzym przyłącza cząsteczki substratu w centrum aktywnym powstaje region, w którym lokalne stężenie substratu jest wysokie; ponadto orientuje to cząsteczki substratu przestrzennie w pozycji dla nich najodpowiedniejszej do oddziaływania, dzięki czemu szybkość reakcji zwiększa się
- kataliza kwasowo-zasadowa - zdolne do jonizacji grupy funkcyjne aminokwasów i grupy prostetyczna działają jako kwasy lub zasady; może być specyficzna (biorą udział tylko protony H3O+ (kataliza specyficzna kwasowa) lub tylko jony OH- (kataliza specyficzna zasadowa); szybkość reakcji wrażliwa na zmiany stężenia protonów, lecz niezależna od stężenia innych kwasów lub zasad w roztworze lub miejscu aktywnym) lub ogólna (kwasowa lub zasadowa; reakcje, których szybkości są wrażliwe na wszystkie obecne kwasy lub zasady)
- kataliza przez odkształcenie cząsteczki substratu - w enzymach katalizujących reakcje lityczne (rozerwanie wiązania kowalencyjnego); enzymy te zwykle przyłączają substraty w konformacji niezbyt dogodnej dla wiązania podlegającego rozerwaniu; utworzone w wyniku tego obszary naprężenia zniekształcają docelowe wiązanie, osłabiając je i czyniąc bardziej podatnym na rozerwanie
- kataliza kowalencyjna - obejmuje tworzenie wiązania kowalencyjnego między enzymem i co najmniej jednym substratem; zmodyfikowany enzym staje się reaktantem (substratem reakcji); kataliza ta wprowadza nowy szlak reakcji, którego energia aktywacji jest niższa niż w szlaku reakcji w homogennym roztworze; chemiczna modyfikacja enzymu jest przejściowa - po zakończeniu reakcji enzym powraca do oryginalnego stanu; szczególnie powszechna w enzymach katalizujących reakcje przenoszenia grup; najczęściej biorą w niej udział reszty aminokwasowe cysteiny i seryny, rzadziej histydyny; najczęściej wykorzystuje mechanizm „ping-pong” (pierwszy substrat jest przyłączany, a jego produkt uwalniany przed przyłączeniem drugiego substratu).
3b. Kataliza enzymatyczna - kinetyka reakcji enzymatycznych:
- zmiana entalpii swobodnej (energii swobodnej Gibbsa, ΔG):
+ określa kierunek przebiegu reakcji chemicznej oraz stężenie substratów i produktów, które będą występowały w stanie równowagi
+ jest równa różnicy między sumą entalpii swobodnych tworzenia produktów reakcji (ΔGp) i sumą entalpii swobodnych tworzenia substratów (ΔGs)
+ ΔGo - określa zmianę entalpii swobodnej, jaka towarzyszy przejściu substratów i produktów od stanu standardowego (stężeń jednomolowych) do stanu równowagi; jest funkcją jedynie początkowych i końcowych stanów reagujących związków, dlatego dostarcza informacji tylko o kierunku i stanie równowagi reakcji (nie dostarcza informacji na temat szybkości, gdyż jest niezależna od mechanizmu reakcji, dlatego reakcja może mieć dużą ujemną wartość ΔGo, lecz zachodzić z niezauważalną szybkością); w biochemii używa się wartości ΔGo', która definiuje ΔGo w standardowym stanie 10-7 M protonów (pH 7,0)
+ jeżeli entalpia swobodna tworzenia produktów jest mniejsza od entalpii swobodnej tworzenia substratów, znak ΔGo i ΔGo' będzie ujemny - reakcja przebiega w kierunku od strony lewej do prawej (reakcje spontaniczne - samorzutne); znak i wielkość zmiany entalpii swobodnej określa, jak daleko będzie zachodziła reakcja
+ ΔGo = -RT lnKeq - zależność między stałą równowagi (Keq) i ΔGo; R - stała gazowa, T - temperatura absolutna [K]
+ Keq równa się iloczynowi stężeń produktów reakcji, podniesionych do potęg o wykładnikach równych współczynnikom stechiometrycznym, przez iloczyn stężeń substratów (również podniesionych do odpowiednich potęg); A + B ↔ P + Q
![]()
+ jeżeli ΔGo ma wartość ujemną, Keq będzie większa od jedności i stężenie produktów w stanie równowagi będzie wyższe niż stężenie substratów; jeśli ma ono wartość dodatnią, Keq będzie mniejsza od jedności i uprzywilejowane będzie tworzenie substratów
- stany przejściowe:
+ tworzy się przejściowy produkt, który nie jest ani wolnym substratem, ani produktem; tzw. kompleks aktywny
