Ciała stałe występujące w wodzie mogą tworzyć trzy układy dyspersyjne:
- układy o dyspersji molekularnej, czyli roztwory rzeczywiste,
- układy o rozdrobnieniu koloidalnym (zole), oznaczające się wymiarami cząstek od 10-3 do
2·10-1 µm,
- zawiesiny (układy grubodyspersyjne) o rozmiarach cząstek powyżej 2·10-1 µm.
Granica pomiędzy rozproszeniem koloidalnym a rozproszeniem molekularnym jest
granicą wybraną w pewnym stopniu dowolnie. Istnieje raczej stopniowe przejście do układów
o rozproszeniu molekularnym do układów o rozproszeniu koloidalnym.
Domieszki występujące w wodzie w stanie koloidalnym nie dają się z niej usunąć
przez filtrowanie lub sedymentację. Zmniejszenie stopnia dyspersji koloidalnych
zanieczyszczeń wody (przejście zolu w żel), można osiągnąć przez zastosowanie procesu
koagulacji.
Koloidy są naturalnymi domieszkami wód powierzchniowych, powodującymi ich
mętność lub zabarwienie. Mogą one być pochodzenia mineralnego (cząstki gliny, iłów) lub
organicznego (produkty rozpadu substancji roślinnych lub zwierzęcych, substancje
próchnicze i humusowe). Koagulacja umożliwia skuteczne usunięcie tych domieszek, a także
bardzo drobnych zawiesin oraz zanieczyszczeń występujących w stanie rozpuszczonym, o
dużych cząsteczkach.
Cząstki koloidów maja charakterystyczną budowę: składają się – z jądra oraz dwóch
warstw: adsorpcyjnej i dyfuzyjnej. Ładunek elektryczny cząstek koloidalnych jest rezultatem selektywnej adsorpcji jonów z roztworu na powierzchni cząstki tj. utworzenia warstwy
adsorpcyjnej. Najlepiej adsorbowane są jony wchodzące w skład struktury cząstki koloidalnej lub takie, których adsorpcja nie narusza ich struktury. Wskutek adsorpcji, na powierzchni
cząstki skupiają się jony dodatnie lub ujemne obecne w roztworze, dzięki czemu cząstka
koloidalna uzyskuje ładunek. Dokoła naładowanej cząstki gromadzą się jony o znaku
przeciwnym, przyciągane siłami elektrostatycznymi. Na skutek przyciągania się
różnoimiennych jonów oraz działania sił energii wewnętrznej powodujących ruch jonów,
zewnętrzna warstwa (dyfuzyjna) uzyskuje charakter obłoku jonów rozrzedzającego się w
kierunku przeciwnym do centrum cząstki. Między warstwą adsorpcyjną a ruchomą częścią
warstwy dyfuzyjnej powstaje różnica potencjałów zwana potencjałem elektrokinetycznym ξ.
Potencjał ten jest zawsze mniejszy od potencjału termodynamicznego Nernsta φ, który
występuje między fazą stałą a ciekłą. Wielkość potencjału elektrokinetycznego ξ stanowi o
stabilności układu koloidalnego – im wyższy potencjał, tym większe siły odpychające, które oddziaływują na cząstki koloidów.
Przeciwnie do sił odpychania działają międzycząsteczkowe siły van der Waalsa, w wyniku
których cząsteczki przyciągają się wzajemnie dążąc do zmniejszenia wielkości powierzchni
granicznej faz.
Inną cechą układów koloidalnych są ruchy Browna. Są one wynikiem zderzeń cząstki
koloidalnej z cząsteczkami wody. Przy dużych rozmiarach cząstek liczba zderzeń ze
wszystkich stron równoważy się, natomiast przy małych wymiarach cząstki koloidalnej
występuje duża fluktuacja w liczbie zderzeń. Zderzenia wzajemne cząstek mogą powodować
zbliżenia się ich na taką odległość, przy której może dojść do trwałego połączenia się cząstek.
Proces ten jest jednak bardzo powolny z uwagi na małą częstotliwość zderzeń prowadzących
do zbliżenia, przy których mogą już działać siły przyciągania.
