


Genetyka
zwierząt
Wydanie II unowocześnione
Krystyna M. Charon
Marek Switoński
WYDAWNICTWO NAUKOWE PWN
WARSZAWA 2004

Autorzy
Prof. dr hab. Krystyna Małgorzata Charon
Katedra Genetyki i Ogólnej Hodowli Zwierząt,
Wydział Nauk o Zwierzętach, SGGW, Warszawa
Prof. dr hab. Marek Świtoński
Katedra Genetyki i Podstaw Hodowli Zwierząt, Wydział
Hodowli i Biologii Zwierząt, Akademia Rolnicza, Poznań
Okładkę i strony tytułowe projektował
Edwin Radzikowski
Redaktor
Krystyna Kruczyńska
Redaktor techniczny
Jolanta Cłbor
Podręcznik akademicki dotowany przez
Ministerstwo Edukacji Narodowej i Sportu
Copyright © by Wydawnictwo Naukowe PWN SA
Warszawa 2000, 2004
ISBN 83-01-14022-4
Wydawnictwo Naukowe PWN SA
00-251 Warszawa, ul. Miodowa 10
tel. (O-prefiks-22) 69-54-321
faks (O-prefiks-22) 69-54-031
e-mail: pwn @pwn.com.pl
http://www.pwn.pl
Wydawnictwo Naukowe PWN SA
Wydanie drugie unowocześnione
Arkuszy drukarskich 20 + 12 stron ilustracji
Druk ukończono w marcu 2004 r.
Skład i łamanie: Fototype, Warszawa
Druk i oprawa: WDG Drukarnia w Gdyni Sp. z o.o.
ul. Sw. Piotra 12, Gdynia


Przedmowa
Coraz większe wykorzystywanie w hodowli zwierząt najnowszych osiągnięć
genetyki sprawia, że uaktualnianie programów nauczania tego przedmiotu
na kierunkach studiów związanych z nauką o zwierzętach staje się
koniecznością. Cytogenetyczna i molekularna identyfikacja mutacji, kontrola
pochodzenia za pomocą markerów związanych z polimorfizmem DNA czy
budowanie map genomów, to tylko niektóre przykłady obrazujące obszary
genetyki istotnie rozbudowane w ostatnich latach. W niniejszym podręczniku
podjęto próbę zaprezentowania, obok podstawowych wiadomości z zakresu
genetyki klasycznej, najnowszych osiągnięć, które znajdują zastosowanie
w hodowli zwierząt.
Podręcznik jest adresowany przede wszystkim do studentów kształcących
się na kierunkach: zootechnika, weterynaria, biotechnologia i biologia.
Mamy także nadzieję, że podręcznik będzie przydatny specjalistom, którzy
profesjonalnie zajmują się hodowlą zwierząt lub prowadzeniem badań
z tego zakresu.
Ważną częścią niniejszego opracowania są oryginalne ilustracje, które
ściśle nawiązują do wybranych zagadnień z cytogenetyki, genetyki mo-
lekularnej czy zmienności klasycznych cech jakościowych (np. umasz-
czenie). W przygotowaniu tej dokumentacji pomogli nam PT Współ-
pracownicy z Katedry Genetyki i Podstaw Hodowli Zwierząt AR w Po-
znaniu: A. Pietrzak, J. Klukowska, A. Pieńkowska, M. Zając, D. Lechniak,
D. Ładoń i J. Sosnowski oraz J. Gruszczyńska, Z. Nowak i M. Gajewska
z Katedry Genetyki i Ogólnej Hodowli Zwierząt SGGW w Warszawie.
Ponadto, kilka zdjęć zostało nam udostępnionych przez E. Słotę i B. Danielak--
Czech z Instytutu Zootechniki w Balicach, H. Geringera z AR we Wroc-
ławiu, K. Jaszczaka i R. Paradę z Instytutu Genetyki i Hodowli Zwierząt
PAN w Jastrzębcu. Niektóre zdjęcia pochodzą z archiwum Redakcji
Przeglądu Hodowlanego. Za okazaną pomoc serdecznie dziękujemy.

Spis treści
1 Wstęp — zarys historii genetyki ..........................
n
2 Chromosomy
i
podziały jądra komórkowego ..
15
2.1. Budowa chromosomu .........................................................................
16
2.2. Barwienie prążkowe chromosomów ....................................................
23
2.3. Mitoza..............................................................................., ......................
31
2.4. Kariotyp ..............................................................................................
33
2.5. Mejoza .................................................................................................
37
2.6. Gametogeneza ......................................................................................
43
3 Gen i jego ekspresja .......................................... 48
3.1. Budowa kwasów nukleinowych ..............................................................
48
3.1.1. DNA .................................................................................................
49
3.1.2. RNA .................................................................................................
50
3.2. Replikacja DNA ....................................................................................
52
3.3. Kod genetyczny ....................................................................................
53
3.4. Transkrypcja ..........................................................................................
54
3.5. Translacja .................................................................................................
56
3.6. Budowa genu...........................................................................................
57
3.7. Regulacja ekspresji genu ....................................................................... .
60
3.7.1. Czynniki transkrypcyjne ..................................................................
61
3.7.2. Geny homeotyczne ..........................................................................
62
3.7.3. Metylacja DNA ....................................................................................
63
3.7.4. Piętno gametyczne .........................................................................
64
3.7.5. Inaktywacja chromosomu X w zarodkach żeńskich ......................
66
4 Metody analizy i modyfikacji genomu ................. 69
4.1. Endonukleazy restrykcyjne .................................................................
69
4.2. Wektory....................................................................................................
72

8
4.3. Rekombinacja molekularna i klonowanie ............................................
76
4.4. Amplifikacja DNA za pomocą łańcuchowej reakcji połimerazy (PCR) . .
78
4.5. Techniki hybrydyzacji i sondy molekularne ...........................................
82
4.6. Polimorfizm fragmentów restrykcyjnych DNA (RFLP) ......................... 86
4.7. Biblioteki genomowe i genowe ............................................................
87
4.8. Sekwencjonowanie DNA .......................................................................
88
4.9. Badanie ekspresji genów ......................................................................
91
4.10. Transgeneza ......................................................................................
91
5 Mutacje .............................................................
95
5.1. Mutacje genomowe ..............................................................................
96
5.2. Mutacje chromosomowe .....................................................................
100
5.3. Mutacje genomowe i chromosomowe zidentyfikowane u zwierząt
hodowanych w Polsce ..............................................................................
111
5.4. Mutacje genowe ...................................................................................
112
5.4.1. Przyczyny i rodzaje mutacji genowych .............................................
112
5.4.2. Skutki mutacji genowych ................................................................... 118
5.4.3. Mutacje genowe wpływające na cechy produkcyjne zwierząt . . .
123
5.4.4. Mutacje genowe wywołujące choroby genetyczne ........................
124
5.4.4.1. Geny letalne, semiletalne i subwitalne .....................................
124
5.4.4.2. Choroby monogenowe wywołujące wrodzone wady rozwojowe
127
5.4.4.3. Choroby monogenowe wywołujące zaburzenia procesów bio
chemicznych ..........................................................................................
132
5.4.4.4. Ograniczanie występowania chorób genetycznych.......................
135
5.4.5. Mutacje genowe odpowiedzialne za odporność/podatność zwierząt
na patogeny ............................................................................................
143
5.5. Mutacje w mitochondrialnym DNA .................................................... 144
6 Podstawowe mechanizmy dziedziczenia cech ..
146
6.1. Dziedziczenie cech warunkowanych jedną parą alleli ...........................
147
6.1.1. Współdziałanie alleli ......................................................................
147
6.1.2. Niezależne dziedziczenie cech (II prawo Mendla) .........................
150
6.1.3. Dziedziczenie cech sprzężonych ................................................... 152
6.1.4. Cechy sprzężone z płcią .................................................................
157
6.1.5. Plejotropia .......................................................................................
160
6.2. Współdziałanie genów z różnych lód w kształtowaniu fenotypu . . .
160
6.2.1. Komplementarność ............................................................................. 161
6.2.2. Epistaza ...........................................................................................
162
6.2.3. Geny modyfikujące ............................................................................ 163
6.2.4. Sumujące działanie genów .................................................................
164
6.3. Dziedziczenie umaszczenia.....................................................................
164
6.4. Penetracja i stopień ekspresji (ekspresywność) genów ...........................
175
6.5. Dziedziczenie pozajądrowe .................................................................
176
7 Zmienność cech.....................................
179
7.1. Rodzaje zmienności i czynniki ją wywołujące ........................................ 179
7.2. Miary zmienności .................................................................................... 182
7.2.1. Miary skupienia .............................................................................
182

7.2.2. Miary rozproszenia ........................................................................
185
7.3. Zmienność cech ilościowych ....................................................................
187
7.3.1. Dziedziczenie cech ilościowych .......................................................
187
7.3.2. Zmienność transgresywna ..............................................................
189
7.3.3. Geny o dużym efekcie ....................................................................
190
8 Podstawy genetyki populacji ............................... 198
8.1. Prawo równowagi genetycznej ............................................................
198
8.2. Frekwencja genów i genotypów w przypadku dominowania ...........
201
8.3. Frekwencja genów i genotypów w przypadku cech uwarunkowanych
szeregiem (serią) alleli wielokrotnych .......................................................
201
8.4. Frekwencja genów ł genotypów w przypadku cech sprzężonych z płcią
204
8.5. Czynniki naruszające równowagę genetyczną ....................................
205
8.6. Wykorzystanie frekwencji alleli do szacowania zmienności genetycznej
wewnątrz i między populacjami .............................................................. .
210
9 Determinacja i różnicowanie płci oraz
interseksualizm .................................................. 210
9.1. Determinacja płci ssaków ....................................................................
216
9.2. Interseksualizm ....................................................................................
221
1 0 Genetyczne podstawy odporności i oporności ..
227
10.1. Genetyczne podłoże różnorodności przeciwciał ...................................
227
10.2. Genetyczne podłoże różnorodności receptorów limfocytów T . . . .
230
10.3. Główny układ zgodności tkankowej — MHC ...................................
231
10.3.1. Struktura i funkcje głównego układu zgodności tkankowej . . .
233
10.3.1.1. Główny układ zgodności tkankowej myszy ...........................
236
10.3.1.2. Główny układ zgodności tkankowej człowieka ......................
237
10.3.1.3. Główny układ zgodności tkankowej zwierząt gospodarskich
239
10.3.2. Identyfikacja cząsteczek głównego układu zgodności tkankowej
243
10.3.3. Znaczenie głównego układu zgodności tkankowej ...................
243
10.3.3.1. Związek MHC z odpornością na choroby ..............................
243
10.3.3.2. Związek MHC z cechami produkcyjnymi i reprodukcyjnymi
244
10.4. Zarys przebiegu reakcji odpornościowej ...........................................
245
10.5. Genetyczna oporność na choroby ......................................................
247
1 1 Markery
genetyczne
............................................. 253
11.1. Antygeny erytrocytarne (grupy krwi) .................................................
254
11.2. Białka surowicy krwi, erytrocytów i leukocytów ...............................
259
11.3. Allotypy immunoglobulin i lipoprotein ...............................................
263
11.4. Białka mleka .......................................................................................
263
11.5. Polimorfizm DNA ..............................................................................
265
11.6. Wykorzystanie markerów genetycznych w hodowli zwierząt...............
268
9

1 2 Mapy genomowe ............................................... 275
12.1. Organizacja DNA jądrowego ................................................................. 275
12.2. Organizacja DNA mitochondrialnego .................................................
277
12.3. Markerowa mapa genomu .................................................................
279
12.3.1. Fizyczna mapa genomu ...............................................................
280
12.3.2. Genetyczna mapa genomu ........................................................
284
12.3.3. Strategia mapowania genomu......................................................... 287
12.3.4. Aktualny stan map genomowych zwierząt gospodarskich . . . .
289
12.3.5. Wykorzystanie markerowych map genomowych w hodowli . . .
292
Literatura uzupełniająca ............................................ 293
Podręczniki ................................................................................................
298
Artykuły przeglądowe .................................................................................... 298
Skorowidz ................................................................... 301

Rozdział
1
Wstęp - zarys historii genetyki
Genetyka stanowi wyodrębnioną dyscyplinę nauk biologicznych, której
przedmiotem badań jest zmienność i dziedziczenie cech organizmów
żywych. Powstanie tej dyscypliny stało się możliwe dzięki intensywnemu
rozwojowi biologii w wieku XIX. Postęp ten znaczony był z jednej strony
poznawaniem struktury i funkcji organizmów, tkanek, komórek i ich
organelli, a z drugiej formułowaniem przełomowych hipotez i teorii, wśród
których część miała epokowe znaczenie dla rozwoju wiedzy o świecie
ożywionym. Opis struktury i funkcji organizmów na poziomie komórkowym
lub wewnątrzkomórkowym jest zależny od dostępnych urządzeń i metod
badawczych. Tym samym rozwój nauk biologicznych opiera się na osiągnię-
ciach fizyków, chemików, konstruktorów, a ostatnio również informatyków.
W wieku XIX sformułowano trzy teorie o ogromnym znaczeniu dla
dalszego rozwoju biologii. Autorami pierwszej z nich — teorii komórkowej
budowy organizmów, ogłoszonej w 1838 roku, byli Schwann i Schleiden.
Główną tezą tej teorii było twierdzenie, że wszystkie rośliny i zwierzęta są
zbudowane z komórek, korę mogą powstawać jedynie przez podział innych
komórek. Okazało się to niezwykle stymulujące dla rozwoju cytologii, czyli
nauki o budowie i funkcji komórek. Dwadzieścia lat później — w 1859
roku — Darwin opublikował dzieło pt. O powstawaniu gatunków drogą doboru
naturalnego, które stało się początkiem nauki o ewolucji. Zgodnie z teorią
Darwina, podstawą ewolucji organizmów żywych jest dobór naturalny,
który faworyzuje osobniki najlepiej przystosowane do środowiska bytowania,
umożliwiając im nie tylko przeżycie, ale także przekazanie tych korzystnych
właściwości potomstwu. W ten sposób powstają organizmy coraz lepiej
przystosowane do danego środowiska. Kilka lat później — w 1866 roku —
Mendel opublikował skromną pracę pt. Badania nad mieszańcami roślin,
stanowiącą zaczątek genetyki. Była ona syntezą kilkuletnich obserwacji
dziedziczenia siedmiu cech u grochu (Pisum sativum), rośliny samopylnej.
Każda z analizowanych cech miała dwie łatwo odróżnialne formy: kwiaty
11

czerwone lub białe, nasiona gładkie lub pomarszczone, nasiona żółte lub
zielone itd. Dokonując krzyżowania między ustalonymi formami przeciw-
stawnymi, Mendel uzyskał pierwsze pokolenie mieszańców (F
1
), które
rozmnażając się w sposób naturalny (samopylnie) dawało drugie pokolenie
mieszańców (F
2
). Bardzo skrupulatna analiza cech w obu pokoleniach dała
podstawy do sformułowania wniosków, że dziedziczenie cech przebiega na
drodze przekazywania zawiązków dziedzicznych (genów) od rodziców do
potomstwa za pośrednictwem gamet, przy czym jedna gameta zawiera
tylko jeden taki element. Spostrzeżenia te ostatecznie pozwoliły na sfor-
mułowanie praw dziedziczenia. Niestety doniosłość tego odkrycia nie
została doceniona przez ówcześnie żyjących biologów.
Wiek XIX przyniósł także ważne odkrycia dotyczące budowy komórki,
w tym struktur związanych z dziedziczeniem. Jądro komórkowe zastało po
raz pierwszy opisane przez Browna już w 1831 roku. Pierwsze doniesienia
o „pałeczkowatych" strukturach, nazwanych w 1888 roku przez Waldeyera
chromosomami, zostały podane przez polskiego botanika Strasburgera
w 1880 roku. Podziały jądra komórkowego zostały opisane pod koniec XIX
wieku. Opis mitozy przedstawił w 1882 roku Flemming, a opis mejozy
powstał w 1883 roku i zawdzięczamy go van Bendenowi i Boveriemu.
Na początku XX wieku podjęto wiele badań, których celem było poznanie
praw rządzących dziedziczeniem cech. Dopiero wówczas zrozumiano
genialność odkryć Mendla. Stało się to za sprawą trzech badaczy: Corrensa,
de Yriesa i Tschermacka, którzy niezależnie od siebie doszli do wniosków,
które kilkadziesiąt lat wcześniej podał Mendel. Następne lata to lawinowy
rozwój nauki nazwanej przez Batesona w 1906 roku genetyką. W ślad za
tym terminem w 1909 roku Johannsen zaproponował określenie „gen" dla
czynnika dziedzicznego warunkującego pojawienie się cechy. W 1909 roku
Hardy i Weinberg stworzyli podstawy genetyki populacji. Badacze ci
niezależnie od siebie sformułowali prawo równowagi genetycznej. Rok
później Morgan ogłosił chromosomową teorię dziedziczenia, którą można
uważać za początek cytogenetyki. W latach 20. Fischer, Wright i Haldane
wprowadzili do genetyki metody statystyczne, które okazały się bardzo
istotnymi narzędziami badawczymi, za pomocą których możliwe stało się
poznawanie mechanizmów odpowiedzialnych za zmienność i dziedziczenie
tzw. cech ilościowych. Ten dział genetyki określa się często jako genetykę
cech ilościowych. Niektórzy autorzy uważają, że zagadnienia genetyki
populacji i genetyki cech ilościowych są na tyle sobie bliskie, że można je
określać tylko jednym terminem — genetyka populacji.
Pomimo ogromnego zaangażowania się wielu naukowców w badania
genetyczne, do końca pierwszej połowy XX wieku niewiele można było
powiedzieć o chemicznej naturze informacji genetycznej. Przez wiele lat
przypuszczano, że nośnikiem informacji genetycznej są białka, o których
różnorodności wiedziano już wówczas dość dużo. Przełom nastąpił w 1944
roku, kiedy to trzej badacze: Avery, MacLeod i McCarty wykazali, że
12

nośnikiem informacji genetycznej jest kwas deoksyrybonukleinowy (DNA).
Do wniosku tego doszli na podstawie wyników eksperymentu, w którym
do niechorobotwórczych szczepów bakterii na drodze transformacji wpro-
wadzili DNA pochodzący z zabitych bakterii wywołujących zapalenie płuc
u myszy. W wyniku tego bakterie niechorobotwórcze nabyły cech zjadliwo-
ści. W doświadczeniu tym można upatrywać początku genetyki molekular-
nej. Odkrycie to nie zostało od razu zaakceptowane przez środowisko
biologów. Początek lat 50. to czas usilnych badań, których celem było
poznanie struktury chemicznej DNA. Model cząsteczki DNA przedstawili
w 1953 roku Watson i Crick. Po tym odkryciu przyszedł czas żmudnych
prac zmierzających do rozszyfrowania kodu genetycznego. Zostały one
zakończone w pierwszej połowie lat 60., a wiodącą rolę w tych badaniach
odegrali między innymi Khorana, Nirenberg i Ochoa. W tym czasie podjęto
również molekularne badania nad regulacją ekspresji genów. Doniosłym
osiągnięciem stało się ogłoszenie w 1960 roku przez Jacoba i Monoda tzw.
teorii operonu, która wyjaśniała mechanizm ekspresji genów u bakterii.
Rozwój genetyki molekularnej i związanych z nią wyrafinowanych
metod badawczych umożliwił przejście od opisu materiału dziedzicznego
do prowadzenia na nim manipulacji i w ten sposób wyodrębniła się nowa
dyscyplina — inżynieria genetyczna. Odkrycie enzymów restrykcyjnych
(1962 r.) oraz enzymu odwrotnej transkryptazy (1970 r.), opracowanie
techniki rekombinacji i klonowania DNA (1972 r.), sekwencjonowania DNA
(1975 r.) czy amplifikacji fragmentów DNA za pomocą łańcuchowej reakcji
polimerazy — PCR (1983 r.) to tylko najważniejsze przykłady współczesnych
narzędzi i metod badawczych wykorzystywanych obecnie powszechnie
w genetyce molekularnej i inżynierii genetycznej. Warto podkreślić, że
molekularne poznanie genomów człowieka, zwierząt, roślin czy mikroor-
ganizmów ma ogromne znaczenie aplikacyjne związane z diagnostyką
i terapią chorób genetycznych, hodowlą zwierząt i roślin, wykrywaniem
1 zwalczaniem patogenów, przetwórstwem żywności, ochroną środowiska
itp. Dążenie do jak najgłębszego poznania informacji genetycznej zwierząt
gospodarskich doprowadziło w ostatnich łatach do uruchomienia interdys
cyplinarnych prpgramów mapowania genomów takich gatunków, jak świnia,
bydło, koń, owca, kura czy pies. Celem tych programów jest wskazanie
położenia genów w chromosomach oraz ustalenie odległości między genami
umiejscowionymi w tym samym chromosomie. W przypadku człowieka
celem jest nie tylko mapowanie genomu, ale również jego sekwencjo-
nowanie, tzn. ustalenie kolejności par nukleotydów w całym DNA ludzkim,
liczącym ponad trzy miliardy par nukleotydów. Program ten został urucho
miony w 1987 roku, a w lutym 2001 roku ogłoszono na łamach dwóch
renomowanych czasopism: Naturę oraz Science zakończenie podstawowego
etapu tych badań. Warto zaznaczyć, że przedsięwzięcie to przez kilka
ostatnich lat było prowadzone przez dwa niezależne zespoły: 1) między
narodowe konsorcjum HUGO (ang. Human Genome Organisation Project)
13

oraz 2) prywatną firmę CELERA. W grudniu 2002 roku ustalono sekwencję
genomu myszy, a we wrześniu 2003 roku ogłoszono wstępną sekwencję
genomu psa. Przewiduje się, że do 2005 roku zostaną zsekwencjonowane
genomy bydła, świni i kury.
Osiągnięcia z zakresu genetyki stały się motorem postępu nauk bio-
logicznych i ich aplikacji, czego jednym z licznych przykładów jest hodowla
zwierząt. Głównymi metodami hodowlanymi są selekcja i krzyżowanie, dla
których podstawą teoretyczną jest wiedza z zakresu genetyki cech iloś-
ciowych oraz genetyki populacji. Istotnym elementem pracy hodowlanej
jest kontrola pochodzenia, którą prowadzi się z wykorzystaniem wiedzy
dotyczącej grup krwi i polimorfizmu białek, a ostatnio także polimorfizmu
DNA. Identyfikacja i eliminacja z populacji hodowlanych nosicieli niepożą-
danych mutacji chromosomowych lub genomowych wymaga zastosowania
metod cytogenetycznych, a w przypadku mutacji genowych — metod
genetyki molekularnej. Wytwarzanie zwierząt mających wprowadzony
sztucznie obcy gen (zwierzęta transgeniczne) związane jest z zastosowaniem
technik inżynierii genetycznej. Ostatnio duże nadzieje są pokładane w wy-
korzystaniu map genomowych do molekularnej identyfikacji genów mają-
cych znaczący wpływ na kształtowanie się cech użytkowych zwierząt.
Wykrycie takich genów stworzy nowe warunki do prowadzenia selekcji,
bowiem zamiast oceny genotypu za pomocą metod statystycznych można
będzie ustalać genotyp osobnika na podstawie badań molekularnych.
Pierwsze osiągnięcia zostały już opublikowane. Ustalono, jakie mutacje są
odpowiedzialne za hipertrofię mięśniową bydła, wysoką plenność owiec
rasy boorola i inverdale, wysoką mięsność świń czy wydajność tłuszczu
w mleku krów.
Obecny poziom wiedzy genetycznej i możliwości jej wykorzystania
wywołują czasami również niepokój. Wydaje się, że za postępującym
w oszałamiającym tempie rozwojem genetyki nie nadąża niestety świado-
mość niektórych twórców i wielu odbiorców tych osiągnięć. Możliwość
ingerowania metodami inżynierii genetycznej i komórkowej w informację
genetyczną drobnoustrojów, roślin, zwierząt, a także człowieka powinna
wywołać głębokie zastanowienie się nad tym, gdzie jest granica, której dla
wspólnego dobra wszystkich organizmów zamieszkujących Ziemię nie
można przekroczyć.

Rozdział
2
Chromosomy i podziały jądra
komórkowego
W podziale systematycznym organizmów żywych wyróżnia się, biorąc
pod uwagę organizację informacji genetycznej i budowę komórki, dwie
podstawowe grupy organizmów: prokarionty (Procaryota) i eukarionty
(Eucaryota).
Prokarionty nie mają wyodrębnionego jądra komórkowego, zdolnego do
podziałów (kariokinezy). Odpowiednikiem jądra komórkowego jest nu-
kleoid, zbudowany z „nagiej" kolistej cząsteczki DNA. Tym samym organi-
zmy te zawierają pojedynczy zestaw genów, czyli są haploidalne. Do
prokariontów zalicza się bakterie i sinice. Niektórzy systematycy zaliczają do
tej grupy także wirusy i riketsje.
Eukarionty zbudowane są z komórek mających jądra komórkowe, które
podlegają podziałom (mitoza, mejoza). W jądrze komórkowym występują
chromosomy, które są zbudowane z kwasów nukleinowych i białek. Podczas
podziału jądra komórkowego chromosomy podlegają procesowi kondensacji
i dzięki temu możliwe jest prowadzenie ich obserwacji za pomocą mikro-
skopu. Chromosomy w stadium metafazy podziału mitotycznego mają
charakterystyczną dla siebie wielkość i morfologię, a za pomocą barwienia
różnicującego możliwe jest ujawnienie wzoru prążków poprzecznych
pojawiających się wzdłuż chromosomu. Do eukariontów zalicza się wszystkie
organizmy komórkowe, oprócz bakterii i sinic.
Odkrycie chromosomów i opis ich zachowania się podczas podziałów
jądra komórkowego oraz wiedza o mechanizmach dziedziczenia cech u roślin
i zwierząt wskazywały już na początku XX wieku na istotne znaczenie
chromosomów w procesie dziedziczenia informacji genetycznej. Komplek-
sowe ujęcie tego problemu zostało dokonane przez Morgana, który w 1910
roku ogłosił chromosomową teorię dziedziczenia. Głównymi tezami tej
teorii było twierdzenie, że: 1) geny zlokalizowane są w chromosomach,
2) każdy gen ma stałe położenie (locus) w chromosomie i 3) loci genów
uszeregowane są liniowo w chromosomie. Należy jednak podkreślić, że nie
15
wiedziano wówczas, z jakich związków chemicznych zbudowany jest
chromosom i co jest rzeczywistym nośnikiem informacji genetycznej oraz
jak przedstawia się struktura wewnętrzna chromosomu.
2.1. Budowa chromosomu
Chromosom jest strukturą jądra komórkowego zbudowaną z kwasu deok-
syrybonukleinowego (DNA), kwasów rybonukleinowych (RNA), białek
histonowych i niehistonowych.
Chromosomy są łatwym obiektem do obserwacji mikroskopowej podczas
podziałów jądra komórkowego. Dzięki kondensacji cząsteczki DNA, która
następuje za pomocą białek histonowych i niehistonowych, chromosom
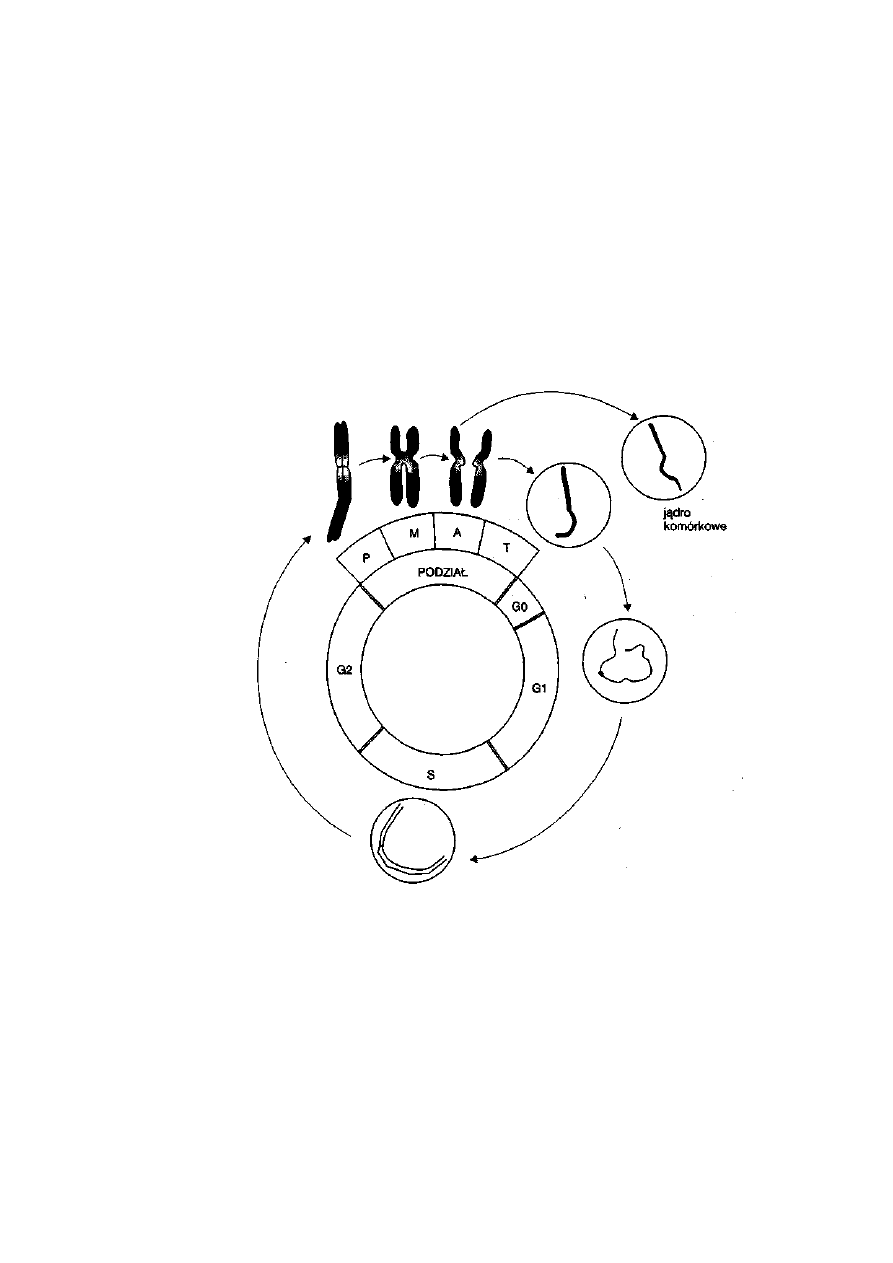
Ryć. 2-1. Fazy (GO, Gl, S, G2 i podział jądra
:
komórkowego) cyklu życiowego komórki
w powiązaniu ze zmianami struktury chromosomu. Oznaczenia: P — profaza,
M — metafaza, A — anafaza i T — telofaza
16

osiąga najmniejsze rozmiary podczas metafazy mitotycznej bądź mejotycznej.
Wówczas to chromosom ma długość rzędu kilku mikrometrów, a jego
morfologia jest bardzo wyrazista i dzięki temu mikroskop świetlny jest
wystarczająco czułym instrumentem pozwalającym prowadzić szczegółową
analizę cytogenetyczną, która obejmuje nie tylko ustalenie liczby chromo-
somów, ale także opis ich morfologii i charakterystycznego układu prążków
poprzecznych. Chromosom w stadium metafazy mitotycznej jest zbudowany
z dwóch chromatyd połączonych w przewężeniu pierwotnym, określanym
jako centromer. O chromatydach mówi się, że są siostrzane, ponieważ
powstały w wyniku replikacji DNA w fazie S cyklu komórkowego (ryć. 2-1).
Chromatydy siostrzane zawierają po jednej cząsteczce DNA o identycznej
sekwencji nukleotydów i tym samym niosą dokładnie taką samą informację
genetyczną. Centromer dzieli chromosom na ramię krótkie (p) oraz ramię
długie (q). Część końcowa ramienia chromosomowego nazywana jest
telomerem. Telomery są zbudowane z powtarzającej się 6-nukleotydowej
sekwencji TTAGGG/AATCCC. Zależnie od położenia centromeru wyróżnia
się cztery typy morfologiczne chromosomów: meta-, submeta-, subtelo-i
akro-(telocentryczny) (ryć. 2-2). Stosunek długości ramienia długiego do
długości ramienia krótkiego jest parametrem określającym morfologię
chromosomu. Wartość tego parametru dla wymienionych wyżej typów
morfologicznych chromosomu mieści się odpowiednio w przedziałach:
(1,00-1,70), (1,71-3,00), (3,01-7,00) i (7,01-oo). Niektóre chromosomy mają,
oprócz przewężenia pierwotnego, także przewężenie wtórne, określane
inaczej jako obszar jąderkotwórczy — NOR (ang. nucleolar organizer
region), który jest odpowiedzialny za uformowanie jąderek podczas inter-
fazy. W jąderkach budowane są podjednostki rybosomów. W obszarze
jąderkotwórczym umiejscowione są geny kodujące rybosomowe kwasy
rybonukleinowe (rRNA) o stałej sedymentacji 5.8S, 18S i 28S. Czwarta
chromarydy
siostrzane
Ryć. 2-2. Morfologia chromosomów obserwowanych podczas metafazy mitotycznej. Typy
morfologiczne są wyodrębniane na podstawie wielkości stosunku długości ramion q: p
17
frakcja rRNA — 5S rRNA jest kodowana przez geny występujące w innych
miejscach genomu. Fragment ramienia chromosomu, który występuje za
przewężeniem wtórnym, nazywa się satelitą.
Chromosomy dzielone są na dwie podstawowe grupy: autosomy i hetero-
somy (chromosomy płci). W grupie autosomów występują identyczne pary
homologiczne zarówno u płci męskiej, jak i żeńskiej. Chromosomy homo-
logiczne mają identyczne uszeregowanie loci tych samych genów. W każdej
parze jeden chromosom pochodzi od matki, a drugi od ojca danego
osobnika. Podobieństwo molekularne występujące w obrębie pary chromo-
somów homologicznych umożliwia ich koniugację podczas profazy I po-
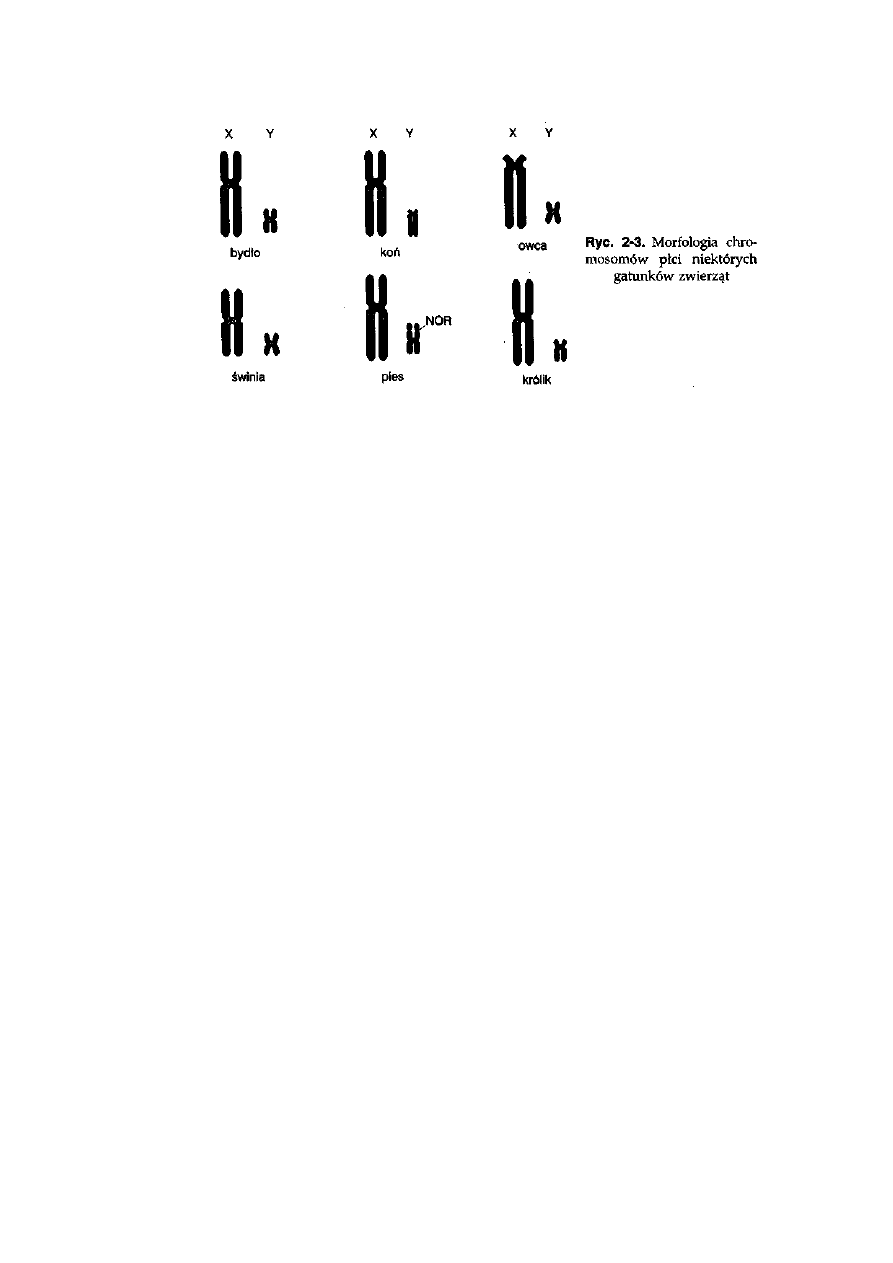
działu mejotycznego. Heterochromosomy, oznaczane u ssaków symbolami
X oraz Y, występują w układzie XX u samic oraz XY u samców. Chromosomy
X i Y różnią się między sobą pod względem długości (chromosom X jest
przeciętnie dwa razy dłuższy niż chromosom Y), morfologii, układu prążków
poprzecznych oraz, co najważniejsze — zestawu loci genów, które są w nich
położone. Chromosom Y jest znacznie mniejszy niż chromosom X i przez
to również uboższy w informację genetyczną (ryć. 2-3). Konsekwencją tych
różnic jest szczątkowa homologia chromosomów X i Y, która ogranicza się
do niewielkiego fragmentu określanego jako obszar pseudoautosomalny.
Oznacza to, że w tym obszarze występuje homologia pozwalająca na
mejotyczną koniugację, podobnie jak występuje to w obrębie par homo-
logicznych autosomów lub pary X-X. Kwestia funkcji heterochromosomów
będzie poruszona w rozdziale: Determinacja i różnicowanie płci oraz
interseksualizm.
Chromosomy podlegają w każdym cyklu komórkowym procesowi
kondensacji — na początku podziału jądra komórkowego oraz dekonden-
sacji — po zakończeniu podziału. W tełofazie i pierwszym okresie interfazy
(faza Gl) chromosomy są zbudowane z jednej chromatydy, która z kolei
zawiera jedną cząsteczkę DNA. Podczas fazy S następuje replikacja —
18
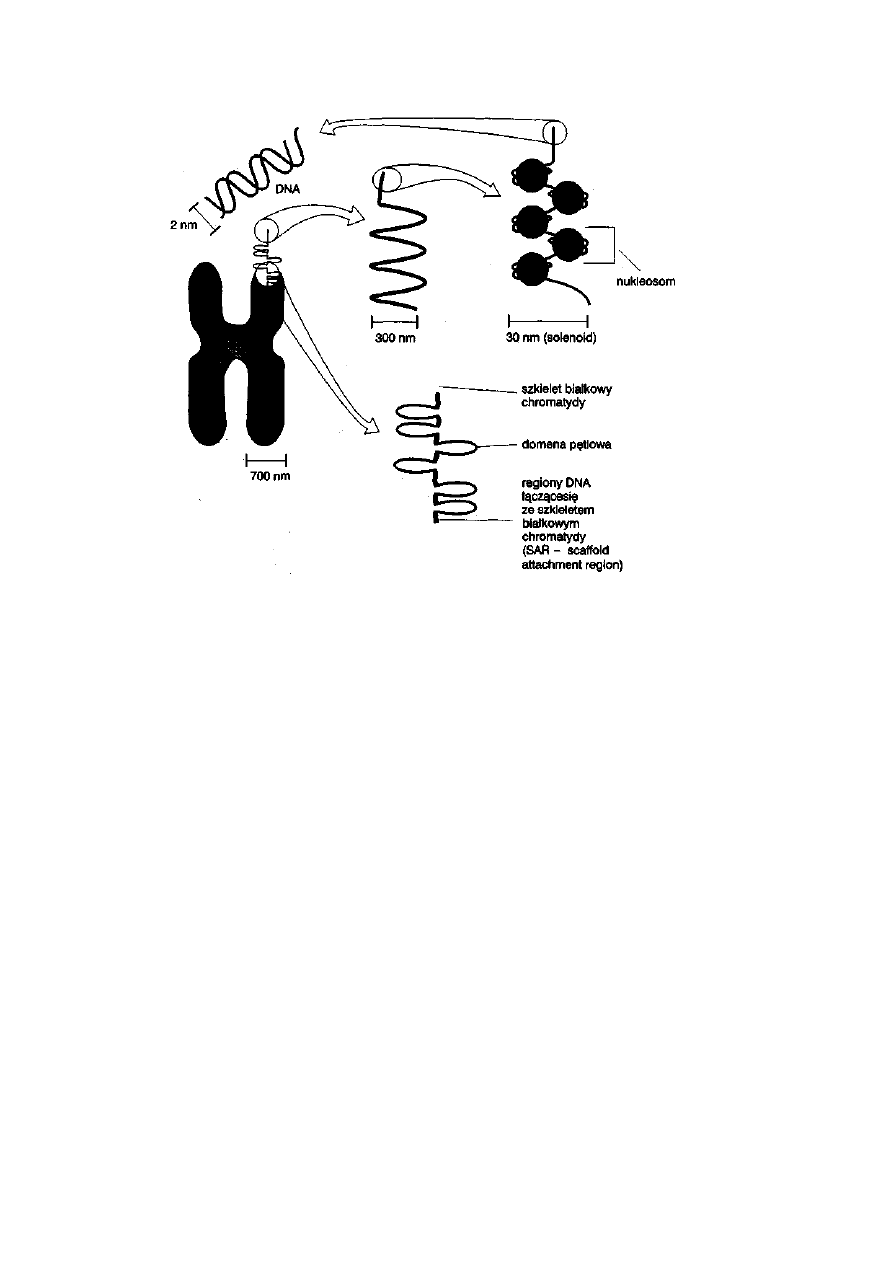
Ryć. 2-4. Struktura wewnętrzna chromosomu metafazowego (wg Biotechnologia zwierząt,
1997, PWN)
samoodtworzenie DNA, która prowadzi do powstania w obrębie chromo-
somu dwóch chromatyd siostrzanych. Wejście komórki w stadium podziału
wiąże się z uruchomieniem procesu kondensacji chromosomu, w wyniku
czego podczas metafazy staje się on strukturą maksymalnie skróconą.
Przyjmuje się, że stosunek długości cząsteczki DNA do długości chromosomu
w stadium metafazy wynosi około 8000:1. Kondensacja i dekondensacja
cząsteczki DNA zachodzi wielostopniowo, a procesy te, ze względu na ich
powtarzalność w kolejnych cyklach życiowych komórki, charakteryzują się
niezwykłą precyzją (ryć. 2-4).
Podstawową jednostką organizacji wewnętrznej chromosomu jest nukleo-
som. Jest on zbudowany z rdzenia białkowego (oktamer histonowy,
zawierający po dwie cząsteczki histonów: H2A, H2B, H3 i H4), na który jest
nawinięty fragment DNA długości od 165 do 245 par nukleotydów, inaczej
par zasad (pz). Fragment DNA owinięty na oktamerze histonowym (1,8
obrotu DNA na oktamerze) jest nazywany rdzeniem nukleosomu i składa
się ze 146 pz. Fragment cząsteczki DNA pomiędzy nukleosomami ma
długość około 60 pz, w zakresie od 0 do 80 pz.
19

Nić nukleosomowa jest z kolei zwinięta spiralnie tworząc solenoid.
W jego powstaniu istotną rolę odgrywają cząsteczki histonu Hl, które
odpowiadają za zbliżenie się sąsiednich nukleosomów. Solenoid podlega
dalszej spiralizacji, a jej ostatecznym etapem jest utworzenie domen
pętlo wych, które są zakotwiczone w rdzeniu białkowym chromatydy,
zbudowanym z białek niehistonowych. Sekwencje DNA leżące u podstaw
pętli są bogate w pary nukleotydów A-T i wiążą się z białkami rdzenia
chromosomu, należącymi do rodziny białek HMG (ang. high mobility
group). Dlatego te sekwencje DNA określane są terminem S AR (ang.
scaffold attachment region), czyli regionami wiążącymi się ze szkieletem
chromosomu. Przedstawiony proces kondensacji chromosomu metafazowego
prowadzi do skrócenia jego długości, z jednoczesnym bardzo wydatnym
zwiększeniem średnicy — z 2 nm w przypadku średnicy cząsteczki DNA do
700 nm w przypadku średnicy chromatydy chromosomu metafazowego.
Dzięki tym zmianom chromosomy są dobrze widoczne w mikroskopie
świetlnym.
W obrębie chromosomu wyróżnia się dwie frakcje chromatyny: euchro-
matynę i heterochromatynę. Euchromatyna podlega cyklicznym procesom
kondensacji i dekondensacji. W niej zlokalizowane są kodujące sekwencje
DNA, czyli geny. Podlega ona replikacji w początkowym okresie fazy S.
Heterochromatyna jest frakcją skondensowaną i dzieli się na dwa typy:
heterochromatynę fakultatywną i heterochromatynę konstytutywną. Hete-
rochromatyna fakultatywna jest częścią chromatyny, która może ulec
trwałej kondensacji; przykładem jest jeden z chromosomów X u osob-
ników żeńskich, który ulega kondensacji i występuje w jądrze inter-
fazowym w postaci tzw. ciałka Barra. Heterochromatyna konstytutywna
jest frakcją chromatyny, która zawsze występuje w stanie skonden-
sowanym. Zbudowana jest z niekodującycK. sekwencji nukleotydowych,
głównie powtarzalnych sekwencji DNA. Jej replikacja występuje w póź-
nym okresie fazy S.
Chromosomy występujące w jądrze interfazowym są zdespiralizowane
i w obrazie mikroskopowym są widoczne w postaci mało zróżnicowanej
chromatyny, rozprzestrzenionej równomiernie na pbszarze całego jądra
komórkowego. Wiadomo jednak, że chromatyna pojedynczych chromo-
somów zajmuje wyodrębnione-przestrzenie — tzw. domeny, przy czym
domeny chromosomów homologicznych nie mają tendencji do łączenia się.
Tendencje takie natomiast wykazują regiony heterochromatyny konstytutyw-
nej różnych chromosomów, które tworzą większe chromocentra. Chromo-
somy interfazowe zachowują strukturę nukleosomowa.
Liczba chromosomów występująca w jądrze prawidłowej komórki
somatycznej jest określana terminem diploidalnej (2n) liczby chromosomów.
Pojawia się ona podczas zapłodnienia, wtedy bowiem haploidalny (n)
zestaw chromosomów pochodzenia ojcowskiego — zawarty w plemniku —
łączy się z haploidalnym zestawem pochodzenia matczynego — obecnym
20
w oocycie drugiego rzędu (komórce jajowej). W wyniku tego w jądrze
komórkowym zygoty oraz w potomnych komórkach somatycznych wy-
stępują pary chromosomów homologicznych, w których jeden chromosom
jest pochodzenia ojcowskiego, a drugi matczynego. Chromosomy homo-
logiczne mają loci tych samych genów, ale mogą zawierać w nich różne allele
21
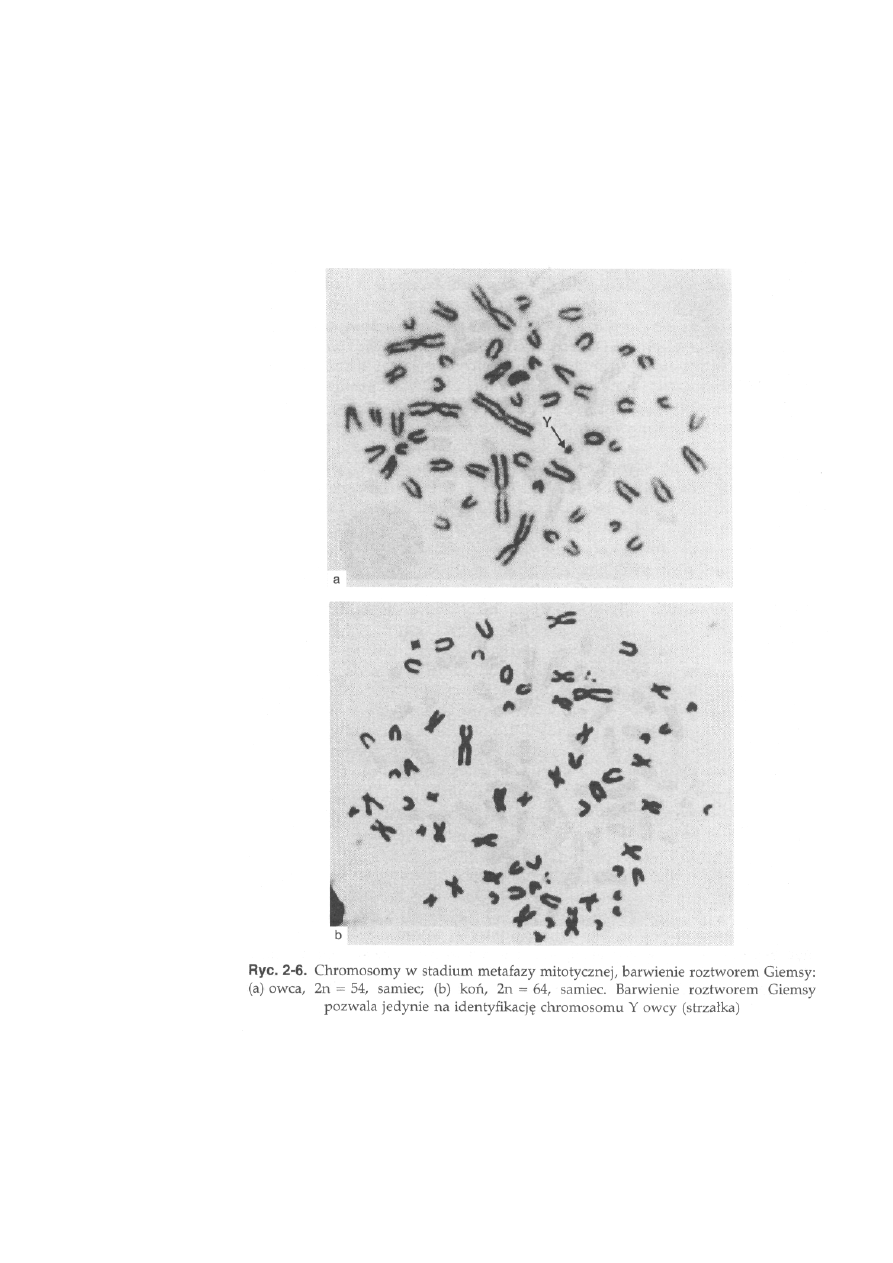
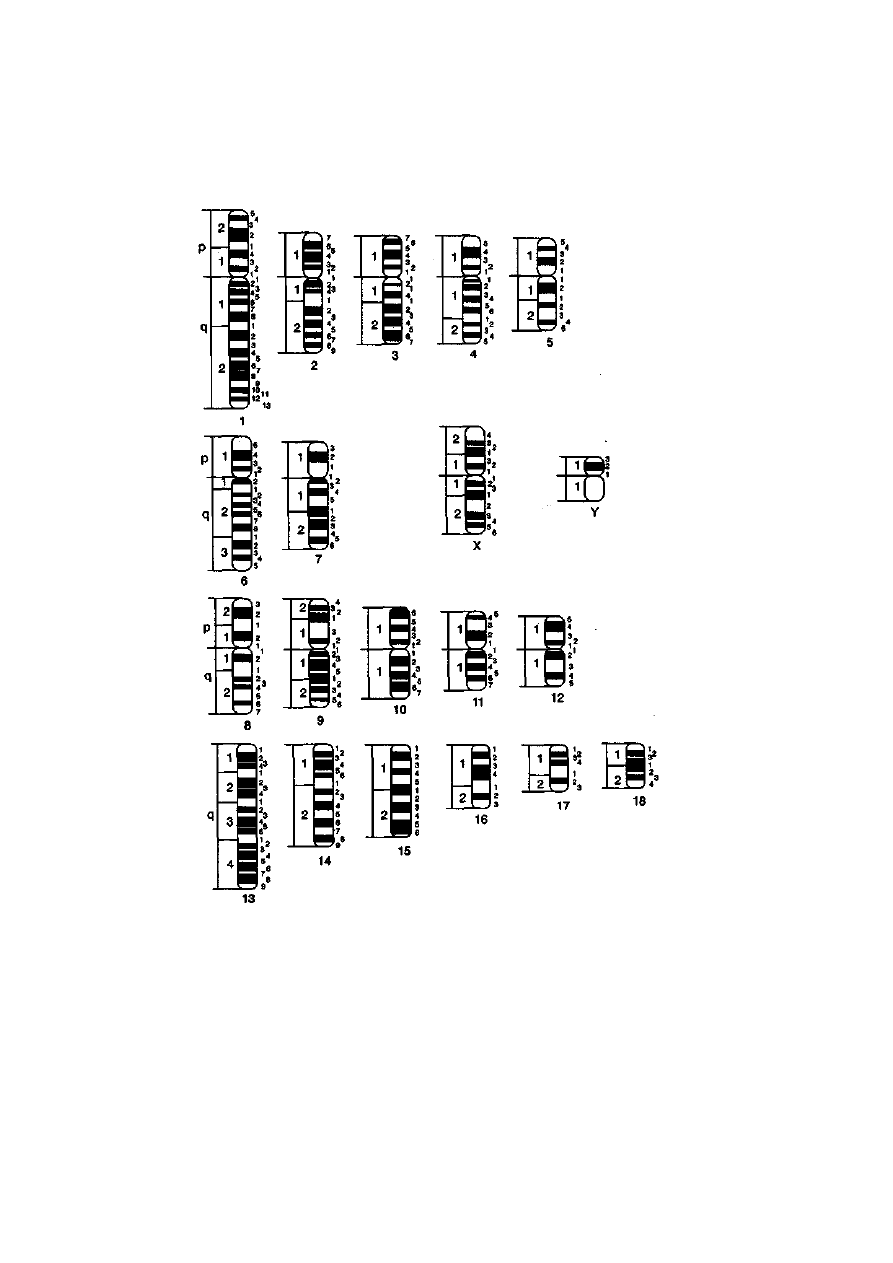
Diploidałna liczba chromosomów u zwierząt użytkowanych przez
człowieka mieści się w szerokim przedziale, od 30 u norki do 78 u psa (tab.
2-1, ryć. 2-5 i 2-6). Należy podkreślić, że czasami liczba chromosomów
obserwowana u danego gatunku nie jest stała. Przykładem takiej sytuacji
jest występowanie zmiennej liczby chromosomów B u lisa pospolitego lub
jenota. Chromosomy B, czyli nadliczbowe względem podstawowego ze-
22

stawu A, charakteryzują się niestabilnym zachowaniem w podziałach
mitotycz-nych i mejotycznych. Efektem tego jest zmienność ich liczby
zarówno między osobnikami, jak i pomiędzy komórkami w obrębie
jednego osobnika. Do tej pory nie poznano funkcji tych chromosomów,
chociaż wiadomo, że często są zbudowane z heterochromatyny
konstytutywnej, czyli można przypuszczać, iż są nieaktywne genetycznie.
Drugim przykładem braku stałości liczby chromosomów w obrębie gatunku
jest szerokie rozprzestrzenienie mutacji chromosomowej, np. fuzji
centrycznej u lisów polarnych. Konsekwencją tego jest występowanie u
lisów osobników, które mogą mieć 50,49 lub 48 chromosomów.
2.2. Barwienie prążkowe chromosomów
Zróżnicowanie wewnętrzne chromosomu, wynikające ze stopnia
spiralizacji nici chromatynowej oraz wzajemnego stosunku ilościowego
par nukleo-tydów G-C oraz A-T, może być uwidocznione za pomocą
technik barwienia różnicującego. Zastosowanie przez Casperssona w
1969 roku kwinakryny do barwienia chromosomów otworzyło nowy
rozdział w cytogenetyce. Okazało się, że różnorodne techniki barwienia
chromosomów pozwalają nie tylko na opis ich wielkości i morfologii, ale
także na ujawnianie na nich charakterystycznych wzorów prążków
poprzecznych. Prążki mają identycz-
23
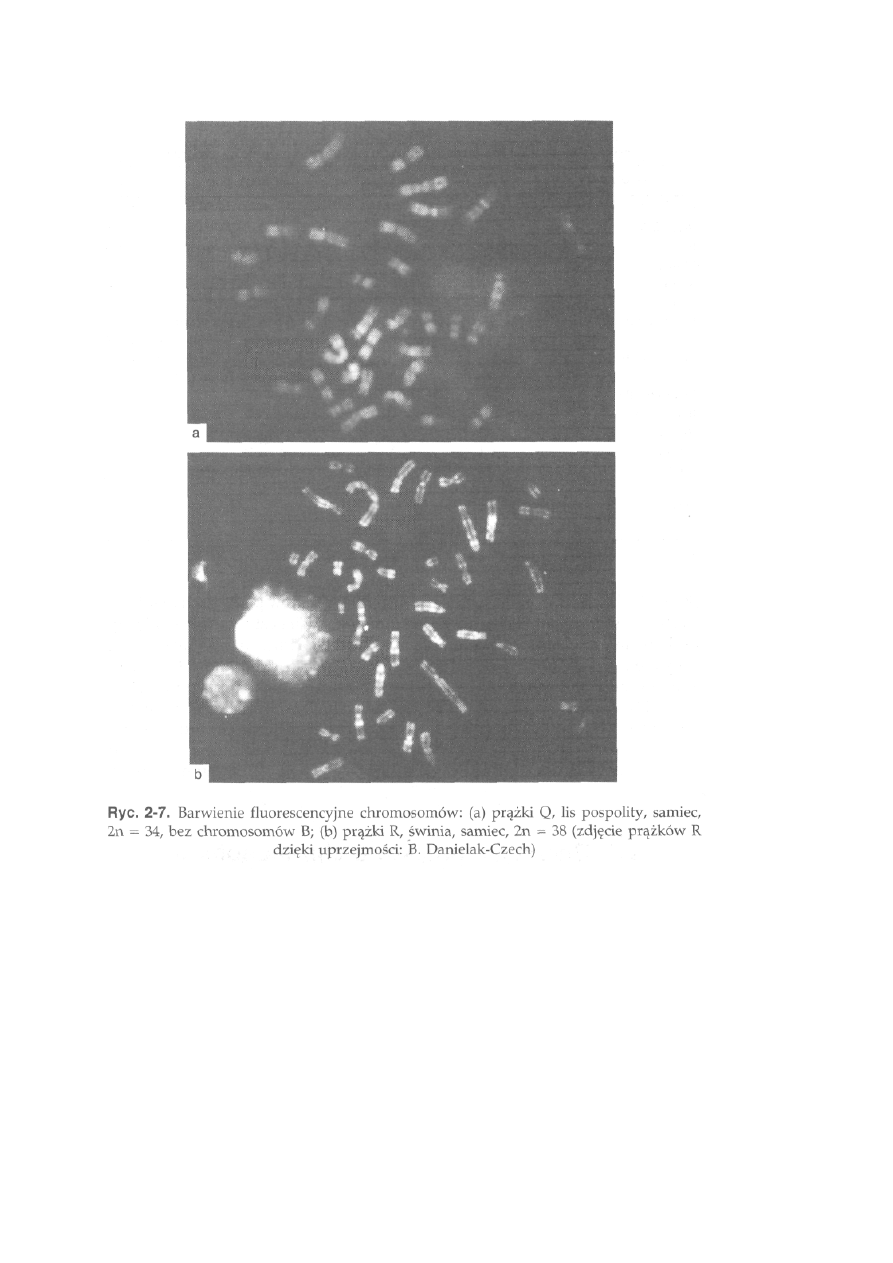
ny układ i wielkość na chromosomach homologicznych i dla danej pary chromosomów są
stałą cechą w obrębie gatunku. Wynika z tego, że gatunek może być charakteryzowany nie
tylko przez typową dla niego diploidalną liczbę chromosomów, ale także układ prążków na
chromosomach. Zależnie od charakteru prążków przypisuje się im odpowiednie symbole.
Prążki Q (QFQ) uwidaczniane są w mikroskopie fluorescencyjnym, po wybarwieniu
chromosomów roztworem fluorochromu — kwinakryny. Na chromosomach pojawiają się
poprzeczne prążki świecące i nie świecące
24
(ryć. 2-7a). Silne wiązanie barwnika kwinakryny następuje w miejscach,
gdzie dominują pary zasad A-T. Podobny efekt uzyskuje się po wybarwieniu
chromosomów barwnikiem DAPI.
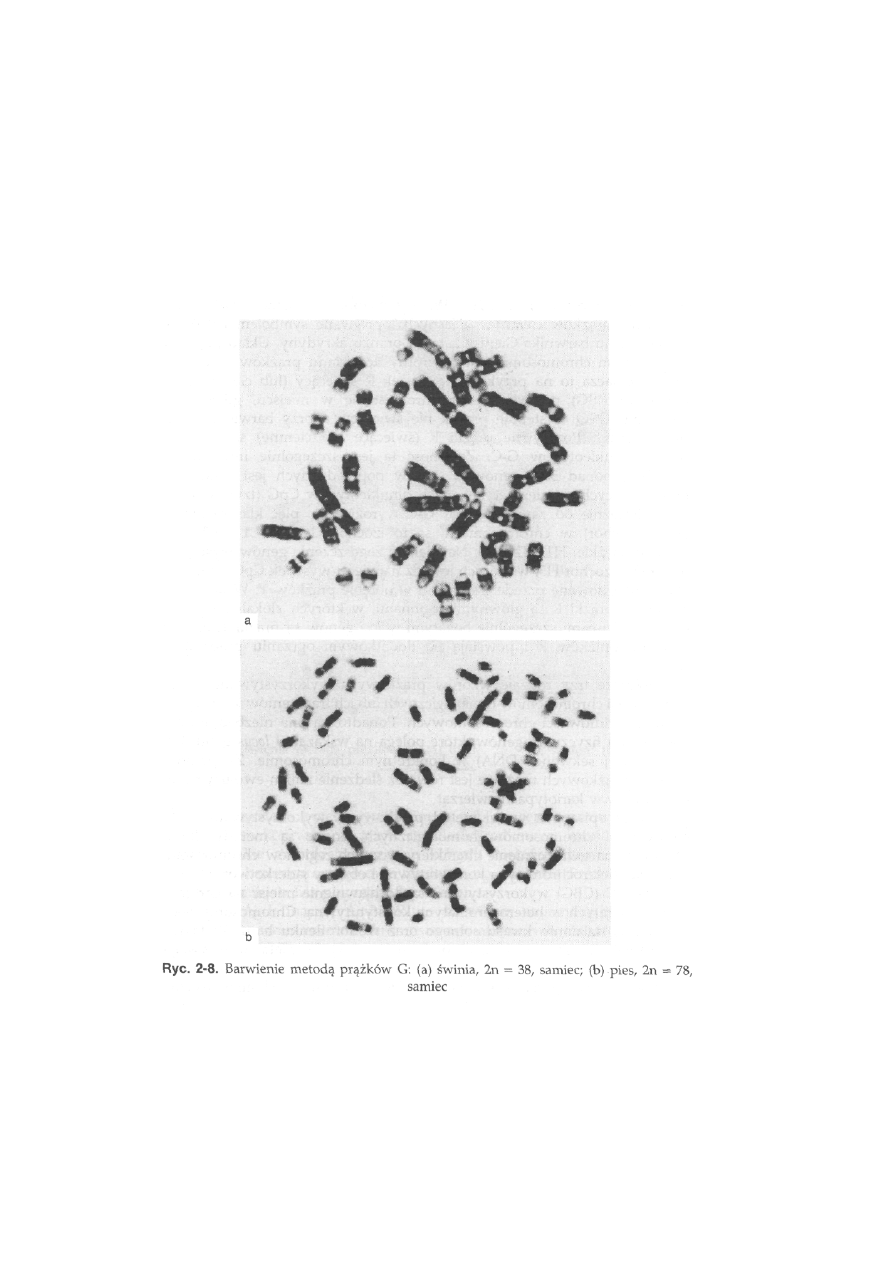
Prążki G (GTG) pojawiają się na chromosomach po przeprowadze-
niu wstępnego trawienia roztworem trypsyny (enzym proteolityczny),
a następnie barwienia roztworem Giemsy. W efekcie na chromosomach
25

pojawiają się prążki ciemne i jasne (ryć. 2-8). Prążki ciemne występują
w tych samych miejscach, gdzie przy barwieniu QFQ obserwowane
są prążki świecące, a prążki jasne odpowiadają nie świecącym prąż-
kom Q.
Prążki R, pojawiające się w układzie prążków świecących i nie
świecących (opisywane symbolem RBA) powstają po wbudowaniu bromo-
deoksyurydyny do DNA chromosomów podczas ich replikacji i barwieniu
oranżem akrydyny (ryć. 2-7b). Wzór ten można również uwidocznić
w postaci prążków ciemnych i jasnych (opisywane symbolem RBG), po
zastosowaniu barwnika Giemsy zamiast oranżu akrydyny. Układ prążków
R na danym chromosomie jest odwrotny "do układu prążków GTG oraz
QFQ. Oznacza to na przykład, że prążek R świecący (lub ciemny przy
barwieniu RBG) pojawia się na chromosomie w miejscu, gdzie przy
barwieniu QFQ występuje prążek nie świecący, a przy barwieniu GTG
prążek jasny. Pozytywne prążki R (świecące lub ciemne) są bogatsze
w pary nukleotydów G-C. Zależność ta jest szczególnie interesująca,
ponieważ ponad 50% genów u ssaków poprzedzonych jest sekwencją
powtarzających się niezmetylowanych dinukleotydów CpG (tzw. wysepki
CpG). Zależnie od zawartości par G-C wyróżnia się pięć klas regionów
DNA (izochor) w chromosomach, są to izochory lekkie: LI i L2 oraz
izochory ciężkie: Hl, H2 i H3. Największe zagęszczenie genów występuje
w obrębie izochor H3, w których jest też najwięcej wysepek CpG. Izochory
H3 są lokalizowane przede wszystkim w obrębie prążków T. Wskazuje to
zatem, że prążki R są głównymi regionami, w których zlokalizowane są
geny, a miejscami szczególnie bogatymi w loci genów są prążki T. Są one
podklasą prążków R i powstają po dodatkowym ogrzaniu preparatów
w 90°C.
Powyższe trzy rodzaje wzorów prążkowych wykorzystywane są do
identyfikacji chromosomów homologicznych lub ich fragmentów w przypad-
ku nieprawidłowości chromosomowych. Ponadto, są one niezbędne przy
mapowaniu fizycznym genów, które polega na wskazaniu locus genu (lub
anonimowej sekwencji DNA) w konkretnym chromosomie. Za pomocą
barwień prążkowych możliwe jest również śledzenie zmian ewolucyjnych,
które zaszły w kariotypach zwierząt.
Oprócz opisanych wyżej metod prążkowych, wykorzystywanych do
identyfikacji chromosomów homologicznych, znane są metody, które
pozwalają na uwidocznienie charakterystycznych regionów chromosomu,
takich jak heterochromatyna konstytutywna i obszary jąderkotwórcze.
Prążki C (CBG) wykorzystywane są do ujawnienia miejsc na chromo-
somach bogatych w heterochromatynę konstytutywną. Chromosomy pod-
dawane są działaniu kwasu solnego oraz wodorotlenku baru. W takich
warunkach znaczna część euchromatyny zostaje usunięta, a pozostają
trudne do usunięcia, silnie skondensowane obszary heterochromatynowe,
które się wybarwiają roztworem Giemsy. Heterochromatyna konstytutywna
26
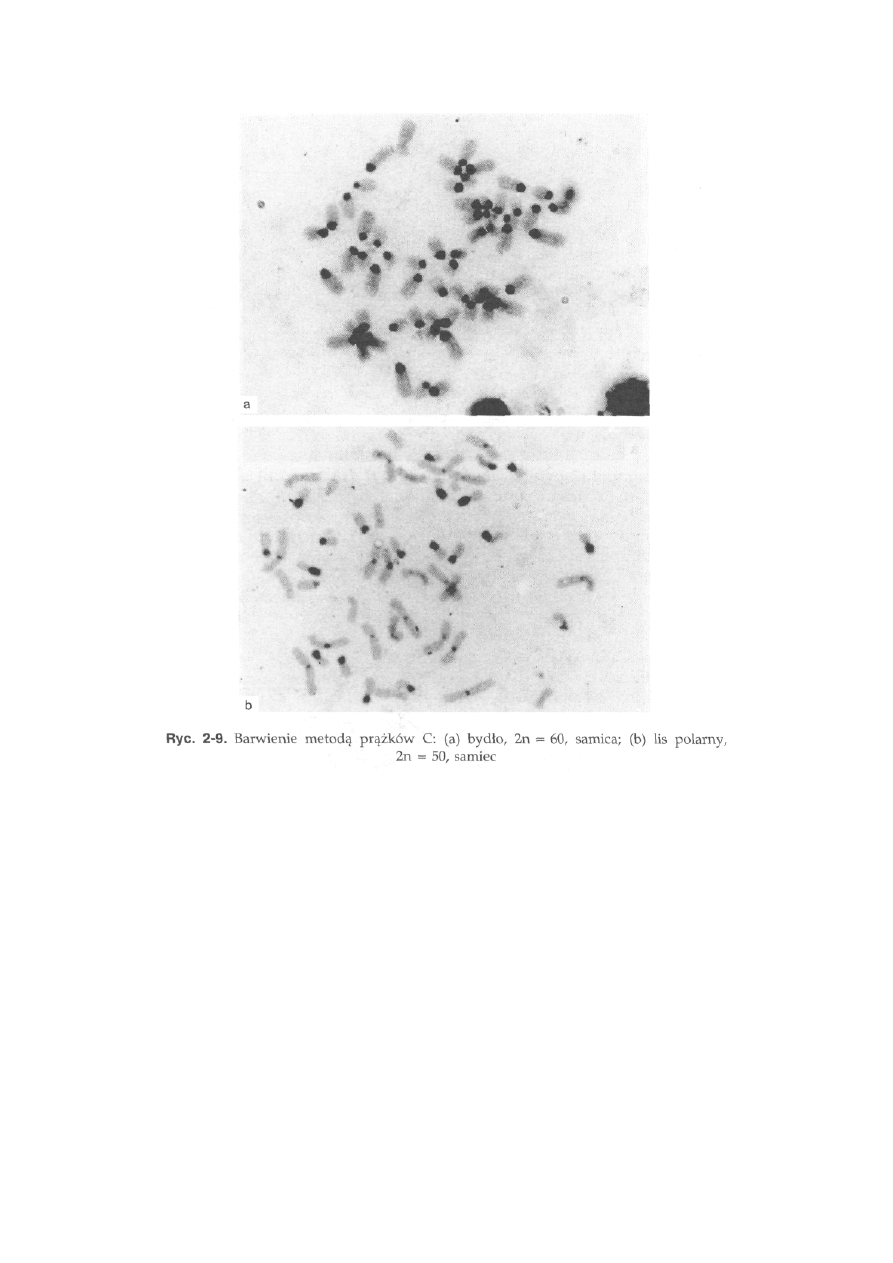
jest zlokalizowana przede wszystkim w pobliżu centromerów (ryć. 2-9a),
a rzadziej na końcach, czy też w częściach środkowych ramion. Znane są
również takie gatunki zwierząt, u których niektóre chromosomy mają
całkowicie heterochromatynowe ramiona, np. ramiona krótkie dziesięciu
par autosomów u lisa polarnego (ryć. 2-9b).
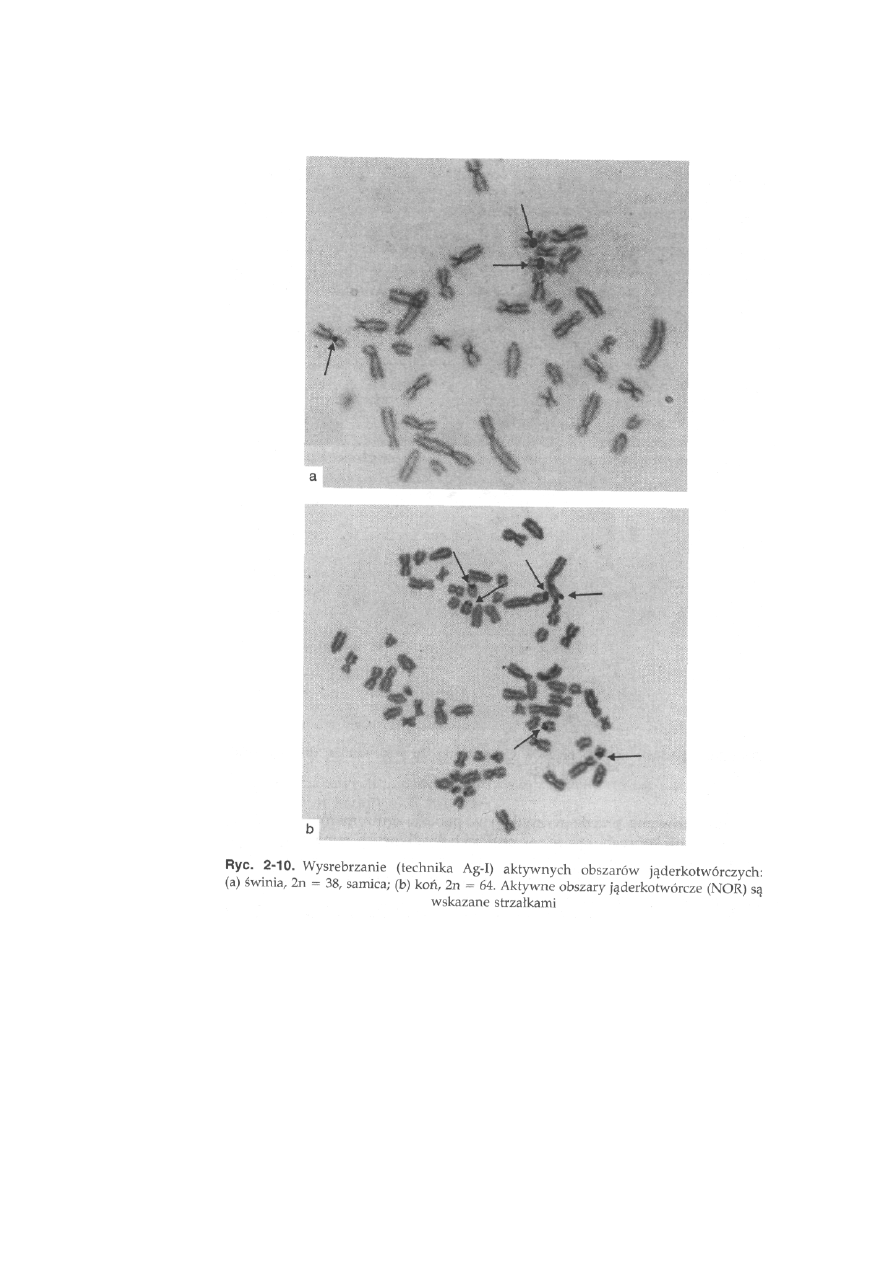
Prążki Ag-I, inaczej Ag-NOR, to barwienie azotanem srebra, które
pozwala na ujawnienie na chromosomach aktywnych obszarów jąderko-
twórczych (NOR). W miejscach tych następuje wytrącenie metalicznego
srebra, które w obrazie mikroskopowym ma wygląd ciemnych plam
(ryć. 2-10).
27
Diploidalna liczba chromosomów oraz ich morfologia i wzory prążkowe
są cechami charakteryzującymi genomy poszczególnych gatunków. Poniżej
przedstawiono podstawowe cechy opisujące chromosomy najważniejszych
gatunków zwierząt domowych.
28

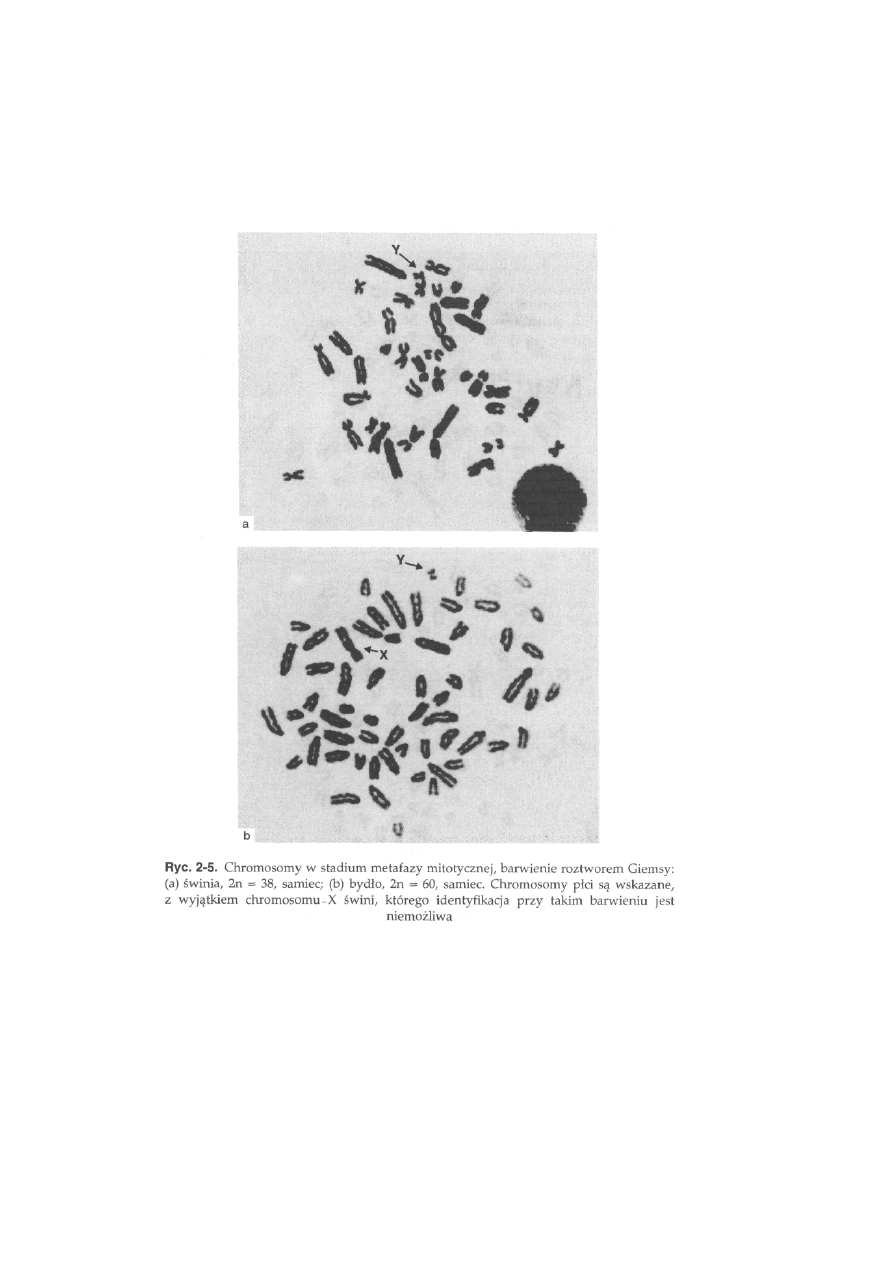
Bydło, 2n - 60 (ryć. 2-5b). Wszystkie autosomy są akrocentryczne. Chromo-
somy płci są łatwe do identyfikacji, ponieważ są to jedyne w kariotypie
chromosomy dwuramienne: chromosom X jest dużym submetacentrykiem,
a chromosom Y jest małym metacentrykiem. Heterochromatyna konstytu-
tywna (prążki C) występuje w okolicach centromeru we wszystkich
autosomach, natomiast w chromosomie X pojawia się jedynie jako blady
prążek interstycjalny w ramieniu długim, a chromosom Y wykazuje
ciemniejsze zabarwienie na całej długości (ryć. 2-9a). Obszary jąderkotwórcze
(NOR) występują w pięciu parach autosomów na końcu ramion długich.
Koza, 2n = 60. Podobnie jak w przypadku bydła, także koza ma wszystkie
autosomy akrocentryczne. Również akrocentryczny jest chromosom X,
a chromosom Y jest małym metacentrykiem. Prążki C pojawiają się w okolicy
centromerowej wszystkich autosomów. W chromosomie X brak jest centro-
merowego prążka C. Obszary jąderkotwórcze występują, podobnie jak
u bydła, w części telomerowej ramion długich pięciu par autosomów.
Owca, 2n = 54 (ryć. 2-6a). Wśród autosomów występują trzy pary dużych
chromosomów metacentrycznych oraz 23 pary akrocentryków. Chromosom
X jest dużym akrocentrykiem, a chromosom Y jest małym metacentrykiem.
W stadium prometafazy, w którym nie jest jeszcze zakończona kondensacja
chromosomów, w chromosomie X widoczne są krótkie ramiona, co odróżnia
go od autosomów akrocentrycznych o podobnej wielkości. Układ prążków
C, a także rozmieszczenie obszarów jąderkotwórczych są podobne do
obserwowanego w kariotypie bydła i kozy.
Świnia, 2n = 38 (ryć. 2-5a). Grupa chromosomów autosomalnych składa się
z 12 par chromosomów dwuramiennych (meta- i submetacentrycznych)
oraz 6 par akrocentrycznych. Chromosom X jest średniej wielkości metacen-
trykiem, trudnym do odróżnienia na podstawie morfologii od autosomów.
Chromosom Y jest najmniejszym chromosomem metacentrycznym. Prążki
C występują w okolicy centromeru wszystkich autosomów i chromosomu
X, natomiast chromosom Y ma całkowicie heterochromatynowe ramię
długie. W grupie chromosomów akrocentrycznych często obserwowane jest
zjawisko polimorfizmu wielkości prążków C. Obszary jąderkotwórcze
występują w dwóch parach autosomów dwuramiennych, w ramionach
krótkich, w pobliżu centromeru (ryć. 2-10a).
Koń, 2n = 64 (ryć. 2-6b). Wśród autosomów można wyróżnić grupę 13 par
chromosomów dwuramiennych oraz grupę 18 par akrocentryków. Chromo-
som X jest dużym chromosomem metacentrycznym, a chromosom Y jest
akrocentrykiem. Chromosomy płci są trudne do odróżnienia od autosomów
o podobnej wielkości i morfologii. Prążki C występują w okolicach centro-
meru wszystkich autosomów i chromosomu X, który ma dodatkowy prążek
29

interstycjalny w ramieniu długim. Chromosom Y jest w znacznej części
heterochromatynowy. Obszary jąderkotwórcze występują w trzech parach
autosomów: w części terminalnej ramienia krótkiego największego chromo-
somu oraz w części przycentromerowej dwóch małych akrocentryków
(ryć. 2-10b).
Kura, 2n = 78. W kariotypie kury, podobnie jak u innych ptaków, występuje
liczna grupa bardzo małych chromosomów, nazywanych mikrochromoso-
mami, dla których określenie morfologii oraz wzoru prążkowego jest
niemożliwe. Dlatego szczegółową analizę cytogenetyczną prowadzi się
jedynie w odniesieniu do dużych chromosomów autosomalnych (makro-
chromosomów) i chromosomów płci Z oraz W. W grupie dużych autosomów
analizuje się najczęściej 8 par, wśród których jest 5 par chromosomów
dwuramiennych i 3 pary akroćentryczne. Chromosom Z jest dużym
metacentrykiem, a chromosom W jest małym metacentrykiem. Obszar
jąderkotwórczy występuje tylko w jednej parze autosomów, z grupy
mikrochromosomów.
Lis pospolity, 2n = 34 +B (ryć. 2-7a). Wszystkie autosomy z grupy A są
chromosomami dwuramiennymi, a chromosomy z grupy B są akroćentryczne
lub subtelocentryczne. Chromosom X jest dużym metacentrykiem, a chromo-
som Y bardzo małym akrocentrykiem, którego na podstawie wielkości bądź
morfologii nie można odróżnić od chromosomów B. Liczba chromosomów
B waha się od O do 7, przy czym najczęściej obserwowano l, 2 lub
3 chromosomy B. Kariotyp lisa pospolitego jest wyjątkowo ubogi w hetero-
chromatynę konstytutywną, którą można zidentyfikować barwieniem CBG
jedynie w trzech parach dużych autosomów, w okolicach centromeru, oraz
w chromosomie Y. W chromosomach B ciemny prążek C nie pojawia się
i dzięki temu barwienie CBG pozwala na odróżnienie chromosomu Y od
chromosomów B. Obszary jąderkotwórcze występują w części telomerowej
ramion długich trzech par autosomów z grupy A.
Lis polarny, 2n = 50, 49 lub 48. W związku z szerokim rozprzestrzenieniem
fuzji centrycznej (translokacji robertsonowskiej) obserwuje się trzy formy
kariotypowe różniące się dipłoidalną liczbą chromosomów. U formy pod-
stawowej (2n = 50) występują 22 pary autosomów dwuramiennych i dwie
pary akrocentryków. Chromosom X jest dużym metacentrykiem, a chromo-
som Y bardzo małym akrocentrykiem. Kariotyp tego gatunku jest bardzo
bogaty w heterochromatynę konstytutywną. Wszystkie chromosomy, łącznie
z chromosomami płci, mają bloki heterochromatynowe w okolicy centro-
meru. Ponadto, 10 par dwuramiennych autosomów ma całkowicie hetero-
chromatynowe ramiona krótkie (ryć. 2-9b). Obszary jąderkotwórcze wy-
stępują w sześciu parach autosomów w części telomerowej krótkich ramion
heterochromatynowych.
30
Pies, 2n = 78 (ryć. 2-8b). Wszystkie autosomy są akrocentryczne. Chromosom
X jest dużym metacentrykiem, a chromosom Y jest małym metacentrykiem.
Heterochromatyna konstytutywna jest umiejscowiona tylko w kilku parach
autosomów. Wyraźne prążki C pojawiają się w okolicach centromerowych
w czterech parach autosomów, chromosomie X (prążek centromerowy i inter-
stycjalny w ramieniu q) i Y. Obszary jąderkotwórcze są obecne w trzech
parach autosomów i chromosomie Y, w części terminalnej ramion długich.
2.3. Mitoza
Mitoza to proces podziału jądra komórkowego, który prowadzi do powstania
dwóch komórek potomnych, mających genotyp identyczny z tym, jaki
miała komórka rodzicielska. Komórki potomne mają nie tylko taką samą
liczbę chromosomów, ale także identyczną sekwencję nukleotydów w DNA.
Podziały mitotyczne są nierozłącznie związane z procesem wzrostu organiz-
mu, odnawianiem niektórych komórek u dorosłych organizmów (np.
komórki krwi, nabłonka), proliferacją limfocytów przy reakcji odpornościowej
organizmu, a także z pierwszymi etapami gametogenezy.
Podział mitotyczny jest jedną z czterech faz występujących w cyklu
życiowym komórki. Fazy Gl, S i G2 tworzą okres międzypodziałowy —
interfazę, a czwartą fazą jest podział. W fazie Gl występuje ekspresja genów
związanych z funkcją danej komórki, a faza S jest związana z procesem
samoodtworzenia się DNA (replikacja). Rozpoczęcie fazy S oznacza, że
komórka zmierza do podziału i dlatego faza G2 jest traktowana jako okres
przygotowywania się komórki do podziału. W cyklu życiowym komórki
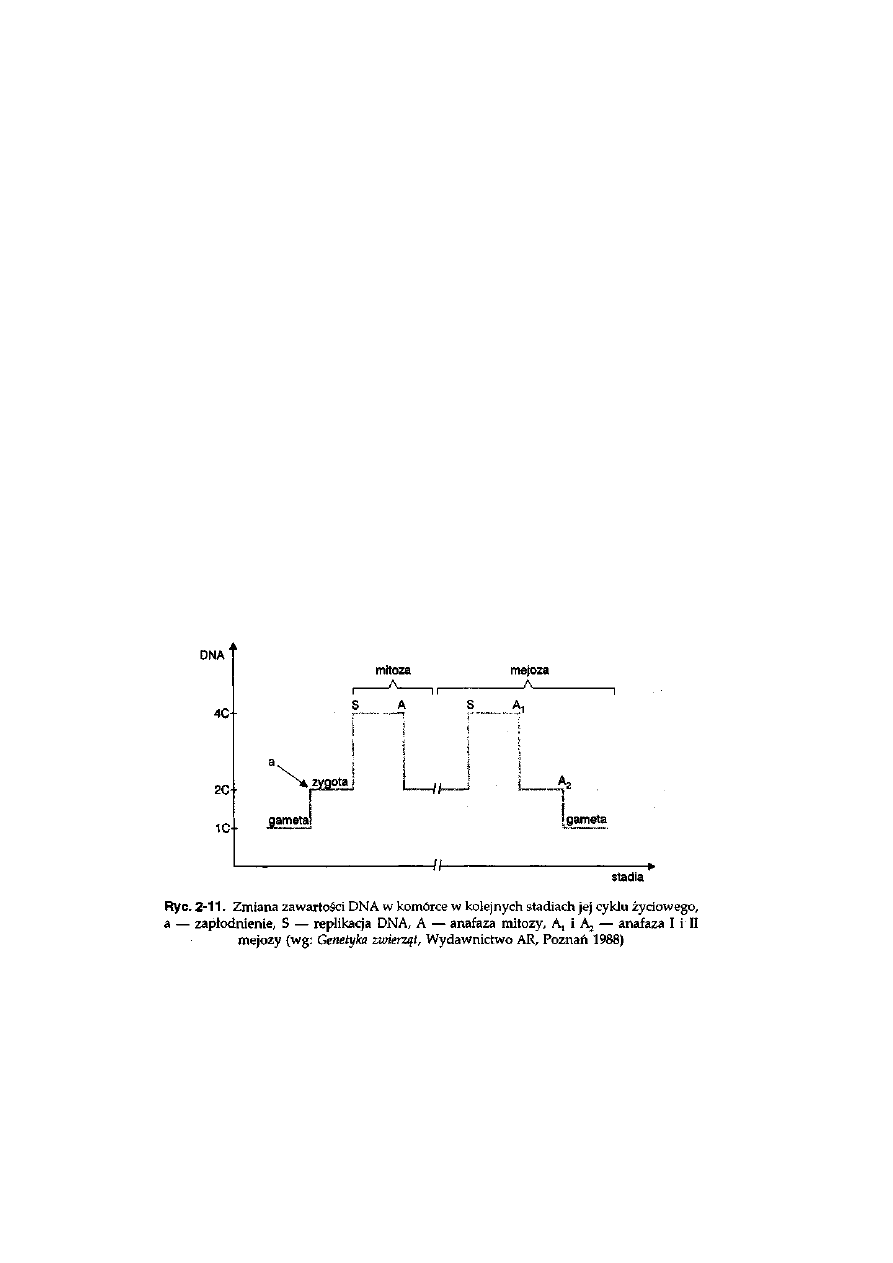
zmianie ulega zawartość DNA w jądrze. Jeżeli ilość DNA wyrazimy za
31

pomocą umownych wartości C, to w jądrze komórkowym po replikacji jest
4C, a począwszy od telofazy do początku fazy S zawartość DNA wynosi 2C.
W trakcie omawiania podziału mej etycznego pokazane zostanie, że naj-
mniejszą zawartość DNA (1C) mają gamety (ryć. 2-11).
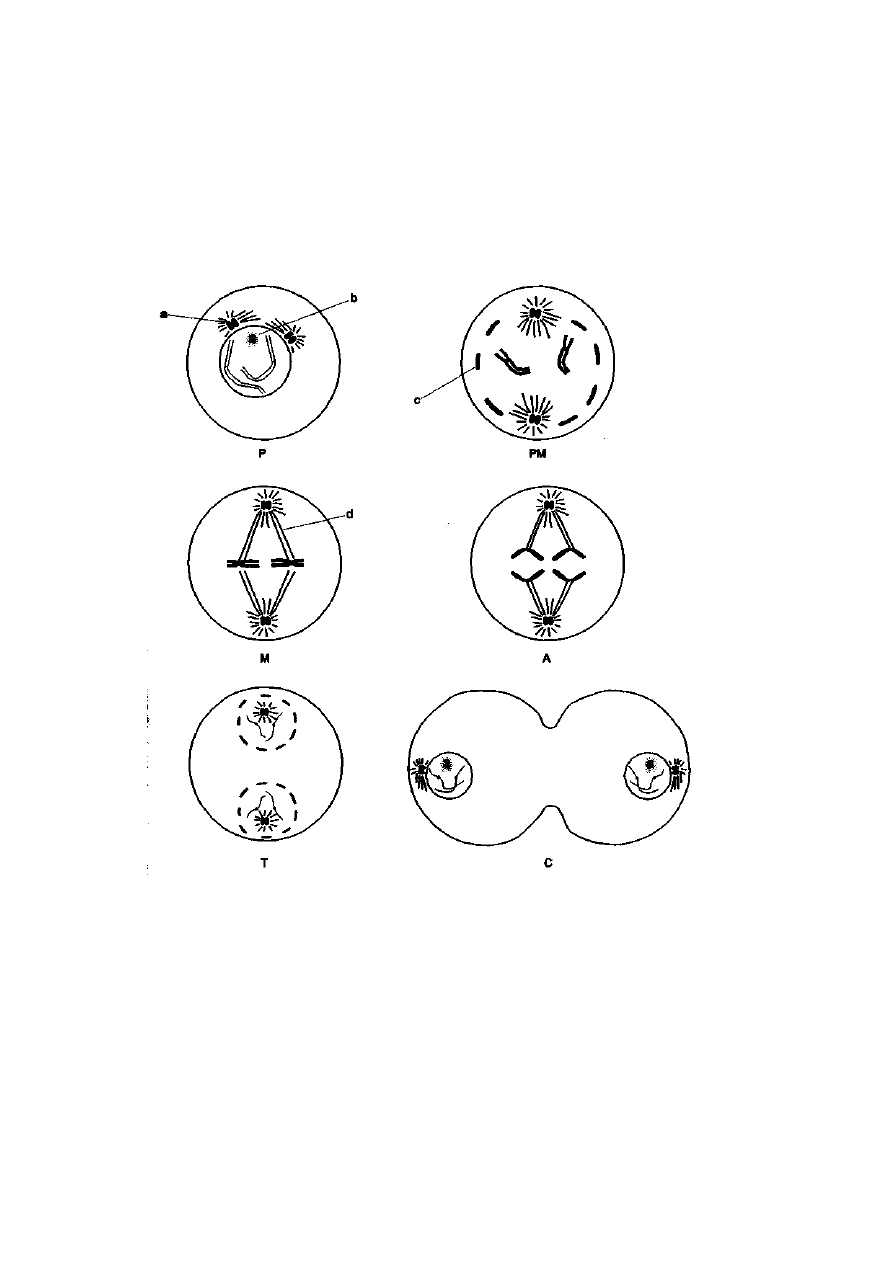
Mitoza składa się z czterech faz: profazy, metafazy, anafazy i telofazy (ryć.
2-12). W profazie rozpoczyna się proces kondensacji chromosomów. Istotnym
zmianom podlegają również inne organelle komórkowe. Centriołe, odpowie-
dzialne za powstanie wrzeciona podziałowego, rozchodzą się do przeciwleg-
łych biegunów komórki. W obrębie jądra komórkowego zanikają jąderka,
organelle komórkowe odpowiedzialne za wytwarzanie rybosomów. W końcu,
dezintegracji ulega błona jądrowa. Po zajściu powyższych procesów i osiągnię-
ciu stanu maksymalnej kondensacji chromosomów następuje metafaza. W tej
fazie chromosomy są rozprzestrzenione w całej komórce. Następuje formowa-
nie się wrzeciona podziałowego, którego włókienka, wychodzące z centroso-
mu (obszar wokół centrioli), łączą się z centromerami chromosomów w taki
sposób, że do każdego chromosomu dochodzą włókienka z dwóch przeciw-
ległych biegunów. Chromosomy w tej fazie są łatwym obiektem do badań
z użyciem mikroskopu świetlnego — są krótkie (ich długość jest rzędu kilku
(im) oraz mają wyraźną morfologię. Chromatydy siostrzane są ze sobą
połączone tylko w centromerze. Po ustawieniu się chromosomów w płaszczy-
źnie równikowej komórki rozpoczyna się proces podziału centromeru wzdłuż
osi podłużnej chromosomu. Z chwilą zakończenia tego procesu rozpoczyna
się anafaza. Po podzieleniu chromosomu chromatydy siostrzane stają się
chromosomami siostrzanymi, które pod wpływem kurczących się włókienek
wrzeciona podziałowego przesuwają się do przeciwległych biegunów
komórki. Po ich osiągnięciu rozpoczyna się ostatni etap kariokinezy
— telofaza. W telofazie zachodzą procesy odwrotne do tych, jakie występują
w profazie. Chromosomy ulegają dekondensacji, odbudowuje się błona
jądrowa oraz jąderka, a centriole dzielą się na dwie parzyste struktury.
W wyniku kariokinezy w komórce powstają dwa jądra potomne, mające
taką samą — diploidalną — liczbę chromosomów jak komórka rodzicielska,
w których każdy z chromosomów jest zbudowany z jednej chromatydy. Po
zakończeniu podziału jądra komórkowego następuje cytokineza, czyli
podział cytoplazmy i rozdział organelli komórkowych. Komórka dzieli się
ostatecznie na dwie komórki potomne przez przewężenie w części środ-
kowej. Po zakończeniu podziału komórki potomne odbudowują do nie-
zbędnego poziomu liczbę organelli komórkowych.
2.4. Kariotyp
Uszeregowanie chromosomów, występujących w jądrze komórki somatycz-
nej, w pary homologiczne określane jest terminem kariotyp. Układanie
kariotypu dla danego osobnika polega na zestawieniu par homologicznych
32
spośród chromosomów w stadium metafazy mitotycznej obecnych w po-
jedynczej komórce. Chromosomy wycinane są ze zdjęcia mikroskopowego
metafazy mitotycznej. Zabieg wycinania jest ostatnio coraz częściej za-
stępowany przez specjalistyczne programy komputerowe pozwalające
na analizę obrazów mikroskopowych przesłanych za pomocą kamery
do pamięci komputera. Do badania kariotypu najczęściej wykorzystuje
się dzielące się w warunkach pozaustrojowych (in vitro) komórki limfo-
cytów.
Być. 2-12. Schemat przebiegu podziału mitotycznego. Oznaczenia: P — profaza, PM
— prometafaza, M — metafaza, A — anafaza, T — telofaza, C — cytokineza, a — centriole,
b— zanikające jaderko, c — dezintegrująca błona jądrowa, d — mikrotubule wrzeciona
podziałowego (wg: Genetyka zwierząt, Wydawnictwo AR, Poznań 1988)
33
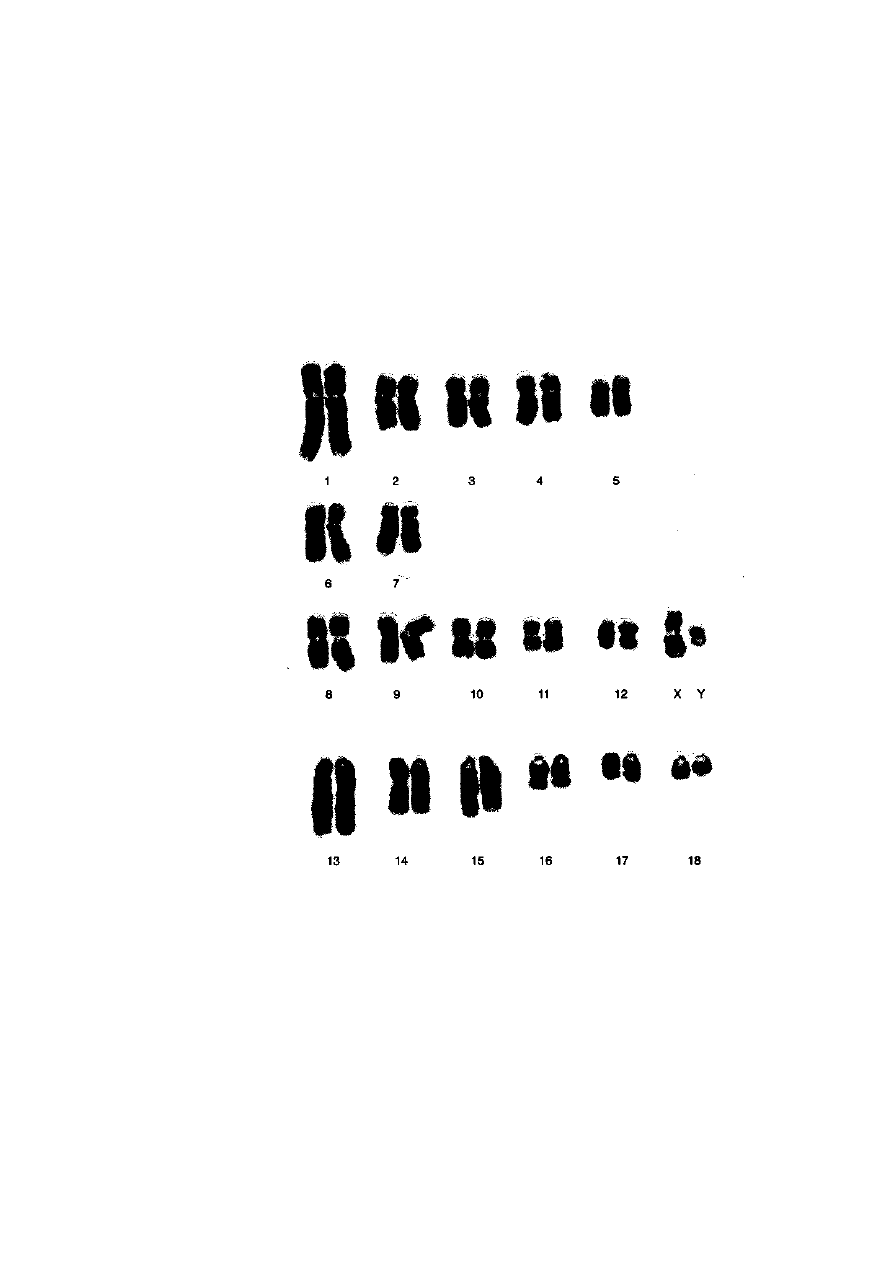
Chromosomy homologiczne rozpoznawane są na podstawie charak-
terystycznych cech morfologicznych, do których zalicza się: długość chromo-
somu, położenie centromeru wyznaczające typ morfologiczny chromosomu
(meta-, submeta-, subtelo- lub akrocentryczny) oraz wzór prążkowy uzys-
kiwany metodami Q, G lub R. Podstawowe znaczenie w układaniu kariotypu
przypisuje się wzorowi prążkowemu, ponieważ posługiwanie się wyłącznie
kryteriami wielkości i typu morfologicznego chromosomu nie pozwala na
precyzyjną identyfikację wszystkich par homologicznych. Jest to spowodo-
wane tym, że często chromosomy z różnych par są podobnej wielkości
i reprezentują ten sam typ morfologiczny. Skrajnym tego przykładem mogą
być zestawy chromosomowe bydła, kozy i psa, w których wszystkie
Ryć. 2-13. Kariotyp knura (barwienie metodą prążków G) uszeregowany zgodnie ze
wzorcem ustalonym przez Komitet ds. Standaryzacji Kariotypu Świni. Chromosomy
pochodzą z płytki metafazowej przedstawionej na ryć. 2-8a
34
chromosomy autosomalne są akrocentryczne i można je usytuować w sze-
regu o stopniowo zmniejszającej się długości, przy czym różnice długości
pomiędzy kolejnymi parami są praktycznie niezauważalne. Nawet w przy-
padku gatunków mających niewielką liczbę diploidalną oraz chromosomy
Ryć. 2-14, Idiogram kariotypu świni (układ prążków G) — wg Komitetu ds. Standaryzacji
Kariotypu Świni (Hereditas 109:151-157,1988)
35

zróżnicowane pod względem morfologicznym, precyzyjne sporządzenie
kariotypu z pominięciem technik prążkowych jest niemożliwe. Przykładem
może być kariotyp świni (2n = 38), w którym rozróżnienie niektórych par
wymaga zastosowania technik prążkowych (ryć. 2-13).
Graficzne przedstawienie kariotypu, obejmujące schematyczne obrazy
poszczególnych chromosomów z zaznaczonym położeniem centromeru
i układem prążków, nazywa się idiogramem (ryć. 2-14).
Opracowanie kariotypu, czasami określanego kariogramem, dla konkret-
nego osobnika polega na uszeregowaniu par chromosomów homologicznych
zgodnie z międzynarodowym wzorcem, przyjętym dla danego gatunku. We
wzorcu poszczególnym parom chromosomowym przyporządkowane są
kolejne numery, w zakresie wyznaczonym przez liczbę par dla danego
gatunku. Każda para homologiczna ma opisaną morfologię, wielkość oraz
wzór prążkowy (najczęściej prążki G lub R). Ponadto, dla poszczególnych
par mogą być wskazane prążki charakterystyczne (ang. landmarks). Prążek
taki (lub prążki) dzieli (-ą) ramię chromosomowe na regiony, które są
numerowane (ryć. 2-14). W obrębie regionów numerowane są także kolejne
prążki, które zależnie od zastosowanej metody barwienia mogą być ciemne
i jasne lub świecące i nie świecące. Tak opracowany idiogram stanowi
bardzo ważne narzędzie służące do opisywania mutacji chromosomowych
oraz mapowania genów w chromosomach.
Pierwsze wzorce kariotypów opracowano podczas Konferencji Standary-
zacyjnej, która odbyła się w 1976 roku w Reading (W. Brytania). Opracowano
wówczas wzorce kariotypów barwionych metodą prążków G dla: bydła, świni,
konia, owcy, kozy i kota. W latach 80. powstały wzorce dla lisa polarnego
i pospolitego oraz opublikowano ulepszone wzorce dla świni, bydła, owcy, kozy
i konia. Natomiast w drugiej połowie lat 90. opracowano częściowo wzorce
kariotypu psa (obejmujący 22 pary spośród 39) i kury (obejmujący 9 najwięk-
Tabela 2-11
Wykaz gatunków, dla których uzgodniono międzynarodowe wzorce kariotypów
Gatunek
Wzory
prążkowe
Źródło
Bydło
G, Q, R
Cytogenetics and Celi Genetks 92:283-299, 2001
Owca
G, Q, R
jw. i 85:317-324, 1999
Koza
R
J
w
-
Świnia
G, R
Hereditas 109:151-157, 1988
Koń
G, R
Chromosome Research 5:433-443, 1997
Lis polarny
G, C, Ag-I
Hereditas 103:33-38, 1985
Lis pospolity
G, C, Ag-I
Hereditas 103:171-176, 1985
Królik
G
Cytogenetics and Celi Genetks 31:240-248, 1981
Pies
G
Chromosome Research 4:306-309, 1996
Kura
G, R
Cytogenetics and Celi Genetks 86:271-276, 1999
36