+ reakcje połówkowe - pierwsza odpowiada tworzeniu (F), a druga rozpadowi (D) kompleksy aktywnego; każdej reakcji połówkowej towarzyszą zmiany ΔGF i ΔGD
+ wiele reakcji przebiega przez różne stany przejściowe, każdy związany z określoną zmianą entalpii swobodnej; w reakcjach tych całkowita zmiana ΔG stanowi sumę wszystkich zmian entalpii swobodnej związanych z tworzeniem i rozpadem wszystkich stanów przejściowych, dlatego na podstawie całkowitej zmiany ΔG nie jest możliwe wnioskowanie liczby lub typu stanów przejściowych, przez które przebiega reakcja (termodynamika nie mówi nic o kinetyce)
+ ΔGF dla większości reakcji chemicznych ma znak dodatni, tworzenie kompleksu aktywnego wymaga więc pokonania barier energetycznych - dlatego ΔGF jest nazywana często energią aktywacji, czyli energią potrzebną na pokonanie danej bariery energetycznej; parametry określające szybkość przebiegu reakcji - wartości ΔGF powstawania stanów przejściowych, poprzez które przebiega reakcja
- kinetyka katalizy enzymatycznej - zmniejszenie bariery energii aktywacji reakcji:
+ wszystkie enzymy przyspieszają reakcje, przyczyniając się do tworzenia kompleksów aktywnych o obniżonej wartości ΔGF
+ jeśli mechanizm lub kolejność etapów chemicznych zachodzących w miejscu aktywnym cząsteczki enzymu są takie same jak dla tej samej reakcji przebiegającej bez udziału katalizatora to środowisko miejsca aktywnego obniża ΔGF stabilizując stan przejściowy; stabilizacja może obejmować grupy kwasowo-zasadowe ustawione w sposób sprzyjający przeniesieniu protonów do lub z kompleksu aktywnego, właściwe ułożenie naładowanych grup lub jonów metalu, co stabilizuje powstałe ładunki lub wymuszenie przestrzennego odkształcenia substratów tak, aby ich geometria stała się podobna do geometrii kompleksu aktywnego
+ kataliza enzymatyczna przebiega według unikatowego mechanizmu, jeśli kompleks aktywny tworzy kowalencyjne wiązanie z enzymem (kataliza kowalencyjna)
- enzymy a Keq - enzymy mogą podlegać przejściowej modyfikacji w procesie katalizy, jednak na końcu reakcji pojawiają się niezmienione; obecność enzymu nie ma żadnego wpływu na ΔGo sumarycznej reakcji (która jest funkcją tylko początkowego i końcowego stanu reagentów)
- czynniki wpływające na szybkość reakcji katalizowanych enzymatycznie:
+ temperatura - podwyższenie temperatury zwiększa szybkość reakcji (zarówno niekatalizowanych, jak i katalizowanych), powodując wzrost energii kinetycznej i częstości zderzeń reagujących cząsteczek; energia cieplna może jednak zwiększać energię kinetyczną enzymu do stanu, w którym zostanie przekroczona bariera energetyczna wystarczająca do rozerwania wiązań niekowalencyjnych utrzymujących przestrzenną strukturę enzymu (denaturacja i utrata aktywności katalitycznej); czynnik Q10 (współczynnik temperaturowy) - charakteryzuje stopień wzrostu szybkości procesu biologicznego przy wzroście temperatury o 10oC (w temperaturach, w których enzymy są stabilne, szybkość większości procesów biologicznych zazwyczaj podwaja się przy takim wzroście temperatury, tj. Q10 = 2)
+ stężenie jonów wodorowych (pH) - zależy od niego szybkość prawie wszystkich katalizowanych enzymatycznie reakcji; większość wewnątrzkomórkowych enzymów wykazuje optymalna aktywność w pH 5-9; zależność aktywności od pH odzwierciedla równowagę między denaturacją enzymu w wysokim i niskim pH i wpływa na ładunek enzymu, substratu lub obu tych elementów
+ stężenie substratu:
- szybkość początkowa (vi) - szybkość reakcji w rozpatrywanym kierunku; w przypadku dużego nadmiaru substratu w stosunku do enzymu jest proporcjonalna do stężenia enzymu
- jeżeli stężenie substratu dla typowego enzymu wzrasta, to vi wzrasta do momentu osiągnięcia maksymalnej wartości Vmax - dalsze zwiększenie stężenia substratu nie powoduje wzrostu vi (enzym jest nasycony substratem)
- w danym momencie tylko cząsteczki substratu, które są połączone z enzymem w kompleks ES mogą być przekształcane w produkt