Koloidy, które adsorbują na sobie warstwę cząsteczek rozpuszczalnika, są bardziej stabilne, gdyż wzajemne zetknięcie się cząstek koloidalnych jest dodatkowo utrudnione. Adsorpcję
cząsteczek rozpuszczalnika na koloidach określa się mianem solwatacji, a w przypadku
roztworu wodnego – hydratacji. Koloidy, które ulegają solwatacji, nazywane są liofilowymi,
natomiast takie, które nie adsorbują cząsteczek rozpuszczalnika – liofobowymi. W
odniesieniu do roztworu wodnego są to odpowiednio koloidy hydrofilowe i hydrofobowe.
Koloidy nadające mętność wodom naturalnym są w większości hydrofobowe, podobnie jak
wodorotlenki glinowe i żelazowe, natomiast związki humusowe powodujące barwę wody
mają charakter zbliżony do koloidów hydrofilowych. Koloidy hydrofilowe mogą tworzyć
warstwy ochronne wokół koloidów hydrofobowych, zwiększając ich trwałość. Koagulację
definiuje się jako proces łączenia cząstek koloidalnych oraz drobnej zawiesiny w zespoły
cząstek (aglomeraty), w wyniku zaistnienia warunków sprzyjających destabilizacji układu.
Zgodnie z teorią DLVO (Derjaguin, Overbeck, Landau, Verwey) [6] o zaistnieniu koagulacji
lub nie – decyduje wypadkowa sił elektrostatycznych i van der Waalsa-Londona. Rys. 1
przedstawia przebieg wypadkowej sił oddziaływań między cząsteczkami. Uwzględnione tu
zostały siły objęte teorią DLVO.
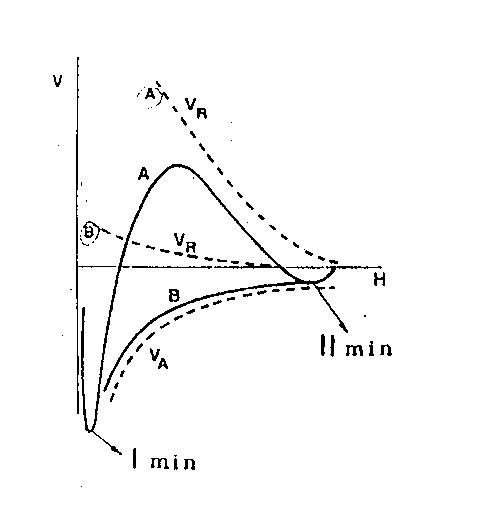
Rys. 1. Zależność energii oddziaływania cząstek (V) od odległości między nimi (H).
VR – energia odpychania,
VA – energia przyciągania,
A,B – krzywe prezentujące całkowitą energię oddziaływania cząstek
W obszarze pośrednim (tj. przy odległościach rzędu grubości podwójnej warstwy
elektrycznej) istnieją dwie możliwości:
- przy dużej wartości VR występuje na krzywej potencjału maksimum zwane „barierą
potencjału”; przy średnich odległościach może także pojawić się drugie minimum, znacznie
płytsze od pierwszego (Rys. 1. Krzywa A),
- przy małych wartościach VR potencjał układu jest zawsze ujemny lub ma wartość zero
(krzywa B, Rys.1)
Jeśli cząstki znajdą się względem siebie w odległości, przy której występuje
minimum, to może dojść do ich koagulacji (w pierwszym minimum) lub flokulacji (w drugim
minimum). Trwałość powstających agregatów zależy m.in. od „głębokości” minimum i
dlatego w przypadku flokulacji jest ona niewielka, a cząstki mogą łatwo zostać
zdyspergowane ponownie, np. przez mieszanie. Toteż, aby nastąpiła trwała agregacja cząstek co może mieć miejsce w pierwszym minimum, konieczne jest pokonanie bariery
energetycznej przez zbliżające się cząstki, np. dzięki ich energii kinetycznej lub obniżenie bądź całkowite zlikwidowanie tej bariery. Agregacja dwóch cząstek może nastąpić tylko po
ich zderzeniu. Liczba zderzeń cząstek koloidalnych ma więc podstawowe znaczenie dla szybkości koagulacji. Nie każde zderzenie jest jednak pod tym względem efektywne tzn.