szych par spośród 39) oraz poprawiono wzorzec kariotypu konia. Obecnie
są prowadzone prace nad uzgodnieniem wzorca kariotypu norki. W tabeli
2-II zestawione są gatunki, dla których opracowano wzorce, z zaznaczeniem
zastosowanych metod prążkowego barwienia chromosomów.
2.5. Mejoza
Podział mejotyczny prowadzi do powstania komórek, w których liczba
chromosomów jest zredukowana o połowę, czyli osobniki o dipoidalnej
liczbie chromosomów wytwarzają gamety z haploidalną ich liczbą. Komórki
po podziale mejotycznym mają zrekombinowaną informację genetyczną, tzn.
w ich genomie powstają unikatowe kombinacje alleli pochodzenia ojcowskie-
go i matczynego. Ponadto, komórki te mają zmniejszoną do poziomu 1C
zawartość DNA. Przedstawiona wyżej ogólna charakterystyka mejozy
wskazuje równocześnie na najważniejsze różnice między mitozą i mejozą.
Mejoza składa się z dwóch podziałów, w których występują takie same
fazy jak w mitozie, tzn.: profaza, metafaza, anafaza i telofaza (ryć. 2-15).
Wejście komórki w podział mejotyczny poprzedzone jest, podobnie jak
w przypadku podziałów mitotycznych, replikacją DNA w fazie S. Komórka
rozpoczynająca podział mejotyczny zawiera 4C DNA.
W profazie pierwszego podziału mejotycznego wyróżnia się pięć stadiów:
leptoten, zygoten, pachyten, diploten i diakinezę. Podczas profazy I zachodzą
ogólne zmiany o charakterze cytologicznym, które występują również
w mitozie. Należą do nich: kondensacja chromosomów, dezintegracja błony
jądrowej, zanik jąderek oraz rozchodzenie się centrioli do przeciwległych
biegunów komórki. Oprócz tego w profazie I zachodzą bardzo ważne
procesy mające podstawowy wpływ na dalszy przebieg podziału. W zygo-
tenie rozpoczyna się koniugacja chromosomów homologicznych, polegająca
na łączeniu się i ścisłym przyleganiu na całej długości chromosomów
homologicznych. Koniugacja chromosomów zachodzi dzięki specjalnej
strukturze białkowej, pojawiającej się w zygotenie i pachytenie profazy I,
która jak spoiwo łączy ze sobą dwa ułożone równolegle chromosomy
homologiczne. Strukturą tą jest kompleks synaptonemalny (ang. synapto-
nemal complex — SC), który składa się z dwóch elementów lateralnych oraz
występującego pomiędzy nimi elementu centralnego (ryć. 2-16 i 2-17).
Między elementami lateralnymi występują guzki (ciałka) rekombinacyjne,
które są zespołem białek odpowiedzialnych za przeprowadzenie crossing
over. Element lateralny przylega do rdzeni białkowych chromatyd sios-
trzanych chromosomu, a element centralny powstaje w miejscu, gdzie
włókienka poprzeczne wychodzące z elementów lateralnych zazębiają się.
Każdy element lateralny zakończony jest tarczką przylegającą, która mocuje
chromosom do wewnętrznej strony błony jądrowej. Elementy lateralne
37
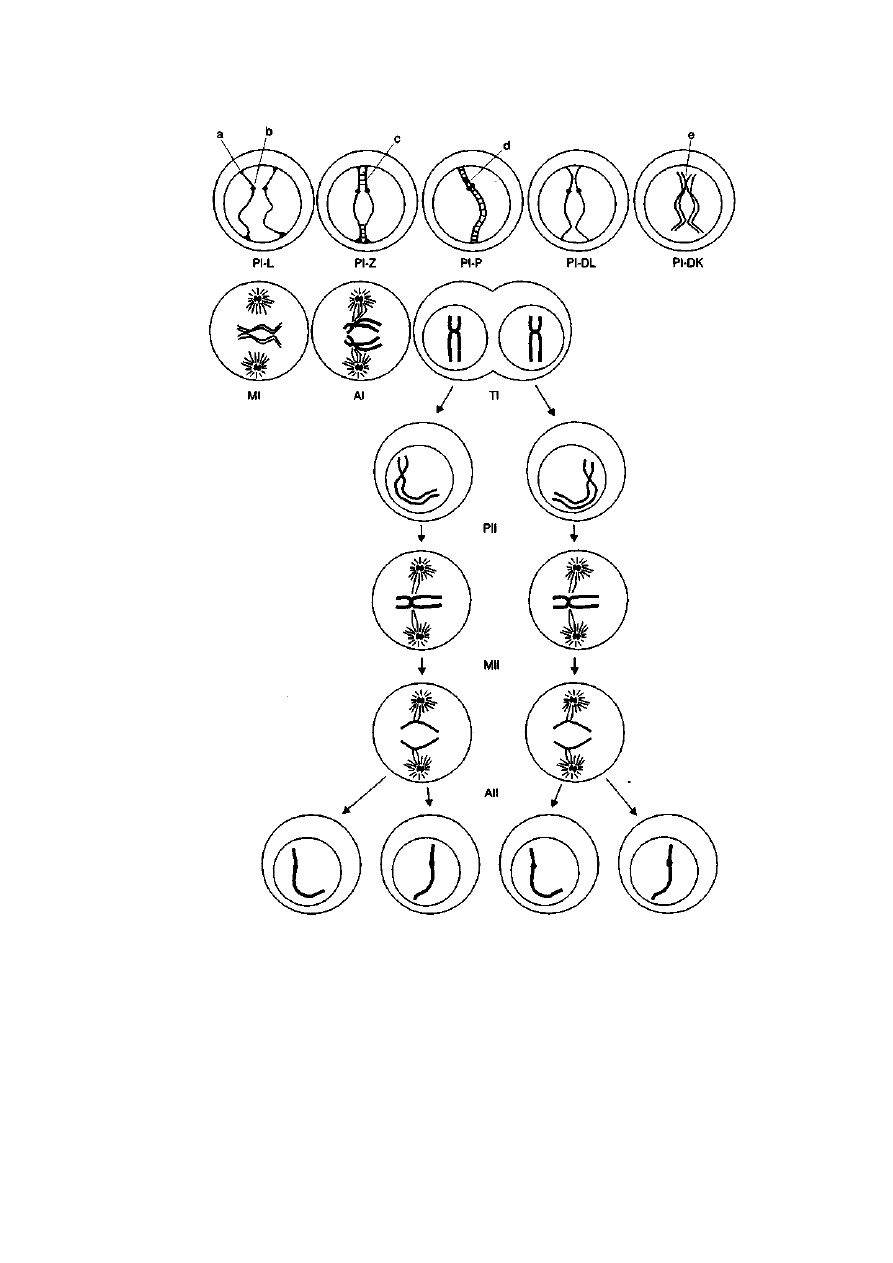
Tli
Ryć. 2-15. Schemat przebiegu podziału mejotycznego. Oznaczenia: PI — profaza I,
L — leptoten, Z — zygoten, P — pachyten, DL — diploten, DK — diakineza,
MI — metafaza I, AI — anafaza I, TI — telofaza I, Pil — profaza II, MII — metafaza II,
Ali — anafaza II, Tli — telofaza II, a — tarczka przylegająca, b — centromer, c — początek
budowania kompleksu synaptonemalnego, d — guzek rekombinacyjny, e — chiazma
(wg: Genetyka zwierząt, Wydawnictwo AR, Poznań 1988)
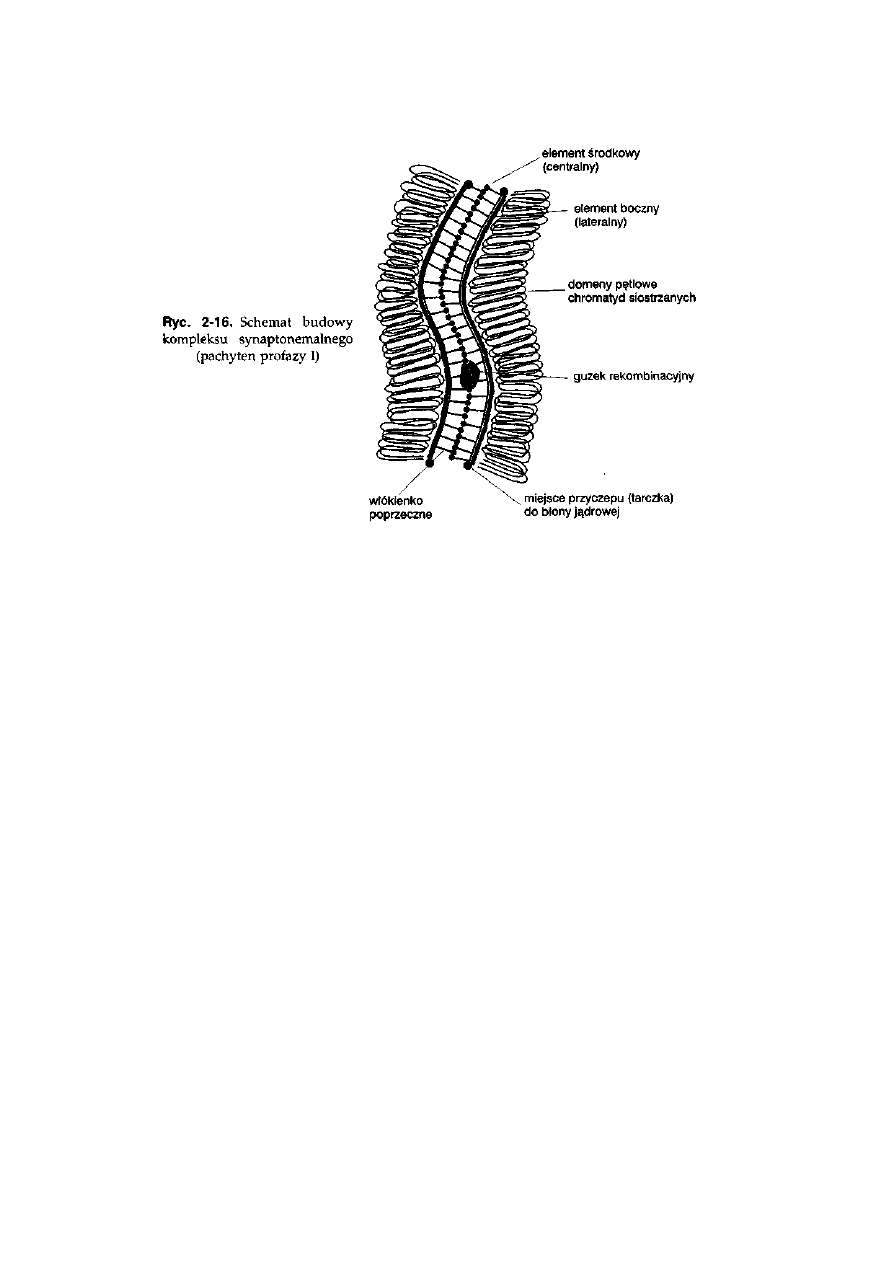
łączą się tylko z niewielką częścią DNA chromosomów (około 1%). Pozostała
część DNA jest ułożona wzdłuż chromosomu w postaci tzw. domen
pętlowych, których długość, zależnie od gatunku, mieści się w granicach od
20 000 pz (inaczej 20 kpz) do 120 000 pz (120 kpz). Wynika z tego, że tylko
niewielka część DNA homologicznych chromosomów kontaktuje się między
sobą za pośrednictwem kompleksu synaptonemalnego. Ponieważ koniugacja
ma charakter homologiczny, to białka kompleksu synaptonemalnego łączą
się ze ściśle określonymi, chociaż jeszcze nie poznanymi, sekwencjami
DNA, występującymi w tej części domen pętlo wy ch, które są zakotwiczone
w rdzeniu białkowym chromatydy. Wiele wskazuje na to, że fragmenty
DNA kontaktujące się z białkami SC są bogate w powtarzające się tan-
demowo sekwencje mikrosatelitarne oraz rozproszone elementy nukleo-
tydowe (LINĘ i SINE). Badania białek SC pokazały, że zdecydowana ich
większość jest syntetyzowana tylko w komórkach dzielących się mejotycznie.
U ssaków dobrze poznano strukturę trzech białek SCP (ang. synaptonemal
complex protein): SCP1, SCP2 i SCP3. Białko SCP1 występuje w obrębie
elementu centralnego i spełnia podstawową rolę w spinaniu struktury
biwalentu. Białka SCP2 i SCP3 uczestniczą w budowie elementów lateral-
nych. Funkcja kompleksów synaptonemalnych była powszechnie łączona
z przeprowadzeniem koniugacji chromosomowej, która z kolei miała
umożliwiać zajście crossing over na początku pachytenu. Ostatnio prze-
prowadzone badania pokazały jednak, że crossing over może zajść, zanim
dojdzie do koniugacji za pośrednictwem SC. Koniugacja chromosomów
39
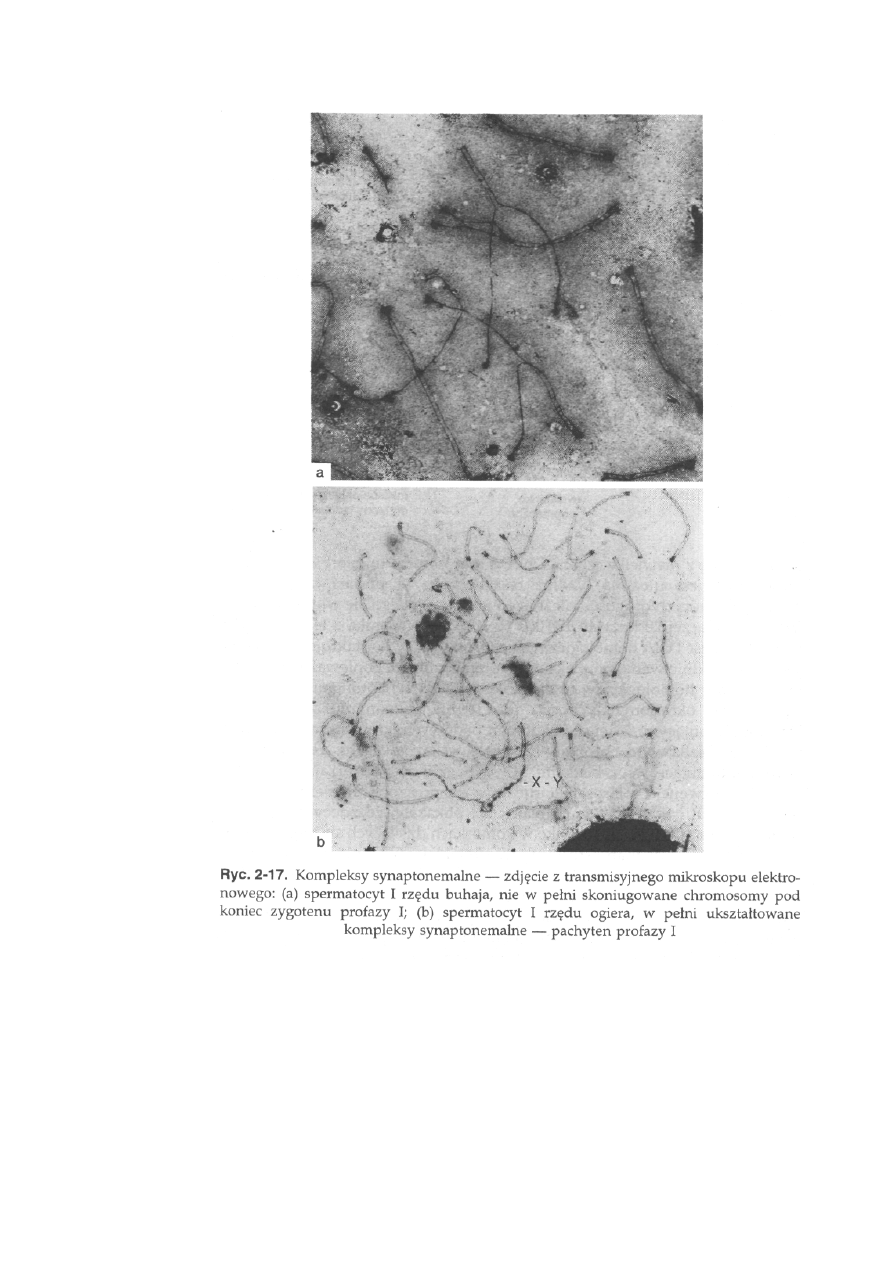
zostaje zakończona na początku pachytenu (ryć. 2-17b). Wynikiem tego
procesu jest pojawienie się haploidalnej liczby biwalentów. Biwalent jest
także formowany przez heterochromosomy X i Y (ryć. na okładce), dzięki
40

niewielkim regionom pseudoautosomalnym (ang. pseudoautosomal region
— PAR), które uruchamiają homologiczną koniugację. Na początku pachy-
tenu występuje zjawisko crossing over, które polega na wymianie fragmen-
tów chromatyd niesiostrzanych w obrębie biwalentu. Zasadniczo crossing
over jest zdarzeniem losowym, tzn. zachodzi w przypadkowym miejscu
biwalentu, przy czym liczba tych zdarzeń waha się od l do 5. Losowość
zachodzenia crossing over jest jednak ograniczona. Wiadomo, że wystąpienie
crossing over w określonym miejscu chromosomu wpływa ograniczająco na
możliwość zajścia crossing over w pobliżu. Zjawisko to określane jest
terminem interferencji. Przyjmuje się, że ogólna liczba crossing over
w komórce dzielącej się mejotycznie jest ograniczona. Potencjalnie mogłoby
istnieć niebezpieczeństwo, że na jednych chromosomach liczba ta byłaby
znaczna, a na innych brak byłoby takich wymian. Miałoby to bardzo
negatywny wpływ na proces segregacji chromosomów w anafazie I,
ponieważ brak crossing over pociąga za sobą brak chiazm. Ponieważ tak się
nie dzieje, dlatego poszukiwano mechanizmu gwarantującego równomier-
ność występowania crossing over. Mechanizmem tym jest wspomniane
wcześniej zjawisko interferencji, a w świetle ostatnich badań można
przypuszczać, że SC jest w to istotnie zaangażowany. Losowość crossing
over naruszona jest również przez to, że zachodzi ono rzadziej w pobliżu
centromeru, a częściej w końcowych częściach ramion chromosomowych —
w obrębie prążków T, które są podklasą prążków R. Należy podkreślić, że
crossing over jest zjawiskiem obligatoryjnym, czyli musi zajść w obrębie
każdego biwalentu. Zasada ta dotyczy także biwalentu X-Y, gdzie crossing
over występuje w obrębie regionu pseudoautosomalnego. Crossing over
prowadzi do powstania nowych układów alleli w obrębie pojedynczej
chromatydy. Chromatyda, która uczestniczyła w wymianie, uzyskuje kombina-
cję alleli pochodzenia ojcowskiego i matczynego, w przeciwieństwie do
sytuacji, jaka była przed crossing over. Wówczas to chromosom, a tym samym
i jego chromatydy, miał zestaw alleli tylko jednego z rodziców, co wynika
z zasady, że w obrębie pary chromosomów homologicznych jeden pochodzi
od ojca, a drugi od matki. Crossing over jest jednym z trzech mechanizmów
odpowiedzialnych za rekombinację genetyczną (mowa jest o tym na końcu
tego podrozdziału), której efektem jest zmienność w świecie ożywionym.
Należy jednak pamiętać, że pierwotnym źródłem zmienności są mutacje,
które — jeśli się pojawią — podlegają rekombinacjom podczas mejozy.
Po zakończeniu pachytenu — w diplotenie, następuje rozkład komplek-
sów synaptonemalnych, poza miejscami, gdzie wystąpiło crossing over,
w których powstają chiazmy, stanowiące cytologiczny dowód zajścia crossing
over. W ostatnim stadium profazy I — diakinezie kontynuowana jest
kondensacja chromosomów oraz rozpoczyna się proces terminalizacji chiazm,
tzn. ich zanikania.
W metafazie I biwalenty, składające się z maksymalnie skondensowanych
chromosomów homologicznych, zmierzają do płaszczyzny równikowej
41
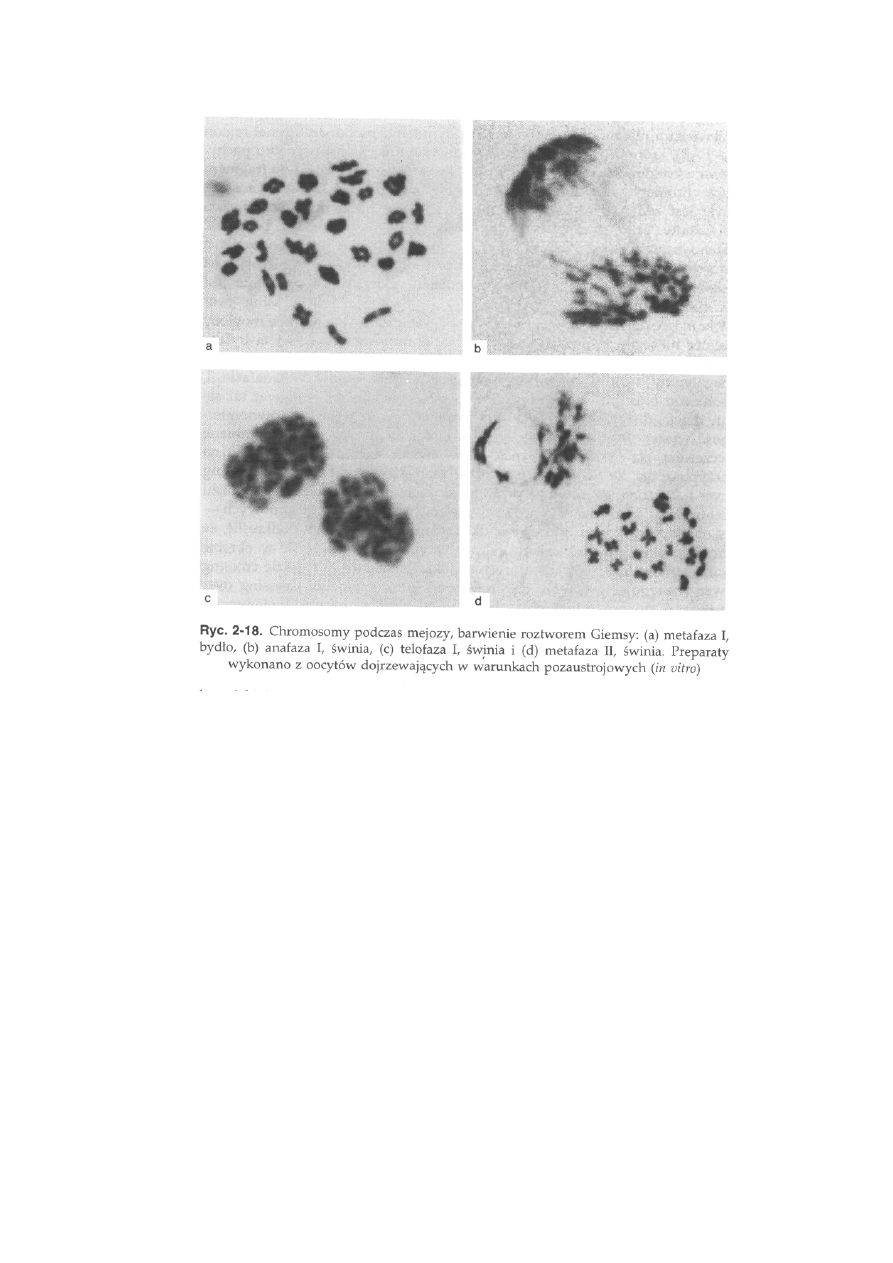
komórki (ryć. 2-18a). Włókna wrzeciona podziałowego rozpoczynają proces
łączenia biegunów komórki z centromerami w taki sposób, że do centromeru
przyłączają się włókna tylko z jednego bieguna, natomiast do centromeru
chromosomu homologicznego wiążą się włókna z bieguna przeciwległego.
Po zakończeniu terminalizacji chiazm rozpoczyna się anafaza I, w której
chromosomy homologiczne rozchodzą się do przeciwległych biegunów
komórki (ryć. 2-18b). Oznacza to, że do każdego z biegunów zmierza po
jednym chromosomie z każdej pary homologicznej, czego wynikiem jest
zmniejszenie o połowę liczby chromosomów w jądrach powstałych po
pierwszym podziale mejotycznym. W telofazie następuje odbudowa jądra
połączona z dekondensacją chromosomów (ryć. 2-18c).
Po zakończeniu pierwszego podziału mejotycznego następuje drugi
podział, który nie jest poprzedzony replikacją DNA. Tym samym jądro
komórkowe rozpoczynające drugi podział mejotyczny zawiera 2C DNA.
42

Drugi podział ma przebieg zgodny z mitozą, z tym jednak że uczestniczy
w nim już tylko haploidalna liczba chromosomów, a ich chromatydy mają
zrekombinowane układy alleli. Po ustawieniu się chromosomów podczas
metafazy II (ryć. 2-18d) w płaszczyźnie równikowej następuje podział
centromeru i rozpoczyna się anafaza II. Włókna wrzeciona podziałowego
rozciągają chromatydy siostrzane do przeciwległych biegunów komórki.
Gdy jednochromatydowe chromosomy siostrzane osiągną bieguny, rozpo-
czyna się telofaza, w której zachodzą procesy odwrotne do tych, jakie
występują w profazie mitotycznej.
Po zakończeniu podziału mej etycznego zawartość DNA w jądrach
komórkowych maleje do poziomu 1C. Chromosomy haploidalnego zestawu są
zbudowane z pojedynczych chromatyd, wśród których jedne są pochodzenia
ojcowskiego, inne matczynego, a niektóre mają zrekombinowany układ alleli.
Rekombinacje genetyczne zachodzące podczas mejozy, rozumiane w sze-
rokim sensie, można podzielić na: chromosomowe, chromatydowe i we-
wnątrzchromatydowe. Podczas pachytenu profazy I w wyniku crossing
over powstają chromatydy składające się z fragmentów o pochodzeniu
ojcowskim i matczynym — jest to rekombinacja wewnątrzchromatydowa
(rekombinacja w ścisłym sensie). W trakcie anafazy I chromosomy homo-
logiczne rozchodzą się losowo do przeciwległych biegunów komórki.
W wyniku tego procesu w telofazie I powstają jądra komórkowe zawierające
kombinacje chromosomów pochodzenia ojcowskiego i matczynego, przy
czym chromatydy tych chromosomów mogą być już zrekombinowane —
jest to rekombinacja chromosomowa. Wreszcie w anafazie II do przeciwleg-
łych biegunów komórki rozchodzą się losowo chromatydy, które mogą być
zrekombinowane lub mieć układ alleli pochodzenia ojcowskiego albo
matczynego. Ten typ określany jest jako rekombinacja chromatydowa.
Trzystopniowa rekombinacja sprawia, że wśród gamet nie występują
dwie o tym samym zestawie alleli. Ten fenomen, obok mutacji, jest
główną przyczyną obserwowanej ogromnej zmienności genetycznej wśród
organizmów.
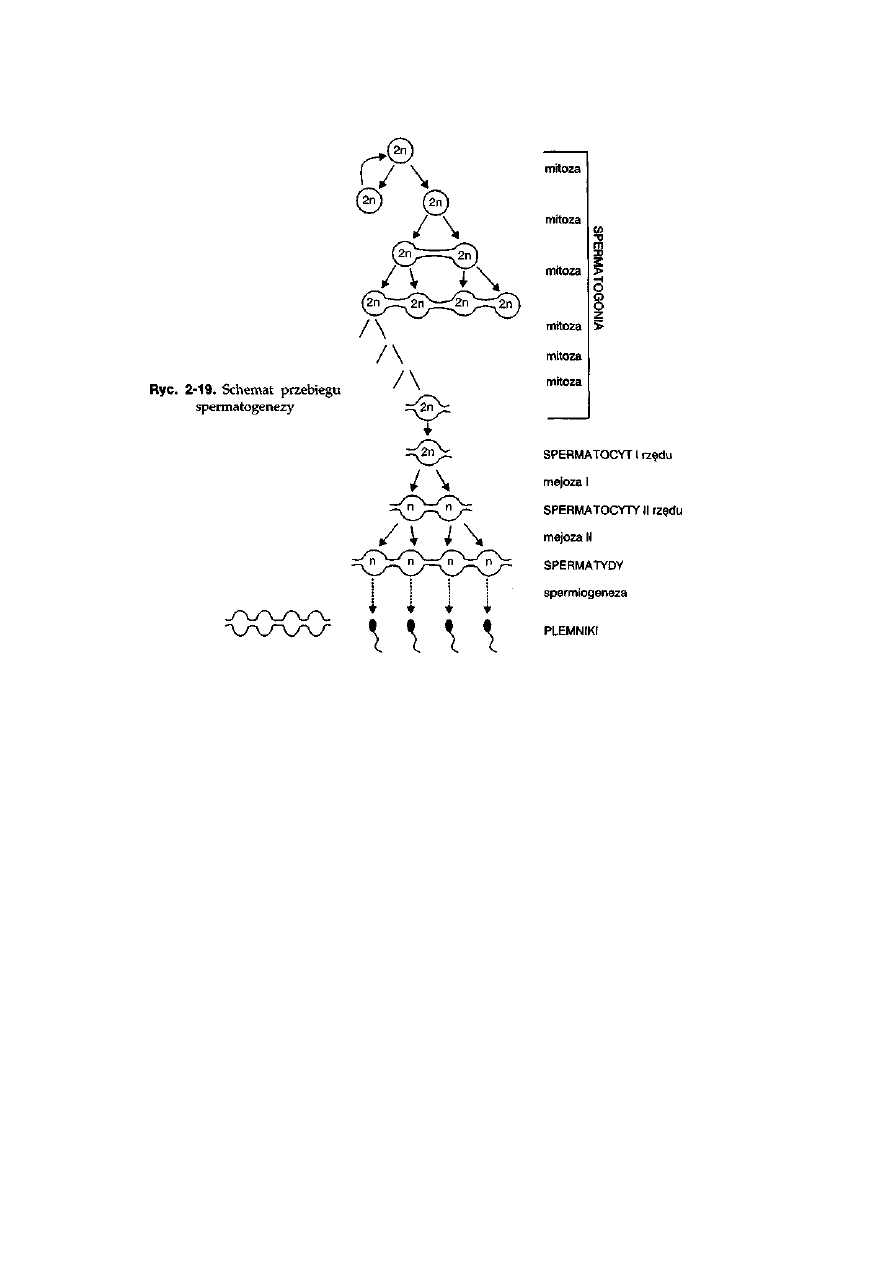
2.6. Gametogeneza
Proces tworzenia gamet jest nierozłącznie związany z podziałem mejotycz-
nym. Przebieg mejozy, a w tym czas i tempo tego podziału oraz procesy
prowadzące do powstania w pełni wykształconych gamet sprawiają, że
pomiędzy gametogenezą męską (spermatogeneza) i żeńską (oogeneza)
występują znaczne różnice.
Gametogeneza męska przebiega w kanalikach plemnikotwórczych gona-
dy męskiej — jądrach. W procesie spermatogenezy można wyróżnić trzy
główne etapy: 1) podziały mitotyczne spermatogonii (spermatogoniogeneza),
2) podział mejotyczny spermatocytów (spermatocytogeneza) oraz 3) spermio-
43

geneza, która prowadzi do powstania z niezróżnicowanych haploidalnych
spermatyd wyspecjalizowanych gamet męskich — plemników (ryć. 2-19).
Intensywne podziały mitotyczne spermatogonii w jądrach dojrzałych
samców umożliwiają masowe wytwarzanie plemników. Pierwszy podział
prowadzi do powstania dwóch spermatogoniów, z których jedna komórka
zajmuje miejsce pierwotnego spermatogonium, a druga podlega dalszym
podziałom. Taki sposób podziału pierwotnego spermatogonium jest typowy
dla komórek pierwotnych (ang. stem cells), czyli komórek nie w pełni
zróżnicowanych, które wykazują zdolność do samoodnawiania. Następne
podziały mitotyczne spermatogoniów, a jest ich najczęściej sześć, nie kończą się
cytokinezą. Podziały te są zsynchronizowane, a powstające kolejne pokolenia
spermatogonii są połączone mostkami cytoplazmatycznymi. Po zakończeniu
cyklu mitoz spermatogonia przekształcają się w spermatocyty I rzędu, które
rozpoczynają podział mejotyczny. Również kolejnym dwóm podziałom
mejozy nie towarzyszy cytokinezą. Ostatecznie, po zakończeniu mejozy,
powstaje grupa licząca kilkaset spermatyd połączonych mostkami
cytoplazmatycznymi, która jest określana jako syncytium. Spermatydy, jako
komórki niezróżnicowane, podlegają głębokim przeobrażeniom podczas
procesu spermiogenezy, w wyniku którego powstają plemniki. Główne
zmiany, jakie zachodzą podczas spermiogenezy, polegają na: 1) przekształceniu
aparatu Golgiego w akrosom, który okrywa górną część jądra komórkowego
znajdującego się w główce plemnika, 2) zgrupowaniu mitochondriów w
pobliżu centrioli, które są umiejscowione po przeciwnej, w stosunku do
akrosomu, stronie jądra komórkowego, 3) wykształceniu na bazie centrioli
włókna osiowego witki, 4) usunięciu prawie całej cytoplazmy obecnej w
spermatydzie i 5) zamianie białek histonowych na białka protaminowe w
chromosomach, czego skutkiem jest utrata struktury nukleosomowej przez
chromatynę jądra plemnika. Dojrzały plemnik zbudowany jest z główki, w
której jest umiejscowione jądro komórkowe i akrosom, wstawki znajdującej się
u podstawy główki, w której zgrupowane są mitochondria, oraz witki, która
stanowi przedłużenie wstawki.
Gametogeneza żeńska u ssaków rozpoczyna się już w życiu płodowym
(ryć. 2-20). Po ukierunkowaniu rozwoju osobniczego na płeć żeńską
niezróżnicowane gonady płodowe przekształcają się w jajniki, w których
komórki linii płciowej lokują się w części korowej gonad. Pierwotne oogonia po
przejściu kilku podziałów mitotycznych przekształcają się w oocyty I
rzędu, które niezwłocznie rozpoczynają podział mejotyczny. W przypadku
bydła następuje to około 60 dnia rozwoju płodowego. Oocyty I rzędu po
osiągnięciu stadium diplotenu profazy I mejozy zatrzymują rozwój. Stan ten
osiągany jest u bydła powyżej 100 dnia rozwoju płodowego. Oocyty
przechodzą w stan spoczynkowy, który jest nazywany diktiotenem. W ten
sposób wszystkie oogonia, które występowały w jajniku, zostają przekształ-
cone w oocyty I rzędu, będące w stadium diktiotenu. Umieszczone są one
zazwyczaj pojedynczo w pierwotnych pęcherzykach jajnikowych.
44
Dalsze etapy oogenezy następują po osiągnięciu przez samicę dojrzałości
płciowej. W kolejnych cyklach płciowych, regulowanych hormonalnie,
pewna część pęcherzyków rozpoczyna wzrost, a w nich rozrasta się również
oocyt, który jest wspomagany przez otaczające komórki pęcherzykowe.
W pęcherzykach, których wzrost był wystarczająco intensywny, przed
owulacją następuje ponowne uruchomienie podziału mejotycznego. Oocyt
I rzędu kończy pierwszy podział mejotyczny, w wyniku którego powstaje
oocyt II rzędu oraz ubogie w cytoplazmę ciałko kierunkowe. Oocyt znajduje
się w stadium metafazy II. Obie komórki umieszczone są w glikoproteinowej
osłonce przejrzystej (zona pellucida) i w takim stadium rozwoju dochodzi do
owulacji. Komórki przemieszczają się w jajowodzie i jeżeli podczas wędrówki
natrafią na plemniki, to może nastąpić dokończenie oogenezy. Tym samym
zapłodnienie jest niezbędnym warunkiem zakończenia oogenezy.
Zapłodnienie oocytu przez plemnik stymuluje dokończenie mejozy
żeńskiej, przy równoczesnym zablokowaniu dostępu innych plemników do
wnętrza oocytu. Dzieje się to na drodze wyrzucenia przez oocyt zawartości
45
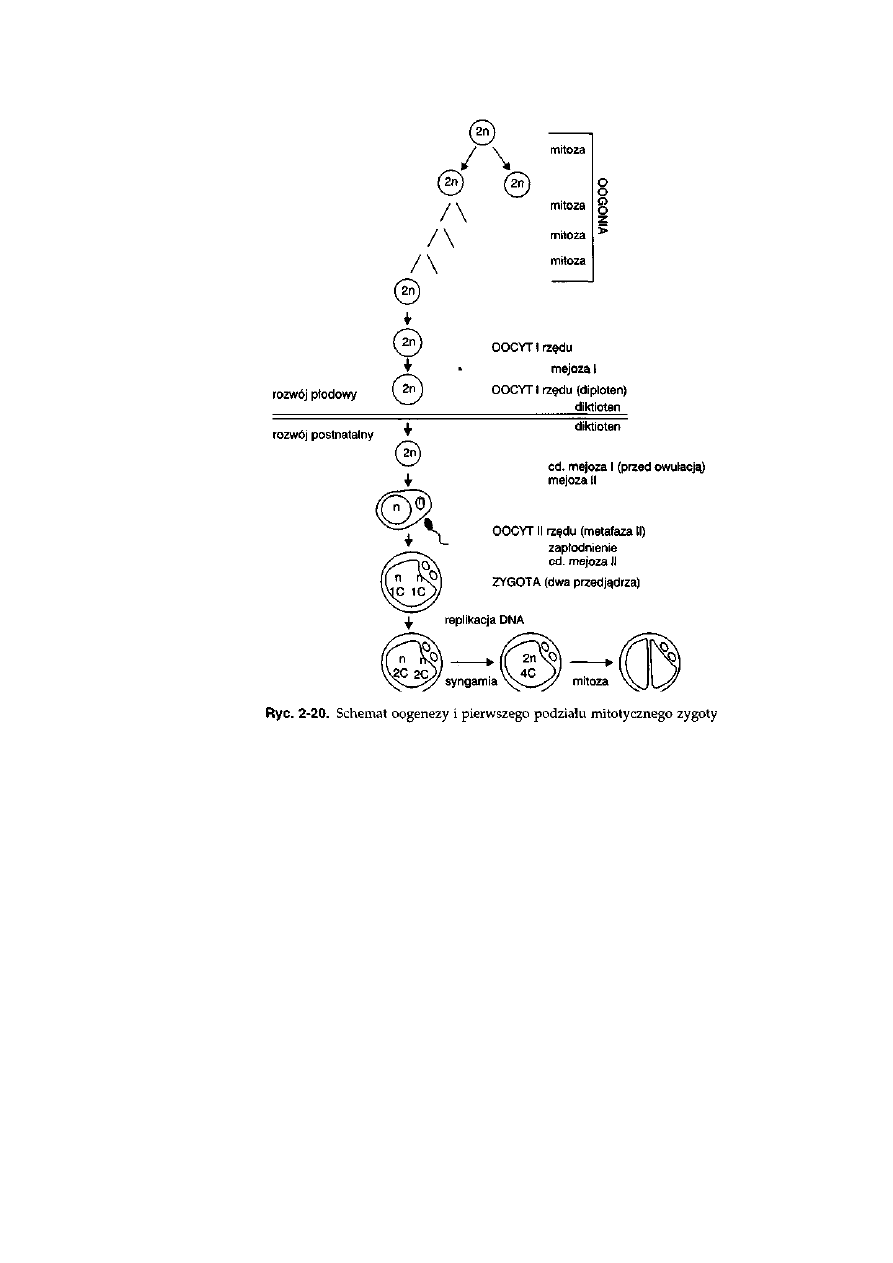
ziaren korowych, znajdujących się pod powierzchnią oocytu, do przestrzeni
pomiędzy błoną oocytu a osłonką przejrzystą. Powoduje to „utwardzenie"
osłonki przejrzystej, która staje się barierą nie do pokonania dla plemników.
Po wniknięciu plemnika oocyt ponownie podejmuje podział mejotyczny.
Rozpoczyna się anafaza II i dochodzi do „wyrzucenia" drugiego ciałka
kierunkowego, po czym następuje telofaza II. Chromosomy pozostałe
w oocycie tworzą przedjądrze żeńskie, a chromatyna plemnika, który
zapłodnił oocyt, podlega dekondensacji i powstaje przedjądrze męskie.
Zapłodniony oocyt II rzędu przekształca się w zygotę, w której występują
dwa haploidalne przedjądrza: jedno powstałe z jądra plemnika, a drugie
reprezentujące zestaw chromosomów żeńskich, które pozostały w oocycie
46

po zakończeniu drugiego podziału mejotycznego. Chromosomy przedjądrza
żeńskiego podlegają dekondensacji i równolegle z chromatyną przedjądrza
męskiego przechodzą proces replikacji DNA. Zawartość DNA wzrasta do
poziomu 4C, co powoduje, że zygota jest gotowa do rozpoczęcia podziału
mitotycznego. Chromosomy ulegają kondensacji i początkowo nadal wy-
stępują jako dwa oddzielne zestawy chromosomów, z tym jednak że każdy
z nich jest zbudowany z dwóch chromatyd siostrzanych. W stadium
metafazy pierwszego podziału mitotycznego zygoty dochodzi do połączenia
się przedjądrzy (syngamii), a dalej następuje anafaza oraz telofaza i powstaje
zarodek zbudowany z dwóch komórek (blastomerów), który dalej będzie
wzrastał na drodze kolejnych podziałów mitotycznych.
Od przedstawionego wyżej schematu oogenezy występują odstępstwa
u niektórych gatunków. Na przykład, profaza I nie zostaje zakończona
w życiu płodowym u psa i niektórych gryzoni. U gatunków tych można
znaleźć oocyty I rzędu w stadium pachytenu w jajnikach nowo urodzonych
osobników żeńskich. Z kolei u królika owulacja jest bezpośrednio in-
dukowana przez akt krycia lub inseminacji.
Poznanie procesów gametogenezy oraz wczesnego rozwoju zarodkowego
umożliwiło rozwój biotechnologii gamet i zarodków. W hodowli zwierząt
na szeroką skalę stosowane są następujące biotechniki wspomagające rozród:
konserwacja nasienia w niskich temperaturach (kriokonserwacja), insemina-
cja, przenoszenie zarodków (stadium moruli lub blastocysty) pozyskanych
od tzw. samic dawczyń do rogów macicy tzw. samic biorczyń, kriokonser-
wacja zarodków i zapłodnienie pozaustrojowe (in vitro). Ostatnio do praktyki
hodowlanej wprowadzane są kolejne techniki, które umożliwiają uzys-
kiwanie potomstwa pożądanej płci. Należy do nich oznaczanie płci zarodków
i segregowanie plemników na frakcję z chromosomem X i frakcję z chromo-
somem Y. Warto zauważyć, że zarówno produkcja zwierząt modyfikowa-
nych genetycznie (zwierzęta transgeniczne), jak i klonowanie zwierząt
również bazują na manipulacjach prowadzonych na oocytach II rzędu,
zygotach, zarodkach, a także hodowanych w warunkach pozaustrojowych
komórkach somatycznych. Jedna z najpowszechniej obecnie wykorzysty-
wanych technik dotyczy manipulacji na oocytach II rzędu, z których
usunięto mikrochirurgicznie wrzeciono podziałowe (chromosomy w stadium
metafazy II). Do takich oocytów, które są określane jako enukleowane,
można wprowadzić techniką elektrofuzji komórkę somatyczną, pochodzącą
z hodowli pozaustrojowej (komórka taka może być zmodyfikowana gene-
tycznie). W taki właśnie sposób sklonowano słynną owcę Doiły, do uzyskania
której wykorzystano komórkę nabłonkową pochodzącą z gruczołu mleko-
wego dorosłej owcy.

Rozdział
3
Gen i jego ekspresja
Znaczenie kwasu deoksyrybonukleinowego (DNA) jako nośnika informacji
genetycznej zostało udowodnione w 1944 roku dzięki eksperymentowi,
który polegał na transformacji niepatogennych bakterii — dwoinek zapalenia
płuc z wykorzystaniem DNA wyizolowanego ze szczepu patogennego.
Efektem tego doświadczenia było nabycie przez niektóre bakterie niepatogen-
ne cech zjadliwości. Badania te przeprowadzili badacze amerykańscy
O.T. Avery, C.M. MacLeod i M. McCarty. W ślad za tym odkryciem
nastąpiły następne. W 1953 roku J.D. Watson i F. Crick przedstawili model
budowy DNA, a przez następne lata rozszyfrowywano kod genetyczny,
przypisując poszczególnym trójkom nukleotydów odpowiednie aminokwasy.
DNA charakteryzuje się trzema właściwościami, które są niezbędne do
pełnienia funkcji nośnika informacji genetycznej. Po pierwsze, w sekwencji
nukleotydów zapisana jest, za pomocą kodu genetycznego, informacja
o pierwszorzędowej strukturze białek, a także o strukturze cząsteczek
rybosomowego (rRNA), transportującego (tRNA) i niekodującego (ncRNA)
kwasu rybonukleinowego. Po drugie, DNA ma zdolność do samood-
twarzania się, czyli replikacji. Po trzecie, DNA może podlegać mutacjom,
czyli procesowi generującemu pierwotną zmienność genetyczną, które
z kolei podlegają rekombinacjom podczas mejozy.
3.1. Budowa kwasów nukleinowych
Zapis informacji genetycznej oraz jej ujawnienie się w postaci cechy
(ekspresja genu) związane są z funkcjonowaniem kwasów nukleinowych.
U zdecydowanej większości organizmów nośnikiem informacji genetycznej
jest DNA. Tylko u nielicznych wirusów (retrowirusy) informacja genetyczna
jest zawarta w kwasie rybonukleinowym (RNA). Z kolei proces ekspresji
48
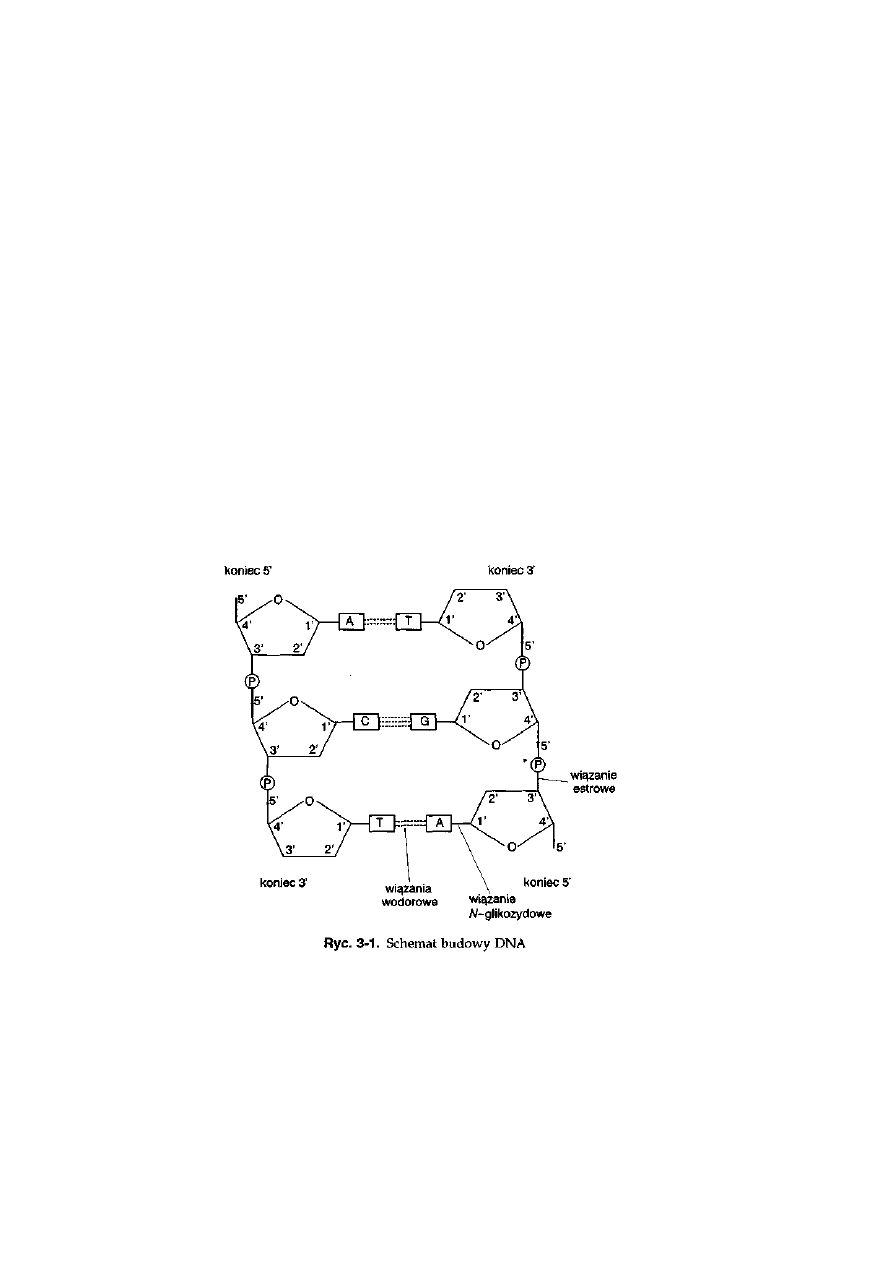
genu jest nierozerwalnie związany z RNA, występującym w trzech pod-
stawowych wariantach: mRNA (informacyjny RNA) — powstający podczas
transkrypcji genu, rRNA (rybosomowy RNA) — stanowiący część struk-
turalną rybosomów, które są organellami komórkowymi odpowiedzialnymi
za przeprowadzenie translacji, i tRNA (transportujący RNA), który jest
zaangażowany w proces translacji poprzez dostarczanie do rybosomów
właściwych aminokwasów.
3.1.1. DNA
Cząsteczka DNA składa się z dwóch owiniętych prawoskrętnie wokół siebie
łańcuchów polinukleotydowych. Struktura ta określana jest w języku polskim
terminem podwójna helisa (ang. helix), a rzadziej podwójny heliks. Pod-
stawową jednostką struktury DNA jest deoksyrybonukleotyd, w którego
skład wchodzą: zasada azotowa, cukier deoksyryboza i reszta kwasu
fosforowego (ryć. 3-1). W DNA występują cztery zasady azotowe, które
zaliczane są do puryn: adenina (A) i guanina (G) lub pirymidyn: ty mina (T)
i cytozyna (C). W rdzeniu każdego łańcucha polinukleotydowego cząsteczki
deoksyrybozy połączone są ze sobą poprzez resztę kwasu fosforowego
wiązaniami fosfodiestrowymi między atomem węgla w pozycji 5' jednej
cząsteczki cukru i atomem węgla 3' sąsiedniej cząsteczki deoksyrybozy.
Cząsteczki zasad azotowych połączone są wiązaniami N-glikozydowymi
49

z atomami węgła występującymi w pozycji l' cząsteczki deoksyrybozy i są
skierowane do wnętrza podwójnej helisy. Ułożenie zasad azotowych
w łańcuchach polinukleotydowych podwójnej helisy DNA oparte jest na
zasadzie komplementarności, tzn. naprzeciw adeniny zawsze występuje
tymina, a naprzeciw guaniny znajduje się cytozyna. Taka organizacja
podwójnej helisy DNA sprawia, że sekwencja nukleotydów w jednej nici
jest różna od sekwencji nukleotydów w drugiej nici. Pomiędzy zasadami
powstają słabe wiązania wodorowe; tak więc w parze AT są dwa wiązania
wodorowe, a w parze GC trzy takie wiązania. Długość cząsteczki DNA
wyraża się liczbą par nukleotydów, lecz częściej stosuje się określenie liczba
par zasad (skrót: pz w jęz. polskim lub bp od słów angielskich base pairs),
bo skierowane do wnętrza podwójnej helisy zasady azotowe stanowią jakby
szczeble drabiny.
Łańcuchy polinukleotydowe DNA są przeciwnie spolaryzowane. Polary-
zacja łańcucha związana jest z występowaniem na jego końcach wolnych
atomów węgla, czyli każdy łańcuch ma koniec 3' i 5'. Przeciwna polaryzacja
polega na tym, że naprzeciw końca 3' jednego łańcucha znajduje się koniec
5' drugiego łańcucha. Polaryzacja łańcuchów ma bardzo ważne znaczenie
dla przebiegu procesu replikacji i transkrypcji, a w przypadku mRNA także
dla procesu translacji. Zwijające się wokół siebie łańcuchy polinukleotydowe
tworzą na powierzchni podwójnej helisy dwie bruzdy biegnące spiralnie.
Ze względu na ich szerokość określa się je jako bruzdę większą i mniejszą.
3.1.2. RNA
Kwasy rybonukleinowe są cząsteczkami zbudowanymi z pojedynczych
łańcuchów polinukleotydowych, w których zamiast deoksyrybozy występuje
ryboza oraz zamiast zasady pirymidynowej — tyminy (T) występuje inna
pirymidyna — uracyl (U).
W ogólnej puli RNA występujących w komórce najliczniej reprezen-
towany jest rybosomowy RNA (rRNA), który wraz z białkami tworzy
strukturę rybosomów i uczestniczy w przeprowadzaniu translacji. W jąderku
zlokalizowane są cztery rodzaje rRNA, które są wyróżniane ze względu na
wielkość cząsteczki, określaną wielkością S — stała sedymentacji. Trzy
rodzaje rRNA: 5.8S, 18S i 28S są kodowane przez geny umiejscowione
w obszarach jąderkotwórczych (ang. nucleolar organizer region — NOR),
inaczej nazywanych przewężeniami wtórnymi. Podczas transkrypcji, pro-
wadzonej przez polimerazę I RNA, powstaje pre-rRNA o stałej sedymentacji
45S, z której po obróbce formowane są wspomniane wyżej trzy rodzaje
rRNA. Przyjmuje się, że w obszarze jęderkotwórczym występuje nawet
kilkadziesiąt powtórzeń genu kodującego prekursorową cząsteczkę 45S
rRNA, którego długość wynosi około 14 000 pz. Liczba tych obszarów jest
cechą charakterystyczną kariotypu danego gatunku, natomiast liczba aktyw-
50
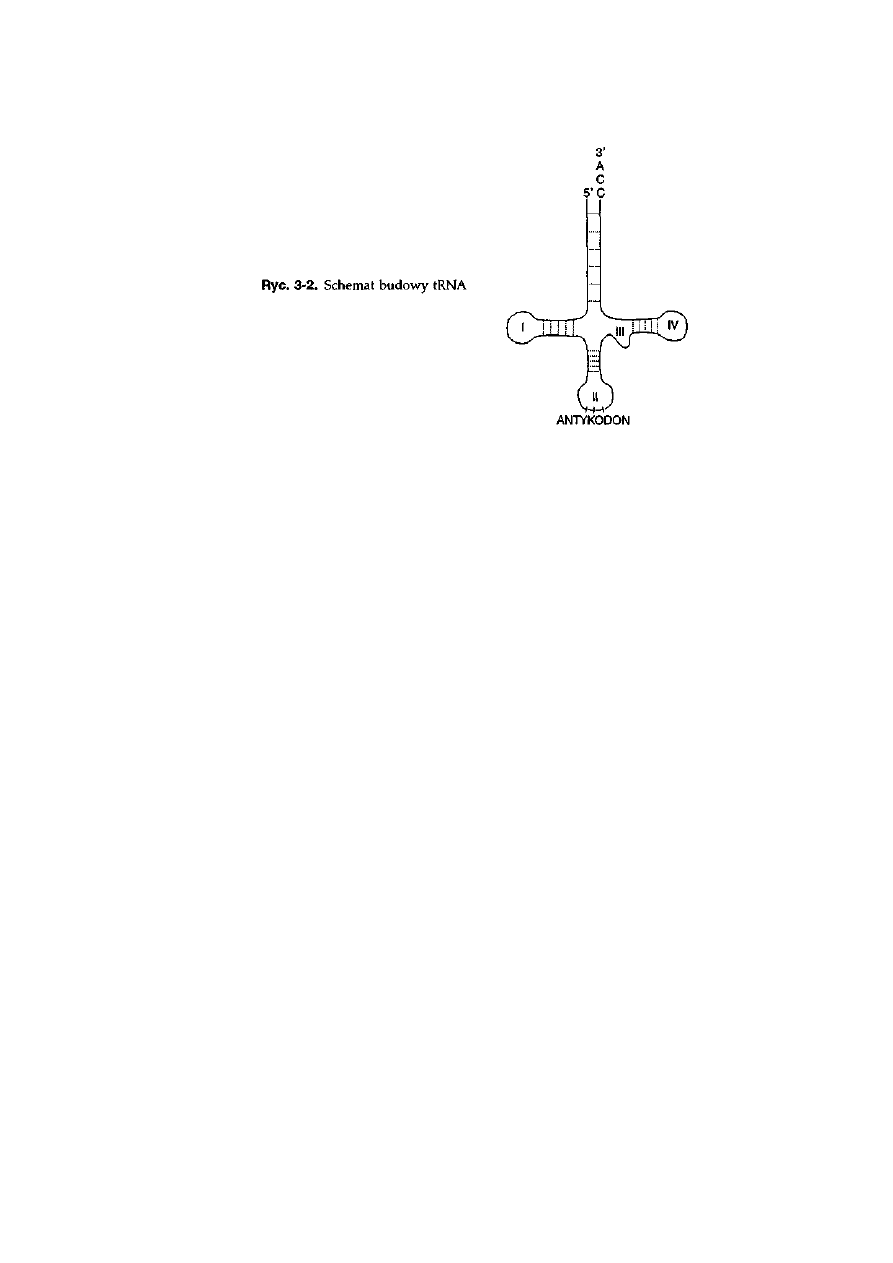
nych obszarów, czyli takich, które podlegają transkrypcji, jest zmienna.
Czwarty rodzaj — 5S rRNA jest kodowany przez gen, występujący w wielu
powtórzeniach w innej części genomu. Geny 5S rRNA są transkrybowane
przez polimerazę III RNA. Wymienione rodzaje rRNA uczestniczą w budo-
wie rybosomu, zatem 18S rRNA występuje w podjednostce małej (40S)
rybosomu, a trzy pozostałe rodzaje, tzn. 5S, 5.8S i 28S, zlokalizowane są
w podjednostce dużej (60S).
Dobrze poznanym rodzajem RNA jest transportujący RNA (tRNA), który
jest zaangażowany w dostarczanie aminokwasów do rybosomów. Tak jak
w przypadku innych cząsteczek RNA także i ten rodzaj zbudowany jest
z pojedynczego łańcucha polinukleotydowego. W nici takiej mogą jednak
występować fragmenty komplementarne, które po połączeniu wiązaniami
wodorowymi między komplementarnymi zasadami azotowymi tworzą struk-
turę przestrzenną takiej cząsteczki. Cząsteczka tRNA jest zbudowana z mniej
niż 100 nukleotydów, a jej struktura jest bardzo charakterystyczna i przypo-
mina liść koniczyny (ryć. 3-2). W strukturze tRNA wyróżnia się cztery pętle:
1) pętla I (licząc od końca 5') — uczestniczy w wiązaniu się z enzymem
syntetazą aminoacylową, która katalizuje związanie tRNA z.właściwym
aminokwasem;
2) pętla II — antykodonowa, zawiera układ trzech nukleotydów, które
są komplementarne do kodonu aminokwasu transportowanego przez tRNA;
3) pętla III — dodatkowa, o mało poznanej funkcji;
4) pętla IV — zaangażowana jest w wiązanie się kompleksu aminoacylo-
-tRNA z rybosomem.
Na końcu 3' cząsteczki tRNA występuje sekwencja CCA, w której do
rybozy nukleotydu adenylowego jest przyłączany aminokwas. Liczba
różnych cząsteczek tRNA występujących w komórce zawiera się w przedziale
od 40 do 60, co oznacza, że przekracza liczbę znanych aminokwasów.
51
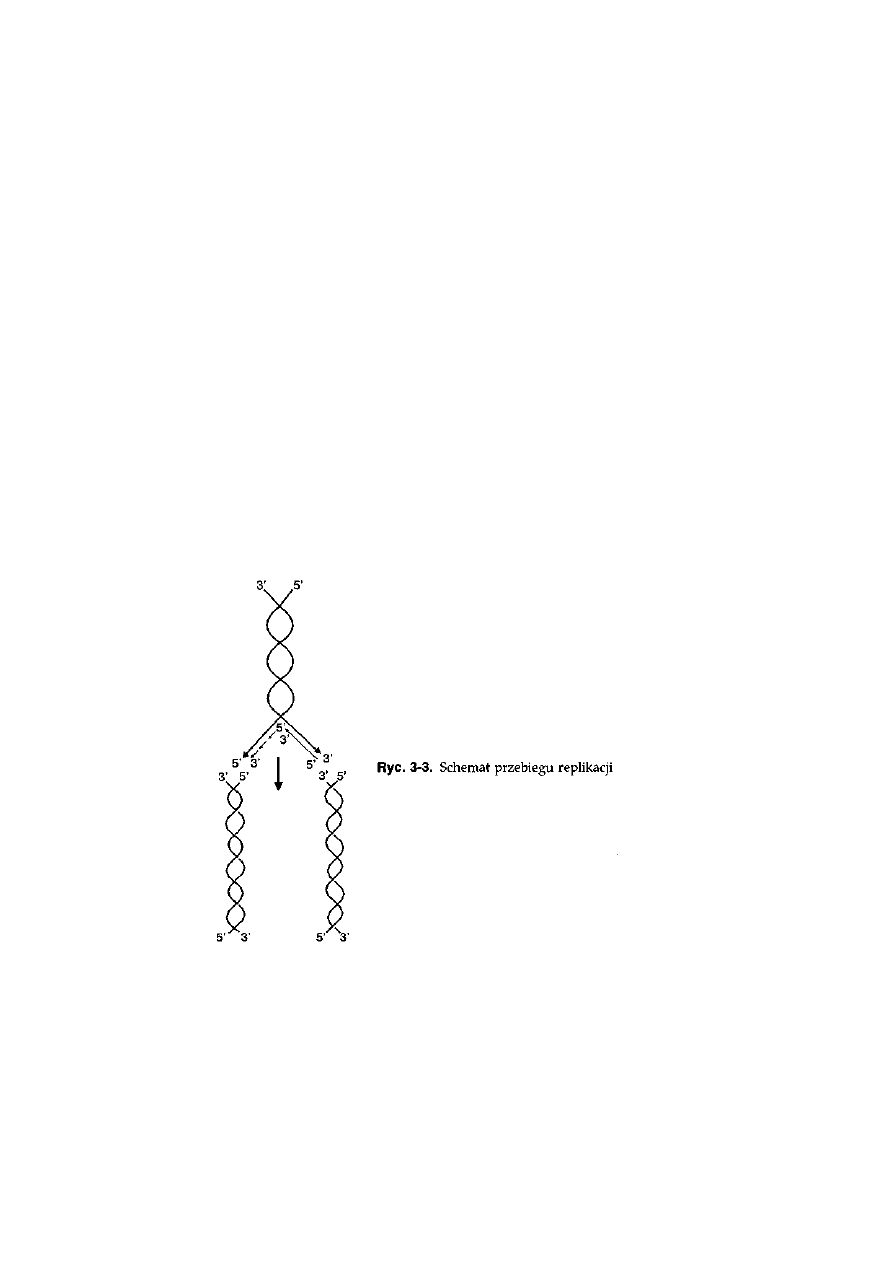
Informacyjny RNA (mRNA) powstaje podczas transkrypcji genów
ulegających ekspresji w komórce i z tego względu jest to najbardziej
zróżnicowana klasa kwasów rybonukleinowych. Struktura cząsteczek mRNA
jest opisana w części dotyczącej procesu transkrypcji.
Na uwagę zasługuje jeszcze jedna klasa RNA, określana terminem
niekodujący RNA (ncRNA — ang. noncoding RNA). Ostatnie badania
wykazują, że jest to bardzo ważna grupa RNA, która bierze udział w procesie
wycinania intronów z pre-mRNA, modyfikacji nukleotydów, a także regulacji
ekspresji genów.
3.2. Replikacja DNA
Proces samoodtwarzania się (replikacji) DNA przebiega w fazie S cyklu
życiowego komórki i jest niezbędnie potrzebny do przeprowadzenia
podziału jądra komórkowego, czyli mitozy, bądź mejozy. Każdy podział
mitotyczny musi być poprzedzony replikacją. Podobnie jest w przypadku
mejozy, z tym jednak że przed drugim podziałem mejotycznym nie
dochodzi do replikacji. Z powodu ogromnej długości cząsteczek DNA
zawartych w chromosomach replikacją rozpoczyna się równocześnie w wielu
miejscach, które określane są jako replikony. Replikon zawiera miejsce
rozpoczęcia i zakończenia replikacji. Na przykład, liczba replikonów w ge-
nomie ssaków szacowana jest na około 25 000, a średnia wielkość jednego
replikonu wynosi około 150 000 par nukleotydów.
52

Replikacja obejmuje dwa zasadnicze etapy: rozplecenie podwójnej helisy
DNA i dobudowanie komplementarnych łańcuchów polinukleotydowych.
W ten sposób powstają tzw. widełki replikacyjne (ryć. 3-3). Rozplecione
łańcuchy polinukleotydowe stanowią matryce, na których w sposób komple-
mentarny syntetyzowane są drugie łańcuchy. Wynika z tego, że po replikacji
dwie nowo powstałe cząsteczki DNA składają się z jednego starego łańcucha
i jednego nowego. Taki sposób replikacji nazywany jest semikonserwatyw-
nym (półzachowawczym). Przyłączanie kolejnych nukleotydów katalizowane
jest przez enzym polimerazę DNA, który odpowiada za powstanie wiązania
estrowego między atomem węgla 3' (wolna grupa 3'-OH) wydłużanego
łańcucha i nowym nukleotydem (trifosforanem nukleozydu). Oznacza to, że
kierunek budowania komplementarnego łańcucha jest ściśle określony
i postępuje od końca 5' w kierunku 3' (5'
→3'). W efekcie tylko na jednym
łańcuchu matrycowym replikującej cząsteczki DNA dobudowywanie nowego
łańcucha przebiega w sposób ciągły. Na drugim łańcuchu dobudowywane są
krótkie fragmenty (tzw. fragmenty Okazaki), które następnie są łączone przez
enzym ligazę w ciągłą nić. Nić syntetyzowana w sposób ciągły jest nazywana
prowadzącą, a nić powstająca w sposób nieciągły opóźnioną.
3.3. Kod genetyczny
Informacja genetyczna jest zapisana w DNA za pomocą kodu genetycznego,
mającego charakter trójkowy, tzn. trzy kolejne nukleotydy (kodon, tryplet)
w DNA zawierają informację o aminokwasie, który ma być wbudowany
w procesie biosyntezy białka. Ponieważ każdy kodon składa się z trzech
spośród czterech nukleotydów, to ogółem możliwe jest utworzenie 64 trójek
(tab. 3-1). Wśród nich jest 61 trójek sensownych oraz trzy kodony nonsen-
sowne, tzn. takie, które nie kodują żadnego aminokwasu, a ich wystąpienie
powoduje zatrzymanie procesu biosyntezy polipeptydu. Tryplet AUG, który
odpowiada za wbudowanie metioniny w łańcuchu polipeptydowym, roz-
poczyna część kodującą genu. Komplementarne ułożenie nukleotydów
w dwóch łańcuchach polinukleotydowych cząsteczki DNA sprawia, że
sekwencje nukleotydów w tych łańcuchach, w obrębie danego odcinka
DNA, są różne. Tym samym tylko jeden z tych łańcuchów zawiera informację
i nić ta nazywana jest sensowną. Drugi, komplementarny łańcuch polinu-
kleotydowy pełni funkcję matrycy (nić matrycowa) w procesie transkrypcji.
Kod genetyczny ma kilka cech o podstawowym znaczeniu dla jego
funkcjonowania. Po pierwsze, kod ma charakter trójkowy, o czym była już
mowa wyżej. Po drugie, jest on uniwersalny, tzn. w całym świecie
ożywionym funkcjonuje ten sam system kodowania informacji genetycznej.
Od tej zasady są tylko nieliczne wyjątki, które dotyczą niektórych kodonów
w mitochondrialnym DNA, np. kodon UGA w kodzie uniwersalnym jest
53
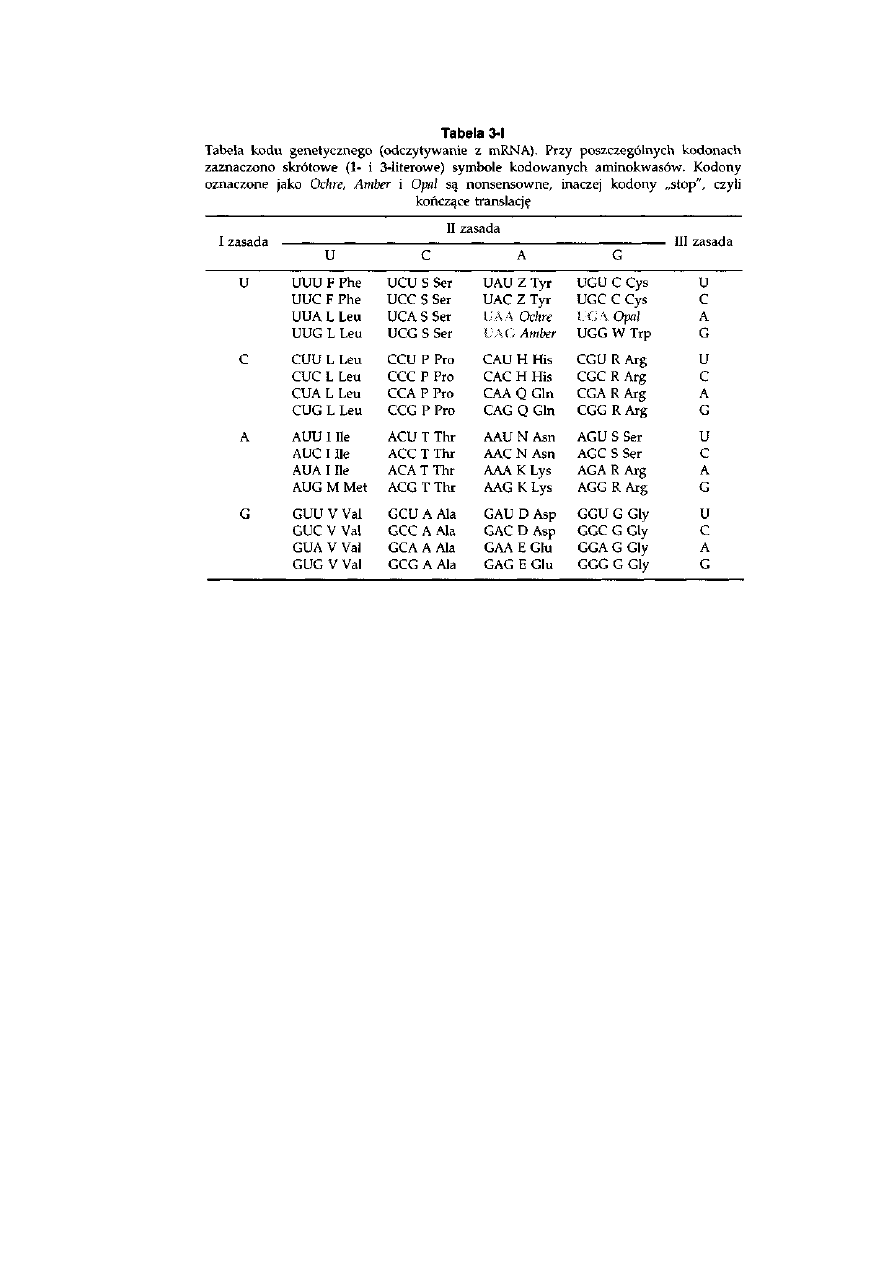
kodonem stop, a w mitochondrialnym DNA ssaków odpowiada za włączenie
tryptofanu do łańcucha polipeptydowego. Po trzecie, kod genetyczny jest
zdegenerowany, co wynika z faktu znacząco większej liczby kodonów niż
aminokwasów. W rezultacie prawie każdy aminokwas, z wyjątkiem metio-
niny i tryptofanu, jest kodowany przez więcej niż jedną trójkę (maksymalnie
przez sześć kodonów, w przypadku leucyny i argininy). Po czwarte, kod
genetyczny jest nie zachodzący na siebie i bezprzecinkowy, czyli kodony są
ułożone kolejno jeden za drugim bez nukleotydów je rozdzielających.
W efekcie podczas translacji sekwencja nukleotydów w mRNA jest od-
czytywana w sposób ciągły.
3.4. Transkrypcja
Pierwszym etapem procesu ekspresji genu jest transkrypcja, która polega na
syntezie informacyjnego kwasu rybonukleinowego (mRNA) na matrycowej
nici DNA. Transkrypcja rozpoczyna się od rozplecenia podwójnej helisy
DNA i przyłączenia polimerazy RNA do sekwencji promotora, która
poprzedza część strukturalną genu. Związanie się polimerazy RNA z pro-
54
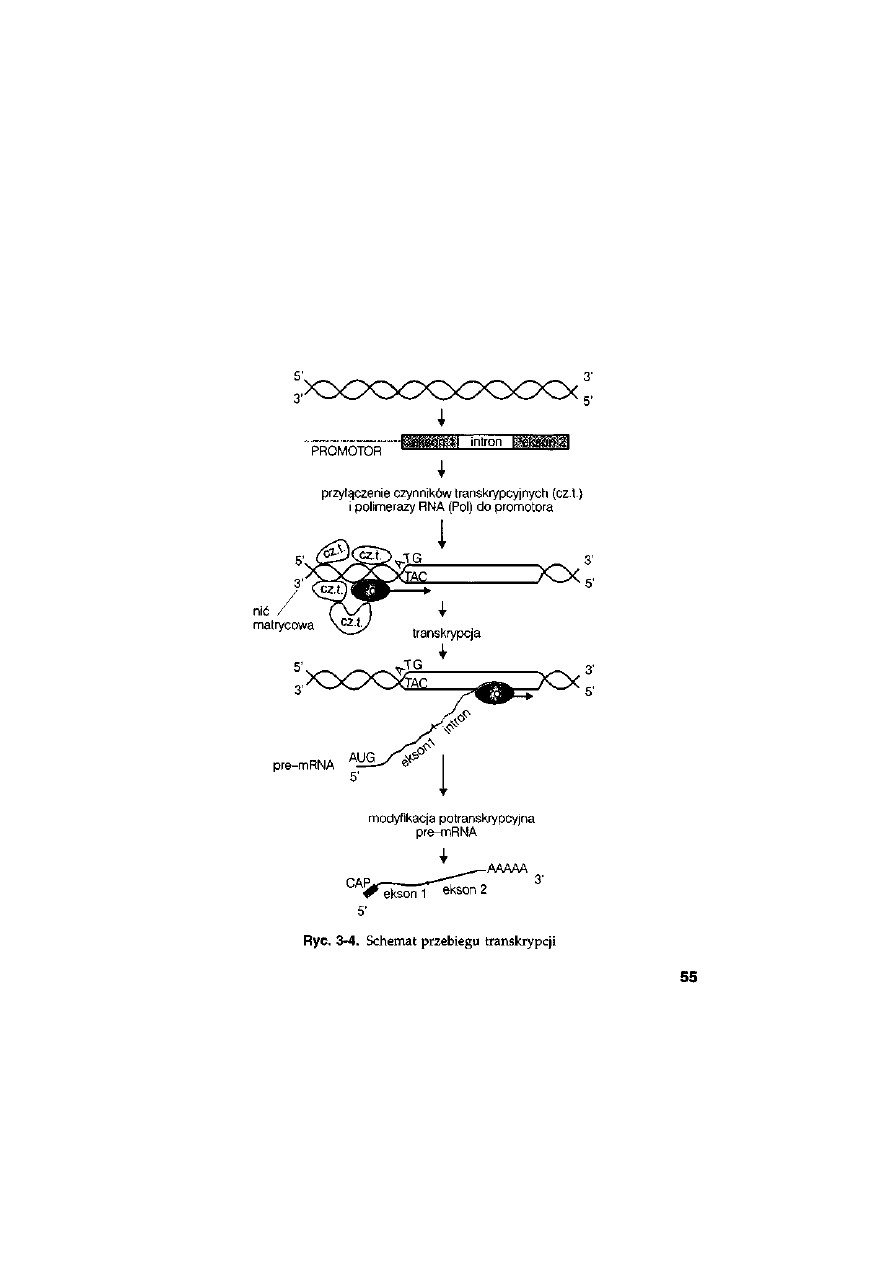
motorem jest uzależnione od równoczesnego przyłączenia się w tym miejscu
zespołu białek (czynniki transkrypcyjne), które kontrolują przebieg transkryp-
cji. Łańcuch mRNA jest syntetyzowany w kierunku od końca 5' do końca 3'
(5'
→3'), czyli kierunek tej reakcji jest taki sam jak przy replikacji. Z kolei
matrycowa nić DNA (komplementarna do nici sensownej) jest odczytywana
w kierunku 3'
→5'. Dzięki temu powstający mRNA ma sekwencję nukleoty-
dów zgodną z sekwencją nici sensownej DNA (ryć. 3-4). Ponieważ u organiz-
mów eukariotycznych geny zbudowane są w części strukturalnej z fragmen-
tów kodujących (eksony) i niekodujących (introny), to z pierwotnego
transkryptu FINA (pre-mRNA) muszą być usunięte introny. Proces polegający
na ich wycinaniu nazywany jest składaniem RNA (ang. splicing) i stanowi
istotną część tzw. obróbki potranskrypcyjnej pre-mRNA, która jest drugim
etapem ekspresji genu. Poza wycięciem intronów, na końcu 3' mRNA

dobudowana jest sekwencja poliA, a na końcu 5' dołączona zostaje 7-metylo-
guanozyna (tzw. czapeczka lub inaczej kapturek, ang. cap). Podczas transkryp-
cji powstają obok cząsteczek mRNA, pełniących funkcję matrycy, na której
syntetyzowany jest łańcuch polipeptydowy, także inne cząsteczki kwasów
rybonukleinowych, mających zasadniczą rolę w translacji (tRNA i rRNA).
3.5. Translacja
Po zakończeniu obróbki potranskrypcyjnej mRNA przechodzi do cytoplazmy i
tam łączy się z rybosomami, co rozpoczyna kolejny etap ekspresji genu,
jakim jest translacja. W procesie tym na matrycy mRNA budowany jest
łańcuch polipeptydowy (ryć. 3-5). Synteza łańcucha polipeptydowego
rozpoczyna się zawsze od aminokwasu metioniny, który jest kodowany
przez trójkę nukleotydów AUG znajdującą się na końcu 5' mRNA. Metionina
łączy się z tzw. inicjatorowym tRNA dla metioniny i taki kompleks jest
wiązany z rybosomem, a antykodon tego tRNA wiąże się komplementarnie
z pierwszym kodonem na końcu 5', jakim jest zawsze AUG. Następnie do
rybosomu dostarczany jest kolejny aminokwas, przez właściwy tRNA,
którego antykodon jest komplementarny do drugiego kodonu mRNA. Po
wprowadzeniu drugiego aminokwasu dochodzi do powstania wiązania
peptydowego między nim i metioniną w taki sposób, że nie związana grupa
karboksylowa pierwszego aminokwasu — metioniny łączy się z nie związaną
grupą aminową drugiego aminokwasu. Wynika z tego, że polipeptyd jest
wydłużany od końca NH
2
do końca COOH. Zsyntetyzowany dipeptyd
jest związany z tRNA dla drugiego aminokwasu, a rybosom przesuwa się
o jeden kodon w kierunku końca 3' uwalniając jednocześnie inicjatorowy
tRNA dla metioniny. Synteza łańcucha polipeptydowego postępuje w ten
sposób do momentu, kiedy rybosom obejmie jeden z trzech kodonów
nonsensownych, a więc kodonów stop (UAA, UAG lub UGA). Z chwilą
osiągnięcia kodonu stop następuje zakończenie translacji i uwolnienie
łańcucha polipetydowego. Uwolniony łańcuch polipeptydowy podlega
modyfikacjom potranslacyjnym, w wyniku których zostaje uformowana
ostateczna postać białka. Modyfikacje potranslacyjne obejmują zmiany
dotyczące wiązań chemicznych (np. rozerwanie wiązania peptydowego)
w obrębie polipeptydu, modyfikacje końca aminowego lub karboksylowego
polipeptydu oraz modyfikacje łańcuchów bocznych aminokwasów wcho-
dzących w skład polipeptydu. Ostatnia z wymienionych kategorii modyfi-
kacji potranslacyjnych jest najczęściej spotykana i polega na przyłączaniu
różnych grup chemicznych w wyniku np.: glikozylacji (przyłączanie reszt
cukrowych), fosforylacji (przyłączanie grup fosforanowych, głównie reszty
kwasu fosforowego), metylacji (przyłączanie grupy metylowej), acetylacji
(przyłączanie grupy reszty acetylowej) itp.
56
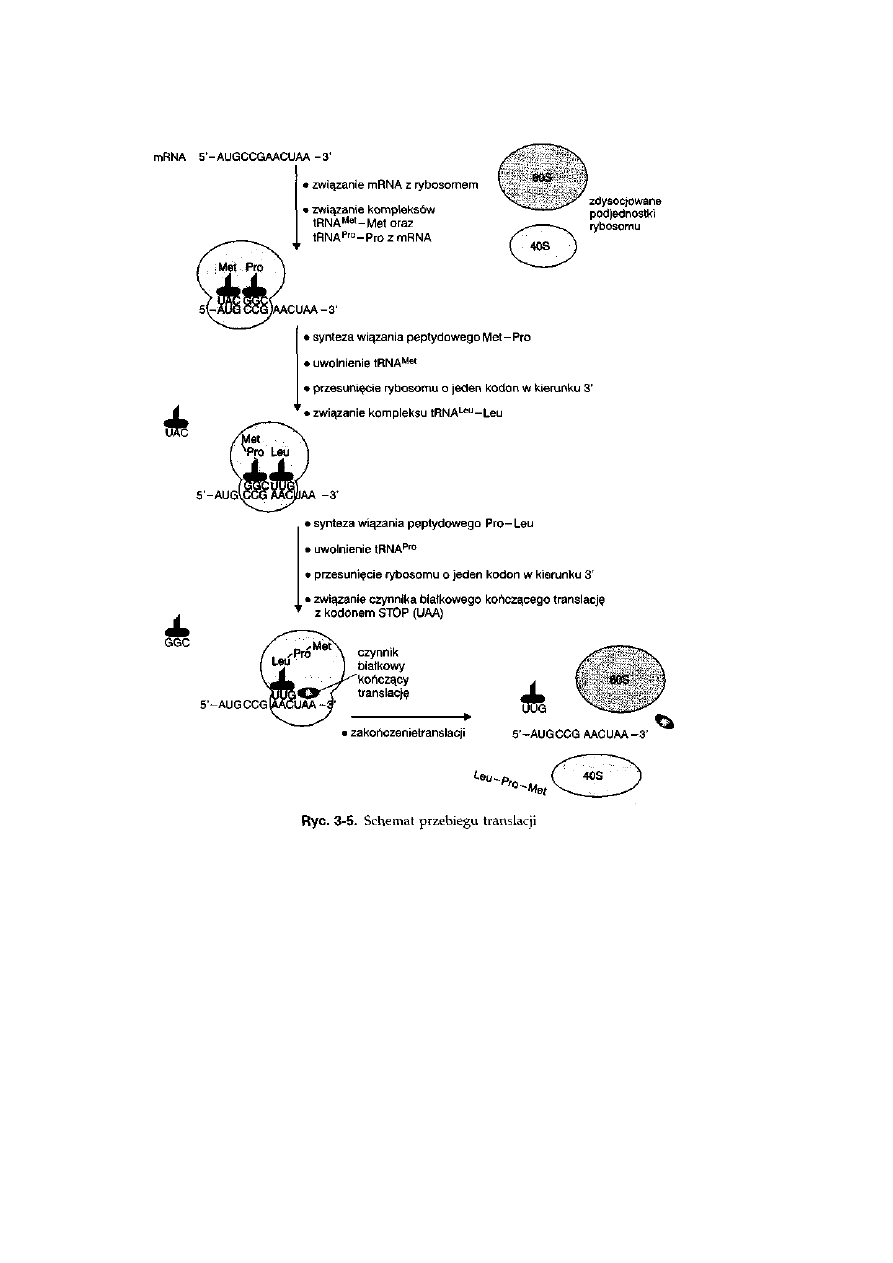
3.6. Budowa genu
Gen jest podstawową jednostką dziedziczności i zajmuje ściśle określone
miejsce (locus) w chromosomie lub też występuje poza jądrem komórkowym,
wewnątrz mitochondriów i chloroplastów (dotyczy roślin). Zmiany w obrębie
genu — mutacje genowe prowadzą do powstania form allelomorficznych,
57

czyli alleli. Zatem, cechą genu jest także podleganie mutacjom, które
mogą się ujawniać fenotypowo. Należy jednak zaznaczyć, że nie każda
mutacja przejawia się fenotypowo. Jeśli mutacja nie zmieni sensu zapisu
genetycznego, np. zmutowany kodon odpowiada za włączenie tego
samego aminokwasu co kodon niezmutowany (kod genetyczny jest
zdegenerowany) lub mutacja wystąpi w obrębie intronu (introny są
wycinane w trakcie obróbki potranskrypcyjnej pre-mRNA), to ujawnienie
fenotypowe nie nastąpi. Mutacja nie wywołująca zmiany w budowie
kodowanego produktu doprowadzi jedynie do powstania allelu, którego
obecność może być wykryta poprzez bezpośrednie badanie DNA. W takiej
sytuacji można mówić o polimorfizmie DNA danego genu. Gen jest
również podstawową jednostką uczestniczącą w rekombinacji genetycznej
podczas podziału mejotycznego. Pod względem molekularnym gen
jest regionem DNA, który podlega transkrypcji. W ramach genu ro-
zumianego jako jednostka transkrypcyjna należy wyróżnić część struk-
turalną genu, czyli ten fragment DNA, w którym zawarta jest informacja
genetyczna o produkcie (białko, rRNA, tRNA), oraz sekwencje regu-
latorowe, które odpowiadają za uruchomienie transkrypcji (ryć. 3-6).
Część strukturalna genu składa się z eksonów, czyli sekwencji, które
po transkrypcji uczestniczą w translacji, oraz intronów, które są wycinane
z pre-mRNA podczas składania RNA (ang. splicing) i nie uczestniczą
w translacji. Oprócz tego z obu stron części strukturalnej (na końcu
5' i 3') występują sekwencje oskrzydlające (ang. untranslated region —
UTR) części kodujące pierwszego i ostatniego eksonu. Sekwencje oskrzy-
dlające nie podlegają translacji. Zgodnie z wcześniej opisanym fenomenem
polarności cząsteczki DNA należy podkreślić, że początek genu znajduje
58

się na końcu 5', a koniec genu na końcu 3'. Pierwszy ekson (koniec 5')
rozpoczyna sekwencja ATG, która koduje metioninę (kodon AUG w mRNA)
— pierwszy aminokwas każdego polipeptydu. Z kolei na końcu 3' genu, po
sekwencji „stop" (kodony TAA, TAG lub TGA), pojawia się sekwencja
AATAAA, która koduje sygnał rozpoczęcia poliadenylacji pre-mRNA
podczas obróbki potranskrypcyjnej, a za nią znajduje się sekwencja (GT)n
odpowiedzialna prawdopodobnie za zakończenie transkrypcji.
Część regulatorowa genu obejmuje sekwencje promotora, wzmacniacza
(ang. enhancer) i wyciszacza (ang. silencer). Promotor bezpośrednio
poprzedza część strukturalną genu i pełni kluczową rolę w rozpoczęciu
transkrypcji. W obrębie promotora występują dwie sekwencje o szcze-
gólnym znaczeniu. W odległości około 30 pz od miejsca inicjacji trans-
krypcji występuje sekwencja TATA (ang. TATA-box), której obecność
jest niezbędna do przyłączenia polimerazy II RNA. Przyłączenie polimerazy
wymaga równoczesnego powiązania grupy białek (czynniki transkryp-
cyjne) z promotorem. Sekwencja CAAT znajduje się około 70-90 pz
przed miejscem rozpoczęcia transkrypcji. Przypuszcza się, że również
ta sekwencja bierze udział w procesie regulacji transkrypcji. Warto
wreszcie zwrócić uwagę, że większość genów (około 56%) zawiera
w obrębie promotora sekwencję składającą się z licznych powtórzeń
dinukleotydu CG, które są nazywane wysepkami CpG. Wysepki CpG
towarzyszą wszystkim genom ulegającym powszechnej ekspresji, tzn.
we wszystkich komórkach. Są to geny pełniące funkcję gospodarza
komórki (ang. housekeeping genes), czyli ich ekspresja jest konieczna
do funkcjonowania komórki.
Sekwencje regulujące typu wzmacniacza i wyciszacza mogą być położone
w znacznej odległości od genu, którego ekspresja znajduje się pod ich
kontrolą, dzięki oddziaływaniu na nie czynników transkrypcyjnych. Wzmac-
niacz jest odpowiedzialny za nasilenie transkrypcji genu, a wyciszacz za
spowolnienie tego procesu. Położenie tych sekwencji względem genu jest
bardzo zróżnicowane i może występować przed lub za genem, a nawet
w jego obrębie, w jednym z intronów.
Wielkość genu, a konkretnie jego części strukturalnej, mieści się w bardzo
szerokich granicach od kilkuset par nukleotydów (pz) do ponad dwóch
milionów pz. Przykładem bardzo małego genu jest gen SR
Υ
, który zawiera
tylko jeden ekson, składający się z 669 nukleotydów. Produktem tego genu
jest białko o długości 223 aminokwasów, odpowiedzialne za ukierunkowanie
rozwoju niezróżnicowanej gonady płodowej w jądro. Gigantycznym genem
jest ludzki gen DMD kodujący białko dystrofinę, które jest zbudowane
z 3685 aminokwasów. Gen ten jest zbudowany z około 2,5 min pz i w jego
skład wchodzi 79 eksonów i 78 intronów, ale tylko 0,6% długości tego genu
obejmuje część kodującą. Mutacje w tym genie odpowiedzialne są za rozwój
groźnej choroby genetycznej człowieka — dystrofii mięśniowej Duchenne'a,
która prowadzi do zaniku mięśni.
59

3.7. Regulacja ekspresji genu
W genomie ssaków występuje około 35 tysięcy genów. Wśród nich znaczną
grupę stanowią geny pełniące tzw. funkcje gospodarza komórki (ang.
housekeeping genes). Produkty kodowane przez te geny są zaangażowane
w podstawowe procesy życiowe komórki, takie jak: przemiany energetyczne,
replikacja DNA, budowa struktur komórkowych itp. Geny z tej grupy
podlegają ekspresji we wszystkich komórkach. Warto pamiętać, że w komór-
ce somatycznej każdy gen (oprócz genów położonych w chromosomach
płci u samców) reprezentowany jest przez dwa allele, które mogą wy-
stępować w układzie homo- lub heterozygotycznym, co ma oczywiście
związek z ekspresją genów.
Podczas rozwoju ontogenetycznego, obok genów ulegających powszech-
nej ekspresji, jest włączana i wyłączana ekspresja różnych genów, zależnie
od tego, w jakim kierunku różnicuje się dana komórka. Z drugiej strony,
w zróżnicowanej komórce ekspresja niektórych genów może być okresowo
włączana lub wyłączana. Jest to bardzo złożony proces, którego całościowe
poznanie wymaga jeszcze wielu lat badań. Niemniej znanych jest już wiele
mechanizmów, które są zaangażowane w kontrolę ekspresji genów, czyli
ich fenotypowe ujawnianie się w postaci odpowiedniej cechy. Ekspresja
genu obejmuje procesy: transkrypcji, potranskrypcyjnej modyfikacji mRNA,
translacji i potranslacyjnej modyfikacji białka. Ukształtowane w ten sposób
białko można zidentyfikować i analizować metodami laboratoryjnymi (np.
elektroforeza białek, badania serologiczne — grupy krwi, hybrydyzacja ze
znakowanym przeciwciałem itp.) lub też można obserwować właściwość
fenotypową organizmu będącą efektem obecności określonego białka (np.
umaszczenie, obecność rogów, podatność na stres itp.). W przypadku cech
uwarunkowanych przez dużą liczbę genów o nie poznanej jeszcze funkcji
(tzw. poligeny, wśród których geny o działaniu sumującym mają podsta-
wowe znaczenie) fenotyp opisuje się za pomocą estymatorów statystycznych.
Regulacja ekspresji genów zachodzi najczęściej na poziomie transkrypcji,
zatem poznanie mechanizmów odpowiedzialnych za podjęcie transkrypcji
przez dany gen ma znaczenie podstawowe. Należy jednak pamiętać, że
kontrola ekspresji może również polegać na potranskrypcyjnej modyfikacji
mRNA, regulacji stabilności mRNA i jego transporcie do cytoplazmy oraz
potranslacyjnej modyfikacji białek. Modyfikacja mRNA, oprócz zmian
opisanych w podrozdziale poświęconym transkrypcji, obejmuje także inne
procesy. Podczas wycinania intronów możliwe jest zróżnicowane składanie
ostatecznego transkryptu, w skład którego będą wchodziły różne kombinacje
eksonów. Mechanizm ten nazywa się alternatywnym składaniem mRNA.
Budowa mRNA może być modyfikowana również przez wybór miejsca
terminacji transkrypcji. Zakończenie transkrypcji związane jest z sekwencją
AAUAAA, która wskazuje, że w odległości około 10-30 nukleotydów od
tego miejsca powinna nastąpić poliadenylacja transkryptu, czyli dobudowa-
60

nie sekwencji poliA (tzw. ogonek poliA) na końcu 3' mRNA. Okazuje się, że
niektóre geny mogą mieć kilka miejsc poliadenyłacji i dlatego na tej samej
matrycy DNA mogą powstawać różne transkrypty. Przykładem takiej sytuacji
jest występowanie dwóch miejsc poliadenyłacji w genie łańcucha ciężkiego
immunoglobulin. Forma rozpuszczalna tego białka powstaje na matrycy
mRNA, który jest krótszy o dwa eksony w porównaniu z mRNA dla formy
zakotwiczonej w błonie plazmatycznej limfocytów B. Wreszcie mRNA
powstały podczas transkrypcji może ulegać, przed przejściem do cytoplazmy,
modyfikacjom polegającym na zmianie sekwencji nukleotydów. Proces ten
nazywa się redagowaniem mRNA. Efektem tego może być na przykład
pojawianie się w transkryptach, powstających w niektórych tkankach,
dodatkowego kodonu stop. Funkcjonują wówczas dwa rodzaje mRNA,
powstałe na bazie tego samego matrycowego DNA, odpowiedzialne za
syntezę polipeptydów o różnej długości. Taką formę modyfikowania mRNA
opisano m.in. w przypadku niektórych genów kodowanych przez mito-
chondrialny DNA (mtDNA). Intensywność procesu biosyntezy łańcuchów
polipeptydowych zależy od liczby kopii mRNA wędrujących z jądra do
cytoplazmy oraz od czasu trwania mRNA w cytoplazmie, czyli od jego
stabilności.
3.7.1. Czynniki transkrypcyjne
Czynnikami transkrypcyjnymi nazywane są białka, które wiążąc się z sek-
wencjami regulatorowymi genu umożliwiają przyłączenie polimerazy RNA
i rozpoczęcie transkrypcji. Białka te w swojej strukturze zawierają dwa
istotne obszary. Jeden odpowiada za wiązanie się czynnika transkrypcyjnego
z właściwą sekwencją nukleotydów w obrębie promotora lub wzmacniacza,
a drugi obszar zaangażowany jest w łączenie się z polimerazą RNA lub
innymi czynnikami transkrypcyjnymi. Czynniki transkrypcyjne, zależnie od
ich struktury trzeciorzędowej, są zaliczane do następujących grup: białka
zawierające tzw. palce cynkowe (ang. zinc finger), białka o strukturze typu
heliks-skręt-heliks (ang. helix-turn-helix) i białka o strukturze określanej
terminem zamek leucynowy (ang. leucine zipper). Wymienione struktury
wchodzą w kontakt z nicią DNA.
Ekspresja genów kodujących czynniki transkrypcyjne może zależeć od
innych czynników transkrypcyjnych lub pozostawać pod kontrolą stymula-
torów pozakomórkowych, takich jak hormony. Sposób oddziaływania
hormonów na ekspresję genów jest zróżnicowany (ryć. 3-7). W przypadku
hormonów steroidowych regulacja ekspresji genów przebiega z udziałem
receptorów wewnątrzkomórkowych. Hormon steroidowy po wniknięciu do
komórki łączy się z właściwym białkiem receptorowym. Kompleks hor-
mon-receptor wnika do jądra komórkowego i tam wiąże się z sekwencjami
regulatorowymi genu docelowego lub genu kodującego odpowiedni czynnik
61
Ryć. 3-7. Schemat regulacji ekspresji genów za pomocą hormonów. Oznaczenia:
HP — hormon peptydowy, HS — hormon steroidowy, R — receptor hormonu pep-
tydowego, RS — receptor hormonu steroidowego, T
1
T
2
— przekaźniki wewnątrzkomór-
kowe, P
l
P
2
— promotory genów, Gj, G
2
— części strukturalne genów
transkrypcyjny uczestniczący w regulacji ekspresji kolejnego genu. Receptor
hormonu steroidowego ma domenę wiążącą DNA oraz domenę wiążącą
hormon. Domena wiążąca DNA jest aktywna tylko wówczas, gdy do
receptora jest przyłączony hormon steroidowy. Hormony peptydowe (np.
hormon wzrostu, insulina itp.) nie wnikają do wnętrza komórki, a jedynie
wiążą się z receptorami błonowymi. Wiązanie takie wywołuje wiele reakcji.
W ich wyniku sygnał zostaje przeniesiony od zaktywowanego receptora
błonowego do jądra komórkowego za pomocą wewnątrzkomórkowych
przekaźników, których mechanizm działania jest jeszcze słabo rozpoznany,
lub też z udziałem przekaźników modyfikujących białka w cytoplazmie, co
można uznać za regulację ekspresji genów na etapie potranslacyjnym.
3.7.2. Geny homeotyczne
Ukształtowanie się wielokomórkowego organizmu z jednokomórkowej
zygoty zależy z jednej strony od intensywnych podziałów komórkowych,
a z drugiej strony od różnicowania się komórek. Poznanie genów od-
62

powiedzialnych za rozwój zarodkowy i płodowy ma podstawowe znaczenie
dla zrozumienia podłoża licznych wrodzonych wad rozwojowych. Postęp
w tym obszarze wiedzy osiągnięto dzięki badaniom prowadzonym na muszce
owocowej. Rozwój muszki owocowej przebiega pod kontrolą genów, które
odpowiadają za poszczególne etapy morfogenezy. Geny te funkcjonują
w układzie hierarchicznym, tzn. ich ekspresja następuje w ściśle określonej
kolejności. Najpierw uaktywniają się geny odpowiedzialne za orientację
zarodka na część przednią i tylną. Następnie kilka grup genów kieruje
ukształtowaniem się segmentów ciała wzdłuż osi podłużnej. Wreszcie za
różnicowanie się poszczególnych segmentów odpowiadają geny homeotyczne,
które w genomie muszki owocowej występują w dwóch kompleksach, ANT-C
i BX-C, położonych w jednym chromosomie. Pierwszy segment zawiera geny
odpowiedzialne za różnicowanie głowy i przedniej części tułowia, a drugi za
różnicowanie tylnych segmentów tułowia i segmentów odwłoka. W obrębie
kompleksu loci genów ułożone są w porządku włączania ich ekspresji.
Ekspresja kolejnych genów odpowiada za różnicowanie się kolejnych segmen-
tów ciała. Struktura tych genów wykazuje zaskakujące podobieństwo. Okazało
się, że mają one sekwencję o długości około 180 pz (tzw. homeoboks), która
koduje fragment białka (tzw. domena białkowa) o długości 60 aminokwasów.
Domena ta jest odpowiedzialna za utworzenie struktury charakterystycznej dla
czynników transkrypcyjnych typu heliks-skręt-heliks.
Geny o podobnej budowie i funkcji zidentyfikowano także u kręgowców
i nazwano je genami HOX. U ssaków geny te, w liczbie około 30, zgrupowane
są w czterech kompleksach: HOX1, HOX2, HOX3 i HOX4.
3.7.3. Metylacja DNA
Ważnym procesem wpływającym na ekspresję genów jest modyfikacja
struktury DNA, która polega przede wszystkim na przyłączaniu grupy
metylowej do cytozyny. Metylacja wywołuje zmianę powinowactwa DNA
do czynników transkrypcyjnych oraz reorganizację nici nukleosomowej,
czego skutkiem jest przejście DNA w stan nieaktywny. Oznacza to, że
metylacja DNA w sekwencjach regulatorowych może mieć istotne znaczenie
dla ekspresji genu. Prawie cały DNA zawiera zmetylowaną cytozynę,
a wyjątek stanowią dinukleotydy CpG (wysepki CpG) występujące w obrębie
promotorów w genach aktywnych transkrypcyjnie. Do promotorów mają-
cych niezmetylowaną cytozynę w wysepkach CpG mogą się przyłączyć
czynniki transkrypcyjne niezbędne do ekspresji genu. Trzeba jednak
podkreślić, że nie wszystkie geny mają w obrębie promotora sekwencje
CpG — szacuje się, że około 56% genów zawiera te sekwencje. Wynika
z tego, że brak metylacji wysepek CpG nie jest uniwersalnym sposobem
uaktywniania ekspresji genów. Obok wielu genów, których transkrypcja
jest hamowana poprzez metylację cytozyny w nukleotydach występujących
63

w części poprzedzającej gen, czyli ze strony końca 5', znane są liczne
geny, dla których nie obserwuje się zależności między transkrypcją i me-
tylacją DNA.
Metylacja cytozyny nie narusza komplementarności wiązania z guaniną,
czyli nie wpływa na prawidłowość replikacji wysepek CpG. Po replikacji
nowo wbudowane nukleotydy zawierające cytozynę podlegają metylacji,
wówczas gdy w matrycowej cząsteczce DNA naprzeciw guaniny występowa-
ła metylowana cytozyna. Stan metylacji DNA badany jest z użyciem dwóch
enzymów restrykcyjnych. Jeden z nich -— HpaII przecina sekwencję CCGG,
w której cytozyny nie są metylowane, a drugi — MspI przecina tę sekwencję
niezależnie od tego, czy cytozyna jest metylowana, czy niemetylowana.
3.7.4. Piętno gametyczne
W klasycznym podejściu do zagadnienia ekspresji genów przyjmuje się
równoważność alleli w parze, tzn. genotyp Aa jest równoważny genotypowi
aA, czyli nieistotne jest, od którego z rodziców pochodzi allel A, a od którego
allel a. Jednak w przypadku niektórych genów stwierdza się zróżnicowaną
ekspresję, zależną od tego, czy allel pochodzi od ojca, czy od matki. Zjawisko
to niekiedy dotyczy całych regionów chromosomowych, które mogą być
nieaktywne, gdy pochodzą np. od matki, lub aktywne, jeśli pochodzą od ojca.
Piętnowanie alleli czy całych regionów chromosomu związane jest prawdo-
podobnie z metylacją DNA i występuje podczas gametogenezy (ang. gametic
imprinting). Piętno utrzymuje się podczas rozwoju zarodkowego i płodowe-
go, a także w komórkach somatycznych organizmu dorosłego. Dopiero
podczas gametogenezy dotychczasowe piętno gametyczne zostaje zniesione
i ustala się wzór piętna, typowy dla danej płci (ryć. 3-8).
Dowód na to, że do rozwoju osobniczego niezbędny jest zarówno
genom ojca, jak i genom matki, a nie tylko diploidalna liczba chromosomów,
znaleziono przy okazji obserwacji rozwoju partenogenetycznych zarodków
myszy. Powstałe na drodze manipulacji zarodki ginogenetyczne, tzn.
zawierające dwa genomy żeńskie, rozwijały się dość dobrze, z tym jednak
że struktury wywodzące się z trofoblastu, czyli uczestniczące w formowaniu
łożyska, były zdecydowanie niedorozwinięte. Natomiast zarodki androge-
netyczne, tzn. mające dwa genomy męskie, dobrze rozwijały struktury
trofoblastu, niedorozwinięty zaś był sam zarodek. W obu przypadkach
dochodziło do obumarcia zarodka. Jedynie zarodki zawierające zarówno
genom żeński, jak i męski rozwijały się prawidłowo.
Piętno gametyczne prowadzi do zróżnicowanego fenotypowo ujawnienia
się niektórych mutacji chromosomowych, w które zaangażowane są frag-
menty podlegające piętnowaniu. Przykładem niech będzie delecja fragmentu
przycentromerowego ramienia długiego chromosomu 15 (delecja 15ql-q3)
u ludzi. Jeśli pacjent odziedziczył tak uszkodzony chromosom od ojca, to
64
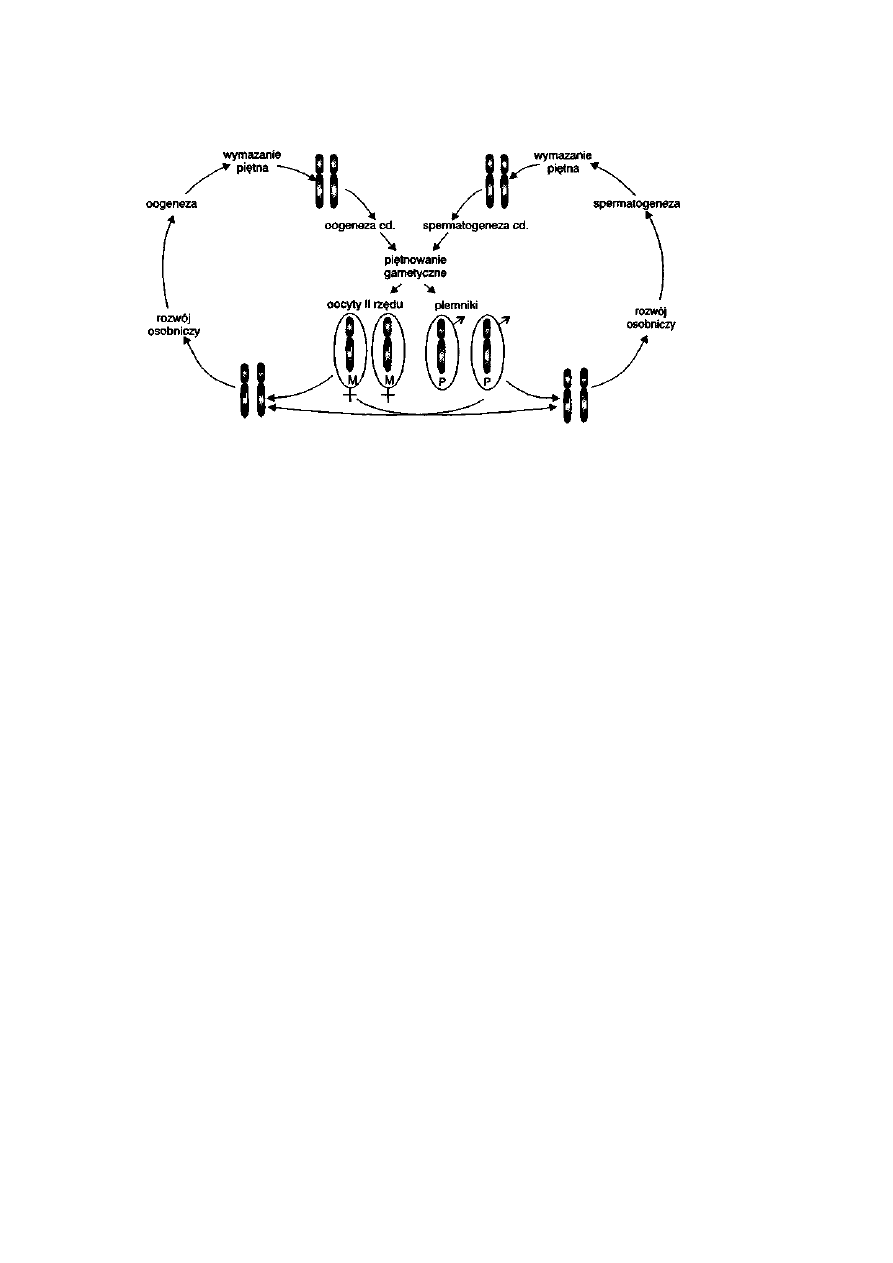
M
r
M P
ZYGOTA ZYGOT-A
2ENSKA
MĘSKA
Ryć. 3-8. Piętnowanie gametyczne. Fragment podlegający piętnowaniu w spermatoge-
nezie jest zaciemniony, a fragment piętnowany w oogenezie jest zaznaczony kolorem
czerwonym. P — chromosom pochodzący od ojca, M — chromosom pochodzący od matki
wówczas rozwija się zespół zmian fenotypowych (m.in. otyłość i niedorozwój
umysłowy), który nosi nazwę zespołu Pradera-Willego. Natomiast odziedzi-
czenie takiej samej mutacji od matki wywołuje zespół zmian (m.in. głębokie
upośledzenie umysłowe, brak mowy, drgawki) określanych jako zespół
Angelmana. Przyjmuje się, że fragment chromosomu 15 podlega piętnowaniu
gametycznemu. W zespole Pradera-Willego stan jest następujący: brakuje
fragmentu w chromosomie odziedziczonym od ojca, a odpowiedni homo-
logiczny fragment w chromosomie pochodzącym od matki nie podlega
ekspresji. W efekcie pacjent nie ma żadnej aktywnej kopii genów wy-
stępujących w części chromosomu objętego delecją. Podobna sytuacja
powstaje, gdy paq"ent ma dwie kopie tego fragmentu chromosomowego
pochodzące od matki (matczyna disomia). W zespole Angelmana brakuje
fragmentu chromosomu 15 pochodzenia matczynego, a ekspresja od-
powiadającego fragmentu chromosomu ojcowskiego jest wyłączona lub
pacjent ma dwie ojcowskie kopie tego fragmentu chromosomowego (disomia
ojcowska). Wiadomo, że w zespole Pradera-Willego występuje utrata
ekspresji wielu genów pochodzenia ojcowskiego, podczas gdy w zespole
Angelmana dotyczy to właściwie tylko jednego genu (UBE3A — ang.
ubiquitin protein ligase E3) pochodzenia matczynego. Wynika z tego, że
w krytycznym regionie 15ql-15q3 inny fragment jest piętnowany w game-
togenezie męskiej, a inny w gametogenezie żeńskiej. Tak więc, tylko jeden
gen — UBE3A nie podlega ekspresji, jeśli pochodzi od ojca, natomiast kilka
genów (m.in. gen SNRPN — ang. smali nuclear ribonucleoprotein polypep-
65

tide N), nie ulega ekspresji, jeśli są położone w chromosomie pochodzącym
od matki.
W zygocie oraz we wszystkich potomnych pokoleniach komórek soma-
tycznych stan napiętnowania genu (-ów) jest utrzymywany. Może się
jednak zdarzyć, że gen napiętnowany podczas gametogenezy będzie
powtórnie piętnowany podczas rozwoju osobniczego. Nałożenie się obu
rodzajów piętnowania oraz obecność tkankowe specyficznego białka (czyn-
nik transkrypcyjny) regulującego ekspresję genu prowadzi do ostatecznego
ukształtowania się wzoru ekspresji genu w kolejnych stadiach rozwojowych
i odpowiednich tkankach. Przykładem może być gen Igf2 (ang. insulin
growth factor type 2) u myszy, który podlega piętnowaniu podczas
gametogenezy żeńskiej. W genie tym występują dwa regiony, które
podlegają metylacji. W stadium przedimplantacyjnym zarodka oba allele
(ojcowski i matczyny) podlegają ekspresji, a w zarodkach starszych oraz
komórkach osobnika dorosłego występuje monoalleliczna ekspresja genu
matczynego. Sytuacja ta jest spowodowana tym, że piętnowanie w gameto-
genezie żeńskiej prowadzi do metylacji miejsca, do którego nie może się
przyłączyć białko (represor), wywołujące zahamowanie transkrypcji. W efek-
cie oba allele są transkrybowane. Podczas wtórnego piętnowania dochodzi
do metylacji fragmentu promotora genu ojcowskiego, co oczywiście unie-
możliwia przyłączenie czynników transkrypcyjnych, niezbędnych do pod-
jęcia transkrypcji. Powoduje to wstrzymanie ekspresji allelu ojcowskiego.
Szczegółowe badania przeprowadzone u myszy ujawniły obecność wielu
regionów chromosomowych, które są objęte zjawiskiem piętnowania game-
tycznego. Na przykład, duże fragmenty chromosomu 7, a także 11 i 12
podlegają piętnowaniu. Obecnie znane są 34 geny, które podlegają pięt-
nowaniu gametycznemu u myszy.
3.7.5. Inaktywacja chromosomu X w zarodkach żeńskich
Szczególnym przykładem trwałej inaktywacji dużej części chromatyny jest
fenomen związany z inaktywacją jednego z chromosomów X w zarodkach
żeńskich. W komórkach męskich występuje tylko jeden chromosom X, obok
chromosomu Y. Stan taki określany jest terminem hemizygotyczności.
W komórkach żeńskich występują dwa chromosomy X, ale podobnie jak
w komórkach męskich tylko jeden chromosom X wykazuje aktywność
transkrypcyjną. Okazało się, że inaktywacją chromosomu X jest formą
piętnowania, które zachodzi podczas wczesnego rozwoju zarodkowego.
W momencie przejęcia kontroli nad rozwojem zarodka przez jego własny
genotyp oba chromosomy X są aktywne w zarodkach żeńskich (układ XX).
W momencie przechodzenia ze stadium moruli do stadium blastocysty
następuje losowa inaktywacją jednego z chromosomów X, czyli aktywny
pozostaje albo chromosom pochodzenia ojcowskiego, albo matczynego.
66
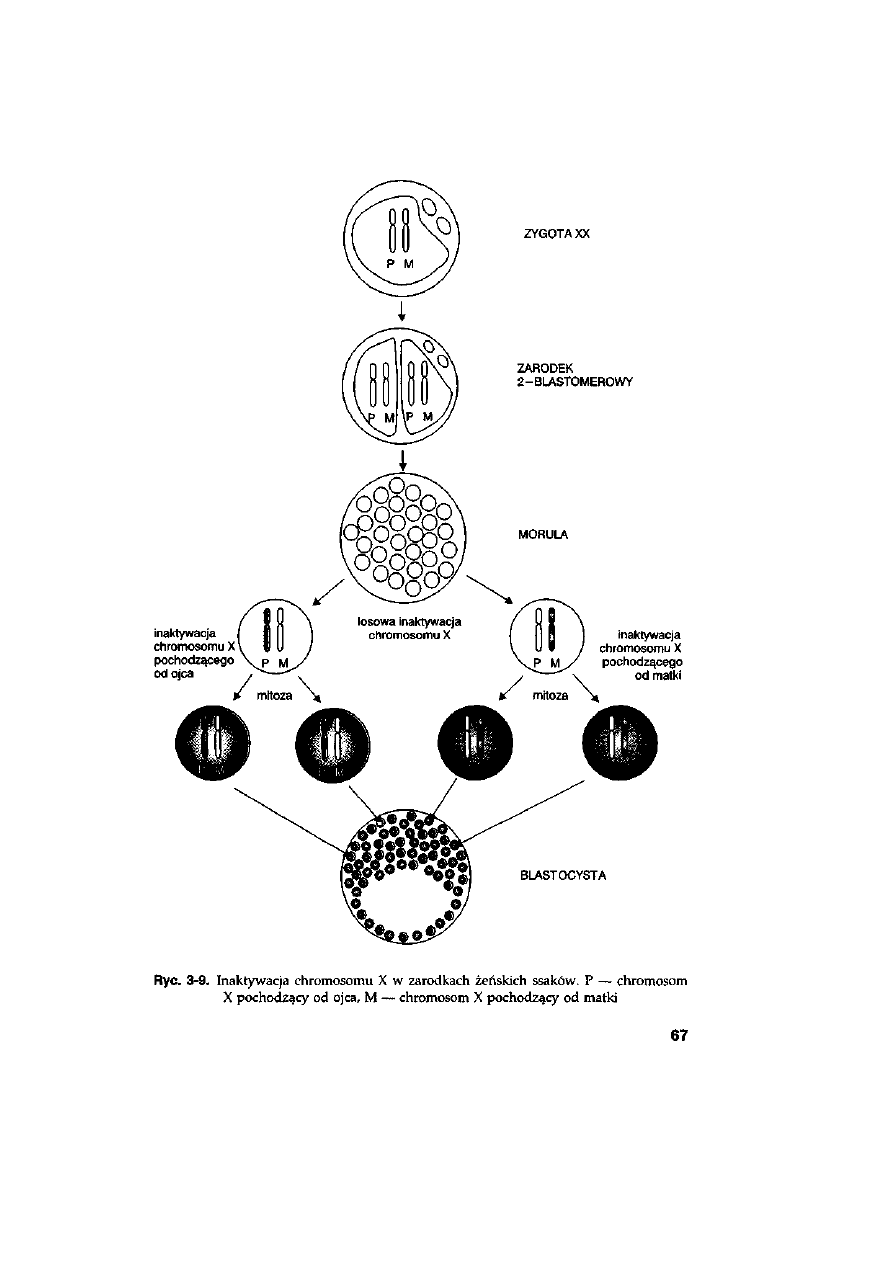

Wzór inaktywacji jest następnie utrzymany we wszystkich komórkach
potomnych. W konsekwencji organizm żeński jest swoistą mozaiką, w której
część komórek ma aktywny chromosom pochodzenia ojcowskiego, a część
pochodzenia matczynego (ryć. 3-9). Inaktywowany chromosom X podlega
kondensacji (heterochromatynizacji) w jądrze interfazowym, a jego obecność
ujawnia się w postaci tzw. ciałka Barra. W cyklu życiowym komórki
chromosom ten podlega typowym przemianom związanym z okresową
kondensacją (przed i w trakcie podziału jądra komórkowego) i niepełną
dekondensacją (podczas interfazy). Rozróżnienie aktywnego i nieaktywnego
chromosomu X w metafazie mitotycznej jest możliwe po zastosowaniu
metody barwienia prążkami R, które odzwierciedlają tempo replikacji
poszczególnych regionów chromosomowych. Barwienie to pokazuje, że
nieaktywny chromosom X jest replikowany w późnym okresie fazy S.
Proces inaktywacji rozpoczyna się od miejsca określanego jako centrum
inaktywacji chromosomu X. Wiadomo, że w ludzkim chromosomie X cent-
rum to występuje w ramieniu długim w obrębie prążka Xql3. W centrum
inaktywacji zidentyfikowano locus Xist (ang. X inactive specific transcript),
który pełni główną rolę w uruchomieniu inaktywacji. Początkowo ekspresji
podlega tylko gen Xist położony w chromosomie ojcowskim i to odbywa się
jeszcze przed rozpoczęciem procesu inaktywacji chromosomu X. Później,
ekspresja genu Xist pojawia się losowo, albo w chromosomie ojcowskim,
albo matczynym. W ślad za ekspresją genu Xist w jednym z chromosomów
X przebiega stopniowo inaktywacja kolejnych regionów tego chromosomu.
Inaktywacja jest związana z metylacją DNA danego chromosomu, który to
stan jest utrzymywany w komórkach potomnych. Warto zaznaczyć, że
produktem locus Xist jest niekodujący RNA (ncRNA). Spełnia on funkcję
regulacyjną, w zakresie zmiany struktury chromatyny inaktywowanego
chromosomu X.