- stała równowagi reakcji tworzenia kompleksu ES nie jest nieskończenie duża, dlatego nawet jeśli substrat występuje w nadmiarze to tylko część enzymu może tworzyć kompleks
- w warunkach nasycenia vi zależy tylko od szybkości, z jaką produkt oddysocjowuje od enzymu, aby enzym mógł się łączyć z nadmiarem substratu
- enzymy działające z maksymalną szybkością nie mają możliwości reagowania na wzrost stężenia substratu, a jedynie na nagły spadek jego stężenia; dlatego w przypadku większości enzymów wewnątrzkomórkowe stężenie ich substratów odpowiada w przybliżeniu wartości Km - dzięki temu zmiany stężenia substratu wywołują odpowiednie zmiany przepływu metabolitów (stanowi to bierny sposób koordynacji przepływu metabolitów
+ stężenie enzymu
- równanie Michaelisa-Menten:
+ wyraża matematyczną zależność między początkową szybkością reakcji vi i stężeniami substratu [S]
![]()
+ stała Michaelisa (Km) - odpowiada stężeniu substratu, przy jakim vi stanowi połowę szybkości maksymalnej osiąganej przy danym stężeniu enzymu
![]()
+ jeśli [S] < Km to Km + [S] jest prawie równe Km; początkowa szybkość reakcji jest wprost proporcjonalna do [S] (ponieważ Vmax i Km są stałymi)
![]()
+ jeśli [S] > Km to Km + [S] jest prawie równe [S]; szybkość reakcji jest maksymalna (Vmax) i nie ma na nią wpływu dalszy wzrost stężenia substratu
+ jeśli [S] = Km to vi = 1/2Vmax; Km odpowiada stężeniu substratu, przy jakim szybkość początkowa stanowi połowę szybkości maksymalnej
- wykres Lineweavera-Burka (wykres podwójnych odwrotności) - liniowe przekształcenie równania Michaelisa-Menten, wykorzystywane do określenia Km i Vmax
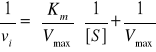
- powinowactwo enzymu do substratu jest równe odwrotności stałej dysocjacji (Kd) kompleksu ES; mniejsza tendencja kompleksu ES do dysocjacji wskazuje na większe powinowactwo enzymu do substratu; Km często jest bliska Kd (ale nie w każdym przypadku)
- podsumowanie - enzymy mogą na kilka różnych sposobów zmniejszać swobodną energię aktywacji Gibbsa (ΔG):
+ obniżanie energii aktywacji przez tworzenie środowiska, w którym następuje stabilizacja stanu przejściowego (np. zniekształcenie cząsteczki substratu - dzieje się to przez utrwalenie konformacji stanu przejściowego pomiędzy substratem, a produktem oraz zniekształcenie przez enzym wiązań w cząsteczce substratu, dzięki czemu zmniejsza się suma energii wymaganej do zakończenia przejścia)
+ obniżanie energii stanu przejściowego przez likwidację niekorzystnych energetycznie oddziaływań ze środowiskiem i ich zamianę na korzystne energetycznie oddziaływania z centrum aktywnym (np. oddziaływania elektrostatyczne ładunków o przeciwnych znakach)
+ wykorzystanie alternatywnego szlaku przejścia, np. tymczasowa reakcja substratu do pośredniego kompleksu enzym-substrat (ES), która nie byłaby możliwa w nieobecności enzymu
+ zmniejszanie entropii reakcji poprzez usytuowanie dostarczanych razem substratów w poprawnej orientacji, niezbędnej do zajścia reakcji
- stabilizacja stanu przejściowego - enzymy w obsługiwanych reakcjach obniżają energię aktywacji (ΔG) poprzez stabilizację stanu przejściowego - jest to możliwe poprzez niezwykle efektywne wykorzystanie efektów oddziaływań elektrostatycznych pomiędzy cząsteczkami w stanie przejściowym, a otaczającym je środowiskiem. W reakcji niekatalizowanej kompleks stanu przejściowego oddziałuje z otaczającą wodą w sposób mało uporządkowany, co zmusza system do pokonania dużej bariery energetycznej; enzym, zapewniając ściśle dopasowaną kieszeń centrum aktywnego, jest w stanie zagwarantować optymalne energetycznie oddziaływanie kompleksu stanu przejściowego z otaczającym je środowiskiem (np. zapewniając mu kontakt z resztami aminokwasowymi centrum aktywnego dokładnie pasującymi elektrostatycznie do rozkładu ładunków na powierzchni kompleksu stanu przejściowego); samo zbliżenie do siebie substratów, a także zapewnienie odpowiednich donorów/akceptorów elektronów zachodzącej reakcji, obniża koszty energetyczne jej przebiegu, co nie ma miejsca dla reakcji zachodzącej spontanicznie w wodzie