agregacja nie musi nastąpić w wyniku każdego zderzenia. Dla tej samej częstości zderzeń o
ich efektywności decydują przede wszystkim własności powierzchni cząstek taki jak: rodzaj i wielkość ładunku, szorstkość powierzchni, stopień hydrofobowości. Po wprowadzeniu do
roztworu substancji adsorbujących się na powierzchni własności te mogą ulec zmianie. W ten sposób skuteczność zderzeń może się bardzo zmienić, gdyż ulegnie zmianie wysokość bariery
energetycznej. Jeśli bariera energetyczna będzie odpowiednio niska, można osiągnąć stan, w którym wszystkie lub prawie wszystkie zderzenia powodują agregację. W tym przypadku o
szybkości koagulacji decyduje tylko część zderzeń. Dalsze dodawanie substancji
modyfikujących własności powierzchniowe nie wywoła już zmiany skuteczności zderzeń, a w
efekcie szybkości koagulacji. Zjawisko to nosi nazwę szybkiej koagulacji. Jeśli natomiast nie wszystkie zderzenia są efektywne, proces określa się mianem powolnej koagulacji, a o jego szybkości decyduje zarówno liczba zderzeń jak i ich skuteczność.
Siłami, które stabilizują układy koloidalne są zatem siły elektrostatyczne odpychania,
jednoimiennie naładowanych cząstek koloidów.
Do koagulacji można doprowadzić przez:
-
dodanie do koloidalnego roztworu elektrolitu obniżającego potencjał
elektrokinetyczny, np. soli metali wielowartościowych, polielektrolitów, surfaktantów,
mocnych kwasów lub zasad;
-
wytworzenie wodorotlenków metali, na których absorbują się kolidy i cząstki
zawiesinowe, (np. wodorotlenek żelaza lub glinu);
-
stworzenie warunków do wzajemnego przyciągania się i aglomeracji cząstek dzięki
neutralizacji ładunków lub wytworzeniu ładunków powierzchniowych o różnym
znaku, (np. przez zmianą pH);
-
mechaniczne mieszanie lub wstrząsanie;
-
stężanie lub rozcieńczanie zolu
-
ogrzewanie;
-
zamrażanie;
-
naświetlanie promieniami rentgenowskimi, krótkofalowymi lub ultrafioletem;
-
naświetlanie radiochemiczne;
-
wyładowania elektryczne;
-
ultradźwięki.
W technologii oczyszczania wody i ścieków największe zastosowanie znalazły metody,
których wykorzystanie wymaga dodawania do oczyszczanej wody elektrolitów lub związków
o odmiennym ładunku.
Koagulacja koloidów liofobowych za pomocą elektrolitów
Wrażliwość roztworów koloidalnych na działanie elektrolitów znana była już
pierwszym badaczom w tej dziedzinie: Selmiemu, Grahamowi, Faradayowi. Następnie w
1900 roku Hardy jako pierwszy stwierdził, że swą trwałość koloidy zawdzięczają
elektrostatycznym odpychaniom cząstek. Stwierdzono, że gdy potencjał ζ maleje wskutek
wzrostu stężenia elektrolitu, to szybkość koagulacji zwiększa się i przy wystarczająco małym potencjale ζ (potencjale krytycznym), zaczyna się szybka koagulacja. W większości
przypadków potencjał krytyczny nie zależy od rodzaju użytego elektrolitu. Siłę koagulującą elektrolitu charakteryzuje się stężenie minimalne Ckr , które pozwala na osiągnięcie szybkiej koagulacji. Ponieważ jednak przejście od powolnej do szybkiej koagulacji zachodzi
stopniowo w miarę wzrostu stężenia elektrolitu, wartość Ckr nie jest bardzo ostra. Siła
koagulacyjna jonów nieorganicznych jest tym większa (tzn. tym mniejsze sa wartości Ckr) im wyższa jest ich wartościowość. Zdolność koagulacyjna kationów dwuwartościowych jest
około 80 razy, a trójwartościowych 640 razy większa niż jonów metali alkalicznych (reguła
Schutzego-Hardy’ego). Siła koagulacyjna kationów organicznych wzrasta w miarę
wydłużania się łańcucha.