Rozdział
4
4.1. Endonukleazy restrykcyjne
Analiza materiału genetycznego może być przeprowadzana bezpośrednio
w komórkach (tzw. metody in situ) bądź w wyizolowanym DNA lub RNA.
Większość metod wymaga fragmentacji (pocięcia; strawienia) DNA, co
umożliwia np. wyizolowanie określonego odcinka DNA lub podzielenie
cząsteczki DNA danego organizmu na krótsze fragmenty do dalszych
szczegółowych badań. W celu przecięcia cząsteczki DNA w ściśle określonym
miejscu stosowane są izolowane z bakterii endonukleazy restrykcyjne,
nazywane inaczej enzymami restrykcyjnymi bądź restryktazami.
Endonukleazy restrykcyjne służą bakteriom do obrony przed inwazją
wirusów, niszcząc niepożądane, obce cząsteczki DNA. W celu ochrony
własnego DNA przed działaniem restryktaz, bakterie wykształciły system
restrykcji-modyfikacji. System ten polega na tym, że bakterie wytwarzają
dwa rodzaje enzymów. Jeden to endonukleazy, rozpoznające i przecinające
(inaczej trawiące) określone sekwencje nukleotydów, drugi to metylotrans-
ferazy (zwane też metylazami), które rozpoznają te same sekwencje
nukleotydów i modyfikują (metylują) je tak, by nie zostały przecięte przez
endonukleazy. Ponieważ obcy, np. wirusowy, DNA nie jest odpowiednio
oznaczony (zmetylowany), niszczony jest przez endonukleazy bakterii.
Obecnie znanych jest już ponad 1000 restryktaz (w handlu dostępnych jest
ponad 200) i tyleż samo metylaz.
Nazewnictwo endonukleaz tworzone jest w ten sposób, że pierwsza
litera stanowi skrót nazwy rodzaju bakterii, druga i trzecia — gatunku,
następnie może być również stosowana litera określająca nazwę szczepu lub
serotypu bakterii, ponadto podawana jest cyfra rzymska oznaczająca numer
kolejny enzymu wyizolowanego z danego gatunku bakterii. Na przykład,
enzym HindIII pochodzi z bakterii Haemophilus influenzae, serotyp d i jest to
trzeci (III) z kolei enzym uzyskany od tego gatunku i serotypu bakterii.
Endonukleazy dzieli się na trzy klasy, w zależności od ich mechanizmu
działania, substratów i produktów, a także kofaktorów. Do klasy I enzymów
69
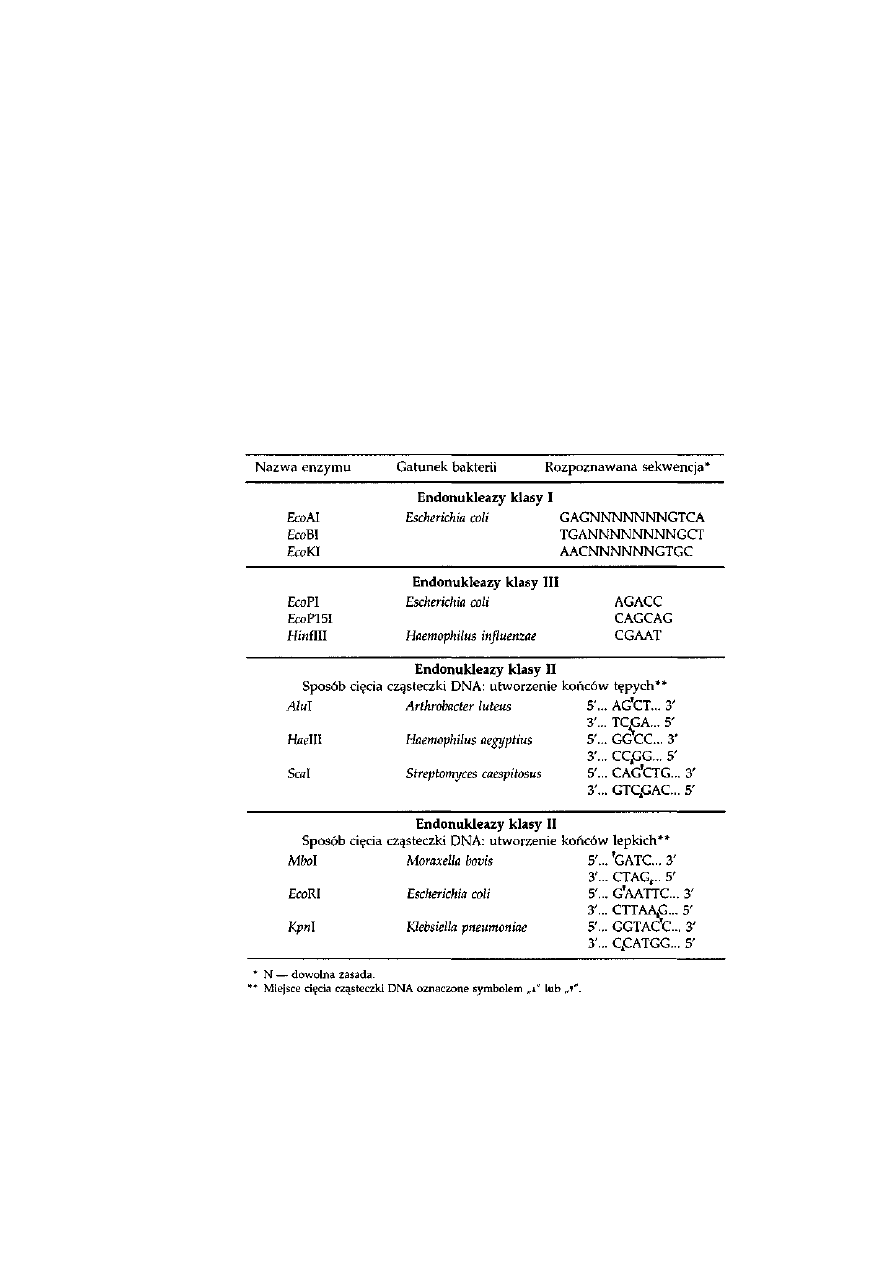
zaliczane są te restryktazy, które wykazują aktywność nukleazy, a ich
działanie zależy od obecności takich kofaktorów, jak ATP, jony magnezu
(Mg
2+
) i S-adenozylometionina. Cechą tych enzymów, utrudniającą ich
wykorzystywanie w analizie DNA, jest to, iż wprawdzie rozpoznają
specyficzne sekwencje nukleotydów, ale przecinają cząsteczkę DNA nie-
specyficznie w pewnej odległości (nawet do 1000 nukleotydów) od rozpo-
znanej sekwencji. Klasę III stanowią restryktazy mające podobne właściwości
do restryktaz klasy I, czyli wymagają obecności ATP i Mg
2+
(obecność S-
adenozylometioniny nie jest niezbędna, ale wzmacnia ich aktywność).
Różnica między tymi dwiema klasami polega na tym, że odległość sekwencji
rozpoznanej od sekwencji przecinanej przez enzym klasy III jest dla danego
enzymu stała i wynosi średnio 25 nukleotydów. Przykłady endonukleaz
klasy I i III zawarte są w tabeli 4-1.
Tabela 4-1
Przykłady enzymów restrykcyjnych
70

Najbardziej przydatne w analizie materiału genetycznego są enzymy
restrykcyjne zaliczane do klasy II (przykłady w tab. 4-1). Enzymy te
rozpoznają najczęściej sekwencje DNA złożone z 4 lub 6, rzadziej 8 nukleo-
tydów, charakteryzujące się dwustronną symetrią (tzw. sekwencje palin-
dromowe). Niektóre enzymy przecinają cząsteczkę DNA w miejscach
znajdujących się naprzeciw siebie, dając fragmenty mające tzw. tępe końce.
Jednak większość endonukleaz przecina cząsteczki DNA w dwóch różnych
miejscach, czego konsekwencją jest powstawanie fragmentów DNA o tzw.
lepkich końcach. Końce te ułatwiają łączenie fragmentów DNA ze sobą
w procesie rekombinacji molekularnej.
Fragmenty o tępych i lepkich końcach wyglądają następująco:
Enzym Thal (pochodzący z bakterii Thermoplasma acidophilum) rozpoznaje
i tnie sekwencję:
Niektóre enzymy, mimo że pochodzą z różnych szczepów bakterii,
rozpoznają te same sekwencje DNA. Są to izoschizomery. Jednak miejsca,
w których enzymy przecinają te sekwencje, mogą być różne.
Izoschizomery Smal (Serratia marcescens) i Xmal (Kanthomonas mafaacearum)
przecinają rozpoznaną sekwencję w różnych miejscach:

od rozpoznawanej sekwencji. Na przykład enzym MboII (Moraxella bovis)
tnie cząsteczkę DNA następująco:
5'...GAAGA(N)
8
'...3'
3'...CTTCT(N)
7
...5'
podobnie enzym FokI (Flavobacterium okeanokoites):
5'...GGATG(N)
9
'...3'
3'...CCTAC(N)
13
...5'
Ponadto większość endonukleaz klasy II przecina dwuniciowe cząsteczki
DNA, a tylko nieliczne enzymy (np. HinPI) tną jednoniciowy DNA.
Warunki optymalne dla aktywności poszczególnych enzymów restryk-
cyjnych różnią się przede wszystkim temperaturą, w której mieszanina
DNA z enzymem jest inkubowana, oraz składem buforu. Niektóre enzymy
potrzebują takich samych buforów. Dokładne informacje o tych warunkach
dostarczane są wraz z enzymami przez firmy biotechnologiczne.
Wykrycie bakteryjnych enzymów restrykcyjnych miało ogromne znacze-
nie dla rozwoju technik molekularnych. Są one podstawowym narzędziem,
wykorzystywanym m.in. do sporządzania map genomowych, sekwencjo-
nowania DNA, izolacji i identyfikacji genów, klonowania molekularnego
i rekombinacji genów czy fragmentów genomu, diagnostyki chorób genetycz-
nych (wykrywanie mutacji genowych) lub w analizie polimorfizmu długości
fragmentów restrykcyjnych DNA (RFLP).
4.2. Wektory
Wektory są podstawowym narzędziem klonowania DNA. Wprowadzenie
(transformacja) fragmentu DNA do komórki biorcy następuje za pośrednic-
twem wektora, do którego fragment ten został uprzednio włączony (rekom-
binacja molekularna). W zarysie klonowanie polega na namnożeniu frag-
mentu DNA w komórkach mikroorganizmów (głównie bakterii). Wektor
(np. plazmidowy, fagowy, drożdżowy) jest zdolny do autonomicznej
replikacji.
Wektory charakteryzują się następującymi cechami:
• są to niewielkie cząsteczki DNA, opisane pod względem fizycznym
i genetycznym, mające zdolność do samodzielnej replikacji w cytoplazmie
gospodarza,
• mają miejsca rozpoznawane przez różne endonukleazy restrykcyjne
(uzyskanie lepkich końców),
• miejsce rozpoznania przez enzym restrykcyjny nie powinno się
znajdować w takim rejonie cząsteczki, którego uszkodzenie powodowałoby
zaburzenie możliwości jej replikacji,
• powinny mieć jeden lub kilka silnych promotorów tak, aby wstawiając
72

w ich sąsiedztwo fragment obcego DNA można było uzyskać efektywną
ekspresję wprowadzonych genów,
• zawierają znaczniki (markery), za pomocą których można je ziden
tyfikować, np. geny oporności na antybiotyki,
• nie są zdolne do przeżycia poza komórką gospodarza,
• ze względów etycznych wektor nie powinien mieć genów, korę mogą
stanowić zagrożenie ekologiczne.
RODZAJE WEKTORÓW
Wektorami najczęściej używanymi do klonowania DNA w komórkach
prokariotycznych są bakteriofagi (fagi), plazmidy i kosmidy.
Wektory plazmidowe
Plazmidami są występujące u wielu gatunków bakterii kowalentnie zamk-
nięte koliste cząsteczki DNA. Plazmidy stosowane w technice klonowania
jako wektory są specjalnie konstruowane z fragmentów naturalnych plaz-
midów bakteryjnych. Wektor plazmidowy musi zawierać region inicjacji
replikacji DNA — replikon oznaczony symbolem ori, gen oporności na
antybiotyk, umożliwiający bakteriom transformantom rozwój na pożywce
zawierającej antybiotyk. Większość wektorów plazmidowych ma wstawiony
syntetyczny oligonukleotyd, „polilinker" (miejsce wielokrotnego klonowania),
którego sekwencje są rozpoznawane przez wiele enzymów restrykcyjnych.
Dzięki temu w wektor taki można wstawiać odcinki klonowanego DNA
uzyskane przez trawienie odpowiednimi enzymami. Do niektórych wek-
torów wprowadzono sekwencję umożliwiającą łatwą identyfikację zrekom-
binowanych bakterii. Najczęściej jest to gen kodujący enzym, rozkładający
barwny substrat, np. gen
β-galaktozydazy oznaczony symbolem lacZ. Metoda
selekcji rekombinantów zostanie omówiona w podrozdziale: Rekombinacja
molekularna i klonowanie. Przykładowy wektor plazmidowy pUC19, który
ma długość 2686 par zasad, przedstawiony jest na rycinie 4-1.
Kosmidy
Kosmid jest plazmidem zmodyfikowanym przez dodanie do niego sekwencji
cos z faga
λ. Dzięki temu kosmidy łączą zalety wektorów plazmidowych i
fagowych. Sekwencja cos umożliwia upakowanie DNA faga w otoczkę
białkową. Kosmidy służą do klonowania większych cząsteczek DNA, długości
10-40 kpz, i dlatego są wykorzystywane w badaniach wielu genów
eukariotycznych oraz do tworzenia bibliotek genomowych.
Wektory fagowe
Spośród wektorów fagowych największe znaczenie ma fag
λ. Za pomocą
modyfikacji molekularnych (np. eliminacja miejsc rozpoznawanych przez
enzymy restrykcyjne) otrzymano wiele pochodnych tego faga. Podobnie jak
73
Ryć. 4-1. Schemat wektora plazmidowego pUC19. Zaznaczono miejsca restrykcyjne dla
niektórych endonukleaz
w przypadku wektorów plazmidowych, do wektorów fagowych wprowa-
dzono polilinkery, geny markerowe i silne promotory. Wektory fagowe są
stosowane do klonowania fragmentów DNA mniejszych niż w wektorach
plazmidowych, ale efektywność tego działania jest większa.
Sztuczny chromosom bakteryjny (BAC)
Do klonowania długich fragmentów DNA (100-300 kpz) jest wykorzysty-
wany sztuczny chromosom bakteryjny BAĆ (ang. bacterial artificial chromo-
some) skonstruowany na bazie plazmidowego czynnika F bakterii E. coli.
Do klonowania fragmentów DNA w organizmach eukariotycznych (np.
drożdżach, komórkach ssaków) stosowane są:
Wektory — pochodne wirusów
Pochodne niektórych wirusów są wektorami stosowanymi do wprowadzania
DNA do komórek roślinnych i zwierzęcych. W przypadku komórek
zwierzęcych są to pochodne wirusów SV40, Papilloma, krowianki, bakulowi-
rusów, adenowirusów i retrowirusów. Najwcześniej był stosowany wirus
SV40, którego zaletą jest duża wydajność uzyskiwania zrekombinowanego
genu, wadą natomiast organiczona długość wbudowanego obcego DNA.
Obecnie coraz częściej stosuje się retrowirusy zapewniające dużą efektyw-
ność transfekcji (transfekcja jest to wprowadzanie fragmentów DNA do
komórek eukariotycznych). Są one również stosowane w somatycznej
terapii różnych schorzeń.
Wektory drożdżowe
Wektory te wykazują zdolność replikacji tak w komórkach drożdży, jak
74
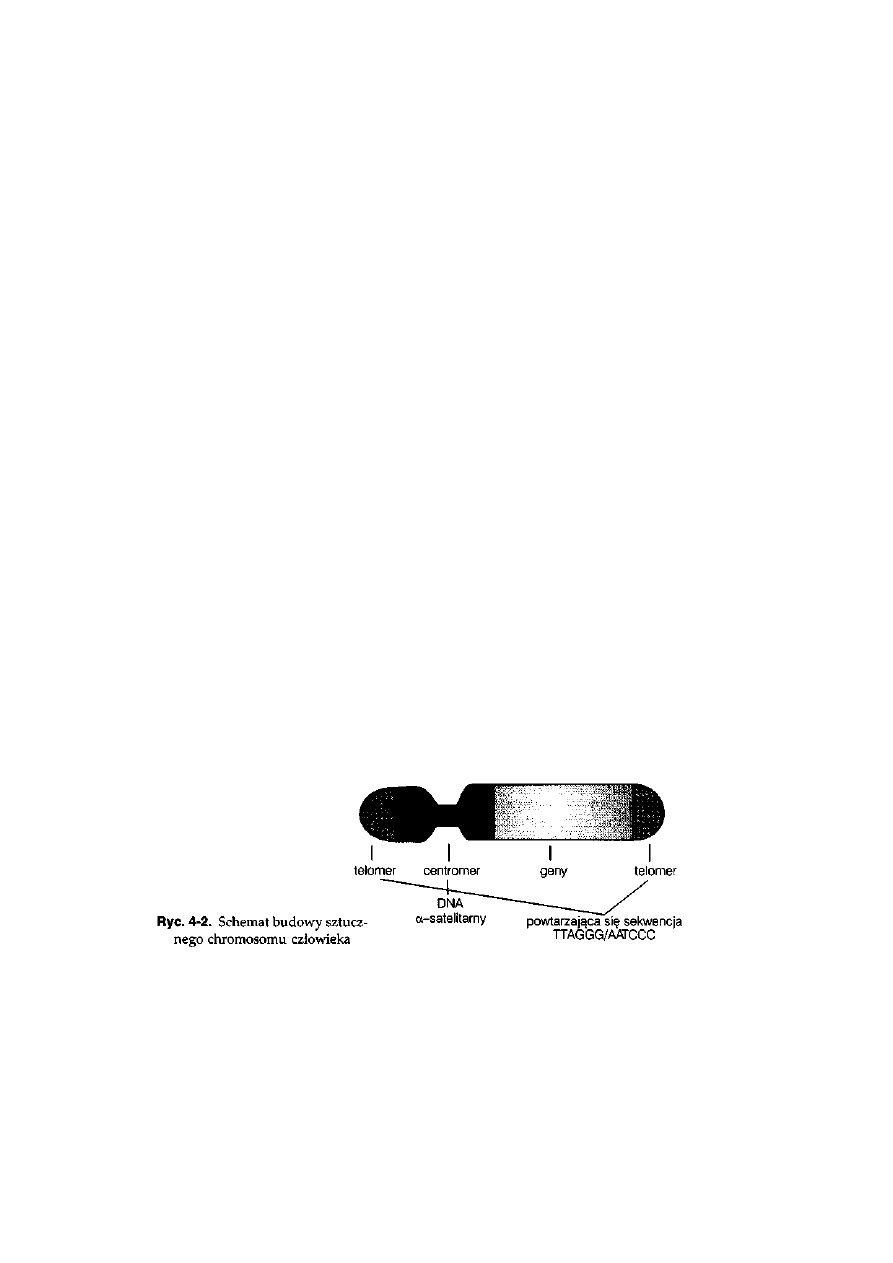
i komórkach bakteryjnych. Zdolność ta wynika z występowania odrębnych
sekwencji startu replikacji i genów markerowych. Typowy wektor droż-
dżowy jest skonstruowany z części prokariotycznej (zawiera sekwencję ori
i marker selekcyjny) i eukariotycznej (zawiera sekwencję ori rozpoznawaną
przez polimerazę eukariotycznego gospodarza, marker selekcyjny i sekwen-
cje umożliwiające ekspresję genu). Taka konstrukcja pozwala na klonowanie
genów w E. coli, a następnie badanie ich ekspresji w drożdżach. Do
wektorów tych należą także sztuczne chromosomy drożdżowe — YACs
(ang. yeast artificial chromosomes), skonstruowane po raz pierwszy w 1983
roku. Są one stosowane do klonowania bardzo długich fragmentów DNA,
do 5 milionów par zasad. Są także wykorzystywane m.in. w budowie map
genomowych. Oprócz sekwencji typowych dla wektorów drożdżowych,
sztuczne chromosomy drożdżowe mają sekwencje umożliwiające replikację
chromosomów i ich segregację w trakcie podziałów mitotycznych.
Sztuczne chromosomy ludzkie
Po skonstruowaniu sztucznego chromosomu drożdży rozpoczęto inten-
sywne prace nad konstrukcją ludzkiego sztucznego chromosomu — HAC
(ang. human artificial chromosome). Było to możliwe dzięki poznaniu
sekwencji telomerowych chromosomów i sekwencji, od których zaczyna
się replikacja cząsteczki DNA. O wiele większym problemem okazało
się wydzielenie części chromosomu nazwanej centromerem. Dotychczasowe
badania zaowocowały skonstruowaniem sztucznego chromosomu ludzkiego
złożonego z telomerowego DNA, fragmentów DNA rozpoczynających
replikację oraz, zamiast centomerowego DNA,
α-satelitarnego DNA (se-
kwencja powtarzająca się, długości 171 par zasad) (ryć. 4-2). Wszystkie
te fragmenty DNA zostały wprowadzone do komórek ludzkiej linii no-
wotworowej, hodowanych in vitro. Komórki te, w przeciwieństwie do
komórek prawidłowych, mają zdolność nieograniczonej liczby podziałów.
Uzyskany dotychczas sztuczny chromosom, nazwany mikrochromosomem,
różni się znacznie od chromosomu funkcjonalnego. Jeżeli konstrukcja
sztucznego chromosomu ludzkiego zakończy się powodzeniem, chromo-
somy te (HACs) będą mogły być użyteczne do badania ekspresji genów
oraz jako wektory stosowane w terapii genowej (dostarczanie genów
do komórek somatycznych in vivo).
75

4.3. Rekombinacja molekularna i klonowanie
Rekombinacja molekularna, będąca podstawową metodą inżynierii genetycz-
nej, polega na łączeniu ze sobą różnych cząsteczek DNA lub RNA. Metoda
rekombinacji DNA umożliwia uzyskanie za pomocą klonowania bardzo
dużej liczby kopii wybranego fragmentu DNA. Klonowaniem molekularnym
nazywamy replikację w komórkach biorcy (np. bakterii) zrekombinowanych
wektorów zawierających DNA dawcy, uzyskany za pomocą jednej z na-
stępujących metod:
• wycięcie odpowiedniego fragmentu DNA dawcy za pomocą enzymów
restrykcyjnych,
• wyizolowanie z komórek dawcy określonego rodzaju mRNA, stano
wiącego transkrypt pojedynczego genu, a następnie zastosowanie reakcji
odwrotnej transkrypcji (przepisanie sekwencji mRNA na komplementarną
sekwencję mRNA
→ cDNA),
• chemiczna synteza fragmentu DNA.
Ligacja. Fragment DNA, który zamierzamy sklonować, musi być połą-
czony z odpowiednim wektorem. By połączenie to było możliwe, konieczny
jest wytwarzany przez bakterie enzym nazwany ligazą DNA.
In vivo ligazą bierze udział w procesie replikacji DNA oraz naprawie
uszkodzeń DNA, katalizując powstawanie wiązań fosfodiestrowych między
nukleotydami. Dzięki swym właściwościom ligazą bierze udział w tworzeniu
wiązań między cząsteczkami DNA pochodzącymi od -różnych organizmów.
Do rekombinowania DNA stosowana jest ligazą, którą uzyskuje się z komó-
rek Escherichia coli zakażonych fagiem T4 (ligazą jest kodowana przez gen
tego faga). Ligazą ma zdolność łączenia ze sobą fragmentów o końcach
zarówno lepkich, jak i tępych, jednak wydajność tego procesu jest większa
w przypadku końców lepkich. Z reguły do przecięcia wektora używany jest
ten sam enzym restrykcyjny, którym był trawiony DNA.
Transformacja. W celu wprowadzenia zrekombinowanych cząsteczek
DNA do komórki bakteryjnej wykorzystywana jest naturalna zdolność
wielu bakterii do pobierania cząsteczek DNA z podłoża. Właściwość tę
odkrył w 1928 roku Griffith w doświadczeniu na myszach zarażanych
dwoinką zapalenia płuc. Bakteria E. coli, która jest często używana w biologii
molekularnej, nie ma zdolności pobierania cząsteczek DNA z podłoża.
Dlatego wprowadzenie cząsteczek DNA do jej wnętrza musi być dokonane
na drodze indukcji chemicznej (destabilizowanie błony komórkowej jonami
wapnia — inkubacja komórek z CaCl
2
w temperaturze bliskiej 0°C) bądź
elektroporacji (działanie impulsów pola elektrycznego w urządzeniu zwanym
elektroporatorem). Wydajność transformacji zależy od stosowanej metody,
w pierwszym przypadku (działanie jonami wapnia) możliwe jest uzyskanie
około 10
6
transformantów z l
μg zrekombinowanego DNA, w drugim
natomiast znacznie więcej, maksymalnie do 10
10
.
76
Wprowadzenie zrekombinowanych cząsteczek DNA do komórek euka-
riotycznych nosi nazwę transfekcji. Jest kilka metod transfekcji, m.in.
mikroiniekcji DNA, elektroporacji oraz indukcji chemicznej fosforanem
wapnia lub dekstranem.
Selekcja transformantów (bakterii, które zawierają zrekombinowany
wektor). Jedna z metod selekcji transformantów wykorzystuje zjawisko
α-
komplementacji. Jest ona możliwa do stosowania w przypadku wektora
plazmidowego z serii pUC. Właściwie można mówić o dwustopniowej
selekcji komórek bakterii po transformacji. Pierwsza zachodzi podczas
hodowli bakterii na pożywce zawierającej antybiotyk. Ponieważ wektor
plazmidowy ma gen oporności na antybiotyk, uzyskuje się wzrost kolonii
bakterii, które mają wektor. Drugi etap selekcji umożliwia odróżnienie
komórek zawierających zrekombinowany wektor od komórek z wektorem
niezrekombinowanym (ryć. 4-3). Wektory z serii pUC mają promotor genu
β-galaktozydazy (enzym ten jest kodowany przez gen oznaczony symbolem
lacZ) i sekwencję kodującą N-końcową część tego białka (fragment alfa
β-
galaktozydazy). Z kolei stosowane do transformacji komórki bakterii
dostarczają C-końcową sekwencję genu lacZ (fragment omega). W wyniku
połączenia, po pobraniu przez bakterię wektora, obu sekwencji genu lacZ
(zjawisko
α-komplementacji) (ryć. 4-3) powstaje funkcjonalny gen β-galak-
tozydazy. Dzięki syntezie tego enzymu kolonie bakterii zawierających
plazmid, hodowane na podłożu z chromogenem X-gal, będą miały barwę
Ryć. 4-3. Zasady
α-komplementacji (połączenie fragmentów genu lacZ z plazmidu
i bakterii) — synteza funkcjonalnego genu enzymu
β-galaktozydazy
77

niebieską. Jeżeli do wektora (w obręb polilinkera znajdującego się w części
„plazmidowej" genu lacZ) zostanie wprowadzony obcy fragment DNA,
synteza N-końcowej części (
β-galaktozydazy zostanie zablokowana, co
spowoduje brak syntezy aktywnego białka i białe zabarwienie kolonii
bakterii zawierających zrekombinowany wektor.
4.4. Amplifikacja DNA za pomocą łańcuchowej
reakcji polimerazy (PCR)
Łańcuchowa reakcja polimerazy — PCR (ang. polymerase chain reaction)
jest podstawową techniką badawczą i diagnostyczną wykorzystującą zdol-
ność DNA do replikacji. Reakcja ta jest katalizowana, podobnie jak to się
dzieje w komórkach, przez polimerazy DNA, zwane także replikazami.
Łańcuchowa reakcja polimerazy znana jest od ponad 10 lat, a jej twórcą był
Kary Mullis, który za swoje badania otrzymał Nagrodę Nobla w roku 1993.
O ogromnym znaczeniu tej metody decyduje to, iż umożliwia ona analizę
DNA nawet w przypadku bardzo małej jego ilości (np. w kryminalistyce —
ślady krwi). PCR jest enzymatyczną metodą amplifikacji (powielania)
określonych sekwencji DNA, a jej czułość pozwala na zwielokrotnienie
(uzyskanie wielu milionów kopii) ściśle określonych sekwencji DNA, mimo
obecności innych sekwencji. Można amplifikować poszukiwane odcinki
DNA, dysponując DNA wyizolowanym nawet z pojedynczych komórek.
Jednak najlepszy DNA do celów diagnostycznych uzyskujemy, gdy pochodzi
z minimum 10
3
komórek. Z reguły amplifikacja określonego fragmentu
DNA jest możliwa wtedy, gdy znana jest jego sekwencja nukleotydowa, na
podstawie której syntetyzowane są krótkie, zazwyczaj około dwudziesto-
nukleotydowe oligonukleotydy, tzw. startery (ang. primer), komplementarne
do badanego DNA (jeden starter do jednej, drugi do drugiej nici).
Aby mogła zajść łańcuchowa reakcja polimerazy, niezbędne są następują-
ce warunki: obecność jednoniciowej matrycy DNA (badany dwuniciowy
DNA jest rozdzielany na dwie nici przez denaturację termiczną, proces ten
nazywany jest inaczej topnieniem), wspomnianych już starterów, trifosfora-
nów deoksynukleotydów (dATP, dCTP, dGTP i dTTP) oraz enzymu
polimerazy DNA. Konieczny jest również bufor reakcyjny uzupełniający
niezbędne do aktywności enzymu jony metali (potasu i magnezu). Trifosfora-
ny w trakcie amplifikacji uczestniczą w wydłużaniu sekwencji starterów
zgodnie z regułą komplementarności zasad azotowych względem amplifiko-
wanej nici DNA. Enzym katalizujący reakcję — Taq polimeraza DNA —
pochodzi z bakterii Thermus aquaticus, żyjącej w gorących źródłach. Dlatego
polimeraza (nazwana od skrótu gatunku bakterii Taq polimeraza) zachowuje
swą aktywność nawet w bardzo wysokiej temperaturze (> 90°C). Zanim
wykryto ten enzym, przeprowadzenie reakcji amplifikacji było ogromnie
78
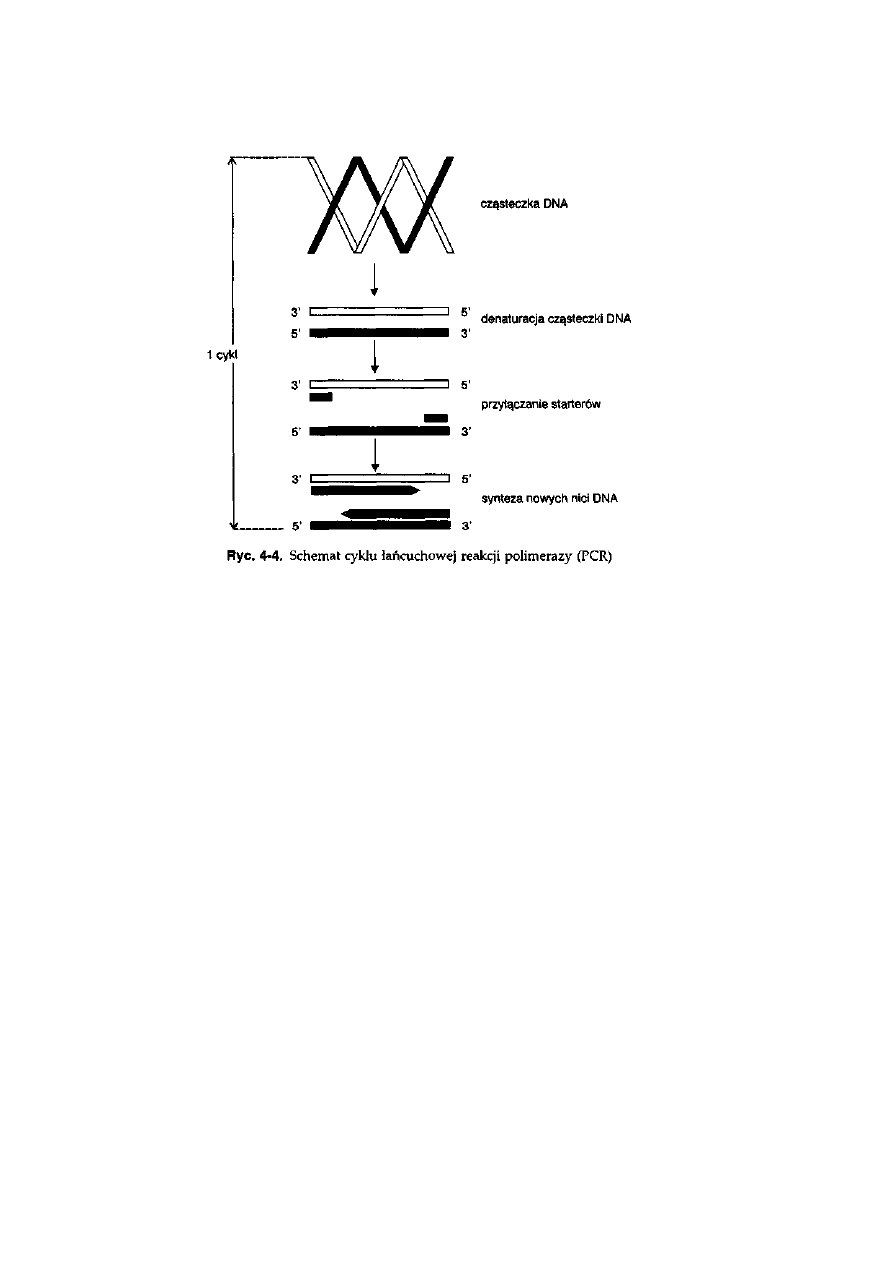
pracochłonne i wymagało wielokrotnego dodawania enzymu. Obecnie
może być stosowana także inna polimeraza, PrimeZyme, pochodząca z innej
termofilnej bakterii Thermus brockianus.
Reakcja amplifikacji DNA składa się z powtarzających się cykli, a każdy
z nich przebiega w trzech etapach (ryć. 4-4):
1 — denaturacja dwuniciowego DNA (ang. denaturation), zachodząca
w temperaturze 92-96°C (zależnie od rodzaju probówki i termocyklera,
którym jest termostat z automatycznie zmieniającą się temperaturą);
2 — przyłączenie (ang. annealing) starterów do komplementarnych
sekwencji na zdenaturowanej matrycy DNA, w temperaturze 37-72°C.
Temperatura przyłączania starterów zależy głównie od zawartości w tych
starterach nukleotydów G i C. Można ją w przybliżeniu określić, wykorzys
tując wzór (Mazaras i wsp., NAR 19:4783, 1991):
T = 69,3°C + 41 x (G + C)/L - 650/L
gdzie:
(G + C) — łączna liczba nukleotydów guanylowych i cytydylowych
L — łączna liczba wszystkich nukleotydów;
3 — wydłużanie starterów (ang. elongation) polegające na dołączaniu,
począwszy od wolnego końca 3'-OH startera, nukleotydów komplementar
nych względem amplifikowanej nici DNA; proces ten zachodzi w tem-
peraturze 72°C.
79

W wyniku wydłużenia starterów powstają syntetyzowane „de novo" nici
DNA, które zawierają miejsce wiązania dla startera w następnym cyklu.
Zatem każda nowa nić DNA staje się matrycą w kolejnym cyklu amplifikacji,
co prowadzi do zwiększania się liczby amplifikowanych fragmentów
w postępie logarytmicznym. Po 30 cyklach tej reakcji jest już 2
30
, czyli
1073 741 824, powielonych fragmentów DNA. Długość powstających frag-
mentów determinują końce 5' starterów. Denaturacja DNA w pierwszym
i wydłużanie starterów w ostatnim cyklu przebiegają w nieco dłuższym
czasie niż w cyklach pozostałych. Dla każdego fragmentu DNA, który ma
być zamplifikowany, należy dobrać właściwe warunki reakcji, przede
wszystkim temperaturę przyłączania starterów.
Całkowity czas trwania łańcuchowej reakcji polimerazy wynosi z reguły
najwyżej kilka godzin. Po zakończeniu reakcji zamplifikowany fragment
DNA, czyli tzw. produkt PCR, może być przechowywany w temperaturze
4°C, do momentu następnego etapu badań.
Metoda PCR ma wiele zastosowań, między innymi do:
• wykrywania obecności w genomie określonych odcinków/genów oraz
identyfikacji nosicieli mutacji powodujących choroby genetyczne
• wykrywania obecności w organizmie wirusowego lub prowirusowego
DNA
• zwiększenia ilości wybranych odcinków DNA do późniejszego sek-
wencjonowania lub tworzenia biblioteki genów.
Opracowane zostały modyfikacje łańcuchowej reakcji polimerazy:
Wewnętrzna (ang. nested) PCR. Stosowana jest w przypadku braku
pewności, że zamplifikowany został właściwy fragment DNA. Metoda ta
polega na amplifikacji mniejszego odcinka DNA, zawartego w obrębie
wcześniej zamplifikowanego dłuższego fragmentu. Oczywiście w tej drugiej
reakcji stosowane są inne startery. Za pomocą tej metody możemy także
zwiększyć czułość reakcji, dzięki dodatkowej amplifikacji produktów wcześ-
niejszej reakcji.
RAPD (ang. random amplified polymorphic DNA). Jest to metoda
losowej amplifikacji fragmentów DNA, w której, w odróżnieniu od metody
PCR, stosowany jest tylko jeden starter. Również warunki reakcji są nieco
inne, niższa jest temperatura przyłączania starterów do matrycowego DNA,
a liczba cykli reakcji jest większa (40-50). Przykład wzoru prążkowego
(losowo zamplifikowanych fragmentów DNA) przepiórki japońskiej uzys-
kanego w wyniku reakcji RAPD przedstawiono na rycinie 4-5.
LCR (ang. ligase chain reaction). Ligazowa reakcja łańcuchowa; zbliżona
jest do PCR. Różnice polegają m.in. na tym, że w LCR oligonukleotydy są
dłuższe (50-60 nukleotydów) i tak przygotowane, by po hybrydyzacji,
odpowiednio do końca 5' i 3' matrycowego DNA, nie pozostała między nimi
80
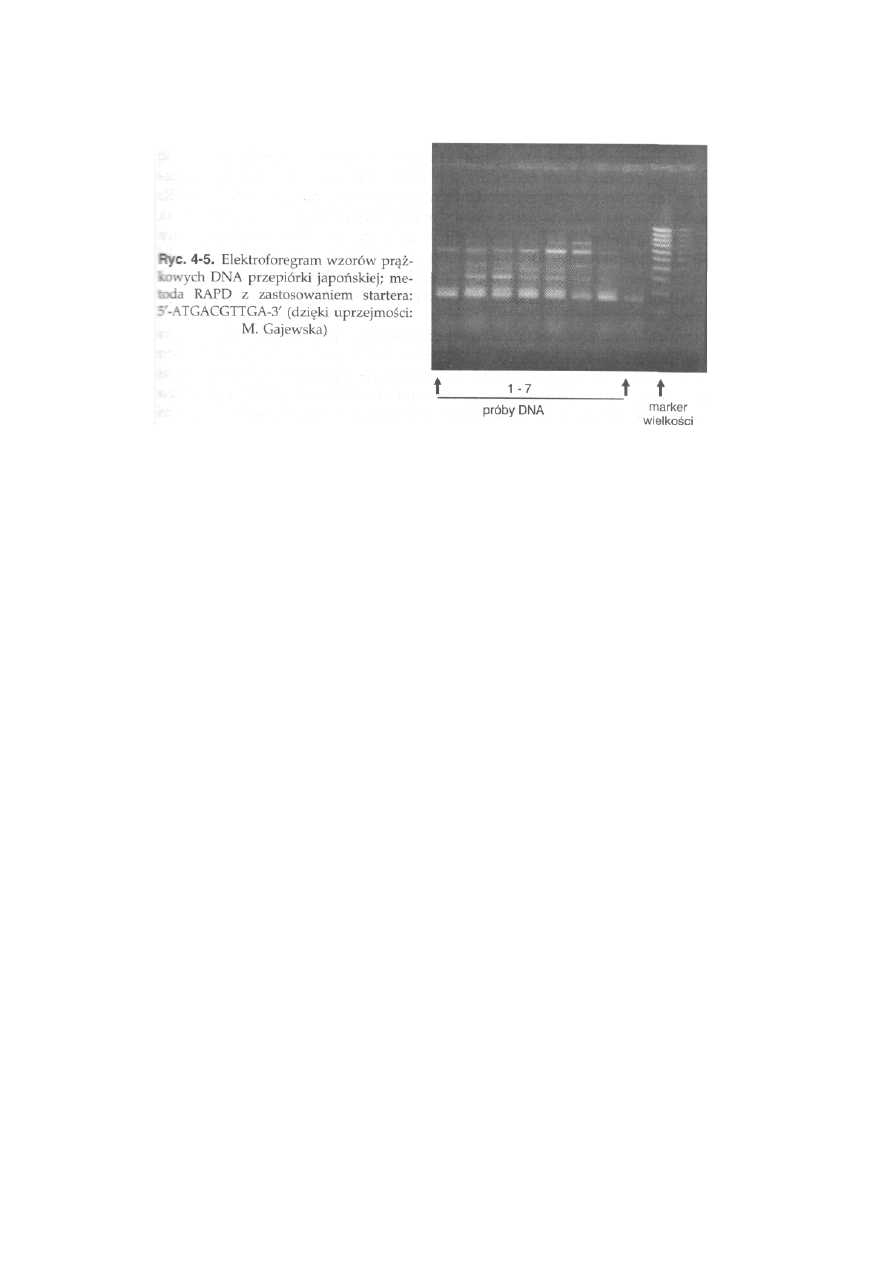
przerwa. Enzymem używanym do tej reakcji jest termostabilna ligaza, która
łączy specyficznie dwa przyległe oligonukleotydy, gdy są one zhybrydyzo-
wane z komplementarną matrycą. Oligonukleotydy są amplifikowane
podczas cykli termicznych w obecności drugiej pary starterów.
W pierwszym etapie reakcji matryca — DNA jest poddawana de-
naturacji (rozdzielana na dwie nici) przez działanie wysoką temperaturą
(94°C). Ochłodzenie mieszaniny reakcyjnej do 60°C powoduje, że komple-
mentarne do normalnego allelu cztery diagnostyczne startery hybrydyzują
z komplementarnymi sekwencjami na matrycy DNA. Jeśli jest idealna
komplementarność między matrycą i zhybrydyzowanymi z nią oligo-
nukleotydami (starterami), wówczas termostabilna ligaza tworzy kowa-
lentne wiązanie między oligonukleotydami. Te dwa etapy są powtarzane,
a produkty ligacji stają się matrycami do hybrydyzacji w kolejnych
cyklach, amplifikując badany gen (odcinek DNA). Jeśli nie ma komple-
mentarności w miejscu połączenia starterów z matrycą DNA (np. mutacja
punktowa), wówczas nie tworzy się wiązanie między starterami i nie
ma produktu amplifikacji.
Reakcja ta stosowana jest do wykrywania mutacji punktowych, co jest
szczególnie istotne z medycznego punktu widzenia (diagnostyka chorób
monogenowych).
W celu identyfikacji produktu LCR stosowane są nieradioaktywne
metody znakowania barwnego, np. z zastosowaniem biotyny. O znakowaniu
biotyną będzie jeszcze mowa w podrozdziale: Techniki hybrydyzacji i sondy
molekularne.
RT-PCR (ang. reverse transcription-polymerase chain reaction). Odwrot-
na transkrypcja-PCR. Łańcuchowa reakcja polimerazy umożliwia amplifika-
cję jedynie kwasu deoksyrybonukleinowego. Nie może być natomiast
81

wykorzystana w badaniach ekspresji genów, w których określana jest ilość
mRNA, jak również do wykrywania wirusów, których materiałem genetycz-
nym jest RNA. W tych przypadkach może być stosowana metoda RT-PCR,
polegająca na tym, iż łańcuchowa reakcja polimerazy jest poprzedzona
odwrotną transkrypcją, podczas której na matrycy mRNA syntetyzowany
jest komplementarny DNA (nazwany cDNA). Reakcja ta przebiega dzięki
zastosowaniu enzymu odwrotnej transkryptazy. Końcowym etapem jest
amplifikacja uzyskanego cDNA na drodze łańcuchowej reakcji polimerazy.
SSCP (ang. single stranded conformation polymorphism). Polimorfizm
konformacji jednoniciowych łańcuchów DNA; jest to technika często
wykorzystywana do wstępnej identyfikacji mutacji punktowych w badanym
fragmencie DNA. Badanie polega na amplifikacji techniką PCR wybranego
fragmentu DNA, a następnie jego denaturacji i elektroforezie. Efektem
denaturacji jest uzyskanie jednoniciowych łańcuchów DNA, które przyjmują
określoną konformację przestrzenną. Konformacja ta zależy od sekwencji
nukleotydów, bowiem pomiędzy niektórymi sekwencjami wchodzącymi
w skład łańcucha będą powstawały wiązania wodorowe między komple-
mentarnymi zasadami azotowymi. Dzięki temu szybkość migracji jedno-
niciowych łańcuchów DNA podczas elektroforezy zależy nie tylko od ich
wielkości/ale także konformacji przestrzennej. Zdenaturowany DNA allelu
zmutowanego może przyjmować inną konformację niż allelu prawidłowego
i w konsekwencji fragmenty te będą migrowały podczas elektroforezy
z różną szybkością. Jeśli badany DNA pochodzi od nosiciela zmutowanego
genu, to wówczas na elektroforegramie pojawiają się prążki (fragmenty)
reprezentujące allel prawidłowy oraz allel zmutowany. Stwierdzenie takiej
formy polimorfizmu wskazuje jedynie na to, że testowany osobnik jest
heterozygotą w obrębie amplifikowanego fragmentu DNA. Nie można
jednak na tej podstawie zidentyfikować typu mutacji ani określić miejsca
mutacji. Dlatego szczegółowa analiza mutacji wymaga zastosowania dodat-
kowych metod, np. RFLP lub sekwencjonowania, które jest oczywiście
najbardziej precyzyjne.
Poszukiwanie określonego fragmentu DNA lub RNA może być prze-
prowadzane także bezpośrednio w komórkach i tkankach za pomocą
metody PCR in situ.
4.5. Techniki hybrydyzacji i sondy molekularne
Hybrydyzacja DNA jest możliwa dzięki zdolności kwasów nukleinowych
do rozdziału na pojedyncze nici komplementarne i ponownego łączenia się
w struktury dwuniciowe. Jak wiadomo, między nićmi DNA:DNA,
RNA: RNA i RNA: DNA istnieją wiązania wodorowe, które ułatwiają ten
82

proces. Zjawisko to stało się podstawą metod analizy kwasów nukleinowych,
takich jak hybrydyzacja Southern blotting (DNA), northern blotting (RNA)
czy PCR (hybrydyzacja starterów z matrycą).
Ogromną zaletą hybrydyzacji jest to, że można ją stosować nawet
wówczas, gdy nie jest dokładnie znana cała sekwencja nukleotydowa
badanego fragmentu DNA.
Southern blotting jest metodą przenoszenia odpowiednio przygo-
towanego DNA na filtr nitrocelulozowy lub nylonowy. Przygotowanie
DNA polega na trawieniu jednym lub kilkoma enzymami restrykcyjnymi
i rozdzieleniu elektroforetycznym uzyskanych fragmentów w żelu aga-
rozowym (dłuższe, czyli cięższe fragmenty) lub poliakryloamidowym
(krótsze, lżejsze, do 500 par zasad). By była możliwa hybrydyzacja
sondy z badanym DNA, kwas nukleinowy znajdujący się w żelu musi
być zdenaturowany (rozdzielony na pojedyncze nici poprzez działanie
roztworem kwasu solnego). Przeniesienie fragmentów DNA z żelu na
filtr następuje przez bufor transferowy, który przenikając przez żel
zabiera ze sobą fragmenty DNA. Fragmenty te są następnie zatrzymywane
na filtrze. Przeniesiony DNA należy utrwalić na filtrze działając wysoką
temperaturą bądź promieniami UV. Tak przygotowany DNA można
nawet kilkakrotnie hybrydyzować z sondami molekularnymi. Sondy
molekularne są to odcinki DNA mające sekwencję nukleotydów kom-
plementarną do badanej. Sondami molekularnymi mogą być fragmenty
DNA bezpośrednio pochodzące z genomu (np. wirusów lub organizmów
wyższych), klonowane sekwencje lub syntetyczne oligomery stosowane
w PCR.
Technika northern blotting, stosowana w hybrydyzacji kwasu rybonu-
kleinowego, który jest nicią pojedynczą, nie wymaga etapu denaturacji.
Znakowanie sond. Sonda musi być odpowiednio wyznakowana, by było
możliwe wykrycie powstałej hybrydy. Sposób znakowania sond można
podzielić na dwie główne grupy:
1. Znakowanie izotopowe (radioaktywne) — przez wprowadzenie do
cząsteczki sondy izotopu promieniotwórczego. Sygnał jest wykrywany
metodą autoradiografii przez ekspozycję błony fotograficznej na działanie
promieniowania powstałej hybrydy. Znakowanie izotopowe charakteryzuje
się dużą czułością, jednak niewielką rozdzielczością. Wadą sond znakowa-
nych izotopem jest również to, iż muszą być stosowane wkrótce po ich
wyznakowaniu. Najczęściej stosowanymi izotopami są:
• fosfor radioaktywny (
32
P), który emituje wysokoenergetyczne promie-
niowanie
β (energia promieniowania E = 1,71 MeV — megawoltów), czas
półtrwania 14,3 dnia. Izotop ten jest stosowany do znakowania DNA i RNA,
a jego podstawowym źródłem są radioaktywne nukleotydy wyznakowane
w pozycji
α: dATP, dCTP i UTP a
32
P,
83

• izotop siarki (
35
S), emitujący promieniowanie
β, które jest około 10
razy słabsze niż fosforu (energia promieniowania E = 0,167 MeV),
okres półtrwania 87,1 dnia. Siarka radioaktywna jest stosowana do
znakowania kwasów nukleinowych, głównie DNA. Atom tlenu w grupie
fosforanowej
α i γ P może być zastąpiony przez S, tak powstają
pochodne nukleotydów,
• tryt (
3
H), który emituje promieniowanie
β (energia promieniowania
E = 0,018 MeV), okres półtrwania 12,26 roku, używany jest do
znakowania kwasów nukleinowych, głównie DNA. Podstawowym
źródłem trytu jest trytowana tymidyna.
2. Znakowanie nieizotopowe (nieradioaktywne) przez dołączenie do
sondy lub wbudowanie w nią znacznika fluorescencyjnego (np. biotyny,
digoksygeniny). Ten rodzaj znakowania wymaga dodatkowego
mechanizmu detekcji (immunologicznego lub fluorescencyjnego), w
zależności od zastosowanego znacznika. Detekcja immunologiczna jest
bardzo dobra do hybrydyzacji na filtrach. Znakowanie nieizotopowe
zapewnia dobrą rozdzielczość, ale daje mniejszą czułość detekcji.
Znacznik (izotopowy lub nieizotopowy) może być równomiernie roz-
mieszczony w całej sekwencji sondy bądź sonda może być znakowana na
końcach (terminalnie). Znakowanie sond można przeprowadzać za pomocą
kilku różnych metod.
Hybrydyzacja na filtrach. Hybrydyzację z wyizolowanym DNA można
przeprowadzać, gdy jest on unieruchomiony na filtrze. Zaletą tej metody
jest to, iż stosunkowo duża ilość kwasu nukleinowego może być
związana z filtrem i wyeksponowana na działanie sondy. Ponadto
niezhybrydyzowana sonda jest odpłukiwana po inkubacji.
Hybrydyzacja jest poprzedzona prehybrydyzacją, której celem jest
wysycenie miejsc na filtrze mogących niespecyficznie związać sondę.
Roztwór prehybrydyzacyjny ma taki sam skład jak hybrydyzacyjny, z wyjąt-
kiem obecności sondy. Sam proces hybrydyzacji polega na długotrwałej
inkubacji (na przykład przez całą noc) badanego DNA w buforze zawiera-
jącym wyznakowaną sondę. Podczas tej inkubacji zachodzą wielokrotnie
procesy asocjacji i dysocjacji sondy z próbką DNA. Kinetyka tych procesów
w dużej mierze zależy od różnicy między temperaturą topnienia — Tm
(jest to temperatura, w której 50% długości nici jest zdysocjowane)
hybrydy a temperaturą, w której przebiega hybrydyzacja. Przyjmuje się,
że różnica ta powinna wynosić 20-25°C. Hybrydyzację przeprowadza
się z reguły w temperaturze 68°C. Na wydajność hybrydyzacji wpływają
także inne czynniki, jak ilość kwasu nukleinowego związanego z filtrem
(nie mniej niż 10
μg), stężenie sondy molekularnej w roztworze
hybrydyzacyjnym (optimum to 50-1200 ng/ml) i stężenie jonów Na
+
. Po
hybrydyzacji nadmiar sondy jest odpłukiwany (najczęściej w temperaturze
65-70°C), a detekcja sygnału jest poprzez autoradiografię (ryć. 4-6).
84
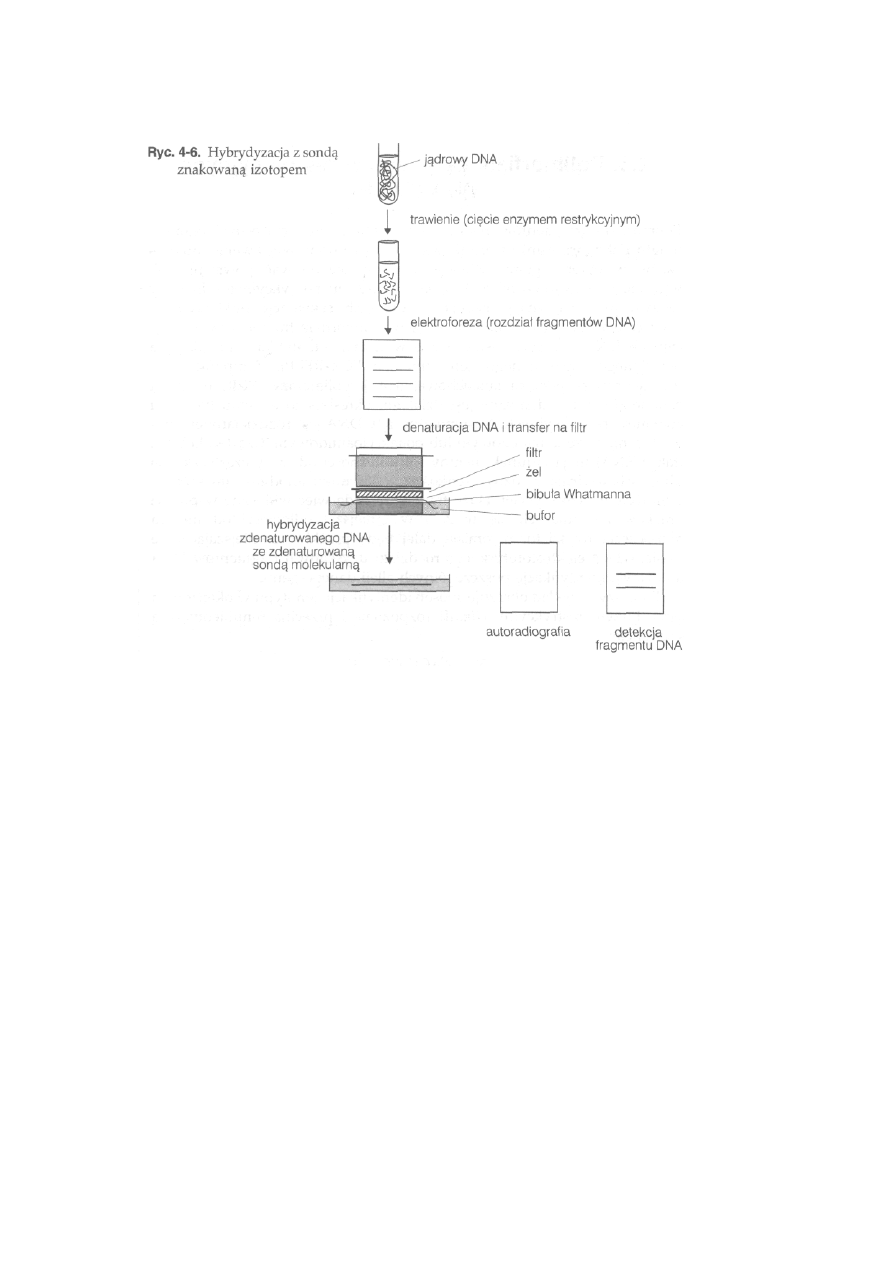
Wykorzystanie metody hybrydyzacji. Metoda hybrydyzacji służy m.in. do
wykrywania ściśle określonych fragmentów DNA (np. w diagnostyce
chorób). W metodzie zwanej „odcisk palca" DNA (ang. fingerprint) stosowa-
na jest sonda molekularna komplementarna do sekwencji minisatelitarnej
(metoda ta jest opisana w rozdziale: Markery genetyczne). Metoda „odcisku
palca" DNA jest stosowana w kontroli pochodzenia oraz do oceny zmienności
genetycznej wewnątrz i między rasami/populacjami. Podobnie jak metoda
PCR, również hybrydyzacja może być przeprowadzana na preparatach
chromosomowych. Metoda ta nosi nazwę hybrydyzacji in situ (ISH). Jej różne
warianty są opisane w rozdziale: Mapy genomowe.
Hybrydyzacja w zminiaturyzowanej postaci, określana jako technika
mikromacierzy DNA, pozwala ustalać genotyp albo analizować ekspresję
setek lub tysięcy genów. Na niewielkiej płytce (ok. l cm
2
) umieszczone są
sondy oligonukleotydowe dla poszczególnych mutacji (genotypowanie) lub
genów (ekspresja). Detekcja hybrydyzacji sondy z wyizolowanym DNA
(genotypowanie) lub z DNA (ekspresja) jest zautomatyzowana.
85

4.6. Polimorfizm fragmentów restrykcyjnych
DNA (RFLP)
Polimorfizm fragmentów restrykcyjnych DNA (ang. restricted fragment
length polymorphism) uwarunkowany jest różnicami w sekwencji nukleo-
tydów w obrębie genu. Mutacje te mogą powodować powstanie lub
likwidację istniejącego miejsca cięcia dla enzymu restrykcyjnego. Enzymy
restrykcyjne rozpoznają specyficzne dla nich sekwencje nukleotydowe
i przecinają DNA w określonym miejscu. Polimorfizm fragmentów restryk-
cyjnych DNA znajduje zastosowanie w testach identyfikacji nosicielstwa
określonego allelu danego genu (metoda PCR-RFLP). Zamplifikowany
(powielony) za pomocą łańcuchowej reakcji polimerazy (PCR) fragment
badanego genu poddawany jest działaniu określonego enzymu lub kilku
enzymów restrykcyjnych. Następnie pocięty DNA jest rozdzielany elektro-
foretycznie w żelu agarozowym lub poliakryloamidowym. Cząsteczki DNA
(także RNA) mają ładunek ujemny i w zależności od masy cząsteczkowej
(długości) wędrują z różną prędkością w kierunku anody — im krótsze,
czyli lżejsze, tym szybciej. Duże fragmenty będą więc widoczne w postaci
prążków znajdujących się w żelu w mniejszej odległości od miejsca
rozpoczęcia rozdziału, natomiast dalej będą prążki o zmniejszającej się
masie. Obraz elektroforetycznego rozdziału uzyskanych fragmentów DNA
umożliwia identyfikację poszczególnych alleli danego genu.
Poniższy przykład obrazuje sposób identyfikacji genotypu w określonym
locus. Enzym restrykcyjny Hindlll rozpoznaje i przecina 6-nukleotydową
sekwencję:
5'...A
T
AGCTT ...3'
3'...TTCGA,A-..5'
Dowolny zamplifikowany za pomocą łańcuchowej reakcji polimerazy (PCR)
odcinek DNA poddany działaniu tego enzymu (trawiony) zostanie pocięty
na fragmenty różnej długości. Odcinek DNA przed trawieniem ma na-
stępującą sekwencję (pogrubionym drukiem zaznaczono motyw rozpo-
znawany przez enzym Hindlll):
GTCGGCAAGCTTAGGTCAGGAGCGGACTAAAGCTTCCGATA
CAGCCGTTCGAATCCAGTCCTCGCCTGATTTCGAAGGCTAT
Po trawieniu (cięciu) DNA enzymem otrzymujemy fragmenty DNA:
GTCGGCA'AGCTTAGGTCAGGAGCGGACTAA'AGCTTCCGATA
CAGCCGTTCGA,ATCCAGTCCTCGCCTGATTTCGAA,AGGCTAT
86

Po rozdziale elektroforetycznym fragmenty DNA ułożą się w kolejności 23
pz, 11 pz i 7 pz.
Metoda PCR-RFLP jest wykorzystywana w hodowli zwierząt do iden-
tyfikacji genotypów pod względem genów, których budowa molekularna
jest znana; wśród nich są geny determinujące cechy jakościowe, a także
ilościowe (ang. quantitative trait loci — QTLs) oraz odpowiedzialne za
rozwój chorób genetycznych czy odporność na niektóre patogeny. Przykłady
takich cech są przedstawione w podrozdziałach: Mutacje genowe, Geny
o dużym efekcie, Interseksualizm.
4.7. Biblioteki genomowe i genowe
Biblioteką (bankiem) genomową nazywamy zbiór klonów bakterii zawiera-
jących zrekombinowane wektory niosące fragmenty DNA. Biblioteka geno-
mową powinna się składać z fragmentów DNA tworzących razem cały
genom określonego gatunku. Natomiast biblioteka genowa (inaczej biblioteka
lub bank cDNA) jest to zbiór sekwencji kodujących białka, wprowadzonych
za pomocą wektorów do bakterii. Klony bakteryjne zawierające fragmenty
DNA lub cDNA mogą być przechowywane w stanie zamrożonym do
momentu ich wykorzystania. Do tworzenia bibliotek (banków) genomowych
i genowych wykorzystywana jest, omówiona wcześniej, metoda rekombinacji
molekularnej.
Tworzenie biblioteki genomowej
Materiałem wyjściowym jest DNA wyizolowany z tkanek i strawiony
(pocięty) enzymem restrykcyjnym. Tak powstałe fragmenty DNA są wpro-
wadzane za pomocą wektora do bakterii. Proces ten jest opisany w części
dotyczącej klonowania DNA. W zależności od wielkości fragmentów DNA
używane są różne wektory. Dla prokariontów i niższych eukariontów
stosowane są wektory plazmidowe lub fagowe, natomiast dla wyższych
eukariontów niezbędne są bardziej pojemne wektory, jak kosmidy, sztuczny
chromosom bakteryjny (BAĆ) lub sztuczny chromosom drożdżowy (YAC).
Biblioteki dużych genomów najczęściej składają się z bibliotek dla po-
szczególnych chromosomów (biblioteki chromosomowe).
Tworzenie biblioteki genowej (cDNA)
Tworzenie biblioteki genowej jest znacznie trudniejsze niż genomowej.
Materiałem wyjściowym w tym przypadku jest RNA. Po wyizolowaniu
RNA z tkanek należy oddzielić mRNA. Kolejnym krokiem jest „przepisanie"
za pomocą reakcji odwrotnej transkrypcji (katalizowanej przez enzym
odwrotną transkryptazę) informacji z mRNA na DNA. Tak otrzymany
cDNA zawiera tylko sekwencje kodujące, czyli eksony. Jest on amplifikowany
87

metodą PCR, a następnie rekombinowany z wektorem i wprowadzany do
bakterii. W ten sposób każdy klon bakterii ma cały określony gen.
Wykorzystanie określonego rodzaju biblioteki zależy od celu badań. Jeśli
zamierzamy poznać organizację całego genu łącznie z intronami i promoto-
rem, wykorzystuje się bibliotekę genomową. Biblioteka genomowa jest
również przydatna w poszukiwaniu genu, o którym niewiele wiadomo.
Stosowana jest wówczas metoda zwana „spacerem po chromosomie" (ang.
chromosome walking), której pierwszym krokiem jest znalezienie w biblio-
tece klonu zawierającego gen bądź marker sprzężony z poszukiwanym
genem. Fragment końcowy sekwencji sprzężonej traktowany jest jako
sonda, która jest wykorzystywana do poszukiwania klonu zawierającego
sąsiadujący, nieznany jeszcze fragment. Postępujemy tak do momentu, gdy
w którymś z kolejnych odcinków znajdziemy poszukiwany gen.
Do badania ekspresji genu wykorzystuje się bibliotekę cDNA. Najczęściej
stosowaną metodą analizy bibliotek jest hybrydyzacja z sondami molekular-
nymi (metoda hybrydyzacji DNA została już opisana wcześniej). Zasad-
niczym elementem tych badań jest odnalezienie klonu zawierającego
interesujący nas gen.
Inną metodą poszukiwania określonego genu jest zastosowanie reakcji
immunologicznej, czyli reakcji antygen-przeciwciało. W tym celu należy
doprowadzić do syntezy przez bakterie białka kodowanego przez ten gen.
Surowica zawierająca przeciwciało identyfikujące określone białko (antygen)
służy jako swego rodzaju „sonda" do przeglądania zbioru biblioteki.
4.8. Sekwencjonowanie DNA
Sekwencjonowanie DNA, polegające na określeniu sekwencji nukleotydów,
jest stosowane dość często w genetyce molekularnej, przede wszystkim do
analizy sklonowanych fragmentów DNA lub produktów łańcuchowej reakcji
polimerazy. Metoda ta umożliwia także wykrycie miejsc mutacyjnych
i opracowanie testu molekularnej identyfikacji genotypów zwierząt pod
kątem nosicielstwa określonych alleli.
Sekwencjonowanie można przeprowadzać za pomocą dwóch różnych
metod: enzymatycznej i chemicznej.
Metoda enzymatyczna została opracowana w 1977 roku przez F. Sangera.
Podstawą tej metody jest enzymatyczna (przeprowadzana przez polimerazę
DNA) synteza komplementarnej nici na zdenaturowanej (jednoniciowej)
matrycy DNA. Ponieważ w nici DNA są cztery rodzaje nukleotydów (różniące
się zasadą), przeprowadzane są cztery oddzielne reakcje syntezy DNA. Każda
z czterech probówek zawiera matrycowy DNA, starter, mieszaninę deoksynuk-
leotydów, bufor reakcyjny, polimerazę DNA oraz jeden z dideoksynukleoty-
dów (ddNTP): ddATP, ddCTP, ddGTP lub ddTTP (ryć. 4-7). Rolą tych
dideoksynukleotydów (tzw. terminatorów) jest zakończenie syntezy nici
88
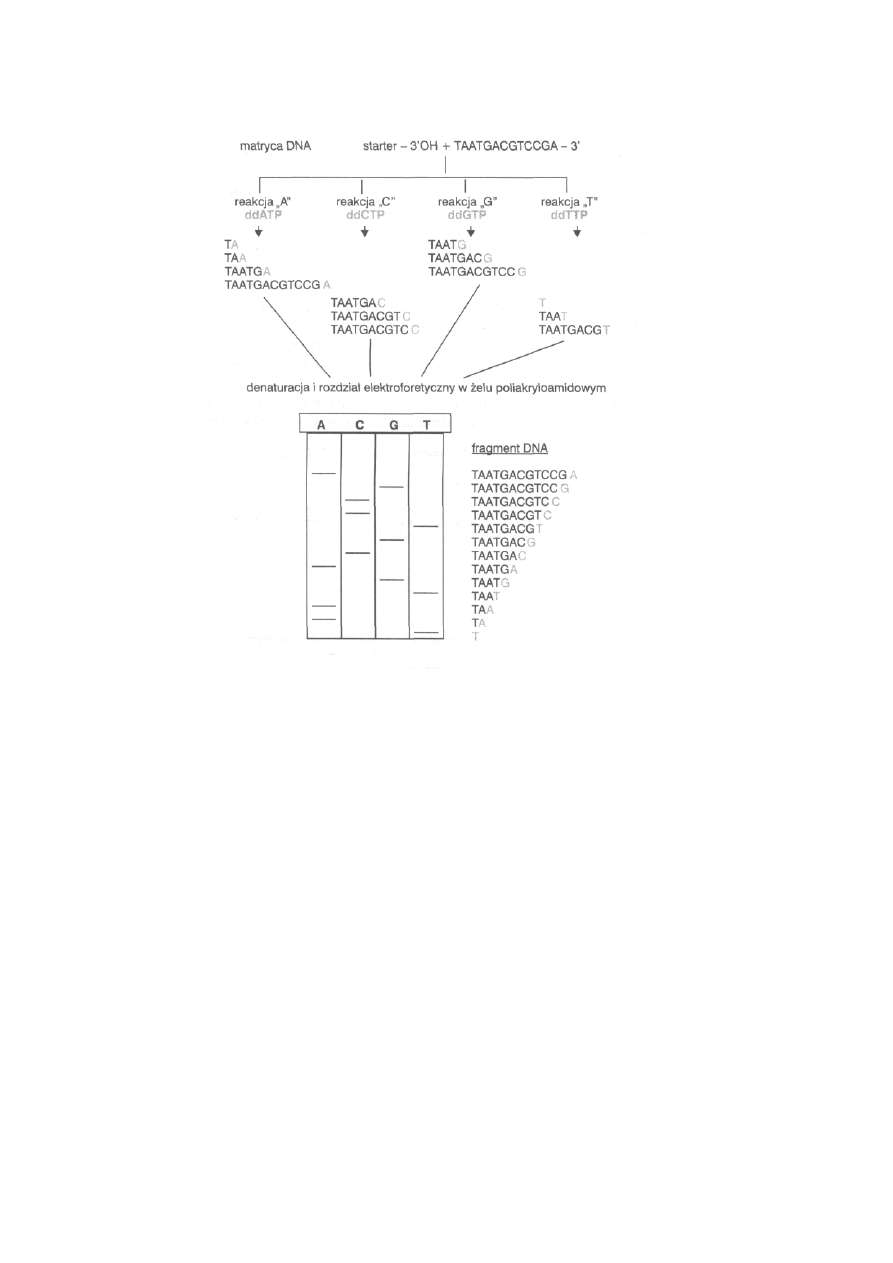
autoradiogram żelu sekwencyjnego
Ryć. 4-7. Schemat sekwencjonowania DNA metodą enzymatyczną Sangera; kolorem
szarym zaznaczono nukleotyd serii dideoksy (wg: Nuć i wsp., Postępy Biologii Komórki,
25 supl. 10 :219-237, 1998)
komplementarnej w miejscu włączenia określonego dideoksynukleotydu.
W wyniku każdej z czterech reakcji otrzymujemy fragmenty DNA o różnej
długości, kończące się odpowiednim nukleozydem, zależnie od użytego
w reakcji dideoksynukleotydu. Zatrzymanie syntezy komplementarnej nici
następuje po powstaniu pełnej sekwencji jednej cząsteczki, ponieważ stosunek
dideoksynukleotydów (ddNTP) do deoksynukleotydów (dNTP) jest tak
dobrany, by tylko część syntetyzowanych fragmentów DNA uległa terminacji.
Po zakończeniu reakcji syntezy powstały DNA jest denaturowany i
rozdzielany w drodze elektroforezy wysokonapięciowej (około 2000 V) w
żelu poliakryloamidowym (długości 40-100 cm, grubości 0,2-0,6 mm). W
celu detekcji prążków DNA, metodą autoradiografii, w reakcji wykorzys-
tywane są wyznakowane izotopowe (izotop fosforu lub siarki) deoksynu-
89
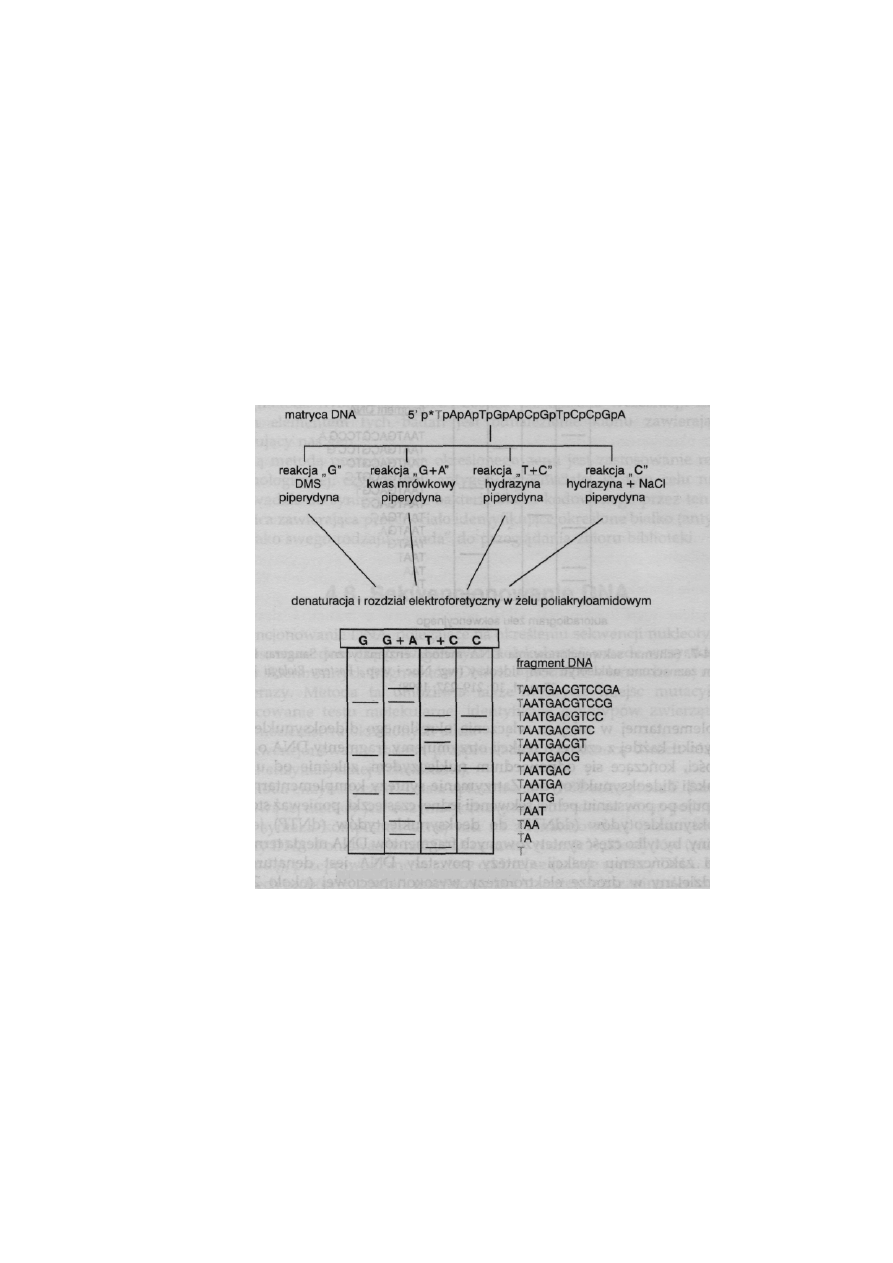
kleotydy lub wyznakowany starter. Można również zastosować detekcję
fluorescencyjną lub wyznakowanie biotyną.
Enzymatyczna metoda sekwencjonowania ma tę zaletę, że jest szybka,
ma bardzo dużą rozdzielczość i pozwala na sekwencjonowanie odcinka
DNA długości około 500-700 nukleotydów.
Metoda chemiczna, opracowana w 1977 roku przez Maxama i Gilberta, polega
na degradacji sekwencjonowanego fragmentu DNA poprzez chemiczne roz-
szczepienie poszczególnych nukleotydów. Do rozszczepiania wykorzystywane są
hydrazyna, siarczan dimetylu (DMS) i kwas mrówkowy, które mają zdolność
modyfikacji zasad w cząsteczce DNA. Do mieszaniny reakcyjnej dodawana jest
także piperydyna, która powoduje przerwanie łańcucha DNA w miejscu
zmodyfikowanym przez jeden z wymienionych związków chemicznych.
Sekwencjonowanie metodą chemiczną przeprowadza się, podobnie jak
przy metodzie enzymatycznej, w czterech oddzielnych probówkach. Przebie-
gają w nich reakcje rozszczepień prowadzonych w 4 wariantach (ryć. 4-8):
autoradiogramżelusekwencyjnego
Ryć. 4-8. Schemat sekwencjonowania DNA metodą chemiczną Maxama-Gilberta; kolorem
szarym zaznaczono znakowany nukleotyd (wg: Nuć i wsp., Postępy Biologii Komórki,
25 supl. 10 :219-237, 1998)
90

W wyniku tych reakcji otrzymujemy fragmenty DNA zaczynające się
zawsze od wspólnego radioaktywnego końca, których długość zależy od
miejsca rozszczepienia łańcucha DNA. Następnym etapem, podobnie jak
w metodzie enzymatycznej, jest rozdział elektroforetyczny tych fragmentów
w żelu poliakryloamidowym, autoradiografia i odczytanie sekwencji nukleo-
tydów. Metoda ta umożliwia sekwencjonowanie fragmentów DNA długości
około 250-300 nukleotydów.
Sekwencjonowanie kwasów nukleinowych zostało zautomatyzowane,
kilka firm oferuje specjalistyczną aparaturę.
Najnowsza metoda sekwencjonowania, tzw. pyroseąuencing, znacznie
różni się od powyższych metod. Wykorzystuje ona bowiem enzymatyczne
(z udziałem 4 enzymów i specyficznych substratów) dobudowywanie nici
komplementarnej do badanego fragmentu DNA, przy czym przy włączeniu
każdego nukleotydu powstaje sygnał świetlny. Reakcja ta przebiega na
mikropłytkach (96 dołków na płytce) po dodaniu substratów i enzymów do
dołka zawierającego badaną próbę DNA. Sekwencja badanego fragmentu
DNA jest odczytywana przez specjalną kamerę połączoną z komputerem (z
oprogramowaniem do analizy tych danych) na podstawie sygnału świetl-
nego, zamienionego na pik proporcjonalny do liczby nukleotydów włączo-
nych do nici komplementarnej do matrycowego DNA (badana próba).
4.9. Badanie ekspresji genów
Ekspresja genu może być badana na etapie transkrypcji (hybrydyzacja in
situ na skrawkach tkanek; northern blotting; RT-PCR) i translacji (western
blotting). Jedna z wymienionych metod, hybrydyzacja in situ na skrawkach
tkanek, pozwala także na wyróżnienie grup komórek, które morfologicznie
nie różnią się od sąsiednich, ale są inne pod względem biochemicznym.
Przykładem mogą być komórki nowotworowe badane bezpośrednio po
transformacji. Różnią się one od innych komórek ekspresją pewnych
genów, nieaktywnych w komórkach zdrowych.
4.10. Transgeneza
Transgenezą nazywamy technikę modyfikowania genomu metodami in-
żynierii genetycznej. Zwierzęta transgeniczne to takie, które w swoim
genomie zawierają zintegrowany DNA pochodzący od innego osobnika.
Jednym z pierwszych eksperymentów transgenezy było wprowadzenie na
początku lat 80. do zygoty myszy szczurzego genu hormonu wzrostu.
Głównym efektem tego zabiegu było szybsze tempo wzrostu i wyraźne
wydłużenie ciała myszy transgenicznych. Samce transgeniczne przekazywały
swojemu potomstwu obcy gen, natomiast samice były niepłodne. Podobne
91

rezultaty otrzymano po wprowadzeniu myszom ludzkiego genu czynnika
uwalniającego hormon wzrostu.
Materiał genetyczny wprowadza się do komórki zazwyczaj za pomocą
jednej z następujących technik:
• bezpośrednia mikroiniekcja genu lub fragmentu DNA do przedjądrza,
najczęściej męskiego, zapłodnionej komórki jajowej. Po mikroiniekcji DNA
dokonywany jest transfer zarodków do hormonalnie przygotowanych
biorczyń. Jest to główna metoda uzyskiwania transgenicznych zwierząt
gospodarskich;
• transfer materiału genetycznego przez zakażanie zrekombinowanymi
retrowirusami. Wielkość fragmentu DNA, około 8 kpz, który może być
wprowadzony tą metodą, ogranicza jej stosowanie. Ponadto istnieje pewne
niebezpieczeństwo replikacji wirusa, które można zmniejszyć przez stosowanie
zmodyfikowanych wirusów zdolnych do jednego tylko cyklu replikacyjnego;
• zastosowanie pierwotnych komórek zarodkowych — ESC (ang.
embryonic steam cells). W metodzie tej wykorzystuje się zdolność pierwot
nych komórek zarodkowych do wzrostu in vitro. Zrekombinowany fragment
DNA wprowadzany jest do hodowanych w warunkach in vitro ESC, które
wcześniej zostały pozyskane z węzłów zarodkowych blastocysty. Następnie
komórki te są wprowadzane ponownie do enukleowanych oocytów lub
pęcherzy trofoblastycznych (tzn. blastocyst pozbawionych węzła zarodkowe
go). Jak dotychczas opisana technika jest stosowana z sukcesem u myszy.
Transformację komórek można też wykonać na drodze elektroporacji
komórki biorcy lub lipofekcji (dostarczenie do komórki obcego DNA
w liposomach).
Dotychczas stosowane metody wprowadzania transgenu nie dają moż-
liwości sterowania tym procesem. Na razie nie mamy żadnego wpływu na
to, w którym miejscu genomu włączy się transgen. Skutki tego procesu
w zależności od miejsca inkorporacji transgenu mogą być różne, nawet
niekorzystne.
Należy jednak zauważyć, że w procesie transgenezy wprowadzane są
konstrukty genowe, które zazwyczaj złożone są z części strukturalnej
wprowadzanego genu i części promotorowej innego genu, umożliwiającej
ukierunkowaną ekspresję transgenu (np. w gruczole mlecznym).
Wykorzystanie techniki transgenezy
1. Doskonalenie zwierząt gospodarskich
Transgeneza stwarza duże możliwości poprawy cech użytkowych zwie-
rząt gospodarskich poprzez modyfikację ich genomu. Technika ta może być
stosowana do:
• uzyskania zwierząt o przyspieszonym tempie wzrostu (wzrost produk
cji mięsa) przez wprowadzenie do ich genomu genów kodujących polipep-
tydy wpływające na wzrost, np. gen struktury hormonu wzrostu (ang.
growth hormone — GH);
92
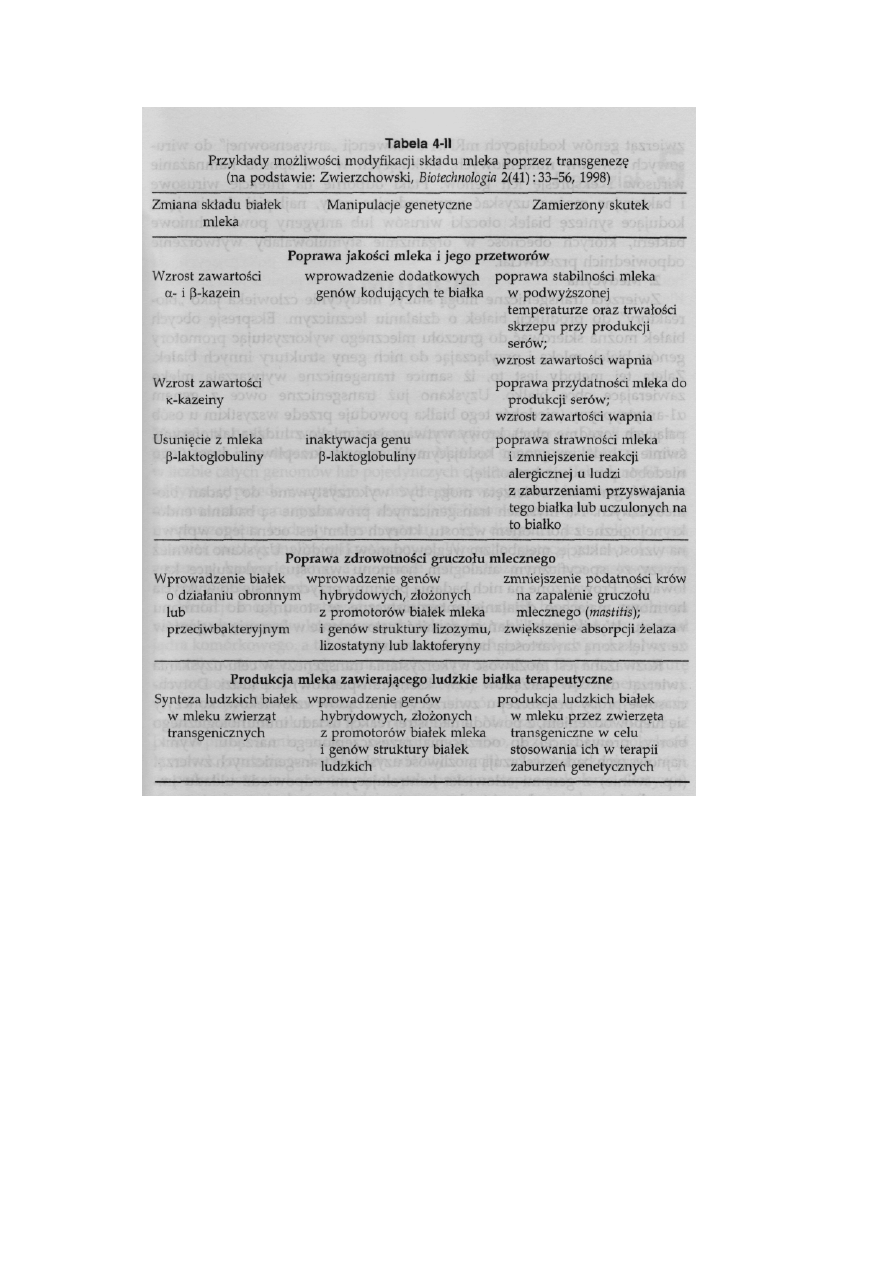
• modyfikacji składu mleka; duże nadzieje wiąże się z poprawą jakości
białek mleka. W tabeli 4-II przedstawione są niektóre możliwości modyfikacji
składu mleka w drodze transgenezy;
• modyfikacji cech biochemicznych u zwierząt, na przykład poprawy
jakości i tempa wzrostu wełny, poprzez wprowadzenie do genomu owiec
genów białek keratynowych.
Duże nadzieje wiąże się z uzyskiwaniem transgenicznych zwierząt
wykazujących tolerancję lub odporność na patogenne wirusy i bakterie.
Odporność na choroby można uzyskać przez wprowadzenie do komórek
93

zwierząt genów kodujących mRNA o sekwencji „antysensównej" do wiru-
sowych kwasów nukleinowych, blokujących w ten sposób namnażanie
wirusów i ekspresję ich genów. Ptaki odporne na infekcje wirusowe
i bakteryjne można uzyskać wprowadzając geny, najlepiej dominujące,
kodujące syntezę białek otoczki wirusów lub antygeny powierzchniowe
bakterii, których obecność w organizmie stymulowałaby wytworzenie
odpowiednich przeciwciał.
2. Medycyna
Zwierzęta transgeniczne mogą służyć medycynie człowieka jako „bio-
reaktory" do produkcji białek o działaniu leczniczym. Ekspresję obcych
białek można skierować do gruczołu mlecznego wykorzystując promotory
genów białek mleka i przyłączając do nich geny struktury innych białek.
Zaletą tej metody jest to, iż samice transgeniczne wytwarzają mleko
zawierające obce białko. Uzyskano już transgeniczne owce z genem cd-
antytrypsyny (niedobór tego białka powoduje przede wszystkim u osób
palących rozedmę płuc), krowy wytwarzające mleko z ludzką laktoferyną,
świnie z ludzkim genem kodującym IX czynnik krzepliwości krwi (jego
niedobór powoduje hemofilię).
Transgeniczne zwierzęta mogą być wykorzystywane do badań bio-
medycznych. Na myszach transgenicznych prowadzone są badania endo-
krynologiczne z hormonem wzrostu, których celem jest ocena jego wpływu
na wzrost, laktację, metabolizm węglowodanów i lipidów. Uzyskano również
myszy ze specyficznym analogiem hormonu wzrostu, wykazujące kar-
łowatość. Prowadzone na nich badania powinny przyczynić się do wykrycia
hormonu mającego działanie antagonistyczne w stosunku do hormonu
wzrostu. Wyniki tych badań mogą mieć zastosowanie w leczeniu pacjentów
ze zwiększoną zawartością hormonu wzrostu.
Rozważana jest możliwość wykorzystania transgenezy w celu uzyskania
zwierząt dawców narządów (tzw. ksenotransplantów) dla ludzi. Dotych-
czasowe próby przeszczepu zwierzęcych narządów człowiekowi kończyły
się niepowodzeniem, z powodu nadostrej reakcji układu immunologicznego
biorcy, prowadzącej do odrzucenia przeszczepionego narządu. Wyniki
najnowszych badań wskazują możliwość uzyskania transgenicznych zwierząt
(np. świnie) z genami człowieka kontrolującymi odpowiedź układu im-
munologicznego na obce gatunkowo antygeny.
W doświadczeniach żywieniowych zwierzęta transgeniczne mogą służyć
do badań ekspresji genów różnych czynników metabolicznych.
3. Badania naukowe
Obiecujące są wyniki badań regulacji ekspresji genów prowadzone na
zwierzętach transgenicznych. Szczególnie ważne dla badań biomedycznych
jest tworzenie nowych modeli zwierzęcych, które wykazują nadekspresję
określonych genów, ekspresję genu w niewłaściwej tkance lub ekspresję
genów zmutowanych.