- kinetyka - podsumowanie:
+ by znaleźć maksymalną szybkość reakcji katalizowanej enzymatycznie, określa się ilość tworzonego produktu w funkcji czasu, przy stopniowym zwiększaniu stężenia substratu, ale jednoczesnym zachowaniu innych warunków, do momentu, w którym dalszy wzrost jego stężenia nie powoduje dalszego przyśpieszania reakcji; szybkość początkowa w takim eksperymencie (V0), rośnie do wartości maksymalnej (Vmax) i nie przekracza jej mimo dalszego wzrostu stężenia substratu
+ w chwili osiągnięcia stanu równowagi (reakcji katalizowanej ze stałą szybkości k2 i reakcji do niej przeciwnej zachodzącej ze stałą szybkości k-2) nie ma zmiany netto stężenia substratów i produktów
+ krzywa Michaelisa-Menten - przedstawia zależność szybkości reakcji od stężenia substratu dla prostej reakcji wg kinetyki Michaelisa-Menten, przy założeniu, że kompleks ES jest koniecznym etapem pośrednim procesu katalitycznego
+ wysycanie enzymu substratem (zbliżanie się do szybkości maksymalnej) oznacza, że maleje liczba wolnych cząsteczek enzymu, gdyż rośnie ilość ES; osiągana asymptotycznie Vmax reakcji enzymatycznej, oznacza, że praktycznie wszystkie miejsca aktywne enzymu (wolne enzymy) zostają wysycone substratem, a wówczas suma ilości (stężenie) kompleksów ES jest miarą ilości samego enzymu
+ stałe:
- Vmax
- Km - ilość substratu (stężenie) potrzebne do osiągnięcia ustalonej szybkości reakcji; jest takim stężeniem substratu przy którym szybkość reakcji osiąga połowę swojej maksymalnej wartości; każdy enzym ma swoją charakterystyczną wartość Km dla danego substratu, która charakteryzuje również siłę wiązania substratu w miejscu aktywnym enzymu
- kkat (szybkość katalizy) - jest liczbą obrotów jednego miejsca aktywnego enzymu w czasie jednej sekundy (liczba reakcji przeprowadzana przez jedno miejsce aktywne w czasie jednej sekundy)
+ jednostki używane do opisu aktywności enzymów:
- jednostka enzymu (U) - taka ilość enzymu, która katalizuje przemianę 1 μmola substratu w czasie 1 minuty w temperaturze 30 °C i określonym pH
- katal (kat) - ilość enzymu katalizująca przemianę 1 mola substratu w czasie 1 sekundy
+ gdy stężenie substratu znacznie przewyższa Km, szybkość katalizy jest równa kkat, liczbie obrotów; w warunkach fizjologicznych większość enzymów nie jest wysycona substratem, zatem stosunek [S]/Km mieści się w granicach od 0,01 do 1,0:
+ gdy [S] jest dużo mniejsze od Km ([S]<<Km), wówczas szybkość katalizowanej reakcji jest znacznie mniejsza od kkat, ponieważ większość miejsc aktywnych nie jest zajęta; do scharakteryzowania kinetyki enzymów w tych warunkach służy równanie V0=kkat/Km[E][S]; stężenie wolnego enzymu jest niemal równe całkowitemu stężeniu enzymu - w tych warunkach stosunek kkat/Km jest stałą szybkości oddziaływania S i E i może być użyty jako miara wydajności katalitycznej; ponieważ stosunek kkat/Km odzwierciedla zarówno powinowactwo chemiczne, jak i zdolność katalityczną, jest używany do porównywania różnych enzymów względem siebie lub preferencji jednego enzymu do różnych substratów
+ w sytuacji, gdy szybkość tworzenia produktu (kkat) jest znacznie szybsza niż szybkość dysocjacji kompleksu ES, wartość kkat/Km zbliża się do wartości stałej tworzenia kompleksu ES - wynika z tego, że górna granica wartości kkat/Km jest ograniczana przez szybkość tworzenia kompleksu ES, która nie może być większa niż częstość z jaka spotykają się enzym i jego substrat na skutek dyfuzji. Częstość ta mieści się w granicach od 108 do 109 (M-1 s-1) i jest to jednocześnie górna granica kkat/Km. W tym punkcie, każde zderzenie enzymu z jego substratem będzie powodowało katalizę i szybkość formowania produktu nie jest limitowana przez szybkość reakcji ale przez szybkość dyfuzji - enzymy posiadające tą własność to enzymy katalitycznie perfekcyjne lub kinetycznie perfekcyjne.