Ogólnie można stwierdzić:
-
koagulację powoduje jon o ładunku przeciwnym niż ładunek cząstki koloidalnej
-
szybkości koagulacji powolnej są zbliżone w przypadku podobnych wartości
potencjału ζ ,
-
siła koagulacyjna jonów zwiększa się w miarę wzrostu ich wartościowości, przy czym
wzrost ten (zgodnie z teorią podwójnej warstwy elektrycznej) powoduje w przypadku
jednakowych stężeń większe obniżenie potencjału ζ ,
-
jony organiczne silnie adsorbujące się, które według teorii Sterna powodują
szczególnie silne zmniejszenie potencjału ζ , wykazują dużą siłę koagulacyjną.
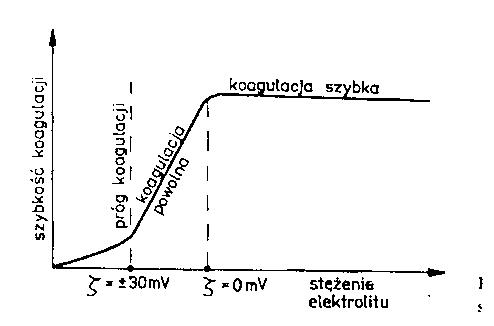
Rys. 2. Zależność szybkości koagulacji od stężenia elektrolitu.
Na rys. 2 przedstawiono zależność szybkości koagulacji od stężenia elektrolitu. Potencjał ζ
stabilnych koloidów wynosi około ± 70 mV. Przy zmniejszeniu potencjału do ± 30 mV
rozpoczyna się koagulacja powolna cząstek koloidalnych, a szybka koagulacja ma miejsce
gdy potencjał ζ zbliża się do 0. Dalsze zwiększanie stężenia elektrolitu nie zmienia szybkości koagulacji, co wskazuje na konieczność stosowania optymalnej dawki koagulantu.
Dstosowanie większej niż wymagana, ilości koagulanta, nie tylko nie poprawia efektów
koagulacji, a wręcz powoduje powstanie stabilnego układu koloidalnego o znaku ładunku
elektrycznego odpowiadającym ładunkowi stosowanego koagulantu.
Trwałość układu koloidalnego można naruszyć wprowadzając do niego jony lub
cząstki koloidów o ładunku przeciwnym do znaku ładunku układu, który chcemy
zdestabilizować. Koagulacja przez dodanie elektrolitu polega na zwiększeniu stężenia jonów o znaku przeciwnym niż znak ładunku cząstek koloidu. Warstwa dyfuzyjna ulega wówczas
ściśnięciu, a potencjał elektrokinetyczny maleje tak, że może nastąpić zetknięcie i
aglomeracja cząstek. Połączenie cząstek następuje wtedy, gdy siły Van der Waalsa są większe niż elektrostatyczne siły odpychania. Koagulacja może również przebiegać w wyniku
zobojętnienia ładunku na powierzchni cząstki przez dobrze adsorbujące się jony przeciwnego znaku. Zdolność koagulacyjna jonów zależy od wielkości ich ładunku, a więc od
wartościowości.
Koagulację można wywołać przez wprowadzenie układu koloidalnego do drugiego
układu o przeciwnym znaku. Powoduje to wzajemne rozładowanie się cząstek koloidalnych i
umożliwia tworzenie się większych aglomeratów. Warunkiem koniecznym wzajemnej
koagulacji jest równość lub zbliżenie do równości bezwzględnych wartości różnoimiennych
ładunków obu zoli. W przypadku nadmiaru lub niedostatecznej ilości jednego z zoli,
koagulacja może przebiegać tylko częściowo lub w ogóle nie dojść do skutku. Koloidy o
przeciwnych ładunkach wzajemnie się adsorbują, przy czym niezobojętniony ładunek pozostaje na nowo utworzonej cząstce.
Trwałość cząstek koloidalnych może także wynikać z utworzenia na ich powierzchni
otoczek drobin rozpuszczalnika (hydratacja cząstek). Otoczki te działają buforująco,
przeciwdziałając wzajemnemu zetknięciu się cząstek. Koagulacja tego typu koloidów (tzw.
hydrofilowych) następuje pod wpływem czynników, które działają odwadniająco.