Rozdział
Mutacje
Mutacje to nagłe i trwałe zmiany w informacji genetycznej. Dzieli się je na:
genomowe, chromosomowe i genowe. Mutacje genomowe dotyczą zmian
w liczbie całych genomów lub pojedynczych chromosomów. Ich identyfikacja
odbywa się przede wszystkim na drodze obserwacji mikroskopowej. Mutacje
chromosomowe, nazywane też aberracjami chromosomowymi, związane są
z naruszeniem budowy chromosomu, a ich diagnoza jest dokonywana
również metodą analizy mikroskopowej. Wreszcie mutacje genowe dotyczą
zmian w budowie genu, a poznanie ich podłoża wymaga zastosowania
metod molekularnych.
Mutacje mogą powstawać samoistnie, jako wynik błędów w przebiegu
niektórych procesów komórkowych, takich jak replikacja DNA czy podział
jądra komórkowego, a także naprawa uszkodzeń powstałych pod wpływem
czynników mutagennych, czyli takich, które uszkadzają DNA lub strukturę
chromosomu (np. promieniowanie jonizujące, niektóre związki chemiczne).
W przypadku powstania mutacji w komórce somatycznej jej trwanie
ograniczone jest okresem życia organizmu. Należałoby zatem rozróżnić
proces przenoszenia mutacji, powstałych de novo, przez kolejne pokolenia
komórek (mutacja w komórkach somatycznych) od dziedziczenia mutacji,
za pośrednictwem komórek płciowych, przez następne pokolenia. Oczywiś-
cie mutacja może być odziedziczona przez potomstwo jedynie wówczas,
gdy jej skutkiem nie jest śmierć lub niepłodność nosiciela.
Mutacje są pierwotnym źródłem zmienności genetycznej i dlatego są
nierozerwalnie związane ze zmianami ewolucyjnymi, obserwowanymi wśród
organizmów żywych. Ponieważ mutacja ma charakter przypadkowy i bez-
kierunkowy, dlatego większość z nich jest niekorzystna dla organizmów.
Długotrwała ewolucja pozwalała na utrwalenie tylko tych mutacji, które
były korzystne lub obojętne dla organizmu. Hodowla zwierząt jest oczywiście
procesem nieporównywalnie krótszym w stosunku do ewolucji, a ponadto,
cel hodowlany jest realizowany zazwyczaj tylko przez kilka najbliższych
95

pokoleń. Dlatego można bez większych zastrzeżeń przyjąć, że mutacje są
zjawiskiem ogólnie niepożądanym w populacjach hodowlanych, a hodowca
jest zazwyczaj zainteresowany eliminacją nosicieli nowo powstających
i niektórych utrwalonych mutacji. Dotyczy to przede wszystkim mutacji
genomowych i chromosomowych. W przypadku mutacji genowych kwestia
ta nie jest już tak oczywista, ponieważ obok mutacji szkodliwych (np.
choroby genetyczne) znanych jest wiele przykładów pokazujących, że
hodowca jest zainteresowany utrzymaniem mutacji w populacjach zwierząt.
Klasycznym przykładem są mutacje odpowiadające za barwę okrywy
włosowej, pojawiające się od czasu do czasu na fermach zwierząt futer-
kowych. Znane są i inne mutacje, które wpływają korzystnie na cechy
produkcyjne zwierząt, takie jak: hipertrofia mięśniowa u niektórych ras
bydła mięsnego (mutacja genu miostatyny), wysoka plenność u niektórych
ras owiec (np. mutacja w genie BMPR-IB) itp.
W dalszej części tego rozdziału duży nacisk położymy na omówienie
negatywnych skutków mutacji, z jakimi spotykamy się w hodowli zwierząt
gospodarskich.
5.1. Mutacje genomowe
Mutacje genomowe dzieli się na dwie kategorie: euploidie i aneuploidie.
Euploidia jest mutacją prowadzącą do zmiany liczby genomów w komór-
ce. Na przykład, zamiast dwóch genomów (2n) w komórce pojawiają się
trzy (3n — triploidia) albo cztery (4n — tetraploidia) genomy lub tylko jeden
genom (n — haploidia, monoploidia). Jeżeli powstają układy o większej
liczbie genomów niż dwa, to stan ten określa się jako poliploidalność.
Powstawanie w zygocie mutacji genomowych typu euploidii może być
wynikiem różnych nieprawidłowości, wśród których do najważniejszych
zalicza się (ryć. 5-1):
1. Zapłodnienie haploidalnego oocytu II rzędu przez więcej niż jeden
plemnik, co daje początek zygocie triploidalnej (zapłodnienie przez dwa
plemniki) czy tetraploidalnej (zapłodnienie przez trzy plemniki). Zdarzenia
takie określa się mianem połispermii.
2. Zaburzenie przebiegu mejozy, głównie żeńskiej, prowadzące do
powstania oocytów II rzędu o niezredukowanej (2n) liczbie chromosomów.
Zapłodnienie takiego oocytu przez haploidalny plemnik daje początek
triploidalnej zygocie. Zakłócenie przebiegu mejozy może nastąpić podczas
anafazy I i wówczas nie dochodzi do wyrzucenia pierwszego ciałka kierunko
wego, a oocyt II rzędu ma diploidalną liczbę chromosomów. Do zaburzenia
wyrzucenia ciałka kierunkowego może dojść również podczas anafazy II,
która odbywa się już po wniknięciu plemnika do oocytu. Także w takim
przypadku przedjądrze żeńskie będzie miało diploidalną liczbę chromoso
mów. Po replikacji i zlaniu przedjądrzy powstaje zygota triploidalna.
96
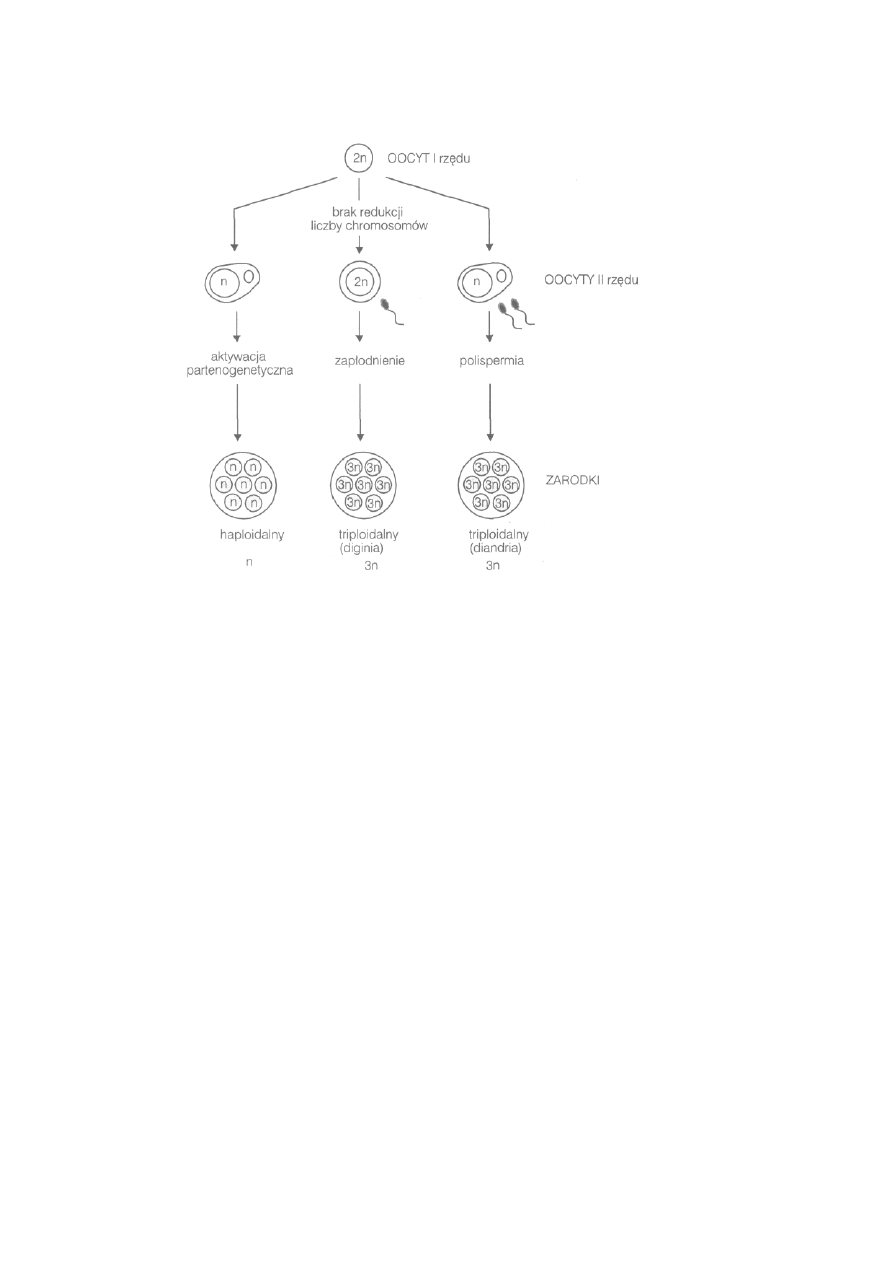
Ryć. 5-1. Niektóre mechanizmy powstawania mutacji genomowych typu euploidii
3. Podjęcie podziału mejotycznego przez spermatogonium lub oogonium
o nieprawidłowym poziomie ploidalności, np. tetraploidalne oogonium,
które powstało w wyniku zakłócenia cytokinezy po mitotycznym podziale
jądra komórkowego. Z komórki takiej po zakończeniu mejozy i zapłodnieniu
powstaje triploidalna zygota.
4. Aktywacja partenogenetyczna polegająca na podjęciu podziałów
mitotycznych przez niezapłodniony oocyt (ginogeneza), czego skutkiem jest
rozwój haploidalnego zarodka.
W trakcie rozwoju zarodkowego może dojść do uformowania układu
miksoploidalnego, to znaczy takiego, gdzie w obrębie zarodka występują komórki
różniące się poziomem ploidalności, np. 2n i 4n czy 2n i 3n itd. Jest to skutek
zakłóceń w pierwszych podziałach mitotycznych zarodka, takich na przykład jak
brak cytokinezy po podziale jądra komórkowego. Ponieważ komórki te wywodzą
się z jednej zygoty, układ taki klasyfikowany jest jako mozaicyzm.
Euploidie są mutacjami letalnymi, co oznacza, że prowadzą one do
śmierci osobnika, zazwyczaj we wczesnym okresie rozwoju zarodkowego.
Tym samym każdy przypadek takiej mutacji zalicza się do kategorii nowo
powstałych (de novo). Praktycznie nie spotyka się tego rodzaju mutacji
w okresie pourodzeniowym. Mutacje te są jedną z przyczyn niepowodzeń
97

w rozrodzie zwierząt. Trudno jednak określić, jaka jest skala tego problemu.
Z badań prowadzonych na zarodkach uzyskiwanych metodą zapłodnienia
in vitro wynika, że co najmniej kilkanaście procent tak uzyskanych zarodków
może być obarczonych mutacjami typu poliploidii lub haploidii.
Aneuploidia to druga kategoria mutacji genomowych, która związana
jest ze zmianą liczby chromosomów w pojedynczych parach homologicz-
nych. Tak więc brak chromosomu nazywa się monosomią (2n — 1), a nadmiar
chromosomów to trisomia (2n +1). Jeśli zmiany wystąpią równocześnie
w dwóch parach chromosomów, to stan taki określany jest jako podwójna
monosomią (2n —l —1) lub podwójna trisomia (2n + l + l). Jeśli natomiast
występuje brak obu chromosomów w parze, to stan taki określany jest jako
nullisomia (2n —2), a obecność czterech chromosomów homologicznych
nazywany jest tetrasomią (2n + 2).
Aneuploidie są spowodowane nieprawidłowościami w przebiegu po-
działów komórkowych. W mejozie momentem krytycznym jest segregacja
chromosomów do przeciwległych biegunów komórki podczas anafazy I.
Nieprawidłowa segregacja w jednym biwalencie, gdy oba chromosomy
zmierzają do jednego bieguna komórki, prowadzi do powstania gamet
o liczbie chromosomów n + 1 oraz n —1. Jeżeli gamety takie będą uczest-
niczyły w zapłodnieniu, to powstaną zarodki obarczone trisomia (2n +1) lub
monosomią (2n —1). Również podczas podziałów mitotycznych (lub drugiego
podziału mejotycznego) może dojść do powstania komórek z nieprawidłową
liczbą chromosomów. Gdy podczas anafazy mitotycznej chromosomy
siostrzane nie rozejdą się symetrycznie do przeciwległych biegunów komórki,
to w komórkach potomnych powstaną układy 2n + l (oba chromosomy
siostrzane po podziale centromeru przeszły do jednego bieguna) oraz 2n — l
(do bieguna nie przeszedł żaden z chromosomów siostrzanych). Błąd
segregacji chromosomów siostrzanych podczas podziału komórek somatycz-
nych (np. zarodka) prowadzi do mozaicyzmu.
Zdecydowana większość aneuploidii jest letalna. Aneuploidie tylko
niektórych chromosomów nie prowadzą do śmierci nosiciela, jednak
powodują powstanie licznych wad rozwojowych. Wyjątek stanowią aneu-
ploidie chromosomów płci, którym na ogół nie towarzyszą wyraźne
zaburzenia w rozwoju somatycznym. Wśród zidentyfikowanych u zwierząt
gospodarskich aneuploidii dominują właśnie przypadki dotyczące chromoso-
mów płci. Zazwyczaj mutacje te stwierdzano w układzie mozaikowym,
w którym obok linii komórkowej z prawidłowym zestawem chromo-
somowym występowała jedna lub więcej linii aneuploidalnych. Wskazuje
to, że aneuploidie w postaci czystej prowadzą prawdopodobnie do śmierci
osobnika. Równocześnie wynika z tego, że podczas rozwoju zarodkowego
dochodzi do nieprawidłowej segregacji chromosomów siostrzanych. Aneu-
ploidie chromosomów płci prowadzą zazwyczaj do niepłodności.
Aneuploidia najczęściej opisywaną u zwierząt jest monosomią chromo-
somu X u klaczy (ryć. 5-2a). Klacz obarczona tą mutacją ma kariotyp
98
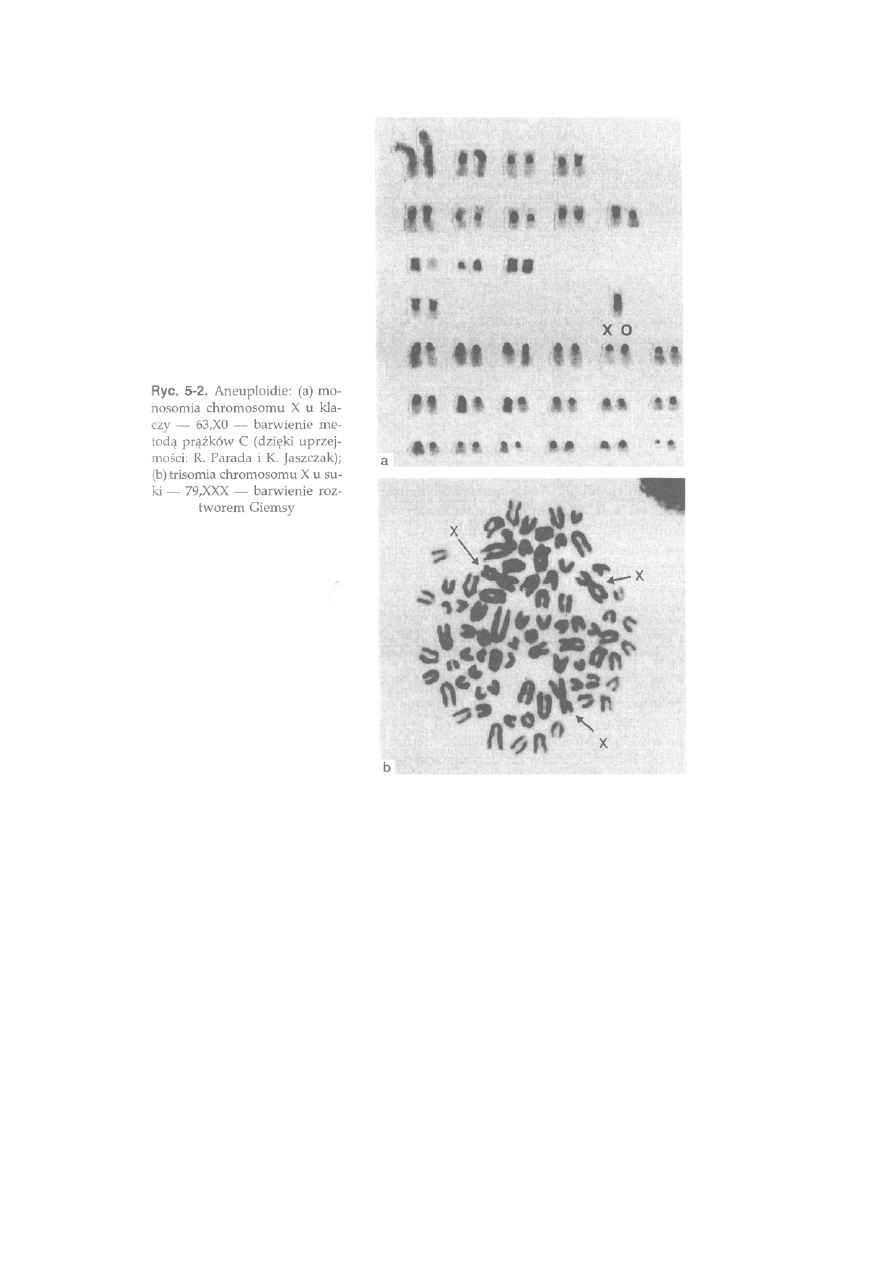
2n = 63,X0 („0" oznacza brak chromosomu X) lub częściej jest to kariotyp
mozaikowy 63,XO/64,XX. Zwierzęta takie mają zasadniczo normalny eksterier,
zazwyczaj nie wykazują objawów rui i są bezpłodne ze względu na
niedorozwój jajników. W związku z tym hodowca nie może znaleźć
przyczyny jałowości takiej klaczy, dopóki nie zdecyduje się na prze-
prowadzenie badania cytogenetycznego. Znane są również przypadki
trisomii XXY u buhajów. Osobniki takie mają diploidalną liczbę chromo-
somów 61,XXY lub występuje u nich układ mozaikowy 60,XY/61,XXY.
99

Również w przypadku tej chromosomopatii nosiciel dotknięty jest bezpłod-
nością, z tym jednak że zazwyczaj towarzyszy temu wolniejsze tempo
wzrostu somatycznego oraz widoczny niedorozwój jąder, co może być
stosunkowo łatwo zauważone przez hodowcę czy lekarza weterynarii.
Stwierdzenie wymienionych zaburzeń jest wystarczającą podstawą wy-
brakowania takiego osobnika. Przypadki trisomii chromosomu X opisywano
znacznie rzadziej. Przykładem może być niepłodna suka o prawidłowym
eksterierze (ryć. 5-2b). W przeciwieństwie do monosomii X0 oraz trisomii
XXY dość często się zdarza, że osobniki mające w swoim kariotypie trzy
chromosomy X są płodne. W potomstwie takich samic mogą się pojawić
zwierzęta obarczone trisomią XXY lub XXX. Jest to skutek segregacji
chromosomów w anafazie I, w wyniku której pojawia się około 50% gamet
z chromosomem X oraz około 50% gamet z układem XX. Zapłodnienie tych
ostatnich prowadzi do powstania zarodka z trisomią.
Przedstawione wyżej prawidłowości wskazują, że aneuploidie ilekroć się
pojawiają, to zazwyczaj mają pochodzenie de novo, gdyż skutkiem ich
wystąpienia jest albo śmierć nosiciela, albo głębokie zaburzenia rozwojowe
lub bezpłodność. Dzięki temu, poza nielicznymi wyjątkami, nie ma ryzyka
przeniesienia tej mutacji na następne pokolenia, a zatem i szerokiego
rozprzestrzenienia w populacji. Niemniej jednak, niezidentyfikowanie
nosiciela wśród zwierząt hodowlanych może być przyczyną dużych strat
ekonomicznych hodowcy, wynikających z kosztów odchowu oraz utrzyma-
nia zwierzęcia o upośledzonych funkcjach rozrodczych. Sytuacja taka jest
szczególnie dotkliwa dla hodowców koni, ze względu na dość częste
występowanie u klaczy monosomii chromosomu X, której zdiagnozowanie,
bez zastosowania metod cytogenetycznych, jest niemożliwe.
5.2. Mutacje chromosomowe
Pierwotną przyczyną zmian w budowie chromosomu jest jego uszkodzenie
(pęknięcie). Jeżeli pęknięcie nie zostanie naprawione, przez odpowiedzialne
za to systemy naprawy komórkowej, to nastąpi utrata fragmentu chromoso-
mu (delecja). Możliwe jest również nieprawidłowe przeprowadzenie napra-
wy uszkodzenia. Wówczas dochodzi do przegrupowania fragmentów w obrę-
bie jednego chromosomu (inwersja) lub kariotypu (fuzja centryczna, tandem
fuzja, translokacja wzajemna). Mogą powstać także inne nietypowe struktury,
takie jak izochromosomy (są zbudowane z dwóch identycznych ramion) lub
chromosomy koliste. Za przyczynę powstania izochromosomów uznaje się
nieprawidłowy podział centromeru w płaszczyźnie prostopadłej do osi
chromosomu, podczas podziału komórkowego. Z kolei chromosom kolisty
może powstać w wyniku dwóch pęknięć na przeciwległych końcach chromo-
somu, a następnie połączenia tych miejsc z równoczesną utratą małych
fragmentów na obu końcach chromosomu. Przyczyną mutacji chromosomo-
wej może być również niezrównoważona wymiana fragmentów chromatyd
100

niesiostrzanych podczas crossing over, prowadząca do powstania chromoso-
mów z brakiem (delecja) lub z podwojeniem (duplikacja) jakiegoś fragmentu.
Mutacja chromosomowa może wystąpić w układzie zrównoważonym,
jeżeli w komórce nie nastąpiła zmiana ilości informacji genetycznej, lub też
w układzie niezrównoważonym, gdy ilość informacji genetycznej w komórce
została zmieniona. Pewne mutacje chromosomowe są ze swojej natury
klasyfikowane już od momentu powstania do grupy niezrównoważonych
(delecje, duplikacje, izochromosomy). Inne — tzn. translokacje wzajemne,
tandem fuzje, fuzje centryczne, inwersje, występując pierwotnie w układzie
zrównoważonym, mogą prowadzić poprzez zakłócony przebieg segregacji
chromosomowej w anafazie I do powstania gamet niezrównoważonych,
a dalej do powstania zygot o zmienionej zawartości materiału genetycznego.
Dziedziczenie mutacji chromosomowych możliwe jest oczywiście jedynie
wówczas, gdy występuje ona w komórkach linii płciowej.
Mutacje chromosomowe, których skutkiem jest przegrupowanie infor-
macji genetycznej w kariotypie, prowadzą do zmiany sąsiedztwa genów.
Zmiana taka może pociągnąć za sobą naruszenie procesów kontroli ekspresji
genów. Zdarzyć się może, że przegrupowanie w pobliżu jakiegoś genu
może wywołać jego ekspresję lub jej zanik. Ma to związek zarówno
z funkcjonowaniem sekwencji poprzedzających gen (promotor, wzmacniacz,
wyciszacz), jak i ze strukturą samego genu. Znane są liczne przykłady
u ludzi, a także i zwierząt laboratoryjnych wpływu takich przegrupowań
chromosomowych na aktywację onkogenów lub inaktywację antyonkoge-
nów, czego skutkiem jest uruchomienie procesu nowotworowego.
U zwierząt gospodarskich najczęściej diagnozowane są mutacje chromo-
somowe w układzie zrównoważonym. Wynika to przede wszystkim z tego,
że mutacje chromosomowe niezrównoważone, podobnie jak aneuploidie,
są letalne lub prowadzą do poważnych, zauważalnych zmian fenotypowych.
Przypadki takie, odnotowywane jako martwe urodzenie, padnięcie po
urodzeniu lub ubój z konieczności, bardzo rzadko są zgłaszane przez
hodowców czy lekarzy weterynarii do badań cytogenetycznych i dlatego
można przyjąć, że zdecydowana większość tych przypadków nie jest
identyfikowana. Z kolei mutacje zrównoważone, nie wywołując zazwyczaj
efektu w postaci zauważalnych zmian fenotypowych, mają najczęściej
negatywny wpływ na płodność nosiciela mutacji. Jeżeli dotknie to osobnika
o dużej wartości genetycznej, to szansa na wykrycie nosicielstwa mutacji
jest znacznie większa, bo osobniki takie z urzędu mogą być objęte badaniami
cytogenetycznymi (np. buhaje), ponadto hodowcy są bardziej zainteresowani
poznaniem przyczyny zmniejszonej płodności.
W dalszej części tego rozdziału będzie mowa o mutacjach chromo-
somowych, które w momencie powstania mają charakter zrównoważony.
Zaliczane są do nich: fuzje centryczne, inaczej centromerowe (translokacje
robertsonowskie), fuzje tandemowe, translokacje wzajemne i inwersje,
wśród których wyróżniane są inwersje peri- i paracentryczne. Szczególna
101
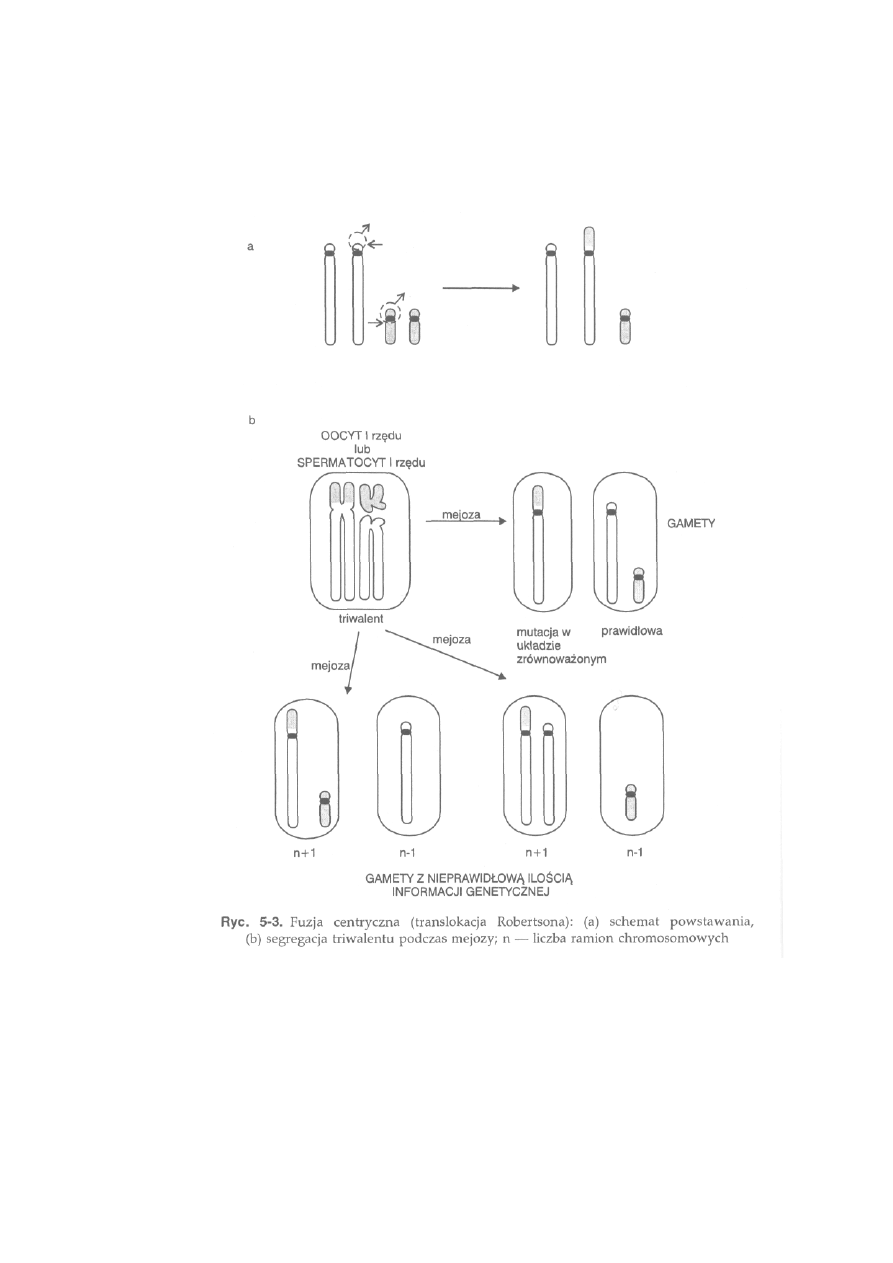
uwaga będzie poświęcona mechanizmom powodującym zmniejszenie płod-
ności u nosicieli tego typu mutacji.
Fuzja centryczna (translokacja robertsonowska) powstaje wówczas,
gdy dwa chromosomy akrocentryczne pękną w pobliżu centromeru,
a następnie uszkodzenie to zostanie niewłaściwie naprawione (ryć. 5-3a,
102
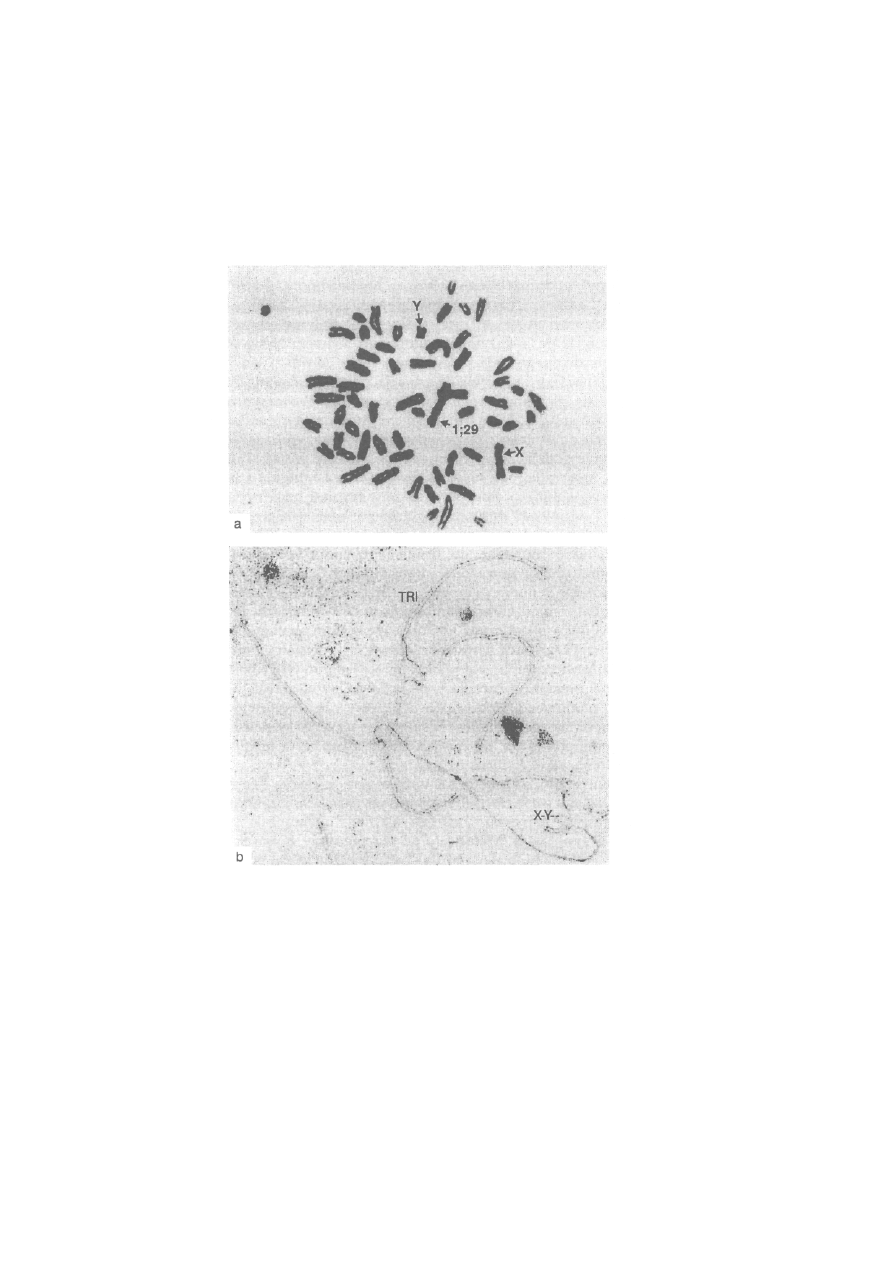
5-4a). Duże fragmenty, reprezentujące prawie cale ramiona długie chromo-
somów, zostają połączone w centromerze i w efekcie powstaje jeden duży
dwuramienny chromosom, natomiast małe fragmenty okołocentromerowe
zostają zagubione podczas następnych podziałów komórkowych (nie wcho-
dzą do jąder komórkowych odbudowujących się podczas telofazy). Utrata
małych fragmentów z okolicy centromeru, który zawiera przede wszystkim
niekodujące sekwencje powtarzalne, nie odbija się negatywnie na nosicielu.
Ryć. 5-4. Translokacje robertsonowskie: (a) rob(l;29), buhaj, barwienie roztworem Giemsy;
chromosom translokacyjny oraz chromosomy pici są wskazane; (b) rob(5;7), kozioł,
kompleksy synaptonemalne obserwowane w transmisyjnym mikroskopie elektronowym;
triwalent oraz biwalent X-Y są wskazane
103

Skutkiem tej mutacji jest zmniejszenie liczby chromosomów o jeden, z rów-
noczesnym zachowaniem nie zmienionej liczby ramion chromosomowych.
Jeżeli fuzji uległy chromosomy homologiczne, to podczas anafazy I
mejozy nastąpi nieprawidłowa segregacja chromosomów. W pachytenie
profazy I nowo powstały chromosom dwuramienny pozostanie jako uniwa-
lent, natomiast w anafazie I do jednego bieguna przejdzie tenże dwu-
ramienny chromosom, podczas gdy na drugim biegunie zabraknie jednego
chromosomu. Prowadzi to oczywiście do tego, że połowa gamet będzie typu
n —l (układ powstający na biegunie, do którego nie przeszedł chromosom
dwuramienny), a druga będzie miała n chromosomów, z tym jednak że
wśród nich będzie chromosom dwuramienny, co w rzeczywistości od-
powiada układowi n + 1. W przypadku zapłodnienia powstają zygoty
aneuploidalne, które z dużym prawdopodobieństwem zginą już podczas
rozwoju embrionalnego bądź płodowego.
Zupełnie inaczej przedstawia się sytuacja, gdy fuzja obejmie chromosomy
wywodzące się z różnych par homologicznych. Wówczas w komórce
powstaje układ składający się z jednego chromosomu dwuramiennego oraz
po jednym chromosomie z dwóch par akrocentrycznych. Jeżeli komórka
taka rozpocznie podział mejotyczny, to w pachytenie profazy I uformuje się
triwalent, w którym chromosomy akrocentryczne koniugują z homologicz-
nymi ramionami chromosomu powstałego w wyniku fuzji (ryć. 5-3b, 5-4b).
W anafazie I zazwyczaj dochodzi do zrównoważonej segregacji, podczas
której chromosom dwuramienny przechodzi do jednego bieguna, a oba
chromosomy akrocentryczne do drugiego bieguna komórki. W efekcie
powstają gamety o zrównoważonej informacji genetycznej, mimo iż połowa
z nich (niosąca dwuramienny chromosom) będzie miała n —l chromosomów.
Jednakże liczba ramion chromosomowych w obu rodzajach gamet będzie
taka sama. W niewielkiej części komórek w stadium anafazy I może dojść
do niewłaściwej segregacji triwalentu i wówczas powstaną gamety, a dalej
zygoty o nieprawidłowej liczbie ramion chromosomowych, czyli ukształtuje
się układ aneuploidalny. Zarodki takie będą ginęły w okresie życia zarod-
kowego lub płodowego. Hodowca będzie wówczas odnotowywał zmniej-
szoną płodność takiego osobnika. Należy zaznaczyć, że nosiciele fuzji mają
prawidłowy fenotyp i nie można ich zidentyfikować w inny sposób, jak
poprzez badanie cytogenetyczne.
Fuzje centryczne są często identyfikowane u bydła, szczególnie ras
mięsnych. Opisano wiele różnych fuzji, w których uczestniczyły chromosomy
z różnych par, ale najpowszechniejsza jest fuzja obejmująca chromosomy
z pary l i 29 (ryć. 5-4a). Mutację tę po raz pierwszy opisał w 1964 roku
Gustavsson u czerwono-białego bydła szwedzkiego. Pięć lat później ten sam
badacz wykazał, że jej nosiciele mają zmniejszoną płodność o około 5%.
Fuzje 1/29 zidentyfikowano w ponad 50 rasach bydła, na wszystkich
kontynentach, a w tym również w Polsce. Najwięcej przypadków fuzji
stwierdza się w rasach mięsnych (montbelliard, blonde d'Aquitaine itp.),
104
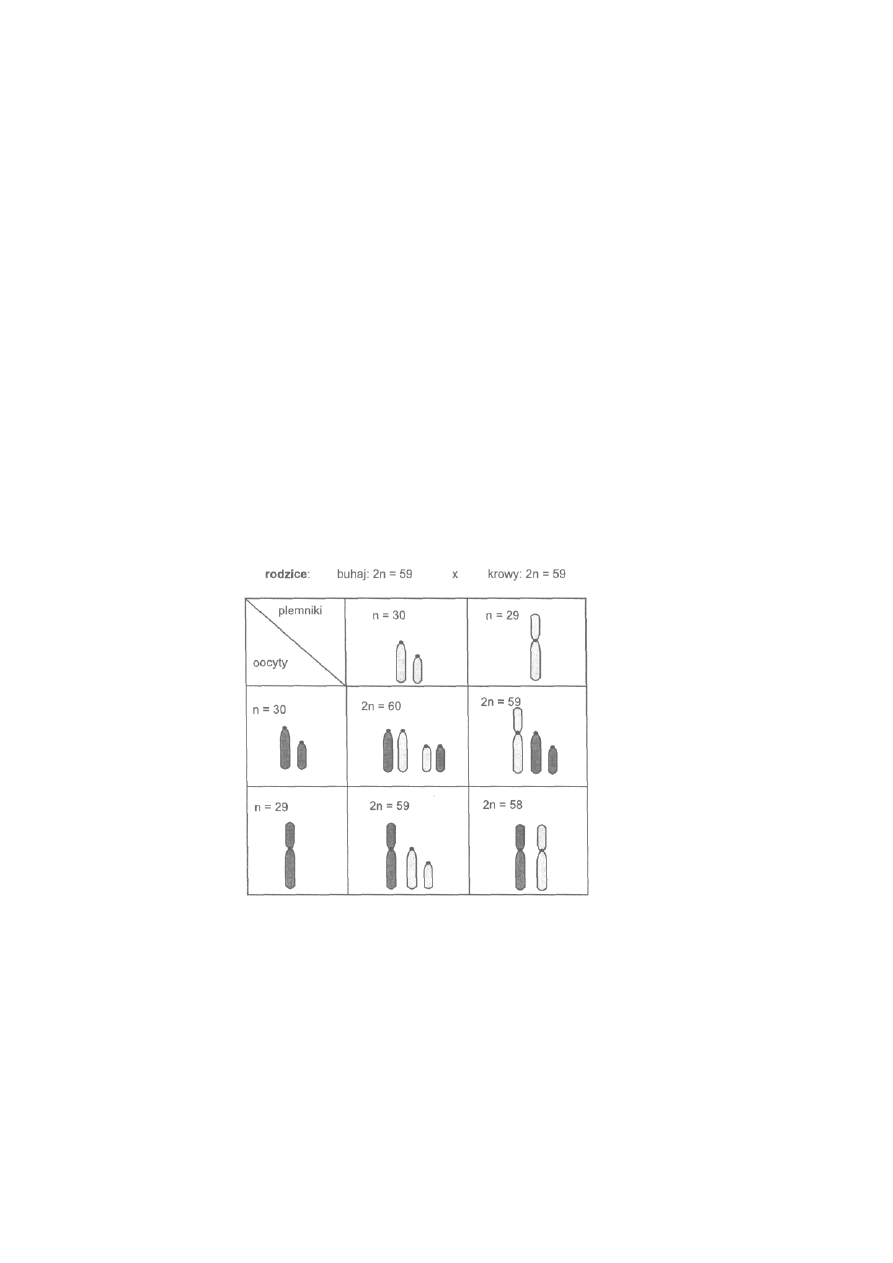
a niekiedy także w małych izolowanych rasach lokalnych (np. białe bydło
rrytyjskie, włoskie bydło rasy podolian czy bydło korsykańskie). Obser-
wowane zmniejszenie płodności jest skutkiem wczesnej zamieralności
zarodków o niezrównoważonym kariotypie. Oprócz fuzji 1;29 zdiagnozo-
wano wiele innych przypadków, w których zaangażowane były chromosomy
z innych par. Przykładem może być fuzja między chromosomami z pary
5 i 22, którą opisano w Polsce.
Szerokie rozprzestrzenienie fuzji centrycznych u bydła i związane z tym
zmniejszenie płodności stało się główną przyczyną wprowadzenia w wielu
krajach do praktyki hodowlanej obowiązkowych badań cytogenetycznych
buhajów przed ich dopuszczeniem do użytkowania rozpłodowego. Szwecja
była pierwszym krajem, który wprowadził taką regulację. W Polsce, począw-
szy od 1989 roku, obowiązkowymi badaniami objęte są wszystkie młode
buhaje, przed ich wprowadzeniem do stacji produkcji nasienia.
Gatunkiem, u którego fuzja centryczna występuje bardzo często, jest lis
polarny. W tym przypadku można wręcz mówić o polimorfizmie kariotypo-
wym, bowiem około 50% zwierząt ma kariotyp heterozygotyczny ze względu
na fuzję (dotyczy ona, jedynych w kariotypie lisa, dwóch par akrocentryków,
nr 23 i 24), a około 30% ma kariotyp podstawowy — 2n = 50 oraz pozostałe
około 20% ma kariotyp homozygotyczny ze względu na fuzję — 2n = 48.
Ciekawe, że u tego gatunku nie stwierdza się zmniejszonej płodności
zwierząt o 2n = 49, natomiast niektóre opracowania wskazują na zwiększoną
plenność samic o kariotypie 2n = 48.
Ryć. 5-5. Dziedziczenie translokacji robertsonowskiej na przykładzie bydła. Chromosomy
biorące udział w fuzji przedstawiono schematycznie
105

Należy podkreślić, że fuzja centryczna jest dziedziczona przez połowę
potomków nosiciela. Jeżeli na przykład u bydła zostaną skojarzeni ze sobą
nosiciele tej samej fuzji centrycznej, to u potomstwa możemy się spodziewać
25% osobników o normalnym kariotypie 2n = 60, 50% nosicieli fuzji
o 2n = 59 i 25% osobników będących homozygotami ze względu na fuzję,
0 2n = 58 (ryć. 5-5). Przedstawione obserwacje wskazują, że szerokie
rozprzestrzenienie fuzji centrycznych w populacjach zwierzęcych (głównie
bydło, lisy polarne, a w mniejszym stopniu także owce i kozy) jest skutkiem
ich dziedziczenia, a nie częstego powstawania de novo. Pokazuje to jedno
cześnie, jak łatwo mutacja taka może zostać rozprzestrzeniona w populacji,
szczególnie wówczas, gdy rozród jest oparty na sztucznej inseminacji.
Translokacja wzajemna powstaje w wyniku niewłaściwego
połączenia podczas procesu naprawy pęknięć nici chromatynowych,
pochodzących z dwóch różnych chromosomów. Jeżeli mutacji takiej będą
podlegały chromosomy homologiczne i wymienione zostaną nierówne
fragmenty chromosomów, to podczas mejozy dojdzie do powstania gamet,
które będą obarczone bądź delecją, bądź duplikacją. Skutek takiej
translokacji będzie zazwyczaj letalny, tak jak to najczęściej obserwuje się
przy mutacjach niezrównoważonych.
Jeżeli translokacją wzajemną będą objęte chromosomy niehomologiczne
(ryć. 5-6a, 5-7), to podczas pachytenu profazy I zostanie uformowany
tetrawalent (ryć. 5-6b, 5-8), który zazwyczaj będzie segregował w anafazie
1 w układzie 2:2, tzn. dwa chromosomy przejdą do jednego bieguna i dwa
chromosomy do drugiego bieguna komórki. Segregacja symetryczna (2:2)
nie gwarantuje jeszcze, że powstaną gamety o zrównoważonej informacji
genetycznej. Wśród trzech możliwych segregacji symetrycznych tylko jedna
warunkuje powstanie biegunów o zrównoważonej ilości informacji genetycz
nej. Jest to segregacja naprzeciwległa, w wyniku której do jednego bieguna
wędrują po jednym nie zmienionym (prawidłowym) chromosomie z obu
par, a do drugiego przechodzą chromosomy, które uczestniczyły w trans
lokacji. Należy zaznaczyć, że suma informacji genetycznej zawarta w dwóch
chromosomach, które podlegały translokacji, jest taka sama jak w dwóch nie
zmienionych chromosomach. W ten sposób segregacja naprzeciwległa
odpowiada za powstanie gamet o zrównoważonej ilości informacji genetycz-
nej. Dwa pozostałe rodzaje segregacji, określane jako sąsiednie, prowadzą
do uformowania niezrównoważonej (delecje, duplikacje) ilości informacji
genetycznej w gametach. Na niezrównoważenie ilości informacji genetycznej
wpływa również crossing over, jeśli zajdzie na odcinku między punktem
pęknięcia i wymiany fragmentów chromosomowych a centromerem. Oczy
wiście segregacja niesymetryczna (3: l lub 4:0) również powoduje utworze
nie gamet genetycznie niezrównoważonych.
Podobnie jak w przypadku fuzji centrycznej, także translokacją wza-
jemna jest mutacją dziedziczną, jeśli oczywiście wystąpi w linii komórek
płciowych. Przyjmuje się, że około 50% gamet wytwarzanych przez
106
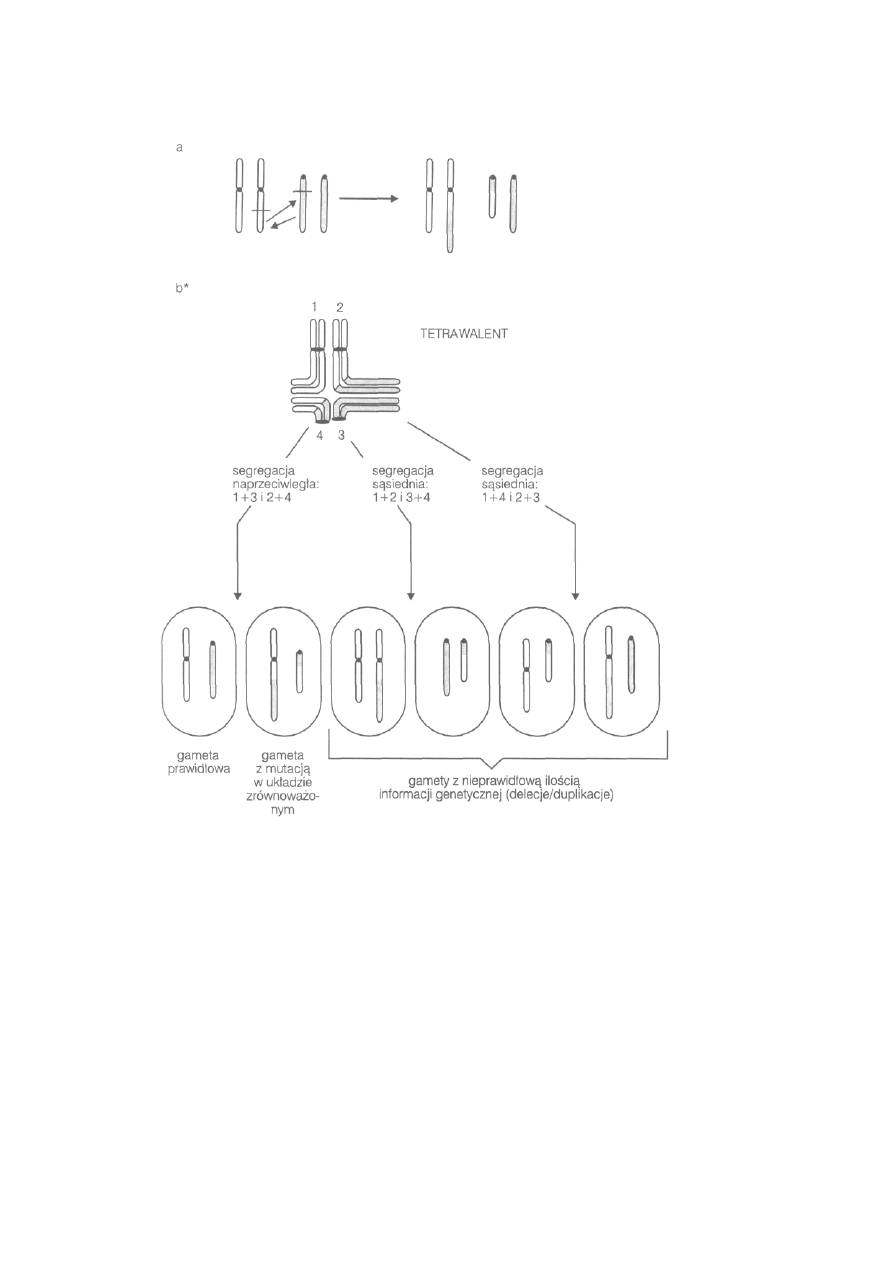
*Schemat nie uwzględnia segregacji tetrawalentu, w którym crossing over
wystąpiło między centromerem i punktem pęknięcia/wymiany
Ryć. 5-6. Translokacja wzajemna: (a) schemat powstawania; (b) segregacja tetrawalentu
podczas mejozy
nosiciela translokacji jest niezrównoważonych genetycznie i dlatego jeżeli
będą one uczestniczyły w zapłodnieniu, to doprowadzą do powstania
zygot, które będą ginęły zazwyczaj w życiu płodowym. Hodowca odnotuje
107
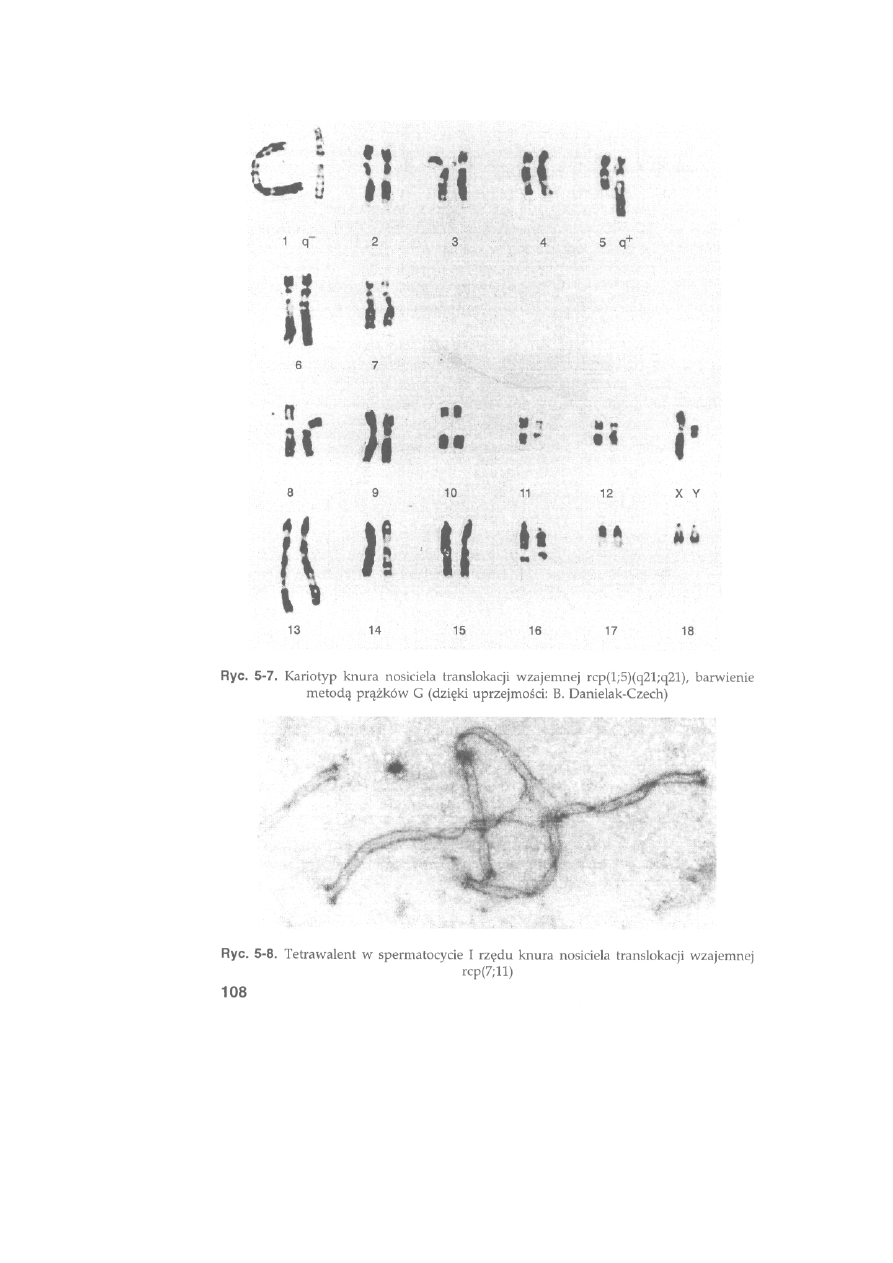

wówczas około 50-procentowy spadek płodności (lub plenności) u takiego
osobnika. Druga połowa gamet będzie miała zrównoważoną ilość informacji
genetycznej, ale wśród nich połowa będzie miała układ, w którym wystąpią
dwa translokowane chromosomy. Właśnie te gamety są odpowiedzialne za
przenoszenie translokacji wzajemnej do następnego pokolenia. W ostatecz-
nym rozrachunku po nosicielu skojarzonym z osobnikami o prawidłowym
kariotypie uzyska się potomstwo, w którym polowa będzie miała prawid-
łowy kariotyp, a połowa będzie nosicielami translokacji. Potomstwo o nie-
zrównoważonym kariotypie zazwyczaj ginie w okresie życia płodowego lub
wkrótce po urodzeniu.
Na krótkie omówienie zasługuje specjalna kategoria translokacji wzajem-
nych, która prowadzi do bezpłodności samców nosicieli. Stan taki towarzyszy
nosicielstwu translokacji wzajemnej, w którą zaangażowany jest chromo-
som X (translokacja typu X-autosom). Bezpłodność samców nosicieli jest
wynikiem zablokowania procesu spermatogenezy we wczesnych stadiach
mejozy (pachyten profazy I). Przypuszcza się, że ma to związek z przed-
wczesnym uaktywnieniem chromosomu X podczas spermatogenezy. U samic
nosicielek stwierdza się jedynie zmniejszenie płodności, podobnie jak
w przypadku translokacji typu autosom-autosom.
Translokacje wzajemne najczęściej są rozpoznawane u świń (ryć. 5-7).
Do tej pory zidentyfikowano na świecie, w rozmaitych rasach, ponad 90
różnych translokacji wzajemnych, które były rozprzestrzenione w popu-
lacjach w różnym stopniu.
Trzeba podkreślić, że nosiciele translokacji (kariotyp zrównoważony)
mają prawidłowy fenotyp i niczym nie odróżniają się od osobników
o prawidłowym kariotypie. Knury nosiciele mają prawidłowe libido, a obraz
nasienia (koncentracja, morfologia czy ruchliwość plemników) nie różni się
w porównaniu z nasieniem knurów o prawidłowym kariotypie. Podobnie
normalny fenotyp mają lochy nosicielki. Jedyną wskazówką sugerującą
nosicielstwo takiej mutacji są obniżone wskaźniki płodności lub plenności.
Oczywiście stan taki może być spowodowany różnymi czynnikami. Niemniej
obserwacje takie powinny być traktowane jako sygnał skłaniający do
przeprowadzenia badań cytogenetycznych. Na przykład we Francji badania
cytogenetyczne wykonywane są u knurów, po których uzyskano mioty
o obniżonej liczebności (poniżej 8 prosiąt w miocie).
Fuzja tandemowa jest mutacją podobną do fuzji centrycznej. Jej
powstanie związane jest z nieprawidłową naprawą pęknięć w dwóch
chromosomach. Jedno z nich występuje w części końcowej ramienia
w chromosomie jedno- lub dwuramiennym, a drugie tuż pod centromerem
w ramieniu długim chromosomu akrocentrycznego. W efekcie duże fragmenty
chromosomowe zostają połączone tandemowo — jeden za drugim, a małe
fragmenty (okołocentromerowy z chromosomu akrocentrycznego i dystalny
z drugiego chromosomu) zostają zagubione, podobnie jak w przypadku fuzji
centrycznej. Jeżeli tandem fuzja zajdzie między dwoma homologicznymi
109

chromosomami akrocentrycznymi, to podczas mejozy, podobnie jak przy
fuzji centrycznej, dojdzie do powstania aneuploidalnych gamet.
Powstanie tandem fuzji między chromosomami niehomologicznymi
wywołuje analogiczne skutki jak fuzja centryczna, tzn. może powodować
nieznaczne zmniejszenie płodności z powodu zamierania zarodków, które
powstały na bazie gamet o niezrównoważonej ilości informacji genetycznej.
Mutacja ta jest dziedziczona przez połowę potomków nosiciela, analogicznie
do schematu przedstawionego dla translokacji robertsonowskich. Tandem
fuzja była rzadko opisywana u zwierząt gospodarskich. Znane są nieliczne
przypadki tej mutacji u bydła, chociaż w rasie czerwonej duńskiej jej
rozprzestrzenienie w latach 70. było dość duże. Oceniano wówczas, że
zmniejszenie płodności wywołane przez tę mutację było rzędu 10%.
Inwersja jest mutacją zachodzącą w obrębie jednego chromosomu,
w którym wystąpiły dwa pęknięcia, a następnie zostały one niewłaściwie
naprawione w taki sposób, że fragment pomiędzy punktami pęknięć został
odwrócony o 180°. Jeżeli w obrębie odwróconego fragmentu występuje
centromer, to wówczas inwersja jest określana jako pericentryczna. Jeżeli
odwrócony fragment nie obejmuje centromeru, to nieprawidłowość jest
określana jako inwersja paracentryczna. Inwersja pericentryczna zazwyczaj
zmienia morfologię chromosomu, z wyjątkiem sytuacji, gdy pęknięcia wystąpiły
w równych odległościach od centromeru w obu ramionach chromosomowych.
Przebieg koniugacji podczas pachytenu profazy I, w obrębie fragmentu
objętego inwersją na jednym z chromosomów, może przebiegać w trojaki
sposób: 1) chromosomy tworzą pętlę inwersyjną, która zapewnia zachowanie
homologicznej koniugacji wzdłuż całego biwalentu, 2) chromosomy częś-
ciowo pozostają nieskoniugowane — w części, gdzie na jednym z chromo-
somów występuje odwrócony fragment, oraz 3) pełna koniugacja, która
częściowo jest niehomologiczna, tam gdzie fragment odwrócony koniuguje
z fragmentem prawidłowym. Najmniej korzystny, z punktu widzenia
skutków gametogenezy, jest pierwszy sposób koniugowania, podczas którego
dochodzi do uformowania pętli inwersyjnej. Jeżeli w obrębie pętli zajdzie
crossing over, to wówczas powstaną gamety obarczone delecją lub duplika-
cją, z jednoczesną możliwością powstania chromosomów dicentrycznych
(mających dwa centromery) oraz acentrycznych (pozbawionych centromeru).
Taki przebieg mejozy prowadzi oczywiście do powstania gamet o niezrów-
noważonej ilości informacji genetycznej i ostatecznie do zmniejszenia
płodności nosiciela mutacji, spowodowanej zamieraniem zarodków o nie-
zrównoważonym kariotypie. Brak koniugacji lub koniugacja niehomologicz-
na uniemożliwia zajście w tym obszarze crossing over i tym samym nie ma
ryzyka powstania gamet obarczonych wymienionymi nieprawidłowościami.
Inwersje dość rzadko opisywano u zwierząt gospodarskich. Niewątpliwie
jest to częściowo wynikiem trudności w ich rozpoznawaniu. Jeżeli zajściu
inwersji nie towarzyszy widoczna zmiana morfologii, to bardzo łatwo
można ją przeoczyć podczas obserwacji mikroskopowej. Uwaga ta dotyczy
110
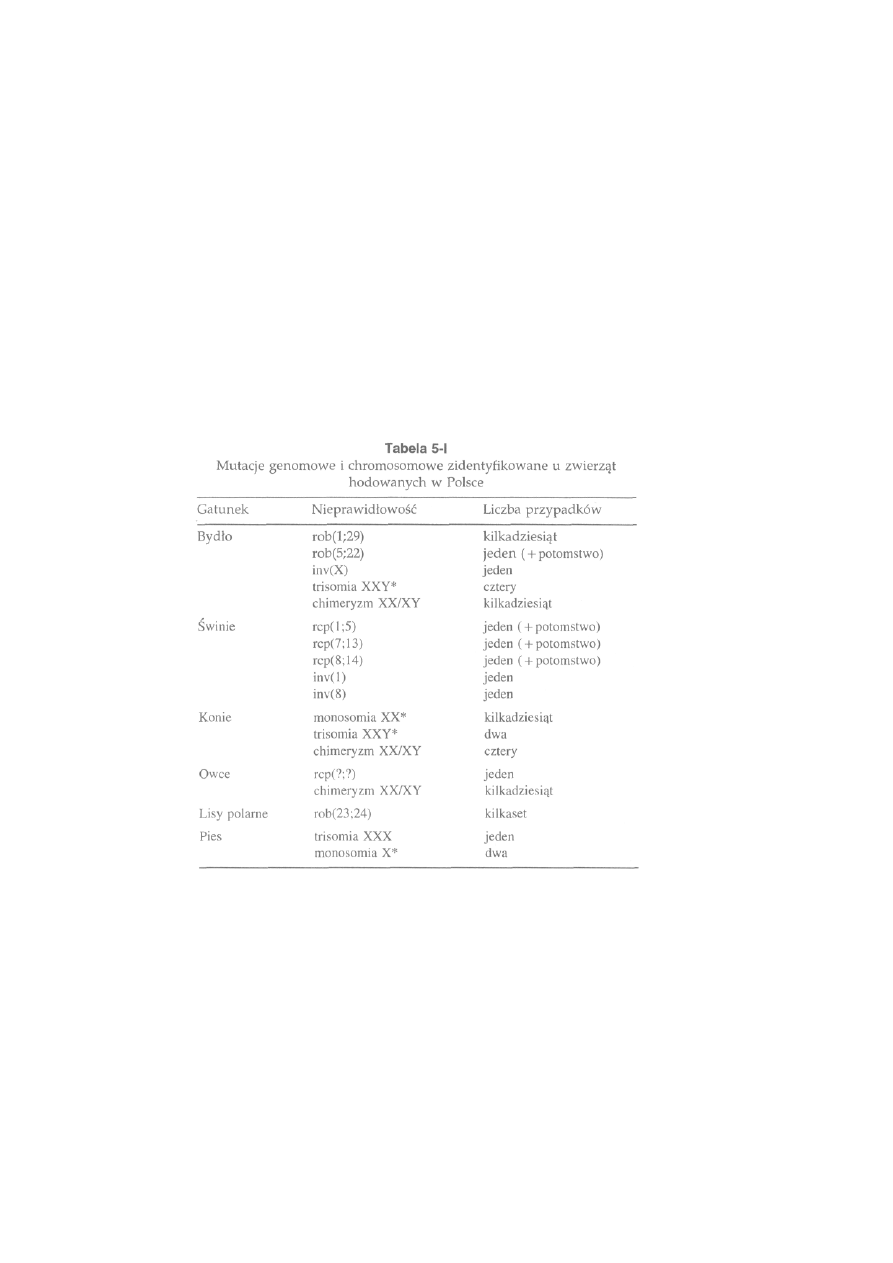
wszystkich inwersji paracentrycznych, ale także niektórych przypadków
pericentrycznych. Dotychczas zdiagnozowano na świecie co najmniej siedem
przypadków inwersji pericentycznych u bydła i trzy u świń oraz tylko jeden
przypadek inwersji paracentrycznej u świni. W przypadku dwóch inwersji
u
świni analizowano proces formowania kompleksów
synaptonemalnych i w żadnym z nich nie stwierdzono obecności pętli
inwersyjnej. Nie wiązano również jednoznacznie tych mutacji ze zmniejszoną
płodnością ich nosicieli.
5.3. Mutacje genomowe i chromosomowe
zidentyfikowane u zwierząt hodowanych w Polsce
Badania cytogenetyczne zwierząt gospodarskich mają w Polsce długą
tradycję. W okresie ostatnich 25 lat zdiagnozowano wiele przypadków
nieprawidłowości chromosomowych. Były wśród nich mutacje typu aneu-
Oznaczenia: rób — translokacja robertsonowska; rep — translokacja wzajemna
(w nawiasach wskazano numery chromosomów zaangażowanych w mutację); rcp(?;?) —
translokacja wzajemna, dla której nie ustalono, jakie chromosomy w niej uczestniczyły;
inv — inwersja; chimeryzm — występowanie dwóch linii komórkowych XX oraz XY
w limfocytach (patrz: frymartynizm); * — część przypadków ma kariotyp mozaikowy,
np. XX/XO lub XXY/XY
111

ploidii chromosomów płci, a także mutacje chromosomowe, takie jak
fuzje centryczne, translokacje wzajemne i inwersje. Oprócz tych typowych
chromosomopatii zidentyfikowano także liczną grupę zwierząt będących
nosicielami chimeryzmu w komórkach krwi, polegającego na wystąpieniu
linii żeńskiej XX i linii męskiej XY. Nieprawidłowość tę szczegółowo
omówiono w podrozdziale: Interseksualizm. Sumaryczne zestawienie
wyników przeprowadzonych badań przedstawione jest w tabeli 5-1.
Zgromadzona wiedza o występowaniu mutacji genomowych i chro-
mosomowych w krajowej populacji bydła sprawiła, że już w 1989 roku
wprowadzono obowiązkowe badania cytogenetyczne młodych buhajów
przed ich dopuszczeniem do użytkowania w stacjach produkcji nasienia.
Aktualne regulacje (1999 r.) prawne obowiązkiem takim objęły także
rozpłodniki innych gatunków wykorzystywane w inseminacji: knury,
ogiery, tryki i kozły. Dzięki temu można skutecznie zapobiegać roz-
przestrzenianiu się nieprawidłowości chromosomowych.
5.4. Mutacje genowe 5.4.1.
Przyczyny i rodzaje mutacji genowych
Mutacje genowe są to zmiany sekwencji nukleotydów w obrębie genu.
Mutacje te mogą zachodzić samoistnie, głównie na skutek błędów w procesie
replikacji DNA, rzadziej na drodze modyfikacji chemicznych zasad azoto-
wych w DNA. Mutacje te noszą nazwę mutacji spontanicznych. W odróż-
nieniu od nich mutacje indukowane są powodowane działaniem mutagen-
nych czynników chemicznych lub fizycznych. Czynniki te mogą powstawać
w wyniku przemian metabolicznych zachodzących w komórkach lub mogą
pochodzić ze środowiska zewnętrznego.
Do najsilniej działających czynników mutagennych należą:
• promieniowanie jonizujące (np. Roentgena), które powoduje zmiany
struktury zasad lub rozrywanie mostków wodorowych między zasadami
w DNA. Wrażliwość organizmów na promieniowanie jonizujące zależy od
posiadanych przez nie mechanizmów naprawy. Na przykład, niektóre
rodzaje bakterii czy gatunki owadów są znacznie bardziej odporne na
promieniowanie jonizujące niż człowiek i zwierzęta;
• promieniowanie nadfioletowe (UV), intensywnie pochłaniane przez
DNA, indukujące zmiany w zasadach pirymidynowych, np. ich dimeryzację
(dimery pirymidynowe powstają w wyniku wytwarzania kowalencyjnych
wiązań między leżącymi obok siebie w łańcuchu DNA zasadami pirymidy-
nowymi) lub rozrywanie podwójnej helisy DNA;
• hydroksyloamina wywołująca zmianę cytozyny na pochodną uracylu,
co prowadzi do tranzycji C
→T;
112

• kwas azotawy powodujący deaminację zasad w DNA i RNA. Na
rrzykład na skutek deaminacji cytozyny powstaje uracyl, który podczas
replikacji przyłącza adeninę zamiast guaniny, komplementarnej do cytozyny;
• związki alkilujące, które dzięki posiadanym grupom etylowym,
metylowym lub bardziej złożonym powodują alkilację (metylowanie lub
etylowanie) zasad w kwasach nukleinowych;
• barwniki akrydynowe (np. proflawina, oranż akrydynowy, akryfla-
wina), które wnikają między zasady w łańcuchu DNA, powodując deforma
cje podwójnej helisy DNA;
• analogi zasad (np. 5-bromouracyl, analog tyminy oraz 2-aminopuryna,
analog adeniny), które mogą być wbudowywane w DNA.
Pomimo dużej różnorodności czynników mutagennych, mogą one
powodować te same typy mutacji w różnych organizmach. Dzieje się tak
dlatego, iż wszelkie typy uszkodzeń w DNA wywołane mutagenami
stanowią tylko zmiany premutacyjne, które w wyniku procesów naprawy
mogą zostać zlikwidowane. W przypadku nieskutecznej naprawy dochodzi
do utrwalenia zmian mutacyjnych.
Niekorzystne skutki działania czynników mutagennych są łagodzone
przez enzymatyczne procesy naprawy. Analizując procesy naprawy można
wyróżnić naprawę bezpośrednią (przez wycinanie) i pośrednią (porep-
likacyjną, zwaną też naprawą w nici potomnej).
Naprawa bezpośrednia — NER (ang. nucleotide excision repair) polega
na wycinaniu nukleotydu (usuwane są duże uszkodzenia) bądź zasady,
uszkodzonej przez promieniowanie jonizujące i czynniki alkilujące. W upro-
szczeniu proces ten przebiega tak, iż po rozpoznaniu uszkodzenia specyficz-
ny enzym (zależna od ATP endonukleaza) nacina nić DNA po obu stronach
uszkodzenia, następnie jednoniciowy, kilku- lub kilkunastonukleotydowy
fragment z uszkodzeniem jest usuwany. Powstała przerwa jest wypełniana
przez polimerazę, a nić DNA jest scalana przez ligazę. Przypuszczalnie
w komórce funkcjonują dwa rodzaje NER, jeden polega na naprawie nici
transkrybowanej genów aktywnych transkrypcyjnie (ang. transcription--
coupłed repair), drugi natomiast polega na naprawie pozostałej części
genomu (ang. global genome repair), czyli nici kodującej i DNA nieaktyw-
nego transkrypcyjnie. Zwykle uszkodzenia nici transkrybowanej usuwane
są dwa lub trzy razy szybciej niż uszkodzenia nici kodującej.
Mechanizmy naprawy są dokładnie zbadane u organizmów prokariotycz-
nych, natomiast proces usuwania uszkodzeń w komórkach zwierzęcych jest
nadal słabo poznany. Wiadomo jednak, iż w komórkach zwierzęcych rodzaj
i umiejscowienie uszkodzeń zależą nie tylko od sekwencji nukleotydów
w nici DNA, ale również od struktury chromatyny. Wyższy poziom
organizacji chromatyny zwiększa dostępność DNA dla niektórych czynników
mutagennych. Na przykład, promieniowanie jonizujące wytwarza więcej
wiązań krzyżowych między białkiem i DNA w chromatynie czynnej
w porównaniu z nieczynną.
113

Naprawa pośrednia zachodzi po zreplikowaniu uszkodzonego DNA.
U bakterii E.coli znany jest kompleks enzymatyczny, który rozpoznaje
zmiany konformacyjne nowo replikowanego DNA, będące efektem nie-
komplementarności zasad. Kompleks ten odróżnia nowo zsyntetyzowaną
nić DNA od nici matrycowej i dokonuje wycięcia niewłaściwie wstawionych
nukleotydów oraz uzupełnia powstałe braki komplementarnymi nukleoty-
dami. Niestety analogiczny proces w komórkach zwierzęcych nie jest
jeszcze dokładnie poznany.
Mutacje genowe zachodzące w komórkach rozrodczych powodują trwałe
zmiany przekazywane z pokolenia na pokolenie. Mutacje te mogą zachodzić
w genach kodujących strukturę białka lub rzadziej w genach kodujących
strukturę tRNA lub rRNA.
Mutacje genowe zachodzące w komórkach somatycznych nie są dziedzi-
czne. Mutacje te w zależności od okresu rozwoju zarodka mogą dotyczyć
wszystkich komórek lub tylko pewnych części komórek dorosłego organiz-
mu. W fenotypie osobnika z mutacją somatyczną można zaobserwować
tzw. mozaikowość, polegającą na miejscowym występowaniu zmienionej
cechy, np. czarny królik mający niebieskie plamki czy kura o jednej nodze
żółtej, a drugiej białej.
Znacznie większe znaczenie mają mutacje somatyczne, zachodzące już
po urodzeniu, u osobników w różnym wieku. Są one zazwyczaj indukowane
przez czynniki mutagenne. Jeżeli mutagen taki odpowiada za uruchomienie
transformacji nowotworowej, to określany jest jako karcynogen. Mutacje
takie, prowadząc do defektów lub zaburzeń ekspresji genów związanych
z proliferacją komórek czy naprawą uszkodzeń DNA, powodują nie
kontrolowany, inwazyjny wzrost tych komórek oraz zasiedlanie przez nie
innych, nawet odległych narządów.
Mutacje genowe spontaniczne charakteryzuje odwracalność i powtarzal-
ność. Odwracalność procesu mutacji polega na tym, że allel, który powstał
dzięki mutacji genu, może zmutować wstecznie do formy wyjściowej tego
genu. Graficznie można to przedstawić następująco:
przy czym A i a są allelami, natomiast u i v oznaczają częstość mutacji w obu
kierunkach.
Potwierdzeniem odwracalności mutacji jest bezrożność i rogatość u owiec.
Zarówno owce bezrogie w rasie dorset horn, jak również owce rogate
w rasach bezrogich mogą być wynikiem mutacji w tym samym locus.
Lepiej poznane są zmiany normalnego allelu w kierunku mutanta, ale
mutacje odwrotne — zmutowanego allelu do normalnego również są
obserwowane.
114

Mutacja wsteczna nie zawsze jest jednak wynikiem powrotu do pierwot-
nego zapisu genetycznego, może ona być następstwem tzw. mutacji
supresorowej, zachodzącej w obrębie tego samego genu, w którym nastąpiła
mutacja pierwotna, lub w zupełnie innym genie.
Istotą powtarzalności mutacji genowej jest to, iż określona mutacja może
wystąpić ponownie, dając zbliżone, a często nawet takie same skutki
fenotypowe. Przykładem tego może być recesywna achondroplazja, wy-
stępująca u kilku ras bydła.
Podatność na mutacje nie jest jednakowa wśród wszystkich genów. Mając
to na uwadze, można podzielić geny na mniej i bardziej labilne lub stabilne.
Do genów labilnych można zaliczyć gen czynnika VIII krzepliwości krwi.
Wśród chorych na hemofilię aż 1/3 przypadków tej choroby jest wynikiem
mutacji de novo. Z kolei genem bardzo stabilnym jest gen choroby (pląsawicy)
Huntingtona, ciężkiego neurodegeneracyjnego zaburzenia dziedzicznego,
pojawiającego się z częstością l: 10000 osób w większości populacji pocho-
dzenia europejskiego. Choroba ta ujawnia się najczęściej w wieku powyżej
40. roku życia i prowadzi do śmierci. Została ona opisana w 1872 roku przez
Johna Huntingtona, stąd jej nazwa, znana była jednak od XVII wieku.
Pojawiła się w Europie i przez emigrantów została przeniesiona na inne
kontynenty. Dotychczas nie wiadomo, czy były jakieś inne ogniska początku
tej choroby. Wynika z tego, że diagnozowane przypadki choroby Huntingto-
na są efektem dziedziczenia mutacji w genie huntingtyny, a nie jej
pojawiania się de novo.
Częstość, z jaką występują mutacje w różnych loci, jest niejednakowa.
W przypadku niektórych genów wykazano, iż większość spontanicznych
mutacji zachodzi w kilku miejscach charakterystycznych dla danego genu,
nazwanych gorącymi miejscami (ang. hot spots). Badania genu hemofilii
A u ludzi potwierdziły, iż takimi gorącymi miejscami są sekwencje dinuk-
leotydowe CG, zwane wyspami CpG. Więszość mutacji zarejestrowanych
w obrębie genu hemofilii A polegało na zamianie dinukleotydowej sekwencji
CG na TG, rzadziej CG na CA. Również w obrębie genu DMD, zlokalizo-
wanego w chromosomie X u ludzi, warunkującego chorobę zwaną dystrofią
mięśniową Duchenne'a, stwierdzono istnienie dwóch regionów (gorące
miejsca), w których najczęściej występują mutacje. Pierwszy region znajduje
się w odległości 0,5 miliona par zasad od promotora, w regionie
DXS164/DXS206, drugi natomiast w części centralnej genu, tj. 1,2 miliona
par zasad od promotora.
Na częstość mutacji genowych spontanicznych mogą mieć wpływ
również czynniki dziedziczne. U muszki owocowej (Drosophila melanogaster)
wykryto gen MuF, zlokalizowany w chromosomie 2 pary, warunkujący
większą podatność innych genów na mutacje.
Większość mutacji jest dla organizmu szkodliwa, a nawet letalna. Czasem
jednak mutacja zmienia właściwości organizmu tak, że zyskuje on cechy
korzystne. Zdarzają się również mutacje zupełnie lub niemal zupełnie
115

obojętne dla organizmu. W wyniku takich mutacji powstały, na przykład,
serie alleli wielokrotnych, które są omówione w rozdziale: Podstawowe
mechanizmy dziedziczenia cech.
Rodzaje mutacji genowych i ich skutki można analizować na poziomie
DNA lub na poziomie polipeptydu. Rozpatrując mutację na poziomie DNA
można wyróżnić następujące zmiany w sekwencji nukleotydów w danym
genie (ryć. 5-9):
1) tranzycja — zamiana jednej zasady purynowej na drugą purynową
lub pirymidynowej na inną pirymidynową (w podwójnej helisie będą
zamiany par zasad A/T
←→G/C),
2) transwersja -- zamiana zasady purynowej na pirymidynową i od
wrotnie pirymidynowej na purynową; w podwójnej helisie będą to zamiany:
3) delecja — wypadnięcie (utrata) jednej lub kilku par nukleotydów,
4) insercja — wstawienie jednej lub kilku par nukleotydów.
Przyczyną samorzutnej zamiany jednej pary nukleotydów na inną,
według hipotezy Watsona i Cricka, mogą być tautomeryczne przekształ-
cenia zasad azotowych, zachodzące pod wpływem działania czynników
zewnętrznych. Przekształcenia te polegają na zmianie położenia prze-
strzennego poszczególnych atomów oraz zmianie rozkładu elektronów
Ryć. 5-9. Mutacje genowe — zmiany sekwencji nukleotydów
116

i protonów w cząsteczce zasady. W dwuniciowej helisie DNA zasady
komplementarne tworzą wiązania między grupami aminowymi (—NH
2
)
i ketonowymi (—CO). Przekształcenie tautomeryczne powoduje zamianę
grupy aminowej na iminową (=NH), zaś ketonowej na enolową (=C—OH), a
w konsekwencji zmianę konfiguracji połączeń wodorowych występujących
między zasadami purynowymi i pirymidynowymi. Adenina w formie keto-
nowej łączy się z tyminą dwoma mostkami wodorowymi, natomiast w formie
enolowej łączy się trzema mostkami z cytozyną. Z kolei, guanina w normalnej
formie (ketonowej) łączy się z cytozyną, a w formie tautomerycznej z tyminą.
Tautomeryczne formy enolowe są nietrwałe i po pewnym czasie wracają
samorzutnie do formy ketonowej, odzyskując zdolność łączenia się z zasada-
mi komplementarnymi dla tych form. Skutkiem przemian tautomerycznych
może być błąd włączania bądź błąd replikacji. Błąd włączania polega na tym,
że enołowa forma tyminy łączy się z guanina. Tautomeryczna forma tyminy
powraca do formy ketonowej i w następnej replikacji łączy się z adeniną.
Błąd replikacji zachodzi wówczas, gdy na przykład w DNA podczas replikacji
tyminą ulega przekształceniu z formy ketonowej w enołowa i zamiast
z adeniną łączy się z guanina. Z kolei, w następnej replikacji do guaniny
przyłączy się komplementarna do niej cytozyną. Skutkiem tego będzie
pojawienie się pary G-C w miejscu, gdzie przed błędem replikacji była para
Oba rodzaje błędów występują także po przekształceniach tautomerycznych
innych zasad.
Mutacje genowe mogą być także następstwem zakłóceń w splicingu,
czyli tzw. obróbce mRNA, nazwanej dojrzewaniem. Splicing polega na
wycinaniu intronów z pierwotnego transkryptu (pre-mRNA), w wyniku
którego powstaje mRNA zawierający tylko sekwencje kodujące (eksony).
Przykładem takiej mutacji jest delecja w genie a
s1
kazeiny mleka, prowadząca
do powstania alleli: F (delecja 3 eksonów) i D (delecja l eksonu).
Od niedawna znany jest bardzo specyficzny rodzaj mutacji genowych —
mutacje dynamiczne. Dotychczas wykryto je u ludzi w 12 loci, z których 10
powoduje choroby genetyczne, między innymi chorobę Huntingtona, zespół
kruchego chromosomu X, dystrofię miotoniczną. Mutacje te związane są
z tzw. niestabilnymi sekwencjami DNA, należącymi do powtarzających się
sekwencji mikrosatelitarnych i polegają na tym, iż z pokolenia na pokolenie
zwiększa się (znacznie rzadziej zmniejsza) liczba powtórzeń tych sekwencji.
Im większy stopień zwielokrotnienia danej sekwencji, tym wcześniej
rejestrowany jest początek objawów chorobowych. Na przykład choroba
Huntingtona jest wynikiem zwielokrotnienia liczby powtórzeń sekwencji
CAG w eksonie l genu kodującego białko huntingtynę, zlokalizowanego
w chromosomie 4. U ludzi zdrowych liczba powtórzeń wynosi 10-36,
przekroczenie liczby 36 powtórzeń powoduje wystąpienie choroby. Z zabu-
rzeniami tymi wiąże się zjawisko zwane antycypacją. W przypadku choroby
Huntingtona (HD) i dystrofii miotonicznej młodociana postać choroby jest
związana z dziedziczeniem ojcowskim (antycypacja odojcowska). Inną
117

cechą charakterystyczną mutacji dynamicznej w genie HD jest tzw. piętno
gametyczne. Jeśli allel zmutowany pochodzi od matki, różnica w liczbie
powtórzeń u potomka w stosunku do liczby powtórzeń u matki może
wynieść do 4. Natomiast jeśli ojciec przekazuje zmutowany allel, różnica
w długości sekwencji może dochodzić do 50%. Przypuszcza się, iż jest to
spowodowane niestabilnością tej sekwencji w spermatogenezie. Zjawisko to
jest charakterystyczne także dla innych zaburzeń wywołanych mutacjami
dynamicznymi.
5.4.2. Skutki mutacji genowych
Mutacje genowe nie zawsze powodują zmiany w składzie aminokwasowym
polipeptydu. Taka sytuacja jest możliwa, gdyż spośród 20 aminokwasów aż 18
jest kodowanych przez 2 do 6 trójek nukleotydowych (kodonów, trypletów).
Jedynie metionina (AUG) i tryptofan (UGG) są kodowane przez pojedyncze
kodony. Na przykład tranzycja U
→C powoduje zmiany aminokwasu w
łańcuchu polipeptydowym, gdyż kodon AAC również koduje asparaginę.
Należy podkreślić, iż mutacje genowe powodujące zauważalny efekt
fenotypowy stanowią jedynie małą część mutacji zachodzących w DNA.
Przeważająca część mutacji genowych nie powoduje żadnych zmian
w produkcie genowym, dlatego nazywane są cichymi podstawieniami (ang.
substitution at silent site). Są one wykorzystywane w analizie polimorfizmu
DNA, identyfikacji markerów genetycznych przydatnych w programach
mapowania genomów.
Jednym ze skutków mutacji genowych, polegających na pojedynczej
zmianie sekwencji nukleotydów w określonym genie, mającym szczególne
znaczenie dla molekularnej analizy DNA, jest polimorfizm fragmentów
restrykcyjnych — RFLP (ang. restriction fragment length polymorphism).
Polimorfizm ten jest wynikiem mutacji punktowych zarówno w obrębie
eksonu, jak i intronu. Mutacje w obrębie intronu nie wpływają zazwyczaj na
funkcję genu, ani kodowanego przez ten gen białka. Również mutacja
w obrębie eksonu może nie powodować zmiany produktu genowego.
Natomiast jej skutkiem może być powstanie miejsca rozpoznawanego
przez enzym restrykcyjny lub utrata miejsca restrykcyjnego dla danego
enzymu. Dzięki temu RFLP określonych loci może być wykorzystywany
jako marker genetyczny.
Technika RFLP opisana w rozdziale: Metody analizy i modyfikacji geno-
mu znajduje również zastosowanie w ustalaniu genotypów pod względem
zmutowanych alleli genów głównych (QTL) kontrolujących cechy jakościowe
(np. białka mleka) czy odpowiedzialnych za powstanie choroby genetycznej.
Analizując skutki mutacji genowych na poziomie polipeptydu można
wyróżnić następujące typy mutacji:
118

1. Mutacje typu zmiany sensu - - na skutek tranzycji lub transwersji
kodon dla jakiegoś aminokwasu zostaje zamieniony na kodon dla innego
aminokwasu.
Na przykład, jeśli kodon GAU dla kwasu asparaginowego w wyniku
tranzycji A
→G zostanie zmieniony na kodon GGU kodujący glicynę, skutki
tego mogą być dla organizmu letalne. Konsekwencją takiej mutacji w 383
nukleotydzie genu kodującego podjednostkę CD18 p
2
-integryny jest choroba
BLAD (ang. bovine leukocyte adhesion deficiency), przejawiająca się
zmniejszeniem odporności cieląt, której następstwem jest śmierć przed
ukończeniem 1. roku życia osobników homozygotycznych pod względem
zmutowanego genu. Szerzej mutacja ta i inne tego typu zostaną omówione
w części dotyczącej molekularnej diagnostyki chorób.
Jak już wcześniej wspomniano, mutacje genowe nie zawsze mają
niekorzystny skutek. Wynikiem mutacji typu zmiany sensu są niektóre geny
warunkujące umaszczenie czy polimorficzne białka. Przykładem allelu
powstałego w drodze tego rodzaju mutacji jest allel E
d
w locus E (Extension)
u bydła, warunkujący czarne umaszczenie. Mutacja ta jest przedstawiona
na rycinie 5-10. Allel dominujący E
d
jest wynikiem tranzycji tyminy
w cytozynę w kodonie 99 allelu dzikiego E
+
, która w łańcuchu peptydowym
powoduje zamianę aminokwasu leucyny na prolinę.
Innym przykładem mutacji typu zmiany sensu, odnoszącym się do
polimorficznych białek, jest locus
β-laktoglobuliny (białko mleka). Różnica
między allelem A i B
β-laktoglobuliny polega na zamianie A-»G, w wyniku
której w pozycji 64 łańcucha polipeptydowego typu A zamiast glicyny jest
kwas asparaginowy, oraz T
→C (zapis dla DNA; w mRNA: U→ęcej
informacji o genetycznym uwarunkowaniu i znaczeniu polimorficznych
białek surowicy krwi, mleka, nasienia i jaj zawiera rozdział: Markery
genetyczne.
2. Mutacje typu nonsens (mutacja stop) - na skutek tranzycji lub
transwersji kodon dla jakiegoś aminokwasu (kodon sensowny) zostaje
zamieniony na jeden z trzech kodonów nonsensownych (terminacyjnych) -
Ryć. 5-10. Mutacje typu zmiany sensu i zmiany fazy odczytu w locus E u bydła
119

amber (UAG), ochrę (UAA) lub opal (UGA), które nie kodują aminokwasów,
lecz stanowią sygnał zakończenia translacji. Wynikiem tej mutacji jest
zmiana długości polipeptydu kodowanego przez dany gen; powstaje krótszy
N-końcowy fragment tego polipeptydu.
Przykładem następstwa mutacji typu nonsens jest recesywna wada
metaboliczna - cytrulinemia, występująca u bydła rasy holsztyńsko-fry-
zyjskiej. Tranzycja cytozyny na tyminę w 86 kodonie genu syntetazy
argininobursztynianowej (enzym biorący udział w cyklu mocznikowym)
powoduje zamianę kodonu CGA dla argininy na kodon TGA kończący
translację. W jej konsekwencji, zamiast aktywnego enzymu składającego się
z 412 aminokwasów, powstaje nieaktywny peptyd złożony z 85 amino-
kwasów. Cielęta homozygotyczne pod względem tego zmutowanego genu
padają w pierwszym tygodniu życia na skutek zaburzeń w wydalaniu
mocznika z organizmu. Cytrulinemia występuje również u ludzi, przy czym
dotychczas stwierdzono 20 mutacji w genie kodującym syntetazę arginino-
bursztynianową.
Podobna mutacja polegająca na zamianie cytozyny na tyminę w 405
kodonie genu syntazy urydynomonofosforanowej, czego następstwem jest
zamiana kodonu CGA (dla argininy) na kodon TGA kończący translację
białka, występuje u bydła. Następstwem tego jest skrócenie łańcucha
polipeptydowego (utrata większości C-końcowych 76 aminokwasów) i utrata
przez enzym funkcji katalitycznych. Jest to schorzenie nazwane DUMPS
(ang. deficiency uridine monophosphate synthase). Jest ono przyczyną
zamierania zarodków, a w konsekwencji zmniejszenia płodności krów.
3. Mutacje zmiany fazy odczytu — na skutek delecji lub insercji jednej
lub kilku par nukleotydów (liczba par nie może być podzielna przez 3)
następuje przesunięcie fazy odczytu, a powstający polipeptyd ma od
miejsca mutacji do końca C nieprawidłowo włączone aminokwasy. Mutacje
te wynikają z cechy bezprzecinkowości kodu genetycznego.
Podobnie jak mutacje typu zmiany sensu, również i ten rodzaj mutacji
może mieć skutki obojętne dla organizmu. We wspomnianym już locus
E u bydła allel recesywny e, kodujący u zwierząt homozygotycznych ee
umaszczenie czerwone, powstał w drodze mutacji typu zmiany fazy odczytu
spowodowanej delecją jednej zasady. Począwszy od kodonu 104 (GGT) dla
glicyny, w którym delecją guaniny (oznaczona na schemacie symbolem
∇)
powoduje przesunięcie fazy odczytu, w procesie translacji przyłączane są
inne aminokwasy. Schemat tej mutacji przedstawiono na rycinie 5-10.
Bydło holsztyńskie ma umaszczenie z reguły czarno-białe, ale są także
zwierzęta czerwono-białe pojawiające się w wyniku kojarzenia czarno-białych
nosicieli recesywnego allelu czerwonego umaszczenia. Opracowano test
molekularny identyfikacji nosicielstwa genu czerwonego umaszczenia,
wykorzystujący to, iż recesywny allel nie ma miejsca restrykcyjnego dla
enzymu Mspl, natomiast allel dominujący ma takie miejsce. Zamplifikowany
za pomocą PCR fragment genu jest trawiony enzymem, następnie roz-
120

dzielany elektrofor etycznie. Uzyskany ełektroforegram (obraz prążków) -
wkazuje obecność jednego prążka długości 531 pz w przypadku allelu
recesywnego, dwóch prążków (201 pz i 331 pz) w przypadku allelu
dominującego.
4. Mutacje typu delecja lub insercja aminokwasu — delecja lub
insercja trzech par nukleotydów stanowiących kodon dla aminokwasu
(lub wielokrotności liczby 3) w łańcuchu DNA powoduje najczęściej
utratę lub dodanie jednego bądź kilku aminokwasów w białku kodowanym
przez dany gen. W przypadku delecji trójki nukleotydów może nastąpić:
utrata dwóch aminokwasów w łańcuchu peptydowym, a na ich miejsce
pojawi się inny aminokwas (jeśli „wypadną" zasady z dwóch kolejnych
kodonów); utrata jednego aminokwasu (delecja kodonu) lub powstanie
kodonu „stop". Insercja trójki może spowodować: dodanie aminokwasu –
w białku, jeśli trójka nukleotydów zostanie wstawiona między kodony;
zamianę fazy odczytu, jeśli trójka „rozerwie" istniejący dotychczas kodon;
możliwe jest także pojawienie się kodonu „stop". Mutacje te mogą
prowadzić do poważnych zaburzeń, a nawet śmiertelnych chorób gene-
tycznych.
Przykładem mutacji typu delecja aminokwasu jest mukowiscydoza
u ludzi, polegająca na zakłóceniu transportu jonów chlorkowych. U chorych
na mukowiscydozę przewody wyprowadzające z narządów wewnętrznych
zostają zaczopowane przez bardzo gęstą wydzielinę. Dotyczy to przede
wszystkim trzustki, płuc i oskrzeli. Częstość występowania tej wady,
prowadzącej do przedwczesnej śmierci, wynosi l: 2500 osób. Prawidłowy
gen warunkuje syntezę białka będącego regulatorem przewodnictwa błono-
wego, zwanego CFTR (ang. cystic fibrosis transmembrane conductance
regulator), pełniącego funkcję kanału chlorkowego. W obrębie tego genu
wykryto wiele mutacji, przy czym najczęściej jest stwierdzana mutacja
w eksonie 10, oznaczona symbolem
Δ
F508 (ryć. 5-11). Mutacja ta prowadzi
121

do utraty jednego aminokwasu — fenyloalaniny w 508 pozycji kodowanego
przez ten gen łańcucha polipeptydowego. Podczas procesu translacji zamiast
kodonu ATC dla izoleucyny (w 507 pozycji łańcucha polipeptydowego)
i następujących po nim kodonów TTT i GGT (fenyloalanina — 508 póz.
i glicyna — 509 póz. łańcucha) odczytywany jest kodon ATT (inny tryplet
dla izoleucyny), a następnie sekwencja GGT dla glicyny.
Przykładem mutacji typu insercja aminokwasu jest wspomniana już
wcześniej choroba Huntingtona u ludzi. Zwielokrotnienie powtarzającej się
sekwencji CAG, kodującej aminokwas glutaminę, powoduje powstanie
domeny glutaminowej, złożonej z takiej liczby glutamin, jaka jest zakodo-
wana w sekwencji (CAG)
n
.
5. Mutacje zachodzące podczas obróbki potranskrypcyjnej, zwanej
składaniem (ang. splicing) pre-mRNA --w wyniku zaburzeń w procesie
wycinania intronów powstaje nieprawidłowy mRNA, czego konsekwencją
jest synteza zmienionego białka. Białko takie może mieć inne właściwości
katalityczne niż białko prawidłowe, na przykład białko determinowane
przez locus Albino (C). Gen C warunkuje wytwarzanie enzymu tyrozynazy.
W następstwie nieprawidłowej obróbki potranskrypcyjnej mRNA powstają
allele zmutowane. Częstość tych mutacji wynosi 10-40%. Również niektóre
allele genu determinującego warianty białka mleka
α
s1
-kazeiny u kóz
powstały w wyniku zaburzeń w procesie obróbki pre-mRNA, o czym pisano
już wcześniej. W przypadku allelu D podczas wycinania intronów został
dodatkowo wycięty ekson kodujący 11 aminokwasów, natomiast w allelu
F - 3 eksony kodujące 37 aminokwasów.
Fenotypowy efekt mutacji genowych może być zauważalny wkrótce
po urodzeniu osobnika, na przykład wielopalczastość, skrócenie krę-
gosłupa u bydła i psów, oklapnięte uszy u kotów szkockich zwisłouchych
czy skrócenie ogona u kotów bobtail (ryć. 5-12 wkładka kolorowa),
a także umaszczenie zwierząt i inne, ale może być również rozpo-
znawalny w okresie późniejszym, na przykład w pierwszym tygodniu
życia (cytrulinemia) lub pierwszym roku życia (BLAD). W wyniku więk-
szości mutacji genowych następuje utrata lub modyfikacja zdolności
organizmu do wytworzenia specyficznego białka, najczęściej enzymu
koniecznego do prawidłowego przebiegu jakiegoś procesu. Często są
to zaburzenia przebiegu szlaku metabolicznego (tzw. blok metaboliczny),
w którym produkt jednej przemiany enzymatycznej stanowi substrat
dla następnej przemiany. Jednym z najlepiej poznanych bloków me-
tabolicznych u ludzi są nieprawidłowości w przemianie aminokwasów
fenyloalaniny i tyrozyny (ryć. 5-13). Mutacje jednego z genów wa-
runkujących określony enzym tego bloku powodują następujące scho-
rzenia:
A. Fenyloketonuria — brak lub niedobór enzymu hydroksylazy fenylo-
alaninowej, przekształcającego fenyloalaninę w tyrozynę; powoduje uszko-
dzenie układu nerwowego oraz niedorozwój umysłowy i fizyczny;
122
B. Kretynizm tarczycowy — brak enzymu katalizującego przemianę
tyrozyny w hormon tarczycy (tyroksynę i trijodotyroninę), powodujący
różny stopień nasilenia niedorozwoju umysłowego;
C. Tyrozynoza -- niedobór enzymu hydroksylazy kwasu p-hydroksy-
fenylopirogronowego, biorącego udział w przemianie tego kwasu w kwas
homogentyzynowy, nie powodujący wyraźnie złych skutków dla organizmu;
D. Alkaptonuria -- brak oksydazy kwasu homogentyzynowego, prze
kształcającego ten kwas w kwas maleiloacetooctowy; kwas homogen
tyzynowy jest odkładany w stawach, chrząstkach i skórze wywołując stany
zapalne, a jego nadmiar jest wydalany z moczem, powodując jego ciemnienie
na powietrzu;
E. Albinizm, czyli bielactwo - - brak enzymu tyrozynazy, przekształ
cającego 3,4-dihydroksyfenyloalaninę (DOPA) w barwnik skóry — melaninę;
osobniki albinotyczne mają bardzo jasną skórę, białe włosy, brwi i rzęsy,
tęczówka oka nie jest zabarwiona, a przeświecające przez nią naczynia
krwionośne nadają jej różowe zabarwienie. Albinizm występuje także
u zwierząt.
Bloki metaboliczne mogą dotyczyć również enzymów związanych
z przemianami innych związków, np. cukrów czy lipidów.
5.4.3. Mutacje genowe wpływające na cechy
produkcyjne zwierząt
Mutacje genowe mogą mieć znaczący wpływ na efektywność hodowli
zwierząt. Jednym z genów, którego polimorficzne formy wykazują związek
z różnymi cechami użytkowymi zwierząt gospodarskich, jest gen hormonu
wzrostu — somatotropiny (ang. growth hormone — GH).
123

Inne mutacje genowe nie mają tak wszechstronnych skutków jak
mutacje w genie hormonu wzrostu. Wykryte dotychczas mutacje u świń
wpływające na jakość mięsa dotyczą: receptora rianodyny (RYR1) —
mutacja powodująca gorączkę złośliwą oraz locus PRKAG3 — mutacja
powodująca tzw. kwaśne mięso (RN).
U bydła i owiec wykryto mutacje powodujące hipertrofię mięśniową.
U bydła cecha ta warunkowana jest zmutowanym allelem genu miostatyny
(locus w chromosomie 2). U owiec natomiast hipertrofia mięśniowa (CLPG)
jest determinowana mutacją locus kodującego ncRNA.
Opłacalność hodowli zwierząt w dużej mierze zależy od cech reproduk-
cyjnych. U trzody chlewnej stwierdzono związek między polimorfizmem
genu receptora estrogenu (ang. estrogen receptor gene — ESR) a wielkością
miotu. Poszczególne allele tego genu warunkują wzrost liczebności miotu
o 1,15 prosięcia u świń bardzo plennej rasy meishan i 0,42 prosięcia
u wielkiej białej.
Spośród wykrytych dotychczas mutacji, wpływających na liczbę jagniąt
rodzonych przez owcę w jednym miocie, największe znaczenie ma mutacja
w genie BMPR-IB wykryta u owiec rasy merynos booroola. W pojedynczej
kopii allel zmutowany zwiększa liczebność miotu o l jagnię. Inne mutacje
to: gen FecJ u owiec jawajskich, gen Thoka u owiec islandzkich i gen Belle-Ile
u owiec utrzymywanych w północnej Francji. Pojedyncza kopia każdego
z tych genów powoduje wzrost liczebności miotu o około 0,6 jagnięcia.
Podobny efekt fenotypowy ma gen FecX' sprzężony z płcią, wykryty
u owiec rasy romney.
Szerzej mutacje te są opisane w rozdziale: Zmienność cech (podrozdział:
Geny o dużym efekcie).
5.4.4. Mutacje genowe wywołujące choroby genetyczne
Mutacje genowe często prowadzą do powstania chorób genetycznych, które
mogą się przejawiać zaburzeniami rozwojowymi o charakterze morfologicz-
nym lub upośledzeniem różnych funkcji organizmu, np. metabolicznych,
odpornościowych, endokrynologicznych, psychicznych itp. Należy jednak
pamiętać, że rozwój niektórych chorób genetycznych zależy również od
wpływu czynników środowiskowych. Stąd poznanie podłoża wielu chorób
genetycznych, a szczególnie wrodzonych wad rozwojowych, sprawia dużo
trudności. O podłożu molekularnym niektórych chorób metabolicznych
wspomniano już w podrozdziale: Skutki mutacji genowych.
5.4.4.1. Geny letalne, semiletalne i subwitalne
Wpływ szkodliwego działania zmutowanych genów jest różny. Zależnie od
stopnia penetracji tych genów można je podzielić na:
124

• geny letalne, powodujące śmierć ponad 90% osobników, w których
genotypach wystąpią w dawce aktywnej (np. homozygota recesywna),
• geny semiletalne, powodujące w aktywnej dawce śmierć od 50 do
90% osobników,
• geny subwitalne, które osłabiają wydolność fizjologiczną i mogą
powodować śmierć od 10 do 50% osobników.
Geny letalne warunkują nieprawidłowości rozwojowe i zakłócenia
w procesach fizjologicznych. Niektóre z nich, z nie wyjaśnionych dotąd
przyczyn, powodują śmierć osobnika, w którego genotypie wystąpią.
Również okres w życiu osobnika, w którym dany gen przejawia działanie
letalne, może być różny. Jedne geny letalne powodują śmierć zygoty we
wczesnym stadium życia embrionalnego, inne zaś później, w okresie
pourodzeniowym lub u dorosłych osobników.
Geny letalne dominujące ujawniają swoje działanie niekorzystne już
w układzie heterozygotycznym; powodując śmierć nie tylko homo-, ale
także i heterozygotycznych osobników same się eliminują i mutacja taka nie
utrwala się w populacji.
Geny letalne o niezupełnej dominacji sygnalizują swoją obecność
w pojedynczej dawce, nie ograniczając na ogół przeżywalności czy spraw-
ności życiowej. Geny te działają letalnie w układzie homozygotycznym,
natomiast u osobników heterozygotycznych nie są letalne, ale dają zauważal-
ny efekt fenotypowy. Przykładem jest gen platynowego ubarwienia u lisów,
gen siwej barwy (sziras) wełny u owiec rasy karakuł czy gen achondroplazji
u bydła i kur.
Gen platynowej barwy u lisów (W
p
) jest wynikiem mutacji genu
w, warunkującego srebrzystą barwę włosa. Innym genem, będącym również
wynikiem mutacji genu
ω
, jest gen W, warunkujący umaszczenie tzw.
białopyskie. Oba zmutowane geny w układzie homozygotycznym (W
p
W
p
,
WW) działają letalnie, również heterozygoty W
p
W nie są zdolne do
życia i giną przed urodzeniem. Kojarzenie między sobą lisów
platynowych bądź białopyskich powoduje zmniejszenie plenności wskutek
zamierania zarodków homozygotycznych. Z punktu widzenia
ekonomicznego korzystniejsze są kojarzenia heterozygotycznego osobnika
platynowego z homozygota pod względem genu srebrzystego ubarwienia,
uzyskamy wówczas lisy zdolne do życia, w stosunku l platynowy: l
srebrzysty (ryć. 5-14 wkładka kolorowa).
Gen siwej barwy karakułów powoduje śmierć osobników homozygotycz-
nych w wieku 4-9 miesięcy wskutek zaburzeń w funkcjonowaniu przy-
współczulnego układu nerwowego, powodujących poważne zakłócenia
procesu trawienia.
Gen achondroplazji stwierdzony po raz pierwszy u bydła irlandzkiej
rasy kerry, który jest letalny w podwójnej dawce, w układzie hetero-
zygotycznym zaznacza swą obecność skróceniem odnóży (krótkonogie
bydło nazwane dexter). U kur gen achondroplazji (Cp) w układzie hetero-
125

zygotycznym (Cpcp) warunkuje krótkonożność. Zarodki o genotypie CpCp
zamierają w okresie od 3 do 4 dnia inkubacji.
Przykładem genu letalnego o niezupełnej dominacji sprzężonego z płcią
jest gen oznaczony symbolem A31, warunkujący pasmowy brak owłosienia
u bydła rasy holsztyńsko-fryzyjskiej. Zmienność tej cechy u krów jest duża.
U krów heterozygotycznych nieowłosione pasma są ledwie dostrzegalne,
natomiast u homozygotycznych — bardzo wyraźne. Krowy takie są wrażliwe
na zimno i źle znoszą czyszczenie. Cecha ta występuje tylko u samic.
U samców gen ten jest letalny. Z kojarzenia krowy homozygotycznej pod
względem tego genu z buhajem normalnym w potomstwie uzyskuje się
jedynie samice. Kojarzenie to przedstawia schemat:
W przypadku gdy matka jest heterozygotą, w potomstwie występuje
stosunek samców do samic l: 2, co obrazuje schemat:
Większość genów letalnych ma charakter recesywny autosomalny.
Nosiciele tych genów (heterozygoty) fenotypowo niczym się nie różnią od
osobników normalnych. Zakres działania tych genów jest różny, od
niewielkich zmian (np. miejscowy brak nabłonka u bydła ayrshire) do
rozległych zaburzeń, jak achondroplazja recesywna. Stopień fenotypowego
przejawienia się genu letalnego zależy zarówno od współdziałania danego
genu z innymi genami, jak też od interakcji ze środowiskiem.
Niektóre geny letalne mogą także powodować śmierć matki. Przykurcz
mięśni kończyn i szyi (cecha recesywna) płodów owiec, bydła i świń
powoduje częste padanie matek na skutek utrudnionych porodów.
Geny semiletalne często jest trudno odróżnić od genów letalnych, gdyż
ich działanie zależy między innymi od czynników środowiskowych. Te
same geny mogą być letalne dla swych nosicieli przebywających w złych
warunkach środowiskowych, nieszkodliwe zaś dla osobników znajdujących
się w dobrych warunkach. Wrodzona katarakta u bydła lub brak gałek
ocznych oraz wiele innych podobnych nieprawidłowości powodują, iż
zwierzęta obarczone nimi mogą żyć tylko w szczególnych warunkach
środowiskowych i pod troskliwą opieką hodowców.
Ponadto niektóre z tych genów mogą być letalne w stosunku do
jednych zwierząt, natomiast u innych mogą tylko zmniejszać sprawność
fizjologiczną.
126

Geny subwitalne powodują odchylenia od normy w budowie anatomicz-
nej i procesach fizjologicznych, a jednocześnie zmniejszają odporność
zwierząt na choroby i niekorzystne czynniki środowiskowe. Niektóre z tych
genów są przyczyną zaburzeń metabolicznych, powodując tzw. bloki
metaboliczne. Przykład takiego bloku podano już wcześniej.
Wiele wad determinowanych genami subwitalnymi dotyczy układu
rozrodczego, powodując niepłodność czy utrudniając akt kopulacji. Na
przykład buhaje dotknięte spastycznym niedowładem kończyn (wada ta ma
dwie postacie — występuje do 8 tygodnia życia lub dopiero w wieku 2-3
lat) niechętnie kryją i są eliminowane z rozpłodu.
Ponieważ większość wad dziedzicznych jest uwarunkowana genami
letalnymi, następny podrozdział dotyczy wad wrodzonych, ich rodzajów,
sposobów dziedziczenia i ograniczania występowania tych wad w hodowli
zwierząt gospodarskich.
5.4.4.2. Choroby monogenowe wywołujące
wrodzone wady rozwojowe
Wrodzonymi wadami rozwojowymi nazywamy wszelkie nieprawidłowości
budowy ciała, powstające w okresie płodowym. Niektóre z wad są zauważal-
ne już w chwili przyjścia na świat osobnika, inne ujawniają się w później-
szym okresie życia (np. epilepsja u bydła).
Teratologia, nauka zajmująca się zaburzeniami rozwojowymi, wyodrębnia
trzy grupy tych zaburzeń:
1) potworności — są to bardzo rozległe zmiany w wyglądzie ciała,
obarczone nimi zwierzęta nazywane są potworkami,
2) wady — są to pewne zaburzenia w prawidłowej budowie,
3) braki (defekty czy usterki) — są to drobne zmiany nie mające
większego znaczenia dla życia zwierzęcia.
Przyczyny powstawania wad wrodzonych mogą być dwojakiego rodzaju:
1) genetyczne — będące wynikiem mutacji genowych, chromosomowych
lub genomowych,
2) środowiskowe — powstałe na skutek działania niekorzystnych czyn
ników środowiskowych na rozwijający się płód, wady te nie dziedziczą się.
Nie każda wada wrodzona jest zatem uwarunkowana genetycznie.
W niektórych przypadkach na powstanie takiej nieprawidłowości mają
wpływ równocześnie czynniki środowiskowe i genetyczne. Większość wad
wrodzonych dziedzicznych ma określony obraz fenotypowy. Zdarza się
jednak, że niektóre anomalie, mimo że są pochodzenia pozagenetycznego,
do złudzenia przypominają nieprawidłowości uwarunkowane założeniami
genetycznymi. Wady takie, przypominające mutanty, a powstałe pod
wpływem czynników środowiskowych, noszą nazwę fenokopii. W przypad-
ku prowadzenia kontroli wrodzonych wad dziedzicznych i ograniczania ich
występowania istotnym problemem jest odróżnienie fenokopii od wady
mającej podłoże genetyczne. Można to uczynić biorąc pod uwagę:
127

1) zgodność wyglądu danej anomalii z opisem genetycznie uwarun
kowanych wad wrodzonych w danej rasie,
2) wystąpienie wady u noworodka pochodzącego z kojarzeń krew-
niaczych zarówno bliskich, jak i dalszych,
3) ujawnienie się tej samej wady u potomstwa jednego rozpłodnika,
4) pojawienie się podobnej anomalii u zwierząt spokrewnionych z ro
dzicami noworodka.
OPIS NIEKTÓRYCH WRODZONYCH WAD ROZWOJOWYCH
Wady układu kostnego
Jednym z najwcześniej poznanych zaburzeń rozwojowych była achondro-
plazja, czyli zahamowanie rozwoju układu kostnego. Nieprawidłowość ta
występuje głównie u bydła (po raz pierwszy została opisana w Irlandii, dała
początek nowej rasie bydła — dexter). Badania genetyczne wykazały, że płody
dotknięte tą wadą, homozygotyczne pod względem genu niezupełnie
dominującego, ulegają poronieniu zwykle przed ósmym miesiącem (ryć. 5-15
wkładka kolorowa). W 2002 roku w Australii wykryto odpowiedzialną za tę
wadę mutację, polegającą na insercji 4 nukleotydów w genie, którego
lokalizacja nie została ujawniona (zapewne test molekularny zostanie opatento-
wany). Mutacja ta powoduje powstanie kodonu terminacyjnego, skutkiem
czego krótszy jest o 1/3 łańcuch polipetydowy, kodowany przez ten gen.
Achondroplazja może być także uwarunkowana genem recesywnym i w takim
przypadku objawia się głównie zaburzeniami w rozwoju kości głowy lub
deformacją szkieletu i kończyn. Nosicielstwo allelu achondroplazji, oznaczone-
go symbolem bd (ang. bulldog disease), stwierdzono u francuskiego buhaja rasy
holsztyńsko-fryzyjskiej, charakteryzującego się wybitną wartością hodowlaną.
Poszczególne wady wrodzone często występują w połączeniu z innymi
nieprawidłowościami. Do anomalii towarzyszących tego rodzaju zaburzeniom
rozwojowym należą: skrócenie części twarzowej głowy, skrócenie lub brak
żuchwy, przepuklina mózgowa i pępkowa oraz rozszczepienie podniebienia.
U drobiu achondroplazja jest warunkowana genem niezupełnie domi-
nującym Cp (ang. creeper — pełzacz). Gen ten jest letalny u większości
homozygot między 3 a 4 dniem inkubacji. Osobniki heterozygotyczne
charakteryzują się skróconymi odnóżami.
Do mutacji wpływających na cały kościec należy karłowatość. U bydła
wada ta występuje u ras mięsnych (np. u herefordów). Jest to prosta cecha
recesywna. Gen karłowatości u osobnika heterozygotycznego, powodując
tylko nieznaczne skrócenie kończyn, wpływa pośrednio na zwięzłość
budowy. U japońskiego bydła mięsnego, u którego występuje recesywna
karłowatość chondrodysplastyczna (ang. chondrodysplastic dwarfism), ce-
chująca się skróceniem kończyn, japońscy badacze wykryli dwie mutacje
(substytucja zasad i delecja) w genie zlokalizowanym w chromosomie
6 i oznaczonym symbolem LBN (LIMBLIN).
128

Zaburzeniem, które występuje u większości ras bydła, a także u owiec,
świń, a nawet ludzi jest tzw. amputacja kończyn (akroteriaza) (ryć. 5-16
wkładka kolorowa). Anomalia ta uwarunkowana jest genem recesywnym.
U osobnika dotkniętego tą wadą odnóża kończą się blisko stawu łokciowego,
pęcinowego lub kolanowego. Wadzie tej często towarzyszą: skrócenie
żuchwy, rozdwojenie podniebienia i brak większości zębów.
U owiec niektórych ras mięsnych (suffolk, hampshire) w Stanach
Zjednoczonych w latach 80. pojawiła się wada budowy kośćca określana
mianem zespołu pajęczego (ang. spider syndrome). Jest ona wynikiem
mutacji (transwersja adeniny na tyminę, której następstwem jest zmiana
aminokwasu waliny na kwas glutaminowy w pozycji 700 łańcucha polipepty-
dowego) w genie receptora czynnika wzrostu fibroblastów (ang. fibroblast
growth factor receptor 3 -- FGFR3). W układzie homozygotycznym allel
zmutowany powoduje kątową deformację kończyn, deformację grzbietu,
żeber i mostka oraz części pyskowej (garbaty nos). Natomiast osobniki
heterozygotyczne charakteryzuje delikatna budowa kośćca z wyraźnym
wydłużeniem kości długich. Śmiertelność wśród jagniąt dotkniętych ze-
społem pajęczym jest bardzo wysoka, a u tych, które przeżyły, stwierdza się
osłabione zdolności rozrodcze, a nawet niepłodność.
Anomalią układu kostnego, która zależnie od gatunku, a nawet płci
wykazuje różny stopień letalności, jest autosomalna recesywna cecha -
skrócenie kręgosłupa. Jest ona letalna u bydła i indyków. U bydła mlecznego
formą tej wady jest wrodzone zniekształcenie kręgów — CVM (ang. central
vertebral malformation). U psów skrócenie kręgosłupa nie jest letalne dla
suk, rozmnażają się one prawidłowo. Ze względu na zmieniony sposób
siedzenia suki te nazwane są „pawianowatymi". Natomiast samce praw-
dopodobnie nie osiągają dojrzałości. Możliwe więc, że jest to cecha letalna
dla jednej płci. Szczenięta obarczone tą wadą cechuje mniejsza żywotność,
bezpośrednio po urodzeniu można je rozpoznać po skróconym ogonie.
Do wrodzonych wad w budowie kończyn należą: zesztywnienie stawów
i wielopalczastość. Ogólne zesztywnienie stawów notowane jest u bydła,
świń i owiec. Genetycznym podłożem tej wady jest autosomalny gen
recesywny. U jagniąt, cieląt i prosiąt sztywność stawów przy urodzeniu jest
połączona z dziedzicznym przykurczem mięśni.
Do najczęściej spotykanych anomalii w budowie głowy zalicza się
przepuklinę mózgową uwarunkowaną recesywnym genem letalnym. Wada
ta, obserwowana głównie u bydła, polega na niezrośnięciu się kości czaszki,
któremu towarzyszy twór różnych rozmiarów wypełniony przeważnie
płynem. Anomalia ta często występuje w połączeniu z zespołem wad typu
achondroplastycznego.
Prostą autosomalna cechą recesywna u bydła jest skrócenie żuchwy.
Wada ta jest oddzielną jednostką w odróżnieniu od podobnej anomalii
występującej w przypadku tzw. amputacji kończyn. Może ona natomiast
towarzyszyć: wodogłowiu, przepuklinie mózgowej, brakowi gałek ocznych
129

lub zesztywnieniu stawów. U owiec obserwowana jest również cecha
recesywna — brak żuchwy. W pewnym sensie odpowiednikiem skrócenia
żuchwy u bydła jest typ dziobu „kaczor Donald" u kurcząt. Anomalia ta
polega na skróceniu dolnej części dzioba. Wyniki badań wskazują, że wada
ta ma charakter semiletalny i jest rezultatem działania autosomalnego
recesywnego genu oznaczonego symbolem dek (ang. duck).
Wady układu powłokowego
Wady układu powłokowego odnoszą się nie tylko do skóry, ale i takich jej
wytworów, jak włosy i pióra. Jedną z najpoważniejszych letalnych wad
dziedzicznych skóry jest rybia łuska u bydła. Nienormalność ta jest prostą
cechą recesywna, która powstaje w wyniku wytwarzania nadmiernej ilości
keratyny i w następstwie — nieprawidłowego rogowacenia nabłonka. Skóra
nowo narodzonego cielęcia składa się jakby z rogowych płytek przypomina-
jących duże łuski. Płytki te są porozdzielane rowkami, w których rosną włosy.
Normalna skóra i włosy pokrywają jedynie okolice nadgarstka i stepu.
Opisana wada układu powłokowego (rybia łuska) występuje także u ludzi.
Zaburzeniem wykazującym dużą zmienność u różnych ras bydła jest
cecha recesywna określana jako miejscowy brak nabłonka. Miejsca nie
pokryte nabłonkiem najczęściej występują na kończynach powyżej pęcin
i na głowie za śluzawicą. Schorzenie to przybiera najostrzejszą formę,
prowadzącą do śmierci płodów lub noworodków, u bydła rasy Jersey,
natomiast najłagodniejszą, u bydła ayrshire. Jednakże i u cieląt rasy ayrshire
miejsca chore mogą ulegać zakażeniu, a to z kolei może spowodować śmierć
w pierwszych tygodniach życia.
Inną wadą dziedziczną układu powłokowego jest brak owłosienia.
U bydła, królików i kotów anomalia ta jest determinowana recesywnym
genem autosomalnym. U bydła wada ta może występować samodzielnie
lub w połączeniu z innymi nieprawidłowościami wrodzonymi, jak wodo-
głowie, skrócenie szczęki, niedorozwój części twarzowej czaszki lub
cyklopia (dwie gałki oczne w jednym oczodole). Cielęta dotknięte tą wadą
w warunkach naturalnych giną. U psów brak owłosienia uwarunkowany
jest genem dominującym. Na podstawie szczegółowych badań można
sądzić, że wszystkie bezwłose psy (tzw. psy tureckie, chińskie, perskie lub
meksykańskie) są heterozygotami, gdyż gen bezwłosowości w podwójnej
dawce powoduje tak duże nieprawidłowości, że osobniki homozygotyczne
rodzą się martwe.
U drobiu wadą układu powłokowego jest nagość, uwarunkowana
recesywnym genem sprzężonym z płcią, oznaczonym symbolem n (ang.
naked). Prawie połowa piskląt dotkniętych tym zaburzeniem zamiera w ciągu
ostatnich 2-3 dni inkubacji. Ptaki, które przeżyją i będą odchowywane
w temperaturze wyższej niż zwykle, w wieku 4-5 miesięcy pokryją się
rzadkim upierzeniem. Nieśność kur obarczonych tą wadą jest znacznie
zmniejszona.
130

Wady układu pokarmowego
Najczęściej obserwowanymi wadami związanymi z układem pokarmowym
są brak odbytu i niedrożność jelit. Brak odbytu jest recesywną cechą letalną,
powodującą wczesne zejście śmiertelne lub prowadzącą do uboju z koniecz-
ności. Niedrożność jelit zaś, to prosta autosomalna cecha recesywną
występująca zarówno u bydła, jak i u koni. U bydła nienormalność ta
dotyczy jelita biodrowego, u koni natomiast okrężnicy.
Wady układu nerwowego
Wady dziedziczne układu nerwowego stanowią około 16% zaburzeń rozwojo-
wych u bydła. Anomalie układu nerwowego wyrażają się między innymi
bezmózgowiem lub niedorozwojem móżdżku (hipoplazja móżdżku). Obie
nienormalności warunkowane są genem recesywnym. Bezmózgowie występuje
na ogół w połączeniu z rozszczepieniem kręgosłupa i zaburzeniami w rozwoju
narządów wewnętrznych. Niedorozwój móżdżku najlepiej został poznany
u bydła i owiec, u których jest prostą cechą recesywną. Zwierzęta dotknięte tą
wadą rodzą się żywe, ale nie mogą stać. Badania anatomiczne wykazują znaczne
zmniejszenie móżdżku lub patologiczne zmiany w warstwie korowej. Podobna
nieprawidłowość obserwowana u kur jest warunkowana recesywnym genem
sprzężonym z płcią. Schorzenie to nazywane jest drżeniem i ujawnia się
w wieku od 18 do 35 dni. U drobiu występuje również recesywne, sprzężone
z płcią zaburzenie zwane paroksyzmem. Kurczęta w wieku około 2 tygodni lub
starsze reagują na hałas i raptowne oświetlenie biegiem, następnie upadkiem,
usztywnieniem nóg oraz drżeniem ciała. Atak taki trwa około 10 sekund.
Większość z tych kurcząt ginie przed 15 tygodniem życia.
Cechą dziedziczną u wielu gatunków zwierząt jest epilepsja, deter-
minowana u bydła genem dominującym, natomiast u królików — recesyw-
nym. U większości cieląt homozygotycznych pod względem genu warun-
kującego epilepsję objawy pojawiają się w wieku od 3 do 6 miesięcy.
Prostą autosomalna cechą występującą u bydła, świń i drobiu jest
wodogłowie wewnętrzne. Cielęta dotknięte tą wadą rodzą się zazwyczaj
żywe, wykazują powiększenie czaszki, nie potrafią utrzymać się na nogach
i giną w ciągu 2 dni. Z punktu widzenia anatomicznego schorzenie to jest
spowodowane nadmiarem płynu nagromadzonego w komorach mózgowia
lub niekiedy w przestrzeniach między oponą pajęczą i miękką. U świń gen
warunkujący tę wadę wykazuje działanie plejotropowe, wpływa bowiem na
umaszczenie, powodując jego rozjaśnienie. Prosięta z wodogłowiem we-
wnętrznym padają do drugiego dnia życia. U drobiu wodogłowie uwarun-
kowane jest niezupełnie dominującym genem Cr (ang. crest), przy czym
u kur gen ten nie jest letalny (np. rasy houdan i polskie czubatki), podczas
gdy u białych kaczek (crested white) osobniki homozygotyczne giną,
natomiast heterozygoty mają piękny czub, pod którym czaszka jest silnie
wysklepiona wskutek ciśnienia powiększonych półkul mózgowych.
U indyków i kur notowane jest wrodzone zaburzenie równowagi, warun-
131

kowane recesywnym genem letalnym lo (ang. loco). Pisklęta obarczone tą
wadą kłują się dobrze, ale nie mogą jeść, pić i giną w ciągu kilku dni.
Anomalią o dużej częstości występowania, szczególnie u buhajów,
powstającą w wyniku zaburzeń ośrodkowego układu nerwowego jest
spastyczne porażenie kończyn tylnych. Fenotypowym obrazem tej wady
jest spionizowanie stawu skokowego w wyniku skurczu mięśnia brzuchate-
go. Wada ta czasami ujawnia się dopiero w wieku 10-12 lat.
Zaburzeniem układu nerwowego, związanym z niedorozwojem przodo-
mózgowia, jest paraliż i przykurcz mięśni kończyn i szyi, występujący u bydła,
świń i owiec. Jest to prosta, autosomalna cecha recesywna, której objawy mogą
być dwojakie: paraliż wszystkich kończyn bądź tylko kończyn tylnych.
Wady układu rozrodczego
Dość często występujące, a równocześnie dobrze poznane są anomalie
budowy i czynności układu rozrodczego, takie jak: niedorozwój (hipoplazja)
jąder, nasieniowodów, jajników, macicy, niedrożność jajowodów czy wnęt-
rostwo. U białych jałówek rasy belgijskiej błękitnej i shorthorn bezpłodność
wynika z niedorozwoju pochwy i macicy. Anomalia ta jest prawdopodobnie
wynikiem plejotropowego działania genu białego umaszczenia w połączeniu
z nieznaną liczbą genów z innych loci.
Niepłodność obserwowana jest także u buhajów. Plemniki buhajów
dotkniętych tą nieprawidłowością cechuje znaczne zgrubienie akrosomu,
czyli szczytowej części główki, dlatego nazwano je „gałkowatymi". Ten typ
niepłodności występuje u buhajów homozygotycznych pod względem
recesywnego genu kn (ang. knurl).
Zaburzeniem rozwojowym układu rozrodczego spotykanym najczęściej
u psów, ale także u knurów, ogierów i samców innych ssaków jest
wnętrostwo, czyli niezstąpienie jednego lub obu jąder przez kanał pach-
winowy do moszny. Nienormalność tę można usunąć zabiegiem chirurgicz-
nym. Obustronne wnętry są niepłodne również po takim zabiegu. W przy-
padku jednostronnego wnętrostwa osobnik jest płodny i może przekazywać
tę wadę potomstwu. Dlatego konieczne jest usuwanie z hodowli rozpłod-
ników obarczonych wnętrostwem.
Inne przykłady zaburzeń rozwoju cech płciowych są omówione w roz-
dziale: Determinacja i różnicowanie płci oraz interseksualizm.
5.4.4.3. Choroby monogenowe wywołujące zaburzenia
procesów biochemicznych
Wpływ genów na budowę morfologiczną i anatomiczną zwierząt jest znacznie
łatwiejszy do rozpoznania niż ich oddziaływanie na procesy fizjologiczne.
Podłoże genetyczne zaburzeń natury biochemicznej, jak na przykład brak lub
niedobór pewnych enzymów czy hormonów, jest trudne do identyfikacji.
Trudność tę powiększa to, iż objawy pojawiają się u zwierząt w różnym wieku.
132