4. Czynniki aktywujące i inhibitory enzymów:
- inhibitory kompetycyjne:
+ inhibitor i substrat współzawodniczą o miejsce aktywne cząsteczki enzymu; najczęściej inhibitor przyłącza się do części miejsca aktywnego enzymu wiążącej substrat i blokuje dostęp dla substratu (kompleks EI jest nieaktywny); struktury większości klasycznych inhibitorów kompetycyjnych przypominają struktury substratu (są tzw. analogami substratów)
+ działanie inhibitora kompetycyjnego polega na zmniejszeniu liczby wolnych cząsteczek enzymu zdolnych do przyłączenia substratu
+ zazwyczaj inhibitor kompetycyjny jest strukturalnie bardzo podobny (lub ma podobny motyw bezpośrednio wiążący się do centrum aktywnego) do prawdziwego substratu dla określonego enzymu
+ przyłączenie substratu usuwa wolne cząsteczki enzymu mogące się połączyć z inhibitorem, więc zwiększenie [S] obniża stężenie kompleksu EI i zwiększa szybkość reakcji; stężenie substratu odpowiednie do całkowitego zniesienia hamowania zależy od stężenia inhibitora, jego powinowactwa do enzymu (Ki) oraz wartości Km enzymu dla danego substratu
+ inhibitor kompetycyjny nie ma wpływu na Vmax, ale zwiększa Km' (pozorną Km dla substratu); Vmax może być osiągnięta poprzez zwiększenie stężenia substratów, które przezwycięży inhibicję
+ przykład:
- hamowanie dehydrogenazy bursztynianowej przez malonian
- hamowanie reduktazy dihydrofolianu przez metotreksat
- inhibicja akompetycyjna:
+ inhibitor nie może się wiązać do wolnego enzymu, a jedynie do kompleksu enzym-substrat (ES), tworząc kompleks enzym-inhibitor-substrat (EIS)
+ zmniejszone jest stężenie kompleksu ES, zwiększa to pozorne powinowactwo enzymu do substratu (wartość Km jest mniejsza)
+ kompleks EIS jest enzymatycznie nieaktywny i reakcja nie może być kontynuowana, dopóki miejsce aktywne enzymu nie zostanie zwolnione, co obniża Vmax w porównaniu do reakcji nieinhibowanej
+ występuje dość rzadko i może dotyczyć niektórych enzymów multimerycznych
- inhibitory niekompetycyjne:
+ mogą się wiązać do wolnego enzymu, jednak nigdy do jego miejsca aktywnego, wobec tego nie konkurują z substratami; przyłączenie inhibitora nie ma wpływu na przyłączenie substratu, dlatego możliwe jest tworzenie kompleksów zarówno typu EI, jak i EIS (oba są enzymatycznie nieaktywne)
+ kompleks EI może przyłączać substrat, jednak jego zdolność przekształcania substratu w produkt (wyrażana wartością Vmax) jest mniejsza
+ inhibitory te przyłączają się w miejscach odmiennych od miejsc przyłączania substratu i zasadniczo nie wykazują żadnego (lub małe) podobieństwa do substratu
+ w prostym niekompetycyjnym hamowaniu E i EI mają identyczne powinowactwa do substratu i kompleks EIS tworzy produkt z bardzo małą, pomijalną szybkością; bardziej złożone niekompetycyjne hamowanie zachodzi wtedy, kiedy przyłączenie inhibitora ma wpływ na pozorne powinowactwo enzymu do substratu
+ wiązanie inhibitora jest całkowicie niezależne od substratu, co oznacza, że większe stężenie substratu nie wpływa na obniżenie oddziaływań z inhibitorem - zmienia się tylko wartość Vmax a wartość Km pozostaje stała
+ inhibicja może zostać zatrzymana po usunięciu inhibitora ze środowiska
- inhibicja mieszana - przypomina inhibicję niekompetycyjną, z wyjątkiem występowania dodatkowo kompleksu enzym-inhibitor-substrat (EIS), mającego znikomą aktywność enzymatyczną; zmiana Km i Vmax