Połączone w wyniku koagulacji cząstki ulegają dalszemu zlepianiu się i narastaniu
(flokulacji) – powstają w ten sposób aglomeraty o masie i rozmiarach pozwalających na
przeprowadzenie procesów osadzania i filtracji. Tworzące się kłaczkowate zawiesiny mają
bardzo rozwiniętą powierzchnię; dzięki przebiegającej równocześnie sorpcji odbywa się
dalsze usuwanie z wody domieszek i zanieczyszczeń.
Koloidy występujące w wodach naturalnych mają zazwyczaj ładunek ujemny.
Destabilizację takiego układu osiąga się przez wprowadzenie do wody uzdatnianej substancji zwanych koagulantami. Koagulanty ulegają w wodzie dysocjacji elektrolitycznej i procesom
hydrolizy. Prowadzi to do utworzenia jonów lub układów koloidalnych o ładunku dodatnim.
Koagulacja jest skomplikowanym procesem fizyko-chemicznym, na który wywiera wpływ
wiele różnorodnych czynników. Wśród nich największe znaczenie mają:
- właściwości wody surowej; jej skład (w tym barwa, mętność), odczyn pH uzdatnianej wody
ma zasadniczy wpływ na przebieg hydrolizy koagulantu. Bardzo często istnieje konieczność
korekty odczynu wody do takiej wartości pH, w którym produkt hydrolizy wykazuje
najmniejszą rozpuszczalność. Jednocześnie odczyn wody musi zapewniać optymalne warunki
koagulowania zanieczyszczeń zawartych w uzdatnianej wodzie.
- rodzaj i dawka koagulantu
Optymalna dawka koagulantu jest to najmniejsza jego ilość, niezbędna do przeprowadzenia
prawidłowej koagulacji. Szybkość kłaczkowania (flokulacja), jak i wielkość kłaczków,
pozwalają wówczas na szybkie usunięcie większości zanieczyszczeń wody. W praktyce dąży
się do tego, aby jak największą ilość powstających w procesie koagulacji zawiesin
kłaczkowatych można było wydzielić z wody przez sedymentację, a tylko pozostałą resztę
przez odfiltrowanie.
Warto podkreślić, że jeśli koagulacja przebiega dzięki oddziaływaniu na siebie dwóch
układów koloidalnych, to nadmiar koagulantu doprowadza do przewagi cząstek o odmiennym
ładunku; tworzy się wówczas nowy układ koloidalny.
Jeśli koagulację powodują jony powstające w wyniku dysocjacji koagulantu, to ich
zbyt duże stężenie (po przekroczeniu pewnej optymalnej dawki) jest niepożądane ze
względów ekonomicznych, chociaż efekt koagulacji nie pogarsza się.
- temperatura
Im niższa temperatura wody, tym szybkość hydrolizy i flokulacji jest mniejsza. Ujemny
wpływ temperatury można częściowo zmniejszyć stosując większą dawkę koagulantu lub
podwyższając pH; stosuje się także dodatek flokulantów.
- sposób mieszania wody z koagulantem
W procesie koagulacji można wyróżnić dwa etapy: szybkie i wolne mieszanie. Mieszanie
szybkie ma na celu równomierne rozprowadzenie koagulantu w całej masie wody, natomiast
wolne mieszanie pozwala na właściwy przebieg kłaczkowania.
Jako koagulanty stosuje się w technologii wody z reguły sole glinu lub żelaza. Najczęstsze zastosowanie znajdują: siarczan glinowy, glinian sodowy, siarczan żelazowy, siarczan
żelazawy i chlorek żelazowy. Do korygowania odczynu koagulowanej wody bardzo często
służy wapno w postaci wody wapiennej.
Koagulacja siarczanem glinowym
Siarczan glinowy ulega w rozcieńczonych roztworach wodnych hydrolizie:
Al2(SO4)3 + 6H2O →2 Al.(OH)3↓+3H2SO4
Następnie zachodzi reakcja między kwasem powstającym w wyniku hydrolizy a naturalną lub
sztucznie wywołaną zasadowością wody:
3H2SO4 + 3 Ca(HCO3)2 → 3CaSO4 + 6CO2 + 6H2O
3H2SO4 + 3 Ca(OH)2 → 3 CaSO4 + 6H2O
Przy określonym odczynie pH roztworu powstający wodorotlenek glinowy wykazuje
najmniejszą rozpuszczalność i najmniejszy potencjał elektryczny układu; wodorotlenek
przybiera wówczas postać koloidalną i łatwo powoduje rozładowanie cząstek koloidalnych o
przeciwnym znaku. Koagulacja przebiega wówczas najszybciej i daje bardzo dobre wyniki.