Typową nieprawidłowością natury biochemicznej jest porfiria występu-
jąca u bydła i świń. U bydła na skutek zaburzeń metabolicznych powstaje
czerwony barwnik porfiryna, wytwarzany w nadmiernych ilościach i od-
kładany we krwi, kościach, zębach i innych częściach ciała. Schorzenie to
wiąże się z zahamowaniem enzymatycznym tworzenia hemu, składnika
hemoglobiny. Zwierzęta dotknięte porfiria są bardzo wrażliwe na promienie
słoneczne, reagują powstawaniem pęcherzy na skórze, a w skrajnych
przypadkach — wrzodów. Genetycznym podłożem tej wady u bydła jest
gen recesywny. U świń porfiria jest prawdopodobnie warunkowana genem
dominującym i schorzenie to ma inne podłoże niż u bydła, gdyż świnie
obarczone tym defektem nie są uczulone na światło słoneczne.
Uczulenie na promienie słoneczne występuje również u owiec, lecz jest
ono jednak innej natury niż porfiria u bydła. Wątroba jagniąt dotkniętych tą
anomalią nie wydziela fikoerytryny, która jest produktem rozkładu chlorofilu.
Fenotypowym obrazem tej wady jest egzema na pysku i uszach, powodująca
śmierć w dwa do trzech tygodni po pojawieniu się pierwszych objawów. Ta
nieprawidłowość metaboliczna ujawnia się u osobników homozygotycznych.
Schorzeniem notowanym u ludzi i psów, a także owiec, charakteryzującym
się zmniejszoną krzepliwością krwi na skutek braku jednego z czynników
biorących udział w tym procesie jest hemofilia, wywoływana działaniem genu
recesywnego sprzężonego z płcią. Są dwa rodzaje tego schorzenia: hemofilia
A — cechuje się brakiem czynnika VIII (globulina przeciwhemofilowa), który
przyspiesza przemianę protrombiny w trombinę, i hemofilia B, mająca
łagodniejszy przebieg, a dotycząca czynnika IX (czynnik Christmas), niezbędne-
go do wytwarzania tromboplastyny osocza. U szczeniąt chorych na hemofilię
A pierwsze objawy pojawiają się w wieku od 6 tygodni do 3 miesięcy. Często
nawet niewinna zabawa szczeniąt może spowodować śmierć osobnika chorego
na hemofilię. U owiec hemofilia A jest chorobą śmiertelną dla większości
obarczonych nią zwierząt. Locus kontrolujący syntezę czynnika VIII u owiec
znajduje się w długim ramieniu chromosomu X w regionie Xq24-q33.
Omówione wyżej wady są uwarunkowane allelami powstałymi w wyni-
ku mutacji genowych. Również innego rodzaju mutacje — aberracje chromo-
somowe mogą być przyczyną wad bądź zmniejszenia wartości cech użytko-
wych. Zagadnienie to jest omówione w podrozdziale: Mutacje chromosomo-
we oraz rozdziale: Determinacja i różnicowanie płci oraz interseksualizm.
Oprócz ogromnej liczby wrodzonych wad dziedzicznych, których pod-
łoże genetyczne jest dobrze poznane, istnieje wiele anomalii, które są
niewątpliwie dziedziczne, ale mechanizm ich dziedziczenia nie jest jeszcze
wyjaśniony. Należą do nich przypadki wielokończynowości, zroślaki (zroś-
nięte dwa płody), wynicowce (niezrośnięcie powłok brzusznych lub klatki
piersiowej, a w konsekwencji tego wydostanie się zawartości jamy brzusznej
lub klatki piersiowej na zewnątrz) oraz przepukliny.
Zestawienie ważniejszych chorób monogenowych dziedzicznych jest
zawarte w tabeli 5-II.
133

134

5.4.4.4. Ograniczanie występowania chorób genetycznych
Częstość występowania genetycznie warunkowanych wad rozwojowych
u zwierząt gospodarskich jest różna w różnych krajach. Tam gdzie prowa-
dzona jest dokładna rejestracja przypadków wad wrodzonych połączona
z ograniczeniem ich występowania, częstość jest niewielka. W Polsce,
niestety, kontrola wad wrodzonych nie jest jeszcze dobrze zorganizowana.
Istnieją jednak akty prawne dotyczące ograniczania występowania tych
zaburzeń.
Opisane wyżej wady są dziedziczone w prosty sposób (jedna para alleli),
ale jedynie te, które są uwarunkowane genami dominującymi (o zupełnej
lub niepełnej dominacji), są zauważalne i łatwe do eliminacji. Większość
wad jest jednak uwarunkowanych genami recesywnymi i trudno je
wyeliminować z populacji hodowlanych.
Zapobieganie rozprzestrzenianiu się wad dziedzicznych w pogłowiu
zwierząt odbywa się przez usuwanie ich nosicieli. Terminem nosiciel
określamy osobnika, który jest fenotypowo normalny, ale w swoim genotypie
135

ma gen letalny (czy semiletalny) w układzie heterozygotycznym. Ujawnienie
się jakiejś wady w populacji u 1% osobników oznacza, że aż 18% osobników
jest nosicielami niepożądanego allelu. Wyjaśnienie tego wyliczenia znajduje
się w rozdziale: Podstawy genetyki populacji.
W przypadku genów sprzężonych z płcią wykrycie nosicielstwa tych
genów nie stwarza większych trudności. Nosicielami mogą być tylko osobniki
płci homogametycznej (u ssaków są to samice, u ptaków samce). Na
przykład od koguta nosiciela recesywnego genu letalnego sprzężonego
z płcią, skojarzonego z normalnymi kurami, będą pochodzić wszyscy
synowie fenotypowo normalni, jednakże połowa z nich będzie homo-
zygotami dominującymi, a połowa nosicielami niepożądanego genu. Nato-
miast wśród jego córek połowa będzie normalnych, a u połowy ujawni się
wada dziedziczna. Jeśli będzie to gen letalny powodujący zamieranie
zarodków, to po wykluciu stosunek liczbowy płci u potomstwa będzie
odbiegał od oczekiwanego 1 : 1 i będzie wynosił 2:1.
Znaczne trudności sprawia wykrywanie nosicieli autosomalnych genów
letalnych o charakterze recesywnym. Geny te, jak już wspomniano, fenotypo-
wo ujawniają swą obecność wówczas, gdy występują w stanie homozygoty-
cznym. Zatem nosicielstwo niepożądanego genu może być potwierdzone lub
wykluczone poprzez zastosowanie odpowiednich kojarzeń. Postępowanie to
ma na celu wykrycie nosicielstwa u rozpłodników, głównie wykorzystywa-
nych w sztucznej inseminacji (np. buhaje).
Znane są trzy klasyczne metody testowania rozpłodników na nosicielstwo
genów recesywnych:
1. Kojarzenie testowanego samca z samicami o genotypie homozygoty
recesywnej. Testowanie takie jest możliwe jedynie wtedy, gdy homozygoty
recesywne wykazujące wadę żyją i są płodne. Schemat takiego kojarzenia
jest następujący:
Jeżeli testowany osobnik jest nosicielem allelu recesywnego, to około
50% jego potomków będzie homozygotami recesywnymi.
2. Kojarzenie samca (domniemanego nosiciela) z dużą liczbą samic,
wśród których można się spodziewać obecności nosicielek recesywnego
allelu.
136

Pojawienie się w potomstwie pochodzącym z takich kojarzeń osobnika
obarczonego wadą świadczy o obecności poszukiwanego allelu recesywnego
w genotypach testowanego rozpłodnika oraz matki noworodka.
3. Dokładną metodą identyfikacji nosicielstwa recesywnego genu, lecz
trudną do zaakceptowania ze względów hodowlanych, jest kojarzenie
samca z jego własnymi córkami. Jeśli przyjmiemy, że samice matki tych
córek są homozygotyczne (AA), a samiec jest heterozygotyczny, to wśród
córek połowa będzie miała genotyp AA, a połowa Aa. Wówczas schemat
kojarzenia testowego jest następujący:
Kojarzenie rozpłodnika (ewentualnego nosiciela) z jego własnymi córkami
powinno dać w potomstwie rozszczepienie w stosunku 7 normalnych do l
obarczonego wadą.
W hodowli bydła młode buhaje, zakupione przez Stacje Hodowli i Una-
sieniania Zwierząt, kojarzone są z losowo wybranymi samicami. Celem
zasadniczym jest ocena wartości genetycznej rozpłodnika w zakresie cech
użytkowości mlecznej na podstawie potomstwa (metoda ta jest opisana
w podręcznikach z zakresu metod hodowlanych). Przeprowadzone kojarze-
nie może umożliwić także wykrycie obecności w genotypie zwierząt recesyw-
nych genów letalnych (tzw. test automatyczny). Jeśli oboje rodzice mają
genotyp heterozygotyczny w danym locus, w potomstwie może się ujawnić
z prawdopodobieństwem równym 1/4 niepożądana cecha recesywna.
Całkowita eliminacja z populacji genów letalnych o nieznanej budowie
molekularnej jest niemożliwa. Zatem jednym ze sposobów unikania w hodo-
wli masowej strat powodowanych dziedzicznymi wadami wrodzonymi jest
niedopuszczanie do kojarzeń krewniaczych. Ponadto należy rejestrować
i zgłaszać w zakładach unasieniania lub w okręgowych stacjach hodowli
zwierząt wszystkie przypadki rodzenia się zwierząt żywych lub martwych
z wadami wrodzonymi. Rejestracja ta z jednej strony daje obraz częstości
występowania tych wad, z drugiej natomiast umożliwia usuwanie z hodowli
nosicieli (zwłaszcza męskich).
137

Rozwój genetyki molekularnej pozwala coraz częściej na identyfikację
nosicielstwa zmutowanych alleli poprzez testy oparte na analizie re-
strykcyjnej (RFLP) zamplifikowanych metodą PCR fragmentów DNA,
w których występuje gen odpowiedzialny za powstanie dziedzicznej
wady wrodzonej czy choroby genetycznej. Zagadnienie to opisano niżej.
DIAGNOSTYKA MOLEKULARNA NIEKTÓRYCH CHORÓB GENETYCZNYCH
Większość chorób genetycznych zwierząt gospodarskich jest spowodowana
mutacjami genowymi, które prowadzą do zmiany sekwencji kodującej
skład aminokwasowy określonego białka. Ostatnie lata przyniosły postęp
w poznawaniu podłoża molekularnego chorób dziedzicznych. W diagnostyce
tych chorób stosowane są techniki biologii molekularnej, głównie PCR-RFLP,
która polega na ustalaniu długości fragmentów restrykcyjnych DNA przez
cięcie (trawienie) zamplifikowanego DNA enzymami restrykcyjnymi (en-
donukleazy), a następnie rozdział tych fragmentów za pomocą elektroforezy
(patrz rozdział: Metody analizy i modyfikacji genomu).
Najbardziej rozpowszechnioną chorobą genetyczną w hodowli bydła
rasy holsztyńsko-fryzyjskiej jest BLAD (ang. bovine leukocyte adhesion
deficiency) — zmniejszona odporność cieląt, spowodowana wrodzonym
niedoborem leukocytarnych cząsteczek adhezyjnych. Jest to choroba auto-
somalna recesywna, występująca również u ludzi pod nazwą LAD (ang.
leukocyte adhesion deficiency). Jest ona warunkowana zmutowanym
recesywnym allelem (oznaczonym symbolem D128G) genu ITGB2, który
koduje strukturalną podjednostkę CD18 p
2
-integryny. Białko to odgrywa
istotną rolę w odporności organizmu. Pewnym niebezpieczeństwem roz-
przestrzeniania tej choroby jest to, iż przypuszczalnie geny korzystnych
cech ilościowych są istotnie sprzężone z locus genu ITGB2. Osobniki
heterozygotyczne (nosiciele niekorzystnego zmutowanego allelu) cechuje
jednocześnie wysoka wartość hodowlana.
Na początku lat 90. w USA frekwencja nosicieli niekorzystnego allelu
D128G wynosiła około 15% wśród buhajów i 6% wśród krów.
U bydła zespół BLAD był znany od kilkunastu lat, po raz pierwszy
opisano go w 1983 roku. Analiza rodowodowa wykazała, że mutacja genu
ITGB2 wystąpiła u buhaja urodzonego w USA w 1952 roku. Jednak
molekularna identyfikacja allelu D128G (BLAD) nastąpiła dopiero w 1992
roku. W latach 70. rozpoczął się intensywny import do Polski bydła rasy
holsztyńsko-fryzyjskiej w celu poprawy wartości genetycznej rodzimego
bydła czarno-białego, tym samym wprowadzono do puli genowej naszego
bydła niekorzystny allel D128G. Ponieważ jest to gen recesywny, szczególne
znaczenie ma identyfikacja jego nosicieli (osobniki heterozygotyczne, feno-
typowo zdrowe). Stwierdzona w pozycji 383 nukleotydu mutacja, powodu-
jąca zamianę w pozycji 128 łańcucha polipeptydowego kwasu asparagino-
138

wego na glicynę, jest podstawą testu molekularnego umożliwiającego
wykrywanie nosicielstwa.
Za pomocą analizy fragmentów restrykcyjnych określa się, jaki allel
występuje w badanym DNA. Wykorzystuje się do tego celu istnienie
różnych miejsc cięcia dla enzymów restrykcyjnych Haelll i Tacjl zależnie od
normalnych i zmutowanych sekwencji nukleotydowych. W allelu prawid-
łowym znajduje się miejsce restrykcyjne rozpoznawane przez enzym Haelll
i miejsce restrykcyjne dla enzymu TaqI. Natomiast w allelu zmutowanym
D128G mutacja punktowa zmienia sekwencję rozpoznawaną przez enzym
TaqI i w następstwie tego enzym nie przecina cząsteczki DNA. Z kolei, ta
sama mutacja punktowa powoduje powstanie drugiego miejsca restrykcyj-
nego dla enzymu HaeIII. Po zamplifikowaniu fragmentu genu
β
2
-integryny
produkt PCR długości 58 par zasad poddaje się trawieniu jednym z wymie-
nionych enzymów, a następnie uzyskane fragmenty rozdziela elektro-
foretycznie (metoda RFLP). Obraz elektroforetycznego rozdziału wygląda
następująco:
W obrębie genu kodującego podjednostkę
β
2
-integryny stwierdzono także
drugą mutację, tzw. ciche podstawienie 775 nukleotydu, które nie wpływa
na sekwencję aminokwasową białka kodowanego przez ten gen.
Inną chorobą genetyczną bydła rasy holsztyńsko-fryzyjskiej jest cyt-
rulinemia, opisana po raz pierwszy u bydła mlecznego w Australii w 1986
roku. W 1993 roku stwierdzono jej występowanie również u bydła tej rasy
w USA. Mutacja ta została już przedstawiona wcześniej. Jej podłożem
genetycznym jest zmutowany, recesywny allel genu warunkującego syntezę
enzymu syntetazy argininobursztynianowej, który bierze udział w cyklu
mocznikowym. Nosiciele cytrulinemii mają genotyp ASAS/asas i fenotypowo
są normalni, natomiast cielęta homozygotyczne asas/asas padają 5-6 dnia po
urodzeniu z powodu upośledzenia zdolności usuwania mocznika z organiz-
mu. Ze względu na możliwość wprowadzenia tego niekorzystnego allelu do
genotypów bydła w innych krajach, podejmowane są środki prewencyjne.
Opracowany test diagnostyczny cytrulinemii z zastosowaniem techniki
PCR-RFLP wykorzystuje to, iż zmutowany allel nie ma miejsca restrykcyj-
139

nego dla enzymu Avall w kodonie 86. Wynikiem testu molekularnego dla
osobnika homozygotycznego pod względem allelu zmutowanego jest jeden
prążek długości 194 pz, dla heterozygotycznego trzy prążki: 194 pz, 118 pz
i 72 pz, a dla normalnej homozygoty dwa prążki 118 pz i 72 pz.
W obrębie genu syntetazy argininobursztynianowej stwierdzono także
tranzycję cytozyny na tyminę, powodującą polimorfizm w 175 kodonie dla
proliny (CCC
→CCT), nie dającą żadnych skutków fenotypowych.
Kolejną, również wcześniej przedstawioną mutacją letalną, występującą
u czarno-białego i czerwono-białego bydła holsztyńsko-fryzyjskiego, jest
recesywna mutacja genu syntazy urydynomonofosforanowej. Zmutowany
allel genu UMPS, określony symbolem DUMPS (ang. deficiency uridine
monophosphate synthase), powoduje zamieranie homozygotycznych zarod-
ków około 40 dnia ciąży, dlatego nazwano go „genem zamieralności
zarodków". Osobniki heterozygotyczne są fenotypowo normalne, ale wyka-
zują obniżony poziom aktywności enzymu. Konsekwencją tego jest podwyż-
szony poziom kwasu orotowego w mleku i moczu krów heterozygotycznych.
Z drugiej jednak strony, krowy takie charakteryzuje większa wydajność
mleka i zawartość białka w mleku, natomiast buhaje mają wyższą wartość
hodowlaną w zakresie cech użytkowości mlecznej. Aby nie dopuszczać do
kojarzeń nosicieli tego niekorzystnego genu, których skutkiem byłoby
zmniejszenie płodności krów (zamieralność zarodków homozygotycznych),
konieczna jest identyfikacja genotypów heterozygotycznych (nosicieli). Po-
dobnie jak w przypadku poprzednich mutacji, do identyfikacji nosicielstwa
zmutowanego allelu stosowana jest metoda PCR-RFLP. Mutacja ta w kodonie
405 genu UMPS znosi miejsce restrykcyjne dla enzymu Aval. Prawidłowy gen
syntazy urydynomonofosforanowej będzie przez ten enzym trawiony, nato-
miast allel zmutowany (DUMPS) — nie będzie. Zamplifikowany fragment
genu trawiony enzymem Avol da następujący obraz rozdziału elektroforetycz-
nego:
osobniki homozygotyczne (zdrowe) — dwa prążki: 53 pz i 36 pz
osobniki heterozygotyczne (nosiciele) — trzy prążki : 53 pz, 36 pz i 89 pz
Ze względu na letalny efekt allelu DUMPS w układzie homozygotycznym,
niemożliwa jest analiza DNA osobników homozygotycznych. Prowadzone
są jednak badania blastocyst przewidzianych do embriotransferu pochodzą-
cych z kojarzeń osobników heterozygotycznych.
U koni dotychczas poznano molekularne tło dwóch chorób genetycznych.
Pierwsza z nich to okresowy paraliż, nazwany HYPP (ang. hyperkalemic
periodic paralysis), pojawiający się u koni „ąuarter" w USA. Odpowiedzialna
za tę chorobę jest mutacja w genie kanału sodowego mięśni, polegająca na
transwersji C
→G w transbłonowej domenie IVS
3
podjednostki a genu
kanału sodowego (zamiana fenyloalaniny na leucynę w regionie między
1358 a 1514 aminokwasem w białku). Objawami tej choroby są ataki drżenia
mięśni i paraliżu, występujące po zmianie diety, okresie głodzenia, za-
140

działaniu czynnika stresogennego lub spożyciu siana z lucerny bogatej
w potas. Okresowy paraliż występuje u koni cennych pod względem
wartości genetycznej, dobrze umięśnionych. Selekcja koni na podstawie
wyników gonitw na torze wyścigowym może prowadzić do wzrostu
frekwencji tego niekorzystnego allelu. Mutacja HYPP dziedziczy się
z niezupełną dominacją, u koni homozygotycznych objawy są silniejsze
niż u heterozygotycznych. Diagnostyka molekularna nosiciela zmutowane-
go ałlelu prowadzona jest za pomocą metody PCR-RFLP, z zastosowa-
niem enzymu restrykcyjnego Tagi. Podobna choroba od 50 lat znana jest
u ludzi.
Druga choroba genetyczna o znanym podłożu molekularnym wy-
stępująca u koni to ciężki złożony brak odporności (ang. severe combined
immunodeficiency disease — SCID). Choroba ta charakteryzuje się
brakiem limfocytów T oraz B, co powoduje załamanie się układu im-
munologicznego. Źrebięta o genotypie homozygoty recesywnej padają
wkrótce po urodzeniu wskutek rozwoju różnorodnych infekcji. Za rozwój
tej choroby odpowiedzialna jest mutacja polegająca na delecji 5 nu-
kleotydów w genie kodującym katalityczną podjednostkę kinazy biał-
kowej zależnej od DNA (DNA-PK). Mutację tę stwierdzono u koni
czystej krwi arabskiej hodowanych w USA, natomiast badania prze-
prowadzone w Polsce wykazały, że nie występuje ona w polskiej
populacji koni arabskich.
Omawiając choroby genetyczne nie sposób pominąć tzw. chorób prio-
nowych. Wprawdzie są one wywoływane przez nietypowy czynnik zakaźny,
którym jest białko, jednak predyspozycje do tych chorób mogą być
warunkowane mutacją genową. Białkowe cząsteczki infekcyjne, nazwane
prionami, namnażają się w ten sposób, iż przekształcają prawidłowe
cząsteczki białka w szkodliwe, indukując zmiany ich konformacji. Znane
choroby prionowe powodują często ubytki w mózgu i w konsekwencji
prowadzą do śmierci. Z tego powodu określane są gąbczastymi encefalopa-
tiami (ang. spongiform encephalopathies).
Przypuszcza się, iż białko prionowe i białko komórkowe (normalne) są
kodowane przez ten sam gen, a różnica między nimi wynika z zakłóceń
w wycinaniu intronów tego genu (splicing mRNA). Gen kodujący białko
komórkowe oznaczono symbolem PrP
P
c
, jego zmutowany allel, warunkujący
białko prionowe — symbolem PrP
Sc
. Gen ten u człowieka znajduje się
w krótkim ramieniu chromosomu 20, na granicy prążka 12 i 13 (20pl2-pl3),
natomiast u bydła i owiec w długim ramieniu chromosomu 13, przy czym
u bydła w pozycji 13ql7, u owiec — 13ql7-ql8.
Jedną z najdawniej, bo około 250 lat temu, poznanych chorób prionowych
u zwierząt jest trzęsawka (ang. scrapie — drapać) u owiec. Choroba
ta jest śmiertelna i charakteryzuje się długim okresem inkubacji, trwającym
miesiące lub lata bez żadnych objawów. W czasie jej rozwoju przekształcone
cząsteczki białka gromadzą się w mózgu i niektórych tkankach, jak
141

śledziona czy węzły chłonne. Diagnozowanie choroby jest oparte na
pośmiertnym klinicznym badaniu mózgu. W ciągu ostatnich 20 lat czyniono
próby ograniczenia występowania scrapie przez selekcję owiec odpornych
na nią i brakowanie podatnych. Metoda ta okazała się jednak zbyt droga
i długotrwała, a jej rezultaty nie były zadowalające. Znacznie większe
możliwości eliminacji scrapie stwarzają techniki biologii molekularnej.
W obrębie genu PrP
c
u owiec szczególnie istotne są trzy mutacje punktowe
w następujących kodonach: 136 (kodon dla waliny zmieniony w kodon dla
alaniny; oznaczenia literowe V/A), 154 (arginina/histydyna; R/H) oraz 171
(arginina/glutamina; R/Q) (tab. 5-III). Jednak duża zmienność rasowa
w podatności na scrapie ogromnie utrudnia określenie genotypu „podat-
nego" i „odpornego". Wydaje się, że najbardziej odporne są owce o genotypie
AA
136
RR
154
RR
171
, a najbardziej podatne owce o genotypie W
136
RR
154
17
j
(genotyp ten występuje jednak bardzo rzadko).
U bydła chorobą prionową jest rozmiękczenie mózgu (ang. bovine
spongiform encephalopathy — BSE). Pierwszy przypadek tej choroby
zanotowano w 1985 roku w Wielkiej Brytanii, rok później została opisana
jej patologia. Jest to choroba ośrodkowego układu nerwowego. Jej charak-
terystyczną cechą jest tworzenie i gromadzenie białka amyloidowego
(odpornego na działanie enzymów proteolitycznych) w mózgu zakażonych
zwierząt. Czynnikiem zakaźnym BSE jest to samo białko prionowe, które
wywołuje scrapie u owiec. Białko to ma około 250 aminokwasów. W genie
PrP
c
u bydła (w chromosomie 13 —BTA13ql7) wykryto polimorfizm,
związany z różnicą w liczbie ośmiopeptydowych powtórzeń (5 lub 6 po-
wtórzeń) (ryć. 5-17). Dotychczas stwierdzono, że częściej występuje 6 po-
wtórzeń. Spośród trzech możliwych genotypów (6:6, 6:5 i 5:5) najrzadziej
występuje genotyp homozygotyczny — pięć powtórzeń. Ponadto tylko ten
genotyp nie występował u krów z BSE. Prowadzone są intensywne badania
na myszach transgenicznych z bydlęcym genem PrP
c
, które powinny
przyczynić się do wyjaśnienia genetycznego tła tej choroby.
Ostatnie badania wykazały, że w pobliżu genu białka prionowego
znajduje się gen Prnd (ang. prion-doppel). Gen ten koduje białko określane
jako Dpl (ang. downstream prion-like protein) lub doppel, które wykazuje
25% homologii w składzie aminokwasowym z białkiem prionowym. Nadek-
spresja tego genu w mózgu może powodować inną chorobę neurodegene-
racyjną. Wykryto już kilka mutacji w tym genie u owiec, kóz i bydła.
142
5.4.5. Mutacje genowe odpowiedzialne za
odporność/podatność zwierząt na patogeny
Gen odporności na kleszcze
W niektórych krajach, głównie w północnej Australii i innych regionach
tropikalnych, jak również w Europie, bydło jest szczególnie podatne na
inwazje kleszczy. Z kolei, bydło indyjskie jest bardzo odporne na kleszcze.
Tę odporność można przenosić na mieszańce poprzez krzyżowanie z rasami
podatnymi, ale jednocześnie powoduje to zmniejszenie produkcyjności
potomstwa krzyżówkowego. Australijscy badacze wykryli u linii pochodzą-
cych z krzyżowania bydła mięsnego ras hereford i shorthorn gen o dużym
efekcie, determinujący odporność na kleszcze. Wykorzystanie tego genu
stwarza możliwość stosunkowo szybkiego zwiększenia odporności na
kleszcze różnych ras bydła.
Gen K88 i F18
U świń odporność na niektóre choroby infekcyjne jest kontrolowana
przez pojedynczy gen. Należą do nich biegunki okresu noworodkowego
u prosiąt, w przeważającej mierze wywoływane enterotoksycznymi szcze-
pami Escherichia coli, powodujące rocznie padnięcie około 10 min prosiąt
na świecie. Podatność na te biegunki zależy od obecności receptorów
na powierzchni komórek nabłonka jelita cienkiego u zwierząt, umożli-
wiających adhezję bakterii i ich rozwój. Z kolei, występowanie tych
143

receptorów warunkowane jest genem głównym (o dużym efekcie). Loci
receptorów dla E. coli K88 zlokalizowano w chromosomie 13, blisko locus Tf
(transferyny). Receptory te prawdopodobnie pełnią także jakieś funkcje
fizjologiczne, stwierdzono bowiem, iż zwierzęta mające te receptory charak-
teryzują się zwiększonymi dobowymi przyrostami masy ciała w przedziale
wagowym od 24 do 100 kg.
U prosiąt w późniejszym wieku (4-12 tygodni) występuje choroba
obrzękowa, której objawami są m.in. ataksja, konwulsje i paraliż oraz
biegunka poodsadzeniowa (kolibacilloza jelitowa), schorzenia wywoływane
przez enterotoksyny wytwarzane przez zasiedlający powierzchnię jelita
cienkiego szczep F18 E. coli (bakterie te mają fimbrie). Odporność/podatność
na adhezję tej bakterii jest kontrolowana przez locus ECF18R (locus receptora
dla E. coli F18), znajdujący się w chromosomie 6 i wchodzący w skład
halotanowej grupy sprzężeniowej. Podatność jest cechą dominującą (allel B),
a odporność recesywną (allel b). Niedawno stwierdzono sprzężenie między
locus ECF18R a dwoma loci (FUT1 i FUT2) dla
α-l,2-fukozylotransferazy.
W jednym z nich (FUT1) wykryto dwie mutacje, z których jedna, oznaczona
symbolem M307, polegająca na tranzycji G-»A w kodonie 307 (w łańcuchu
peptydowym zamiana alaniny na treoninę), wykazuje ścisłe sprzężenie
z genem ECF18R. Mutacja ta likwiduje miejsce trawienia DNA dla enzymu
CfoI. Analiza polimorfizmu DNA przeprowadzona metodą RFLP wykazuje
obecność dwóch prążków (93 pz i 328 pz) u osobników homozygotycznych
MSOZ
A/A
(allel zmutowany), trzech prążków (93, 241 i 87 pz) u homozygot
M307
G/G
(allel normalny) oraz wszystkich czterech prążków u zwierząt
heterozygotycznych. W badaniach szwajcarskich wykazano 100% sprzężenie
allelu ECF18R
1
', warunkującego odporność na infekcje E. coli F18, z ałlelem
zmutowanym M307
A
, oraz takie samo sprzężenie allelu ECF18R
B
(podatność
na biegunki i chorobę obrzękową) z ałlelem normalnym M307
G
. Stwarza to
szansę wczesnego wykrywania prosiąt podatnych na infekcje E. coli F18 na
podstawie testu diagnostycznego dla mutacji M307 w locus
α-fukozylotrans-
ferazy. Badania przeprowadzone w Polsce wykazały wysoką frekwencję
allelu odporności w rasie złotnicka pstra.
5.5. Mutacje w mitochondrialnym DNA
Mutacje w mitochondrialnym DNA (mtDNA) zachodzą znacznie częściej
niż mutacje w DNA jądrowym. Przyczyną tego może być brak w mitochon-
driach mechanizmów naprawy uszkodzeń DNA.
Mutacje te mogą być dziedziczone po matce, mogą także powstawać
de novo w komórce jajowej lub w zarodku we wczesnym stadium roz-
wojowym. Część mutacji w mitochondrialnym DNA zachodzi w ciągu
całego życia osobnika, a ich charakterystyczną cechą jest to, iż mogą
144

dotyczyć całych tkanek, pojedynczych komórek lub nawet poszczególnych
cząsteczek mtDNA tej samej komórki.
Miejsce i zasięg występowania danej mutacji powodują, iż w tkance
występują razem gen prawidłowy i jego zmutowany allel bądź dana
mutacja może występować w każdej cząsteczce mtDNA we wszystkich
tkankach. Pierwszy rodzaj mutacji nazywany jest mutacją heteroplaz-
matyczną, drugi natomiast mutacją homoplazmatyczną.
Mutacje w mitochondrialnym DNA są przyczyną bardzo poważnych
chorób genetycznych u ludzi. Należą do nich przede wszystkim choroby
ośrodkowego układu nerwowego i układu mięśniowego. Niestety brak
jest efektywnych metod leczenia chorób wywoływanych tymi mutacjami.
Cechą charakterystyczną chorób powodowanych mutacjami w mtDNA
jest to, iż mutacja jest przekazywana przez komórkę jajową od chorej
matki zarówno córkom, jak i synom, ale dalszym pokoleniom cechę
tę przekazują tylko córki.
Mutacje w mitochondrialnym DNA, podobnie jak w DNA jądrowym,
polegają na substytucjach nukleotydów (głównie tranzycja), insercji bądź
delecji nukleotydów. Skutkiem tranzycji są takie choroby u człowieka, jak
dziedziczna neuropatia nerwu wzrokowego — zespół Lebera (LHON)
i neuropatia obwodowa mięśni połączona z ataksją i barwnikowym zwyrod-
nieniem siatkówki (NARP). Podstawienia nukleotydów w genach kodujących
tRNA powodują m.in. mitochondrialną miopatię, padaczkę miokloniczną
i tzw. poszarpane czerwone włókna (MERRF), dziedziczną miopatię i kar-
diomiopatię (MMC) oraz zespół chorobowy oznaczony skrótem MELAS
(encefalomiopatia mitochondrialną, kwasica mleczanowa i napady udaropo-
dobne). Z kolei mutacje typu insercja-delecja są przyczyną większości
miopatii ocznych (np. chroniczny postępujący paraliż mięśni oka — CEOP)
i zespołu Pearsona, polegającego na zaburzeniach funkcjonowania szpiku
kostnego u dzieci i niewydolności trzustki.
Większość mutacji w mtDNA cechuje specyficzna relacja między geno-
typem a fenotypem. Określona choroba może być wywołana mutacjami
w różnych genach i odwrotnie — dana mutacja może być przyczyną
różnych chorób. Przykładem pierwszej sytuacji jest wspomniana już choroba
LHON, której podłożem genetycznym są mutacje w 6 różnych podjednost-
kach dehydrogenazy NADH lub mutacja w genie apocytochromu b. Z kolei,
mutacja w pozycji 8993 genu syntazy ATP warunkuje chorobę NARP oraz
zespół Leigha (śmiertelna choroba wieku dziecięcego).
Badania występowania i skutków mutacji genów mitochondrialnych
u zwierząt są nieliczne. Wiadomo na przykład, że niektóre allele genu
karboksylazy fosfoenolopirogronianowej (enzym biorący udział w szlaku
glukoneogenezy) występują u kurcząt podatnych na chorobę Mareka,
wywoływaną przez wirus, inne natomiast u odpornych.
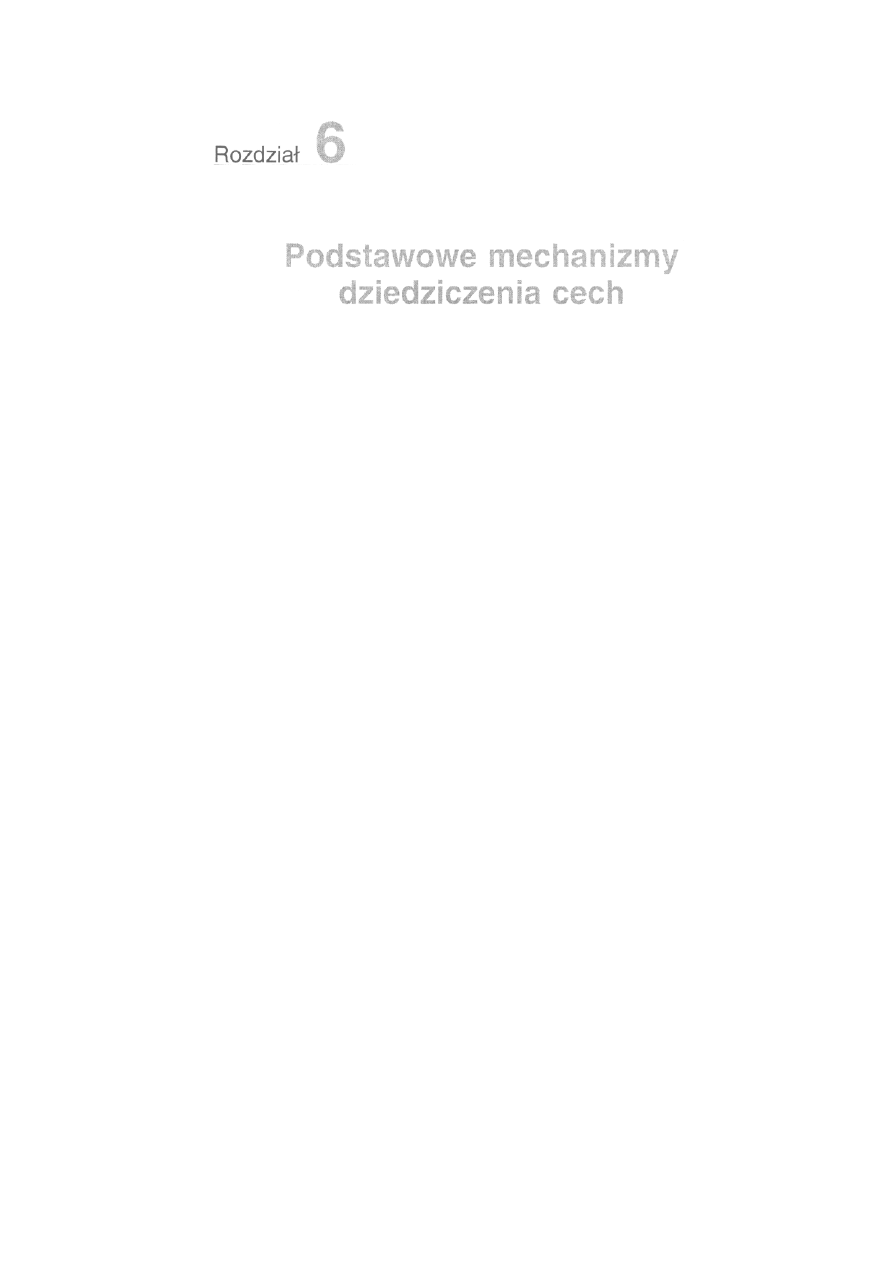
Genetyka klasyczna (mendlowska) jako cechę określa właściwość or-
ganizmu, która jest łatwa do odróżnienia w zespole innych właściwości
(np. barwa umaszczenia, posiadanie/brak rogów). Dzięki osiągnięciom
genetyki biochemicznej powstała hipoteza jeden gen — jeden enzym.
W myśl tej hipotezy synteza każdego enzymu jest kontrolowana przez
określony gen. Dalsze badania spowodowały uściślenie tej hipotezy:
jeden gen — jeden łańcuch polipeptydowy. Oznacza to, że jeden gen
warunkuje sekwencję aminokwasów w polipeptydzie.
W opisie mechanizmów dziedziczenia łatwiej jest posługiwać się mend-
lowskim pojęciem cechy jako pewnej właściwości. Ogólnie cechy można
podzielić na jakościowe i ilościowe. Cechy jakościowe są warunkowane
jedną, rzadziej kilkoma współdziałającymi ze sobą parami genów. Natomiast
podłoże genetyczne cech ilościowych jest bardziej złożone, bowiem na ich
ekspresję wpływa kilkanaście, a nawet kilkadziesiąt par genów. Ponadto,
w przypadku wielu cech ilościowych ich wartość jest modyfikowana przez
czynniki środowiskowe.
Mechanizm dziedziczenia cech ilościowych jest omówiony w rozdziale:
Zmienność cech.
Do cech jakościowych zalicza się takie, jak: umaszczenie zwierząt,
kolor upierzenia u drobiu; rogatość lub bezrożność bydła, owiec i kóz;
układy grupowe krwi itd. Wspólną właściwością tych cech jest to, że
identyfikacja różnych wariantów fenotypowych danej cechy jest względnie
prosta.
Mechanizm dziedziczenia cech jakościowych jest zróżnicowany, a ich
ujawnienie się zależy przede wszystkim od założeń genetycznych. Wpływ
czynników środowiskowych na ekspresję tych cech jest niewielki i dotyczy
tylko nielicznych przypadków, np. umaszczenia. Znane są również przy-
kłady, kiedy jeden gen wpływa na kształtowanie się kilku cech. Zjawisko to
nazywa się plejotropią.
146

Zestaw genów występujących w komórkach jednego osobnika nazywamy
genotypem, a zespół cech warunkowanych przez genotyp i czynniki środo-
wiskowe — fenotypem.
Genotyp jest ustalany na podstawie fenotypu osobnika lub jego krewnych
bądź przez bezpośrednie badanie DNA. Fenotyp jest opisywany na pod-
stawie obserwacji lub pomiaru cechy oraz analiz molekularnych (identyfika-
cja białek).
Jak już wspomniano, cechy jakościowe mogą być warunkowane jedną
lub kilkoma parami genów współdziałających ze sobą. Pierwszy rodzaj
współdziałania nosi nazwę allelicznego (1 para genów), drugi natomiast —
nieallelicznego (kilka par genów).
Para genów warunkujących powstanie określonej cechy zajmuje okreś-
lone miejsce (locus) w dwóch homologicznych chromosomach. Geny te
nazywane są parą alleli lub allelomorfów. Allelomorficzną parę genów
oznacza się tą samą literą — wielką lub małą (np. A-a). Wybór litery zwykle
jest związany z angielską nazwą pierwszego wariantu allelu wykrytego
w danym locus.
Zygota, która zawiera dwa jednakowe geny z danej pary alleli, nazywa
się homozygotą (AA lub aa), natomiast mająca dwa różne geny — hetero-
zygotą (Aa). Kojarzenie jednakowych homozygot między sobą daje zawsze
potomstwo jednolite pod względem interesującej nas cechy. W przypadku
kojarzenia heterozygot w potomstwie wystąpi rozszczepienie cech, dając
wszystkie możliwe formy danej cechy.
Podstawowe reguły dziedziczenia cech zawarte są w prawach Mendla.
I prawo Mendla, zwane prawem czystości gamet, mówi o tym, że
gameta zawiera tylko jeden gen z danej pary alleli. W procesie game-
togenezy allele z danej pary rozchodzą się do gamet. Podczas zapłodnienia
dochodzi do losowego łączenia się alleli w pary. Potomek dziedziczy
zatem jeden allel z danej pary od ojca, drugi natomiast od matki.
6.1. Dziedziczenie cech warunkowanych jedną
parą alleli
6.1.1. Współdziałanie alleli
Cechy jakościowe kontrolowane przez jeden gen są wynikiem współ-
działania między parą alleli tego genu. Można wyróżnić następujące rodzaje
tego współdziałania:
1) dominowanie całkowite — typ Pisum
2) dominowanie niezupełne (dziedziczenie pośrednie) — typ Zea
3) kodominowanie
4) naddominowanie.
147

Przykładem całkowitej dominacji może być bezrożność u bydła. Stwier-
dzono, iż cecha bezrożności/rogatości jest kontrolowana przez locus polled
(znajdujący się w l chromosomie), który ma dwa allele: P — dominujący
allel bezrożności i p — recesywny gen rogatości. Wszystkie zwierzęta
(bydło) rogate są homozygotyczne pod względem recesywnego allelu p.
Allel ten u osobników heterozygotycznych nie ujawnia się w fenotypie,
gdyż gen dominujący P maskuje jego ekspresję. Para rodziców, będących
homozygotami alternatywnymi (PP i pp) pod względem locus polled, zawsze
da potomstwo heterozygotyczne, wykazujące dominującą formę cechy.
Kojarząc te osobniki między sobą uzyskamy w następnym pokoleniu 3/4
zwierząt bezrogich (PP i Pp) i 1/4 rogatych (pp).
Innym przykładem jest biały pas u czarno umaszczonych świń rasy
hampshire, kontrolowany przez allel dominujący Be
w
z locus Be. Z kolei
u świń rasy cornwall, również czarno umaszczonych, w locus tym występuje
allel be, jednolitego umaszczenia. Krzyżowanie międzyrasowe świń obu
tych ras przedstawiono na rycinie 6-1.
Przedstawiony na przykładzie sposób dziedziczenia z dominowaniem
całkowitym nazywany jest również dziedziczeniem typu Pisum. Do cech
dziedziczących się z dominowaniem całkowitym należą także: bezrożność
(cecha dominująca) i rogatość kóz, jednolite umaszczenie (dominuje)
i łaciatość u bydła, czarne umaszczenie u bydła, dominujące nad czerwonym
i inne.
Dominowanie niezupełne genów z jednej pary alleli polega na tym, że
u osobników heterozygotycznych pod względem danej pary alleli występuje
pośrednia forma cechy. Ilustracją tego typu dziedziczenia, nazywanego
również dziedziczeniem typu Zea, jest poniższy przykład.
U bydła rasy shorthorn obok zwierząt czerwono umaszczonych zdarzają
się osobniki o umaszczeniu białym oraz zwierzęta mające umaszczenie
pośrednie — mroziate. Kojarząc zwierzęta umaszczone czerwono ze zwie-
rzętami białymi, otrzymuje się potomstwo (pokolenie F
l
) mroziate. Natomiast
z kojarzenia osobników mroziato umaszczonych uzyskuje się potomstwo
(pokolenie F
2
), z którego część ma umaszczenie czerwone, część mroziate,
część zaś białe. Jeżeli obserwacje będą prowadzone na dużej grupie zwierząt,
to stosunek liczbowy osobników czerwonych, mroziatych i białych w poko-
leniu F
2
wyniesie 1:2:1.
Charakterystyczną cechą tego typu dziedziczenia jest to, że fenotyp
zwierzęcia odzwierciedla jego genotyp. Wiele cech zwierząt domowych
dziedziczy się tak jak cecha umaszczenia u shorthornów. Są to na przykład:
szurpatość u kur (pióra nastroszone i pozwijane), upierzenie kur andaluzyj-
skich (czarne, białe i stalowoniebieskie), długość uszu u owiec rasy karakuł
i inne.
W przypadku kodominowania w fenotypie ujawniają swą obecność
obydwa geny z określonej pary alleli. Są one zatem w stosunku do siebie
równorzędne. Przykładem tego typu współdziałania allelicznego jest grupa
148
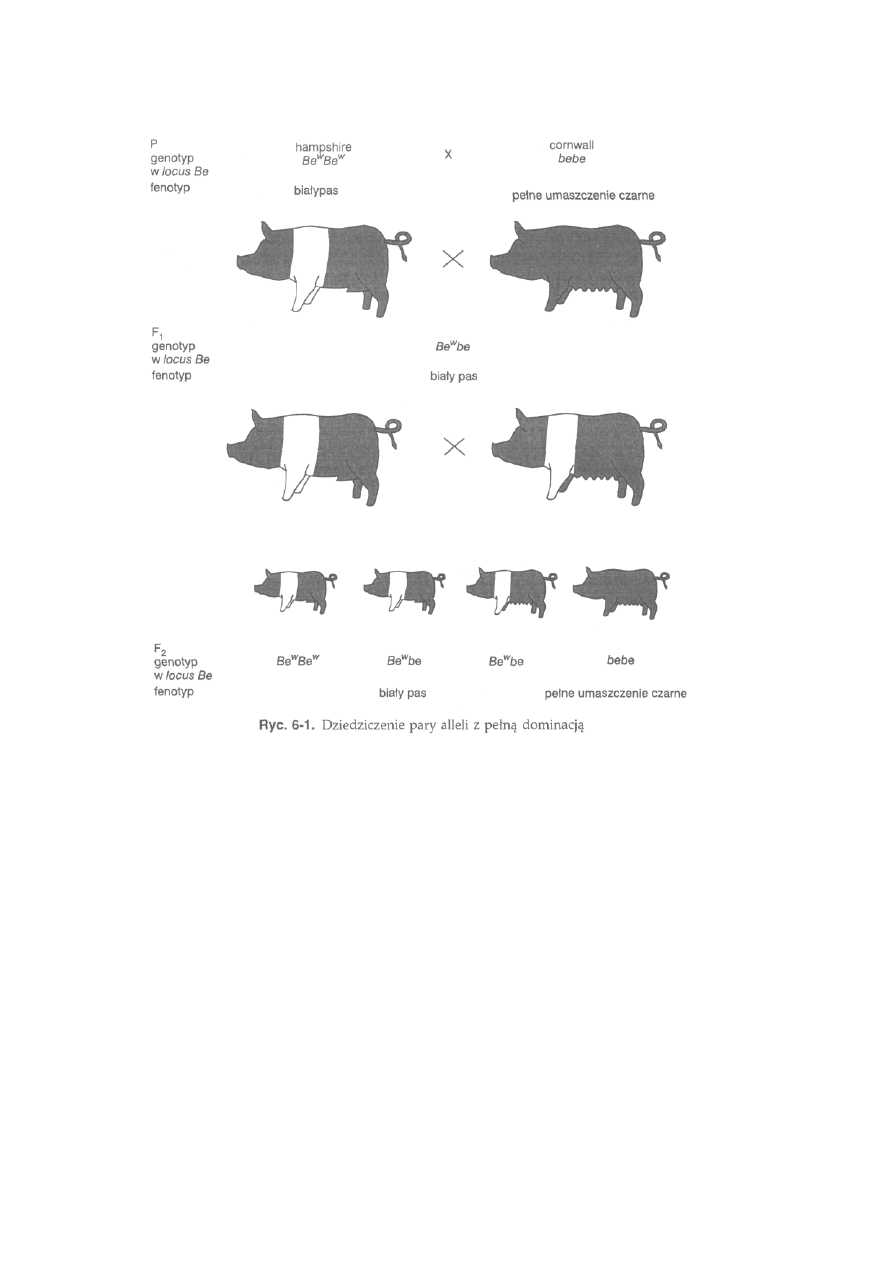
krwi AB w układzie ABO u ludzi, w której oba allele L
A
i L
B
ulegają ekspresji
w jednakowym stopniu.
Naddominowanie polega na tym, że genotyp heterozygotyczny (Aa)
warunkuje większą ekspresję cechy niż genotyp homozygotyczny (AA lub
aa).
Przedstawione dotychczas rodzaje współdziałania dotyczyły genów
występujących w postaci par alleli mających tylko dwie alternatywne formy.
W przypadku niektórych loci, w wyniku mutacji genowych (patrz rozdział:
Mutacje), obok dotychczasowej pary alleli pojawia się allel trzeci, który
warunkuje tę samą cechę co allele pierwotne, dając jedynie inną jej formę
lub natężenie. Zdarzają się również takie loci, w których u różnych osobników
znajdują się więcej niż trzy allele. Układ taki, w którym w jakiejś populacji
149

występują więcej niż dwa allele, nazywamy szeregiem (serią) alleli wielo-
krotnych.
Allele wielokrotne charakteryzują się tym, iż:
• warunkują tylko jedną cechę
• każdy osobnik może mieć tylko dwa allele z danego szeregu
• wśród zwierząt stanowiących populację można napotkać wiele geno
typów zarówno homozygotycznych pod względem któregokolwiek allelu
z tego szeregu, jak i heterozygotycznych pod względem jakichkolwiek
dwóch alleli
• liczba różnych genotypów w populacji zależy od liczby alleli w danym
szeregu. Jeśli szereg składa się z n liczby alleli, to liczba genotypów będzie
się równała:
Przedstawione przykłady dziedziczenia dotyczyły tylko jednej cechy
kontrolowanej przez jedną parę alleli. Jeśli rozpatrywane jest dziedziczenie
dwóch lub więcej takich cech równocześnie, to mogą się one dziedziczyć
niezależnie od siebie lub zależnie (cechy sprzężone).
6.1.2. Niezależne dziedziczenie cech (II
prawo Mendla)
Niezależne dziedziczenie cech stwierdził Grzegorz Mendel, badając m.in.
sposób dziedziczenia barwy kwiatów i kształtu nasion u groszku. Na
podstawie uzyskanych wyników sformułował prawo (II prawo Mendla),
mówiące o niezależnym dziedziczeniu się cech. Oznacza to, że podczas
mejozy segregacja alleli jednej pary jest niezależna od alleli drugiej pary.
Warunkiem niezależnego dziedziczenia się cech jest umiejscowienie deter-
minujących je genów w różnych parach chromosomów. Również gdy loci
genów znajdują się w tym samym chromosomie w dużej odległości od
siebie, to ich segregacja jest praktycznie niezależna.
Niezależne dziedziczenie cech obrazuje przykład krzyżowania zwierząt
różniących się dwiema cechami — umaszczeniem (czarne, czerwone),
warunkowanym parą alleli E
d
-e (z locus Extension umiejscowionego w chro-
mosomie 18) i bezrożnościa/rogatością (allele P-p z locus polled w chromo-
somie 1).
Wszystkie gamety osobnika aberdeen-angus (bydło czarne, bezrogie)
zawierają dominujące geny E
d
i P, natomiast gamety osobnika rasy shorthorn
(czerwone, rogate) — allele recesywne e i p. Zwierzęta z pokolenia Fj będą
mieć genotyp E
d
ePp, czyli będą czarne, bezrogie. Każdy osobnik z tego
pokolenia wytwarza 4 różne rodzaj gamet: E
d
P, E
d
p, eP, ep. Dzieje się tak
150
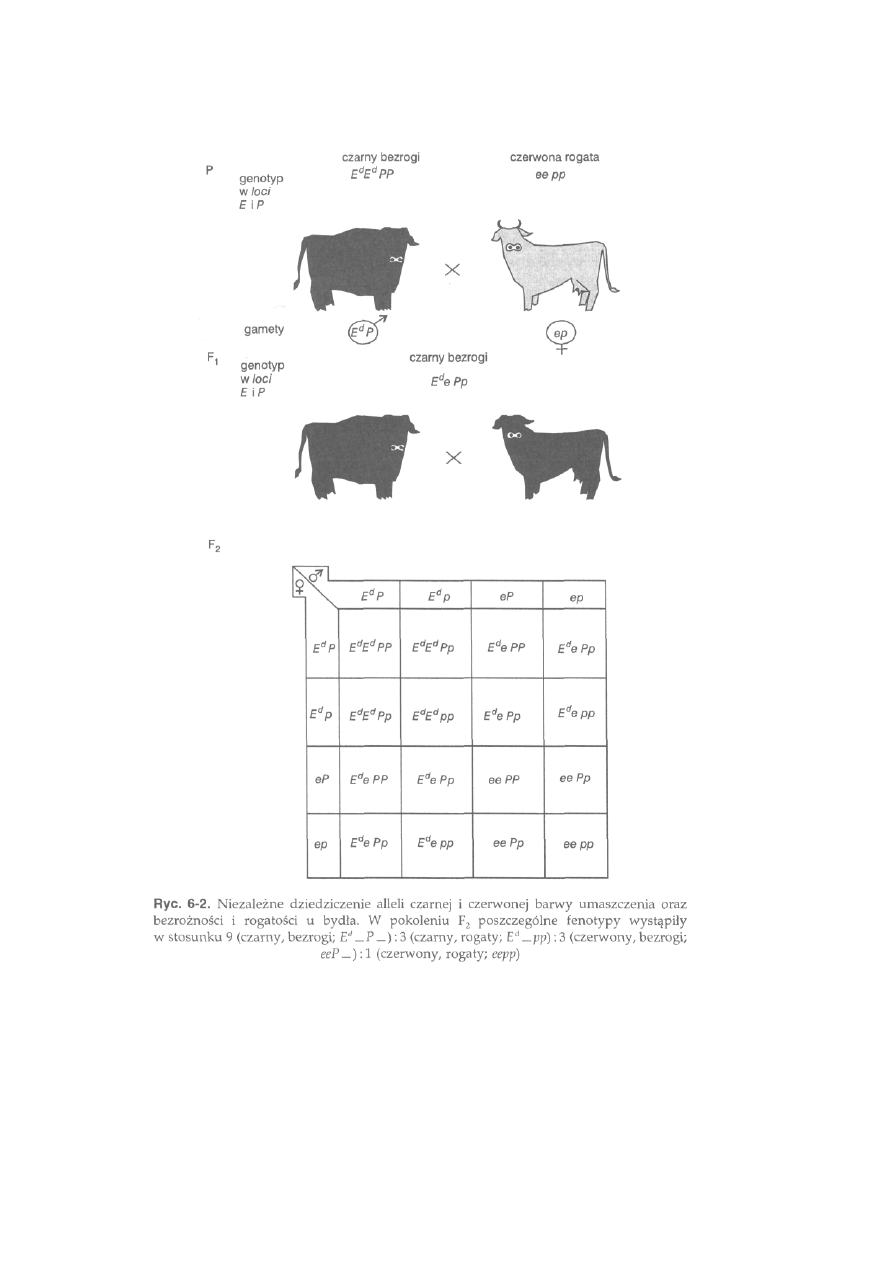
151

dlatego, że allele E
d
-e przechodzą do komórek rozrodczych niezależnie od
alleli P-p, gdyż te dwie pary genów znajdują się w innych parach
chromosomów homologicznych. Ponieważ każdy osobnik wytwarza 4 ro-
dzaje gamet, to w wyniku losowego połączenia się gamety męskiej i żeńskiej
może powstać 4 x 4 = 16 możliwych kombinacji.
Najłatwiej przedstawić wszystkie możliwe genotypy w pokoleniu F
2
,
sporządzając szachownicę genetyczną Punneta. Spośród 16 możliwych
kombinacji genów w pokoleniu F
2
uzyskuje się tylko 4 różne fenotypy
w następującym stosunku liczbowym: 9 (czarne, bezrogie): 3 (czarne,
rogate): 3 (czerwone, bezrogie): l (czerwone, rogate) (ryć. 6-2). Najliczniejszą
grupę potomstwa stanowią osobniki o genotypie, w którym występuje
przynajmniej jeden gen dominujący z każdej pary, a najmniej liczną grupę
(tylko l na 16 możliwych) stanowią osobniki podwójnie recesywne homo-
zygotyczne (ee pp — czerwony, rogaty).
Liczbowy stosunek rozszczepienia cech w pokoleniu F
2
(9:3:3:1) wystąpi
tylko wtedy, gdy obie cechy dziedziczą się z dominowaniem całkowitym.
W przypadku obu cech dziedziczących się z dominowaniem niezupełnym
(typ Zea) należy spodziewać się nie 4, lecz 9 różnych fenotypów. Natomiast
gdy jedna cecha dziedziczy się według typu Pisum, a druga według typu
Zea, istnieje możliwość ujawnienia się 6 różnych fenotypów. Jeśli będzie
rozpatrywana większa liczba cech równocześnie, zwiększy się liczba moż-
liwych fenotypów.
Liczbę gamet wytwarzanych przez osobniki heterozygotyczne pod
względem n par alleli, liczbę oczekiwanych fenotypów i genotypów
w pokoleniu F
2
można obliczyć ze wzorów:
liczba różnych gamet = 2
n
liczba fenotypów = 2
n
liczba genotypów = 3
n
6.1.3. Dziedziczenie cech sprzężonych
Podstawę II prawa Mendla o niezależnym dziedziczeniu się cech stanowi
to, iż geny umiejscowione w różnych chromosomach zachowują się
niezależnie podczas mejozy. W tym przypadku można mówić o niezależnej
segregacji chromosomów. Jednak organizmy żywe mają tysiące, dziesiątki
tysięcy różnych genów, a tylko kilka do kilkudziesięciu chromosomów.
Zatem każdy chromosom zawiera wiele genów. Podczas procesu gametoge-
nezy geny znajdujące się w określonym chromosomie będą przekazane do
powstającej komórki rozrodczej łącznie, inaczej mówiąc, cechy kontrolowane
przez te geny będą się dziedziczyć razem. Cechy takie nazywamy sprzężo-
nymi. Podany wyżej wzór na obliczanie liczby różnych gamet (2
n
) nie może
152

być stosowany do tych cech. Sposób segregacji genów znajdujących się
w tym samym chromosomie zależy od odległości między nimi. W rozdziale:
Chromosomy i podziały jądra komórkowego wspomniano o tym, iż
w profazie I podziału mejotycznego zachodzi koniugacja chromosomów
i powstają biwalenty, złożone z homologicznych chromosomów. Dochodzi
wówczas do wymiany odcinków między niesiostrzanymi chromatydami,
zwanej crossing over. W wyniku crossing over u części osobników potom-
nych, tzw. rekombinantów, pojawią się kombinacje cech w innym układzie
niż u rodziców. Częstość tej wymiany jest tym większa, im dalej od siebie
znajdują się geny. Prawidłowość ta została wykorzystana w opracowywaniu
map genetycznych ukazujących względne położenie poszczególnych loci
w chromosomach.
Sposób określania sprzężenia (odległości) loci przedstawia poniższy
przykład.
U muszki owocowej (Drosophila melanogaster) w drugim chromosomie
znajdują się między innymi geny warunkujące czarną barwę ciała (gen
recesywny b — black) i kształt skrzydeł, skrzydła szczątkowe (gen recesywny
v — vestigal). Normalny fenotyp barwy ciała muszki to barwa jasnobrunatna,
normalne skrzydła są dłuższe od odwłoka. Przyjmuje się symbol „ + " na
oznaczenie allelu normalnej formy (tzw. dzikiej) każdej cechy u muszki.
Jednak w celu lepszego zrozumienia zostaną przyjęte symbole B (barwa
ciała dzika — dominująca) i V (kształt skrzydeł dziki — dominujący).
W odróżnieniu od cech segregujących niezależnie, genotyp dla cech
sprzężonych jest zapisywany w postaci ułamka, w którym licznik oznacza
jeden chromosom, mianownik drugi chromosom z tej pary. Dla muszki
dzikiej genotyp pod względem dwóch rozpatrywanych cech jest na-
stępujący:
Natomiast genotyp podwójnej homozygoty recesywnej:
W celu określenia, jakie allele u osobnika heterozygotycznego (dominu-
jące czy recesywne) z dwu loci sprzężonych są przenoszone przez ten sam
chromosom, została przyjęta nazwa faza sprzężenia. Geny dominujące
z tych loci mogą się znajdować w tym samym chromosomie i wtedy
mówimy, że są one w fazie przyciągania (cis), bądź w różnych chromosomach i
jest to faza odpychania (trans).
153
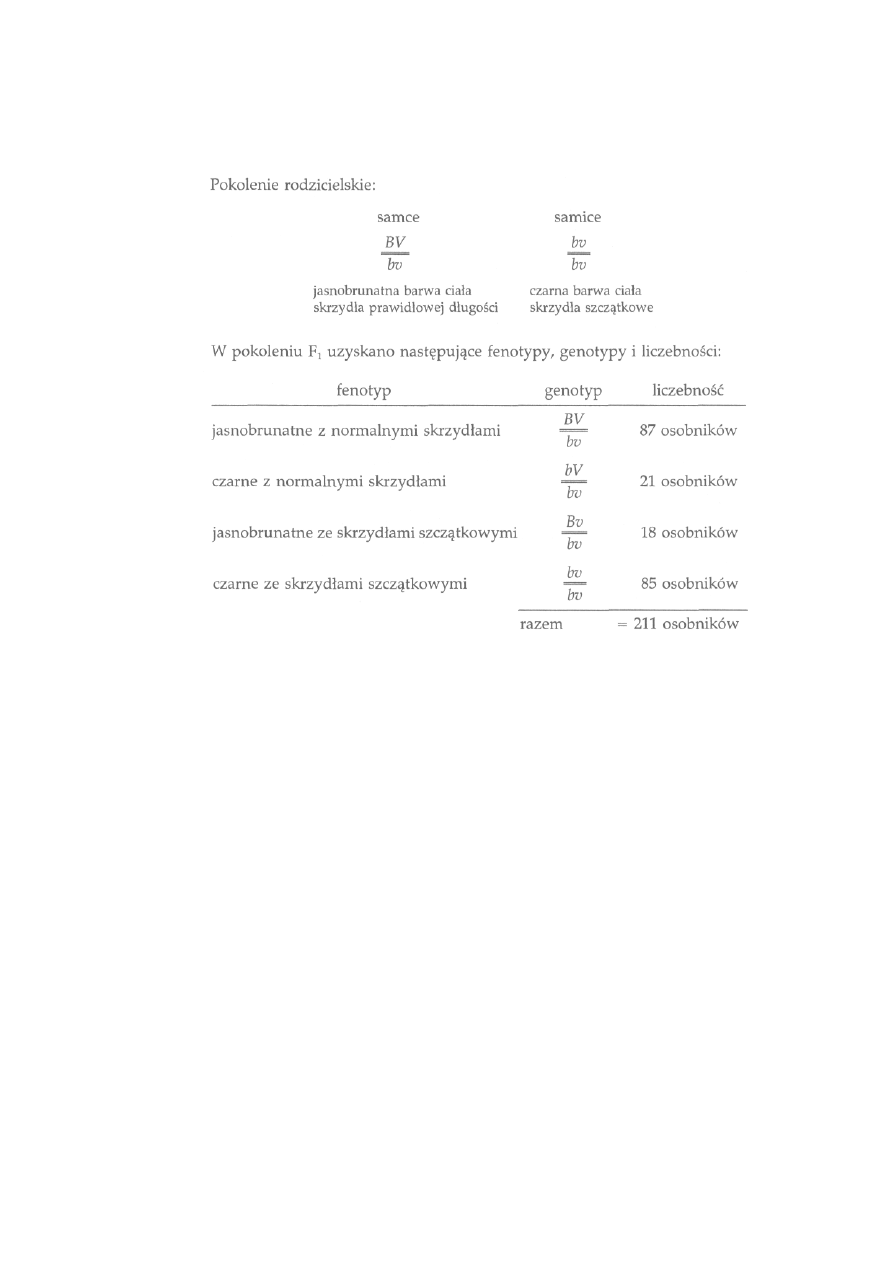
Ewentualne sprzężenie cech najłatwiej jest sprawdzić krzyżując osobniki
heterozygotyczne z homozygotami recesywnymi — jest to klasyczne
krzyżowanie testowe.
Zatem rodzicielski układ alleli wystąpił u 172 osobników (są to typy
rodzicielskie fenotypu), natomiast pozostałe 39 miało genotypy rekom-
binacyjne (osobniki takie są nazywane rekombinantami). W danym przy-
kładzie rekombinanty stanowią około 18,5% całego potomstwa z tej krzyżów-
ki. Procent rekombinacji przyjęto za jednostkę odległości między genami,
której symbolem jest cM (centymorgan, od nazwiska twórcy chromosomowej
teorii dziedziczenia Tomasza Morgana), czyli l cM = 1% rekombinacji.
W naszym przykładzie odległość między loci B i V wynosi 18,5 cM.
Doświadczalnie zostało udowodnione, że cechy sprzężone dają w krzyżów-
kach zawsze zbliżony procent rekombinantów. Wynika to z tego, iż częstość,
z jaką crossing over zachodzi między sprzężonymi loci, jest stała.
W przedstawionym wyżej przykładzie nie została uwzględniona moż-
liwość zajścia podwójnego crosing over. Podwójny crossing over zachodzi
znacznie rzadziej niż pojedynczy. Można jednak przewidzieć częstość
występowania podwójnego crossing over, jest ona iloczynem częstości
przypadków pojedynczych crossing over. Na przykład, jeśli pojedynczy
154

crossing over w obszarze I zachodzi z częstością 0,02 (2%), a w obszarze
II — 0,05 (5%), to częstość występowania podwójnego crossing over
wynosi 0,02x0,05 = 0,001, czyli 0,1%. Jest to jednak wartość przybliżona,
gdyż w naturze jest ona niższa. Przyczynę stanowi to, iż zajście crossing
over w jakimś odcinku chromosomu zmniejsza prawdopodobieństwo
drugiego crossing over w pobliżu tego odcinka. Zjawisko to nosi nazwę
interferencji.
Rekombinanty są osobnikami mającymi nowe kombinacje genów sprzę-
żonych. Jeśli jednak zajdzie podwójny crossing over, układ genów będzie
taki, jak przed tą wymianą i nie będzie można odróżnić rekombinantów.
Skutkiem tego będzie zmniejszona liczba rekombinantów i błąd w obliczeniu
odległości między loci sprzężonymi. By tego uniknąć, stosuje się tzw.
krzyżówki trzypunktowe, umożliwiające dokładne ustalenie położenia loci
względem siebie. Jeśli przyjmiemy, że kolejność ułożenia loci w chromosomie
jest następująca: A-B-C, to obliczona odległość między loci A i C powinna
być sumą odległości między A i B oraz B i C.
Sposób ustalenia sprzężenia między trzema genami przedstawiony
zostanie na omawianym przykładzie muszki owocowej. Trzecią cechą
rozpatrywaną będzie kolor oczu. U muszki owocowej dominuje czerwony
kolor oczu (dziki), a gen recesywny c (cinnabar), warunkuje kolor cynobrowy.
Również w tej krzyżówce, w celu lepszego uwidocznienia efektu crossing
over, allele dominujące będą oznaczone literami alfabetu, a nie za pomocą
przyjętego dla nich symbolu „ + ".
Jeśli skrzyżujemy osobniki różniące się allelami w trzech loci sprzężo-
nych, to na podstawie fenotypów potomstwa wykryjemy wszystkie
przypadki pojedynczych i podwójnych crossing over między skrajnymi loci.
Ponadto możliwe będzie ustalenie kolejności ułożenia trzech rozpatry-
wanych loci w chromosomie.
W pokoleniu rodzicielskim jedna płeć musi mieć genotyp potrójnie
heterozygotyczny:
Natomiast drugi rodzic ma genotyp:
W wyniku pojedynczego crossing over w obszarze I między genami
B i C powstaną gamety bCV i Bcv, natomiast skutkiem pojedynczego
crossing over w obszarze II między genami C i V będą gamety BCv i bcV.
Z kolei, podwójny crossing over w obszarze I i II da gamety o następującym
zestawie alleli: bCv i BcV.
155

W potomstwie uzyskano następujące genotypy i liczebności:
Obliczanie odległości między loci sprzężonymi:
Odległość między genami B i C jest stosunkiem sumy rekombinantów
powstałych w wyniku crossing over w I obszarze i rekombinantów
powstałych po podwójnym crossing over do wszystkich fenotypów, wyra-
żonym w procentach:
Analogicznie do tego odległość między loci C i V wynosi:
Odcinek chromosomu 2 Drosophila melanogaster zawierający rozpatrywane
loci wygląda następująco:
Dla każdej grupy genów sprzężonych można ustalić odległości między
nimi oraz ich wzajemne położenie w chromosomie. W ten sposób tworzone
są mapy genetyczne. Odległości mapowe są obliczone z częstości rekom-
binacji między analizowanymi genami i kolejno sumowane. W celu okreś-
156

lenia, w którym chromosomie znajduje się dana grupa genów sprzężonych,
należy zastosować inne metody badania (cytogenetyczne). Powstaje wówczas
mapa fizyczna. O mapach genetycznych i fizycznych oraz ich zastosowaniu
jest mowa w rozdziale: Mapy genomowe. W rozdziale tym pokazano m.in.,
że odległości genetyczne między sprzężonymi loci szacuje się za pomocą
funkcji Kosambiego.
6.1.4. Cechy sprzężone z płcią
Cechy, których geny są umieszczone w chromosomach płci, nazywamy
cechami sprzężonymi z płcią. Najczęściej zjawisko to odnosimy do położenia
genu w chromosomie X ssaków lub chromosomie Z ptaków. Dzieje się tak
dlatego, że chromosomy te mają nieporównywalnie więcej loci genów niż
chromosom Y (ssaki) i chromosom W (ptaki). Sposób ich dziedziczenia jest
inny niż cech autosomalnych. U zwierząt płci heterogametycznej wszystkie
cechy sprzężone z płcią zależą od jednego genu, a nie — jak u płci
homogametycznej — od pary genów.
U ssaków samice są homogametyczne (XX), samce zaś heterogametyczne
(XV). Natomiast u ptaków płcią homogametyczną są samce (określane dla
odróżnienia od ssaków — ZZ), a heterogametyczną samice (ZW).
Sposób dziedziczenia cechy sprzężonej z płcią obrazuje poniższy przykład.
Cecha zmniejszonej krzepliwości krwi (hemofilia), występująca u ludzi,
psów i niektórych zwierząt gospodarskich, uwarunkowana jest recesywnym
genem h. Dominujący allel z tej pary (H) warunkuje prawidłową krzepliwość
krwi.
Wszystkie możliwe fenotypy i genotypy pod względem tego locus
można zapisać symbolami w dwojaki sposób:
Fenotyp
Samice Samce
Samice Samce
Jak wynika z tego zestawienia, samce nie mogą być heterozygotami
(nosicielami genu hemofilii), ponieważ genotyp cechy sprzężonej z płcią
wyznaczany jest u nich tylko jednym genem.
Układ genetyczny polegający na tym, iż u osobnika diploidalnego
występuje tylko jeden gen z danej pary, nazywamy hemizygotycznym,
a osobnika — hemizygotą. Gdy płeć heterogametyczną ma cechę dominującą,
a pleć homogametyczną cechę recesywną, obserwujemy zjawisko dziedzi-
czenia na krzyż (córki dziedziczą po ojcu, a synowie po matce). Gdy jest
odwrotny układ (cecha dominująca u osobnika homogametycznego), cecha
będzie się dziedziczyć tak jak prosta cecha autosomalna.
157
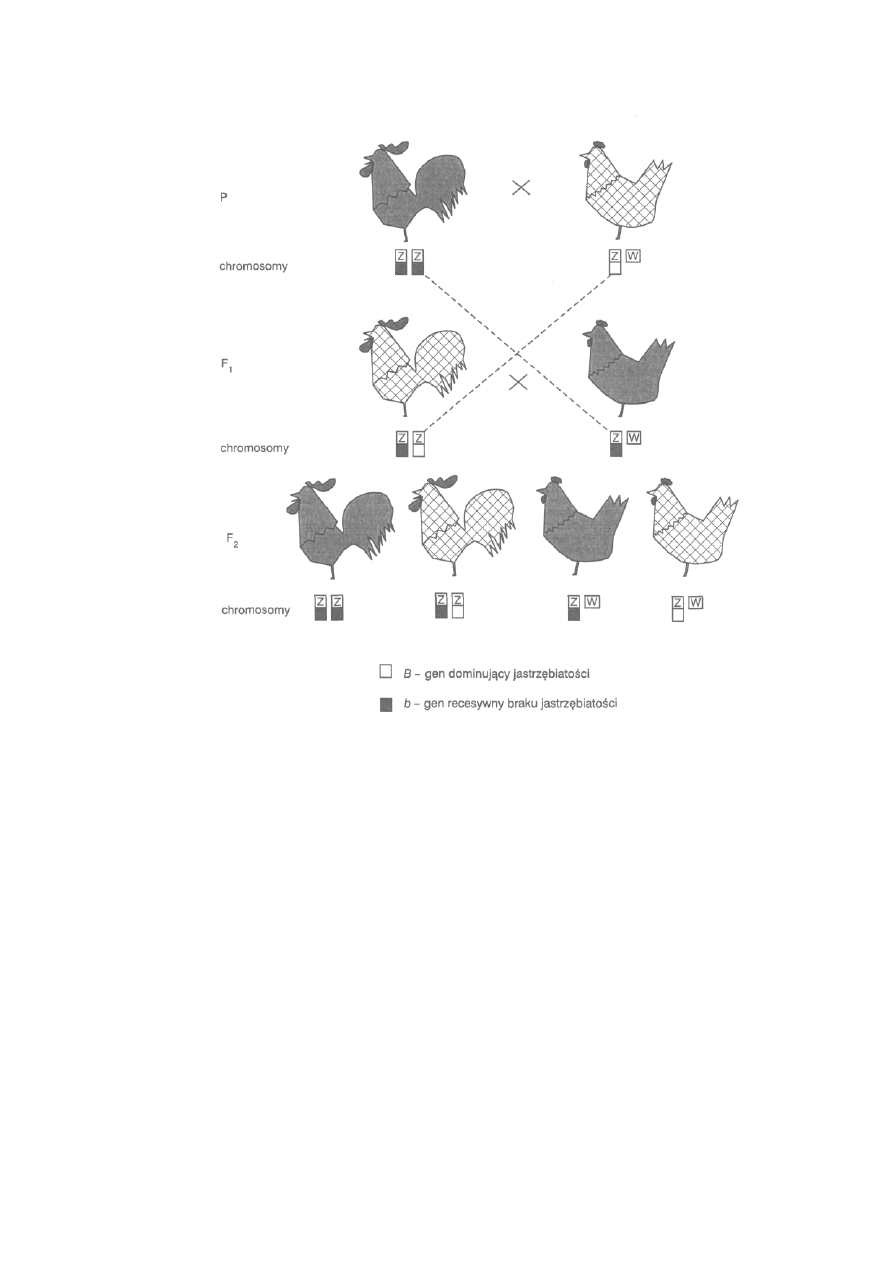
Ryć. 6-3. Dziedziczenie barwy upierzenia u kur — cecha sprzężona z płcią, w pokoleniu
F, „dziedziczenie na krzyż"
Dziedziczenie na krzyż przedstawiono na rycinie 6-3 na przykładzie
jastrzębiatości upierzenia u kur:
Gen B, warunkujący cechę jastrzębiatości upierzenia, dominuje nad jego
allelem b (gen czarnej barwy upierzenia). Gen ten hamuje okresowo
odkładanie się czarnego pigmentu podczas wzrostu pióra. Skutkiem tego jest
obecność czarnych i jasnych prążków w chorągiewce. Obecność w genotypie
dwóch kopii genu B zahamowuje odkładanie pigmentu na dłuższy okres niż
obecność jednego genu. Dlatego koguty homozygotyczne pod względem tego
genu są jaśniejsze niż hemizygotyczne kurki (ryć. 6-4 wkładka kolorowa).
Cechy sprzężone z płcią wykorzystano w hodowli kur do wytworzenia
ras lub mieszańców autoseksingowych, tj. takich, u których zaraz po
158

wykluciu można rozróżnić płeć. Rasą autoseksingową są polbary, wy-
tworzone przez polską badaczkę Laurę Kaufman, mające gen jastrzębiatego
upierzenia. Do produkcji mieszańców autoseksingowych można wykorzystać
geny srebrzystości (np. rasa sussex) i złocistości upierzenia (rhode island
red), geny kontrolujące tempo opierzania się skrzydeł i ogona (K-k) czy gen
karłowatości (dw, ang. dwarf — karłowaty).
Gen karłowatości u homozygotycznych (dwdw) samców wpływa na
zmniejszenie masy ciała osobnika dorosłego o około 40% w porównaniu
z ptakiem dorosłym. Natomiast kury mające ten gen (dw-) zużywają około
25% mniej paszy niż kury o genotypie Dw-. Potomstwo (brojlery) pochodzące
z kojarzenia koguta DwDw z kurą dw- będzie miało masę ciała tylko o 3%
mniejszą niż brojlery po normalnych rodzicach. Taki dobór par rodzicielskich
jest opłacalny z ekonomicznego punktu widzenia.
Ciekawym przykładem cech sprzężonych z płcią u kotów jest czarna
i ruda barwa sierści, warunkowana przez locus O (ang. orange), zlokalizo-
wany w chromosomie X. Samce mogą być tylko czarne (o-) lub rude (O-).
U samic heterozygotycznych występuje umaszczenie szylkretowe — część
włosów czarnych, inne — rude (genotyp Oo) (ryć. 6-5 wkładka kolorowa).
Jest ono następstwem tego, iż allele czarnego i rudego umaszczenia ujawniają
się fenotypowo niezależnie od siebie, co wynika z losowości inaktywacji
chromosomu X pochodzenia ojcowskiego lub matczynego podczas rozwoju
zarodkowego.
U zwierząt domowych jest wiele cech sprzężonych z płcią, część z nich
przedstawiono w rozdziale: Mutacje.
Znane są przypadki cech sprzężonych z płcią warunkowanych szeregiem
alleli wielokrotnych. Barwa upierzenia u gołębi jest kontrolowana przez
szereg złożony z trzech alleli: B
A
(popielatoczerwone), B (niebieskie -
dzikie) i b (czekoladowe).
Nie wszystkie cechy typowe dla danej płci są sprzężone z chromosomem
X. Niektóre z nich to tzw. cechy związane z płcią. Geny warunkujące te
cechy należą do autosomalnych (umieszczone w autosomach), ale ich
ekspresja jest uzależniona od płci. Osobniki męskie i żeńskie, mając taki sam
genotyp, mogą się różnić fenotypowo. Przykładem takiej cechy jest rogatość
owiec rasy dorset horn. Zwierzęta homozygotyczne pod względem domi-
nującego genu bezrożności będą bezrogie niezależnie od płci, natomiast
osobniki homozygotyczne recesywne będą rogate. Różnice w fenotypie
wystąpią u zwierząt heterozygotycznych — samce będą rogate, samice zaś
bezrogie. Różna ekspresja fenotypowa tego samego genotypu u osobników
obu płci jest następstwem oddziaływania hormonów wytwarzanych przez
daną płeć. Innymi przykładami cech związanych z płcią są: mahoniowo-białe
i czerwono-białe umaszczenie bydła rasy ayrshire oraz broda u kozłów.
Wpływ płci na fenotypowa ekspresję genów jest także obserwowany
w przypadku cech, których założenia genetyczne znajdują się u osobników
obu płci, ale ujawniają się tylko u jednej płci. Cechy te nazywane są cechami
159

ograniczonymi płcią lub ograniczonymi do płci. Należą do nich: zdolność
wydzielania mleka u samic ssaków, nieśność samic ptaków, wnętrostwo czy
przepuklina mosznowa u samców.
6.1.5. Plejotropia
O plejotropii mówimy wówczas, kiedy jeden gen oddziałuje na powstanie
kilku cech. Plejotropia może być właściwa lub rzekoma. Plejotropia właściwa
występuje wtedy, gdy gen plejotropowy oddziałuje na kilka odrębnych
ośrodków. Przykładem jest gen warunkujący platynową barwę u lisów. Lisy
platynowe w przeciwieństwie do osobników umaszczonych standardowo
są mniej żywotne i bardziej pobudliwe. Osobniki homozygotyczne pod
względem tego genu nie są zdolne do życia i zamierają w okresie
prenatalnym. Podobnie gen warunkujący siwą (sziras) barwę włosów
u karakułów powoduje śmierć jagniąt homozygotycznych w wieku 4-9
miesięcy. Również u koni niektóre geny umaszczenia wykazują działanie
plejotropowe. Na przykład u około 1/4 potomstwa pochodzącego z kojarze-
nia między sobą koni srokatych (overo) występuje zespół „białych źrebiąt",
opisany w podrozdziale: Dziedziczenie umaszczenia. W podrozdziale tym
omówiony jest także plejotropowy efekt genów umaszczenia u innych
gatunków zwierząt.
Przyczyny plejotropii właściwej mogą mieć podłoże biochemiczne. Na
przykład gen warunkuje syntezę enzymu, który bierze udział w syntezie
jakiegoś związku (np. hormonu). Z kolei ten związek ma różnorodne
funkcje fizjologiczne, wpływa zatem na inne właściwości organizmu.
W przypadku plejotropii rzekomej gen kontroluje jakąś cechę, która z kolei
rzutuje (w zależności od wpływów środowiska) na zróżnicowanie innych cech.
Na przykład gen warunkujący szurpatość (zmiany w budowie piór) u drobiu
wpływa także m.in. na tempo przemiany materii, pracę serca i aktywność
procesów trawiennych. Zmiany te są jednak następstwem nieprawidłowego
opierzenia, które nie chroni ptaka przed nadmierną utratą ciepła.
6.2. Współdziałanie genów z różnych loci w
kształtowaniu fenotypu
W omawianych dotychczas przykładach jedna para współdziałających ze
sobą genów allelomorficznych warunkowała powstanie jednej cechy. Jed-
nakże nie tylko geny należące do tej samej pary mogą współdziałać ze sobą
w wytworzeniu cechy. W przypadku wielu cech geny z różnych par alleli,
w wyniku łącznego działania, powodują pojawienie się nowej formy cechy
jakościowej.
160
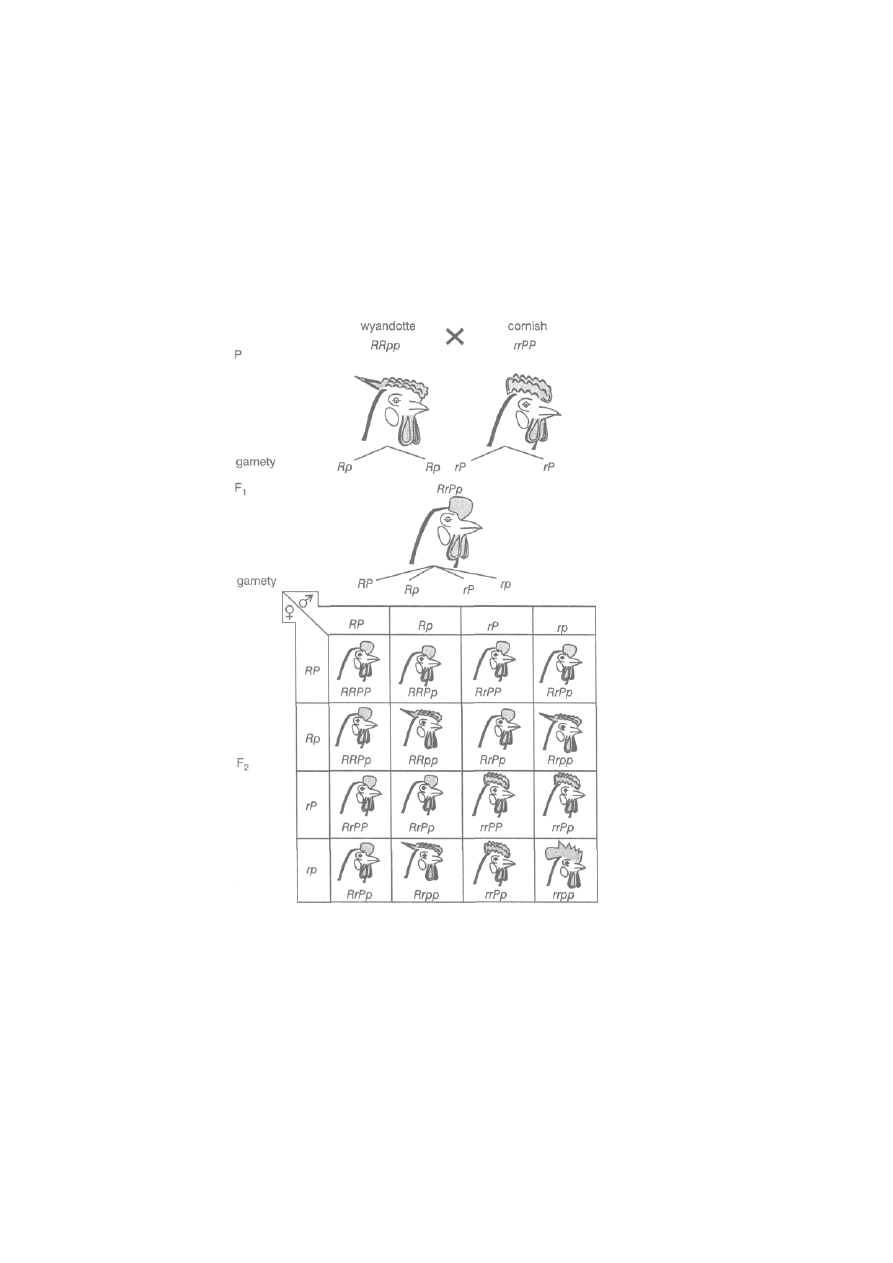
Współdziałanie między genami z różnych par alleli w kształtowaniu
fenotypu nosi nazwę współdziałania nieallelicznego. Jest kilka rodzajów
tego współdziałania.
6.2.1. Komplementarność
Jednym z najprostszych przykładów współdziałania genów nieallelicznych
jest dziedziczenie kształtu grzebienia u kur (ryć. 6-6a). W wykształceniu
czterech form tej cechy biorą udział geny z dwóch różnych /ócz: R-r oraz P-p.
Ryć. 6-6a. Współdziałanie dopełniające dwóch par genów w przypadku krzyżowania
kur z grzebieniem różyczkowym i groszkowym
161

Gen R warunkuje kształt różyczkowy grzebienia, a jego allel r — pojedynczy,
natomiast gen P powoduje powstanie grzebienia groszkowego, jego allel
p — grzebienia pojedynczego (ryć. 6-6b wkładka kolorowa). Typ grzebienia
pojedynczego jest zatem recesywny zarówno w stosunku do grzebienia
groszkowego, jak i różyczkowego, warunkujący go genotyp to podwójna
homozygota recesywna — rrpp. W wyniku współdziałania genów R i P po-
wstaje czwarta forma cechy — grzebień orzeszkowy, której genotyp może
mieć kilka różnych wariantów, gdyż wystarczy obecność jednego genu
dominującego z każdej pary alleli (R_P_).
Z krzyżowania osobników o ustalonej cesze kształtu grzebienia, tj.
z grzebieniem różyczkowym (Rrpp) i groszkowym (rrPP), w pokoleniu
Ej wszystkie ptaki będą podwójnymi heterozygotami RrPp i będą miały
grzebień orzeszkowy. Natomiast w pokoleniu F
2
nastąpi rozszczepienie
cech w stosunku 9 orzeszkowych : 3 różyczkowe : 3 groszkowe : l pojedyn-
czy. Różnice między wynikami tego krzyżowania a krzyżowania uwzględ-
niającego dwie niezależnie dziedziczące się cechy polegają na tym, że
w pokoleniu F
l
pojawia się nowy fenotyp, całkowicie różny od obojga
rodziców, natomiast w pokoleniu F
2
, w którym wystąpią 4 fenotypy
w stosunku 9:3:3:1, pojawia się również nowy typ — grzebień pojedynczy,
którego nie było w dwóch pokoleniach przodków.
Dwa dominujące geny z różnych par alleli, współdziałające razem
i wytwarzające odmienną formę cechy niż ta, która powstaje w wyniku
działania każdego z nich osobno, nazywamy genami komplementarnymi
lub dopełniającymi się.
6.2.2. Epistaza
Kolejnym rodzajem współdziałania genów nieallelicznych jest epistaza,
charakteryzująca się tym, że od obecności genu z określonej pary alleli
zależy ekspresja innej pary alleli. Gen, który hamuje ujawnienie się genu
z innej pary, nazywany jest genem epistatycznym, natomiast gen hamowany
to gen hipostatyczny.
Zjawisko epistazy występuje na przykład w dziedziczeniu umaszczenia
u świń niektórych ras biało umaszczonych. Gen dominujący z locus I
(inhibitor) hamuje ujawnienie się barwy kolorowej mimo obecności w ge-
notypie genów barwnego umaszczenia.
Zjawisko epistazy może wyjaśnić poniższy przykład. Kury niektórych
ras (np. wyandotte, plymouth rock) mają upierzenie białe, gdyż brak w ich
genotypach genu C, warunkującego wytwarzanie pigmentu (melaniny)
w piórach. Ich genotyp pod względem tej pary alleli jest homozygota
recesywna (cc), która blokuje syntezę tyrozynazy — enzymu biorącego
udział w wytwarzaniu melaniny. U białych leghornów jest inna sytuacja,
podobna do wspomnianej wyżej u świń. Ptaki te są również białe, ale mają
162

Ryć. 6-7. Różny stopień białej plamistości spowodowany działaniem genów modyfiku-
jących
w swych genotypach gen C. Działanie tego genu jest jednak hamowane
przez inny dominujący gen z locus I, który w układzie homozygotycznym
całkowicie uniemożliwia syntezę melaniny w piórach. Genotyp leghornów
pod względem barwy upierzenia jest następujący: IICC. W potomstwie F
2
,
którego dziadkowie należeli do dwóch ras białych — leghornów i wyandotte
(iicc), oprócz ptaków białych pojawią się osobniki o upierzeniu barwnym
w stosunku 13 białych : 3 barwnych.
Podobnie umaszczenie białe u świń jest wynikiem epistatycznego
działania genów z locus L Genotyp biało umaszczonych świń (np. rasy
wielkiej białej i landrace pod względem trzech podstawowych loci jest
następujący: aa II E
P
E
P
, natomiast czarno umaszczonych (np. rasy cornwall):
aa ii EE. Allele z locus E i A są opisane w dalszej części tego rozdziału,
dotyczącej dziedziczenia umaszczenia.
Gen epistatyczny nie zawsze jest dominujący w swojej parze. W przypad-
ku albinizmu układ epistatyczny w stosunku do genów barwy stanowią
dwa geny recesywne cc (z tego samego locus co gen C). Inne przykłady
epistatycznego współdziałania genów nieallelicznych przedstawione są także
w podrozdziałach: Dziedziczenie umaszczenia i Antygeny erytrocytarne.
Przedstawiony wcześniej rodzaj współdziałania komplementarnego jest
także formą oddziaływania epistatycznego genu dominującego jednej pary
wobec układu homozygotycznego recesywnego drugiej pary. Jednak nie
zawsze jedna para genów maskuje ekspresję tylko jednej innej pary genów.
Geny epistatyczne mogą tłumić działanie wielu innych par.
6.2.3. Geny modyfikujące
Istnieje pewna grupa genów, które modyfikują przejawianie się jakiejś
prostej cechy, warunkowanej zwykle jedną parą genów. Geny te, zwane
modyfikatorami, powodują duże zróżnicowanie cechy u osobników o jed-
nakowych założeniach warunkujących tę cechę. Na przykład geny mo-
dyfikatory wpływają na zasięg i rozmieszczenie białych plam u bydła
(ryć. 6-7), psów i kotów, mimo iż cecha łaciatości warunkowana jest
inną parą alleli.
163

6.2.4. Sumujące działanie genów
Inną formą współdziałania nieallelicznego genów jest polimeria. Zjawisko
to polega na tym, iż wiele (nawet kilkadziesiąt) genów z różnych loci
warunkuje jedną cechę, powodując różne jej nasilenie, zależne od sumują-
cego się ich działania. Geny te nazywane są genami polimerycznymi,
addytywnymi lub poligenami. Ich sumujące się działanie jest podstawą
dziedziczenia cech ilościowych (np. wydajność mleczna, nieśność itd.).
Należy pamiętać, że kształtowanie się tych cech zależy również od wpływu
czynników środowiskowych. Mechanizm działania genów połimerycznych
omówiony jest w podrozdziale: Zmienność cech ilościowych.
6.3. Dziedziczenie umaszczenia
Umaszczenie zwierząt zależy przede wszystkim od pigmentu zawartego we
włosach i piórach. Synteza, rozmieszczenie i wielkość ziarenek tego pigmentu
są determinowane genetycznie przez wiele różnych loci, ale czynniki
środowiskowe mogą modyfikować umaszczenie, wpływając na przykład na
zmianę ilości wytwarzanego pigmentu. U wielu zwierząt ubarwienie sierści
zmienia się w zależności od pory roku (np. biała w zimie, a czarna lub
brązowa w lecie). Zmiany te spowodowane są działaniem niektórych
hormonów, takich jak gonadotropiny, kortykotropina i hormon melano-
tropowy — MSH (ang. melanocyte stimulating hormone).
U ptaków większość kolorów jest związana z obecnością pigmentów
karoteinowych, natomiast u ssaków pigmentacja zależy od syntezy i roz-
mieszczenia melanin w rdzeniu włosa i korze włosowej oraz naskórku skóry
właściwej.
Wyróżnia się dwa podstawowe typy melanin — eumelaniny (kolor
brązowy lub czarny), syntetyzowane z metabolitów DOPA-chromu, i feo-
melaniny (kolor czerwony lub żółty), wytwarzane z metabolitów cysteinylo-
-DOPA-chinonu.
Enzymem włączonym w syntezę obu typów pigmentu jest tyrozynaza.
Jest to enzym zawierający miedź i katalizujący trzy różne reakcje w procesie
biosyntezy melaniny (ryć. 6-8):
1) hydroksylację tyrozyny do 3,4-dihydroksyfenyloalaniny (DOPA)
2) oksydację DOPA do DOPA-chinonu
3) oksydację 5,6-dihydroksyindolu (DHI) do indolochinonu.
Niski poziom tyrozynazy prowadzi do wytwarzania feomelaniny, natomiast
wysoki poziom tyrozynazy powoduje produkcję eumelaniny.
Do niedawna uważano, iż tyrozynaza jest jedynym enzymem niezbęd-
nym do wytwarzania pigmentu. Ostatnie badania wykazały, że przypusz-
czalnie również inne geny specyficzne dla procesu syntezy pigmentu mogą
modulować pigmentację.
164
Ryć. 6-8. Schemat biosyntezy melaniny
Aktywność tyrozynazy jest regulowana przez hormon melanotropowy
(MSH) i receptor MSH. Hormon melanotropowy, zwany również in-
termedyną lub melanotropiną, jest wytwarzany przez część pośrednią
części gruczołowej przysadki. Hormon ten stymuluje produkcję poszcze-
gólnych melanin na przemian. Reakcja na działanie tego hormonu jest
kontrolowana przez locus A (Agouti). Natomiast receptor hormonu me-
lanotropowego (MSH) kontroluje poziom tyrozynazy wewnątrz melanocytu.
Receptor ten jest determinowany przez locus E (Extension). Następstwem
działania tego receptora jest wzrost poziomu tyrozynazy i produkcja
eumelaniny. Główną jego funkcją natomiast jest regulacja syntezy czarnego
pigmentu (eumelanina), a hamowanie produkcji feomelaniny wewnątrz
melanocytów.
Melaniny są wytwarzane w melanocytach. Melanocyty pochodzą
z rdzenia nerwowego, a podczas rozwoju embrionalnego wędrują do
powstającej skóry. Ta wędrówka melanocytów znajduje się pod ścisłą
kontrolą genów.
W determinację koloru jest włączonych wiele loci. Wiele genów, jeśli nie
wszystkie związane z wytwarzaniem pigmentu, ma działanie plejotropowe
na rozwój i różnicowanie organizmu.
Wytwarzanie melanin u ssaków jest kontrolowane przez geny działające
na różnych poziomach: tkankowym, komórkowym i subkomórkowym.
REGULACJA NA POZIOMIE TKANKOWYM
Regulacja genetyczna na poziomie tkankowym polega na kontroli liczby
i rozmieszczenia melanocytów. Jest wiele genów, które współdziałają ze
sobą w sposób kompleksowy. Geny, które zostały już sklonowane, warun-
kujące pigmentację ssaków i działające na poziomie tkankowym to przede
wszystkim: gen srokatości (Piebaldism) i gen nabytego bielactwa (Vitiligo).
Badania genetycznego tła umaszczenia są najbardziej rozwinięte w od-
165

niesieniu do myszy. Do loci działających na poziomie tkankowym,
zmapo-wanych w genomie myszy należą także loci: Splotch (Sp) - -
plamisty, Dominant White Spotting (W) - - dominujący biały nakrapiany i
Steel (Sl) - stalowy.
REGULACJA NA POZIOMIE KOMÓRKOWYM
Geny regulujące wytwarzanie pigmentu na poziomie komórkowym od-
działują na strukturę i/lub funkcje istniejących melanocytów, co wpływa na
koloryt (układ barw). Należą do nich geny z loci D (Dilute — rozjaśniony),
E (Extension - - pełna barwa), A (Agouti — dziki), Pa (Pallid — blady)
i P (Pink-eyed dilution — gen rozcieńczonych różowych oczu). Mutacje
w tych loci wpływają wyraźnie na kolor oczu, w jednolity typowy sposób,
przez działanie na mechanizmy włączone w funkcję melanocytu. Większość
z tych mutacji ma wpływ plejotropowy na rozwój.
Locus D koduje strukturalne białko, które wydaje się istotne dla rozdziału
ziaren melaniny do peryferyjnych melanocytów. U osobników ze zmutowa-
nym allelem w locus D dendryty melanocytów są istotnie mniej rozwinięte
niż u osobników normalnych (typu dzikiego), a konsekwencją tego jest
ograniczenie rozdziału melanosomów i rozjaśniony kolor włosów.
Locus E (Extension) koduje przede wszystkim receptor MSH, pełniący
istotną rolę w interakcji melanocytów z hormonem melanotropowym. Locus
ten kontroluje ilość eumelaniny.
Locus A (Agouti) kontroluje syntezę białka będącego antagonistą receptora
MSH i mającego zdolność blokowania działania tego receptora. Następstwem
działania tego białka jest niski poziom tyrozynazy i wytwarzanie feomela-
niny. Na przykład u bydła zwierzęta homozygotyczne ee będą umaszczone
żółto lub czerwono.
U osobników typu dzikiego, nazywanych agouti, melanocyty wytwarzają
w różnym czasie eumelaninę lub feomelaninę, oba typy melanin nie są więc
syntetyzowane jednocześnie.
Locus Agouti działa wewnątrz mikrośrodowiska mieszków włosowych,
podczas gdy locus Extension działa w melanocytach.
REGULACJA NA POZIOMIE SUBKOMÓRKOWYM
Melanogeneza jest regulowana także na poziomie subkomórkowym (en-
zymatycznym), gdzie krytyczną rolę odgrywa synteza i ekspresja różnych
melanogenicznych enzymów i inhibitorów. Geny, które działają na pigmen-
tację ssaków na poziomie subkomórkowym, wpływają bezpośrednio na
ilość i typ wytwarzanej melaniny. W wyniku ich działania może nastąpić
istotny wzrost lub zmniejszenie ilości pigmentu tworzonego w melanocytach
(np. brak syntezy melaniny w przypadku albinizmu), lub też mogą wystąpić
zmiany typu syntetyzowanej melaniny (np. odwracalna zamiana wy-
twarzania czarnej eumelaniny na produkcję żółtej feomelaniny).
166

Najważniejsze z loci działających na poziomie subkomórkowym to
Albina (C) i Brown (B).
Locus C kontroluje liczbę i intensywność ziaren pigmentu. Historycznie był
on uważany za locus strukturalny dla tyrozynazy, gdyż białkiem kodowanym
przez ten locus jest enzym tyrozynaza, której funkcja jest decydująca dla
wytwarzania pigmentu. Tyrozynaza przejawia swą ekspresję w melanocytach.
Mutacje w locus C powodują brak pigmentu we włosach, skórze
i tęczówce oka, ale mutacje te mają także wpływ plejotropowy jeszcze
dokładnie nie poznany. Jednym ze znanych efektów plejotropowego
działania mutacji w tym locus jest zaburzenie czucia.
Interesujące jest to, iż mutacja w genie tyrozynazy u myszy jest
pojedynczą mutacją punktową, natomiast u ludzi stwierdzono ponad 30
mutacji powodujących albinizm, ale są to mutacje typu nonsens, zmiany
sensu i mutacje zmiany fazy odczytu. Niekiedy mutacje w locus Albino
powstają podczas składania mRNA (splicingu), czyli wycinania intronów,
a konsekwencją tego jest synteza białka o zmienionej sekwencji amino-
kwasowej. Takie białka nie są kompetentne katalitycznie, ale dotychczas nie
stwierdzono, czy pełnią one jakąś rolę (np. inhibitora) w melanocytach.
Badania poszczególnych mutacji w locus Albino u królików, warun-
kujących umaszczenie himalajskie i szynszylowate, wykazały, że melanocyty
mutanty wytwarzają tyrozynazę z istotnie zmienioną aktywnością katalitycz-
ną. Mutacja umaszczenia himalajskiego (c
h
) polega na zmianie w glikozylacji,
która powoduje uwrażliwienie efektu fenotypowego na temperaturę. U szyn-
szyla (c
ch
) natomiast zwiększona jest wrażliwość na inaktywację proteolitycz-
ną, prowadząca do osłabienia funkcji enzymu. Geny z locus C u królików
tworzą szereg alleli wielokrotnych, w którym występuje dominacja w na-
stępującej kolejności: C, c
ch
, c
h
, c (ryć. 6-9 wkładka kolorowa).
Tyrozynaza jest najistotniejszym enzymem w wytwarzaniu melaniny,
natomiast inne geny regulują typ formowanego pigmentu, jeszcze inne —
sposób, w jaki pigment ma być formowany.
Locus B (Brown) — struktura i organizacja genu z tego locus jest podobna
do genu Albino, a białko kodowane przez ten gen ma wszystkie cechy
charakterystyczne dla tyrozynazy. Specyficzna funkcja tego białka nie jest
jeszcze poznana. Fenotypowym efektem mutacji w genie Brown jest wytwa-
rzanie brązowej mełaniny, ale mechanizm działania tego genu dotąd nie jest
znany. Wiadomo jednak, iż kolor brązowy wynika z punktowej mutacji
w obrębie genu Brown, która prowadzi do wymiany cysteiny na tyrozynę.
Mutacja ta zachodzi w bogatej w cysteinę pierwszej domenie białka.
Mechanizm dziedziczenia umaszczenia u zwierząt futerkowych
na przykładzie lisa
Istnieją dwa gatunki lisa — lis pospolity (Vulpex vulpex) i lis polarny (Alopex
lagopus). Umaszczenie u obu gatunków determinowane jest głównie genami
z podstawowych loci — A, B i E. Warunkiem ekspresji genów z tych loci jest
167
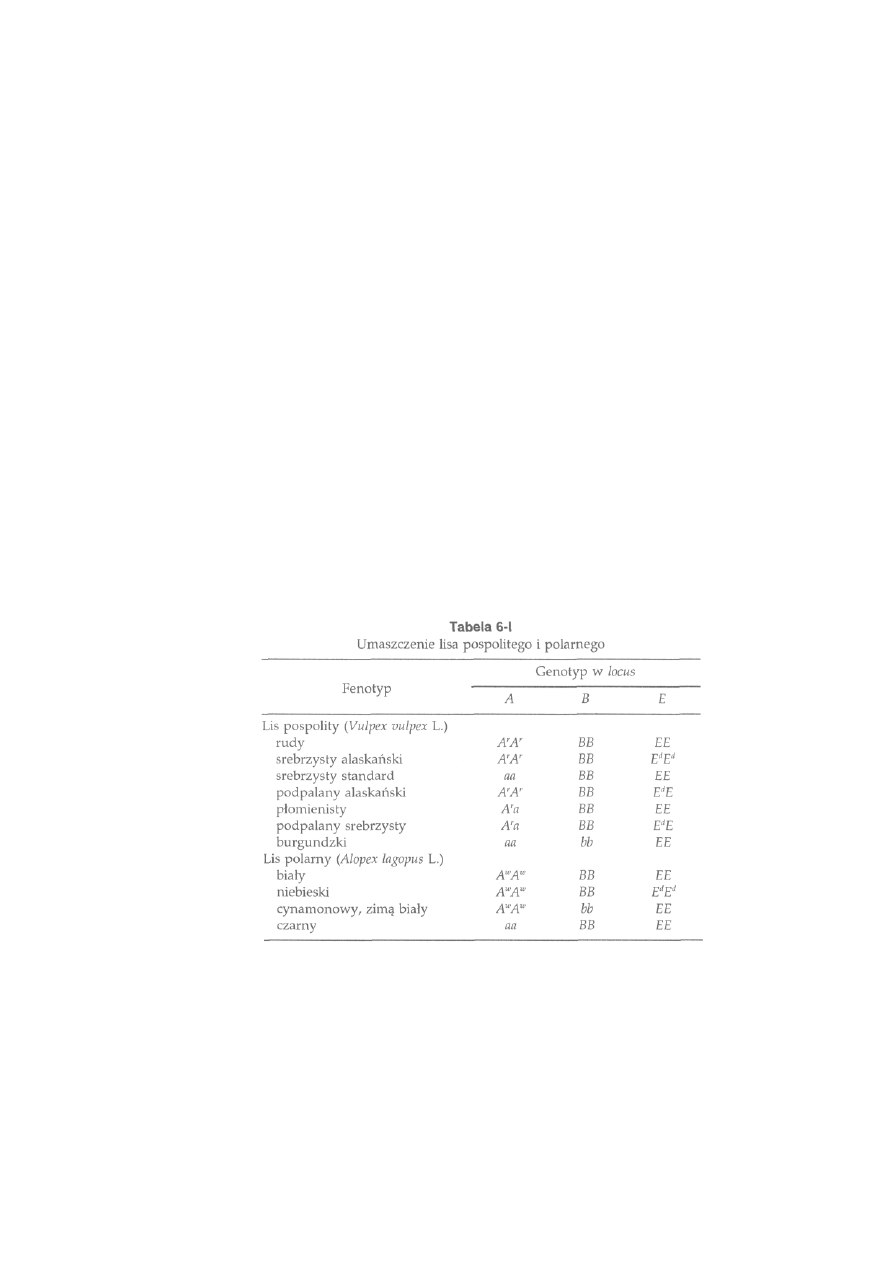
oczywiście obecność odpowiednich genów z locus C. W przypadku genotypu
homozygoty recesywnej cc będzie fenotyp albinotyczny, bez względu na to,
jakie są geny z innych loci.
Genotypy umaszczenia różnych odmian lisa pospolitego i polarnego są
przedstawione w tabeli 6-1.
U lisa pospolitego w locus A są dwa allele: A
r
— dominujący i a -
recesywny. Allel A
r
warunkuje wstrzymanie wytwarzania eumelaniny. Jego
alternatywny allela w podwójnej dawce warunkuje umaszczenie czarne lisa
srebrzystego. Natomiast u lisa polarnego zamiast allelu A jest allel A". Allel
ten odpowiedzialny jest za eliminację eumelaniny. Feomelanina, która
z reguły zajmuje miejsce eumelaniny, u lisa polarnego jest rozcieńczona lub
usunięta w ogóle. Lis polarny o genotypie aa jest czarny, ale genotyp ten
występuje bardzo rzadko (np. w populacji lisa polarnego w Islandii).
W locus B również są dwa allele — dominujący B (czarny pigment) i b,
w układzie homozygotycznym -- pigment czekoladowobrązowy. Jedynie
odmiana burgundzka lisa pospolitego jest homozygotyczna pod względem
allelu b.
W locus E, warunkującym wytwarzanie czarnej/czekoladowobrązowej
eumelaniny, u obu gatunków są dwa allele — E i E
d
. Allel E w układzie
homozygotycznym nie ma wpływu na umaszczenie, w tym przypadku
kolor jest determinowany allelami z locus A. U obu gatunków lisa allel E
d
cechuje się niezupełną dominacją.
Oprócz omawianych loci u lisa pospolitego występuje szereg innych loci,
jak G, P, S, R, W, warunkujących różne odmiany umaszczenia. We wszystkich
tych loci, z wyjątkiem locus W, efekt fenotypowy dają tylko genotypy
homozygotyczne recesywne: u lisa burgundzkiego -- genotyp gg, u lisa
168

srebrzystego: genotyp pp - - perłowe umaszczenie oraz genotyp ss —
umaszczenie perłowe Mansfield i rr — umaszczenie Radium. W locus W są
następujące allele: w, W (białopyski), W (platynowy), W
3
(biały Georgian)
i W
M
(gen marble), przy czym genotyp ww występuje u lisa rudego
i srebrzystego, natomiast gen marble daje inny efekt fenotypowy u tych
odmian. Mutacje dominujące, takie jak W i W, w układzie homozygotycz-
nym mają efekt letalny. Letalny jest także genotyp WW.
U lisa polarnego na wiele odmian umaszczenia wpływają allele recesywne
(w homozygocie) z loci: D (dd — biały polarny, DD — niebieski), F (ff — szafir)
i G (gg — arktyczny niebieski) oraz u lisa niebieskiego allele dominujące z loci
l (L — Laponia), s (trzy allele: S, S
J
i S
H
) i t (T). Mutacja w locus s (Shadow)
charakteryzuje się heterochromią, której skutkiem może być różny kolor oczu
u tego samego osobnika, np. jedno oko niebieskie, a drugie brązowe.
Genetyczne podłoże umaszczenia u bydła
Wyniki wielu badań sugerują, że u bydła umaszczenie czarne, brązowe
i czerwone jest determinowane przez allele z dwóch loci: Extension (E) i
Agouti (A).
Locus E koduje syntezę białka MSH-R (ang. melanocyte stimulating hormon
receptor), złożonego z około 350 aminokwasów. W locus tym, umiejscowionym
w chromosomie 18, są 3 allele: E
d
warunkujący umaszczenie czarne dominują-
ce, recesywny allel e, który w układzie homozygotycznym warunkuje
umaszczenie czerwone, oraz allel E
+
umożliwiający ekspresję alleli z locus A.
Allele te powstały w wyniku mutacji punktowych, opisanych w rozdziale:
Mutacje. Allel dominujący E
d
, kodujący czarny kolor, jest homologiczny do
allelu E
S
0 u myszy. W łańcuchu polipeptydowym kodowanym przez allel E
S
0
nastąpiła zamiana trzech kolejnych cząsteczek leucyny na aminokwasy
leucyna-prolina-leucyna, natomiast u bydła ten sam fragment alłelu E
rf
koduje
następującą kolejność aminokwasów: prolina-leucyna-leucyna.
Locus A koduje syntezę białka, będącego antagonistą białka MSH-R.
W locus tym są dwa allele: dominujący A
+
(synteza pigmentu brązowego)
i recesywny a. Allele z tego locus mogą wykazać ekspresję tylko wtedy, gdy
w locus E jest genotyp E
+
_. U zwierząt z genotypem E
+
E
+
lub E
+
e allel A
+
koduje umaszczenie brązowe, natomiast allel a w układzie homozygotycz-
nym — recesywne czarne.
Fenotypy czarny dominujący (determinowany przez allel E'') i czarny
recesywny są nieodróżnialne. Niestety dotychczas nie przeprowadzano
badań molekularnych locus A.
Wyniki badań porównawczych wskazują na homologię działania alleli z locus
Extension (E) u bydła i myszy. Wszystkie allele dotychczas zidentyfikowane
w locus E u bydła mają odpowiadające im allele u myszy, natomiast w locus
A tylko w przypadku allelu recesywnego a jest analogiczny allel u myszy. Z kolei
allel A
+
u bydła wydaje się wywoływać efekt różny od działania większości
innych alleli z locus Agouti (A) opisanych dotychczas u ssaków.
169