- inhibitory nieodwracalne:
+ nieodwracalnie reagują z enzymem; powodują chemiczną modyfikację enzymu, które obejmują zazwyczaj tworzenie lub rozkład wiązań kowalencyjnych z resztami aminokwasowymi odpowiadającymi za przyłączenie substratu, katalizę lub utrzymanie funkcjonalnej konformacji enzymu
+ enzym poddany działaniu takiego inhibitora (np. atom metalu ciężkiego lub związek acylujący) pozostaje zahamowany nawet po usunięciu z otaczającego środowiska pozostałych cząsteczek inhibitora - następuje trwałe unieczynnienie cząsteczki enzymu
+ przykład:
- eflornityna - lek stosowany w śpiączce afrykańskiej
- penicylina i aspiryna - inhibitory te wstępnie (jeszcze nie kowalencyjnie), wiążą się z miejscami aktywnymi enzymów, po czym dopiero aktywność enzymu przekształca je w reaktywne formy, które wiążą się nieodwracalnie z jedną lub więcej resztami aminokwasowymi miejsca aktywnego enzymu, blokując je
- aktywatory enzymów:
+ proenzym - nieczynna postać enzymu; centrum aktywne zablokowane rpzez inhibitory (najczęściej białkowe lub polipeptydowe); aktywatory uczynniają proenzym
+ substancje wzmagające lub umożliwiające działanie enzymów; aktywatory sprzyjają uformowaniu właściwej konformacji centrum aktywnego (działają przez efekty allosteryczne)
+ niektóre aktywatory enzymatyczne chronią enzymy przed działaniem czynników chemicznych, inne odszczepiają od nieczynnych proenzymów blokujące je grupy chemiczne
+ przykłady:
- trypsyna aktywująca chymotrypsynę
- jony chlorkowe aktywujące α-amylazę ślinową
- enterokinaza - aktywator pepsyny (odłącza peptydy od pepsynogenu).
5. Enzymy jako markery chorób człowieka:
- enzymy wskaźnikowe (indykatorowe, nekroenzymy) - pojawiają się w dużych ilościach po uszkodzeniu narządów; zawartość enzymu wskaźnikowego jest zależna od jego ilości w uszkodzonej tkance, liczby uszkodzonych komórek i stopnia ich uszkodzenia, a także rozmieszczenia enzymów w różnych narządach; swoiste i nieswoiste narządowo; diagnostycznie liczy się ich górna granica normy
- enzymy pełniące funkcje niezbędne dla życia komórki są obecne we wszystkich tkankach organizmu; inne enzymy lub izoenzymy ulegają ekspresji tylko w specyficznych typach komórek, w pewnych okresach rozwoju lub w odpowiedzi na specyficzne fizjologiczne lub patofizjologiczne zmiany; analiza obecności i dystrybucji enzymów, których ekspresja jest normalnie tkankowo-, czasowo- lub warunkowospecyficzna często pomaga w diagnozowaniu
- enzymy osocza:
+ funkcjonalne - enzymy, proenzymy i ich substraty obecne cały czas w krążeniu zdrowych ludzi i pełniące funkcje biologiczne (np. lipaza lipprotein, pseudocholinesteraz, proenzymy krzepnięcia krwi i fibrynolizy); większosć z nich jest syntetyzowana i wydzielana przez wątrobę
+ niefunkcjonalne - pełnią nieznane fizjologicznie funkcje we krewi; pochodzą ze zwykłego, prawidłowego rozpadu erytrocytów, leukocytów i innych komórek; uszkodzenie tkanki lub nekroza są zazwyczaj połączone ze wzrostem poziomu wielu z tych enzymów
+ główne enzymy osocza wykorzystywane w diagnostyce klinicznej (wiele nie jest specyficznych dla podanych chorób):
- aminotransferazy:
+ asparaginianowa (AST, SGOT) - zawał serca; narządowo niespecyficzny enzym biorący udział w przemianach białek; przenosi grupy aminowe z aminokwasów na α-ketokwasy
+ alaninowa (ALT, SGPT) - wirusowe zapalenie wątroby
+ przyczyny wzrostu aktywności AST i ALT:
- martwica mięśnia serca (zawał mięśnia sercowego, pourazowe uszkodzenie serca np. po zabiegach kardiochirurgicznych)
- choroby wątroby (zapalenia wątroby niezależnie od etiologii)
- uszkodzenie mięśni szkieletowych (urazy, dystrofia mięśniowa)
- mononukleoza zakaźna
- hipoksja
- niewydolność krążenia
- zapalenie trzustki
+ wskaźnik de Ritisa - stosunek ASTT do ALT; w warunkach prawidłowych wskaźnik ten jest większy od jedności (wartość AST nieco wyższa od ALT), natomiast spadek poniżej 0,9 silnie wskazuje na chorobę miąższu wątroby; nie wylicza się Go w warunkach prawidłowych (gdy aktywność AST i ALT mieści się w normie)
- amylaza - ostre zapalenie trzustki; także urazy ślinianek, alkoholizm, niektóre nowotworów złośliwych (rak oskrzela, tarczycy, wątroby), zapalenie otrzewnej, świnka, niewydolność nerek
- ceruloplazmina - uczestniczy w transporcie żelaza; obniżone stężenie - zwyrodnienie wątrobowo-soczewkowate (choroba Wilsona), choroby wątroby; podwyższone stężenie - ciąża, stany zapalne, nowotwory
- fosfokinaza kreatynowa - choroby mięśni, zawał serca
- transpeptydaza γ-glutamylowa - enzym związany z przemianą aminokwasów, wydalany głównie z żółci; różne choroby wątroby (żółtaczka zastoinowa, ucisk na przewód żółciowy przez zmienioną zapalanie trzustkę, stosowanie leków przeciwpadaczkowych, nadużywanie alkoholu, nowotwory wątroby)
- dehydrogenaza mleczanowa (izoenzymy) - zawał serca
- lipaza - ostre zapalenie trzustki
- fosfataza kwasowa - katalizuje defosforylację różnych estrów fosforanowych; rak gruczołu krokowego z przerzutami, masaż gruczołu krokowego, choroby kości (w tym nowotwory przerzutowe, osteoporoza), nadmierny rozpad erytrocytów lub trombocytów (różne choroby)
- fosfataza zasadowa (izoenzymy) - nadmierna aktywność osteoblastów (nadczynność przytarczyc, nowotwory pierwotne i przerzutowe kości, niedobór witaminy D), upośledzenie wydalania żółci (zamknięcie dróg żółciowych przez kamień lub nowotwór), objaw paranowotworowy przy wzmożonej produkcji przez niektóre nowotwory.
- izoenzymy dehydrogenazy mleczajowej i ich rola w wykrywaniu zawałów serca:
+ dehydrogenaza L-mleczanowa - enzym tetrameryczny, którego cztery podjednostki występują w dwóch izoformach określonych symbolami H (serce) i M (mięśnie); podjednostki te mogą się łączyć w celu wytworzenia aktywnych izoenzymów dehydrogenazy mleczajowej (I1-I5)
+ podjednostki H i M są kodowane przez odmienne geny, których ekspresja jest różnie regulowana w poszczególnych tkankach (w sercu ekspresji ulega prawie wyłącznie podjednostka H, dominuje izoenzym I1; w wątrobie dominuje izoenzym I5, zbudowany wyłącznie z podjednostek M)
+ w stanie prawidłowym w osoczu występują niewielkie ilości dehydrogenazy mleczajowej; po zawale lub w chorobie wątroby tkanki uwalniają charakterystyczne izomerazy do krwi; podwyższony poziom I1/I5 jest wykrywany przez badanie aktywności katalitycznej oddzielonych elektroforetycznie różnych oligomerów dehydrogenazy mleczajowej
- enzymy mogą być użyteczne w rozpoznaniu chorób genetycznych (wykorzystanie polimerazy w PCR, enzymy w RFLP).
Wyszukiwarka