Wodorotlenek glinowy wykazuje jednak wrażliwość na zmiany pH, co powoduje konieczność
stałej kontroli procesu.
W środowisku zbyt alkalicznym powstający wodorotlenek glinowy (posiadający
właściwości amfoteryczne) tworzy rozpuszczalne gliniany:
Al.(OH)3 + OH → Al(OH)4
Natomiast w środowisku kwaśnym powstają rozpuszczalne sole glinu:
Al.(OH)3 + 3H+ → Al3+ + 3H2O
Dla koagulacji siarczanem glinowym wartość odczynu odpowiadająca punktowi
izoelektrycznemu waha się od pH = 6,4 do 7,8 w zależności od pochodzenia i skłądu
chemicznego wody naturalnej. Podany powyżej zakres optymalnego pH należy traktować
jako orientacyjny.
Koagulacja solami żelazowymi
Siarczan żelazowy i chlorek żelazowy wykazują zdolność do wytwarzania kłaczków w
procesie koagulacji w szerszym zakresie pH niż siarczan glinowy. Hydroliza siarczanu
żelazowego i chlorku żelazowego:
FeSO4 + 2H2O → 2Fe(OH)3↓ + 3 H2SO4
FeCl3 + 3 H2O → Fe (OH)3↓ + 3 HCl
Oraz reakcje wtórne przebiegają analogicznie jak w przypadku stosowania siarczanu
glinowego.
Koagulacja siarczanem żelazawym
Zdolność koagulacyjna jonów dwuwartościowych jest znacznie mniejsza niż jonów
trójwartościowych, a rozpuszczalność powstającego w wyniku hydrolizy wodorotlenku
żelazawego jest większa niż wodorotlenku żelazowego. Wynika stąd konieczność utleniania
żelaza dwuwratościowego do trójwartościowego. Reakcja utleniania jonów Fe2+ do Fe3+
przebiega z dobrym skutkiem tylko przy wartościach pH powyżej 8,5. Powstający w wyniku
hydrolizy siarczanu żelazawego wodorotlenek żelazawy utlenia się do wodorotlenku
żelazowego pod wpływem tlenu zawartego w wodzie:
FeSO4 + 2H2O → Fe(OH)2 + 2H2SO4
4Fe(OH)2 + O2 + 2H2O → 4Fe(OH)3↓
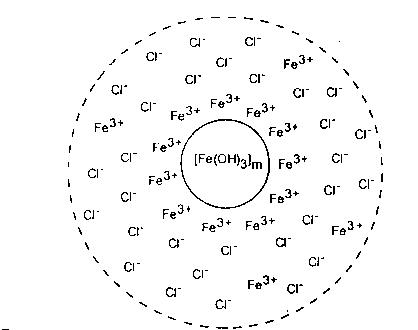
Rys. 3. Budowa miceli wodorotlenku żelazowego.
Odpowiedni odczyn wody ustala się przez alkalizację wapnem:
2 H2SO4 + Ca(OH)2 → CaSO4 + 2H2O
Proces utleniania przyspiesza stosowanie chloru:
6FeSO4 + 3 Cl2 → 2 Fe2(SO4)3 + 2 FeCl3
Substancje wspomagające koagulację
Celem stosowania środków wspomagających koagulację jest przyspieszenie
powstawania kłaczków i ich sedymentacji, zwiększenie powierzchni właściwej kłaczków, a
więc ich pojemności sorpcyjnej, zmniejszenie ujemnego wpływu niskich temperatur wody,
rozszerzenie optymalnego dla koagulacji zakresu pH i zmniejszenie dawki koagulantu.
Substancjami wspomagającymi koagulację mogą być substancje nieorganiczne lub
organiczne. Dawki substancji wspomagających są mniejsze niż koagulantów. Część ze
stosowanych substancji działa jako obciążniki kłaczków, inne wspomagają flokulację.