Wiele innych loci bierze udział w determinacji różnych odmian umasz-
czenia. Na przykład, cecha jednolitego umaszczenia lub łaciatości znajduje
się pod kontrola genów z locus S (Self): dominujący S — jednolite
umaszczenie, recesywny s — łaciatość. U ras bydła (np. belgijskie błękitne),
u których allel s jest utrwalony, obserwowana jest zmienność w ekspresji
cechy łaciatości, niektóre zwierzęta są niemal całkowicie pozbawione
pigmentowanych łat. W tym samym locus u bydła tej rasy znajduje się allel
S
c
, warunkujący fenotyp nazwany colorsided — łaciaty, który dominuje nad
s, ale jest recesywny w stosunku do allelu S.
U niektórych ras bydła pewne geny wykazują niekorzystne działanie
plejotropowe, powodując różne zaburzenia. Przykładem jest zmapowany
w 5 chromosomie locus Roan — dereszowate (krasę, mroziate), w którym są
dwa allele: r
+
czarny (u bydła błękitnego belgijskiego) lub czerwony (u bydła
rasy shorthorn) i R biały. U jałówek obu ras locus Roan wpływa istotnie na
występowanie choroby białych jałowic, kora jest efektem plejotropowego
oddziaływania allelu R na płodność. Choroba ta jest opisana w rozdziale:
Determinacja i różnicowanie płci oraz interseksualizm. Zróżnicowanie umasz-
czenia — czerwone lub czarne wynika z tego, że bydło belgijskie błękitne
charakteryzuje duża frekwencja allelu dominującego E w locus Extension, a mała
allelu e (czerwone umaszczenie), natomiast u shorthornów jest odwrotnie.
Genetyczne podłoże umaszczenia u świń
U świń znane są cztery główne loci determinujące umaszczenie: locus
A (Agouti), locus E (Extension), locus I (biały dominujący) i locus Be (Belted -
opasany).
W locus A zidentyfikowano dwa allele: A
w
(agouti biały brzuch) i allel
a - - nieagouti. Allel A
w
jest obecny u dzikich świń i jest dominujący
w stosunku do pozostałych kolorów oraz koloru mangalica, ale recesywny
w stosunku do białego (I). Większość ras świń udomowionych ma allel
recesywny a — nieagouti.
W locus E stwierdzono dotychczas trzy allele: E — umaszczenie jednolite
czarne jak w rasie wielkiej czarnej, cornwall i hampshire, E
r
— umaszczenie
czarne nakrapiane (spotting) jak u berkshire, poland-china i pietrain, e -
umaszczenie jednolicie czerwone (rude) jak u tamworth i duroc-jersey.
Kolejność dominowania tych alleli jest następująca: E-E
p
-e. Badania, w któ-
rych do analizy sprzężeń użyto ponad 230 markerów genetycznych,
wskazują, iż locus ten jest umiejscowiony w chromosomie 6, blisko telome-
rowego regionu ramienia p. Wyniki badań porównawczego mapowania
locus Extension u myszy (chromosom 8), bydła (chromosom 18) i świń
(chromosom 6) wskazują na homologię między tymi gatunkami. Podobnie
jak u myszy i bydła, także u świń receptor hormonu melanotropowego
MSH-R jest determinowany przez geny z locus E.
W locus I są dwa główne allele: 7 (inhibicja koloru), odpowiedzialny za
umaszczenie dominujące białe, i allel i — recesywny (kolorowy). Wśród
170

świń domowych dominujący biały jest przeważający, występuje on w homo-
zygocie (II) w rasie wielka biała i wszystkich odmianach landrace, natomiast
inne rasy są homozygotami recesywnymi (ii). Locus I został zlokalizowany
w chromosomie 8. Dziedziczenie umaszczenia u mieszańców niektórych ras
sugeruje istnienie dalszych trzech alleli w tym locus, oznaczonych symbolem
I
d
(deresz), I
p
(czarne łaty) oraz i
m
(brudny szary).
Biały pas, charakterystyczny dla niektórych ras, jak np. wessex sad-
dleback, hampshire (ryć. 6-10 wkładka kolorowa), hannover-braunschweig,
jest kodowany przez allel Be
w
z locus Be, dominujący w stosunku do
jednolitego umaszczenia. Zmienna szerokość pasa zależy od działania
genów modyfikujących (zmienność poligeniczna). Genotyp tego knura jest
następujący: aa U EE Be
w
Be
w
.
Na umaszczenie świń oprócz wymienionych czterech loci wpływają
także allele z locus C. Na przykład u świń rasy mangalica umaszczenie
brudnobiałe jest prawdopodobnie determinowane przez allel c
e
z locus C.
Również geny z innych loci u różnych ras świń kodują specyficzne wzory
umaszczenia. Na przykład u świń rasy hampshire stwierdzono obecność
genów recesywnych z loci Red-eye (czerwonych oczu) i Dilution (rozjaśniony).
Genetyczne podłoże umaszczenia u koni
Umaszczenie u koni determinowane jest głównie przez geny znajdujące się
w locus E (Extension) i A (Agouti). Rozjaśnienie barwy jest warunkowane
działaniem genów z co najmniej trzech loci: C (Albina), D (Dun — bułany)
i Z (Siher dapple — siwy jabłkowity). Umaszczenie białe, siwe i dereszowate
jest kontrolowane odpowiednio przez allele z loci W (White — dominujący
biały), G (Gray — siwy) i RN (Roan — deresz). Wzory umaszczenia, takie jak
tobiano (TO), overo (O), sabino (SB) czy tarantowate (LP), są determinowane
przez geny z innych loci. We wszystkich wymienionych loci dotychczas
zidentyfikowano po dwa allele, ale prowadzone są badania w celu potwier-
dzenia hipotez o istnieniu innych alleli (np. trzeciego allelu w locus Extension
— E
D
).
Locus E warunkuje wytwarzanie określonego rodzaju melaniny, allel
dominujący E determinuje syntezę eumelaniny, w przypadku jego braku
(genotyp ee) syntetyzowana jest feomelanina. Allele z tego locus wpływają
na ekspresję genów z locus A. Allel recesywny e w podwójnej dawce (ee)
tłumi działanie allelu A. Natomiast obecność w genotypie alleli dominujących
z obu loci (E i A) warunkuje umaszczenie gniade.
W locus C u koni nie występuje allel recesywny c, który w układzie
homozygotycznym u innych gatunków zwierząt warunkuje albinizm.
Rozjaśnienie umaszczenia u koni powodowane jest przez allel C"', wykazu-
jący niepełną dominację. W układzie homozygotycznym (C
cr
C
cr
) allel ten
warunkuje umaszczenie kremowe (ang. cremello).
Umaszczenie białe u koni jest warunkowane przez dominujący allel
W (jest to dominacja całkowita, zatem wystarczy jeden allel W, by było
171
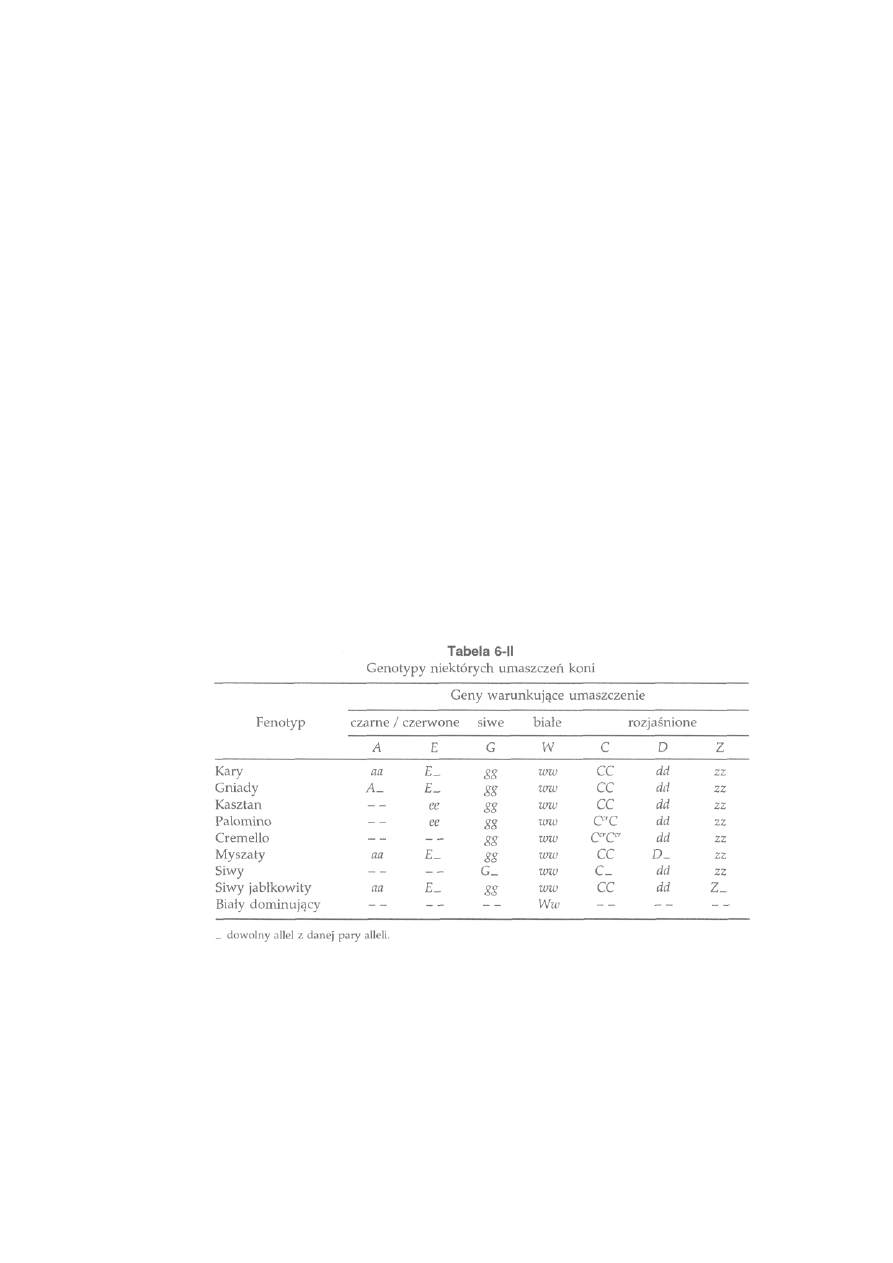
umaszczenie białe), który wykazuje działanie epistatyczne w stosunku do
poznanych dotychczas genów kontrolujących umaszczenie. Wszystkie
rodzaje umaszczenia, z wyjątkiem białego, w locus W mają genotyp
homozygoty recesywnej (ww). Natomiast umaszczenie siwe kontroluje allel
dominujący z locus G, który jest epistatyczny w stosunku do wszystkich
alleli z innych loci, z wyjątkiem allelu W, wobec którego jest hipostatyczny.
Allel G (w homo- i heterozygocie) powoduje redukcję wytwarzania pigmentu
postępującą wraz z wiekiem zwierzęcia.
Locus D kontroluje intensywność wytwarzania eumelaniny i feomelani-
ny. Allel D z locus Dun wykazuje zupełną dominację, osobniki homozygoty-
czne (DD) są nie do odróżnienia na podstawie fenotypu od heterozygot
(Dd). Jego wpływ na rozjaśnienie barwy jest jednak mniejszy niż allelu
C
cr
z locus Albina. Na przykład obecność allelu D w genotypie osobnika
karego powoduje umaszczenie myszate (aaE_CCD_).
Niektóre spośród wymienionych genów umaszczenia działają plejotro-
powo. Należą do nich allele dominujące z loci W, O i RN. Allel W w podwój-
nej dawce (genotyp homozygoty dominującej) powoduje zamieranie zarod-
ków we wczesnym okresie ciąży. Konie umaszczone biało są hetero-
zygotyczne pod względem genów tego locus (Ww). Z kolei większość źrebiąt
homozygotycznych pod względem allelu O jest obarczona zespołem „białych
źrebiąt" - LWFS (ang. lethal white foal syndrome), polegającym na
niemożności wydalenia smółki z powodu braku komórek nerwowych
(zwojów autonomicznych) kontrolujących ruchy perystaltyczne jelita lub,
znacznie rzadziej, na skutek braku części jelita. Natomiast układ homo-
zygotyczny w locus RN (RNRN) jest uważany za letalny.
Genotypy w 7 loci, determinujące wybrane rodzaje umaszczenia koni,
zestawiono w tabeli 6-II.
172

Genetyczne podłoże umaszczenia u owiec
U owiec stwierdzono trzy typy pigmentu: czarna eumelanina, czekoladowo-
brązowa (moorit) eumelanina i brązowoczerwona (tan) feomelanina.
Badania specjalistyczne wykazały, że umaszczenie tan jest warunkowane
wytwarzaniem pigmentu feomelaniny. Przykładem tego jest brązowoczer-
wony kolor wełny karakułów.
Najczęściej występujące umaszczenia u owiec są klasyfikowane zgodnie
z trzema kryteriami: typ pigmentu, wzór koloru i obecność lub brak białych
znaków. Wzory koloru składają się bądź z regularnej mieszaniny jasno
i ciemno ubarwionych powierzchni ciała, bądź mieszaniny jasnych i ciem-
nych włókien wełny.
Owce biało umaszczone są zwykle prawie pozbawione pigmentu, ale
mogą mieć pigment tan, który w fenotypie ujawnia się ze zmienną
intensywnością, od umaszczenia jasnobrązowego do intensywnego czer-
wonawobrązowego.
Umaszczenie owiec jest kontrolowane prawdopodobnie przez geny z 11
loci (tab. 6-III).
Loews Agonii odpowiada za białe lub brązowe (tan) umaszczenie i wzory
umaszczenia. W locus tym stwierdzono ogółem 16 alleli kontrolujących
zmianę syntezy eumelaniny na syntezę feomelaniny. Niektóre z nich
warunkują wytwarzanie pigmentu tylko w poszczególnych partiach ciała,
inne wpływają na typ włókien, bądź też oba efekty występują łącznie.
Allel A
wh
, będący pierwszym w kolejności dominowania z szeregu
alleli z tego locus, warunkuje białe umaszczenie owiec. Głównym efektem
allelu A
wh
jest całkowite zahamowanie wytwarzania eumelaniny, podczas
gdy feomelanina może być syntetyzowana. Konsekwencją tego może
być umaszczenie brązowoczerwone (tan) owiec, w których genotypach
jest allel A
wh
. Ten typ umaszczenia występuje u owiec rasy french solognot i
brązowych (dominujących) karakułów, natomiast o wiele jaśniejsze
umaszczenie brązowoczerwone (tan) występuje u owiec islandzkich i welsh
mountain. Białe umaszczenie owiec ras wełnistych jest wynikiem selekcji
prowadzonej przeciwko umaszczeniu brązowoczerwonemu (tan) u owiec
nosicieli allelu A
wh
.
Następne geny w szeregu alleli z locus Agouti częściowo hamują syntezę
eumelaniny, natomiast allel recesywny A
e
w układzie homozygotycznym
umożliwia pełną syntezę eumelaniny.
Niektóre allele z tego locus mają efekt plejotropowy, np. allel A
wh
zmniejsza
płodność owiec o około 0,15 jagnięcia w miocie i hamuje aktywność seksualną
maciorek islandzkich poza normalnym sezonem rozpłodowym.
Locus Brown determinuje syntezę czarnego i czekoladowobrązowego
(moorit) pigmentu. Allel typu dzikiego, B
w
, warunkuje wytwarzanie czarnej
eumelaniny, natomiast recesywny allel, B
rw
— czekoladowobrązowej eume-
laniny.
173
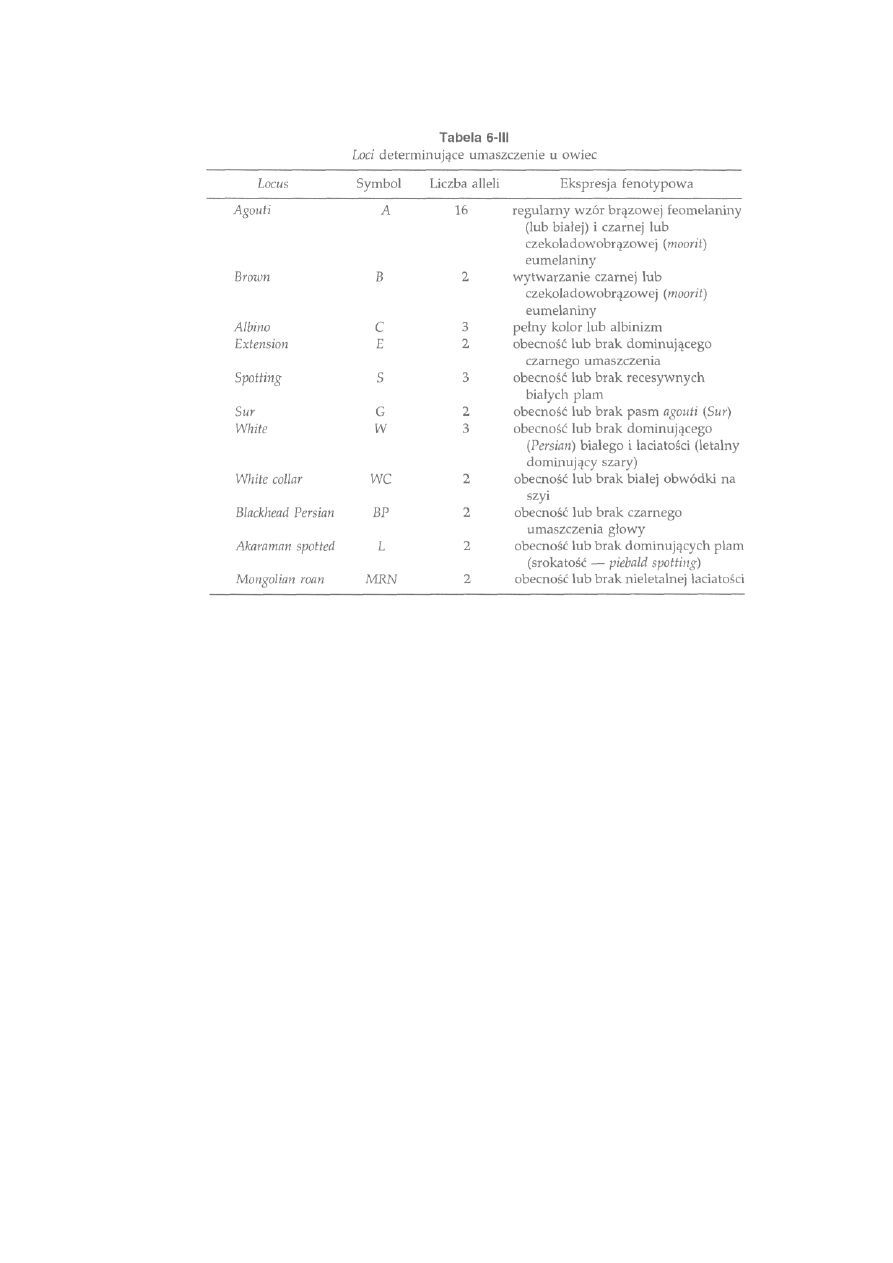
Na umaszczenie u owiec wpływają także allele z innych loci:
Allel recesywny C
a
z locus Albino warunkuje w układzie homozygotycz-
nym całkowity albinizm. Umaszczenie czarne dominujące jest kontrolowane
przez allel dominujący E
db
z locus Extension. Allel ten inaktywuje działanie
alleli z locus Agouti poprzez zahamowanie syntezy feomelaniny. Allel typu
dzikiego, recesywny, E
w
umożliwia normalną ekspresję alleli z locus Agouti.
Allel recesywny S
s
z locus Spotting w podwójnej dawce (S
S
S
s
) determinuje
białe znaki u owiec umaszczonych kolorowo. Owce ras wełnistych, z biało
umaszczoną głową są zwykle homozygotyczne pod względem genu białych
plam (S
S
S
S
), a także homozygotyczne białe, A
wh
A
wh
. U owiec heterozygotycz-
nych pod względem allelu A
1
"
1
' allel S
s
redukuje pigment tan (brązowy),
natomiast homozygoty S
S
S
S
są całkowicie białe.
U umaszczonych kolorowo owiec kożuchowych białe lub złote końce
włosów są warunkowane allelem G
s
z locus Sur. Allel ten jest recesywny lub
hipostatyczny w stosunku do barwy czarnej dominującej, ale epistatyczny
do dominującej brązowej (tan).
Umaszczenie białe dominujące (białe perskie, łaciate afgańskie) jest
kontrolowane przez allel W
a
z locus White. Allel ten jest epistatyczny
174

w stosunku do wszystkich innych genów umaszczenia. Inny allel z tego
locus, W
rn
, warunkuje dominującą szarość u karakułów, która w formie
homozygotycznej jest letalna; również w połączeniu z allelem dominującej
białości allel W
rn
wywołuje w większości przypadków efekt śmiertelny.
6.4. Penetracja i stopień
ekspresji (ekspresywność) genów
Fenotyp rozpatrywany jako całokształt cech jest wynikiem działania wszyst-
kich genów danego osobnika, współdziałania tych genów między sobą oraz
współdziałania ze środowiskiem. Omówione przykłady współdziałania
allelicznego i nieallelicznego świadczą o tym, iż działanie genów może
wywoływać różnorodne efekty.
Często zdarzają się przypadki, kiedy wśród osobników o jednakowych
genotypach niektóre wykazują fenotypową obecność cechy, inne zaś
jej nie mają. Z drugiej strony, ten sam genotyp u różnych osobników
może spowodować różne nasilenie cechy w fenotypie. Pierwsze z tych
zjawisk określane jest jako penetracja genu, natomiast drugie jako eks-
presywność genu, rozumiana także jako stopień ekspresji (przejawiania
się) genu lub wyrazistość. Oba pojęcia — penetracja genu i ekspresywność
genu zostały wprowadzone w 1926 roku przez neuropatologa Oskara
Yogta.
Penetracja, inaczej określana jako przenikliwość, jest to częstość, z jaką
dominujący lub recesywny gen (w homozygocie) albo układ heterozygotycz-
ny ujawnia się w fenotypie nosiciela. Penetracja może być całkowita lub
niezupełna. O penetracji całkowitej mówimy wówczas, gdy u wszystkich
osobników o danym genotypie występuje taki sam fenotyp. Natomiast jeśli
nie wszystkie osobniki, mające taki sam genotyp, wykazują charakterystycz-
ny dla niego fenotyp, mówimy o penetracji niezupełnej. Na przykład, jeżeli
gen dominujący ujawnia swą obecność w fenotypie tylko u 80% osobników,
to mówimy, że jego penetracja wynosi 80%.
Przykładem penetracji niezupełnej u zwierząt gospodarskich może
być wada pojawiająca się u nowo wylężonych kurcząt — wrodzone
drżenie. Natężenie drgań jest bardzo zróżnicowane. Stwierdzono to na
podstawie wyników doświadczenia, w którym po 4 latach prowadzenia
kojarzeń ptaków heterozygotycznych pod względem genu drżenia uzy-
skano jedynie 10% kurcząt dotkniętych tą wadą, zamiast 25% oczekiwanych
zgodnie z klasycznym stosunkiem mendlowskiej segregacji genów.
Penetracja określonego genu może być różna zależnie od płci, a nawet,
w przypadkach krańcowych, ograniczona do jednej płci (patrz podrozdział:
Cechy sprzężone z płcią).
175

Pojęcie stopień ekspresji (ekspresywność, stopień przejawiania się) genu
oznacza poziom zmienności fenotypowej określonej cechy wśród osobników
o tym samym genotypie.
Zmienność ekspresywności genu często jest obserwowana w przypadku
chorób genetycznych i wad dziedzicznych. Przykładem wady dziedzicznej
0 różnym stopniu przejawiania się genu jest miejscowy brak nabłonka
u bydła rasy ayrshire (łagodna forma) i Jersey (forma bardzo ostra) (patrz
rozdział: Mutacje). Obserwowana zmienność stopnia ekspresji genu może
być wywołana działaniem różnych u tych ras genów modyfikujących.
W przypadku chorób genetycznych czy wad dziedzicznych ocenę ekspresy-
wności genu mogą utrudniać takie czynniki, jak: wiek, w którym pojawiają
się pierwsze objawy, czy występowanie fenokopii (patrz podrozdział:
Mutacje genowe wywołujące choroby genetyczne), które nie są dziedziczne.
Zmienność stopnia ekspresji jest dość rozpowszechnioną właściwością
genów. Należy jednak pamiętać o tym, że nie zawsze zmienność fenotypowa
jakiegoś genotypu jest wynikiem różnego stopnia przejawiania się genu.
Geny z różnych loci mogą dawać podobne fenotypy, ujawniające się
w różnym stopniu. Także allele tego samego genu mogą charakteryzować
się podobną ekspresywnością. Pewność, iż mamy do czynienia ze zmienno-
ścią przejawiania się tego samego genu, możemy mieć wtedy, gdy jest ona
obserwowana u osobników spokrewnionych ze sobą.
Pojęcia penetracja i ekspresywność genu są czasem trudne do roz-
dzielenia. Wiele genów o małym stopniu penetracji wykazuje jednocześnie
słabą ekspresywność. Z kolei, geny charakteryzujące się dużą ekspresyw-
nością wykazują wysoki stopień penetracji. Zarówno stopień penetracji,
jak i fenotypowego przejawiania się genu zależą od współdziałania
tego genu z innymi genami oraz od jego współdziałania ze
środowiskiem. Spośród czynników niegenetycznych wpływających na
ekspresję genu ważną rolę odgrywają wpływy matczyne na rozwijający
się płód (w szczególności należy do nich zaliczyć hormony, sole
mineralne oraz inne związki krążące we krwi, docierające przez łożysko
do rozwijającego się płodu). Nie bez znaczenia jest stan zdrowotny
matki, a także położenie zarodka w macicy i nawet obecność innych
płodów.
6.5. Dziedziczenie pozajądrowe
Wszystkie omawiane dotychczas cechy są uwarunkowane genami zawartymi
w DNA jądrowym, a ich sposób dziedziczenia określany jest ogólnym
terminem — dziedziczenie mendlowskie. Już na początku naszego stulecia
zauważono, że niektóre cechy nie podporządkowują się temu terminowi,
dziedziczą się różnie, zależnie od kierunku krzyżowania. Wykrycie obecności
DNA poza jądrem komórkowym — w mitochondriach, a także chloroplas-
176

tach roślin wyjaśniło podłoże tego zjawiska. Powstał nowy termin -
dziedziczenie pozachromosomowe, inaczej pozajądrowe lub cytoplazma-
tyczne.
Mitochondrialny DNA, w przeciwieństwie do DNA jądrowego, jest
przekazywany następnemu pokoleniu wyłącznie przez matki. Wprawdzie
w 1991 roku w doświadczeniu na myszach wykryto u potomstwa cząsteczki
mtDNA pochodzące od ojca, ale częstość ich występowania była minimalna
i wynosiła 10
-4
częstości matczynego mtDNA. Taki sposób dziedziczenia
może być przyczyną powstawania heteroplazmii (równoczesne występowa-
nie zmutowanego i prawidłowego mtDNA u danego osobnika). Jak dotąd,
u żadnego innego gatunku nie potwierdzono przekazywania potomstwu
mtDNA przez ojca. Przyczyną tego, w świetle wyników najnowszych
badań, może być znakowanie (piętnowanie) cząsteczkami białka (zwanego
ubikwityną) mitochondriów w dojrzewających plemnikach podczas procesu
spermiogenezy. Po zapłodnieniu komórka jajowa może rozpoznawać tak
napiętnowane organelle i niszczyć je. Plemnik zawiera od 50 do 100
mitochondriów, natomiast komórka jajowa ma ich około 100 tysięcy. W miarę
rozwoju zarodka liczba mitochondriów ojcowskich maleje.
O mutacjach zachodzących w obrębie mitochondrialnego DNA i ich
skutkach jest mowa w rozdziale: Mutacje.
U zwierząt gospodarskich badania wpływu genów w mitochondrialnym
DNA na cechy produkcyjne najczęściej prowadzone są na bydle mlecznym.
Geny mitochondrialne kodują przede wszystkim enzymy niezbędne do
przebiegu procesów zachodzących w łańcuchu oddechowym. Ponad 90%
energii potrzebnej komórkom sekrecyjnym gruczołu mlecznego jest dostar-
czane przez ATP (adenozynotrifosforan) wytwarzany w mitochondriach.
U bydła rasy holsztyńskiej stwierdzono dodatnią korelację, której wartość
zawiera się w granicach od 0,3 do 0,4, między syntezą ATP w mitochondriach
a wartością genetyczną w zakresie produkcji mleka. Badania wpływu
genotypu w mtDNA krowy na wydajność mleka i tłuszczu wykazały, że
warunkuje on od 2 do 10% zmienności tych cech.
W celu oszacowania wpływu loci mitochondrialnego DNA na cechy
użytkowości mlecznej porównywano średnie wartości tych cech w rodzi-
nach (po określonej matce) z polimorfizmem alleli mtDNA, analizowanego
metodą RFLP. W pewnym doświadczeniu spośród 2713 krów z 131 stad
wybrano rodziny charakteryzujące się wysoką, średnią i niską wydajnością
mleka. Oszacowany dla tych rodzin efekt matki wynosił odpowiednio:
+ 991 ±108 kg, +26 ±99 kg, -879 ±114 kg mleka. Analiza polimorfizmu
fragmentów restrykcyjnych mitochondrialnego DNA wykazała, iż największa
frekwencja alleli występujących we wszystkich grupach wynosiła ^ 0,9.
Natomiast allele charakteryzujące się niską frekwencją (^ 0,1) rozkładały się
nielosowo w grupach wydajności mleka i pochodzeniowych po matce, co
może wskazywać na związek między polimorfizmem mtDNA a wydajnością
mleka.
t l i

Wyniki badań prowadzonych na bydle holsztyńsko-fryzyjskim przez
innych badaczy potwiedzają istnienie tej współzależności. Stwierdzono
bowiem istotny wpływ zamiany (tranzycji - - rodzaj mutacji punktowej
opisany w rozdziale: Mutacje) adeniny na guaninę w 169 nukleotydzie
regionu D-loop (jest to szczególnie ważny region) mitochondrialnego DNA
na zawartość tłuszczu w mleku i wartość energetyczną mleka.
Wpływ matczynego mtDNA na cechy użytkowości mlecznej potomstwa
może być wykorzystany w doskonaleniu bydła. Potencjalne możliwości
stwarzają techniki klonowania oparte głównie na umieszczaniu w enuk-
leowanym (pozbawionym jądra) oocycie jądra komórki somatycznej. Można
zatem wykorzystywać do tego celu oocyty pochodzące od krów mających
szczególnie korzystne allele w mitochondrialnym DNA.
U owiec badania mitochondrialnego DNA, prowadzone za pomocą
technik molekularnych (RFLP), wykazały istotną współzależność między
polimorfizmem fragmentów restrykcyjnych mtDNA a masą jagniąt przy
urodzeniu. Organizacja molekularna mtDNA jest omówiona w rozdziale:
Mapy genomowe.

Rozdział
7.1. Rodzaje zmienności i czynniki ją wywołujące
Zmienność jest to różnorodność wartości lub jakości cech obserwowana
wśród osobników. Zmienność danej cechy może być obserwowana między
osobnikami i jest to zmienność osobnicza, czyli indywidualna. Istnieje także
zmienność grupowa, występująca między osobnikami należącymi do różnych
ras (zmienność rasowa) lub gatunków (zmienność gatunkowa). W przypadku
cech, które ujawniają się periodycznie u tego samego osobnika w kolejnych
sezonach (np. wydajność mleczna w kolejnych laktacjach), mamy do
czynienia ze zmiennością wewnątrzosobniczą.
Wszelkie różnice uzewnętrzniające się (widoczne lub dające się określić
bądź zmierzyć) między zwierzętami określamy mianem zmienności fenoty-
powej. Zmienność ta powstaje w wyniku różnic genetycznych między
zwierzętami (zmienność genetyczna) oraz oddziaływania zróżnicowanych
warunków środowiskowych (zmienność środowiskowa). Na zmienność
fenotypową, zwaną także ogólną, może również wpływać zmienność
wynikająca z wzajemnego współdziałania genotypów z warunkami środo-
wiskowymi, tzw. interakcja genotyp-środowisko. Zmienność oznaczamy
symbolem S
2
, jeśli rozpatrujemy populację zwierząt, lub S
2
w przypadku
próby losowej pochodzącej z tej populacji.
179

Podstawą genetycznego doskonalenia zwierząt jest zmienność genetycz-
na. Jej źródłem są przede wszystkim mutacje i rekombinacje genetyczne
oraz w mniejszym stopniu współdziałanie między genami. Mutacje prowa-
dzą do powstawania nowych genów, innych układów w obrębie chromo-
somu lub między chromosomami. Natomiast rekombinacje, będące wynikiem
segregacji chromosomów i crossing over w czasie mejozy oraz losowego
łączenia się gamet zróżnicowanych genetycznie, są przyczyną powstawania
różnych genotypów.
Głównymi składnikami zmienności genetycznej są: zmienność addytyw-
na, odchylenie dominacyjne i odchylenie epistatyczne.
Zmienność addytywna spowodowana jest niejednakowym sumującym
efektem działania alleli z loci poligenów, które kontrolują występowanie
danej cechy. Ten rodzaj zmienności zostanie omówiony szerzej w podroz-
dziale: Zmienność cech ilościowych.
Zmienność dominacyjna jest to część zmienności genetycznej, która jest
powodowana dominacyjnym współdziałaniem genów warunkujących cechę.
Efekt dominacji przejawia się nieaddytywnym zróżnicowaniem wartości
między osobnikiem heterozygotycznym, w którego genotypie funkcjonuje
jeden gen dominujący, i osobnikami homozygotycznymi — dominującym
i recesywnym. Poniższy schemat krzyżowania bydła obrazuje to odchylenie.
Osobniki fenotypowo identyczne (bezrogie), ale różniące się pod względem
założeń genetycznych (homo- i heterozygotyczne) dadzą potomstwo różniące
się nie tylko pod względem genotypu, ale także fenotypu (bezrogie i rogate).
Zmienność epistatyczna jest składową zmienności genetycznej powodo-
waną nieallelicznym współdziałaniem genów — epistazą. Przykładem
epistatycznego działania genów jednej pary alleli na geny innej pary jest
180

białe upierzenie kur rasy leghorn i whiterock. Barwa upierzenia u tych ras
jest warunkowana dwiema parami alleli: C-c (wystąpienie barwy) oraz I-i
(blokada wytwarzania me/aniny, upierzenie biafe). Z krzyżowania osobników
0 białym upierzeniu, należących do tych ras, uzyskuje się mieszańce biało
upierzone. W pokoleniu F
2
wystąpią natomiast osobniki zarówno biało, jak
1 barwnie upierzone.
W pracy hodowlanej szczególnie ważna jest znajomość udziału zmien-
ności genetycznej, a zwłaszcza jej składnika — zmienności addytywnej,
w zmienności fenotypowej danej cechy ilościowej. Do szacowania tego
udziału stosowane są parametry genetyczne, do których należy odziedziczal-
ność. Szczegółowe informacje dotyczące odziedziczalności i pozostałych
parametrów genetycznych oraz ich wykorzystania w doskonaleniu zwierząt
znajdują się w podręcznikach z zakresu metod hodowlanych.
Zmienność środowiskowa wynika z różnorodnych warunków środo-
wiskowych oddziałujących na zwierzęta stale (np. czynniki klimatyczne)
lub okresowo (żywienie, sposób utrzymania itp.). Do czynników śro-
dowiskowych, mających istotne znaczenie, należy także efekt matki
pre-i postnatalny. Wpływ środowiska matki zostanie omówiony w
dalszej części tego rozdziału.
Zmienność cechy może mieć charakter skokowy lub ciągły w zależności
od jej genetycznego uwarunkowania.
Zmienność skokowa odnosi się przede wszystkim do cech jakościowych,
warunkowanych zwykle jedną parą alleli. Udział pojedynczego genu
w wykształceniu cechy jakościowej jest duży, dlatego gen ten ujawnia się
mimo ewentualnego modyfikującego działania środowiska czy wpływu na
daną cechę innych genów zwanych modyfikatorami. Charakterystyka
zmienności cechy jakościowej polega na określeniu częstości występowania
(frekwencji) genów, genotypów i fenotypów w danej populacji (stadzie).
Zagadnienie to jest szerzej omówione w rozdziale: Podstawy genetyki
populacji.
Zmienność skokowa występuje również w przypadku pewnych cech
ilościowych, których wartość wyrażana jest za pomocą liczb naturalnych,
np. liczba prosiąt w miocie.
Zmiennością ciągłą charakteryzuje się większość cech ilościowych.
Nasilenie tych cech wyrażane jest za pomocą liczb rzeczywistych z przedziału
181

charakterystycznego dla danej cechy, np. wydajność mleka w kilogramach,
wysokość w kłębie w centymetrach, procentowa zawartość tłuszczu w mleku
itp. Charakterystyka zmienności tych cech przedstawiana jest za pomocą
miar zmienności opisanych poniżej.
7.2. Miary zmienności
Określanie zmienności fenotypowej cechy przeprowadzane jest na wybranej
losowo z populacji odpowiednio licznej grupie osobników, zwanej próbą.
Z punktu widzenia statystycznego populacją nazywamy zbiór zwierząt,
którego elementami są poszczególne osobniki.
W celu określenia zmienności cechy musimy ją oznaczyć (np. zmierzyć)
u wszystkich osobników stanowiących próbę. Uzyskany w ten sposób nie
uporządkowany materiał wymaga zestawienia (uporządkowania) np. według
wartości rosnących lub malejących (szereg statystyczny uporządkowany).
Jeśli liczba obserwacji jest duża, a niektóre wartości powtarzają się, dane
zestawia się w tzw. szereg rozdzielczy, w którym na podstawie przyjętych
przedziałów klasowych można wyodrębnić poszczególne klasy. Takie
uporządkowanie danych liczbowych (obserwacji) ułatwia statystyczną
i graficzną analizę zmienności cechy.
Szczegółową charakterystykę populacji można przeprowadzić po osza-
cowaniu odpowiednich parametrów statystycznych, które można podzielić
na dwie grupy:
1) miary skupienia
2) miary rozproszenia (dyspersji).
7.2.1. Miary skupienia
Do miar skupienia należą przeciętne klasyczne (średnia arytmetyczna,
ważona, harmoniczna i geometryczna) oraz przeciętne pozycyjne (wartość
środkowa — mediana i wartość modalna — dominanta).
Średnia arytmetyczna nie odzwierciedla zmienności, ale informuje
o poziomie cechy w danej populacji i obliczana jest ze wzoru:

Średnia arytmetyczna charakteryzuje się następującymi właściwościami:
Przykład (dane z tab. 7-1):
Średnią ważoną obliczamy wówczas, gdy danej wartości cechy od-
powiada kilka obserwacji (tworzy się wówczas przedziały wartości cechy)
lub gdy obliczamy średnią dla populacji na podstawie średnich dla prób.
Wzór na jej obliczanie jest następujący:
Średnia harmoniczna jest stosowana najczęściej do obliczania średnich
wartości otrzymywanych ze wskaźników czasu, np. do obliczania średniej
prędkości.
Wzór na obliczanie średniej harmonicznej jest następujący:
Przykład:
Ważono mleko oddawane przez krowę w kolejnych minutach doju.
W pierwszej minucie uzyskano 4 kg mleka, w drugiej 2 kg, a w trzeciej l kg.
Stosując wzór na obliczanie średniej harmonicznej uzyskujemy wynik
mówiący, jaka była średnia szybkość oddawania mleka:
183

Średnią geometryczną stosuje się wtedy, gdy rozkład zmienności cechy
jest asymetryczny, tzn. odbiega od rozkładu normalnego. Jest ona szczególnie
przydatna do obliczania średniego tempa przyrostu (lub zmniejszania się)
jakiegoś wskaźnika w jednostce czasu. Średnia geometryczna jest pierwiast-
kiem n stopnia z iloczynu n wartości cechy. W celu uproszczenia obliczeń
można się posłużyć formą logarytmiczną:

Przykład:
Określano masę ciała cieląt od urodzenia do 6 miesięcy w odstępach
miesięcznych, a następnie obliczono wskaźnik wzrostu dla kolejnych
miesięcy (wskaźnik wzrostu jest to przyrost masy ciała wyrażony w procen-
tach średniej masy ciała w danym okresie). Wartości zlogarytmowane tego
współczynnika wyniosły: 0,3010; 0,1239; 0,0864; 0,0697; 0,0414; 0,0294,
natomiast ich suma = 0,6518. Chcąc określić tempo wzrostu cieląt w okresie
6 miesięcy należy obliczyć średnią geometryczną:
Średni wskaźnik tempa wzrostu cieląt w okresie 6 miesięcy wyniósł 1,28,
czyli 128%.
Wartość środkowa (mediana) jest to wartość cechy, która dzieli szereg
uporządkowany malejąco lub rosnąco na połowę.
Wartość modalna (średnia modalna) jest to wartość cechy, która najczęś-
ciej powtarza się w szeregu liczbowym.
7.2.2. Miary rozproszenia
Podstawowe miary rozproszenia to wariancja, standardowe odchylenie
i współczynnik zmienności.
Wariancja jest to średni kwadrat odchyleń obserwacji (xi) od średniej
arytmetycznej
Biorąc pod uwagę, że najczęściej średnia arytmetyczna nie jest liczbą
całkowitą, w celu ułatwienia obliczeń stosuje się wzór roboczy, w którym
licznik jest sumą kwadratów odchyleń:
Jeśli próbę stanowi mała liczba obserwacji, w celu uniknięcia błędu
zalecane jest dzielenie nie przez N obserwacji, ale przez N—1.. Za mało
liczną próbę przyjmuje się poniżej 100 obserwacji. Wariancja nie może
mieć wartości ujemnej. Ma ona miano takie jak analizowana cecha,
ale podniesione do kwadratu.
185

Standardowe odchylenie jest pierwiastkiem kwadratowym z wariancji.
Jego wartość informuje o średnim odchyleniu in plus i in minus od średniej
arytmetycznej i jest liczbą mianowaną w jednostkach badanej cechy.
Charakteryzując zmienność cechy podaje się wartość średniej arytmetycznej
i standardowego odchylenia
Współczynnik zmienności obliczany jest ze wzoru:
gdzie:
Współczynnik zmienności wyraża zmienność w procentach, dzięki
czemu znajduje zastosowanie do porównywania zmienności różnych cech
(mierzonych w różnych jednostkach, np. wydajność mleczna [kg] i zawar-
tość tłuszczu w mleku [%]) w tej samej populacji, a także zmienności tej
samej cechy w różnych populacjach (np. wydajność mleczna w stadzie
krów rasy nizinnej czarno-białej i czerwonej polskiej). Współczynnik
zmienności może być również miarą precyzji w doświadczeniu prze-
prowadzanym na kilku grupach zwierząt. Dobór zwierząt do grup
doświadczalnych powinien być losowy, tak więc zmienność badanej cechy
we wszystkich grupach musi być zbliżona. Różnica wartości współczynnika
zmienności w granicach 5-10% oznacza możliwą do przyjęcia precyzję
doświadczenia.
Sposób obliczania miar rozproszenia przedstawiono niżej, wykorzystując
dane liczbowe zawarte w tabeli 7-1, dotyczące masy ciała owiec w wieku 10
miesięcy. Wariancja:

7.3. Zmienność cech ilościowych
7.3.1. Dziedziczenie cech ilościowych
Cechy o znaczeniu gospodarczym w zdecydowanej większości należą do
kategorii cech ilościowych. W przeciwieństwie do cech jakościowych
mechanizm ich dziedziczenia jest złożony i trudny do pełnego wyjaśnienia.
Ponadto zmienność tych cech zależy również od wpływu czynników
środowiskowych.
Cechy ilościowe warunkowane są wieloma genami z różnych loci
(poligeny). Poligeny, nazywane inaczej genami polimerycznymi, addytyw-
nymi lub kumulatywnymi, specyficznie ze sobą współdziałają. Przyjmuje
się, że efekty poszczególnych poligenów sumują się i w ten sposób
warunkują nasilenie cechy. Liczba poligenów kontrolujących cechy ilościowe
jest nieznana. Zakłada się, że każdy poligen ma dwa allele, pomiędzy
którymi zachodzi dziedziczenie pośrednie, przy czym jeden z nich ma
działanie pozytywne (tzn. zwiększające wartość cechy), a drugi neutralne,
czyli obojętne dla wartości cechy. Kolejnym założeniem jest to, że efekty
alleli pozytywnych z różnych loci poligenów są sobie równe i wreszcie
— efekty te sumują się przy kształtowaniu fenotypu. Genetyczne tło cech
ilościowych stwarza możliwość bardzo dużej liczby kombinacji układów
alleli kontrolujących daną cechę. Najczęściej występują genotypy warun-
kujące wartości fenotypowe cechy, które skupiają się wokół wartości średniej,
natomiast zmniejsza się częstość występowania form skrajnych.
Zagadnienie to obrazuje przykład, w którym hipotetycznie przyjęto,
że nieśność kur warunkowana jest trzema parami alleli z różnych
loci. Kury o genotypach „pozytywnych" AABBCC są zdolne do produkcji
240 jaj rocznie, natomiast o genotypach zawierających allele neutralne
aabbcc — do produkcji 120 jaj rocznie. Założono również, że efekty
genów A, B i C są identyczne (A = B = C) i każdy z tych genów
zwiększa nieśność kur o 20 jaj. Koguty o genotypach AABBCC, mające
założenia genetyczne warunkujące nieśność 240 jaj rocznie, skrzyżowano
z kurami o genotypach aabbcc, niosącymi 120 jaj rocznie.
AABBCC x aabbcc
240 120
Uzyskane w pokoleniu F
l
potomstwo było heterozygotyczne (AaBbCc),
czyli miało założenia genetyczne warunkujące nieśność 180 jaj rocznie.
U potomstwa w pokoleniu F
2
nastąpiło rozszczepienie, uzyskano 64
genotypy, warunkujące 7 poziomów nieśności (fenotypów tej cechy).
Szeregując w pionie genotypy warunkujące takie same wartości cechy
i łącząc linią krzywą genotypy znajdujące się najwyżej, uzyskujemy
przedstawiony niżej obraz graficzny:
187

Po zestawieniu wartości fenotypowych cechy, determinowanych przez okreś-
lone genotypy i obliczeniu średniej wartości cechy w pokoleniu F
2
uzyskujemy:
W podanym przykładzie celowo pominięto wpływ czynników środowis-
kowych. Czynniki te oraz ich wpływ na fenotypową ekspresję cech
ilościowych będą omówione nieco później. Znaczenie tych czynników jest
istotne, gdyż zacierają one różnice między fenotypami, co w przypadku
cech ilościowych o zmienności ciągłej powoduje, że graficznym obrazem tej
zmienności jest krzywa rozkładu normalnego, zwana krzywą Gaussa.
Rozkład normalny charakteryzuje się tym, iż:
• jest to rozkład, w którym najwięcej osobników wykazuje wartość
cechy zbliżoną do średniej, a najmniej skrajne wartości, poniżej i powyżej
średniej,
• rozkład ten jest symetryczny, a oś symetrii przechodzi przez punkt,
w którym jest średnia wartość cechy,
188

• mediana i wartość modalna (opisane wcześniej) znajdują się w tym
samym punkcie, co wartość średniej arytmetycznej,
• w przedziale x±S znajduje się ok. 68,2% wszystkich obserwacji,
• w przedziale x ±2 S znajduje się ok. 95,4% wszystkich obserwacji,
• w przedziale x±3 S znajduje się ok. 99,6% wszystkich obserwacji cechy.
Jak już wspomniano, na fenotypowe ujawnienie się każdej cechy
ilościowej wyraźny wpływ wywierają czynniki środowiskowe. Mogą one
maskować oddziaływanie poligenów. Spośród czynników środowiskowych
na szczególną uwagę zasługuje efekt matki (ang. maternal effect). Mimo iż
potomstwo otrzymuje od ojca i od matki taką samą liczbę chromosomów,
wpływ matki na potomstwo jest większy od wpływu ojcowskiego. Wpływ
ten wynika z trzech źródeł:
• pierwszym jest podłoże genetyczne — dodatkowe założenia dziedzicz
ne przekazywane przez matkę poza chromosomami (tzw. dziedziczenie
pozajądrowe, omówione w rozdziale: Podstawowe mechanizmy dziedzicze
nia cech),
• dwa następne źródła wynikają z oddziaływania środowiska matki
w okresie życia płodowego (efekt prenatalny) i po urodzeniu (efekt
postnatalny).
Efekt prenatalny został wykryty przez wykorzystanie transferu zarodków
zwierzęcych i analizę ich rozwoju. Na ten efekt składa się przede wszystkim
wpływ: wielkości macicy, ilości substancji odżywczych dostarczanych płodowi
(lub płodom), wielkości matki oraz jej stanu fizjologicznego i zdrowotnego.
Wielkość prenatalnego efektu matki zależy również od założeń genetycz-
nych przekazywanych potomstwu zarówno w jądrowym, jak i mitochon-
drialnym DNA. Niedawno u bydła mlecznego wykazano istotny wpływ
genów mitochondriałnych na wydajność mleka, procent tłuszczu w mleku
oraz wartość energetyczną mleka.
Efekt postnatalny występuje przede wszystkim u ssaków i został
określony za pomocą eksperymentów krzyżowego podsadzania noworod-
ków innym matkom (np. z innych ras). Efekt ten wynika bowiem z mlecz-
ności samicy i jej zachowania opiekuńczego (tzw. behawior matczyny).
U trzody chlewnej, na przykład, stwierdzono, że wpływ efektu matki jest
najistotniejszy dla wzrostu prosiąt do 21 dnia życia, a więc wówczas gdy
prosięta odżywiają się jedynie mlekiem matki.
Wielkość efektu matki prę- i postnatalnego zależy od gatunku, rasy,
a także wielu czynników środowiskowych.
7.3.2. Zmienność transgresywna
Zwierzęta gospodarskie, nawet ras o ustalonym genotypie, są hetero-
zygotyczne pod względem znacznej liczby loci. W wyniku krzyżowania
takich osobników potomstwo może mieć założenia genetyczne warunkujące
189

wartości cechy takie, jakie obserwowano u rodziców, bądź wartości wyższe
lub niższe niż u rodziców, czyli wykazujące większą zmienność cechy.
Zjawisko to nosi nazwę transgresji. Ilustruje je przykład:
Załóżmy, że wydajność mleczna krów jest kontrolowana przez geny
z 3 loci — D, E i F. Efekty genów D, E i F są identyczne (D = E = F), a
każdy z nich zwiększa produkcję mleka o 400 kg. Genotyp ddeeff
warunkuje wydajność 2400 kg mleka, DDEEFF — 4800 kg, natomiast
heterozygotyczny DdEeFf— 3600 kg mleka.
W potomstwie rodziców heterozygotycznych pod względem genów
kontrolujących produkcję mleka mogą wystąpić różne genotypy (typowe
rozszczepienie przy krzyżowaniu heterozygot), warunkujące różną wydaj-
ność mleczną. Przeważająca część potomstwa będzie się charakteryzować
średnią wartością cechy, równą wartości cechy u rodziców. Pojawią się
jednak także inne wartości cechy — wyższe niż u rodziców (np. genotyp
DDEEFF, warunkujący produkcję 4800 kg mleka) lub niższe (ddeeff 2400 kg
mleka).
Zjawisko transgresji jest dość często obserwowane w hodowli zwierząt
gospodarskich i jest źródłem zmienności transgresywnej, wykorzystywanej
w pracy hodowlanej.
7.3.3. Geny o dużym efekcie
W podanych wyżej przykładach determinacji cech ilościowych zakładano
udział kilku lub kilkunastu par alleli z różnych loci o małym efekcie
addytywnym (dziedziczenie poligeniczne). Cechy takie charakteryzują się
rozkładem normalnym. W przypadku niektórych cech zaobserwowano
zmienność, której graficzny obraz odbiega od krzywej Gaussa i może być
dwumodalny lub przesunięty, spłaszczony albo nadmiernie uwypuklony.
Jest to wynikiem dziedziczenia, w którym obok poligenów działa gen
o dużym efekcie, zwany inaczej genem głównym.
Gen o dużym efekcie jest identyfikowany wówczas, gdy u przeciwstaw-
nych homozygot różnice wartości fenotypowej cech wynoszą co najmniej
jedno standardowe odchylenie. Identyfikacja tych genów poprzez statystycz-
ną analizę rozkładu zmienności cechy w populacji jest trudna. Niektóre geny
o dużym efekcie zostały wykryte przypadkowo. Od niedawna dla genów
o dużym efekcie fenotypowym w zakresie cech produkcyjnych używa się
określenia locus cechy ilościowej — QTL (ang. quantitative trait locus).
Jednym z genów mających duży wpływ na różne cechy produkcyjne
zwierząt gospodarskich jest gen hormonu wzrostu GH (ang. growth
hormone). U bydła locus GH znajduje się w chromosomie l (w regionie
1q23-q25). Wykazano istotny wpływ polimorfizmu w tym locus na wydajność
krów ras mlecznych. W badaniach prowadzonych na simentalerach stwier-
dzono, iż zwierzęta homozygotyczne pod względem allelu B hormonu
190

wzrostu charakteryzowały się o 382 + 18,5 kg większą wydajnością mleka
w porównaniu z osobnikami homozygotycznymi AA. U bydła mięsnego gen
hormonu wzrostu wpływa na cechy użytkowości mięsnej. Wspólnie z genem
czynnika wzrostu insulinopodobnego l (IGF-1) odgrywa on istotną rolę
w kształtowaniu tempa wzrostu i składu tuszy. Hormon wzrostu reguluje
rozwój mięśni, a IGF-1 wspomaga jego działanie. Badania prowadzone na
mieszańcach piedmontese x chianina wskazują na istotną zależność między
polimorfizmem tego genu a długością i obwodem klatki piersiowej. Stwier-
dzono także, iż gen hormonu wzrostu oddziałuje na proces starzenia się
i reprodukcję oraz na odpowiedź immunologiczną organizmu.
Loews GH u świni znajduje się w chromosomie 12pl4 i wykazuje dużą
homologię do genu GH u człowieka, bydła i szczura. Wykazano wpływ
hormonu wrostu na zwiększenie masy mięśni, z równoczesnym zmniej-
szeniem otłuszczenia, a także na poprawę wykorzystania paszy i przyrosty
dzienne masy ciała. Opisano dotychczas około 20 alleli hormonu wzrostu
u tego gatunku. Dalsze badania zmierzają do ustalenia, które allele mają
związek z użytkowością mięsną.
Wpływ polimorfizmu genu hormonu wzrostu na cechy użytkowości
mięsnej i tempo wzrostu jest przedmiotem badań prowadzonych także na
drobiu. Dotychczas wykazano, iż polimorficzne warianty genu hormonu
wzrostu (u kur znanych jest już 16 alleli) są odpowiedzialne za niektóre
zaburzenia rozwojowe, jak karłowatość i akromegalia.
Pozostałe ważniejsze geny o dużym efekcie zostaną przedstawione
z podziałem na gatunki zwierząt, a najnowsze osiągnięcia są omówione
w rozdziale 12.3.5.
Świnie
U trzody chlewnej zidentyfikowano kilka genów o dużym efekcie.
Gen receptora rianodyny (KYR1). Autosomalny recesywny allel (gorączki
złośliwej) genu RYR1 jest najlepiej poznanym genem o dużym efekcie
u trzody chlewnej. Badania cytogenetyczne wykazały, iż jest on zlokalizo-
wany w chromosomie 6 (lokalizacja ta przedstawiona jest w rozdziale:
Mapy genomowe). W wyniku działania tego genu zwiększa się podatność
świń na stresy (zespół PSS — ang. porcine stress syndrome), powodowane
transportem, warunkami termicznymi i niektórymi czynnikami chemicznymi.
Następstwem miopatii stresowych są niekorzystne zmiany fizykochemiczne
w organizmie, prowadzące do pogorszenia jakości mięsa — odbarwienie
i nieprzyjemny zapach. Przypadek takich zmian po raz pierwszy został
opisany w 1957 roku przez Ludwigsena. Jednocześnie gen ten ma dodatni
wpływ na niektóre cechy związane z użytkowością mięsną, jak zawartość
mięsa w tuszy i powierzchnia oka polędwicy. Paramety tych cech są
najkorzystniejsze u zwierząt heterozygotycznych, będących nosicielami
zmutowanego allelu. Zwierzęta takie są więc dobrym materiałem do tuczu.
191
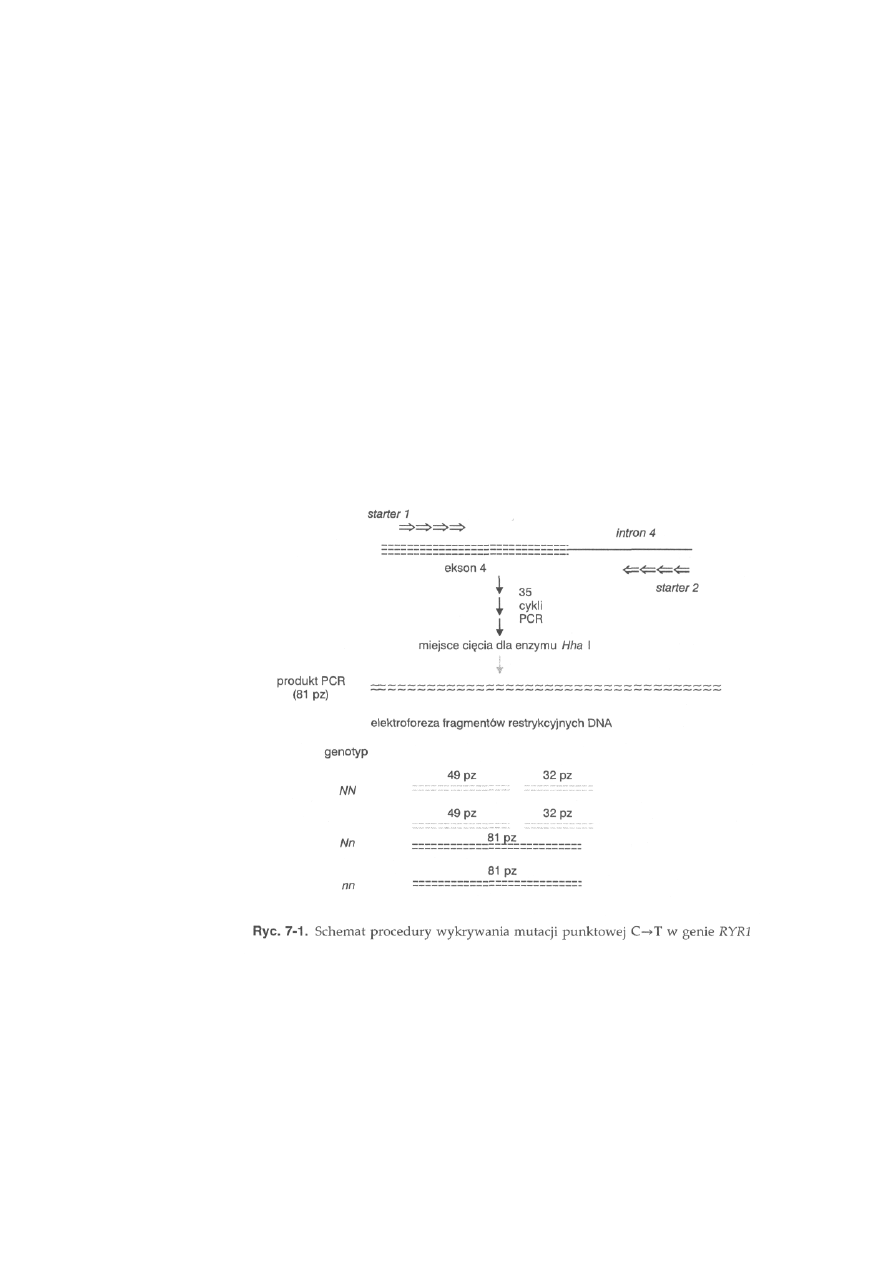
Ponieważ gen gorączki (hipertermii) złośliwej powoduje specyficzną
wrażliwość na gaz używany w anestezji — halotan, początkowo oznaczono
go symbolem Hal
n
. U osobników Hal
n
Hal
n
(homozygoty recesywne) wy-
stępuje nadmierny transport jonów Ca
++
z retikulum endoplazmatycznego
do cytoplazmy komórek. Badania przeprowadzone przez naukowców
kanadyjskich wykazały, że fenotypowo rozpoznany Hal
n
jest mutacją
genu receptora rianodyny (RYR1), jednej z podjednostek kanału wa-
pniowego w mięśniach szkieletowych. Dlatego obecnie gen hipertermii
złośliwej jest oznaczany symbolem RYR1. W obrębie genu RYR1 stwierdzono
18 mutacji punktowych, z których najistotniejsza jest mutacja w 1843
nukleotydzie, powodująca zamianę argininy w cysteinę w pozycji 615
łańcucha polipeptydowego. Mutacja ta likwiduje miejsce trawienia nie-
którymi enzymami, co umożliwiło opracowanie molekularnych testów
diagnostycznych. W jednym z nich stosowane są dwa enzymy restrykcyjne
(HgiAI oraz HinPI). W innym, opatentowanym przez badaczy kanadyjskich,
fragment genu receptora rianodyny zawierający mutację trawiony jest
enzymem Hhal. Procedurę tego testu przedstawiono na rycinie 7-1. Zam-
plifikowany fragment DNA długości 81 par zasad obejmuje rejon wystąpienia
mutacji punktowej C
→T. W normalnym genie enzym HhaI rozpoznaje
sekwencję GCGG i tnie DNA na dwa odcinki, natomiast w zmutowanym
192

allelu zmiana tej sekwencji powoduje, że enzym nie znajduje miejsca cięcia,
czego wynikiem jest uzyskanie nie strawionego fragmentu genu. Obraz
elektroforetycznego rozdziału zamplifikowanego odcinka genu dla osobnika
homozygotycznego (NN) wykaże dwa prążki długości 49 pz i 32 pz, dla
homozygotycznego (nn) - - jeden prążek długości 81 pz, natomiast dla
heterozygoty trzy prążki: 81 pz, 49 pz, 32 pz.
Diagnostyka wrażliwości świń na stres za pomocą testu molekularnego
umożliwia eliminowanie z hodowli osobników podatnych. W pracy hodow-
lanej należy prowadzić kojarzenia tak, by wykorzystać wspomniane już
wcześniej zalety genotypu heterozygotycznego.
Gorączka złośliwa występuje także u innych gatunków zwierząt oraz
u ludzi. Gen hipertermii u ludzi jest zlokalizowany w chromosomie 19,
u bydła w 18, a u koni w 10 chromosomie.
Gen „kwaśnego mięsa" — gen podjednostki
γ
kinazy białkowej ak-
tywowanej przez AMP (PRKAG3). Pierwsze sugestie co do istnienia genu
o dużym efekcie, wpływającego na wzrost zawartości glikogenu w tkance
mięśniowej u świń rasy hampshire pojawiły się w 1986 roku. Loews
odpowiedzialny za cechę kwaśnego mięsa zlokalizowano w chromosomie
15 na podstawie analizy asocjacji przeprowadzonej dla rodzin referencyjnych
z wykorzystaniem markerów genetycznych i oznaczono symbolem RN.
Dalsze badania z zastosowaniem precyzyjnych metod analizy genomu
zostały uwieńczone w 2000 roku wykryciem mutacji w genie PRKAG3 (gen
trzeciej izoformy podjednostki y kinazy białkowej aktywowanej przez
AMP). W genie tym stwierdzono siedem mutacji, ale tylko tranzycja G-»A
w kodonie 200, powodująca zamianę argininy na glicynę w łańcuchu
polipeptydowym, warunkuje cechę kwaśnego mięsa. U nosicieli zmutowa-
nego allelu stwierdza się dwukrotnie większą zawartość glikogenu w mięś-
niach (zwłaszcza w szynce). Test molekularny wykrywający mutację genu
PRKAG3 został opatentowany.
Gen wpływający na wielkość miotu (ESR). Niektóre chińskie rasy świń
charakteryzują się niezwykłą plennością. Badania prowadzone w USA miały
na celu wykrycie genetycznego podłoża tej cechy. Wyniki tych badań
wskazują, iż zlokalizowany w chromosomie l (1p2.5-2.4) locus genu receptora
steroidowego hormonu płciowego estrogenu — ESR (ang. estrogen receptor
gene) może mieć istotny wpływ na wielkość miotu. Stwierdzono, że jeden
z alleli tego genu, występujący w chińskiej plennej rasie meishan, wykazuje
związek z wielkością miotu i może być uważany za gen główny kontrolujący
tę cechę. Lochy mające w swym genotypie dwie kopie tego genu dają mioty
większe o l do 1,4 prosięcia żywo urodzonego w porównaniu z lochami nie
mającymi tego genu. Badania molekularne (z zastosowaniem enzymów
restrykcyjnych) genu receptora estrogenu, prowadzone u świń innych ras
(niemiecka landrace, yorkshire, duroc), pozwoliły na wykrycie dwóch
mutacji punktowych (zamiana A
→T w kodonie 1665 i A→G w kodonie
193

1754). Gen ten nie wykazuje plejotropowego negatywnego działania na
inne, ekonomicznie ważne cechy, jak wzrost i grubość słoniny na grzbiecie.
Polimorfizm tego genu może mieć szczególne znaczenie w pracach zmie-
rzających do utworzenia syntetycznych linii świń, wyprowadzanych z udzia-
łem rasy meishan.
Bydło
Geny białek mleka. Do genów głównych o szczególnym znaczeniu dla
wyników ekonomicznych hodowli bydła należą geny białek mleka:
κ-kazeiny
i
β-laktoglobuliny.
Gen
κ-kazeiny, zlokalizowany w chromosomie 6, ma długość około 13
tysięcy par zasad i zawiera 5 eksonów. W najdłuższym eksonie 4 znajduje
się większość sekwencji kodujących dojrzałe białko
κ-kazeiny. Wykazano
związek między genami kazein a cechami użytkowości mlecznej (wydajność
mleka, białka i tłuszczu). Mleko krów z genem
κ-kazeiny typu B ma większą
zawartość białka oraz lepszą przydatność do produkcji serów niż mleko
z wariantem
κ-kazeiny A.
Allele kontrolujące syntezę poszczególnych wariantów tego białka
powstały w wyniku mutacji punktowej w genie
κ-kazeiny wariantu A.
W następstwie takiej mutacji w pozycji 136 łańcucha polipeptydowego
κ-
kazeiny wariantu A znajduje się aminokwas izoleucyna (kodon ATC),
podczas gdy w wariancie B — treonina (w DNA kodon ACC). Natomiast
w pozycji 148 wariant B
κ-kazeiny ma kwas asparaginowy (kodon GAT),
wariant A alaninę (GCT). Te dwa warianty
κ-kazeiny można rozróżnić za
pomocą analizy polimorfizmu długości fragmentów restrykcyjnych. Pod-
stawienia zasad w nukleotydach powodują powstanie miejsc cięcia dla
enzymu HindIII, natomiast zniknięcie miejsca trawienia przez enzym Hm/L
Dzięki różnicy w składzie nukleotydowym genu możliwe jest określanie
genotypu kontrolującego wariant
κ-kazeiny w mleku za pomocą testu
PCR-RFLP (ryć. 7-2). Opracowana została metoda identyfikacji genotypów
κ-kazeiny, wykorzystywanych jako dodatkowe kryterium selekcji buhajów,
utrzymywanych w zakładach unasieniania. Dotychczas genotyp ojca można
było określić na podstawie segregacji polimorficznych wariantów
κ-kazeiny,
identyfikowanych w mleku córek, przy czym genotypy homozygotyczne
mogły być weryfikowane jako statystycznie bardziej lub mniej praw-
dopodobne.
Gen
β-laktoglobuliny uważany jest za gen o dużym efekcie, gdyż
określony wariant tego białka, podobnie jak
κ-kazeiny, wpływa na wydajność
mleka i jego składników. Mleko zawierające
β-laktoglobulinę B różni się od
mleka z wariantem A większą zawartością suchej masy, tłuszczu i kazein
oraz lepszą przydatnością technologiczną. Gen
β-laktoglobuliny ma długość
około 7800 par zasad, zawiera 7 eksonów. Białko wariantu A różni się od
B dwoma aminokwasami. Jest to konsekwencja różnic w sekwencji nukleo-
194
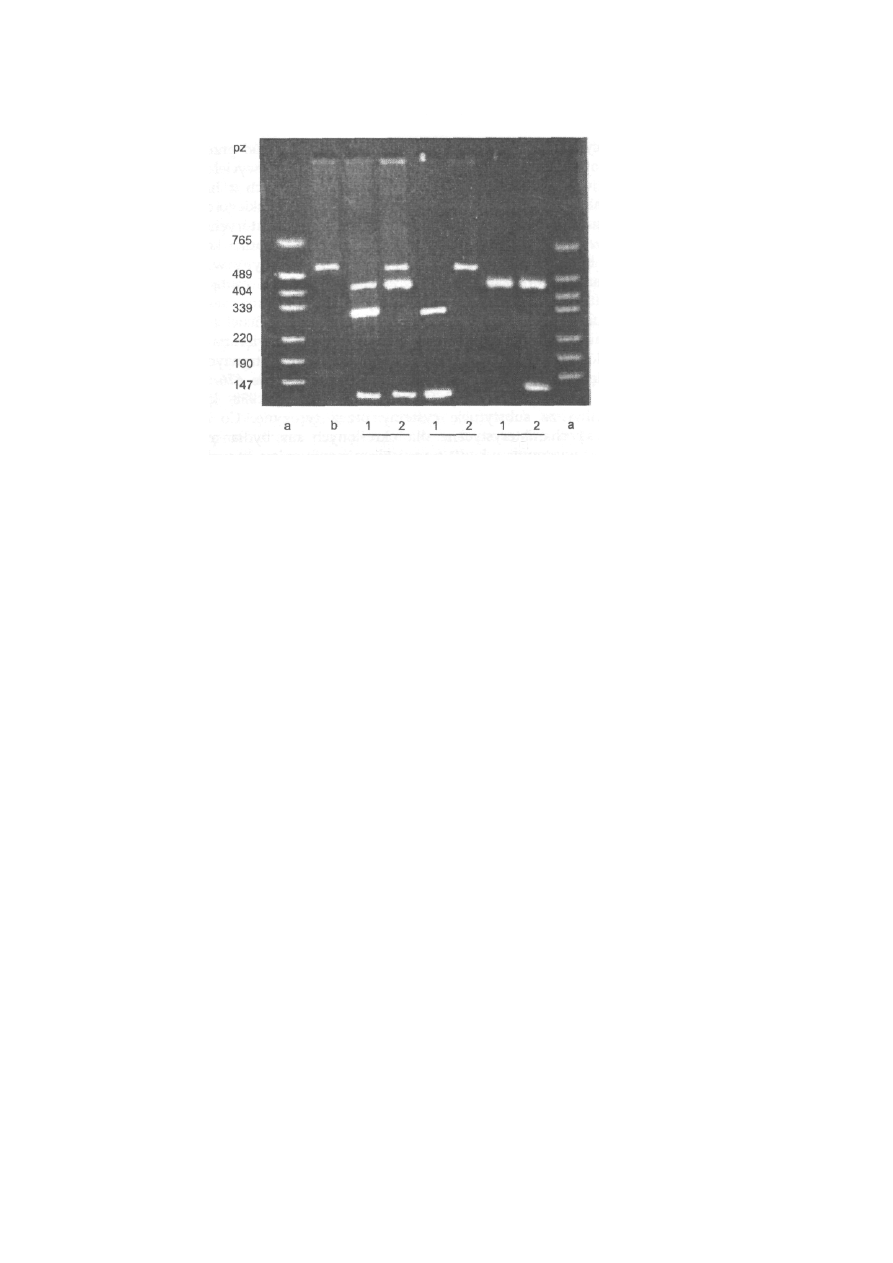
AB AA BB
Ryć. 7-2. Identyfikacja genotypu
κ-kazeiny (wariant A i B) metodą PCR-RFLP.
a — marker, b — produkt nie strawiony, l — trawienie enzymem restrykcyjnym Hinfi,
2 — trawienie enzymem restrykcyjnym Hindlll
tydowej obu alleli. Stosowane są różne warianty molekularnej identyfikacji
genotypów
β-laktoglobuliny z zastosowaniem metod PCR-RFLP i SSCP.
Wyniki oznaczeń są wykorzystywane jako dodatkowe kryterium selekcyjne
w doskonaleniu cech użytkowości mlecznej.
Gen hipertrofii mięśniowej (MH — ang. muscular hypertrophy).
Hipertrofia mięśniowa występująca u niektórych ras bydła mięsnego
(belgijskiego błękitnego i asturiana) determinowana jest zmutowanym
allelem genu miostatyny (locus miostatyny zmapowano w chromosomie 2,
jego lokalizacja jest przedstawiona w rozdziale: Mapy genomowe). Gen
miostatyny należy do rodziny TGF
β (ang. transforming growth factor β).
Zmutowany allel różni się od normalnego delecją 11 nukleotydów między
821 a 831 nukleotydem, dlatego mutacja ta jest oznaczona jako
nt821(del11). Bezpośrednim następstwem ekspresji tego allelu jest
zwiększona zawartość chudego mięsa w tuszy oraz jego lepsze właściwości
dietetyczne, spowodowane zmniejszoną ilością tłuszczu i tkanki łącznej.
Dlatego cecha ta jest preferowana przez hodowców zarówno w hodowli
czystych ras, jak i ich mieszańców. Obecność zmutowanego allelu w
genotypie ma jednak także niekorzystne następstwa, należą do nich:
opóźnienie okresu dojrzałości płciowej u osobników obu płci,
zmniejszona zdolność rozpłodowa — u buhajów mniejsze stężenie
hormonów płciowych w osoczu krwi, mniejszy obwód i masa jąder,
natomiast u krów dłuższa ciąża. Duża masa płodu stwarza wyraźne
trudności naturalnego porodu. Rozwiązaniem
195

ograniczającym śmiertelność okołoporodową cieląt jest poród przez cesars-
kie cięcie. Powoduje ono jednak wydłużenie okresu międzywycieleniowego
o około 20 dni. Pomimo ujemnych następstw związanych z hipertrofią
mięśni, cecha ta ze względów ekonomicznych (koszt cesarskiego cięcia jest
równy tylko 1/10 wartości mięsa cielęcia) jest w niektórych krajach
utrwalana w hodowli bydła typu mięsnego (ryć. 7-3 wkładka kolorowa).
Najnowsze badania struktury molekularnej genu miostatyny w różnych
rasach mięsnych wykazały obecność kolejnych czterech mutacji, które
uszkadzają funkcję kodowanego białka. Trzy z nich powodują przedwczes-
ne pojawienie się kodonu „stop" i w efekcie skrócenie łańcucha polipep-
tydowego miostatyny. Są to: a) insercja 10 nukleotydów powiązana z delecją
7 nukleotydów, począwszy od pozycji 419 nukleotydu, b) tranzycja C
→T
w 610 nukleotydzie, c) transwersja G
→ T w nukleotydzie 676. Czwarta
mutacja d) polega na tranzycji G
→A w nukleotydzie 938, która jest
odpowiedzialna za substytucję cysteiny przez tyrozynę. Co ciekawe,
mutacje te są charakterystyczne dla określonych ras bydła mięsnego.
Mutacje a) i c) występują u bydła francuskiego maine-anjou, b) u charolaise
(Francja) i d) u gasconne (Francja) i piedmontese (Włochy).
Owce
Gen hipertrofii mięśniowej (CLPG). U owiec występuje mutacja powodu-
jąca hipertrofię mięśniową, która jest związana ze wzrostem masy mięśni
o około 32% i zmniejszeniem zawartości tłuszczu o około 8% oraz lepszym
wykorzystaniem paszy. Analiza molekularna DNA przeprowadzona u rodzin
referencyjnych z wykorzystaniem 21 markerów genetycznych bydła umoż-
liwiła zmapowanie genu hipertrofii mięśniowej w 18 chromosomie. Jest to
pojedynczy autosomalny gen, nazwany callipyge (z greckiego calli — piękne,
pyge -- pośladki) -- symbol CLPG. Prowadzone są badania w celu jego
izolacji. Przypuszcza się, że istnieją dwa allele — CLPG i clpg, przy czym
zwierzęta o genotypie CLPG/clpg cechuje hipertrofia, natomiast osobniki
z genotypem clpg/clpg i CLPG/CLPG mają normalny fenotyp. Jak wykazały
ostatnie badania, w przypadku genotypu heterozygotycznego wystąpienie
hipertrofii zależy od tego, czy gen CLPG pochodzi od ojca, czy od matki.
Gen CLPG pochodzący od matki jest nieaktywny. Hipertrofia ujawnia się
u potomka wówczas, gdy dziedziczy on gen CLPG od ojca (piętno game-
tyczne). Ostatnio ustalono, że podłoże molekularne tej cechy związane jest
z mutacją w niekodującym regionie DNA. Jest to substytucja nukleotydowa
A
→G w regionie niekodującym żadnego białka. Przypuszcza się, że locus
ten podlega transkrypcji, a jego produkt to niekodujący RNA (ncRNA -
ang. noncoding RNA), który pełni nieznaną jeszcze funkcję regulacyjną.
Gen wysokiej plenności (BMPR-IB) u owiec rasy merynos
booroola. U owiec wykryto geny główne, warunkujące zwiększony stopień
owulacji, takie jak gen Inverdale (FecX
I
, zlokalizowany w chromosomie
płci X)
196

u owiec rasy romney, „Thoka" u owiec islandskich, gen wysokiej plenności
u owiec rasy olkuskiej w Polsce czy u syntetycznej rasy owiec Cambridge.
Jednak najbardziej znany jest gen wysokiej plenności zidentyfikowany na
początku lat 80. ubiegłego wieku u owiec rasy merynos booroola w Australii
i określony wówczas symbolem FecB (ang. fecundity gene of booroola).
W przeciwieństwie do owiec ras wysokopiennych (owca romanowska, owca
fińska), u których zwiększona plenność jest cechą poligeniczną, uwarun-
kowaną genami o addytywnym efekcie, wysoki stopień owulacji u maciorek
rasy merynos booroola jest wynikiem wpływu genu FecB na rozwój
pęcherzyków Graafa i ciałek żółtych. Ponadto maciorki mające ten gen
w swym genotypie wydzielają mniej hormonów inhibitorów działających
na hormony gonadotropowe.
Ponieważ obecność nawet jednej jego kopii w genotypie maciorki
powoduje znaczny wzrost stopnia owulacji (w konsekwencji zwiększa jej
plenność o około l jagnię w miocie), w niektórych krajach czynione są
próby wprowadzenia tego genu do ras miejscowych i wykorzystania go
w programach intensyfikacji produkcji żywca jagnięcego.
Prowadzone przez badaczy nowozelandzkich badania segregacji cechy
dużej plenności w rodzinach referencyjnych, połączone z badaniami
polimorfizmu w loci markerowych (markery klasy I i II), wskazały lokalizację
tego genu w chromosomie 6 (6q23-q21). Dalsze wieloletnie badania z za-
stosowaniem metod genetyki molekularnej zaowocowały wykryciem w 2001
roku mutacji w genie receptora białka oznaczonego symbolem BMP-IB,
odpowiedzialnej za dużą plenność owiec rasy merynos booroola. W locus
BMPR-IB wykryto mutację w 830 nukleotydzie, polegającą na tranzycji
adeniny na guaninę, której konsekwencją jest zamiana glutaminy na argininę
w pozycji 249 łańcucha polipeptydowego. Opracowany test molekularny
(PCR-RFLP) identyfikacji genotypu w locus BMPR-IB z zastosowaniem
restryktazy AvaII wykorzystuje to, że mutacja powoduje powstanie sekwencji
rozpoznawanej przez ten enzym. Obraz rozdziału elektroforetycznego
zamplifikowanego i strawionego enzymem fragmentu genu BMPR-IB,
zawierającego miejsce mutacyjne, będzie następujący: 2 prążki (110 pz
i 30 pz) dla osobników homozygotycznych pod względem allelu zmutowa-
nego, trzy prążki (110 pz, 30 pz i 140 pz) dla heterozygotycznego i jeden
prążek (140 pz; produkt PCR nie strawiony) dla homozygoty typu dzikiego.

Rozdział
Podstawy genetyki populacji
Przedstawione w poprzednich rozdziałach genetyczne uwarunkowania
zmienności cech i mechanizmy ich dziedziczenia, mimo iż dotyczyły nieraz
grupy zwierząt, w istocie odnoszone były do genotypu czy zwierzęcia jako
jednostki. Wiele zjawisk i procesów nie dotyczy jednak pojedynczych
osobników, lecz zbiorowości określonej jako populacja. Hodowla jest
działalnością genetyczno-populacyjną. Populację stanowią zwierzęta należące
do jednego gatunku, mogące się rozmnażać i występować na danym
obszarze. Suma wszystkich genów występujących w genotypach zwierząt
tworzących populację nazywa się pulą genową.
Szczegółowe omówienie wszystkich zagadnień z zakresu genetyki
populacji zainteresowani znajdą w innych podręcznikach. W rozdziale
tym przedstawiono jedynie zagadnienie struktury genetycznej populacji
oraz wykorzystanie frekwencji alleli w szacowaniu zmienności gene-
tycznej.
Podstawowym prawem genetyki populacji, opisującym częstość wystę-
powania (frekwencję) poszczególnych alleli i genotypów, jest prawo
równowagi genetycznej sformułowane w 1908 roku przez angielskiego
matematyka G.H. Hardy'ego i niemieckego lekarza W. Weinberga. Ponie-
waż badacze ci działali niezależnie od siebie, prawo to nosi nazwę
Hardy' ego-Weinberga.
8.1. Prawo równowagi genetycznej
Struktura genetyczna populacji
Populację tworzy określona, teoretycznie nieskończona liczba osobników.
Rozpatrując daną cechę można powiedzieć, że każdy osobnik ma określony
fenotyp, tak więc w populacji występuje określona liczba fenotypów,
198

z których każdy reprezentowany jest przez pewną liczbę osobników.
Częstość występowania, inaczej frekwencja danego fenotypu, oznacza
stosunek tego fenotypu do całkowitej liczby fenotypów. Frekwencję wyra-
żamy w procentach lub w postaci ułamka dziesiętnego. Suma frekwencji
wszystkich fenotypów równa się l lub 100%. Z kolei stosunek liczby
osobników o danym genotypie do ogólnej liczby osobników występujących
w populacji określamy mianem frekwencji genotypów. Natomiast frekwencja
genów jest to udział liczby loci zajętych przez dany allel względem ogólnej
liczby loci, które ten allel mógłby zająć w badanej populacji. Frekwencja
genotypów i frekwencja genów są elementami składowymi ogólniejszego
pojęcia — struktury genetycznej populacji. Oba te elementy są od siebie
zależne. Frekwencja genów w danym pokoleniu zależy od frekwencji
poszczególnych genotypów, natomiast frekwencja genotypów w pokoleniu
następnym zależy od frekwencji genów w gametach pokolenia poprzedniego
(rodzicielskiego).
Obliczenie frekwencji genów na podstawie fenotypów jest najprostsze
w przypadku cech dziedziczących się pośrednio (z niezupełną dominacją).
Fenotyp osobnika jest bowiem odzwierciedleniem jego genotypu. Sposób
postępowania obrazuje poniższy przykład.
W stadzie kur andaluzyjskich (barwa upierzenia dziedzicząca się z nie-
zupełną dominacją) liczącym 1000 ptaków jest 250 ptaków czarnych, 250
białych i 500 o upierzeniu niebieskim (andaluzyjskim). Frekwencja po-
szczególnych fenotypów i jednocześnie genotypów jest następująca:
Łączna liczba alleli warunkujących omawianą cechę wynosi 2000 (1000
ptaków, każdy ma dwa allele). 250 ptaków czarnych ma w swych genotypach
500 genów A, ptaki niebieskie mają 500 genów A i 500 genów a. W genoty-
pach 250 ptaków białych jest 500 alleli a. Zatem w stadzie tym jest 1000 alleli
A i 1000 a.
Częstość występowania (frekwencja) allelu A = 1000/2000 = 0,5 (50%),
podobnie frekwencja allelu a = 1000/2000 = 0,5 (50%).
Suma frekwencji obu alleli wynosi l (100%). Jeśli frekwencję allelu
dominującego oznaczymy symbolem p, a allelu recesywnego symbolem q, to:
199

Rozpatrywana cecha dziedziczy się z niezupełną dominacją, zatem rozkład
fenotypów i genotypów w tej populacji wynosi:
Suma frekwencji genotypów jest rozwinięciem kwadratu dwumianu (p + q)
2
i wynosi l (100%). Jeśli w populacji będą kojarzenia losowe oraz będzie
jednakowa frekwencja genów u obu płci, wówczas struktura genetyczna
w pokoleniu potomnym będzie następująca:
czyli:
Zatem struktura genetyczna populacji w pokoleniu potomnym jest taka
sama jak w pokoleniu rodzicielskim. Prawidłowość ta zawarta jest w prawie
Hardy 'ego-Weinberga, które mówi o tym, że: w dużej, losowo
kojarzącej się populacji, w której frekwencje genów u obu płci są jednakowe,
a osobniki charakteryzują się równą płodnością i żywotnością,
frekwencje genów i genotypów nie zmieniają się z pokolenia na pokolenie,
jeśli nie działają czynniki naruszające równowagę.
Prawo to rzadko jest spełnione w odniesieniu do wszystkich loci,
bowiem nawet w naturalnych populacjach następuje dobór naturalny
i selekcja naturalna, a' zdarzają się również migracje i mutacje, które
wpływają na zmianę struktury genetycznej populacji. O czynnikach naru-
szających równowagę genetyczną będzie mowa nieco dalej.
200

8.2. Frekwencja genów i genotypów w
przypadku dominowania
W przypadku cechy dziedziczącej się z dominacją zupełną fenotyp domi-
nujący występuje u osobników będących homozygotą dominującą lub
heterozygotą pod względem danego locus. Zatem obliczanie frekwencji
zarówno tych genotypów, jak i genów stwarza pewne trudności. W populacji
rozmnażanej losowo źródłem informacji do obliczeń frekwencji genów jest
częstość genotypów homozygotycznych recesywnych, gdyż w ich przypadku
wiadomo na pewno, że zawierają jedynie allele recesywne. Wiedząc, że
frekwencja homozygot recesywnych wynosi q
2
, łatwo jest wyliczyć frekwen-
cję allelu recesywnego
Z kolei frekwencja allelu dominującego
wynosi p = l— q.
Tok postępowania obrazuje poniższy przykład:
W pewnym stadzie bydła na 1000 krów 910 jest czarno umaszczonych, a 90
czerwonych. W stadzie tym prowadzone są kojarzenia losowe. Frekwencja
genotypów recesywnych (osobniki czerwone — ee) wynosi 0,09. Pozostałe 910
krów ma genotypy homo- (E
d
E
d
) lub heterozygotyczne (E
d
e). Znając frekwencję
genotypu recesywnego (ee) można obliczyć frekwencję allelu czerwonego
umaszczenia e —q, która wynosi
Stąd frekwencja genu
czarnego umaszczenia (E
d
= p) wynosi l —0,3 = 0,7. Wiedząc, że w przypadku
losowych kojarzeń rozkład genotypów jest następujący:
łatwo jest obliczyć frekwencję pozostałych genotypów:
W stadzie tym na 910 krów czarno umaszczonych należy oczekiwać 490
osobników o genotypach homozygotycznych dominujących i 420 hetero-
zygot. Podany sposób obliczania częstości genów i genotypów stosuje się do
populacji będących w stanie równowagi genetyczej.
8.3. Frekwencja genów i genotypów w przypadku
cech uwarunkowanych szeregiem (serią)
alleli wielokrotnych
W przypadku cech warunkowanych szeregiem alleli wielokrotnych
rozkład genotypów w populacji będzie następujący:
201

przy czym p,q, r lub inne litery symbolizują częstość występowania genów
w szeregu alleli wielokrotnych. Suma frekwencji wszystkich alleli tworzących
szereg wynosi l (100%). Sposób obliczania frekwencji poszczególnych
genów i genotypów zostanie przedstawiony na przykładzie cechy warun-
kowanej szeregiem złożonym z trzech alleli.
U królików podstawowe typy umaszczenia warunkowane są szeregiem
alleli wielokrotnych
Umaszczenie dzikie warunkuje gen C,
himalajskie —
i albinotyczne — gen c w układzie homozygotycznym
(cc). W rozmnażającej się losowo populacji złożonej ze 150 królików
stwierdzono 135 osobników dziko umaszczonych (pełna barwa), 13
himalajskich i 2 albinosy. Frekwencja poszczególnych fenotypów wynosi
zatem:
Frekwencję genów oznaczono symbolem p dla genu C, q dla genu
dla
genu c.
W populacji tej, w której są kojarzenia losowe, fenotypy, genotypy i ich
frekwencja są następujące (tab. 8-1):
202

Po dodaniu takich samych genotypów uzyska się rozkład:
Z zestawienia tego wynika, że suma częstości fenotypów umaszczenia
himalajskiego i albinotycznego wynosi:
Zatem suma częstości genów d
1
i c wynosi q + r i równa się pierwiastkowi
kwadratowemu z sumy częstości występowania fenotypów umaszczenia
himalajskiego i albinotycznego. Podstawiając dane liczbowe, (q + r) równa
się:
Frekwencję genu c = r można obliczyć, znając frekwencję fenotypu (jedno-
cześnie genotypu) królików albinotycznych (cc). W danym przykładzie
r
2
= 0,013, stąd r =
= 0,114.
Frekwencję genu
można obliczyć następująco:
W badanej populacji królików frekwencja genów umaszczenia jest więc
następująca:
203

ogółem = 1,000
8.4. Frekwencja genów i genotypów w przypadku
cech sprzężonych z płcią
W odróżnieniu od cech warunkowanych genami znajdującymi się w chromo-
somach autosomalnych, których liczba w genotypach jest jednakowa bez
względu na płeć osobnika, geny warunkujące cechy sprzężone z płcią mogą
występować także w formie hemizygotycznej.
Osobniki płci heterogametycznej mogą mieć jedynie dwa rodzaje
genotypów pod względem cechy sprzężonej z płcią (cecha występuje lub
nie na skutek braku genu, który ją warunkuje). U osobników homo-
gametycznych dodatkowo może wystąpić nosicielstwo genu recesywnego
(osobniki heterozygotyczne).
Rozkład genotypów u osobników płci heterogametycznej nie będzie
zatem rozkładem dwumianowym, lecz przyjmie postać:
przy czym A jest genem dominującym, natomiast a — jego recesywnym allelem.
Natomiast u osobników homogametycznych frekwencja poszczególnych
genotypów będzie taka jak w przypadku cech autosomalnych, czyli zgodna
z rozkładem dwumianowym.
U ludzi jedną z najczęściej występujących cech sprzężonych z płcią jest
daltonizm. Frekwencja genu d warunkującego daltonizm w populacji
ludzkiej wynosi średnio q = 0,08. Na tej podstawie można oszacować
częstość występowania daltonizmu zarówno u kobiet, jak i u mężczyzn.
U kobiet (płeć homogametyczna) frekwencja poszczególnych genotypów
pod względem tej cechy wynosi:
U mężczyzn (płeć heterogametyczna) frekwencja genotypów jest równa
frekwencji genów rozróżniania barw lub daltonizmu:
204

D - = p = 0 , 9 2
d- = q = 0,08
Zatem wśród 10000 kobiet daltonizm (dd) wystąpi u 64, 1472
kobiety będą rozróżniały barwy, ale będą nosicielkami genu daltonizmu
(Dd), pozostałe zaś 8464 nie mają w swych genotypach genu daltonizmu
(DD). W grupie mężczyzn o tej samej liczebności aż 800 mężczyzn
będzie daltonistami (d — ), pozostali natomiast (9200) będą normalnie
rozróżniać barwy (D — ).
8.5. Czynniki naruszające równowagę genetyczną
Do czynników naruszających równowagę genetyczną populacji należą:
mutacje, migracje, selekcja, dobór par do kojarzeń i dryf genetyczny. O ile
mutacje i dryf genetyczny nie zależą od hodowcy, o tyle pozostałe czynniki
są związane z jego świadomą działalnością. Wszystkie te czynniki, z wyjąt-
kiem doboru, wpływają na zmianę frekwencji genów i genotypów. Dobór
zmienia jedynie frekwencję genotypów (będzie o tym mowa dalej).
Mutacje
Mutacje genowe zmieniając frekwencję genów naruszają równowagę
genetyczną populacji. W następstwie tych mutacji nie tylko ulega zmianie
frekwencja alleli istniejących w danym locus, ale także mogą się pojawić
nowe allele. Konsekwencją zmiany frekwencji genów jest zmiana frekwencji
genotypów.
Ekspresja fenotypowa mutacji, która może wpłynąć na zmianę struktury
genetycznej populacji, zależy od genetycznego uwarunkowania danej cechy.
Jeśli zmutuje jeden z genów addytywnych, kontrolujących cechę ilościową,
mutacja ta może być fenotypowo niezauważalna i nie ma wówczas szans
na utrwalenie jej w drodze selekcji. Natomiast mutacja genu warunkującego
cechę jakościową, np. umaszczenie zwierząt futerkowych, powodująca
pojawienie się nowej barwy okrywy włosowej, może przyczynić się do
wytworzenia odmiany z frekwencją genów umaszczenia inną niż w popu-
lacji, w której mutacja wystąpiła.
W rozdziale dotyczącym mutacji podano, iż częstość mutacji allelu
dominującego w recesywny oznaczamy symbolem u, natomiast częstość
mutacji odwrotnej symbolem v. Jeśli zmutuje allel dominujący, jego
frekwencja zmaleje o wartość równą up. Analogicznie, mutacja allelu
recesywnego spowoduje zmniejszenie jego frekwencji o vq. Łączna zmiana
frekwencji allelu recesywnego w przypadku dwukierunkowej mutacji
wyniesie:
205

Frekwencja allelu dominującego zmieni się o wartość:
Na przykład: w pewnym stadzie liczącym 100 krów frekwencja allelu
czarnego umaszczenia (locus E) wynosiła E
d
= p = 0,7, allelu czerwonego
umaszczenia e = q = 0,3. Pula alleli w tym locus składała się z 140 alleli
czarnego i 60 alleli czerwonego umaszczenia. W wyniku mutacji genowej,
której częstość była taka sama dla allelu dominującego i recesywnego
i wynosiła 0,1,14 alleli E
d
zmutowało w kierunku allelu e, a 6 alleli e w kierunku
allelu E
d
. W konsekwencji frekwencja allelu E
d
zmieniła się o wartość:
i wyniosła 0,66, natomiast częstość allelu e zmieniła się o:
czyli wzrosła do 0,34.
Zmianie uległa także frekwencja genotypów. Zgodnie z prawem Har-
dy'ego-Weinberga frekwencja genotypów w tym stadzie była następująca:
przed mutacją: E
d
E
d
= p
2
= 0,49; E
d
e = 2pq = 0,42; ee = q
2
= 0,09, po
mutacji: E
d
E
d
= p
2
= 0,4356; E
d
e = 2pq = 0,4488; ee = q = 0,1156.
Migracje
Migracje zwierząt polegają na wprowadzaniu nowych osobników do stada
lub ich usuwaniu. Wpływ tego czynnika na strukturę genetyczną populacji
zostanie omówiony na przykładzie imigracji (wprowadzania) osobników do
populacji.
Przy krzyżowaniu różnych ras w zależności od tego, ile razy dokonywane
jest to krzyżowanie, wielkość zmian frekwencji genów i genotypów jest różna.
Jeśli jest to zabieg jednorazowy, po pewnym czasie populacja wróci do
równowagi, czyli ustali się nowy układ genetyczny. Natomiast krzyżowanie
uszlachetniające, które polega na kilkakrotnym przekrzyżowaniu pogłowia
prymitywnego z rasą szlachetną, powoduje zdecydowaną zmianę frekwencji
genów istniejących w populacji wyjściowej. Może nawet, w przypadku
wielokrotnego krzyżowania, doprowadzić do całkowitego wyparcia genów rasy
prymitywnej przez geny rasy szlachetnej (jest to tzw. krzyżowanie wypierające).
W hodowli niektórych gatunków zwierząt stosowana jest sztuczna
inseminacja, zatem migracja nie zawsze wiąże się z wprowadzaniem
osobników, może ona dotyczyć wprowadzania nasienia. Sztuczna insemina-
cja może powodować bardzo szybkie rozpowszechnianie genów.
Rozpatrzmy przykład wprowadzenia do populacji (stada) jakiejś grupy
zwierząt. Jeśli stosunek nowo wprowadzonych genotypów do całkowitej
liczby genotypów w populacji po imigracji (jest to intensywność imigracji)
206

oznaczymy literą m, to część osobników rodzimych będzie równa (1 —m).
Z kolei frekwencję genu w stadzie wyjściowym oznaczamy literą p,
a frekwencję genu u osobników wprowadzonych do stada literą p', zatem
nowa frekwencja genu będzie równa:
a zmiana frekwencji tego genu będzie wynosiła:
Załóżmy, że do omawianego wcześniej stada liczącego 100 krów, w którym
frekwencja allelu E
d
wynosiła 0,7, wprowadzono 20 krów, z frekwencją allelu
E
d
wynoszącą 0,9. Frekwencja genotypów w stadzie wynosiła:
E
d
E
d
= p
2
= 0,49; E
d
e = 2pq = 0,42; ee = q= 0,09
natomiast w grupie zwierząt imigrantów:
E
d
E
d
= p
2
= 0,81; E
d
e = 2pq = 0,18; ee = q = 0,01. Nowa
frekwencja allelu E
d
wynosi teraz:
Δ
P
1
= 0,2 • 0,9 + (l -0,2) • 0,7 = 0,74
czyli wzrosła o 0,4. Nowa frekwencja allelu e wynosi (1 — 0,74) = 0,26.
Można to sprawdzić za pomocą wzoru:
Frekwencja poszczególnych genotypów w stadzie po imigracji przedstawia
się następująco:
E
d
E
d
= p
2
= 0,5476; E'e = 2pq = 0,3848; ee = q
2
= 0,0676
Po imigracji w stadzie wzrosła liczba zwierząt z genotypem homozygotycz-
nym E
d
E
d
, zmalała osobników heterozygotycznyh E
d
e i homozygotycznych ee.
Wielkość zmiany frekwencji genotypów w przypadku imigracji zależy od
intensywności imigracji (m) oraz różnicy we frekwencji genów między
zwierzętami w danej populacji i osobnikami wprowadzonymi do niej.
Selekcja
Wybór zwierząt, które hodowca zamierza pozostawić do hodowli, nazywany
jest selekcją. Hodowca preferuje geny korzystne, a eliminuje z populacji
zwierząt geny niepożądane. Selekcjonując zwierzęta najlepsze hodowca
pozostawia w populacji określone genotypy. Równocześnie z selekcją
prowadzone jest brakowanie (usuwanie) zwierząt w związku z ich nieprzy-
datnością wynikającą z wieku, stanu zdrowotnego, obniżenia płodności
i produkcyjności lub niepożądanego genotypu. Zmiany w strukturze genety-
cznej populacji zależą od tego, jakie geny czy genotypy są preferowane.
Zmiany te są trwałe i pozostają nawet po zaprzestaniu prowadzenia selekcji.
207

Siłę oddziaływania selekcji określa się mianem współczynnika selekcji
i oznacza symbolem s. Współczynnik ten może przybierać wartość od O do l,
co oznacza, że wartość O jest wówczas, gdy nie ma w ogóle selekcji, wartość
l współczynnik osiąga, gdy usuwane są wszystkie niepożądane genotypy.
Zmiana frekwencji genu, będąca konsekwencją selekcji, zależy od rodzaju
tej selekcji. Jeśli selekcja w zakresie cechy dziedziczącej się z dominacją
zupełną prowadzona jest przeciwko genotypom recesywnym, zmianę
częstości genu recesywnego, będącą jej konsekwencją, można obliczyć ze
wzoru:
Jeśli natomiast selekcja preferuje genotyp recesywny, wówczas frekwencja
genu tworzącego ten genotyp wyniesie:
Od pewnego czasu w hodowli bydła cecha rogatości jest uważana za
niekorzystną. Cecha ta dziedziczy się z dominacją zupełną, czyli osobniki
bezrogie mają genotyp homozygoty dominującej lub heterozygotyczny pod
względem locus P (polled). W pewnym stadzie, w którym frekwencja allelu
bezrożności wynosi 0,8, prowadzona jest selekcja przeciwko genotypowi
recesywnemu pp. Jeśli współczynnik selekcji będzie wynosić 0,4, to oznacza,
że w każdym pokoleniu będzie usuwanych 40% osobników z genotypem
homozygoty recesywnej. Wówczas korzystając ze wzoru możemy obliczyć
wielkość zmiany częstości genu recesywnego:
Frekwencja allelu recesywnego w stadzie w wyniku selekcji przeciwko
genotypowi recesywnemu zmniejszy się o 0,013 i wyniesie 0,187.
Jeśli selekcję przeprowadzimy tylko w jednym pokoleniu i nie będą
działały inne czynniki naruszające równowagę genetyczną populacji, wów-
czas w pokoleniu potomnym ustali się nowa frekwencja genów oraz
genotypów i populacja wróci do równowagi. Efekt selekcji zostanie za-
chowany.
Dobór par do kojarzeń
W hodowli zwierząt nie zawsze stosowane są kojarzenia losowe. Jeśli celowo
dobierane są zwierzęta w pary rodzicielskie, prowadzi to do naruszenia
równowagi genetycznej populacji poprzez zmianę frekwencji genotypów.
Załóżmy, że w omawianym już wcześniej stadzie kur andaluzyjskich,
złożonym z 1000 ptaków, zastosowano kojarzenia w obrębie takich samych
208

genotypów. Oznacza to, iż kojarzono koguty czarne z kurami czarnymi,
koguty niebieskie (forma pośrednia cechy) z kurami niebieskimi i koguty
białe z kurami białymi. W stadzie tym frekwencja genów jest jednakowa
u osobników obu płci i wynosi p = 0,5 (allel A) i q = 0,5 (allel a). W
pokoleniu rodzicielskim frekwencja genotypów była następująca:
0,25 AA + 0,5 Aa + 0,25 aa = l
Po zastosowaniu kojarzeń „podobnego z podobnym" frekwencja uległa
zmianie
Frekwencja genów pozostała nie zmieniona, pod warunkiem że hodowca
nie prowadził selekcji i nie wprowadzono do stada nowych zwierząt. Aby
się o tym przekonać, można przeprowadzić obliczenia:
Jeżeli hodowca znów zastosuje kojarzenia losowe w stadzie i nadal nie
będzie prowadzić selekcji, stado powróci do stanu równowagi genetycznej.
Przy kojarzeniach osobników o genotypach identycznych wzrasta frek-
wencja genotypów homozygotycznych, maleje natomiast częstość genoty-
pów heterozygotycznych. Odwrotna sytuacja zaistniałaby, gdyby kojarzono
między sobą osobniki będące przeciwstawnymi homozygotami. Wówczas
zwiększyłaby się częstość genotypów heterozygotycznych kosztem homo-
zygotycznych.
Niekiedy, na przykład w celu wzmocnienia (utrwalenia danej cechy)
w populacji, stosowane są kojarzenia osobników spokrewnionych, np.
ojca z córką czy matki z synem. Kojarzenia takie powodują wzrost
częstości występowania homozygot w pokoleniu potomnym. W hodowli
zwierząt laboratoryjnych kojarzenia w bliskim pokrewieństwie (najczęściej
209

brat x siostra) prowadzą do wytworzenia tzw. szczepów wsobnych, w któ-
rych stopień homozygotyczności jest bliski 100%. Szczepy wsobne są
wykorzystywane w badaniach biomedycznych.
Dryf genetyczny
Dryf genetyczny, zjawisko tak nazwane przez Wrighta w 1931 roku, polega
na zmianie frekwencji genów i genotypów w małych populacjach, która jest
wynikiem odchyleń w losowej segregacji genów do gamet i losowego
łączenia się gamet. Dryf genetyczny jest zjawiskiem całkowicie niezależnym
od woli hodowcy. Rozkład genotypów w pokoleniu potomnym zależy od
frekwencji genów u rodziców. Prawidłowość tego rozkładu wynika z praw-
dopodobieństwa losowego połączenia gamet w następstwie kojarzeń loso-
wych. Wszelkie odstępstwa od oczekiwanego rozkładu są konsekwencją
dryfu genetycznego, zwłaszcza w małych populacjach, w których działa on
najsilniej. Dryf przyczynia się do zwiększania się różnic między małymi
populacjami w zakresie struktury genetycznej, jednocześnie zmniejsza się
zmienność genetyczna w obrębie poszczególnych populacji. Niekorzystnym
następstwem dryfu jest także wzrost frekwencji genotypów homozygotycz-
nych kosztem heterozygotycznych. Biorąc pod uwagę, iż allele niekorzystne
z reguły są recesywne, szczególnie niebezpieczny może być wzrost homo-
zygot recesywnych, którego skutkiem będzie zmniejszenie płodności,
żywotności czy ujawnienie się wad dziedzicznych.
Dla zobrazowania tego zjawiska grupę 20 owiec heterozygotycznych pod
względem locus A skrzyżowano z trykiem również heterozygotycznym w tym
locus. Przy losowej segregacji genów do gamet i losowym łączeniu gamet
w zygoty należy oczekiwać, iż w potomstwie uzyskamy 5 osobników homozygot
dominujących, 10 heterozygot i 5 homozygot recesywnych pod względem
rozpatrywanego locus. Może jednak się zdarzyć tak, że jedynie plemniki
zawierające allel dominujący wnikną do komórek jajowych. Wtedy w potomst-
wie będzie 10 osobników (50%) homozygot dominujących i 10 heterozygot. Jeśli
było to jednorazowe zaburzenie, to w następnym pokoleniu ustali się nowa
równowaga genetyczna, w której frekwencja genu dominującego wyraźnie
wzrośnie. Jeżeli zaburzenie kojarzeń losowych będzie się powtarzać, to struktura
genetyczna populacji będzie się zmieniać (dryfować) w różnych kierunkach.
8.6. Wykorzystanie frekwencji alleli do szacowania
zmienności genetycznej wewnątrz i między
populacjami
Zmienność genetyczna ma ogromne znaczenie dla wszystkich gatunków
zwierząt. Im większa jest pula genów w jakiejś populacji, tym silniejsze są
zdolności adaptacyjne tej populacji do zmiennych warunków środowisko-
210

wych. Na skutek długotrwałej selekcji, zwłaszcza w małych populacjach,
może dojść do znacznego ograniczenia puli genów, czego wynikiem będzie
utrata zdolności adaptacyjnych. By temu niekorzystnemu zjawisku zapobiec,
zalecane jest monitorowanie zmian w strukturze genetycznej populacji,
które zachodzą w wyniku pracy hodowlanej. W tym celu szacuje się
zmienność genetyczną w populacji na podstawie frekwencji alleli w loci
markerowych, na przykład warunkujących antygeny erytrocytarne, różne
typy białek surowicy krwi czy enzymy erytrocytarne. Mogą to być loci
kontrolujące syntezę transferyny, albuminy, esterazy, amylazy, ceruloplaz-
miny czy anhydrazy węglanowej, należące do markerów genetycznych
klasy I, opisanych w rozdziale: Markery genetyczne. Od pewnego czasu do
tego celu wykorzystywane są niekodujące sekwencje DNA (polimorfizm
mini- i mikrosatelitarny, czyli markery genetyczne klasy II).
Parametrem charakteryzującym zmienność genetyczną w populacji jest
współczynnik heterozygotyczności. Wartość tego parametru zawiera się
w granicach od O do l, można także przedstawiać ją w procentach. Średnia
heterozygotyczność (H) w danej populacji obliczana jest ze wzoru:
211

Parametrem charakteryzującym stopień zmienności (polimorficzności)
danego locus jest wskaźnik określany symbolem PIĆ (ang. polymorphic
Information content). Jednak najczęściej parametr ten służy do określenia
przydatności danego markera genetycznego do analizy sprzężeń z
innymi loci. O markerach genetycznych jest mowa w innym
rozdziale. By obliczyć wartość PIĆ, należy znać frekwencję wszystkich
alleli w danym locus:
Dystans genetyczny
Zróżnicowanie genetyczne między populacjami można określić
porównując częstość występowania określonych genów w tych
populacjach i szacując tzw. dystans genetyczny na podstawie
polimorfizmu grup krwi i białek lub sekwencji mikrosatelitarnych.
Sekwencje mikrosatelitarne charakteryzują się wyjątkowo wysokim
stopniem polimorfizmu i dzięki temu są bardziej przydatne niż markery
klasy I do szacowania dystansu genetycznego między gatunkami
blisko ze sobą spokrewnionymi, a także między populacjami w obrębie
gatunku.
Szacowanie dystansu genetycznego i wybór ras, które cechuje duża
zmienność genetyczna, służy zachowaniu biologicznej różnorodności
zwierząt gospodarskich. Do szacowania dystansu genetycznego (Dr), tak
na podstawie polimorfizmu sekwencji mikrosatelitarnych, jak i
polimorfizmu białek i antygenów erytrocytarnych, najczęściej stosowany
jest wzór:
Dystans genetyczny może być przedstawiany graficznie w postaci
dendrogramów, obliczonych różnymi metodami, na przykład:
najbliższego sąsiedztwa — SLM (ang. single linkage method),
najdalszego sąsiedztwa — CLM (ang. complete linkage method) lub
skupienia parami — UPGM (ang. unweighted pair group method).
212
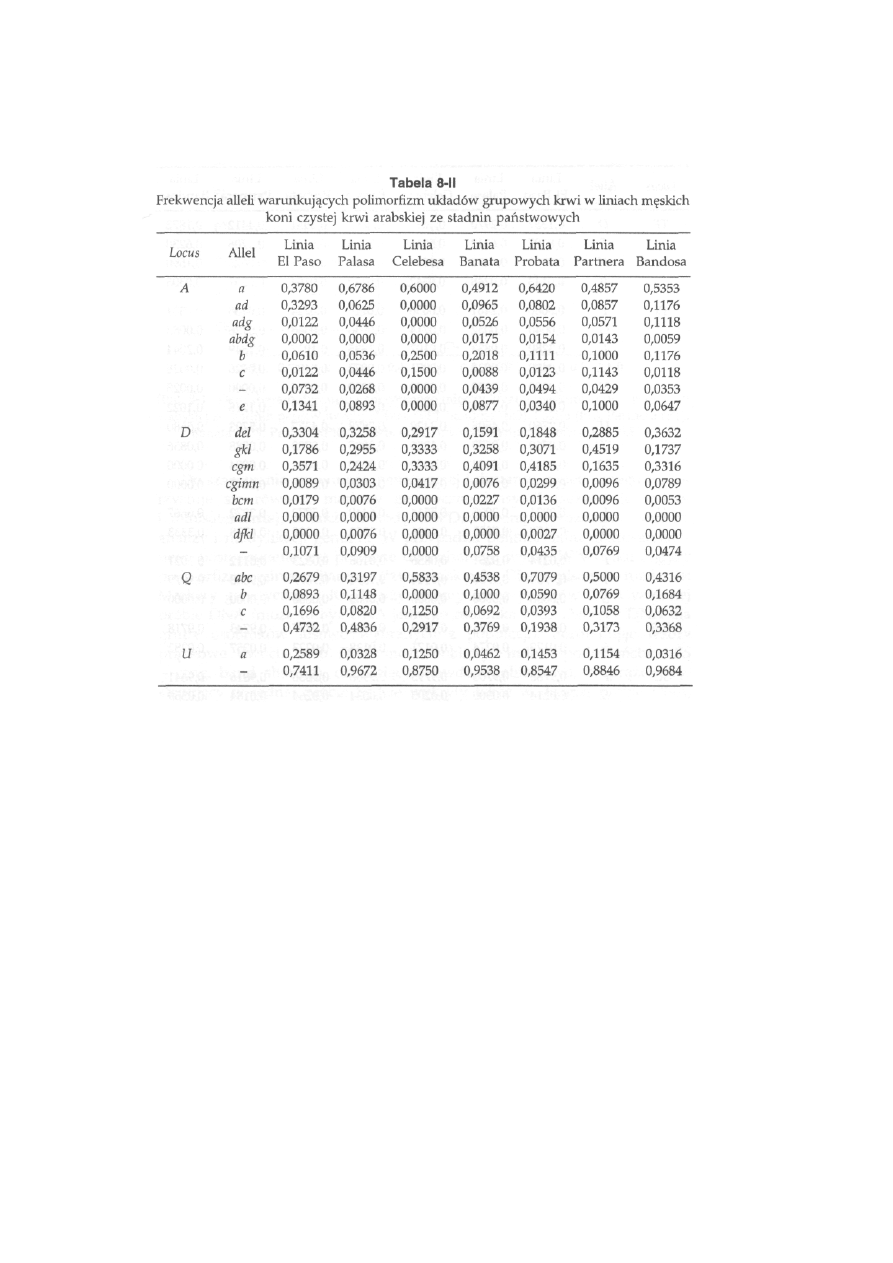
Sposób szacowania dystansu genetycznego obrazuje poniższy przykład
(wg: Niemczewski i wsp., Prace i Materiały Zootechniczne, 50:111-120, 1997).
Na podstawie frekwencji alleli w 14 loci obliczono dystans genetyczny
między liniami męskimi koni czystej krwi arabskiej ze stadnin państwowych.
Częstość występowania poszczególnych alleli zawarto w tabelach 8-II i 8-III.
Analizą objęto 141 koni z linii El Paso, 219 z linii Palasa, 146 z linii Celebesa,
150 z linii Banata, 369 z linii Probata, 146 z linii Partnera i 202 z linii Bandosa.
Do sporządzenia dendrogramu dystansu genetycznego wykorzystano ob-
liczenia wykonane metodą najbliższego sąsiedztwa — SLM. Największy
dystans genetyczny stwierdzono między liniami El Paso i Celebesa, naj-
mniejszy między linią Celebesa a Probata, Palasa a Partnera, a także między
linią Banata a Celebesa i Probata. Na przykład, wartość dystansu genetycz-
nego między liniami Celebesa a Banata wyniosła 0,32, Partnera a Banata 0,39
oraz Palasa a Partnera 0,37 (ryć. 8-1).
213
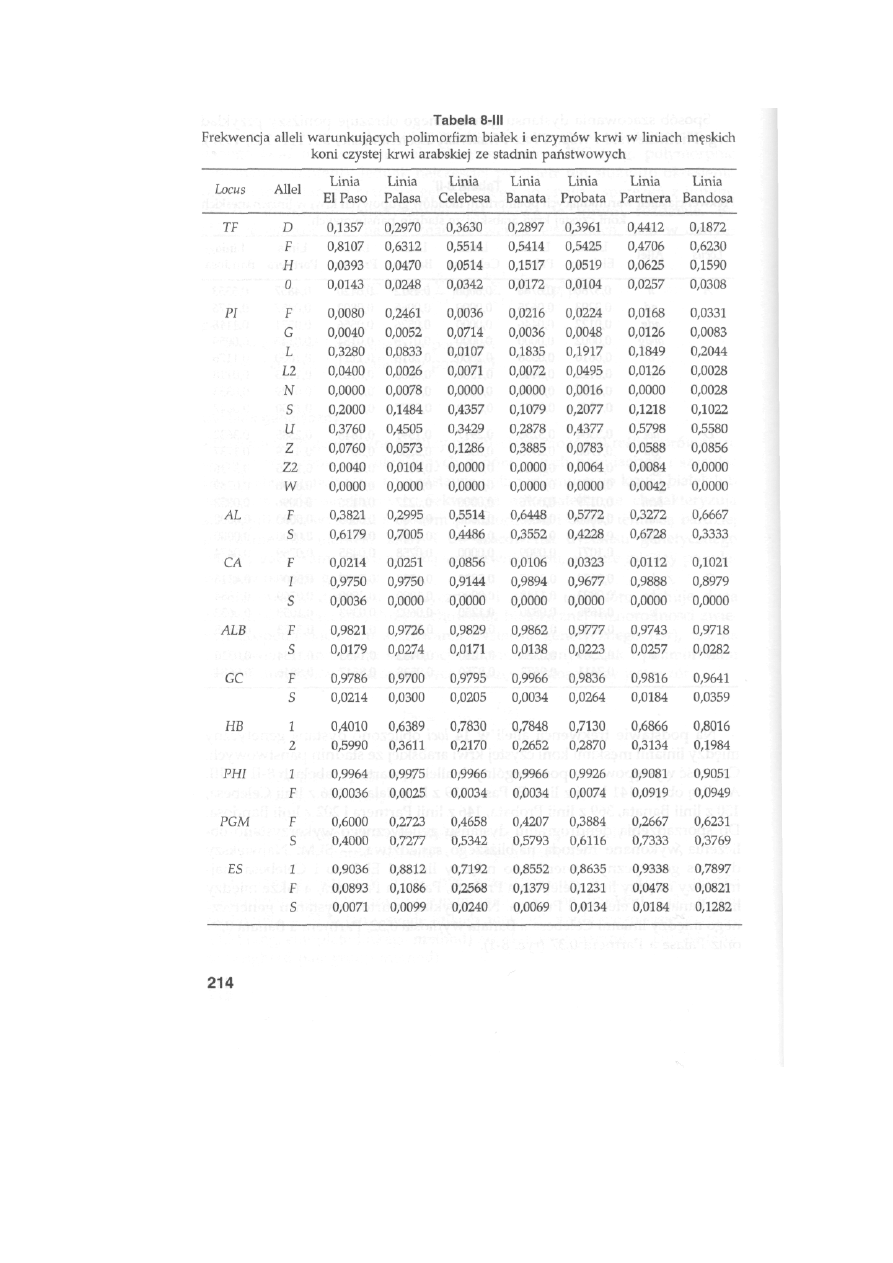
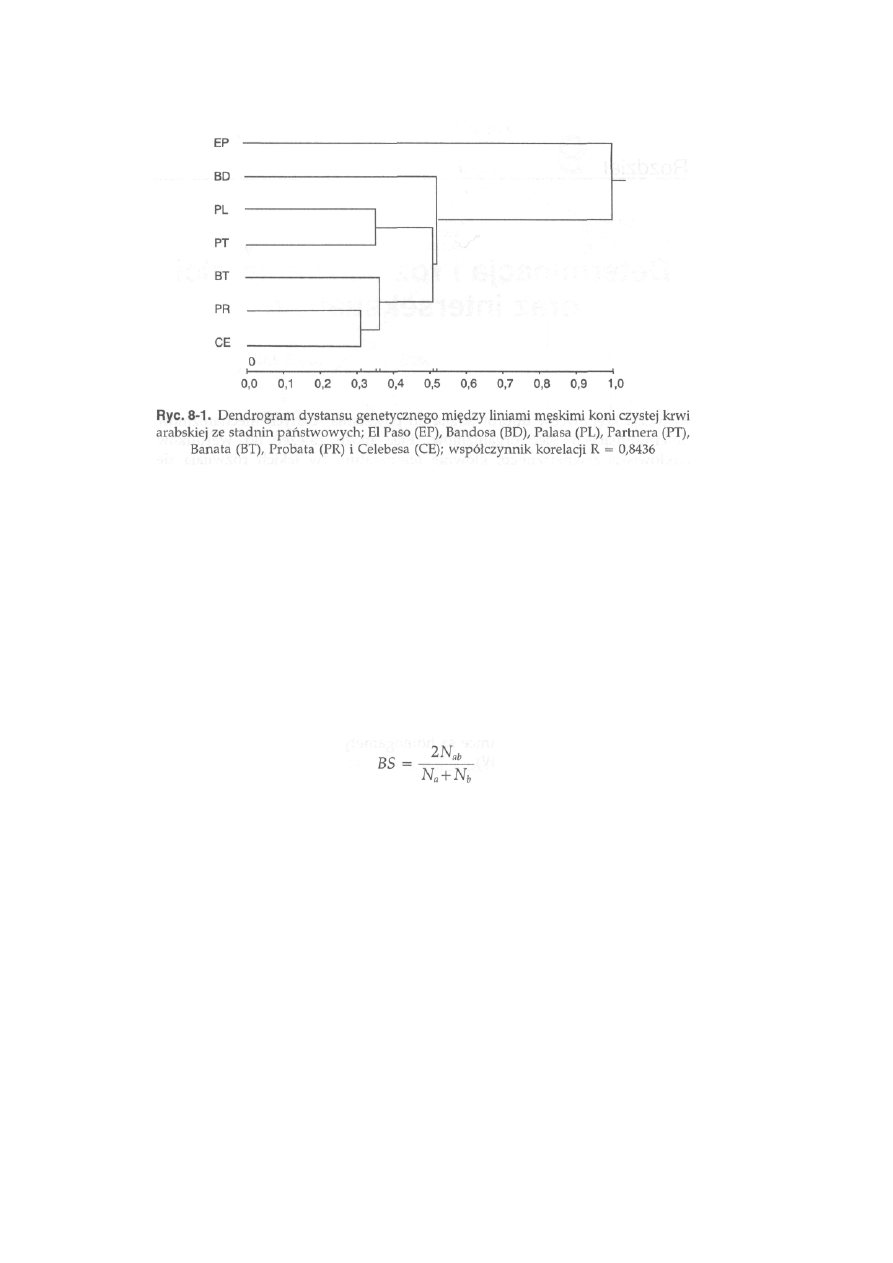
Do szacowania zmienności genetycznej i dystansu genetycznego wyko-
rzystuje się również markery genetyczne klasy II (sekwencje mikro-
i minisatelitarne), a także metodę RAPD, opisaną w rozdziale: Metody
analizy i modyfikacji genomu. W przypadku polimorfizmu mikrosatelitar-
nego postępowanie jest podobne do opisanego wyżej. W wyniku analizy
polimorfizmu minisatelitarnego (tzw. odcisk palca DNA, opisany w rozdziale:
Markery genetyczne) i RAPD uzyskujemy obraz prążków DNA w danej
próbie DNA (może to być DNA jednego osobnika bądź zbiorczy DNA dla
grupy osobników losowo wybranych z populacji). Porównując wzory
prążkowe dwóch prób DNA można obliczyć indeks podobieństwa BS
- ang. band sharing), określający prawdopodobieństwo, że prążek wy-
stępujący w jednej próbie będzie wykryty także w drugiej próbie DNA,
wykorzystując wzór:
gdzie:
N
ab
— liczba identycznych prążków w obu próbach DNA (a i b)
N
a
iN
h
— całkowita liczba prążków odpowiednio w próbie a i próbie b
Wartość tego prawdopodobieństwa zawiera się w granicach 0-1. Im
mniejsza zmienność genetyczna w danej populacji, tym większa wartość
tego współczynnika.