Do obciążników należą gliny, bentonit, ziemia Fullera, pył granitowy, anhydrytowy, drobny
piasek kwarcowy lub mielony węglan wapniowy. Typowymi flokulantami są krzemionka
aktywowana oraz polielektrolity.
Polielektrolity są wysokocząsteczkowymi polimerami organicznymi. Dzieli się je na
naturalne i syntetyczne. Naturalne wytwarzane są ze skrobi i celulozy. Polielektrolity
syntetyczne uzyskuje się w wyniku polimeryzacji monomerów organicznych o
niewysyconych wiązaniach. Zawierają one grupy jonotwórcze, a ich masy cząsteczkowe
wynoszą 1·106 – 4 · 106 u, a stosunek długości łańcucha polimeru do jego szerokości wynosi (1000-3000):1. Z uwagi na rodzaj grup jonotwórczych polielektrolity dzieli się na niejonowe (polialkohokle, polietery, poliamidy, heterocykliczne polimery N-winylowe), anionowe
(polimery karboksylowe i sulfonowe), kationowe (czwartorzędowe polimeryczne związki
anionowe, polimery sulfoniowe i fosfoniowe). Zdolność do dysocjacji zależy od ich budowy i zawartości grup jonogennych (kwasowych lub zasadowych), takich jak:
─ SO3H, ─ COOH, ─ PO3H2, ─ NH2, ==NH. Liniowe polimery mające zarówno grupy
kwasowe jak i zasadowe są związkami amfoterycznymi a ich dysocjacja i ładunek zależą od
pH roztworu. W wyniku dysocjacji w wodzie polielektrolitów kationowych powstają aniony i
polikation, natomiast anionowych kationy i polianion.
Stosowane ilości polielektrolitów wynoszą zwykle 0,1 – 1,0% dawki koagulantu
podstawowego. Należy unikać przedawkowania, ponieważ optymalna ilość flokulanta jest
zazwyczaj znacznie mniejsza niż łączna zdolność powierzchni cząstek stałych do jego
adsorpcji. Uważa się, że optymalna dawka polielektrolitu to taka, która powoduje pokrycie co najmniej 50% powierzchni usuwanych zanieczyszczeń. Polielektrolity powodują
destabilizację usuwanych koloidów w wyniku neutralizacji ładunku oraz działając jako
czynniki mostkujące i sieciujące powodują aglomerację i powstawanie dużych i trwałych
kłaczków. Uważa się, że podczas koagulacji koloidów hydrofobowych dominuje adsorpcja
polielketrolitów na ich powierzchni, a w przypadku koloidów hydrofilowych istotną rolę
odgrywa mechanizm destabilizacji i strącania usuwanych zanieczyszczeń.
Wyszukiwarka
Podobne podstrony:
Koagulacja i sedymentacja jako metody uzdatniania wody i oczyszczania ścieków, Inżynieria Ekologiczn
instrukcja bhp przy magazynowaniu i stosowaniu chloru w oczyszczalni sciekow i stacji uzdatniania wo
morawski sciaga, Studia, Uzdatnianie wody
UZDATNIANIE WODY
Uzdatnianie wody - Odgazowanie (1), Technologia Wody i Ścieków
5.Zastosowanie mas jonowymiennych w technologii uzdatniania wody, pytania dyplomowe
sciaga mor 2 (1), Studia, Uzdatnianie wody
Remont stacji uzdatniania wody
3z3, Inżynieria Środowiska, mgr 1 semestr, Uzdatnianie wody do celów przemysłowych, wykłady, opracow
45-7-BWiS-Ujęcia i urządzenia do uzdatniania wody
Technologia wody - koagulacja, Materiały na IŚ, Projekty, referat itp
sciaga woda, Studia, Uzdatnianie wody
Procesy sorpcji i wymiany jonowej w uzdatnianiu wody i oczyszczaniu ścieków
Przemysłowa regeneranja węgli aktywnych stosowanych do uzdatniania wody
ee15-uzdatnianie wody, OCHRONA ŚRODOWISKA
6 Uzdatnianie Wody Pitnej
od michała j, Inżynieria środowiska, Podstawy Projektowania Stacji Uzdatniania Wody
Projekt koncepcyjny stacji uzdatniania wody
więcej podobnych podstron