Rozdział
9
Determinacja i różnicowanie płci
oraz interseksualizm
Ewolucja kręgowców doprowadziła do powstania różnych mechanizmów
determinujących płeć organizmów. Płeć może zależeć od warunków
środowiska zewnętrznego, głównie temperatury, w jakich rozwijają się
zarodki. Przy takim mechanizmie determinacji zostaje ona ustalona raz
w momencie różnicowania się gonad i nie zmienia się przez całe życie. Ten
typ determinacji płci obserwuje się u wielu gatunków gadów, w tym
u wszystkich krokodyli, wielu gatunków żółwi i niektórych gatunków
jaszczurek. Z kolei u niektórych gatunków ryb zachodzi tzw. behawioralna
determinacji płci. Zwierzęta takie są zazwyczaj obojnakami, tzn. mają
gonady męskie i żeńskie. Zależnie od środowiska społecznego osobnik
przyjmuje rolę męską lub żeńską. Wśród ryb spotyka się również gatunki,
które są hermafrodytami sekwencyjnymi, czyli zmieniają one płeć w ciągu
życia. U ptaków i ssaków płeć osobnika zależy od układu chromosomów
płci, a konkretnie od genotypu w loci odpowiedzialnych za kształtowanie
cech płciowych. W przypadku ssaków wyróżnia się chromosomy X oraz Y,
a w przypadku ptaków Z oraz W. U ssaków płcią heterogametyczną są
samce (układ XY), a płcią homogametyczną są samice (układ XX). U ptaków
sytuacja jest odwrotna — samce są homogametyczne (układ ZZ), a samice
heterogametyczne (układ Z W).
9.1. Determinacja płci ssaków
Ukształtowanie się cech płciowych ssaków obejmuje trzy zasadnicze etapy:
1) powstanie w wyniku zapłodnienia układu chromosomów płci XX lub XY
w zygocie — kształtuje się wówczas tzw. płeć chromosomowa, 2) rozwój
niezróżnicowanych gonad zarodka w kierunku jądra lub jajnika — wy-
kształcenie tzw. płci gonadowej i 3) uformowanie się wewnętrznych
216
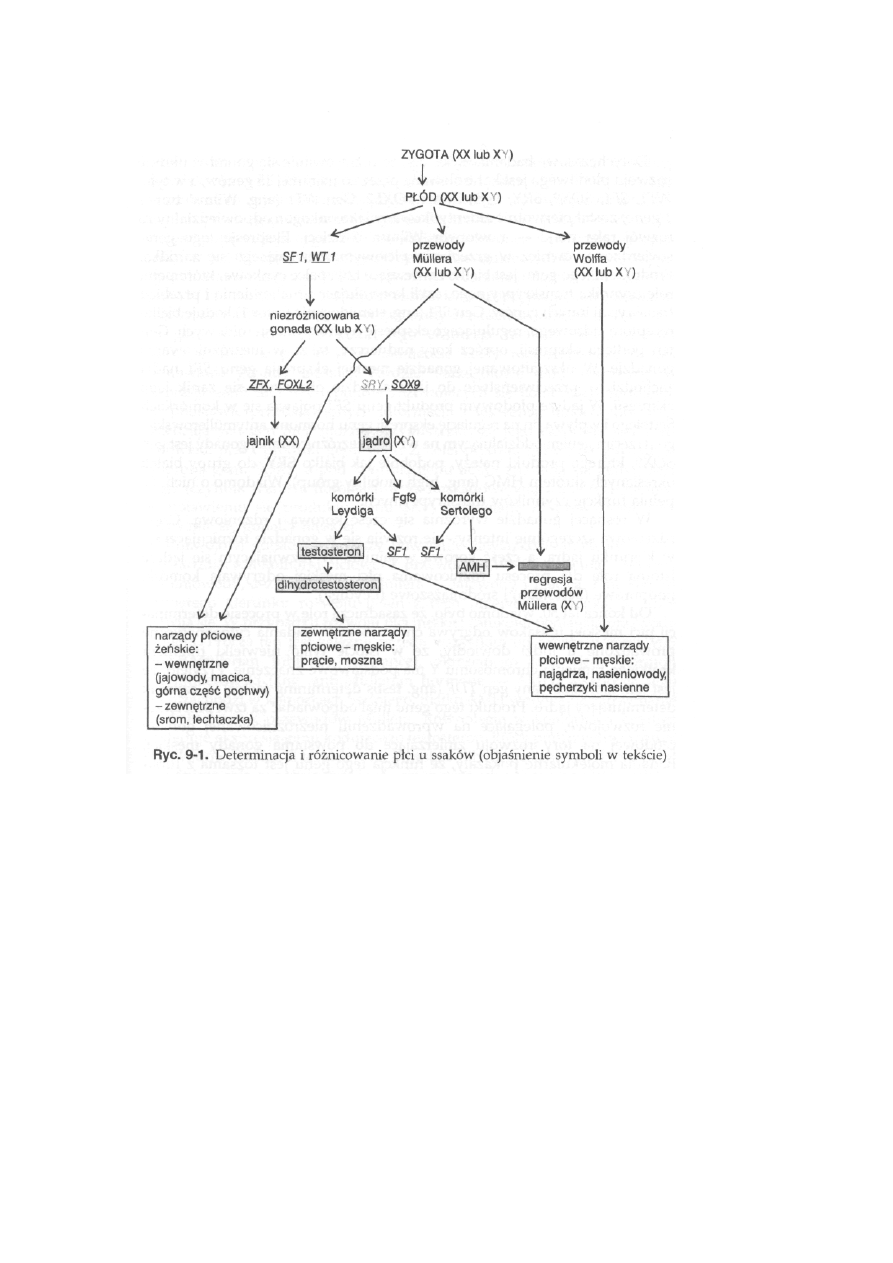
i zewnętrznych cech płciowych — powstanie tzw. pici fenotypowej (ryć. 9-1).
Pleć fenotypowa obejmuje również zmiany powstające w mózgu, które są
odpowiedzialne za ukształtowanie się zachowania samczego bądź samiczego.
W rozwijającym się zarodku zawiązki gonad powstają z komórek
śródnerczowych i w ich kierunku wędrują pierwotne komórki płciowe.
Pojawiają się one w endodermie pęcherzyka żółtkowego, a następnie
ruchem amebowatym migrują między komórkami do grzebienia płciowego
przylegającego do pranerczy. Następnie grzebień płciowy oddziela się
od pranercza i przekształca się w niezróżnicowaną gonadę. Pierwotne
komórki płciowe po zasiedleniu gonad podlegają intensywnym podziałom
mitotycznym.
217

Dotychczasowe badania wykazały, że różnicowanie się gonad w okresie
rozwoju płodowego jest kontrolowane przez co najmniej 15 genów, a w tym:
WT1, SF1, SOX9, SRY, Fgf9, ZFX i FOXL2. Gen WT2 (ang. Wilms' tumor l
gene) został pierwotnie zidentyfikowany jako onkogen odpowiedzialny za
rozwój raka nerki — nowotwór Wilmsa u dzieci. Ekspresję tego genu
stwierdzono również w grzebieniu płciowym rozwijającego się zarodka.
Produktem tego genu jest białko zawierające tzw. palce cynkowe, które pełni
rolę czynnika transkrypcyjnego, czyli kontrolujące uruchomienie i przebieg
transkrypcji innych genów. Gen SF1 (ang. steroidogenic factor 1) koduje białko
receptora jądrowego, regulującego ekspresję hydroksylaz steroidowych. Gen
ten podlega ekspresji, oprócz kory nadnerczy, także w niezróżnicowanej
gonadzie. W ukształtowanej gonadzie męskiej ekspresja genu SF1 nadal
zachodzi, w przeciwieństwie do jajnika, gdzie obserwuje się zanik jego
ekspresji. W jądrze płodowym produkt genu SF1 pojawia się w komórkach
Sertolego i wpływa on na regulację ekspresji genu hormonu antymullerowskie-
go. Trzecim genem oddziałującym na wzrost niezróżnicowanej gonady jest gen
SOX9, którego produkt należy, podobnie jak białko SRY, do grupy białek
określanych skrótem HMG (ang. high mobility group). Wiadomo o nich, że
pełnią funkcję czynników transkrypcyjnych.
W rosnącej gonadzie wyróżnia się część korową i rdzeniową. Część
rdzeniowa szczególnie intensywnie rozwija się w gonadzie różnicującej się
w kierunku jądra, a część korowa w jajniku. W rozwijającym się jądrze
istotną rolę dla procesu różnicowania płci męskiej odgrywają komórki
podporowe (Sertolego) i śródmiąższowe (Leydiga).
Od końca lat 50. wiadomo było, że zasadniczą rolę w procesie determina-
cji płci męskiej u ssaków odgrywa chromosom Y. Badania cytogenetyczne
prowadzone u ludzi dowiodły, że w istocie tylko niewielki fragment
krótkiego ramienia chromosomu Y ma podstawowe znaczenie. Przyjęto, że
jest tam zlokalizowany gen TDF (ang. testis determining factor) — czynnik
determinujący jądro. Produkt tego genu miał odpowiadać za tzw. przełącze-
nie rozwojowe, polegające na wprowadzeniu niezróżnicowanej gonady
płodowej na tory rozwoju zmierzające do powstania gonady męskiej.
Badania molekularne pokazały, że funkcja tego genu jest tożsama z funk-
cją genu SRY (ang. sex determining region Y), którego locus występuje
w chromosomie Y. Gen SRY zbudowany jest z pojedynczego eksonu
i koduje informację o peptydzie składającym się z ponad 200 aminokwasów.
Białko kodowane przez ten gen składa się z trzech regionów (ryć. 9-2).
218

Region środkowy, składający się z 79 aminokwasów, ma strukturę podobną
do białek z grupy HMG, które mają zdolność wiązania się z DNA i dlatego
uważane są za czynniki transkrypcyjne, czyli białka wpływające na urucho-
mienie i przebieg procesu transkrypcji. Bardzo ciekawe jest również to, że
w części poprzedzającej gen SRY znajduje się sekwencja CpG (tzw. wysepka
CpG), w której obrębie występuje sekwencja nukleotydów przyłączająca
czynnik transkrypcyjny WT1, omówiony wcześniej. Białko SRY, tak jak
białka WT1 i SOX9, pełni funkcję czynnika transkrypcyjnego, odpowiedzial-
nego za kontrolę ekspresji genów związanych z różnicowaniem się pierwot-
nej gonady w kierunku jądra. Jego ekspresja zachodzi w grzebieniu
płciowym (listwie płciowej), w momencie kiedy rozpoczyna się proces
formowania gonady męskiej. Bardziej szczegółowe dane wskazują, że
ekspresja genu SRY występuje w różnicujących się komórkach Sertolego.
Na podstawie przedstawionych informacji o sekwencji nukleotydów po-
przedzających gen SRY można przypuszczać, że jego ekspresja jest kon-
trolowana przez produkt genu WTL Z kolei produkt genu SRY zawiaduje
ekspresją genu SOX9, a pod kontrolą genu SOX9 znajdują się geny AMH
i Fgf9 (czynnik wzrostu fibroblastów 9). Kaskadowe włączanie tych genów,
po pojawieniu się produktu genu SRY, odpowiada za prawidłowe róż-
nicowanie się gonady męskiej.
Rozwojowi gonad towarzyszy powstanie zawiązków przewodów wy-
prowadzających komórki płciowe. Z przewodów Wolffa rozwinąć się mogą
nasieniowody, a z przewodów Miillera — jajowody i macica. Zależnie od
przyjętego kierunku rozwoju jeden z tych przewodów zanika, a drugi
rozwija się. W przypadku rozwoju płci męskiej zanikowi podlegają przewody
Miillera. Proces ten jest zależny przede wszystkim od ekspresji genu MIS
(ang. Miillerian inhibitor substance), określanego także jako hormon
antymiillerowski (ang. anti-Mullerian hormone — AMH). Ekspresja tego
genu zachodzi w komórkach Sertolego. Białko AMH wpływa na zatrzymanie
rozwoju i zanik przewodów Miillera. Równolegle w komórkach Leydiga
następuje ekspresja genu kodującego testosteron, który podtrzymuje rozwój
przewodów Wolffa. Z testosteronu powstaje dihydrotestosteron, który pełni
zasadniczą rolę w różnicowaniu się zewnętrznych narządów płciowych.
Gdy gen SRY jest nieobecny, niezróżnicowana gonada płodowa rozwija
się spontanicznie w jajnik. Wiedza na temat kontroli genetycznej róż-
nicowania się jajnika jest dużo mniejsza, aniżeli w odniesieniu do jądra.
Początkowo przypuszczano, że proces ten jest w znacznej mierze kont-
rolowany przez ekspresję genu DAX1. Wiadomo, że jego locus występuje
w chromosomie X, a jego produktem jest jądrowy receptor dla hormonów.
Ekspresja tego genu jest stwierdzana już w niezróżnicowanej gonadzie.
Obecnie większą rolę przypisuje się genowi ZFX (tzw. gen palca cynkowego),
również położonemu w chromosomie X, oraz autosomalnemu genowi
FOXL2 (receptor hormonu stymulującego rozwój pęcherzyków jajnikowych).
W rozwijającym się jajniku nie zachodzi ekspresja genu MIS oraz genu
219

kodującego testosteron. W efekcie następuje spontaniczny rozwój przewo-
dów Miillera w jajowody, macicę i górną część pochwy. Procesowi temu
towarzyszy równoczesny zanik przewodów Wolffa.
Interesujące są wnioski płynące z obserwacji osobników z układem
chromosomów XY, w których genotypie występuje duplikacja locus DSS
(DAX1). U takich osobników następuje rozwój interseksualny
wynikający z niepełnego zróżnicowania gonady w kierunku jądra. Jeśli w
genotypie wystąpi delecja tego locus, to rozwój w kierunku męskim
nie jest zakłócony. Mutacje tego typu (duplikacja lub delecja) nie mają
wpływu na rozwój cech płciowych samic. Zakłada się, że produkt
locus DSS (DAX1) konkuruje z produktem genu SRY w wiązaniu się z
promotorami innych genów odpowiedzialnych za kształtowanie się
gonady męskiej. Jeśli w genotypie osobnika męskiego występują dwie
kopie locus DSS (DAX1), to wówczas dochodzi do zakłócenia procesu
kontroli ekspresji genów przez białko SRY.
Poznanie molekularnych podstaw determinacji płci ma w hodowli duże
znaczenie praktyczne. Wczesna ocena płci zarodków pozwala na uzys-
kiwanie potomstwa o pożądanej płci, jeśli stosuje się nowoczesne biotechniki
rozrodu powiązane z przenoszeniem zarodków do tzw. samic biorczyń.
Zarodki mogą być pozyskiwane od superowulowanych samic dawczyń lub
powstawać podczas zapłodnienia pozaustrojowego (in vitro), czy też klono-
wania. Bardzo przydatną metodą określania płci zarodków jest wykrywanie
sekwencji genu SRY w DNA wyizolowanym z komórek zarodka męskiego.
Zastosowanie znajduje też metoda polegająca na analizowaniu sekwencji
molekularnych genu amelogeniny (AMEL). Gen ten umiejscowiony jest
w obu chromosomach płci, z tym jednak że gen położony w chromosomie
Y obarczony jest delecja (jest krótszy o 63 pary nukleotydów) względem
genu położonego w chromosomie X. Wykrywanie obecności genu SRY oraz
alleli genu amelogeniny prowadzone jest na podstawie amplifikacji techniką
PCR (patrz rozdział: Metody analizy i modyfikacji genomu) wybranego
fragmentu DNA oraz elektroforezy uzyskanych produktów.
Należy również zwrócić uwagę, że w chromosomie Y poza genem SRY,
który pełni zasadniczą rolę w procesie różnicowania się gonady męskiej,
występują także inne geny. Szczególnie istotną częścią chromosomu Y jest
region AZF. Jego nazwa pochodzi od słów angielskich azoospermia factor,
oznaczających czynnik odpowiedzialny za powstanie azoospermii, czyli
brak plemników w ejakulacie. W regionie tym występują geny, których
ekspresja związana jest z przebiegiem spermatogenezy. Delecje w tym
regionie prowadzą do zaburzeń procesu spermatogenezy, takich jak cał-
kowity brak spermatogoniów, lub do zahamowania podziału mejotycznego.
Do tej pory udało się zidentyfikować tylko kilka genów z tego obszaru
chromosomu Y, np. gen RBM (ang. RNA binding motif) biorący praw-
dopodobnie udział w procesie składania (ang. splicing) mRNA. Znane są też
geny położone w innych częściach chromosomu Y, np. gen Ube1Y (ang.
220

ubiąuitin activating enzyme-1), który prawdopodobnie jest zaangażowany
w ubikwitynację histonów w spermatydach, co może z kolei mieć związek
z wymianą histonów na protaminy w chromatynie plemników. Z przed-
stawionych wyżej informacji można wyciągnąć wniosek, że informacja
genetyczna zawarta w chromosomie Y jest bogatsza, niż to wcześniej
zakładano.
9.2. Interseksualizm
Zaburzenie procesu determinacji i różnicowania płci prowadzi do powstania
wrodzonych wad rozwojowych układu rozrodczego, które określa się
terminem interseksualizmu lub obojnactwa. Zaburzenia tego typu wywoły-
wane są przez mutacje chromosomów płci lub mutacje genów zaan-
gażowanych w proces determinacji i różnicowania płci oraz mogą być
skutkiem nieprawidłowego przebiegu ciąży (np. frymartynizm). Precyzyjna
diagnoza przypadków interseksualizmu stwarza niejednokrotnie problemy,
ze względu na bardzo duże zróżnicownie obserwowanych zmian. Kiedy
niemożliwe jest jednoznaczne określenie podłoża obserwowanych zmian,
często takie przypadki klasyfikuje się na dwie kategorie: 1) hermafrodytyzm
prawdziwy, kiedy stwierdza się równoczesną obecność gonad męskich
i żeńskich lub gdy występują struktury złożone typu jajnikojądro (ovotestis)
i 2) pseudohermafrodytyzm męski, kiedy gonadom męskim towarzyszą
zaburzenia w przekształcaniu się przewodów Wolffa i Miillera lub w po-
wstawaniu zewnętrznych narządów płciowych.
Frymartynizm
Frymartynizm jest najczęściej występującą postacią interseksualizmu u zwie-
rząt domowych, głównie przeżuwaczy. Podczas rozwoju ciąży mnogiej
może dojść do powstania połączeń naczyniowych (anastomoz) między
łożyskami rozwijających się płodów. Jeśli połączenie łożysk dotyczy płodów
różnopłciowych, to wówczas rozwój cech płciowych samicy (frymartyna)
jest zaburzony, co w konsekwencji prowadzi prawie zawsze do niepłodności.
Połączenie krwiobiegów bliźniąt różnopłciowych sprawia, że związki hor-
monalne oraz inne czynniki aktywne wytwarzane przez jądra płodowe
docierają do organizmu samicy i wpływają na proces różnicowania się
żeńskich cech płciowych. Rozmiar zaburzeń w ukształtowaniu cech płcio-
wych samicy frymartyna zależy od momentu, w którym powstały połączenia
naczyniowe. W rozwoju embrionalnym różnicowanie się jąder poprzedza
okres, w którym kształtują się jajniki. Jeśli do połączeń tętniczo-żylnych
łożysk doszło w okresie różnicownia się pierwotnej gonady w kierunku
jądra u osobnika męskiego, to wydzielany czynnik transkrypcyjny SRY
może zakłócić proces kształtowania jajników u osobnika bliźniaczego płci
221

żeńskiej w takim stopniu, że rozwinie się nawet gonada męska, a w ślad za
tym drogi płciowe będą miały charakter męski. Jeśli anastomozy powstaną
w chwili gdy jajnik jest już ukształtowany, to wówczas wydzielany przez
jądra płodowe hormon antymiillerowski i testosteron spowodują zakłócenia
w różnicowaniu się dróg wyprowadzających gamety oraz zewnętrznych
narządów płciowych. Możliwe jest również powstanie połączeń, w okresie
gdy przewody Miillera będą już przekształcone w jajowody i macicę.
Wówczas dihydrotestosteron, powstający z wydzielanego przez komórki
Leydiga testosteronu, może zakłócić, w mniejszym lub większym zakresie,
proces kształtowania się zewnętrznych narządów płciowych. Trzeba pod-
kreślić, że prawie we wszystkich przypadkach, niezależnie od stopnia
zakłócenia procesu różnicowania cech płciowych, stwierdza się niepłodność
samic frymartynów. Jednocześnie cechy płciowe męskiego bliźniaka są
rozwinięte prawidłowo. Buhaje takie mogą być wykorzystywane w roz-
rodzie, chociaż parametry określające ich przydatność rozpłodową (np.
obraz nasienia, wskaźnik niepowtarzalności rui itp.) są często obniżone.
Istotną kwestią jest możliwość wykrywania frymartynizmu u samic
hodowlanych, a także wskazywanie samców pochodzących z ciąż bliź-
niaczych różnopłciowych. Okazuje się, że badania kliniczne samic są
niewystarczające, szczególnie wówczas, gdy zewnętrzne narządy płciowe są
prawidłowo uformowane. Bardzo pomocne okazało się badanie cyto-
genetyczne, polegające na ustaleniu zestawu chromosomów płci w leuko-
cytach. Ze względu na wspólną cyrkulację krwi u płodów, których łożyska
są połączone anastomozami, dochodzi do zagnieżdżenia się komórek
macierzystych szpiku kostnego samicy w szpiku kostnym samca i odwrotnie.
Z tego powodu we krwi frymartynów, a także samców będących bliźniętami
frymartynów, stwierdza się obecność dwóch linii komórkowych — jednej
własnej, a drugiej współbłiźniaka. Linie te różnią się układem chromosomów
płci — w jednej występuje układ XX, a w drugiej układ XY. Ponieważ linie
te pochodzą od dwóch różnych genotypów, dlatego stan taki określany jest
jako chimeryzm leukocytarny XX/XY. Chimeryzm dotyczy również ery-
trocytów, które — jak wiadomo — pozbawione są u ssaków jąder komór-
kowych. Chimeryzm erytrocytarny można stwierdzić przez badanie grup
krwi. Postęp genetyki molekularnej umożliwia wykrywanie chimeryzmu
leukocytarnego nie tylko za pomocą metod cytogenetycznych, ale także
molekularnych (np. sekwencje mikrosatelitarne). Identyfikacja sekwencji
typowych dla chromosomu Y (np. sekwencja SRY) w DNA wyizolowanym
z komórek krwi samicy, przy jednoczesnym ich braku np. w komórkach
błony śluzowej, może być wskazówką, że badany osobnik jest frymartynem.
Powstawanie anastomoz jest bardzo często stwierdzane u bydła. Szacuje
się, że w przypadku ponad 90% ciąż bliźniaczych rozwijane są anastomozy.
Znacznie rzadziej zdarza się to u owiec (od l do 10%, zależnie od rasy). Biorąc
jednak pod uwagę, że ciąże mnogie występują bardzo rzadko u bydła
(poniżej 2% ciąż), a często u owiec (od 40 do blisko 100% ciąż, zależnie od
222

rasy), w konsekwencji częstość frymartynizmu u tych gatunków kształtuje
się na zbliżonym poziomie, w zakresie od 0,5% (bydło) do kilku procent
(niektóre rasy owiec, np. merynos booroola). Nieznane jest do tej pory
molekularne podłoże powstawania anastomoz. Z badań przeprowadzonych
na owcach wynika, że skłonność do tworzenia anastomoz może być uwarun-
kowana genetycznie. Wniosek taki wyciągnięto na podstawie pojawiania się
chimeryzmu leukocytarnego XX/XY u zwierząt spokrewnionych.
Mutacje chromosomów płci
Przyczyną rozwoju interseksualnego często są mutacje związane z chromo-
somami płci. Z jednej strony mogą to być aneuploidie, wśród których
najczęściej obserwuje się przypadki monosomii chromosomu X (zespół
chorobowy X0) oraz trisomii chromosomów płci (zespoły chorobowe XXY,
XYY lub XXX). U ludzi monosomia chromosomu X odpowiada za powstanie
zespołu Turnera, a trisomia XXY jest przyczyną rozwoju zespołu Klinefeltera.
Z drugiej strony ważną rolę odgrywają mutacje strukturalne, w które są
zaangażowane chromosomy płci. Przyczyny powstawania tych mutacji
zostały omówione w rozdziale: Mutacje. Aneuploidie chromosomów płci
prowadzą przede wszystkim do upośledzenia funkcji gonad. Zazwyczaj nie
towarzyszą im zaburzenia polegające na równoczesnym rozwoju struktur
typowych dla dwóch płci. Monosomia chromosomu X najczęściej jest
diagnozowana u klaczy, które są fenotypowo prawidłowo rozwinięte.
U nosicielek ruja nie występuje w ogóle lub występuje nieregularnie.
Gonady są niedorozwinięte i brak w nich rozwijających się pęcherzyków
jajnikowych. Zmiany te prowadzą do trwałej bezpłodności samic z mono-
somia chromosomu X. W grupie trisomii chromosomów płci najwięcej
przypadków dotyczy trisomii XXY. Kilkanaście takich przypadków opisano
u buhajów. Charakteryzowały się one gorszym tempem wzrostu, miały
niedorozwinięte jądra, a w nasieniu takich osobników zazwyczaj nie
stwierdzano plemników, co oczywiście prowadziło do bezpłodności. Z kolei
trisomie XXX oraz XYY zazwyczaj nie prowadzą do bezpłodności oraz nie
wywołują zmian fenotypowych.
Zespół niewrażliwości na androgeny
Mutacje genów zaangażowanych w proces determinacji i różnicowania płci
są przyczyną wielu przypadków interseksualizmu. Przykładem może być
zespół niewrażliwości na androgeny (zespół feminizujących jąder). Zwierzęta
obarczone tym zespołem chorobowym mają w swoich komórkach układ
chromosomów płci XY i prawidłowo rozwinięte zewnętrzne narządy płciowe
żeńskie, niedorozwinięte wewnętrzne narządy płciowe żeńskie oraz jądra.
Przyczyną tych zaburzeń jest mutacja zlokalizowanego w chromosomie X
genu kodującego receptor dla androgenów. Konsekwencją występowania
223

nieprawidłowego receptora dla androgenów jest nieodbieranie przez komór-
ki docelowe sygnału związanego z obecnością testosteronu i dihydrotestoste-
ronu, co prowadzi do różnicowania się wewnętrznych i zewnętrznych
narządów płciowych w kierunku żeńskim.
Zespół odwróconej płci u koni
W hodowli koni częstym typem interseksualizmu jest tzw. zespół odwróconej
płci (ang. sex reversal), który polega na niezgodności między płcią fenoty-
pową i chromosomową. Najczęściej zespół ten diagnozowany jest u fenoty-
powych samic, które mają układ chromosomów XY. Zewnętrzne narządy
płciowe są zazwyczaj prawidłowo rozwinięte, chociaż zdarzają się zwierzęta
z powiększoną łechtaczką. Wewnętrzne narządy płciowe wywodzą się
z przewodów Miillera i są prawidłowo wykształcone lub przejawiają oznaki
niedorozwoju. W zdecydowanej większości przypadków stwierdza się
niedorozwinięte gonady żeńskie, a w nielicznych przypadkach opisano
obecność niedorozwiniętych jąder lub jajnikojąder. Badania molekularne
genów położonych w chromosomie Y, z wykorzystaniem techniki PCR,
ujawniały u samic z zespołem odwróconej płci obecność genów charak-
terystycznych dla tego chromosomu (AMEL, ZFY, STS), przy jednoczesnym
braku kluczowego genu SRY. Przyczyny takiego stanu mogą być różne,
a wśród nich należy wymienić: mutację w locus SRY (np. częściowa lub
całkowita delecja), przeniesienie genu SRY podczas nierównoważonego
crossing over do chromosomu X lub translokację chromosomową między
chromosomem Y i autosomem.
U koni występują również przypadki odwrócenia płci u osobników
z układem chromosomów XX. Zewnętrzne narządy płciowe takich zwierząt
mogą być bardzo zróżnicowane, od prawie typowo samczych do samiczych,
ale z powiększoną łechtaczką. Zazwyczaj stwierdza się obecność niefunk-
cjonalnych jąder, w których kanaliki plemnikotwórcze są wyścielone jedynie
komórkami Sertolego. W genotypie tych zwierząt nie ma genu SRY, który
uznawany jest za niezbędny do rozwoju gonady męskiej. Taki typ od-
wrócenia płci spotykany jest również u kóz, świń i psów.
Interseksualizm bezrogich kóz
Ze szczególnym przypadkiem interseksualizmu można się spotkać w hodowli
kóz. Bezrożność kóz uwarunkowana jest przez dominujący allel P, a wy-
stępowanie rogów związane jest wyłącznie z genotypem homozygoty
recesywnej pp. Okazało się, że wśród osobników bezrogich często zdarzają
się zwierzęta obojnacze, które mają układ chromosomów płci XX, a w ich
genotypie nie stwierdza się genu SRY. Osobniki takie powinny być samicami,
tymczasem w procesie różnicowania płci występują zaburzenia dotyczące
zarówno zewnętrznych, jak i wewnętrznych cech płciowych, a stopień
224

nasilenia tych zmian jest bardzo zróżnicowany. Gonady zazwyczaj mają
strukturę jądra, a rzadko jajnikojądra. Jądra takich osobników nie wykazują
jednak aktywności spermatogenetycznej. Usytuowanie gonad może być
pachwinowe lub mogą być położone w worku mosznowym. Zewnętrzne
cechy płciowe mogą mieć nieomal prawidłowy charakter żeński i wówczas
odróżnienie takiego obojnaka od prawidłowej samicy jest praktycznie
niemożliwe. Czasami stwierdza się zwiększoną odległość między odbytem
i sromem lub znaczne powiększenie łechtaczki. W przypadku takich
osobników stosunkowo łatwo udaje się zdiagnozować interseksualizm.
Wreszcie mogą występować również osobniki, które mają zdecydowanie
samczy fenotyp. Są one trudne do odróżnienia od samców o prawidłowej
płci chromosomowej, czyli mających układ chromosomów płci XY, a w ich
genotypie występuje gen SRY. W wyniku przeprowadzonych badań nad
zmapowaniem locus genu bezrożności ustalono, że jest on położony
w chromosomie l — w części dystalnej. Ciekawe jest, że locus genu
bezrożności bydła został umiejscowiony również w chromosomie l, ale
w części proksymalnej (w pobliżu centromeru). Z badań porównawczych
wiadomo, że kariotypy obu gatunków są prawie identyczne, również
w zakresie układu loci w chromosomach. Można z tego wyciągnąć wniosek,
że za występowanie rogów odpowiedzialne są co najmniej dwa loci u tych
gatunków. Co więcej należy podkreślić, że u bydła występowanie rogów nie
wpływa na zakłócenie różnicowania się płci. W 2001 r. ukończono wieloletnie
badania, których celem była identyfikacja locus odpowiedzialnego za oboj-
nactwo bezrożnych kóz (tzw. gen PIS — ang. polledness and intersexuality
syndrome). Okazało się, że odpowiedzialna za to jest delecja 11 700 par
zasad, zlokalizowana w niekodującym fragmencie DNA, co zakłóca ekspresję
dwóch położonych w pobliżu genów: PISRT1 (zaangażowany w determina-
cję płci poprzez oddziaływanie na produkt genu SOX9) oraz FOXL2
(uczestniczy prawdopodobnie w rozwoju jajników w okresie płodowym).
Choroba białych jałowic
Innym przykładem genu o działaniu plejotropowym, który jest odpowie-
dzialny za rozwój interseksualny, jest allel R genu Roan (umaszczenie
dereszowate) u bydła. Allel R odpowiada za powstanie białego umaszczenia,
allel r
+
za umaszczenie czarne (u bydła belgijskiego błękitnego) lub czerwone
(u shorthornów). Allele te prezentują typ dziedziczenia pośredniego, z tym
jednak że genotyp Rr
+
wykazuje niepełną penetrację, tzn. około 9% białych
zwierząt ma genotyp heterozygotyczny i podobna proporcja heterozygot
ma umaszczenie barwne. U obu ras bydła stwierdzono, że allel R zakłóca
różnicowanie się przewodów Miillera w kierunku żeńskich wewnętrznych
narządów płciowych u jałówek. Zaburzenia te polegają na braku lub
niedorozwoju pochwy, szyjki macicy i macicy, natomiast jajniki są rozwinięte
prawidłowo. Zespół tych zmian jest określany jako choroba białych jałowic
225

(ang. white heifer disease), bowiem ponad 90% chorych jałówek ma
umaszczenie białe, a wśród pozostałych 10% przeważają jałówki umaszczone
niebiesko (belgijskie błękitne) lub mroziato (shorthorny). Dotychczasowe
obserwacje dowodzą, że choroba białych jałowic ma złożone podłoże,
w którym oprócz czynników genetycznych (allel R) istotną rolę odgrywają
również nie rozpoznane jeszcze wpływy środowiskowe. Locus genu Roan
umiejscowiono w chromosomie 5 w kariotypie bydła. Przypuszcza się, że
genem roan może być gen kodujący czynnik wzrostu komórek tucznych.
Locus tego genu został wcześniej zmapowany w regionie, w którym ostatnio
umiejscowiono locus genu Roan.
Interseksualizm u świń
Przypadki interseksualizmu u świń obserwuje się z częstością około 0,3%.
Tło genetyczne tych przypadków jest słabo poznane. Badania chromo-
somowe wykazują, że zdecydowana większość zwierząt obarczonych
interseksualizmem ma kariotyp żeński — 38,XX. Wśród nich niejednokrotnie
zdarzają się przypadki, gdzie gonady mają strukturę jądra lub jajnikojądra,
pomimo nieobecności genu SRY, a zewnętrzne i wewnętrzne narządy
płciowe są nieprawidłowo rozwinięte. Sytuacja taka wskazuje, że powstanie
gonady męskiej może nastąpić pomimo braku genu SRY. Zakłada się, że
produkt genu SRY wstrzymuje ekspresję nieznanego genu autosomałnego,
który koduje polipeptyd wpływający na zahamowanie ekspresji innych
genów uczestniczących w różnicowaniu płci męskiej. Zatem rozwój w kie-
runku męskim osobników z chromosomami XX byłby spowodowany tym,
że produkt tego zmutowanego genu nie hamuje rozwoju cech męskich.
W Polsce opisano powtarzające się przypadki takiego właśnie intersek-
sualizmu na fermie tuczu trzody chlewnej. W potomstwie jednego knura
pojawiło się kilkanaście obojnaków, w których kariotypie występowały dwa
chromosomy X. U zwierząt tych stwierdzano obecność jąder, pomimo braku
genu SRY w ich genotypie, macicy i nasieniowodów oraz zewnętrznych
narządów płciowych żeńskich, z tym jednak że łechtaczka była bardzo
powiększona, a srom tworzył z odbytem wspólny otwór (ryć. 9-3 wkładka
kolorowa). Rozwój interseksualizmu u świń stwierdzano także u osobników
z układem chromosomów XY lub chimeryzmem leukocytarnym 38,XX/38,XY.
Badania cytogenetyczne nie dają oczywiście wiążącej odpowiedzi na pytanie
o podłoże genetyczne różnych form interseksualizmu. Można natomiast
przypuszczać, że chimeryzm leukocytarny XX/XY wskazuje na frymartynizm,
a układ chromosomów XY i zewnętrzne narządy żeńskie są oznakami
zespołu niewrażliwości na androgeny.

Rozdział
10
Genetyczne podstawy odporności i
oporności
Układ odpornościowy jest pod względem „inteligencji" drugim po układzie
nerwowym układem komórkowym organizmu. Ma on, podobnie jak układ
nerwowy, zdolność uczenia się, zapamiętywania, reagowania na różne
bodźce i umiejętność oceny tych bodźców pod kątem ich oddziaływania na
organizm. Główną rolą tego układu jest rozróżnianie obcych białek (anty-
genów) i reakcja immunologiczna, której celem jest zwalczenie, usunięcie
tych obcych białek. Zastanawiające jest, iż układ może rozróżnić ogromną
liczbę antygenów i odpowiedzieć na infekcje, mimo że ma tylko pięć klas
przeciwciał. Zdolność ta wynika przede wszystkim ze specyficznego sposobu
genetycznej kontroli syntezy przeciwciał, ale także z posiadania przez
organizmy wyższe unikatowego układu genetycznego, jakim jest główny
układ zgodności tkankowej.
10.1. Genetyczne podłoże różnorodności
przeciwciał
Immunoglobuliny, czyli przeciwciała, wykryte w 1890 roku przez von
Behringa i Shibasaburo Kitasato, są grupą złożonych białek obecnych
w surowicy krwi i płynach tkankowych wszystkich ssaków. Ich wytwarzanie
przez limfocyty B jest stymulowane przez kontakt układu odpornościowego
organizmu z obcą immunogenetycznie cząsteczką (antygenem). Są one
elementem odporności swoistej (nabytej, adaptatywnej), ich zadaniem jest
rozpoznanie i neutralizacja obcych antygenów (np. bakterii czy wirusów).
Cząsteczka przeciwciała ma masę 150 kDa i składa się z dwóch łańcuchów
ciężkich (ang. heavy — H), po 50 kDa każdy, połączonych z dwoma
łańcuchami lekkimi (ang. light — L), o masie po 25 kDa. Jest pięć typów
łańcuchów ciężkich (gr.
α - - alfa, δ - - delta, ε - - epsilon, γ - - gamma
227
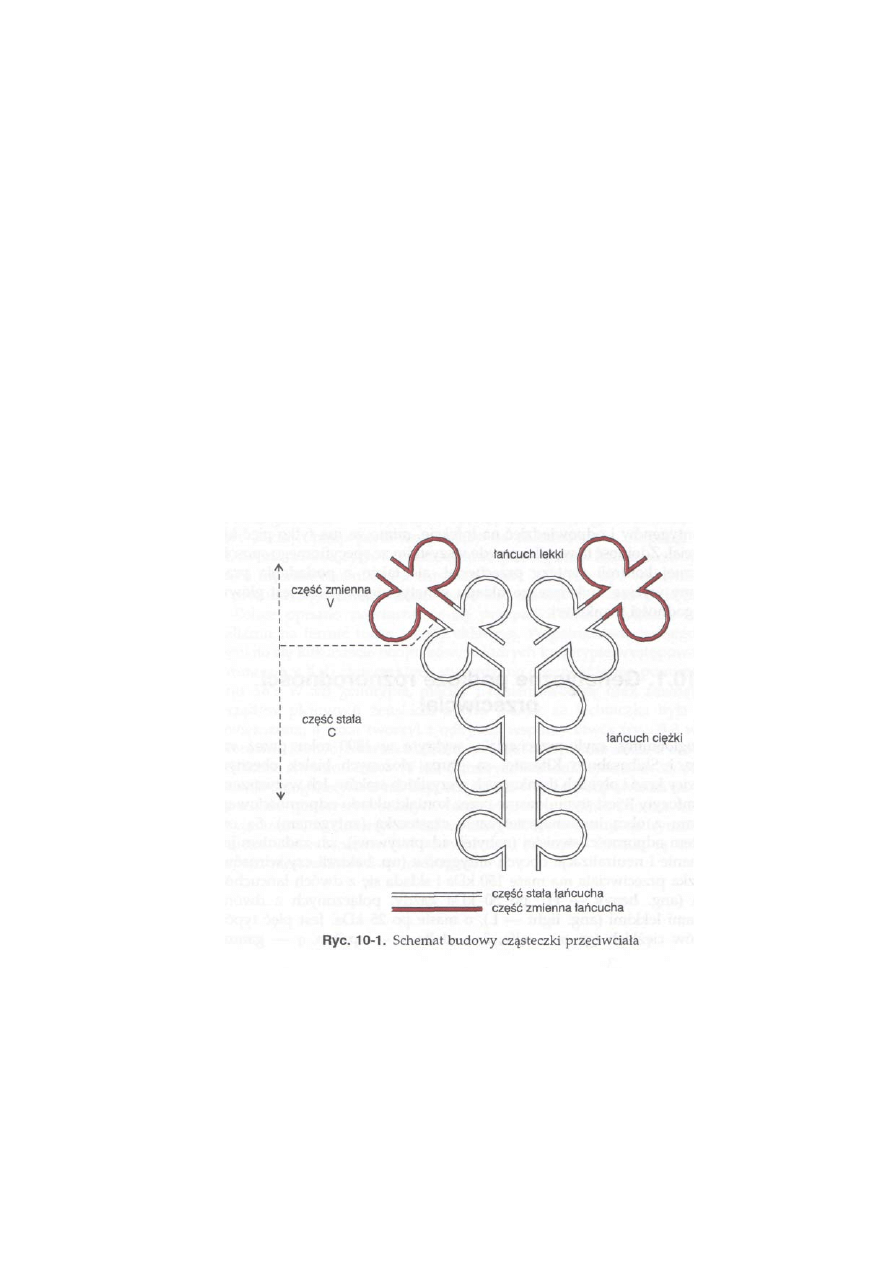
i
μ - - mi) i dwa typy lekkich (κ— kappa i λ. — lambda). Stosunek
przeciwciał, które mają łańcuch lekki typu
κ, do łańcucha
λ
zależy od
gatunku (np. u ludzi wynosi on 70:30, u myszy l: 20, u koni l: 25).
Immunoglobuliny dzieli się na podstawie różnic w budowie łańcuchów
ciężkich na 5 klas : IgA, IgD, IgE, IgG i IgM. Nazewnictwo to pochodzi od:
Ig — skrót od angielskiego słowa immunoglobulin, natomiast A, D, E,
G i M od symboli łańcuchów ciężkich.
Przeciwciało ma kształt litery Y (ryć. 10-1), utworzonej przez łańcuchy
ciężkie, a wzdłuż jej ramion znajdują się łańcuchy lekkie. Łańcuchy ciężkie
połączone są ze sobą wiązaniami dwusiarczkowymi, których liczba i miejsce
zależą od klasy i podklasy immunoglobuliny. Zarówno łańcuchy ciężkie,
jak i lekkie mają części zmienne (ang. variable — V) oraz części stałe (ang.
constant — C). W części zmiennej znajdują się trzy regiony hiperzmienne
(ang. hypervariable), oznaczone HV1, HV2 i HV3. Ponieważ regiony te
determinują swoistość przeciwciał, nazywane są także regionami deter-
minującymi dopasowanie (ang. complementarity determining regions -
CDR). W części stałej łańcucha ciężkiego, między domeną CH1 i CH2,
znajduje się tzw. region zawiasowy (ang. hinge region), w którym są
wiązania dwusiarczkowe łączące oba łańcuchy ciężkie.
Genetyczne podłoże powstawania przeciwciał zostało wyjaśnione przez
Susumu Tonegawa w 1976 roku. Badacz ten odkrył, iż informacja genetyczna
228

immunoglobulin zawarta jest w znajdujących się w różnych chromo-
somach czterech grupach „minigenów", które nazwał segmentami. Róż-
norodność przeciwciał, ich swoistość wynikają z tego, iż dziedziczone
są nie całe geny immunogłobulinowe, ale ich segmenty. Proces syntezy
przeciwciała następuje po skompletowaniu genów, czyli połączeniu
się segmentów po jednym z każdej grupy minigenów. Dopiero po
połączeniu w całość segmenty te tworzą kompletny funkcjonalny gen.
Proces ten zachodzi w poszczególnych limfocytach B podczas ich doj-
rzewania. Za to odkrycie Tonegawa otrzymał w 1987 roku Nagrodę Nobla.
Część zmienna łańcucha lekkiego jest determinowana przez dwa geny
V (ang. variable) i J (ang. joining), natomiast łańcucha ciężkiego przez
trzy geny: V, D (ang. diversity) i J. Część stała obu rodzajów łańcuchów
jest kodowana przez gen C. Geny kodujące łańcuch ciężki znajdują
się u człowieka w 14 chromosomie, natomiast geny łańcucha lekkiego
typu
κ
w 2, typu
λ w 22 chromosomie, u myszy natomiast odpowiednio
w chromosomach 2, 6 i 16.
Różnorodność przeciwciał wynika z wielu przyczyn, przede wszystkim z:
• rearanżacji genów części zmiennych (V) i stałych (C) łańcuchów
immunoglobulin
• rekombinacji między wieloma genami V
• insercji minieksonów
• mutacji somatycznych
Różnorodność ciężkich łańcuchów immunoglobulinowych wynika z re-
kombinacji (rearanżacji) genów V, D, J i C, a łańcuchów lekkich z rekom-
binacji regionów V, ] i C. Teoretycznie jest możliwość powstania do 10
n
różnych immunoglobulinowych sekwencji domen. Jednak rzeczywista liczba
powstałych wariantów przeciwciał wynosi 10
7
-10
8
. Przypuszczalnie jest
wiele genów kodujących region V. Dla łańcucha ciężkiego ich liczba nie jest
dokładnie znana (kilkadziesiąt, może kilkaset), dla lekkiego — kilka. Istnieją
co najmniej 3 rodziny genów (liczące 1-9 genów każda) dla regionu D,
6 genów funkcjonalnych i 3 pseudogeny regionu J, natomiast dla regionu
C > 30 genów u ludzi i > 300 u myszy dla łańcucha lekkiego typu
K
oraz
6 u ludzi i 4 u myszy dla łańcucha typu A,. Ogólnie liczbę genów
determinujących poszczególne regiony łańcuchów immunoglobulin można
określić jako kilkaset genów V, kilkadziesiąt genów D i kilka genów J. Geny
determinujące region C różnią się od pozostałych tym, że wpływają na
funkcję przeciwciała, a nie na jego powinowactwo do antygenu.
Kompletne funkcjonalne geny kodujące cząsteczki przeciwciał powstają
z omówionych wyżej genów, których kolejność łączenia się jest następująca:
gen D z genem J, następnie z genem V; powstały kompleks DJV łączy się
z genem kodującym część stałą łańcucha ciężkiego. W funkcjonalnym genie
łańcucha lekkiego nie ma genu D (ryć. 10-2). Rekombinacyjne łączenie się
genów, zachodzące dzięki aktywności enzymu — rekombinazy, jest głów-
nym źródłem różnorodności przeciwciał.
229
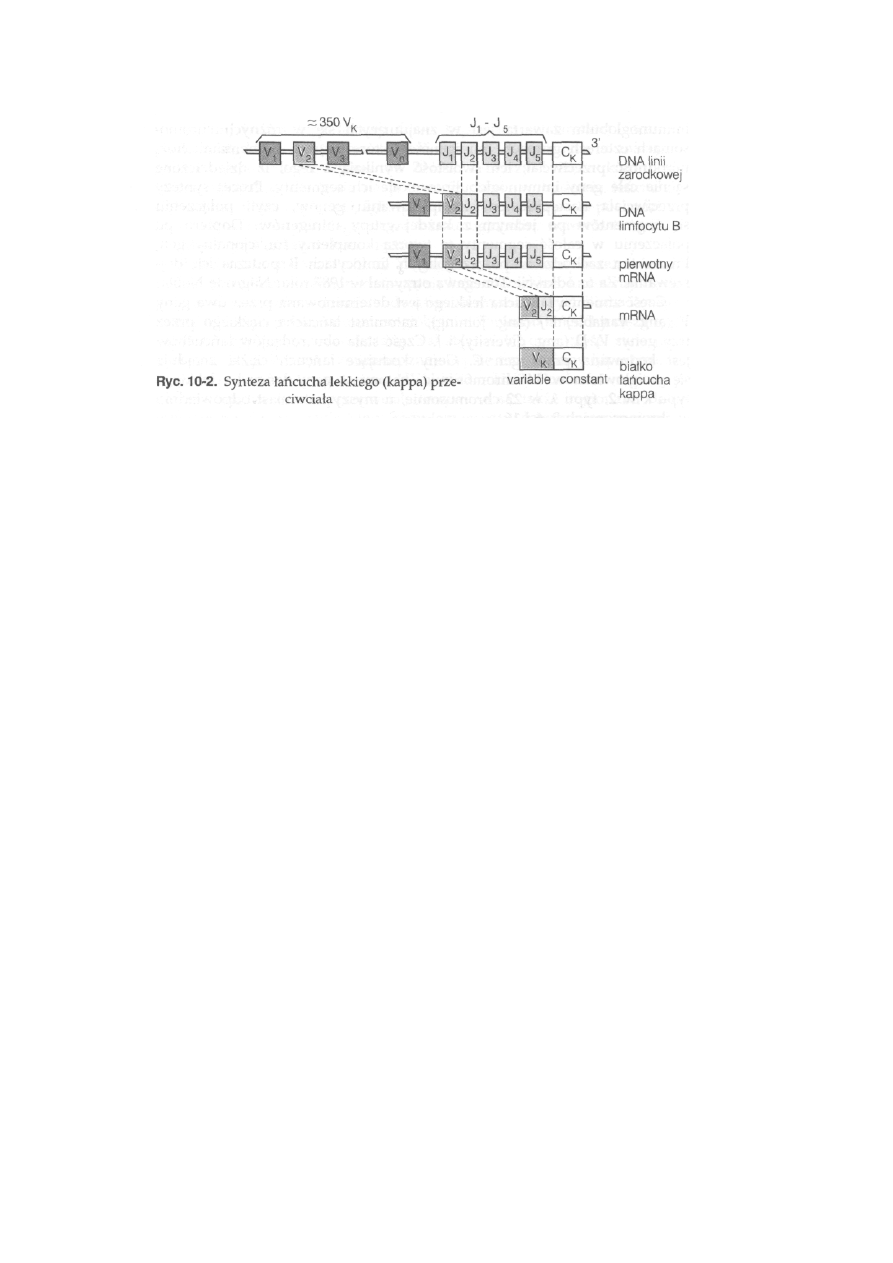
Podczas procesu powstawania genu kompletnego do połączeń między
genami — segmentami (V-J, V-D i D-J) mogą być wstawiane nukleotydy (od
l do 15), bądź też usuwane (do około 20 nukleotydów). Ponieważ są to
nowo powstałe odcinki DNA, prowadzi to do zmienności na złączach
i utworzenia nowego genu, tym samym liczba wariantów genu D] może być
zwiększona około 10 razy, natomiast genu VDJ następne 10 razy.
Dodatkowo na różnorodność sekwencji przeciwciał mają wpływ mutacje
somatyczne zachodzące w rekombinowanym genie VJ (dla łańcucha lek-
kiego) i VDJ (dla łańcucha ciężkiego). Każdy limfocyt B wytwarza tylko
jeden rodzaj łańcucha ciężkiego i jeden lekkiego, które razem stanowią
przeciwciało, będące specyficznym receptorem tego limfocytu. Geny kodu-
jące łańcuchy mają zdolność niezwykle szybkiego mutowania w odpowiedzi
na pobudzenie limfocytu B poprzez związanie obcego białka (antygenu).
Najczęściej są to mutacje punktowe, powodujące zmianę pojedynczego
aminokwasu, rzadziej natomiast delecje, insercje lub konwersje. Pojedyncza
nawet zmiana aminokwasu w części zmiennej łańcucha ciężkiego może
doprowadzić do całkowitej utraty przez przeciwciało zdolności wiązania
antygenu lub też do zwiększenia jego powinowactwa do antygenu.
10.2. Genetyczne podłoże różnorodności
receptorów limfocytów J
W odpowiedzi immunologicznej organizmu biorą udział dwie populacje
limfocytów: limfocyty T i limfocyty B, przy czym limfocyty T uczestniczą
w odpowiedzi typu komórkowego, atakując i niszcząc komórki zakażone
(T
c
— cytotoksyczne limfocyty T), oraz odpowiedzi typu humoralnego
230

(T
h
— pomocnicze limfocyty T), podobnie jak limfocyty B, które odpowiadają
za syntezę przeciwciał.
Dwie subpopulacje limfocytów T (T
c
i T
h
) różnią się receptorami
błonowymi. Limfocyty pomocnicze mają receptory CD4, cytotoksyczne -
CD8. Reaktywność tych receptorów znajdujących się na limfocytach
pomocniczych i cytotoksycznych jest skierowana odpowiednio na
antygeny MHC klasy II i klasy I.
Proces rearanżacji elementów genów receptorowych, który następuje
podczas dojrzewania komórek T w grasicy, zachodzi podobnie jak rekom-
binacja genów przeciwciał. Poprzez proces rearanżacji genów, jeden do
czterech elementów zostaje włączonych do genów kodujących pierwotną
strukturę białek receptora komórki T. Znane są dwa rodzaje receptorów
limfocytów T — receptory składające się z łańcuchów
α i β oraz receptory
składające się z łańcuchów
γ i δ. U człowieka geny kodujące łańcuchy
receptorów znajdują się w chromosomie 7 (dla łańcuchów
β i γ) i 14 (dla
łańcuchów
α i δ). Części zmienne łańcuchów kodowane są przez
geny V i J (łańcuchy a i y) i przez geny V, D i J (łańcuchy p i §).
Na przykład u myszy 100 regionów łańcucha
α
-V i 50 regionów
łańcucha
α-J może tworzyć do 5000 różnych genów dla łańcucha α.
Podobnie, około 500 różnych genów kodujących łańcuch
β może być
utworzonych z 20 genów
β-V, 2 regionów D i 12 genów dla łańcucha β-J.
Oczywiście jest mało prawdopodobne, że wszystkie elementy genów są
włączone w rearanżacje, ale teoretycznie losowe kombinacje łańcuchów
α
i
β mogą dać do 2,5 x 10
6
α-β heterodimerów. Gdyby zostały wzięte pod
uwagę wszystkie mechanizmy biorące udział w tworzeniu różnorodności
struktury receptorów limfocytów T, liczba różnych receptorów wyniosłaby
do 10
15
receptorów
α-β i 10
18
γ-δ. Potencjał dla różnorodności receptorów
komórek T jest istotnie wyższy niż wynosi liczba komórek T obecnych w
organizmie.
10.3. Główny układ zgodności tkankowej — MHC
Główny układ zgodności tkankowej został odkryty w wyniku badań nad
genetycznym tłem przyjmowania lub odrzucania przeszczepu u myszy.
Bodźcem do podjęcia tych badań było odkrycie przez Landsteinera grup
krwi systemu ABO i wyjaśnienie zjawiska aglutynacji krwinek
czerwonych biorcy pod wpływem surowicy krwi dawcy. Prowadzone w
latach 30. przez Petera Gorera poszukiwania innych antygenów
grupowych krwi, odpowiedzialnych za przyjęcie przeszczepu,
doprowadziły do wykrycia grup (klas) antygenów oznaczonych I, II i III.
Przełomowe znaczenie dla poznania i wyjaśnienia roli tych antygenów
miało wytworzenie przez Georga Snella linii kongenicznych myszy
różniących się tylko locus zgodności tkankowej, który kontroluje ich
syntezę. Badania polegające na przeszczepach skórnych
231

między tymi liniami, wykonywanych dla kilkunastu kolejnych pokoleń,
wykazały, iż reakcja na ten zabieg zależy przede wszystkim od locus
głównego układu zgodności tkankowej (ang. major histocompatibility
complex — MHC), a tylko w niewielkim stopniu od innych loci, nazwanych
słabymi antygenami zgodności tkankowej (ang. minor histocompatibility
loci). Za badania nad MHC Snęli wraz z Francuzem Daussetem i Ameryka-
ninem Benacerrafem otrzymali w 1980 roku Nagrodę Nobla.
Różnice między słabymi antygenami zgodności tkankowej a MHC są
zarówno natury jakościowej, jak i ilościowej. Geny słabych antygenów
zgodności tkankowej nie są jeszcze dobrze poznane i, być może dlatego, często
ich efekt jest niedoceniany. Tymczasem niektóre z tych antygenów stymulują
silniejszą odpowiedź transplantacyjną niż antygeny MHC. W zasadzie
antygeny minor H loci nie biorą udziału w reakcji immunologicznej
prowadzącej do syntezy przeciwciał, natomiast mogą być rozpoznawane
przez limfocyty T w połączeniu z antygenami MHC, podobnie jak to
występuje przy rozpoznawaniu antygenów wirusowych. U myszy loci słabych
antygenów zgodności tkankowej (minor H loci) są najlepiej poznane
i wiadomo, że większość z nich znajduje się w chromosomie 2. Do słabych
antygenów transplantacyjnych myszy należą między innymi: H-Y (tylko
u samców), H-l, H-3, H-5 i inne. Antygeny te są istotną barierą podczas
transplantacji tkanek, zwłaszcza transplantacji szpiku kostnego. Nawet
wówczas, gdy biorca i dawca są zgodni pod względem haplotypów MHC,
słabe antygeny transplantacyjne mogą spowodować odrzucenie przeszczepu.
W dziedziczeniu słabych antygenów zgodności tkankowej obserwuje się
pewne charakterystyczne zjawiska. Antygen H-Y występuje tylko u samców,
gdyż gen kodujący go jest zlokalizowany w chromosomie Y. Zatem jest on
przekazywany przez ojca męskim potomkom. Natomiast inny antygen,
nazwany Mta (ang. maternally transmitted antigen), pochodzi tylko od
matki, ponieważ jest on kontrolowany przez gen mitochondrialny (zjawisko
to jest omówione w rozdziale: Podstawowe mechanizmy dziedziczenia cech).
Tabela 10-1
Lokalizacja MHC i nazwa u poszczególnych gatunków
232

Ponieważ główną funkcją genów MHC jest determinowanie antygenów
leukocytarnych (ang. leukocyte antigens — LA), nazewnictwo tego układu
u poszczególnych gatunków stanowi symbol zawierający w pierwszym
członie nazwę gatunku, a drugim podstawową funkcję — LA (tab. 10-1).
Główny układ zgodności tkankowej jest ogromnie konserwatywnym
regionem DNA, jego struktura i funkcje są podobne u ludzi i wszystkich
gatunków zwierząt. Umożliwia to prowadzenie badań molekularnych MHC
u jednego gatunku z zastosowaniem sond molekularnych uzyskanych
z DNA innego gatunku.
10.3.1. Struktura i funkcje głównego układu
zgodności tkankowej
U ludzi i zwierząt MHC jest odcinkiem DNA składającym się ze specyficz-
nych regionów genów kodujących trzy klasy białek — klasę I, II i III,
będących glikoproteinami. U drobiu nie stwierdzono regionu klasy III.
Wykryto natomiast specyficzny region klasy IV, kodujący białka zlokalizo-
wane na krwinkach czerwonych. Z powodu powiązań tego układu z ukła-
dem grupowym krwi B, MHC u drobiu nazwano kompleksem B. Ponieważ
wiele białek MHC zostało zidentyfikowanych metodą serologiczną, często
są one nazywane antygenami MHC. Białka (cząsteczki) MHC tak ze względu
na podobieństwo na poziomie struktury białka, jak i na poziomie genów
zaliczane są do nadrodziny immunoglobulin.
Mechanizm dziedziczenia układu zgodności tkankowej jest taki jak
w przypadku jednej pary alleli. Potomek otrzymuje od każdego rodzica po
jednym chromosomie z każdej pary chromosomów homologicznych, a wraz
z nim — haploidalny genotyp MHC (haplotyp). Ponieważ MHC zawiera
wiele genów oraz, jak już wspomniano, układ ten cechuje ogromny
polimorfizm, w populacji istnieje bardzo duża liczba różnych haplotypów.
Zwłaszcza niektóre geny MHC, należące do klasy I i II (np. regionu K,
D i L u myszy i B, C i A u ludzi), są wysoce polimorficzne. W wielu
przypadkach polimorfizm ten jest wynikiem konwersji genów, rzadziej
mutacji punktowych, crossing over czy rekombinacji wewnątrzgenowych.
W obrębie MHC znajdują się regiony szczególnie często uczestniczące
w rekombinacjach genetycznych.
Polimorfizm alleliczny obserwowany w loci MHC charakteryzuje się
tym, iż:
• liczba alleli w większości loci jest bardzo duża (> 50)
• allele często różnią się dużą liczbą mutacji punktowych typu zmiany
sensu, powodujących podstawienia aminokwasów w cząsteczce MHC (> 20
dla loci klasy II i > 30 dla loci klasy I); w większości innych systemów
genetycznych allele różnią się nielicznymi podstawieniami nukleotydów
233

• polimorfizm występuje głównie w odcinkach istotnych funkcjonalnie,
a więc w tych, które kodują miejsce wiązania peptydów; zjawisko polimor-
fizmu w innych systemach genetycznych częściej obserwowane jest w re
gionach, których znaczenie funkcjonalne jest mniej istotne
• polimorfizm w obrębie MHC, co zostało już udowodnione, wpływa
na możliwość immunologicznego rozpoznania obcych białek.
Mimo iż początkowo uważano, że MHC pełni funkcję tylko w odrzucaniu
przeszczepu, obecnie wiadomo, że białka kodowane przez ten region biorą
udział w mechanizmach rozpoznania immunologicznego, w tym także
w interakcji różnych komórek limfoidalnych oraz limfocytów i komórek
prezentujących antygen.
Cząsteczki MHC klasy I są obecne na powierzchni wszystkich komórek
jądrzastych i warunkują zachowanie się cytotoksycznych limfocytów T,
biorąc tym samym udział w reakcji przeciwko infekcji wirusowej, a także
w odrzucaniu przeszczepu. Cząsteczki te składają się z dwóch łańcuchów
polipeptydowych — lekkiego i ciężkiego (ryć. 10-3). Łańcuch ciężki jest
kodowany przez geny MHC, natomiast związany z nim niekowalencyjnie
łańcuch lekki p
2
-mikroglobuliny (białko o masie 12 kDa) jest determinowany
przez gen znajdujący się poza układem MHC.
Łańcuch ciężki składa się z trzech części:
• Najdłuższy, N-końcowy fragment łańcucha ciężkiego, stanowi około
80% jego długości i znajduje się na zewnątrz komórki. Jest on kodowany
przez geny MHC i ma trzy koliste domeny, oznaczone symbolami
αl,
α2 i α3, każda złożona z około 90 aminokwasów. Domena α3 jest
ściśle sprzężona z
β
2
-mikroglobuliną. Ma ona wewnątrzłańcuchowe
dwusiarczkowe wiązanie i podobną strukturę trzeciorzędową do domeny
immunoglobuliny. Domeny
αl i α2 odznaczają się polimorfizmem,
który jest podstawą różnic między cząsteczkami MHC klasy I
pochodzącymi od różnych osobników. W domenach tych występują
alloantygeniczne miejsca,
234
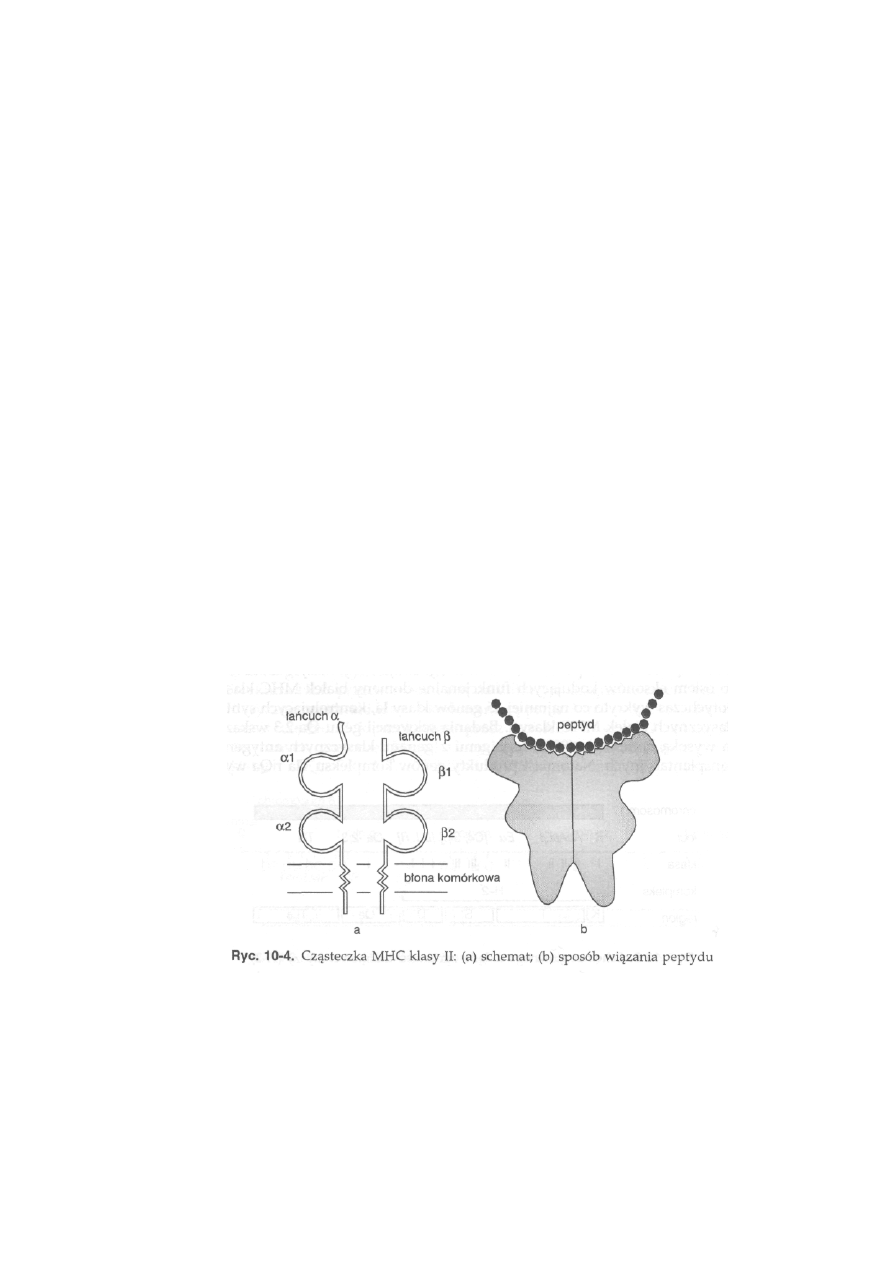
zawierające determinanty specyficzne dla osobników. Fragment
węglowodanowy (CHO) jest dołączony do domeny
α2. Domeny αl i
α2 tworzą rowek osadzony na β
2
-mikroglobulinie i domenie
α3, które
są między nim a błoną komórkową. W rowku tym wiązane są antygeny,
które następnie są prezentowane limfocytom T.
• Druga część łańcucha ciężkiego to zawierający około 20
aminokwasów fragment hydrofobowy, przechodzący przez błonę
komórkową.
• Trzecią częścią jest liczący około 20-40 aminokwasów
fragment hydrofilowy cząsteczki MHC klasy I (C-końcowy), który leży w
cytoplazmie komórki.
Cząsteczki MHC klasy II, stwierdzane tylko na powierzchni
wyspecjalizowanych komórek układu odpornościowego (makrofagi,
komórki den-drytyczne i limfocyty B), regulują immunologiczne
rozpoznanie antygenów za pomocą limfocytów B i T. Cząsteczki te są
zbudowane z dwóch niekowalencyjnie połączonych łańcuchów o
podobnej budowie, oznaczonych symbolami
α i β, kontrolowanych
przez geny zlokalizowane w obrębie MHC (ryć. 10-4). Łańcuchy te,
podobnie jak łańcuch ciężki cząsteczki klasy I, składają się z trzech
części: zewnątrzkomórkowej N-końcowej,
śródbłonowej i
wewnątrzkomórkowej. Oba łańcuchy mają w części N-końcowej po
dwie koliste domeny. Domeny
α2 i α2 są strukturalnie podobne do
domen części stałych łańcuchów ciężkich im-munoglobulin. Domeny
zewnętrzne (
αl i βl) obu łańcuchów tworzą rowek podobny do
tworzonego przez domeny
αl i α2 łańcucha ciężkiego białek MHC
klasy I. Polimorfizm cząsteczek MHC klasy II dotyczy przede wszystkim
domen
αl i βl. Cząsteczki MHC klasy II mają zdolność wytwarzania
dimerów złożonych z dwóch łańcuchów a i dwóch łańcuchów
β.
235

Zarówno białka MHC klasy I, jak i klasy II prezentują obcy peptyd
(antygen) limfocytom T. Jednak cząsteczki klasy I mogą wiązać jedynie
krótki, złożony z kilku aminokwasów peptyd, ponieważ mają zamknięte
miejsce wiążące. Natomiast białka MHC klasy II mogą wiązać peptydy
różnej długości, od kilkunastu do ponad 20 aminokwasów, gdyż ich miejsce
wiążące pozostaje otwarte z obu stron.
Cząsteczki MHC klasy III wchodzą w skład układu dopełniacza
(komplementu). Zadaniem układu dopełniacza jest uzupełnianie (do-
pełnianie) roli przeciwciał w zakresie unieszkodliwiania antygenu. W jego
skład wchodzą białka surowicy i płynów tkankowych, których jest
łącznie z czynnikami regulującymi około 30. Białka te są oznaczane
literą C (ang. complement) i liczbą. Białka C2, C4 i czynnik B (BF)
u ludzi są kontrolowane przez loci MHC klasy III, u myszy natomiast
przez loci regionu S w obrębie H-2.
10.3.1.1. Główny układ zgodności tkankowej myszy
Badania głównego układu zgodności tkankowej były prowadzone począt-
kowo u myszy, a ich wyniki miały ogromne znaczenie dla późniejszych
badań MHC człowieka, a potem również zwierząt gospodarskich.
Pierwotnie w MHC u myszy zidentyfikowano regiony K, I, S i D, które
określono symbolem H-2 (ryć. 10-5). Późniejsze badania ujawniły, że także
geny w regionach sprzężonych Qa i Tlą kodują cząsteczki MHC. Geny
kodujące białka MHC klasy I znajdują się w regionach H-2K i H-2D (tzw.
klasyczne cząsteczki klasy I, oznaczane symbolem la). Geny zlokalizowane
w regionach Qa ł Tlą kodują cząsteczki zaliczane także do klasy I, z tym
jednak że określane są one jako cząsteczki nieklasyczne (Ib), niektóre z nich
też biorą udział w odpowiedzi immunologicznej. Geny klasy I MHC mają
po osiem eksonów kodujących funkcjonalne domeny białek MHC klasy I.
Dotychczas wykryto co najmniej 15 genów klasy la, kontrolujących syntezę
klasycznych białek MHC klasy I. Badania sekwencji genu Qa-2,3 wskazują
na wysoką homologię (80%) tego genu z genami klasycznych antygenów
transplantacyjnych. Natomiast produkty genów kompleksu Tlą i Qa wyka-
236

żują wiele różnic w porównaniu z antygenami transplantacyjnymi. W
regionie Qa-2,3 i Tla zmapowano dotychczas 31 genów.
Gen kodujący łańcuch lekki (
β
2
-mikroglobulinę) cząsteczek klasy I,
bardzo istotny dla ekspresji produktów genów klasy I, znajduje się
poza MHC, u myszy w 2, u człowieka w 15 chromosomie, w grupie
sprzężeniowej z locus dehydrogenazy sorbitolu. Gen ten u ludzi składa
się z ośmiu eksonów, natomiast u myszy z trzech eksonów. Sekwencja
aminokwasowa
β
2
-mikroglobuliny wykazuje wysoką homologię z domeną
immunoglobulin i odpowiednimi domenami antygenów
transplantacyjnych klasy I.
Białka klasy II są determinowane przez geny zlokalizowane w sub-
regionach H2-A i -E, -P, -O/N oraz -M. Każda podklasa ma co najmniej
jeden gen łańcucha
α i β, z wyjątkiem H2-P, który ma pojedynczy
pseudogen oznaczony symbolem H-2Pb. Wzór eksonów i intronów genów
klasy II wskazuje na bliską współzależność między tymi genami i
podobieństwa struktury z genami klasy I oraz genem
β
2
-mikroglobuliny.
Białka klasy III są kontrolowane przez geny regionu S.
Wśród regionów H-2 na szczególną uwagę zasługuje region I (między
regionami K i S), w którym znajdują się geny kontrolujące odpowiedź
immunologiczną (wytwarzanie przeciwciał przeciwko ponad 40 różnym
antygenom), oznaczone symbolem Ir (ang. immune response).
10.3.1.2. Główny układ zgodności tkankowej człowieka
•
U ludzi MHC (HLA) znajduje się w krótkim ramieniu chromosomu 6,
zajmuje łącznie około 4 mpz DNA (4 miliony par zasad), czyli ponad 0,1%
całego genomu. Częstość rekombinacji między skrajnymi końcami tego
kompleksu genów wynosi około 1%.
Struktura HLA przedstawiona jest na liniowym diagramie (ryć. 10-6).
Klasyczne cząsteczki klasy I kodowane są przez 3 loci — HLA-A, -B i -C,
natomiast cząsteczki kontrolowane przez loci HLA-E, -F i -G należą do
nieklasycznych cząsteczek klasy I. Geny klasy II HLA znajdują się w pięciu
podklasach: DQ, DR, DP, DOB/DNA i DM, przy czym trzy
pierwsze
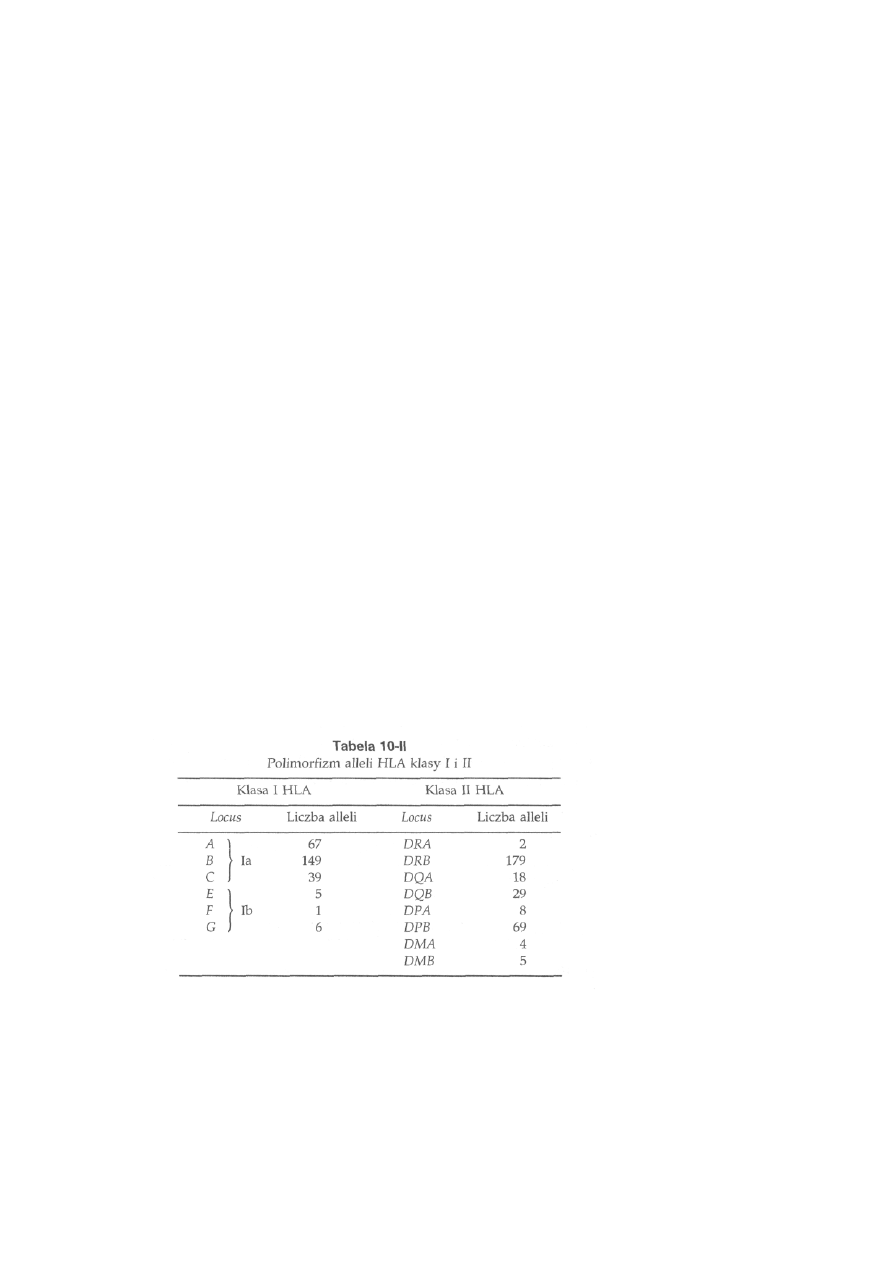
kodują klasyczne cząsteczki klasy II, podczas gdy czwarta podklasa,
DOB/DNA, determinuje syntezę nieklasycznych cząsteczek klasy II. Od-
kryty niedawno locus DM jest nieco inny niż pozostałe, bowiem jego
sekwencja jest w takim samym stopniu podobna do genów klasy I, jak i do
genów klasy II. Cząsteczka MHC kodowana przez ten locus odgrywa
szczególną rolę w prezentacji antygenów, co potwierdza fakt, iż komórki
nie wykazujące ekspresji tej cząsteczki mają upośledzoną zdolność
prezentacji antygenów.
Podobnie jak u myszy, region klasy II HLA zawiera także geny, które
strukturalnie nie należą do genów klasy II. Najbardziej interesujące z im-
munologicznego punktu widzenia są geny włączone w przetwarzanie
własnych białek komórkowych. Biorą one udział w proteolizie białek, m.in.
białek transkrypcyjnych oraz białek nieprawidłowych. Są to geny
TAP1 i TAP2 kodujące peptydowe transportery, przenoszące peptydy
(pocięte białka) do siateczki śródpłazmatycznej w celu prezentacji ich przez
cząsteczki klasy I MHC. Dwa inne geny LMP2 i LMP7 kodują podjednostki
kompleksu białek cytoplazmatycznych nazywanych proteasomami. O
szczególnej roli proteasomu świadczy określanie go jako
„wewnątrzkomórkowego układu immunologicznego".
Dotychczas wykryto 267 allełi klasy I HLA, 314 alleli klasy II i 40
alłeli klasy III. Ta liczba alleli wynika z ogromnego polimorfizmu genów
MHC. Stwierdzono na przykład, że geny HLA-A, -B, -DRB i -DPB
występują w postaci kilkudziesięciu alleli (tab. 10-11). Nieliczne z
genów MHC, na przykład DRA, mają tylko 2 allele. Jest pewnym
paradoksem, iż cząsteczka DR jest kodowana przez wysoko
polimorficzny gen DRB (determinujący syntezę łańcucha
β) oraz
dialleliczny gen DRĄ (kontrolujący syntezę łańcucha
α).
Ponadto w obrębie HLA wykryto także kilkanaście pseudogenów, nie
podlegających transkrypcji (np. HLA klasy II — DPB2 i DPA2) oraz geny
kodujące inne białka, m.in. geny dla 21-hydroksylazy (CYP21), białek
szoku
238

cieplnego 70 (HSP70) i dwa geny dla czynnika martwicy nowotworu (TNFA
i TNFB).
Jak wspomniano na początku tego podrozdziału, główny układ zgodności
tkankowej jest regionem względnie konserwatywnym i istnieje wiele
homologii między MHC u ludzi i różnych gatunków zwierząt. Na przykład
homologia genów klasy II u ludzi i myszy jest następująca:
myszy: Aa A
β
E
α
E
β P O/N M
ludzie: DQ
α DQβ DKα DRβ DP DOB/DNA DM
10.3.1.3. Główny układ zgodności tkankowej
zwierząt gospodarskich
Główny układ zgodności tkankowej zwierząt gospodarskich jest mniej
poznany. Wiadomo jednak, iż homologia między poszczególnymi gatunkami
i człowiekiem jest duża. Porównanie genów znajdujących się w obrębie
MHC u poszczególnych gatunków zwierząt zawarto w tabeli 10-111.
Główny układ zgodności tkankowej bydła
Główny układ zgodności tkankowej bydła zlokalizowano w 23 chromosomie,
w którym znanych jest także ponad 30 genów strukturalnych i kilkanaście
markerów mikrosatelitarnych. Jest to najlepiej poznany chromosom bydła.
Liczba poznanych haplotypów BoLA jest już bardzo długa i stale się
zwiększa. Struktura BoLA przedstawiona jest na rycinie 10-7.
Produkty genów klasy I są to klasyczne cząsteczki MHC. Nie wykryto
dotąd cząsteczek klasy I będących nieklasycznymi cząsteczkami. Geny klasy
I należą do dwóch grup — A i B, różniących się delecją 6 par zasad
w eksonie 5, który koduje krótki fragment transbłonowy łańcucha cząsteczki
239

MHC. W obrębie klasy II obserwowany jest duży polimorfizm genów. Za
pomocą analizy molekularnej (RFLP) zidentyfikowano już 46 haplotypów
DRB, 26 DQB i 31 DQA. W locus DRB wykryto pseudogen (DRB1), różniący
się od genów tego locus kodonem „stop" w eksonie kodującym transbłonowy
fragment łańcucha cząsteczki MHC. Homologia między genami BoLA i HLA
jest stosunkowo duża. Na przykład gen DRB3 wykazuje 85% homologii
z genem HLA — DRB1, natomiast DQA bydła i człowieka — 75% homologii.
Główny układ zgodności tkankowej świni
Główny układ zgodności tkankowej świń został zmapowany w chromosomie
7 ponad ćwierć wieku temu. Regiony klasy I i II znajdują się blisko siebie,
odległość między nimi wynosi 0,4 cM. Natomiast odległość między genami
klasy I i III oraz III i II jest co najmniej o 100 tyś. par zasad mniejsza niż
w MHC u ludzi. Nie jest znana dokładna liczba genów klasy I SLA, szacuje
się, że jest ich około 7-10. Tylko kilka genów regionu klasy I wykazuje
ekspresję. Są to PD1, PD7 i PD14, których homologia wynosi około 80-85%,
i bardziej różniące się od nich (podobieństwo około 50%) PD6 i PD15.
Wydaje się, że region klasy I SLA jest mniejszy od analogicznego regionu
u ludzi i myszy.
Region klasy II zawiera wiele genów, spośród których przypuszczalnie
tylko DR i DQ wykazują ekspresję na poziomie białka. Niektóre geny, DQA,
DQB, DRĄ i DRB, mają dużą homologię, w granicach 75-87%, z od-
powiadającymi im genami HLA. Podobnie jak u innych gatunków, w SLA
240
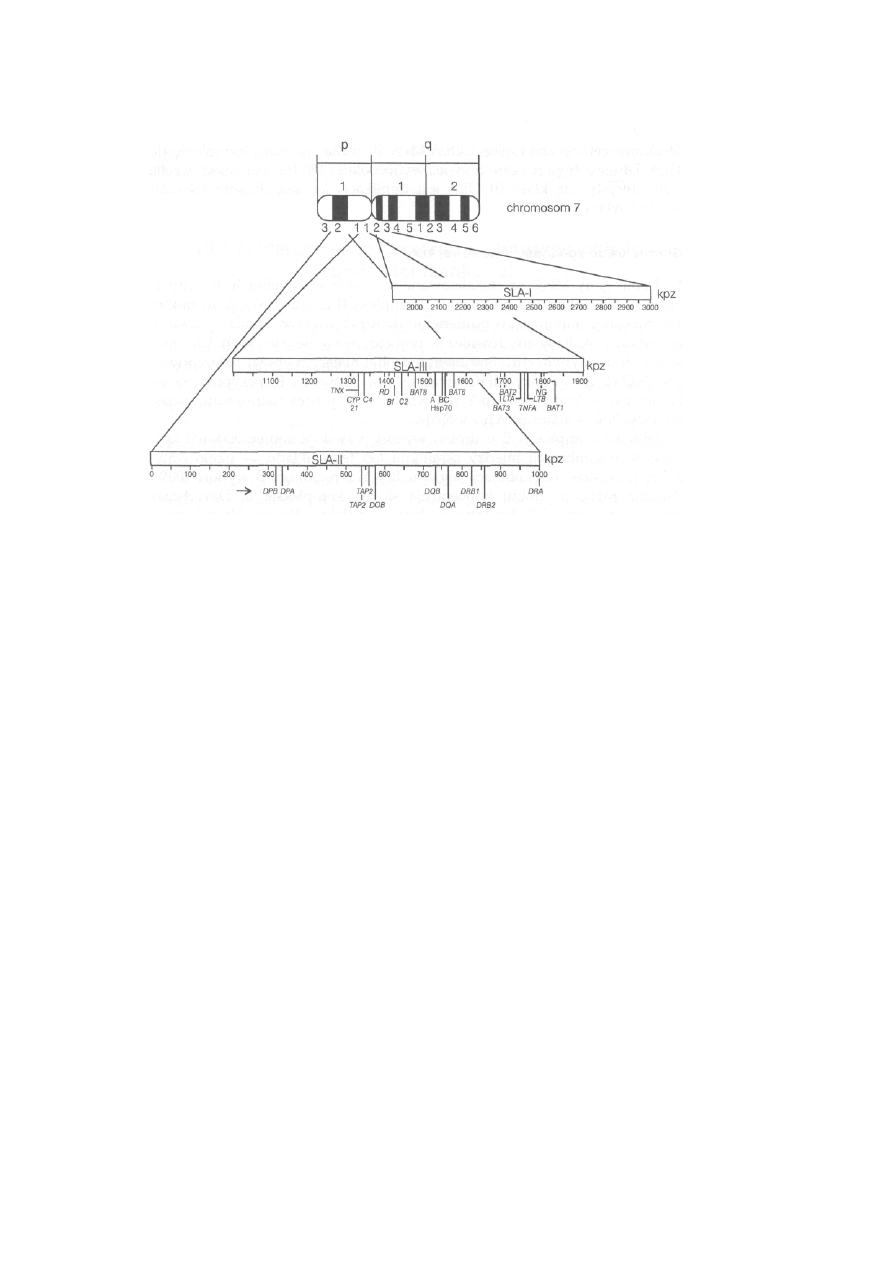
Ryć. 10-8. Schemat głównego układu zgodności tkankowej świni (SLA) (wg: Schook
i Lamont, 1996, zmodyfikowano)
znajdują się także pseudogeny. Jest nim na przykład gen DRB2, w którym
stwierdzono delecję nukleotydu w kodonie 53.
W regionie klasy III, w przeciwieństwie do innych gatunków, znajdują
się po jednym genie CYP22, C2 i C4. W regionie tym są także inne geny
typowe dla tego układu (ryć. 10-8).
Główny układ zgodności tkankowej owcy
Główny układ zgodności tkankowej owiec oznaczony jest skrótem OLA,
ale można również spotkać określenie Ovar (ang. ovine aries). Dotąd nie
sporządzono mapy fizycznej tego układu, ale informacje uzyskane za
pomocą analizy sprzężeń wykazują podobieństwa OLA do MHC u ludzi
i bydła. Mapa sprzężeniowa owczego chromosomu 20, w którym znajduje
się główny układ zgodności tkankowej, ma długość 5,6 cM. W regionie
OLA klasy I zidentyfikowano blisko sprzężone loci: OLA -A i -B, a częstość
rekombinacji między nimi wynosi 0,6%. W regionie klasy II, podobnie jak
u bydła, obok genów MHC znajdują się pseudogeny. Najlepiej poznany
jest pseudogen DRB2. W wyniku mutacji, które wykryto w tym pseudo-
genie, w eksonach 3 i 4 znajdują się kodony „stop", powodujące, iż nie
jest on funkcjonalny. Geny klasy II wykazują duży polimorfizm, przy
czym geny z locus DQB cechuje większy polimorfizm niż z locus DQA i DRĄ.
241
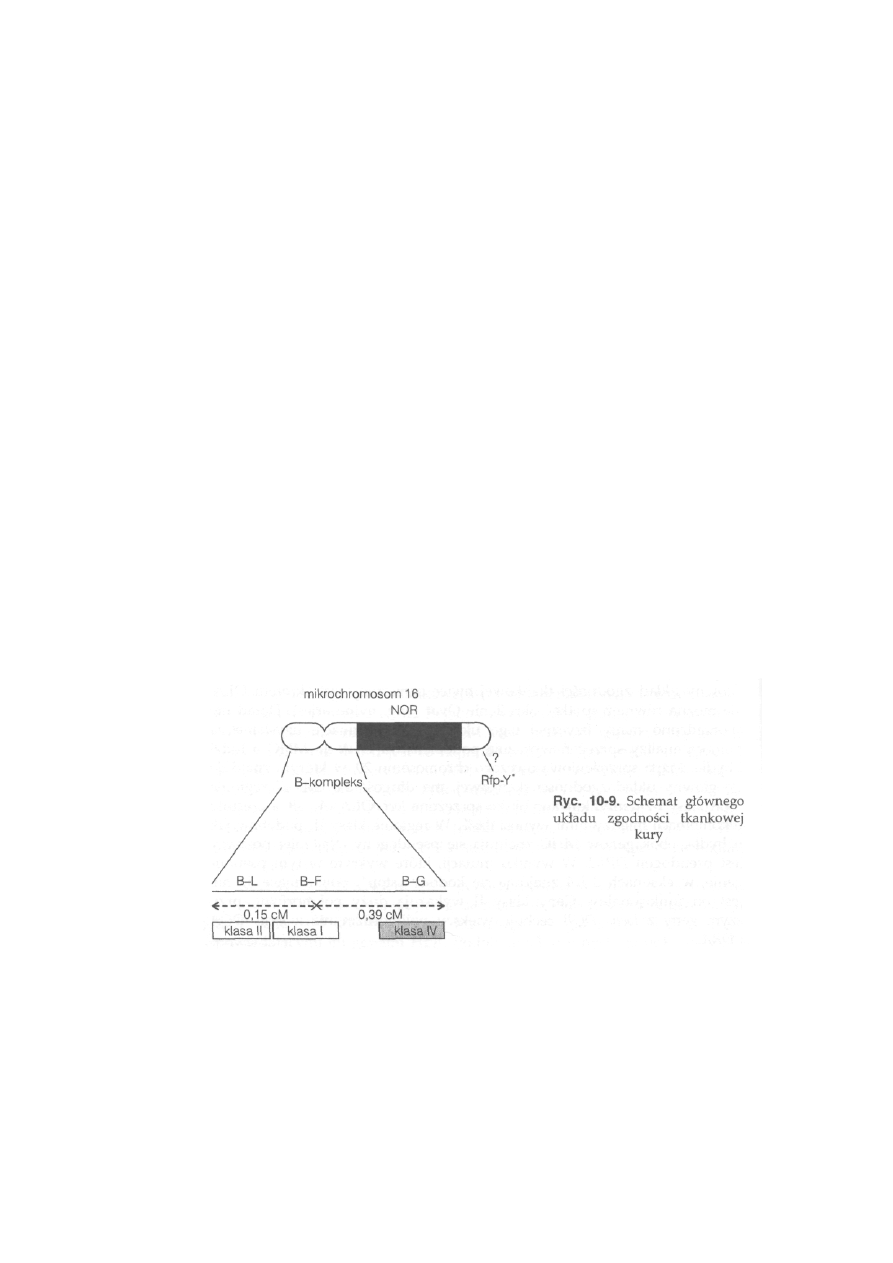
Struktura genetyczna regionu OLA klasy III wykazuje dużą homologię do
HLA. Długość tego regionu u owiec wynosi około 150 tyś. par zasad. Cecha
charakterystyczną klasy III OLA jest duplikacja loci dopełniacza C4 i 21--
hydroksylazy (CYP21).
Główny układ zgodności tkankowej kury
U drobiu geny MHC są zlokalizowane w dwóch kompleksach: B i Rfp-Y,
które są klasyfikowane niezależnie. Kompleks B znajduje się w 16 mikro-
chromosomie, natomiast w odniesieniu do Rfp-Y większość badaczy uważa,
że jest on zlokalizowany również w tym chromosomie (ryć. 10-9). Chromo-
som 16 jest jedynym chromosomem u drobiu mającym obszar jąderkotwór-
czy (NOR). Za typowy MHC uważany jest kompleks B, przypuszcza się
natomiast, że kompleks Rfp-Y jest włączony w proces rozpoznania przez
komórki NK — naturalnych zabójców.
Długość kompleksu B u drobiu wynosi prawdopodobnie 300-400 kpz,
częstość rekombinacji między skrajnymi loci tego układu -- około 0,5%.
Z kolei, częstość rekombinacji regionu Rfp-Y z regionem B wynosi 100%.
Badania MHC u drobiu koncentrują się na kompleksie B. Dotychczas
stwierdzono ponad 30 haplotypów tego kompleksu. Region klasy I ozna-
czony jest symbolem B-F, klasy II — B-L, a klasy IV — B-G. O ile funkcje
regionów B-F i B-L są znane, o tyle funkcja regionu B-G wciąż nie jest
wyjaśniona. U drobiu nie ma regionu klasy III, chociaż niedawno zmapo-
wano w obrębie MHC u drobiu gen G9a należący do klasy III HLA.
Wyniki badań prowadzonych na dużych populacjach referencyjnych
wskazują na możliwość sprzężenia przynajmniej kilku genów klasy III
z MHC drobiu.
Jak już wspomniano przy omawianiu budowy cząsteczki klasy I, gen
łańcucha lekkiego (
β
2
-mikroglobuliny) znajduje się poza głównym układem
242

zgodności tkankowej. U drobiu zlokalizowano go metodą FISH (hybrydyza-
cja fluorescencyjna in situ) w dużym mikrochromosomie (numer 9 lub 10
pod względem wielkości).
10.3.2. Identyfikacja cząsteczek głównego układu
zgodności tkankowej
Określanie genotypu osobnika pod względem układu zgodności tkankowej
(haplotypu) może być przeprowadzane dwoma podstawowymi sposobami:
1) za pomocą metod serologicznych, pozwalających na identyfikację
określonej cząsteczki (białka, antygenu) MHC,
2) poprzez analizę molekularną DNA, dzięki której możliwe jest wykrycie
obecności danego allelu MHC.
Do badań serologicznych wykorzystywane są surowice lub, od pewnego
czasu, przeciwciała monoklonalne. Natomiast bezpośrednia identyfikacja
genów MHC jest możliwa dzięki takim technikom genetyki molekularnej, jak:
• hybrydyzacja z sondą komplementarną do danego allelu (ang. allele
specific oligonucleotides — ASO), poprzedzona amplifikacją danego frag
mentu DNA (PCR),
• metoda RFLP.
Cząsteczki MHC klasy II można również identyfikować w mieszanej
hodowli limfocytów (MLC). Udoskonaleniem tej techniki jest test HTC,
w którym używane są homozygotyczne komórki stymulujące. Zaintereso-
wani tym zagadnieniem znajdą szczegółowe informacje w podręczniku
immunologii.
10.3.3. Znaczenie głównego układu zgodności tkankowej
10.3.3.1. Związek MHC z odpornością na choroby
Najważniejszą funkcją cząsteczek MHC jest udział w odpowiedzi immunolo-
gicznej organizmu na infekcje wewnątrzkomórkowe. Mechanizm ten przed-
stawiony jest w podrozdziale: Zarys przebiegu reakcji odpornościowej.
Rola głównego układu zgodności tkankowej w odporności/podatności na
wiele chorób u ludzi jest przedmiotem intensywnie prowadzonych badań.
Znane są już powiązania antygenów MHC z zapadalnością na różne choroby.
U zwierząt gospodarskich analogiczne badania są znacznie mniej zaawan-
sowane, ale ich wyniki świadczą o wpływie MHC na podatność/odporność na
niektóre choroby.
Na przykład, badania układu zgodności tkankowej u bydła (BoLA)
wykazały jego związek z odpornością na mastitis, białaczkę, tuberkulozę,
inwazję kleszczy i pasożytów oraz trypanosomatozę (tab. 10-IV).
243
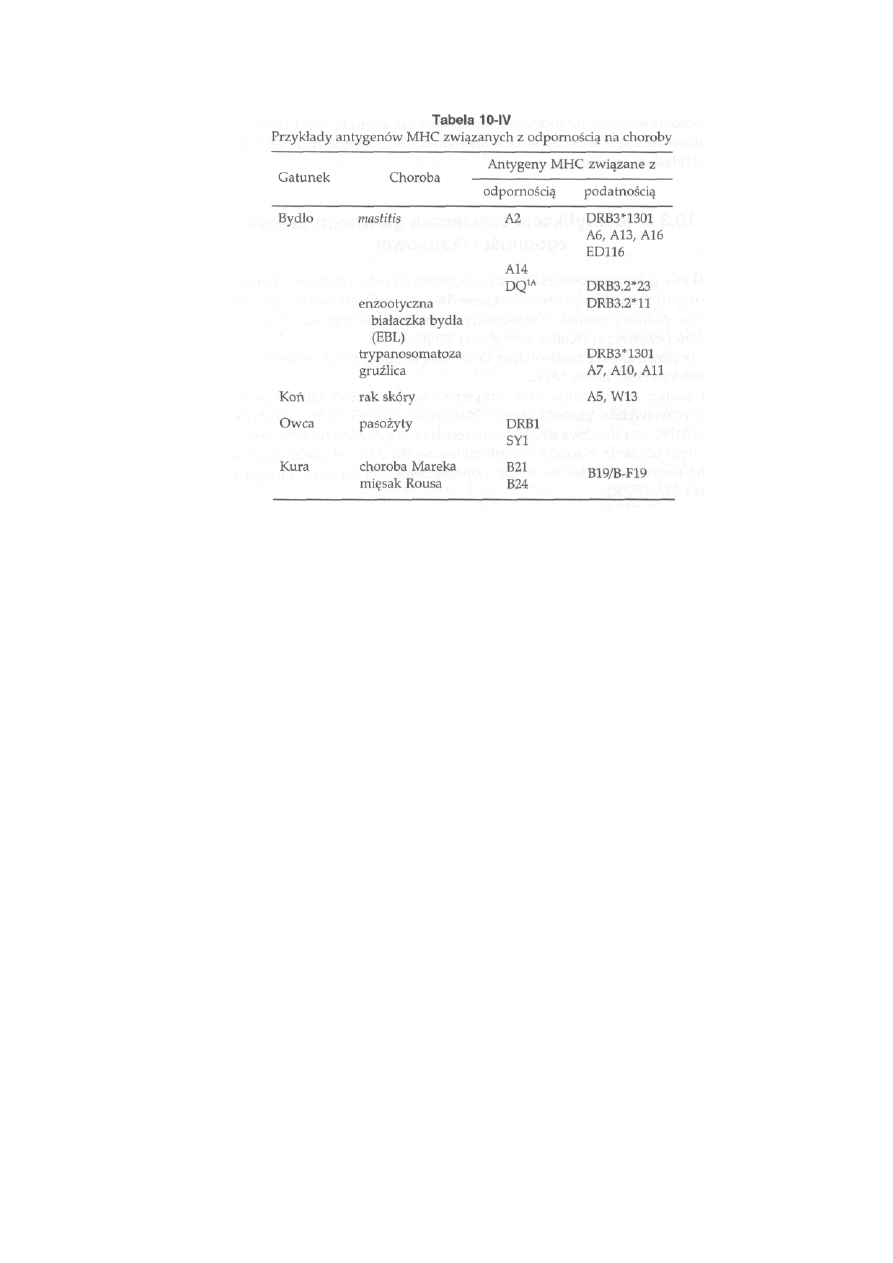
U owiec najbardziej zaawansowane są badania nad zależnością między
podatnością/odpornością na inwazje pasożytnicze a głównym układem
zgodności tkankowej. Podstawowym celem tych badań jest ustalenie
antygenów MHC mogących być wskaźnikami selekcyjnymi odporności
owiec na pasożyty wewnętrzne.
Główny układ zgodności tkankowej u koni (ELA) jest najmniej poznany,
ale stwierdzono już jego związek z odpornością na raka skóry.
U trzody chlewnej geny klasy I są przypuszczalnie głównymi genami
włączonymi w regulację odpowiedzi immunologicznej na inwazję nicieni
Trichinella spiralis.
U kur główny układ zgodności tkankowej jest związany z odpornością
na chorobę Mareka, kokcydiozę, mięsaka Rousa i cholerę ptasią.
Coraz częściej u zwierząt gospodarskich identyfikowane są haplotypy
związane z określoną reakcją na infekcje. Na przykład, haplotypy klasy II bydła
uznane za skorelowane z odpornością/podatnością na białaczkę są następujące:
DQA*3A-DQB*3A-DRB2*2A-DRB3.2*11 — odporność
DQA*12-DQB*12-DRB2*3A-DRB3.2*8 — podatność
10.3.3.2. Związek MHC z cechami produkcyjnymi i reprodukcyjnymi
Główny układ zgodności tkankowej, oprócz szczególnej roli w reakcji
immunologicznej, oddziałuje także na inne cechy (produkcyjne i reproduk-
cyjne). U bydła mlecznego stwierdzono istotne oddziaływanie niektórych
244

cząsteczek MHC klasy I na cechy związane z użytkowością mleczną,
natomiast u bydła ras mięsnych zauważono korelację między przyrostami
masy ciała i przekrojem mięśnia najdłuższego grzbietu a obecnością
określonych antygenów MHC.
Wyniki badań prowadzonych na trzodzie chlewnej świadczą o wpływie
MHC na cechy tuczne i rzeźne świń, przy czym stwierdzono różną
współzależność danego haplotypu z cechami produkcyjnymi zależnie od rasy.
Badania znaczenia głównego układu zgodności tkankowej w reprodukcji
zwierząt gospodarskich prowadzone były przede wszystkim na świniach.
Dotyczyły one wpływu MHC na stopień owulacji, przeżywalność zarodków
i płodów oraz liczebność miotu. Wykazano wyraźny wpływ haplotypu
rodziców na te cechy. Również u bydła antygeny MHC mogą wpływać na
zamieralność zarodków, obniżać stopień przeżywalności i masę ciała cieląt.
Wykazano ponadto istotny związek niektórych antygenów klasy II z za-
trzymaniem łożyska. U koni natomiast stwierdzono, iż geny ELA mogą
wpływać na zmniejszenie płodności przy kojarzeniach ogierów i klaczy
o identycznych haplotypach.
Dokładne poznanie struktury i funkcji głównego układu zgodności
tkankowej zwierząt gospodarskich oraz jego związku z odpornością,
reprodukcją i produkcyjnością powinno w przyszłości umożliwić wykorzy-
stanie cząsteczek (antygenów) MHC jako markerów genetycznych w dos-
konaleniu zwierząt pod względem tych cech.
10.4. Zarys przebiegu reakcji odpornościowej
W odpowiedzi na wniknięcie patogenów (wirusy, bakterie i pasożyty) do
organizmu układ immunologiczny reaguje w różny sposób, zależnie od
rodzaju patogenu i miejsca infekcji. Zasadniczą rolę w ich rozpoznaniu, bez
względu na to, czy są to antygeny zewnątrzkomórkowe czy wewnątrz-
komórkowe, odgrywają cząsteczki głównego układu zgodności tkankowej.
Przebieg reakcji immunologicznej jest niezmiernie skomplikowany i, jak
dotychczas, nie do końca został poznany. Obecna wiedza pozwala na
przedstawienie jedynie w zarysie przebiegu reakcji, zwłaszcza w odniesieniu
do zakażenia zewnątrzkomórkowego.
Cząsteczki MHC klasy I biorą udział w odpowiedzi układu immunolo-
gicznego na wirusową infekcję wewnątrzkomórkową. W zakażonej komórce
białka wirusowe ulegają proteolizie (w cytosolu), czyli są degradowane do
peptydów. Peptydy te są następnie przekazywane do siateczki śródplaz-
matycznej. W siateczce śródplazmatycznej są syntetyzowane cząsteczki
klasy I MHC. Tutaj też następuje związanie cząsteczki MHC z peptydem,
po czym powstały kompleks transportowany jest na powierzchnię komórki,
gdzie zostaje rozpoznany przez obecne w pobliżu limfocyty T cytotoksyczne,
245
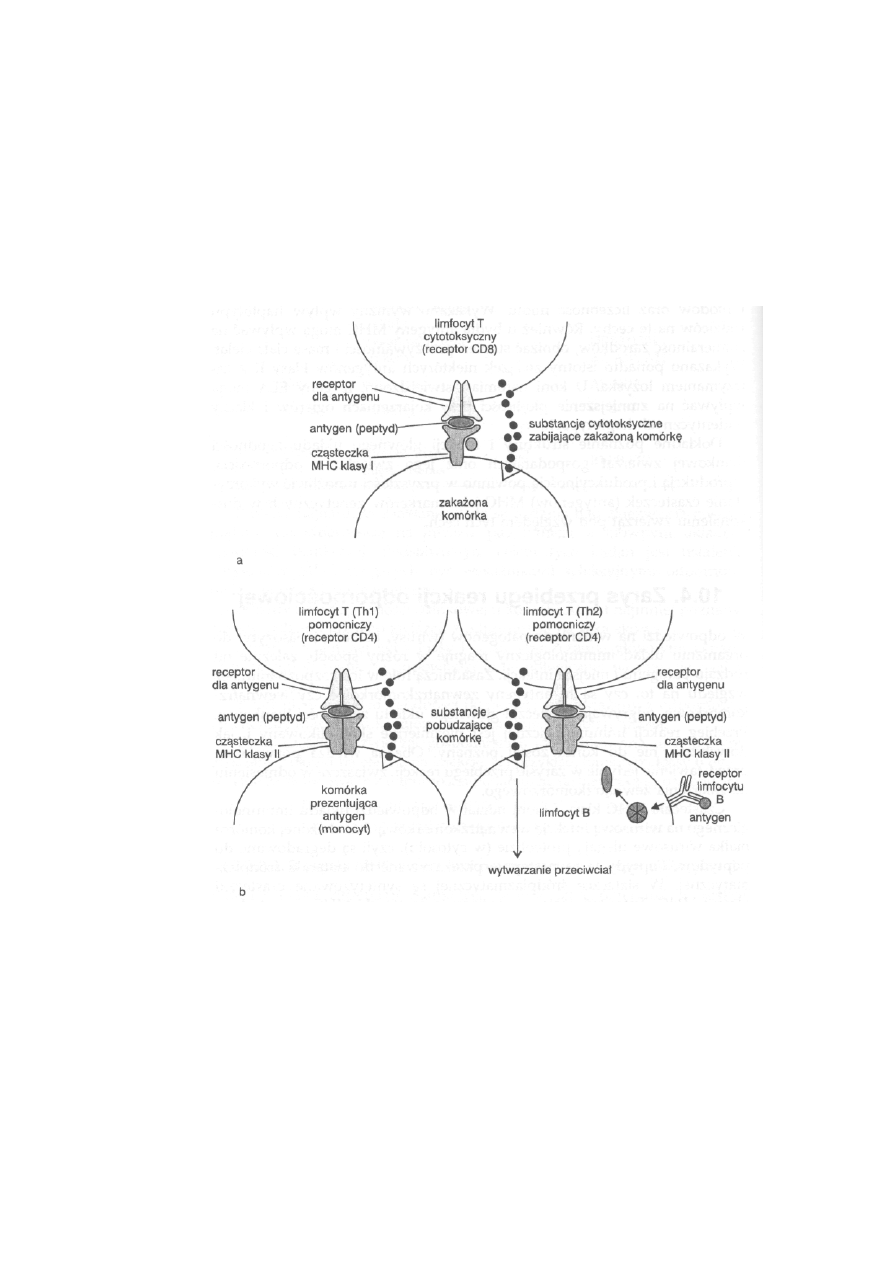
nazywane inaczej zabijającymi, mające na powierzchni cząsteczkę CD8.
Limfocyty te uwalniają cytokiny hamujące replikację wirusów i cytotoksyny,
które zabijają zakażoną komórkę (ryć. 10-10a).
Cząsteczki MHC klasy II uczestniczą w reakcji przeciwko infekcji
zewnątrzkomórkowej, prezentując przede wszystkim obce antygeny, które
zostały pochłonięte przez wyspecjalizowane komórki układu immunologicz-
nego — komórki dendrytyczne, makrofagi lub limfocyty B. Podczas infekcji
bakteryjnej, jeśli bakterie zostaną pochłonięte przez makrofagi, są rozkładane
na peptydy w wakuolach (endosomach). Peptydy te wiążą się z cząsteczkami
MHC klasy II, przedostającymi się z siateczki śródplazmatycznej. Powstały
Ryc. 10-10. Reakcja immunologiczna: (a) prezentacja antygenu przez cząsteczki MHC
klasy I; (b) prezentacja antygenu przez cząsteczki MHC klasy II
246

kompleks jest transportowany na powierzchnię komórki i tutaj roz-
poznawany przez limfocyty T CD4 typu zapalnego (Thl). Limfocyty
te nie mszczą komórek tak jak limfocyty T cytotoksyczne (CD8), ale
uwalniają limfokiny aktywujące komórkę do zwalczenia infekcji (ryć.
10-10b). W określonych przypadkach, gdy jest zbyt mała liczba limfocytów
B zdolnych do rozpoznania antygenu (co może wystąpić przy pierwszym
zetknięciu się organizmu z danym antygenem) bądź gdy fagocytowane
cząsteczki lub bakterie są dużych rozmiarów, makrofagi mogą pełnić
rolę pośrednią, prezentując limfocytom B cząsteczki po fagocytozie i wstę-
pnej fragmentacji. Makrofagi wchłaniają, degradują i prezentują wszystkie
antygeny (ryć. 10-10b).
Jeśli bakterie zostaną pochłonięte przez limfocyty B, w odpowiedzi
immunologicznej biorą udział limfocyty T CD4 pomocnicze, określane jako
Th2. Dojrzały limfocyt B ma na swej powierzchni przeciwciała IgM lub IgM
i IgD, które służą jako receptory antygenów. Obce białko rozpoznane przez
limfocyt B jest przekazywane do wakuoli (endosomów) i tam trawione na
krótkie peptydy. Następnie peptydy te są wiązane przez cząsteczki klasy II,
które dotarły tu z siateczki śródplazmatycznej. Utworzony kompleks, po
przedostaniu się na powierzchnię komórki, jest rozpoznawany przez limfocyt
pomocniczy T. Limfocyt ten daje sygnał limfocytom B do proliferacji
(namnażania) i syntezy przeciwciał. Zaktywowane limfocyty B rozpoczynają
syntezę przeciwciał wtórnych (IgG, IgE lub IgA), które różnią się od
pierwotnych tylko strukturą łańcucha ciężkiego. Tak więc wytwarzanie
przeciwciał przez limfocyty B jest kontrolowane przez cząsteczki MHC
i limfocyty T (ryć. 10-10b).
Zdolność cząsteczek MHC do wiązania peptydów, a raczej ich pojemność
jest różna. Jedna cząsteczka MHC klasy I może w swym rowku związać
około 1000 różnych peptydów, natomiast cząsteczka klasy II może ich
związać dwa razy więcej.
Cząsteczki MHC biorą także udział w usuwaniu własnych białek
pochodzących z komórek, które uległy apoptozie. Apoptoza (gr. apoptosis -
opadanie) jest to programowana śmierć komórki, czyli jej fizjologiczna
śmierć. Podczas tego procesu komórka początkowo kurczy się i oddziela od
sąsiednich komórek, chromatyna ulega kondensacji na obrzeżach jądra,
wkrótce potem jądro, a następnie cała komórka pęka, a jej fragmenty są
trawione przez sąsiadujące komórki.
10.5. Genetyczna oporność na choroby
Zmienność obserwowana u zwierząt dotyczy nie tylko cech użytkowych
(mleczność, nieśność itd.), ale także oporności/podatności na choroby zakaźne
i inwazyjne. Ostatnio coraz więcej uwagi poświęca się genetycznie uwarun-
247
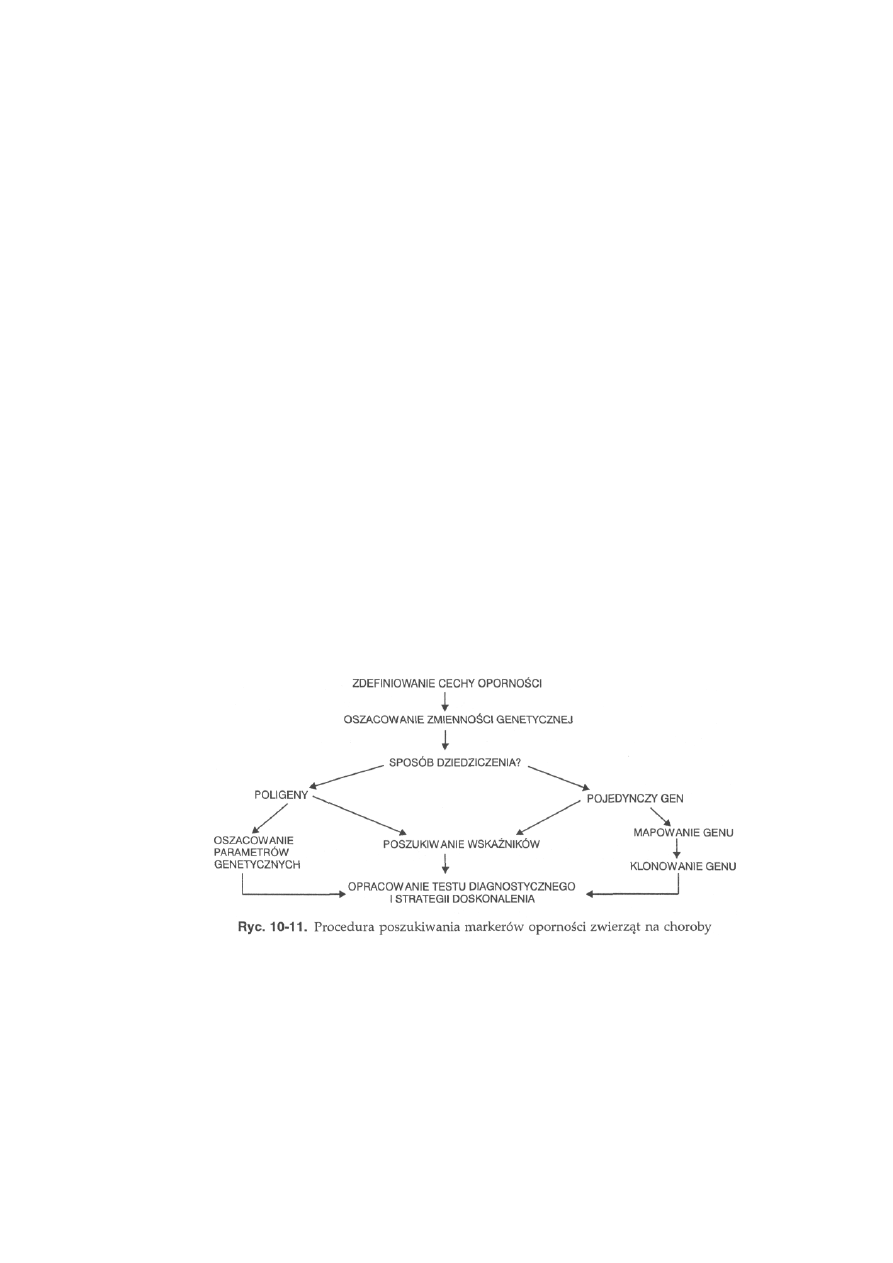
kowanej oporności na choroby. Oporność na choroby jest definiowana jako
właściwość organizmu uniemożliwiająca rozwój choroby pomimo wniknięcia
patogenu. Genetyczna oporność nie obejmuje typowej odpowiedzi im-
munologicznej.
Mechanizm dziedziczenia genetycznej oporności na różne choroby nie
został jeszcze w pełni poznany. Na ogół jest to cecha poligeniczna, chociaż
w przypadku niektórych schorzeń stwierdzono, iż oporność na nie zależy
od pojedynczego genu o dużym efekcie, np. mutacja w genie FUT1
odpowiadająca za oporność prosiąt na chorobę obrzękową. W ośrodkach
naukowych wielu krajów prowadzone są intensywne badania, których
celem jest znalezienie takich genów.
Podstawowe założenia poszukiwania genów odpowiedzialnych za opor-
ność zwierząt na choroby obrazuje schemat (ryć. 10-11). Wybór cechy
będącej wskaźnikiem oporności powinien być przeprowadzony z
uwzględnieniem pewnych kryteriów. Cecha ta powinna:
• charakteryzować się dużą genetyczną zmiennością i odziedziczalnością
• mieć istotną wartość ekonomiczną; może nią być produkt nadający się
do sprzedaży bądź zmniejszający koszty produkcji
• być łatwa do mierzenia bez ponoszenia wysokich kosztów.
Gdy cecha — wskaźnik oporności jest już wybrana, należy oszacować jej
zmienność genetyczną. Umożliwi to poznanie sposobu jej dziedziczenia.
W przypadku gdy oporność na chorobę jest cechą poligeniczna, następnym
krokiem jest oszacowanie parametrów genetycznych: odziedziczalności tej
cechy i jej korelacji genetycznej z innymi cechami istotnymi ekonomicznie.
Informacje te mogą być przydatne w opracowaniu strategii doskonalenia
oporności. Jeśli okaże się, iż cecha oporności jest warunkowana pojedynczym
genem, celowe jest zmapowanie tego genu, czyli określenie jego położenia
w chromosomie, następnie sklonowanie go i poznanie sekwencji nukleo-
tydów.
248

Zasadniczym krokiem w badaniach genetycznej oporności jest po-
szukiwanie sprzężeń między genetyczną opornością, warunkowaną wieloma
lub jednym genem, a markerami genetycznymi, takimi jak antygeny
erytrocytarne czy białka polimorficzne (markery klasy I) lub niekodujące
sekwencje DNA (markery klasy II). Sprzężenia takie są szczególnie korzystne
w selekcji, gdyż umożliwiają określanie oporności/podatności zwierzęcia
bez konieczności zarażania go patogenami.
W opracowaniu strategii doskonalenia oporności genetycznej wykorzys-
tuje się zarówno metody tradycyjne, jak i techniki inżynierii genetycznej.
Tradycyjna praca hodowlana w kierunku zwiększenia oporności zwierząt
może być prowadzona w drodze selekcji pośredniej na podstawie takich
cech, jak:
• cechy eksterierowe (widoczne gołym okiem),
• cechy, które są określane za pomocą testów serologicznych lub
elektroforezy,
• cechy ustalane za pomocą badań molekularnych DNA.
Klasycznymi przykładami cech eksterierowych są: ciemna obwódka
wokół oczu u bydła rasy hereford (bydło z biało umaszczoną głową),
związana z opornością na raka oczu, i barwne upierzenie drobiu, wskazujące
na większą niż u osobników białych oporność na choroby wirusowe.
Znajomość cech określanych metodami serologicznymi, które mogą być
wskaźnikami oporności na choroby, umożliwiałaby prowadzenie selekcji
wśród zwierząt młodych. Wyniki badań prowadzonych na owcach w Au-
stralii i Nowej Zelandii wskazują na zależność między genotypem hemo-
globiny a częstością zarażenia pasożytem Haemonchus contortus. Zwierzęta
z hemoglobiną AA są bardziej oporne niż osobniki z hemoglobiną AB i BB.
Innym markerem biochemicznym mogą być antygeny erytrocytarne grup
krwi. Na przykład, u bydła opornego na białaczkę częściej występują
antygeny erytrocytarne B, Y
2/
D' i P'.
Największe możliwości ograniczania występowania chorób u zwierząt
na drodze hodowlanej stwarzają wyniki badań molekularnych określających
sekwencje nukleotydowe genów, jak również badania genetycznej struktury
głównego układu zgodności tkankowej (MHC). Ustalenie na tej podstawie
oporności (lub podatności) na choroby jest możliwe już u zarodka, co ma
zasadnicze znaczenie w hodowli bydła wobec coraz szerzej stosowanego ich
transferu.
Na zmienność w podatności na choroby infekcyjne i pasożytniczne
składają się zarówno różnice osobnicze, jak i rasowe. Na przykład, rodzime
rasy bydła w Indii są bardziej wrażliwe na brucelozę niż ich mieszańce
z rasami europejskimi. W Kenii zarażenie bydła motylicą wątrobową jest
o wiele częstsze u ras importowanych z Europy niż u ras miejscowych.
Powszechnie znana jest oporność rodzimego bydła afrykańskiego N'Dama
na infekcję świdrowca Trypanosoma brucei brucei, która prowadzi do śpiączki
afrykańskiej (trypanosomatoza). Choroba ta dziesiątkuje populacje bydła
249

afrykańskiego. Różnice rasowe w oporności/podatności na choroby mogą
być wykorzystywane do krzyżowania w celu uzyskania mieszańców opor-
nych na daną chorobę.
Zmienność podatności zwierząt na choroby najlepiej poznano u kur.
Dzięki wysokiej plenności i szybkiej rotacji pokoleń prowadzona u tego
gatunku intensywna selekcja w kierunku oporności na chorobę Mareka
umożliwiła wytworzenie rodów kur całkowicie niewrażliwych na to scho-
rzenie, z zachowaniem ich wysokiej produkcyjności. Mechanizm oporności
na chorobę Mareka przypuszczalnie polega na ograniczaniu rozwoju guzów
u zainfekowanych ptaków, nie zabezpieczając ich przed infekcją. Zjawisko
oporności u kur obserwowane jest także w odniesieniu do kokcydiozy
i salmonellozy. Kokcydioza powoduje obniżony stopień wzrostu, zmniej-
szone wykorzystanie paszy, a także, zwłaszcza u młodych kurcząt, śmiertel-
ność. Za wskaźnik oporności na kokcydiozę przyjęto liczbę produkowanych
oocyst. Badania genetycznych uwarunkowań oporności na kokcydiozę
wskazują na rolę genów głównego układu zgodności tkankowej (MHC)
i genów zlokalizowanych poza tym układem. Kokcydioza jest równocześnie
przykładem choroby, której częstość występowania jest w dużej mierze
stymulowana działaniem czynników środowiskowych, w tym przypadku
jest to temperatura otoczenia. Zależność ta jest następstwem zróżnicowanej
zdolności termoregulacji u młodych ptaków.
Wyniki badań wrażliwości kur na bakterie Salmonella (S. typhimurium, S.
pullorum, S. gallinarum, S. enteritidis) sugerują, że oporność na nie zależy od
funkcji fagocytarnych komórek przede wszystkim śledziony i wątroby.
Ponadto świadczą one, iż oporność na salmonellozę jest dominująca i może
być warunkowana pojedynczym genem.
Wykorzystanie zjawiska genetycznej oporności w celu poprawy zdro-
wotności zwierząt gospodarskich ma szczególne znaczenie w przypadku
chorób trudnych do zwalczania metodami weterynaryjnymi. Należą do
nich stany zapalne gruczołu mlecznego (mastitis), które są przyczyną
poważnych strat ekonomicznych w hodowli bydła mlecznego. Jedną
z dróg ograniczania występowania mastitis u bydła jest wykorzystanie
w pracy hodowlanej różnic w genetycznych skłonnościach do tego scho-
rzenia. Obserwowane są zarówno różnice rasowe, jak i indywidualne.
Na przykład, krowy rasy nizinnej czarno-białej i czerwonej duńskiej
częściej zapadają na zapalenia gruczołu mlecznego niż holsztyńsko-fry-
zyjskiej, simentalery czy szwyce.
Oprócz różnic rasowych w podatności krów na mastitis stwierdzono
także wyraźne różnice w częstości występowania tego schorzenia między
rodzinami (po matkach) i liniami (po ojcach). Ze względu na stosowaną
sztuczną inseminację problem przekazywania przez ojca oporności na
mastitis jest bardzo istotny. Prowadzone są badania nad możliwością
wprowadzenia do oceny wartości hodowlanej buhajów cechy „choroba"
(mastitis). W Norwegii od 1978 roku mastitis jest już włączone jako cecha
250

selekcyjna w programach hodowlanych. Ocena rozpłodników jest dokony-
wana na podstawie zdrowotności gruczołu mlecznego ich potomstwa
żeńskiego. Inną drogą zwalczania mastitis jest selekcja buhajów w kierunku
zmniejszenia zawartości komórek somatycznych w mleku ich córek. Ta
metoda selekcji jest zalecana na przykład w Szwecji. Jednakże badania
niektórych autorów wykazały istotną rolę poszczególnych rodzajów komórek
somatycznych w zabezpieczaniu wymienia przed drobnoustrojami chorobo-
twórczymi. Można zatem przypuszczać, iż selekcja w kierunku zmniejszenia
zawartości komórek somatycznych w mleku może zmniejszyć zdolność
krów do reakcji na infekcje gruczołu mlecznego. Wydaje się, iż byłoby
celowe prowadzenie selekcji pośredniej, opartej na wskaźnikach fenotypo-
wych oporności. Wielu badaczy wiąże skłonność krów do stanów zapalnych
(mastitis) z budową i wielkością wymienia oraz strzyków, która w znacznym
stopniu jest uwarunkowana genetycznie. Stwierdzono na przykład, że
mastitis najczęściej występuje u krów, których wymiona wykazują morfo-
logiczne i fizjologiczne odchylenia od normy, takie jak: nieproporcjonalny
rozwój przednich i tylnych ćwiartek, zbyt duża wielkość i niskie zawieszenie,
za duże lub za małe strzyki. Obecnie w programach hodowlanych kładzie
się wyraźny nacisk na cechy morfologiczne wymion i zdolność wydojową
krów, w celu ograniczania występowania mastitis poprzez selekcję. Pod-
stawowymi kryteriami tej selekcji są, oprócz wydajności, kształt i wielkość
wymienia oraz strzyków, ustawienie strzyków, wysokość zawieszenia
wymienia i szybkość oddawania mleka. Wybór tych cech jest uzasadniony
ich uwarunkowaniem genetycznym i powiązaniem ze stanem zdrowotnym
wymienia. Celem tych działań jest wyselekcjonowanie krów o prawidłowej
budowie wymienia, charakteryzujących się wysoką produkcją mleka i zwięk-
szoną opornością na mastitis.
Innym schorzeniem u bydła, którego leczenie jest bezskuteczne, jest
białaczka limfatyczna. Jej występowanie stwierdzono na wszystkich kon-
tynentach, ale najczęściej atakuje ona bydło w krajach strefy umiarkowanej.
Ponieważ wszelkie metody zwalczania białaczki (poza eliminacją zwierząt
zarażonych) stosowane przez lekarzy weterynarii nie przynoszą oczekiwa-
nych wyników, coraz więcej uwagi poświęca się zagadnieniom wpływu
założeń genetycznych na skłonność do tego schorzenia, jak również
niekorzystnym warunkom środowiska (np. niedobór niektórych pierwiast-
ków, jak: magnez, wapń, kobalt i mangan). Na podstawie licznych badań
można sądzić, że skłonność do białaczki jest przekazywana przez rodziców,
a zwłaszcza przez chore matki. Wydaje się, że większa zapadalność na
białaczki krów od matek chorych jest następstwem nie tylko dziedziczenia
skłonności do tej choroby, ale także zakażenia przez łożysko, siarę i mleko
oraz, co zostało udowodnione, przez uprzednio zarażoną wirusem białaczki
komórkę jajową. Wpływ buhajów na skłonność ich córek do białaczki nie
jest tak silny jak wpływ matek. Wiadomo, iż wirus białaczki limfatycznej nie
jest przenoszony podczas sztucznej inseminacji przez nasienie. Z drugiej
251

jednak strony wielu badaczy stwierdzało, że krowy po niektórych buhajach
znacznie częściej zapadają na tę chorobę, co może być dowodem na
dziedziczenie podatności na białaczkę.
Badania nad genetyczną opornością świń na choroby dotyczą przede
wszystkim biegunek u prosiąt, ale także innych schorzeń, na przykład
schorzeń kończyn. Genetyczne uwarunkowanie oporności prosiąt na bie-
gunki okresu noworodkowego i poodsadzeniowego wywoływane szczepami
K88 E. coli i F18 E. coli jest opisane w podrozdziale: Mutacje genowe. W tym
miejscu należy podkreślić, że praktycznie nie jest możliwe uzyskanie
zwierząt opornych na kilka chorób jednocześnie. Można jedynie mówić
0 doskonaleniu cechy oporności na określone schorzenie.
U owiec stwierdzono zróżnicowaną oporność na pasożyty wewnętrzne.
Stwarza to możliwość wykorzystywania tej właściwości w ograniczaniu
stopnia inwazji pasożytniczych u tego gatunku zwierząt. Oporność owiec
na pasożyty żołądkowo-jelitowe może przejawiać się jako zdolność żywiciela
do kontrolowania występowania pasożyta, przez zmniejszanie jego ilości
1 ograniczanie przeżywalności (ang. resistance), lub też jako zdolność do
współżycia z pasożytem (ang. resilience) poprzez utrzymywanie produkcyj-
ności na nie zmienionym poziomie nawet podczas inwazji. Genetyczna
zmienność zdolności do współżycia żywiciela z pasożytem, w odróżnieniu
od genetycznej zmienności oporności, cieszy się mniejszym zainteresowniem
wśród badaczy, przede wszystkim z powodu trudności w znalezieniu
odpowiedniego wskaźnika tego stanu. Stwierdzono, iż między opornością
owiec na zarażenie nicieniami żołądkowo-jelitowymi (np. Haemonchus
contortus) a „resilience" istnieje dodatnia korelacja genetyczna, której wartość
waha się w granicach 0,5-0,6. Prowadząc selekcję w kierunku oporności na
pasożyty można oczekiwać poprawy skorelowanej z nią cechy zdolności
współżycia z pasożytem.
Rozdział
W klasycznym ujęciu marker genetyczny to cecha jakościowa, uwarun-
kowana w sposób prosty — tzn. przez jedną parę alleli, przydatna do celów
analizy genetycznej. Przydatność markera zależy od tego, czy jest on
polimorficzny, co zachodzi wówczas, gdy w populacji występują przynaj-
mniej dwa allele w locus tego markera. Polimorfizm markerów bada się
zazwyczaj za pomocą metod analitycznych, wśród których dominują metody
serologiczne oraz biochemiczne — oparte na elektroforezie.
Metody serologiczne wymagają stosowania surowic testowych, zawiera-
jących przeciwciała skierowane przeciw ściśle określonej postaci białka,
a dokładniej — rozpoznające konkretną determinantę antygenową. W ten
sposób badane są antygeny erytrocytarne (grupy krwi), antygenowe deter-
minanty białek surowicy krwi (allotypy), antygeny głównego układu
zgodności tkankowej, umiejscowione na niektórych frakcjach leukocytów
(antygeny klasy II) lub innych komórkach mających jądro (antygeny klasy I).
Technikę elektroforezy wykorzystuje się do rozdziału w polu elektrycz-
nym białek oraz fragmentów DNA. Rozdział elektroforetyczny ujawnia
formy różniące się ruchliwością, która zależy od wielkości i ładunku
elektrycznego cząsteczki białka (lub fragmentu DNA) oraz jej konformacji
przestrzennej, a z drugiej strony od parametrów fizycznych rozdziału
(napięcie, temperatura, stężenie nośnika — żelu itp.). W przypadku DNA
rozdział może być poprzedzony trawieniem wyizolowanego (lub zamplifi-
kowanego metodą PCR) DNA z użyciem enzymów restrykcyjnych. Za-
stosowanie enzymów restrykcyjnych pozwala na ujawnienie polimorfizmu
długości fragmentów restrykcyjnych (RFLP) w obrębie genów lub polimor-
fizmu powtarzalnych sekwencji minisatelitarnych, które nie kodują infor-
macji genetycznej. Natomiast bezpośrednia elektroforeza wybranych frag-
mentów DNA, uzyskanych metodą PCR, pozwala na analizę polimorficznych
loci zawierających sekwencje mikrosatełitarne lub też ujawnienie zróż-
nicowanej konformacji pojedynczych łańcuchów zamplifikowanego frag-
253

mentu DNA genu (metoda SSCP — ang. single stranded conformation
polymorphism).
Objęcie badaniami niekodujących sekwencji DNA spowodowało, że
markery zostały podzielone na dwie klasy. Klasa I obejmuje klasyczne
markery, czyli sekwencje kodujące -- geny. Polimorfizm tych markerów
wykrywany jest poprzez analizę produktów genów (metody serologiczne
i technika elektroforezy białek) lub badanie DNA tych genów (metody
RFLP, SSCP). Klasa II markerów to sekwencje niekodujące; wśród nich
najważniejsze miejsce zajmują tandemowo powtarzające się sekwencje
mikrosatelitarne, a w mniejszym stopniu sekwencje minisatelitarne. W tym
przypadku mają zastosowanie tylko badania oparte na elektroforezie DNA.
Oprócz wymienionych wyżej dwóch klas markerów genetycznych można
wyróżnić jeszcze grupę markerów związanych z polimorfizmem chromo-
somowym, który jest wykrywany za pomocą technik prążkowego barwienia
chromosomów. Polimorfizm chromosomowy dotyczy przede wszystkim
wielkości bloków heterochromatyny konstytutywnej (barwienia techniką
CBG) oraz obszarów jąderkotwórczych (technika Ag-I, czyli Ag-NOR).
11.1. Antygeny erytrocytarne (grupy krwi)
Grupy krwi były najwcześniej wykorzystane w analizie genetycznej, np.
dotyczącej kontroli pochodzenia. Do pionierów tych badań należy zaliczyć
m.in. polskiego badacza Ludwika Hirszfelda. Przez grupę krwi należy
rozumieć typ krwi, cechujący się obecnością charakterystycznych białek,
o właściwościach antygenowych, na powierzchni erytrocytów. Identyfikacja
antygenów erytrocytarnych wymaga zastosowania surowic testowych, które
zawierają swoiste przeciwciała skierowane przeciw konkretnemu antygenowi.
Intensywne badania nad polimorfizmem antygenów erytrocytarnych u
zwierząt domowych podjęto na przełomie lat 50. i 60. Ich efektem było
wykrycie wielu układów grupowych. W ich obrębie zidentyfikowano liczne
antygeny i allele je warunkujące. Konkretny układ grupowy krwi jest
warunkowany przez pojedynczy gen (locus), który w populacji jest re-
prezentowany zazwyczaj przez wiele alleli (seria alleli wielokrotnych).
Białka kodowane przez te geny pełnią różnorodne funkcje biologiczne.
Z badań prowadzonych u człowieka wynika, że mogą one być enzymami,
receptorami, cząsteczkami adhezyjnymi czy białkami strukturalnymi. Wspól-
ną ich właściwością jest to, że są związane z błoną plazmatyczną erytrocytów,
a ich obecność można wykryć metodami serologicznymi. Po dodaniu do
surowicy testowej zawiesiny erytrocytów dochodzi do reakcji anty-gen-
przeciwciało. Przeciwciała obecne w surowicy testowej wiążą się z
determinantami antygenowymi. W wyniku reakcji antygen-przeciwciało
dochodzi do aglutynacji lub hemolizy krwinek czerwonych. Jeśli białko
ma tylko jedną determinantę antygenową, wówczas jest rozpoznawane
254
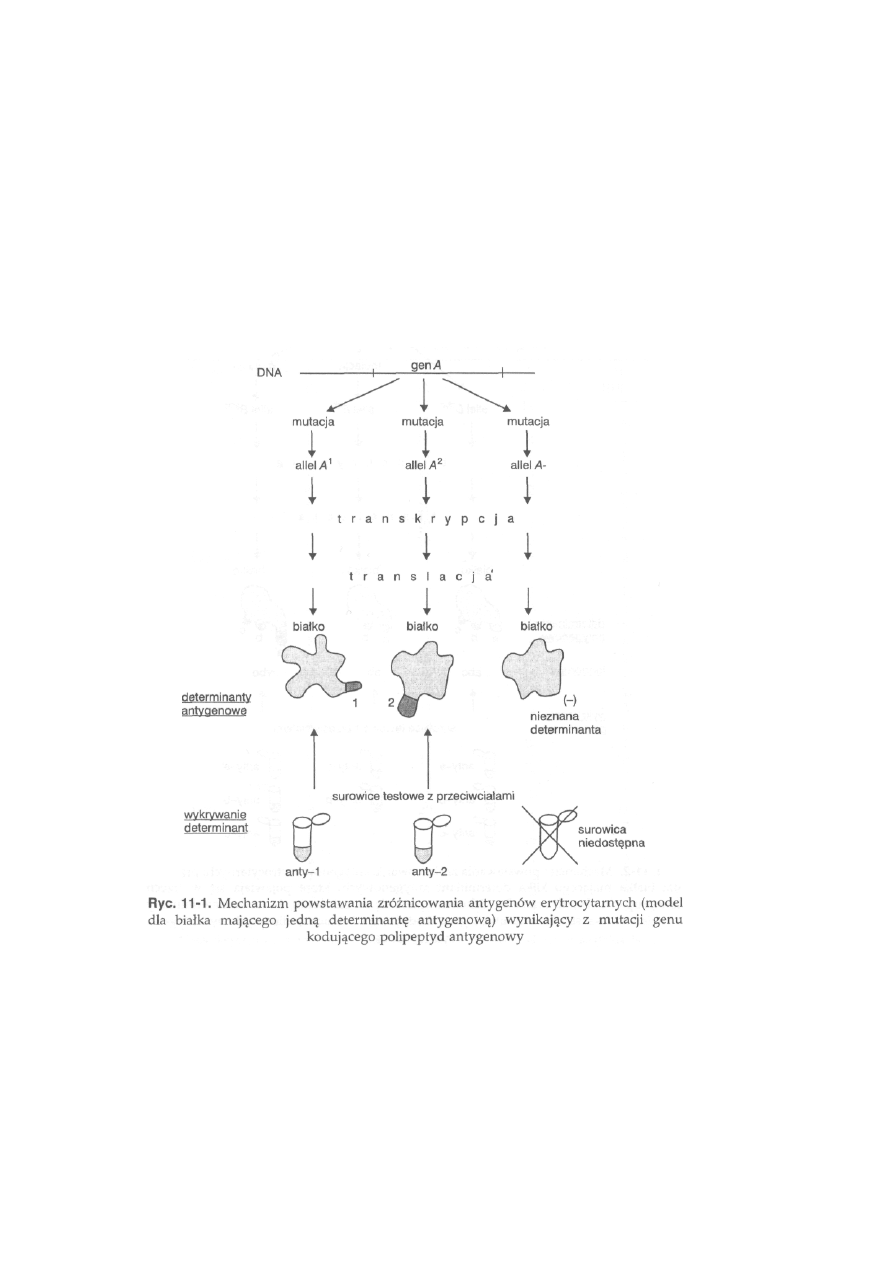
za pomocą jednej surowicy testowej. Jeśli białko ma kilka determinant
antygenowych, to wówczas ta forma białka jest wykrywana po zastosowa-
niu odpowiedniej liczby surowic testowych. Wreszcie może być taka
sytuacja, że białko jest pozbawione determinant wykrywalnych dostępnymi
surowicami testowymi. Występowanie różnych form determinanty antyge-
nowej w danym białku jest skutkiem mutacji punktowych. Mutacja taka
może doprowadzić do zmiany sekwencji aminokwasów, a to z kolei może
wpłynąć na zmianę struktury białka, a w tym jego determinanty anty-
genowej. Struktura białka zależy również od modyfikacji potranslacyjnej
polipeptydu przeprowadzonej przez enzymy kodowane przez inne geny.
Jeśli takie geny mają wiele alleli, może to prowadzić do zróżnicowanej
modyfikacji potranslacyjnej, a przez to do utworzenia (lub zlikwidowania)
255
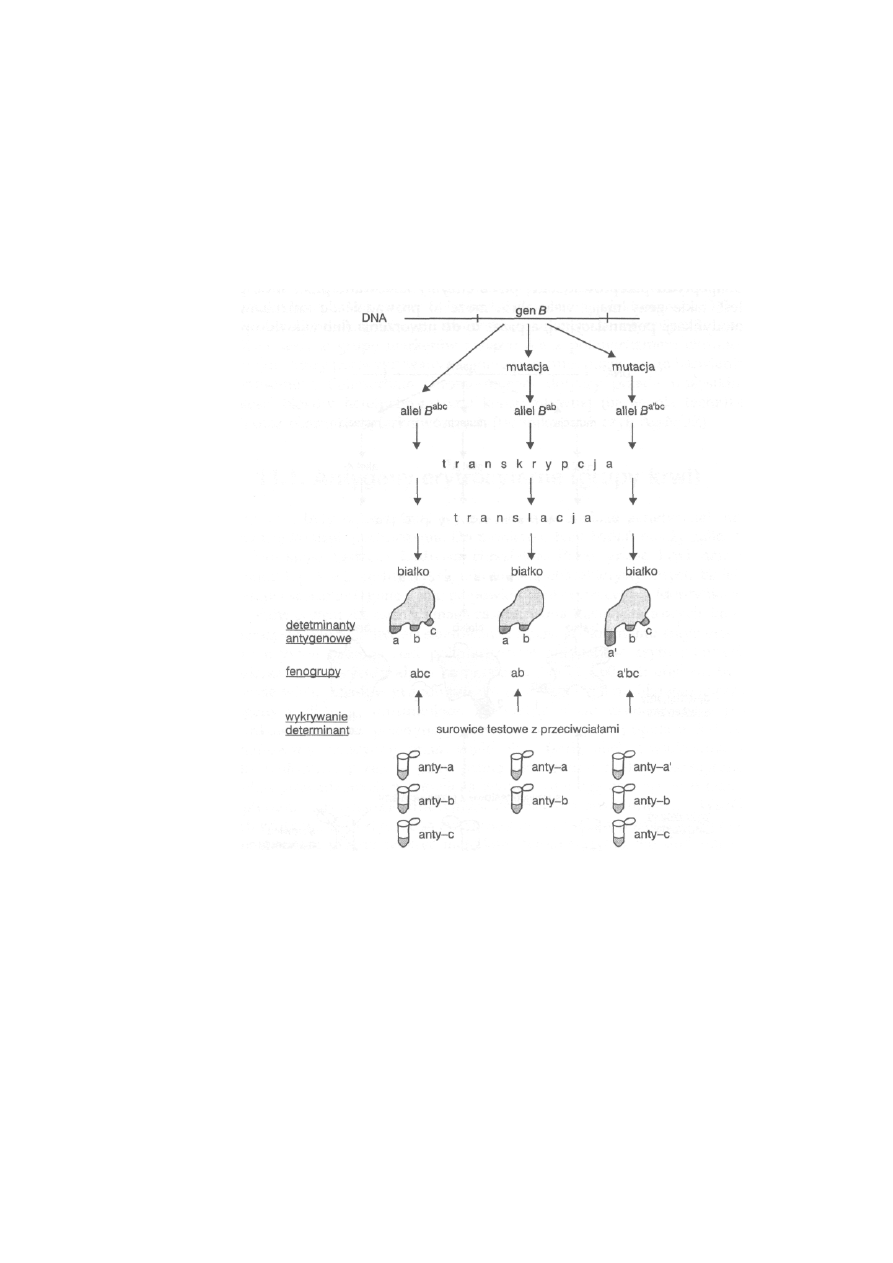
nowej determinanty w antygenie erytrocytarnym. Gdyby dla uproszczenia
przyjąć, że białko ma tylko jedną determinantę antygenową, to wówczas
każda mutacja genu, prowadząca do zmiany budowy białka — rozpozna-
wanej przez surowice testowe — będzie skutkowała powstaniem nowego
allelu (ryć. 11-1). Sytuacja staje się bardziej złożona, gdy białko ma więcej
determinant antygenowych. Wówczas pojedynczy allel jest opisywany za
pomocą kilku surowic testowych. Mówi się wówczas o fenogrupach, czyli
zespole antygenów, które dziedziczą się jak jednostka, a więc są uwarunko-
wane przez jeden allel. W takiej sytuacji mutacja genu zmienia jedną
Ryć. 11-2. Mechanizm powstawania zróżnicowania antygenów erytrocytarnych; przykład
dla białka mającego kilka determinant antygenowych, które pojawiają się w trzech
fenogrupach: abc, ab oraz a’bc). Schemat opiera się na założeniu, że nowe determinanty
antygenowe są skutkiem mutacji genu kodującego łańcuch polipeptydowy antygenu
erytrocytarnego
256
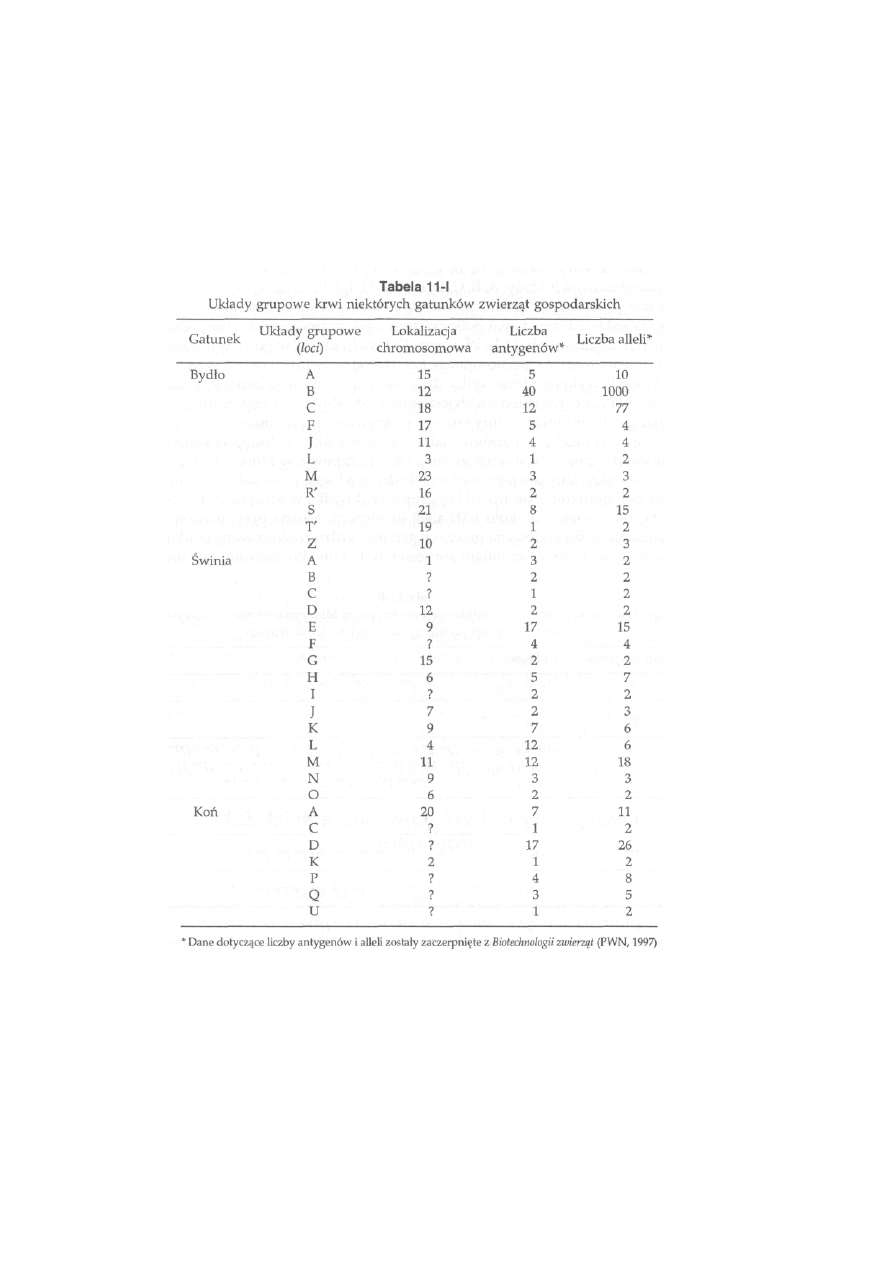
z determinant, ale nie narusza struktury pozostałych (ryć. 11-2). Badania
prowadzone u zwierząt wykazały, że występują obie sytuacje.
Należy jednak zaznaczyć, że istnieje także druga teoria tłumacząca
fenomen fenogrup. Przyjmuje ona, że każdy antygen warunkowany jest
przez jeden gen. Wówczas zespół antygenów dziedziczący się jako jednostka
genetyczna byłby uwarunkowany przez grupę genów bardzo silnie ze sobą
sprzężonych. Przy takim uwarunkowaniu genetycznym może dochodzić do
powstawania rekombinantów, a w konsekwencji do nietypowego dziedzi-
257

czenia antygenów krwinkowych. Przykłady takie opisano u bydła, głównie
w odniesieniu do układu grupowego B. Należy pamiętać, że istnieją białka
zbudowane z podjednostek kodowanych przez różne geny. Zatem możliwe
jest, że w niektórych przypadkach obie teorie będą miały zastosowanie do
wyjaśnienia połimorfizmu antygenów erytrocytarnych.
Liczba znanych układów grupowych (loci) u zwierząt gospodarskich
wynosi: 7 w genomie konia (układy: A, C, D, K, P, Q, U) oraz owcy (układy: A,
B, C, D, M, R, X), 11 w genomie bydła (układy: A, B, C, F, J, L, M, S, Z, T', R'), 13
w genomie kury (układy: A, B, C, D, E, F, G, H, J, K, M, O, P) oraz 15
w genomie świni (układy: A, B, C, D, E, F, G, H, I, J, K, L, M, N, O). Biologiczna
funkcja powyższych białek nie została jeszcze poznana. Natomiast bardzo
bogata jest wiedza na temat połimorfizmu występującego w obrębie poszcze-
gólnych układów (tab. 11-1). W przypadku wielu układów grupowych znane
jest umiejscowienie chromosomowe loci. Wśród poznanych układów są takie,
w których wykryto dotąd tylko dwa allele (np. układ L bydła czy układ
C świni), przy czym jeden z nich jest odpowiedzialny za występowanie białka
mającego determinantę antygenową wykrywaną przez surowicę testową,
a drugi allel (allel „ —") koduje białko, które w reakcji z dostępną surowicą
testową nie daje reakcji serologicznej. Układ grupowy, w którym występuje
allel „ — " określany jest jako otwarty. Na drugim biegunie są układy o olbrzy-
mim polimorfizmie, jak np. układ grupowy B bydła, w którym wykryto 40
antygenów i opisano około 1000 alleli (fenogrup). Można przypuszczać, że
cząsteczka białka kodowana przez ten gen ma bardzo zróżnicowaną strukturę
i przez to ma wiele determinant antygenowych. Ponadto determinanty mogą
258

mieć wiele wariantów, a każdy z nich jest rozpoznawany przez odrębną
surowicę testową. W efekcie wśród alleli będą takie, które warunkują
pojawienie się tylko jednej determinanty (rozpoznawanej przez jedną
surowicę), oraz takie, które odpowiadają za występowanie kilku determinant
(rozpoznawanych przez analogiczną liczbę surowic). Pomiędzy tymi skrajnymi
przykładami są liczne układy grupowe, w których zidentyfikowano kilka
czy kilkanaście alleli. W tabeli 11-11 przedstawione są rozpoznane dotychczas
antygeny i allele u koni.
Na uwagę zasługuje układ grupowy A świni. Jego ekspresja zależna jest
od dodatkowego locus S, który ma działanie epistatyczne w stosunku do
locus układu A. Gen S ma dwa allele: S oraz s. W układzie grupowym
A występują dwa antygeny A oraz O. Pojawienie się antygenu A lub
O uzależnione jest od genotypu w locus S, przy czym działanie epistatyczne
pojawia się przy genotypie homozygoty recesywnej ss.
Jak to już wcześniej wspomniano, badanie grup krwi wymaga stosowania
surowic testowych, które powinny spełniać warunek swoistości. Surowice
takie są produkowane przez laboratoria, które zajmują się badaniami
antygenów erytrocytarnych na potrzeby kontroli pochodzenia zwierząt.
Procedura produkcji oparta jest na immunizacji zwierząt, pozyskaniu od nich
surowicy odpornościowej (na ogół poliwalentnej, czyli zawierającej wiele
różnych przeciwciał), a następnie jej „oczyszczaniu" (absorpcji niespecyficz-
nych przeciwciał) do momentu otrzymania surowicy testowej (monowalent-
nej), to znaczy zawierającej przeciwciała skierowane przeciw konkretnemu
antygenowi erytrocytarnemu. Niezmiernie istotne jest, aby surowice testowe,
wyprodukowane w różnych laboratoriach, charakteryzowały się identyczną
swoistością. Cel ten osiąga się poprzez cykliczne przeprowadzanie, pod
auspicjami Międzynarodowego Towarzystwa Genetyki Zwierząt (ang.
International Society for Animal Genetics — ISAG), międzynarodowych
testów porównawczych (ang. comparison test). Polegają one na tym, że do
laboratoriów uczestniczących w teście przesyłane są zestawy zakodowanych
próbek krwi zwierząt. Za pomocą dostępnych surowic testowych należy
ustalić, jakie antygeny występują na erytrocytach w poszczególnych prób-
kach. Zbiorcze wyniki testu są udostępniane zainteresowanym laboratoriom.
W Polsce produkcją surowic testowych zajmuje się Instytut Zootechniki
(bydło, świnie, konie) oraz Zakład Hodowli Koni AR w Poznaniu (konie).
11.2. Białka surowicy krwi, erytrocytów
i leukocytów
Białka surowicy krwi, erytrocytów i leukocytów są drugą, po grupach krwi,
pod względem liczebności grupą markerów genetycznych klasy I. Poszcze-
gólne warianty tych białek są określane za pomocą technik elektroforetycz-
nych (np. elektroforeza w żelu poliakryloamidowym lub elektroforeza
259
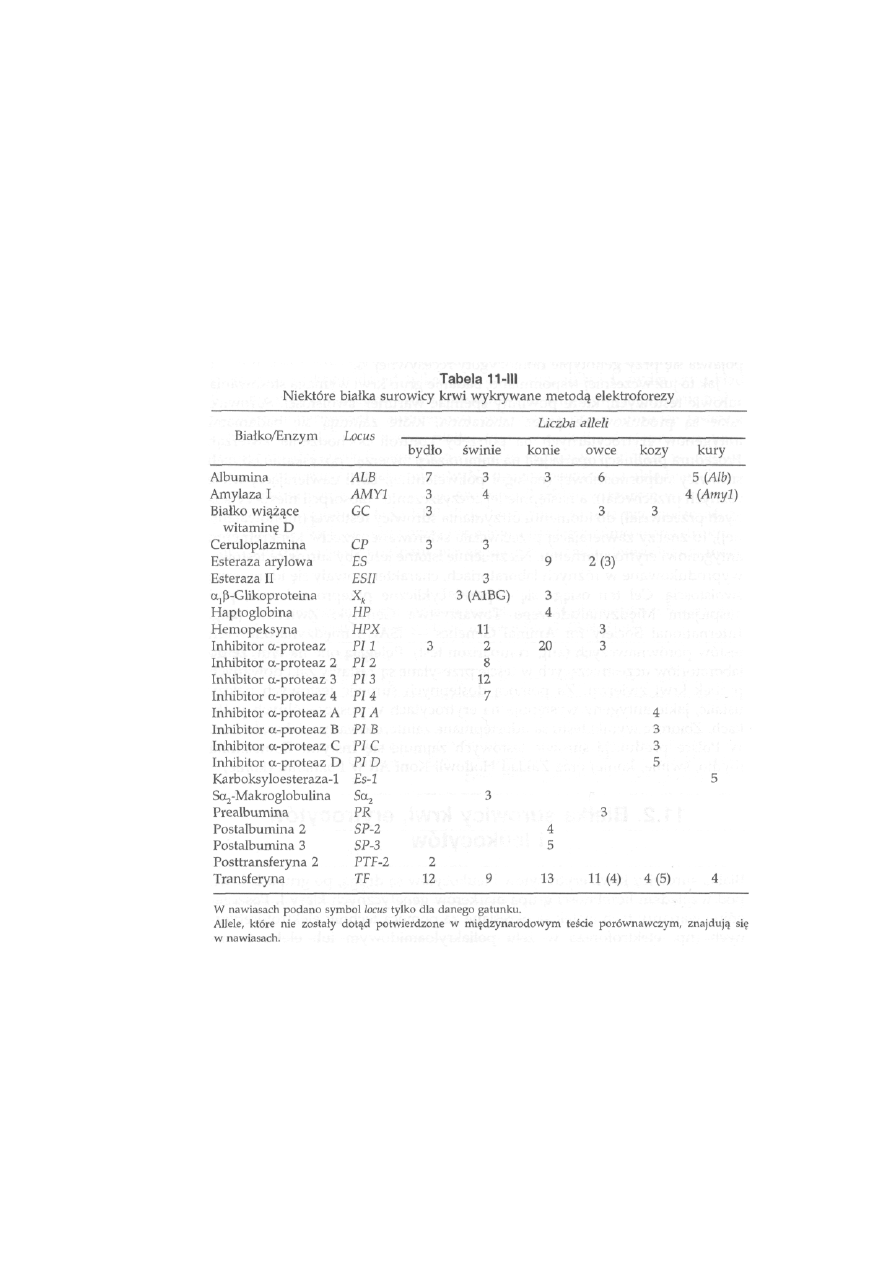
dwuwymiarowa czy rozdział białek w gradiencie pH — tzw. elektrofokusing).
Identyfikacja wariantów tych białek musi być potwierdzona w teście porów-
nawczym (pod kontrolą Międzynarodowego Towarzystwa Genetyki Zwie-
rząt). Na przykład u koni testy takie wykonywane są dla 16 systemów białek.
Polimorfizm białek leukocytów, nazywanych także cząsteczkami lub
antygenami głównego układu zgodności tkankowej (MHC), i ich znaczenie
są omówione szczegółowo w rozdziale: Genetyczne podstawy odporności
i oporności.
Polimorfizm białek surowicy krwi wynika ze zmienności ich form
strukturalnych, będących skutkiem mutacji punktowych w genie kontrolu-
jącym syntezę danego białka lub genach kodujących enzymy odpowiedzialne
za modyfikację potranslacyjną. Niektóre białka i enzymy surowicy krwi
zestawiono w tabeli ll-III. Spośród ponad 25 białek najbardziej polimorficzne
260

są albumina, inhibitor
α-proteaz i transferyna. Liczba rozpoznanych
wariantów elektroforetycznych białek jest jednak specyficzną cechą
gatunkową.
Albuminy współdziałają z globulinami w regulacji równowagi płynów
ustrojowych. Najwięcej wariantów tego białka zidentyfikowano u
bydła. U owiec synteza albumin jest kontrolowana przez locus ALB
umiejscowiony w chromosomie 6 i będący w sprzężeniu z locus białka
wiążącego witaminę D, przy czym jeden z alleli (ALB
S
) występuje u
większości ras. U świń locus albuminy jest umiejscowiony w chromosomie
8ql2.
Inhibitory
α-proteaz cechuje największy polimorfizm u takich
gatunków, jak świnia, koń i koza. U świń są one determinowane przez
ściśle sprzężone loci (Pi1, PO1A, PO1B, PIZ, P/3, PJ4) zajmujące łącznie
region długości około 2 centymorganów (chromosom 7q24-q26). Ze
względu na to, że różne rasy świń mają całkowicie odmienne haplotypy
(czyli układy sprzężonych alleli z loci genów dla inhibitorów
α-proteaz),
są one cennymi markerami genetycznymi. Tak więc, w rasie szwedzkiej
yorkshire oraz landrace zidentyfikowano po około 40 różnych haplotypów.
Z drugiej strony, niektóre allele są charakterystyczne dla danych ras, jak
np. PO1A
B
i PO1B
Y
dla wielkiej białej polskiej.
U koni w jednym poznanym dotychczas locus inhibitora
α-proteaz
wykryto 20 alleli, z kolei u kóz inhibitory
α-proteaz są kontrolowane
przez 4 loci, charakteryzujące się podobnym stopniem polimorfizmu (3-5
alleli).
Transferyna jest
β-globuliną, mającą dwa aktywne centra wiązania
żelaza. Jej podstawową funkcją jest transport żelaza z miejsc
absorpcji w przewodzie pokarmowym do różnych komórek organizmu.
Jest to najbardziej polimorficzne białko zidentyfikowane dotychczas u
podstawowych gatunków zwierząt gospodarskich. U koni jest ono
determinowane przez 13 alleli, przy czym jedynie allele Tf-D i Tf-F
2
występują prawie u wszystkich ras koni. Natomiast u świń allel Tf
B
jest
głównym allelem u ras europejskich i azjatyckich, podczas gdy allel Tf
A
występuje przede wszystkim u yorkshire, hampshire i duroc. Najrzadsze
allele to Tf
E
(tylko u świń rasy hampshire) oraz Tf
1
(u dzika
europejskiego). Locus transferyny u świń znajduje się w chromosomie 13
(13q31), u bydła w chromosomie l, u owiec także w chromosomie l (Iq42-
q45).
Spośród białek i enzymów erytrocytarnych (tab. 11-IV) szczególnie
interesujące są deaminaza adenozyny i hemoglobina.
Deaminaza adenozyny została dotychczas wykryta u trzody chlewnej,
owiec (chromosom 15), kur i bydła, przy czym u bydła jest to enzym
leukocytarny. U świń w locus ADA znanych jest sześć alleli. Polimorficzne
warianty są związane z różną aktywnością tego enzymu. U
osobników o genotypie ADA°ADA° brak jest deaminazy adenozyny w
erytrocytach, natomiast jest ona aktywna w leukocytach i innych
tkankach. Allel ten, określany jako zerowy, występuje szczególnie często
u zwierząt podatnych na stres, które mają skłonność do wytwarzania
mięsa typu DFD (ciemne, twarde i suche).
261
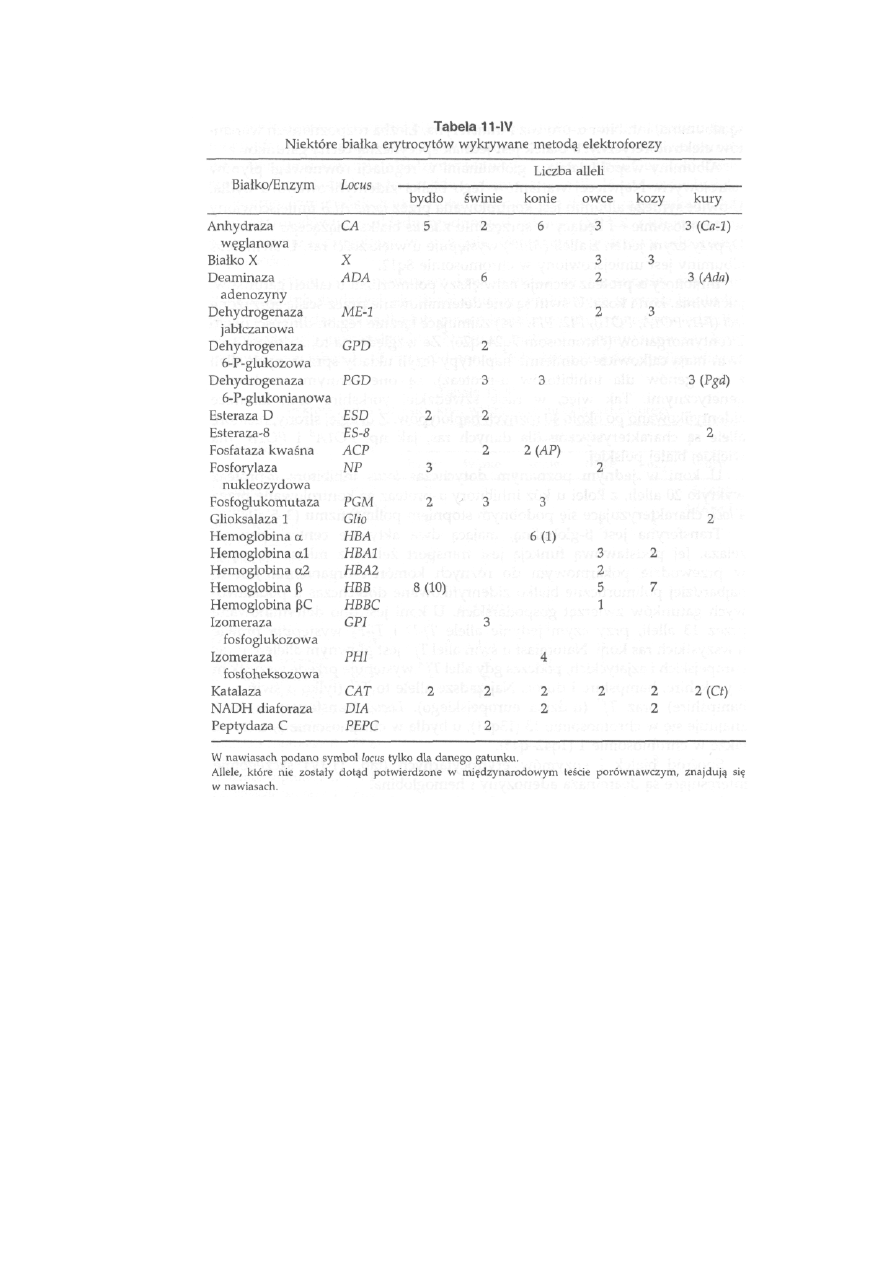
Hemoglobina wykazuje największy polimorfizm u owiec, bydła i kóz.
Warianty tego białka różnią się tylko budową łańcucha
β. Na przykład
u bydła w trzech pozycjach tego łańcucha, kontrolowanego przez locus
znajdujący się w chromosomie 15q22-q27, są różne aminokwasy. Polimorfizm
genu kontrolującego hemoglobinę występuje u niektórych ras bydła, jak np.
u rasy Jersey i niektórych ras mięsnych. Większość ras bydła, wśród nich
bydło czarno-białe i czerwono-białe, jest monomorficzna. U owiec warianty
hemoglobiny kontrolowane są przez loci: HBA (24 chromosom) i HBB (15
262

chromosom). Ciekawe jest, iż łańcuch
β hemoglobiny C (HBB C) jest
wytwarzany tylko w przypadku anemii i tylko u osobników mających allel
HBA, a wielkość tego wytwarzania zależy od nasilenia anemii. Ponadto
wariant A hemoglobiny
β (HBB A) wykazuje większe powinowactwo do
tlenu niż wariant B (HBB B) i związany jest z większą wartością hematokrytu.
11.3. Allotypy immunoglobulin i lipoprotein
Antygenowe właściwości surowicy krwi, określane terminem allotypy, są
to ugrupowania chemiczne (najczęściej jeden lub kilka aminokwasów) wy-
stępujące na cząsteczkach białek i różnicujące osobniki tego samego
gatunku. U większości gatunków zwierząt gospodarskich poznane allotypy
zlokalizowane są przede wszystkim na
α-, β- i γ-globulinach. Stopień
polimorfizmu allotypowego jest różny zależnie od gatunku. U świń
zidentyfikowano dotychczas 16 allotypów we frakcji
β-globulin, 14
allotypów dla
γ-globulin i 3 dla α-globulin. Natomiast u owiec wykryto 11
antygenowych markerów cząsteczek
α-globulin, 10 determinant
antygenowych we frakcji
β-globulin oraz 7 we frakcji γ-globulin.
W przypadku antygenów immunoglobulin u bydła, dotychczas ziden-
tyfikowano 3 allotypy, kontrolowane przez allele bva
l
, bva
2
oraz bvb.
Również lipoproteiny, biorące udział w transporcie cholesterolu, wyka-
zują właściwości antygenowe. W zależności od właściwości fizycznych
wyróżnia się pięć klas tych białek, od lipoprotein o bardzo małej gęstości
(VLDL) do lipoprotein o bardzo dużej gęstości (VHDL). Największe
zróżnicowanie allotypów lipoprotein, oznaczonych symbolem Lp, stwier-
dzono dotychczas u świń, u których najbardziej polimorficzny, bo liczący
ponad 21 allotypów, jest układ Lpb. Większość tych allotypów jest deter-
minowana przez kodominujące allele. W następnych układach (Lpr,
Lpt i Lpu) znane są po dwa allele, a w piątym układzie (Lps) tylko jeden
allel. U pozostałych gatunków zwierząt polimorfizm allotypowy
lipoprotein jest niewielki. U bydła zidentyfikowano 3 allotypy, u owiec 1.
11.4. Białka mleka
Głównymi białkami mleka są kazeiny
α
s1
,
α
s2
,
β i κ
oraz białko serwatkowe
— P-laktoglobulina. U bydła loci kazein znajdują się obok siebie w 6
chromosomie (6q26-q33) i zajmują razem odcinek DNA długości około
200 kpz (tysięcy par zasad). Ze względu na ścisłe sprzężenie loci kazein,
układ sprzężonych alleli nazywany jest haplotypem.
Genotyp zwierzęcia w loci kontrolujących białka mleka można określać
za pomocą metody elektroforezy stosowanej zarówno w przypadku
rozdziału białek (żel skrobiowy lub poliakryloamidowy), jak i analizy na
poziomie DNA (żel agarozowy lub poliakryloamidowy). Określenie
genotypu na podstawie
263

badania białka jest możliwe tylko u samic będących w okresie laktacji.
Natomiast analiza polimorfizmu genów kodujących białka (z wykorzysta-
niem metody RFLP, opisanej w rozdziale: Metody analizy i modyfikacji ge-
nomu) może być przeprowadzana u osobników obu płci i w każdym wieku.
U bydła w locus kazeiny
α
sl
wykryto pięć alleli: A, B, C, D i E, przy czym
największą frekwencją u bydła europejskiego charakteryzuje się allel B,
a u bydła indyjskiego — alleł C.
Białko
α
s2
-kazeiny różni się długością łańcucha od białka
α
s1
-kazeiny,
składają się one bowiem odpowiednio z 207 i 199 aminokwasów. Poszczegól-
ne warianty a
S2
-kazeiny są kontrolowane przez 4 allele: A, B, C i D, z których
jedynie A i D są obecne w genotypie bydła ras europejskich.
β-Kazeina wykazuje większy polimorfizm niż kazeiny
α
s1
i
α
S2
, albowiem
w locus kontrolującym syntezę tego białka stwierdzono 7 alleli. U bydła
europejskiego najczęściej występuje wariant
β-kazeiny A, bardzo rzadko
wariant
β-kazeiny B (prawie wyłącznie stwierdzane są genotypy hetero-
zygotyczne AB).
Dotychczas u bydła wykryto 6 typów
κ-kazeiny, determinowanych
allelami: A, B, C, E, F i G. Różnice między poszczególnymi wariantami
polegają na zmianie aminokwasu, będącej konsekwencją mutacji punktowej
(tranzycji jednej zasady na inną). Tak więc allele A i B różnią się
nukleotydami w dwóch miejscach łańcucha DNA, które powodują
zmianę aminokwasu. W pozycji 136 łańcucha polipeptydowego B jest
treonina, a w wariancie A — izoleucyna, natomiast w pozycji 148
wariant B
κ-kazeiny ma kwas asparaginowy, a wariant A — alaninę.
Molekularna identyfikacja alleli A i B metodą PCR-RFLP jest przedstawiona
w rozdziale: Zmienność cech.
Allel E
κ-kazeiny różni się od A i B podstawieniem A→G (seryna->glicyna
w pozycji 155 łańcucha polipeptydowego). Allel F powstał w wyniku
mutacji punktowej w allelu A, polegającej na tranzycji G
→A, której skutkiem
jest zmiana w pozycji 10 łańcucha polipeptydowego aminokwasu argininy
na histydynę. Podobnie allel G powstał wskutek tranzycji C
→T w allelu A,
powodującej zamianę argininy na cysteinę w pozycji 97 sekwencji amino-
kwasowej białka. Mutacje te tworzą miejsca restrykcyjne dla enzymów HhaI
i MaII, co stwarza możliwość molekularnej identyfikacji poszczególnych
alleli za pomocą metody PCR-RFLP.
Białko serwatkowe mleka bydła,
β-laktoglobulina, występuje w dwóch
wariantach A i B.
β-Laktoglobulina odgrywa rolę w odporności organizmu,
pobierana bowiem przez oseski z mlekiem matki chroni je przed infekcjami.
Mleko zawierające
β-laktoglobulinę B różni się od mleka z β-laktoglobulina
A większą zawartością suchej masy, tłuszczu i kazein oraz lepszą przydat-
nością technologiczną do produkcji serów. Podobnie jak w przypadku
κ-
kazeiny, znane są różnice w sekwencji aminokwasowej obu wariantów
β-
laktoglobuliny, wynikające z polimorfizmu genu kodującego to białko.
Białko
β-laktoglobuliny składa się z 162 aminokwasów. Różnica między
264

wariantem A i B polega na tym, że wariant A w pozycji 64 sekwencji
aminokwasowej zawiera kwas asparaginowy (GAT), natomiast wariant
B — glicynę (GGT). Z kolei w pozycji 118 w wariancie A jest walina (GTC),
zaś w B -- alanina (GCC). Zmiana zasady w kodonie 118 genu stwarza
miejsce restrykcyjne dla enzymu HaeIII (GG/CC) w allelu
β-laktoglobuliny
B. Zostało to wykorzystane w opracowanym teście PCR-RFLP, który także
jest już rutynowo stosowany.
W mleku kóz i owiec, podobnie jak bydła, są cztery frakcje białek.
Najbardziej polimorficzny jest gen kazeiny
α
s1
u kóz (7 alleli — A, B, C, D,
E, F, O). O ile allele A, B, C i E różnią się między sobą kilkoma podstawieniami
aminokwasów, o tyle allele D i F wykazują znacznie większe różnice,
polegające na delecji odpowiednio 11 i 37 aminokwasów. W pozostałych loci
wykryto po 2 allele. Podobnie u owiec, z wyjątkiem locus P-laktoglobuliny
(chromosom 3p28), w którym są 3 allele, w loci kontrolującym główne białka
mleka występują po 2 allele.
11.5. Polimorfizm DNA
W jądrowym DNA zdecydowanie przeważają sekwencje niekodujące,
wśród których dużą przydatność dla potrzeb analizy genetycznej mają
sekwencje powtarzające się tandemowo. W połowie lat 80. opisano po
raz pierwszy sekwencje minisatelitarne. Ich cechą charakterystyczną jest
to, że długość podstawowego motywu powtarzającego się wynosi zazwyczaj
kilkanaście nukleotydów. Do najbardziej znanych sekwencji minisate-
litarnych należą: 33.5 (motyw podstawowy: GGGAGGTGGGCAGGAGG),
33.6 (motyw podstawowy: AGGGCTGGAGG) i 33.15 (motyw podstawowy:
AGAGGTGGGCAGGTGG). Sekwencje te występują w wielu miejscach (loci)
genomu, a w konkretnym locus może być różna liczba powtórzeń motywu
podstawowego. Jeśli strawiony enzymem restrykcyjnym i rozdzielony
elektroforętycznie DNA hybrydyzuje się z wyznakowaną radioaktywnie
sondą zawierającą sekwencję minisatelitarną, to na błonie radiograficznej
pojawia się układ prążków, który reprezentuje fragmenty DNA o różnej
długości. Fragmenty te pochodzą z wielu loci, a ich długość zależy od liczby
powtórzeń motywu podstawowego i także odległości, jaka dzieliła sekwencję
minisatelitarną od miejsca cięcia przez enzym restrykcyjny. Uzyskany układ
prążków, nazywany niekiedy „odciskiem palca" DNA (ang. DNA fingerprint),
przedstawia wysoce charakterystyczny obraz osobniczego DNA, który może
być wykorzystany m.in. do kontroli pochodzenia i identyfikacji śladów
biologicznych. Obok równoczesnej analizy wielu loci możliwe jest także
badanie polimorfizmu sekwencji minisatelitarnej w pojedynczym locus.
Badanie takie wymaga zastosowania sond o dużej specyficzności, to znaczy
zawierających nie tylko sekwencję minisatelitarną, ale także sekwencje ją
otaczające, oraz przeprowadzenia hybrydyzacji zgodnie z zaostrzoną proce-
265

durą (ang. high stringency). Z zebranych do tej pory informacji o wy-
stępowaniu sekwencji minisatelitarnych w różnych genomach wynika, że
ich loci są położone głównie w częściach telomerowych
chromosomów. Z tego względu ich przydatność w budowaniu
markerowych map genomo-wych jest ograniczona.
Wkrótce po odkryciu polimorfizmu sekwencji minisatelitarnych opisano
polimorfizm krótkich sekwencji, bo zawierających motyw podstawowy
długości kilku nukleotydów, także powtarzających się tandemowo. Zostały
one określone terminem sekwencji mikrosatelitarnych. Do tej pory najczęściej
opisywano dwu- (np. CA) lub czteronukleotydowe (np. CATA) motywy
podstawowe. Sekwencje mikrosatelitarne mogą występować jako proste
powtórzenia motywu podstawowego lub mogą mieć układ złożony, pole-
gający na tym, że powtórzenia motywu podstawowego są przedzielone
inną sekwencją — np. (CA)
n
AT(CA)
m
lub też sekwencja przedzielająca jest
otoczona różnymi powtarzającymi się motywami podstawowymi — np.
(GT)
n
GAT(AG)
m
. Wykrywanie polimorfizmu mikrosatelitarnego może być
prowadzone podobnie jak sekwencji minisatelitarnych, tzn. hybrydyzacji
z sondami i wówczas analiza dotyczy równocześnie wielu loci. Szczególne
jednak znaczenie zyskały sekwencje mikrosatelitarne w chwili, gdy za
pomocą techniki PCR, po rozpoznaniu sekwencji otaczających sekwencję
mikrosatelitarną, możliwe stało się analizowanie pojedynczych loci. Na
podstawie wiedzy o sekwencjach otaczających mikrosatelitę syntetyzowane
są oligonukleotydy pełniące funkcję starterów dla polimerazy DNA. Badanie
polimorfizmu sekwencji mikrosatelitarnych w pojedynczym locus polega na
amplifikacji techniką PCR specyficznych fragmentów, pochodzących z DNA
wyizolowanego od danego osobnika, a następnie określeniu ich długości za
pomocą klasycznej elektroforezy (np. żel poliakryloamidowy + barwienie
azotanem srebra) lub w automatycznym sekwenatorze DNA. Zależnie od
genotypu osobnika stwierdza się występowanie jednego fragmentu, w przy-
padku homozygoty, lub dwóch fragmentów — u heterozygoty (ryć. 11-3
wkładka kolorowa). Sekwencje mikrosatelitarne stały się najpowszechniej-
szym źródłem markerów genetycznych, dzięki wysokiemu polimorfizmowi
i równomiernemu rozproszeniu w genomie. Szacuje się, że w genomie
ssaków sekwencje takie występują co 6-10 tysięcy par nukleotydów. Warto
zaznaczyć, że sekwencje mikrosatelitarne mogą występować w intronach
(przykładem są geny apolipoproteiny A oraz B świni) lub nawet w eksonach
(gen kodujący huntingtynę u człowieka).
Wykrywanie polimorfizmu (markerów) DNA możliwe jest również
wówczas, gdy nie są znane ani sondy molekularne, ani sekwencje starterowe
umożliwiające amplifikację techniką PCR konkretnego, polimorficznego
fragmentu DNA. Jedną z częściej stosowanych procedur w takiej sytuacji
jest losowa amplifikacja polimorficznego DNA (ang. random amplified
polymorphic DNA — RAPD). Analiza tego typu polega na amplifikacji
nieznanych fragmentów DNA, za pomocą sekwencji starterowej o losowej
266

kombinacji nukleotydów (zazwyczaj składa się z 10 nukleotydów), w której
dominują nukleotydy C i G. Zamplifikowane fragmenty, reprezentujące
zazwyczaj różne loci w genomie, są poddawane elektroforezie, w wyniku
której uzyskuje się kilka prążków (fragmentów DNA różnej długości).
Badając grupę osobników można stwierdzić różnice w układzie prążków,
które są wynikiem mutacji punktowych tworzących lub znoszących miejsce
rozpoznawane przez losowy 10-nukleotydowy oligonukleotyd starterowy
dla reakcji PCR. Zidentyfikowanie takiego polimorficznego fragmentu
pozwala na dalszą jego analizę, w tym sekwencjonowanie. Po określeniu
sekwencji tego fragmentu możliwe staje się ograniczenie analizy genetycznej
do tego pojedynczego locus. W ten sposób identyfikuje się markery określane
skrótem SCAR (ang. seauence characterized amplified region). Jeśli amp-
lifikowany fragment jest wystarczająco duży, to możliwe jest jego zlokalizo-
wanie w chromosomie (mapowanie fizyczne). Przykładem takiej procedury
jest identyfikacja polimorficznego markera RAPD, amplifikowanego przez
starter 5'-AGAATAGGC-3', charakterystycznego dla psów obciążonych
chorobą genetyczną — postępującym zanikiem siatkówki (ang. progressive
rod-cone degeneration). Polimorficzny fragment, wykryty za pomocą techniki
RAPD, został następnie molekularnie opisany, co doprowadziło do iden-
tyfikacji markera specyficznego SCAR, który zlokalizowano w chromosomie
9 u psa. Należy zaznaczyć, że markery RAPD czy SCAR mogą zawierać
sekwencje kodujące lub niekodujące i dlatego ich jednoznaczna klasyfikacja
do kategorii markerów klasy I lub II jest trudna.
Badanie polimorfizmu DNA jest powszechnie stosowane także dla
markerów klasy I. Analiza taka opiera się najczęściej na wykrywaniu
polimorfizmu długości fragmentów restrykcyjnych (RFLP), a także polimor-
fizmu konformacji jednoniciowych łańcuchów DNA (SSCP). Istotą obu
metod jest identyfikacja mutacji punktowych w obrębie genów. O metodach
tych napisano w rozdziale: Metody analizy i modyfikacji genomu.
Dynamiczny rozwój badań, których celem jest sekwencjonowanie geno-
mów różnych gatunków, zaowocował identyfikacją nowej klasy markerów
genetycznych, określanych symbolem SNP (ang. single nucleotide polymor-
phism). Polimorfizm ten związany jest z występowaniem pojedynczych
zmian sekwencji nukleotydów w homologicznych sekwencjach, np. allel
l - AATCGGC oraz allel 2 — AATGGGC. Mutacja punktowa prowadzi do
powstania miejsca polimorficznego typu SNP, jeśli każdy z alleli występuje
w populacji z częstością nie mniejszą niż 1%. Polimorfizm SNP wiąże się
zazwyczaj z obecnością w puli genowej populacji jedynie dwóch alleli.
Niewątpliwą zaletą polimorfizmu SNP jest jego powszechność w genomach
różnych gatunków. Na przykład, prace nad ustaleniem sekwencji DNA
genomu człowieka ujawniło co najmniej 1,4 mln miejsc SNP, spośród
których większość występuje poza strukturą genów. Wynika z tego, że te
proste podstawienie nukleotydów zazwyczaj ma charakter tzw. cichego
podstawienia, czyli nie wpływa na budowę polipetydu, ale może od-
267

działywać na ekspresję genu. Jednakże niektóre z tych podstawień mogą
wywoływać skutki fenotypowe. Ogromne nasycenie genomów markerami
SNP sprawia coraz powszechniejsze ich wykorzystywanie w analizie
genetycznej. Do identyfikacji alleli SNP może być przydatna technika RFLP
(jeśli mutacja zmienia sekwencję rozpoznawaną przez enzym restrykcyjny)
lub SSCP. Jednak identyfikacja wielu miejsc SNP wymaga odwołania się do
innych technik, w tym sekwencjonowania DNA. Polimorfizm na poziomie
sekwencji nukleotydów może być również związany z brakiem (delecja) lub
wstawieniem (insercja) jednego lub kilku nukleotydów. Tego typu markery
opisywane są terminem InDel (Insertion Deletion polymorphism) i mogą
mieć np. następujące formy: allel l — TCGAATGAATT (insercja AAT) i allel
2 — TCGGAATT (delecja AAT).
11.6. Wykorzystanie markerów genetycznych w
hodowli zwierząt
Markery genetyczne znajdują zastosowanie w pracy hodowlanej między
innymi w kontroli pochodzenia, ocenie wartości genetycznej zwierząt pod
kątem cech użytkowych i selekcji pośredniej, tzw. MAS (ang. marker
assisted selection), ocenie zmienności genetycznej wewnątrz i między
populacjami (liniami) oraz szacowaniu dystansu genetycznego. Informacje
o umiejscowieniu loci markerów genetycznych znajdują się w markerowych
mapach genomu (rozdział: Mapy genomowe).
Kontrola pochodzenia u zwierząt od wielu lat jest prowadzona na
podstawie układów grupowych krwi i polimorficznych białek, czyli mar-
kerów genetycznych klasy I. Podstawą tej kontroli jest to, że potomek
dziedziczy jeden allel z każdego locus od ojca, drugi od matki. Zatem
w przypadku grup krwi nie może on mieć antygenu erytrocytarnego,
którego nie stwierdzono u jednego z rodziców. Do kontroli pochodzenia
najlepsze są układy grupowe, w których występuje wiele antygenów,
u bydła są to układy B (40 antygenów), C (12 antygenów) oraz S (8 anty-
genów), u świń układy M (12 antygenów), E (17 antygenów) i L (12 anty-
genów), natomiast u koni układy A (7 antygenów), D (17 antygenów),
P (4 antygeny) i Q (3 antygeny). Przykład kontroli pochodzenia na podstawie
polimorfizmu antygenów erytrocytarnych oraz białek surowicy krwi i eryt-
rocytów przedstawiono w tabeli 11-V. Porównanie genotypów i fenotypów
źrebaka i klaczy wskazuje na to, że w fenotypie ojca musiały wystąpić
antygeny erytrocytarne A-a, D-cgm, P-ad i Q-b oraz typy białek TF-O,
ALB-A, ES-I, PHI-I. Warunków tych nie spełnia ogier l, można wykluczyć
jego ojcostwo. Spełnia je ogier 2, on zatem może być ojcem źrebięcia.
Ponieważ kontrola pochodzenia na podstawie polimorfizmu antygenów
erytrocytarnych (grup krwi) i polimorficznych białek surowicy krwi nie
zawsze stwarza możliwość jednoznacznego wskazania pary rodzicielskiej,
268
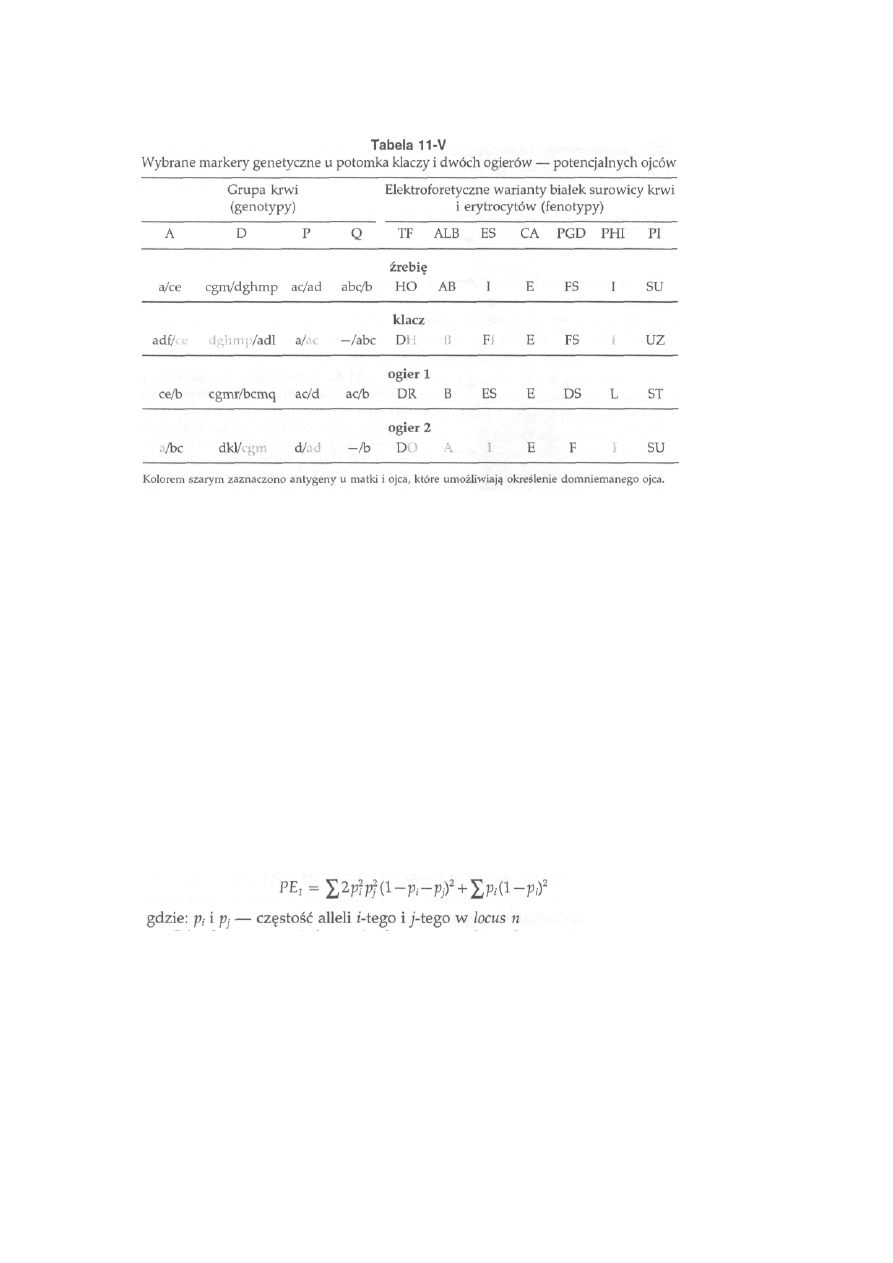
ostatnio coraz częściej są stosowane do tego celu markery genetyczne
klasy II (niekodujące sekwencje DNA). Daje to możliwość wykorzystania
znacznie bardziej precyzyjnych kryteriów analizy genetycznej. Można
wyróżnić dwie podstawowe metody stosowane do kontroli pochodzenia;
jedna jest oparta na polimorfizmie minisatelitarnym, druga na polimorfizmie
mikrosatelitarnym. Polimorfizm minisatelitarny jest podstawą metody „odcis-
ku palca" DNA. W metodzie tej strawiony (pocięty) enzymem restrykcyjnym
i rozdzielony elektroforetycznie DNA jest hybrydyzowany z sondą, którą jest
np. sekwencja minisatelitarna. Uzyskany obraz prążków jest charakterystycz-
ny dla danego osobnika, ale potomek otrzymuje od każdego z rodziców po
jednym prążku („allelu") danej sekwencji (ryć. 11-4). Prawdopodobieństwo
wystąpienia tego samego obrazu prążków u dwóch niespokrewnionych
osobników jest minimalne. Dokładność kontroli pochodzenia można zwięk-
szyć przez użycie w analizie kilku sond molekularnych.
W celu określenia prawdopodobieństwa wykluczenia pochodzenia
danego osobnika po danym rodzicu (-ach) stosowane są odpowiednie
wzory. W przypadku analizy polimorfizmu w jednym locus minisatelitarnym
u potomka i jednego z rodziców (nie zawsze dysponujemy pełnymi danymi
pochodzenia) prawdopodobieństwo wykluczenia obliczane jest ze wzoru:
Gdy dostępne są informacje dotyczące polimorfizmu minisatelitarnego
w DNA obojga rodziców i potomka, prawdopodobieństwo wykluczenia
obliczane jest ze wzoru:
269
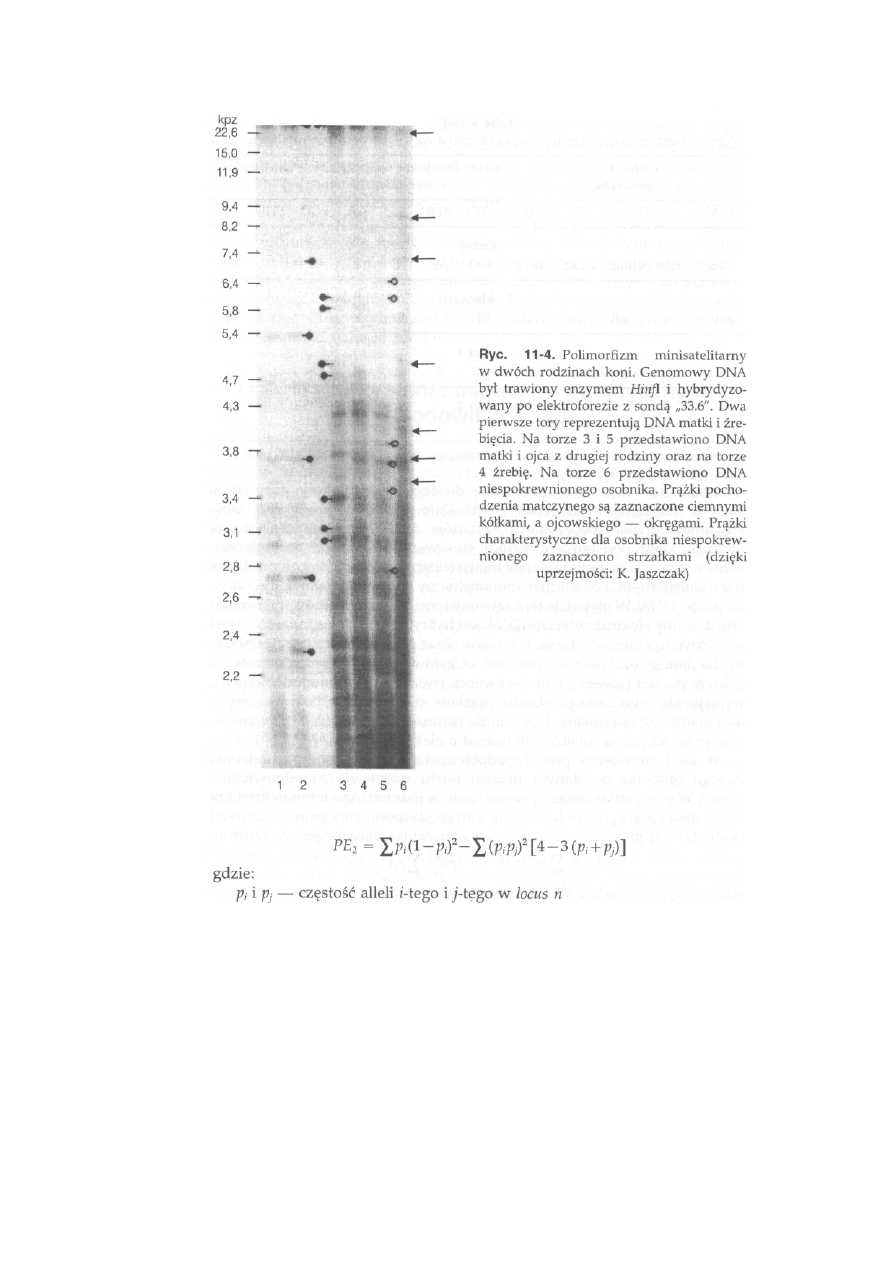
Jak już wspomniano, analiza polimorfizmu w kilku loci minisatelitarnych
równocześnie zwiększa dokładność kontroli pochodzenia. W takim przypad-
ku określa się tzw. łączne prawdopodobieństwo wykluczenia dla wszystkich
270

badanych loci. Wzór na obliczanie tego prawdopodobieństwa ma tę sama
postać dla obu przypadków, dostępności informacji zarówno o jednym,
jak i obojgu rodzicach:
Druga metoda stosowana obecnie do kontroli pochodzenia wykorzystuje
polimorfizm sekwencji mikrosatelitarnych. W przypadku każdej sekwencji
mikrosatelitarnej potomek otrzymuje jeden allel od matki, drugi od ojca,
dokładnie tak, jak to jest przy cechach monogenowych — jakościowych.
Istnieje możliwość kontroli pochodzenia za pomocą PCR multipleks, w której
stosuje się kilka markerów równocześnie. Długość alleli (produktu PCR
— zamplifikowanych sekwencji mikrosatelitarnych) może być oznaczana za
pomocą automatycznego sekwenatora. Przykład kontroli pochodzenia na
podstawie sekwencji mikrosatelitarnej 2319 (powtórzenie 4-nukleotydowe)
u lisów, której długość określono metodą elektroforetycznego rozdziału
w żelu poliakryloamidowym barwionym azotanem srebra oraz za pomocą
automatycznego sekwenatora, przedstawiono na rycinie 11-5 (wkładka
kolorowa). Krzywe 1-5 obrazują genotypy szczeniąt (3-5) oraz ich do-
mniemanych rodziców — ojca (1) i matki (2). Ojciec jest homozygotą pod
względem allelu długości 235 pz, matka natomiast heterozygotą (jeden allel
- 243 pz, drugi — 263 pz). Wszystkie szczenięta są heterozygotami, przy
czym allel długości 235 pz odziedziczyły po ojcu. Dwa z nich otrzymały od
matki allel długości 243 pz, natomiast trzeci — allel 263 pz. Widoczny „pik"
przy długości 270 pz jest to marker wielkości fragmentu DNA, dodawany
do każdej próby DNA analizowanej za pomocą sekwenatora.
W ostatnich latach badacze coraz częściej zastanawiają się nad wy-
korzystaniem do kontroli pochodzenia polimorfizmu pojedynczych nu-
kleotydów, określanego symbolem SNPs (ang. single nucleotide poly-
morphisms). Polimorfizm ten jest wynikiem mutacji punktowych (zamiana
określonej zasady w danym nukleotydzie na inną). Przyczyną odchodzenia
od wykorzystywania sekwencji mikrosatelitarnych w kontroli pochodzenia
są głównie zachodzące w nich mutacje, których częstość szacuje się
na 10
-2
-10
-5
na pokolenie. Zaletą SNPs jest bardzo duża gęstość ich
rozmieszczenia w genomie (na przykład w genomie człowieka występują
one co ok. 1300 par zasad). Również to, że do identyfikacji genotypu
w przypadku polimorfizmu pojedynczych nukleotydów można zastosować
zdecydowanie więcej technik molekularnych niż do identyfikacji genotypu
w loci mikrosatelitarnych, sprawia, iż w niedalekiej przyszłości polimorfizm
pojedynczych nukleotydów zastąpi sekwencje mikrosatelitarne nie tylko
w kontroli pochodzenia, ale także w poszukiwaniach genów ważnych
z hodowlanego punktu widzenia, szacowaniu zmienności genetycznej
i dystansu genetycznego.
271

Badacze niemieccy wybrali na razie 60 SNPs do kontroli pochodzenia
u bydła holsztyńsko-fryzyjskiego, brunatnego szwajcarskiego i simentali.
Identyfikacji genotypu w loci tych markerów daje 99,99% prawdopodobień-
stwa wykluczenia.
Sekwencje mikrosatelitarne są bardzo pomocne w analizie genetycznego
tła cech ilościowych. Cechy użytkowe (ilościowe) są determinowane przez
wiele genów o małym efekcie każdego z nich, dlatego identyfikacja ekspresji
tych genów jest znacznie utrudniona. Prowadzone są badania w celu
znalezienia takich genów, które mają znaczący wpływ na kształtowanie się
cech użytkowych. Analiza asocjacji między markerami mikrosatelitarnymi
a cechami ilościowymi jest pierwszym krokiem do identyfikacji genów
„ważnych", czyli istotnych z hodowlanego punktu widzenia. Znając sprzę-
żenie między określonym markerem genetycznym a np. wydajnością białka
w mleku, można dla młodej jałówki określić, jaka będzie wartość tej cechy
w przyszłości. Wykorzystując markery mikrosatelitarne wykryto u bydła
kilka loci znajdujących się w różnych chromosomach, warunkujących cechy
użytkowości mlecznej (tab. 11-VI). U świń określono także regiony chromo-
somów sprzężone z cechami użytkowości mięsnej i rozpłodowej (tab. 11-VII).
Niektóre cechy użytkowe występują tylko u jednej płci lub nie można
ich zmierzyć przyżyciowe. Cechy takie mogą być doskonalone jedynie
poprzez selekcje pośrednią. Rozwój technik molekularnych przyczynił się
do opracowania metody selekcji pośredniej nazwanej MAS (ang. marker
assisted selection). W metodzie tej kryterium wyboru zwierzęcia jest jego
genotyp w locus markera genetycznego wykazującego asocjację z doskona-
loną cechą użytkową. Większość cech użytkowych o znaczeniu ekonomicz-
nym ujawnia się fenotypowo u zwierząt w określonym wieku (np. cechy
użytkowości mlecznej). W takim przypadku selekcja pośrednia (MAS) jest
znacznie bardziej intensywna i dokładna w porównaniu z selekcją tradycyj-
ną, a postęp genetyczny jest uzyskiwany w krótszym czasie. Wynika to
z możliwości określenia genotypu u bardzo młodych zwierząt.
Badania asocjacji markerów genetycznych z cechami ilościowymi zaowo-
cowały już wykryciem u większości gatunków zwierząt gospodarskich sporej
liczby loci genów ważnych, o istotnym wpływie na cechy użytkowe. Okazało
się, że niektóre geny oddziałujące na wartość cechy ilościowej mają ogromne
znaczenie. Geny takie nazwano głównymi lub genami o dużym efekcie.
Należą do nich między innymi geny warunkujące hipertrofię mięśniową
u bydła i owiec, zwiększoną plenność świń, zwiększoną płodność owiec oraz
geny białek mleka —
κ-kazeiny i β-laktoglobuliny (jest o nich mowa
w rozdziale: Zmienność cech). W przypadku tych cech jest już możliwa
selekcja bezpośrednia, której kryteriami są genotypy zwierząt w danych loci.
Markery genetyczne, głównie klasy II, są również pomocne w doskonaleniu
niektórych cech jakościowych. Przykładem tego jest wykorzystanie
markerów mikrosatelitarnych do identyfikacji obecności w genotypie hetero-
zygotycznym recesywnego allelu rogatości u bydła. Jest to cecha dziedzicząca
272

się z dominacją całkowitą, której fenotyp jest taki sam u osobnika homo-
zygotycznego i heterozygotycznego. Problem eliminacji z populacji genu
rogatości jest dość istotny w krajach zachodnich, w których coraz więcej
uwagi poświęca się dobrostanowi zwierząt. Posiadanie rogów jest cechą
niekorzystną tak w aspekcie dobrostanu (gdyż może być przyczyną uszko-
dzeń ciała), jak również intensywnej hodowli (zasiniaczenia i ukryte
273

uszkodzenia tusz są przyczyną istotnych strat ekonomicznych w produkcji
żywca). Stosowany w stadach bydła mięsnego zabieg usuwania rogów
powoduje cierpienie zwierząt oraz zmniejszenie ich dziennych przyrostów,
ponadto jest zabiegiem kłopotliwym. Aby tego uniknąć, coraz więcej uwagi
poświęca się genetycznemu uwarunkowaniu bezrożności i możliwości
prowadzenia selekcji w tym kierunku. Dotychczas wiadomo jedynie, że
locus polled, kontrolujący bezrożność/rogatość u bydła, występuje w l chromo-
somie. Identyfikacja nosicieli genu p może być przeprowadzana za pomocą
analizy polimorfizmu sprzężonych z tym genem markerów genetycznych.
Znane są już dwie sekwencje mikrosatelitarne, sprzężone z locus polled,
które mogą być wykorzystane do tego celu.
Polimorfizm markerów genetycznych jest wykorzystywany w badaniach
nad odpornością/podatnością zwierząt na różne choroby i zaburzenia
genetyczne, dzięki czemu nosiciele niekorzystnych alleli mogą być wykrywa-
ni, zanim wystąpią objawy chorobowe. Szczególne znaczenie mają badania
związku między antygenami/genami głównego układu zgodności tkankowej
(MHC) a występowaniem chorób, zwłaszcza infekcyjnych. Zagadnienie to
jest opisane w rozdziale: Genetyczne podstawy odporności i oporności.
W badaniach z zakresu genetyki populacji znajdują zastosowanie zarówno
markery genetyczne klasy I, jak i klasy II. Zastosowanie frekwencji alleli
markerów genetycznych klasy I do szacowania zmienności genetycznej
wewnątrz i między populacjami omówione zostało w rozdziale: Podstawy
genetyki populacji. Znaczenie markerów genetycznych w hodowli zwierząt
podkreśla to, że są one wykorzystywane do analizy zmienności genetycznej
w ramach programów Światowej Strategii Zachowania Zasobów Genetycznych
Zwierząt Gospodarskich FAO (Global Strategy for the Farm Animal Genetic
Resources), w której są uwzględnione zasady międzynarodowego porozumie-
nia — Konwencji o Biologicznej Różnorodności. Zmienność genetyczna
w obrębie ras ginących lub uznanych za zagrożone jest określana na podstawie
polimorfizmu markerów genetycznych, przede wszystkim polimorfizmu
antygenów erytrocytarnych (grup krwi) i polimorfizmu DNA mikrosatelitarnego.
Markery genetyczne są także niezwykle przydatne w szacowaniu stopnia
zinbredowania (homozygotyczności) poszczególnych populacji (linii), w ana-
lizie podobieństw i różnic między populacjami i ustalaniu dystansu genetycz-
nego między nimi. Szacowanie dystansu genetycznego i wybór ras, które
cechuje duża zmienność genetyczna, służy zachowaniu biologicznej różno-
rodności zwierząt gospodarskich. Z kolei ocena zmienności genetycznej
między populacjami (liniami) ułatwia wybór optymalnego wariantu krzyżo-
wania, a w krzyżowaniu towarowym uzyskanie maksymalnego efektu
heterozji, ujawniającego się głównie zwiększeniem wydajności cech użyt-
kowych zwierząt. Charakterystyka wybranych markerów genetycznych
pozwala śledzić wpływ pracy selekcyjnej na strukturę genetyczną danej
populacji, jak określone geny ulegają eliminacji równolegle z eliminacją
cech niekorzystnych z hodowlanego punktu widzenia.
274

Rozdział
12
Mapy genomowe
Genom jest pojęciem określającym zbiór informacji genetycznej zawartej
w gamecie. Składa się na nią przede wszystkim DNA jądra komórkowego
oraz DNA występujący w mitochondriach (mtDNA). Ponieważ długość
cząsteczki mtDNA jest niezmiernie mała w porównaniu z DNA haploidal-
nego zestawu chromosomów, dlatego pojęcie genomu odnoszone jest do
DNA jądrowego. Genom może być opisany za pomocą kilku parametrów.
Po pierwsze, jest to haploidalna liczba chromosomów charakterystyczna dla
gatunku. Drugim parametrem jest łączna długość DNA występującego
w haploidalnym zestawie chromosomów. Wielkość ta zawiera się u ssaków
w przedziale między 2 a 3 miliardy par zasad. Wreszcie genom może być
opisany za pomocą całkowitej liczby loci genów. Przyjmuje się, że w genomie
ssaków występuje około 35 tysięcy genów. Odległości między sprzężonymi
ze sobą genami można wyrazić za pomocą jednostki względnej — centy-
morgana (cM), która odzwierciedla częstość zachodzenia crossing over
między loci sprzężonych genów. Całkowita długość genomu u ssaków,
wyrażona w cM, mieści się w przedziale od 1600 cM (mysz) do 3200 cM
(człowiek). U zwierząt gospodarskich wielkość ta oscyluje wokół 2500 cM.
Rozmieszczenie loci genów w genomie jest nierównomierne. Miejscami
silnie nasyconymi genami są regiony występujące na końcach ramion
chromosomowych. Tam też znacznie częściej zachodzi crossing over. Miejsca
te można wybarwić metodą barwienia prążkowego typu T.
12.1. Organizacja DNA jądrowego
W obrębie genomu występują zarówno sekwencje kodujące, czyli geny, jak
i sekwencje niekodujące. Zdecydowanie przeważają sekwencje niekodujące,
które stanowią około 97% całego DNA (ryć. 12-1). Sekwencje powtarzalne
275
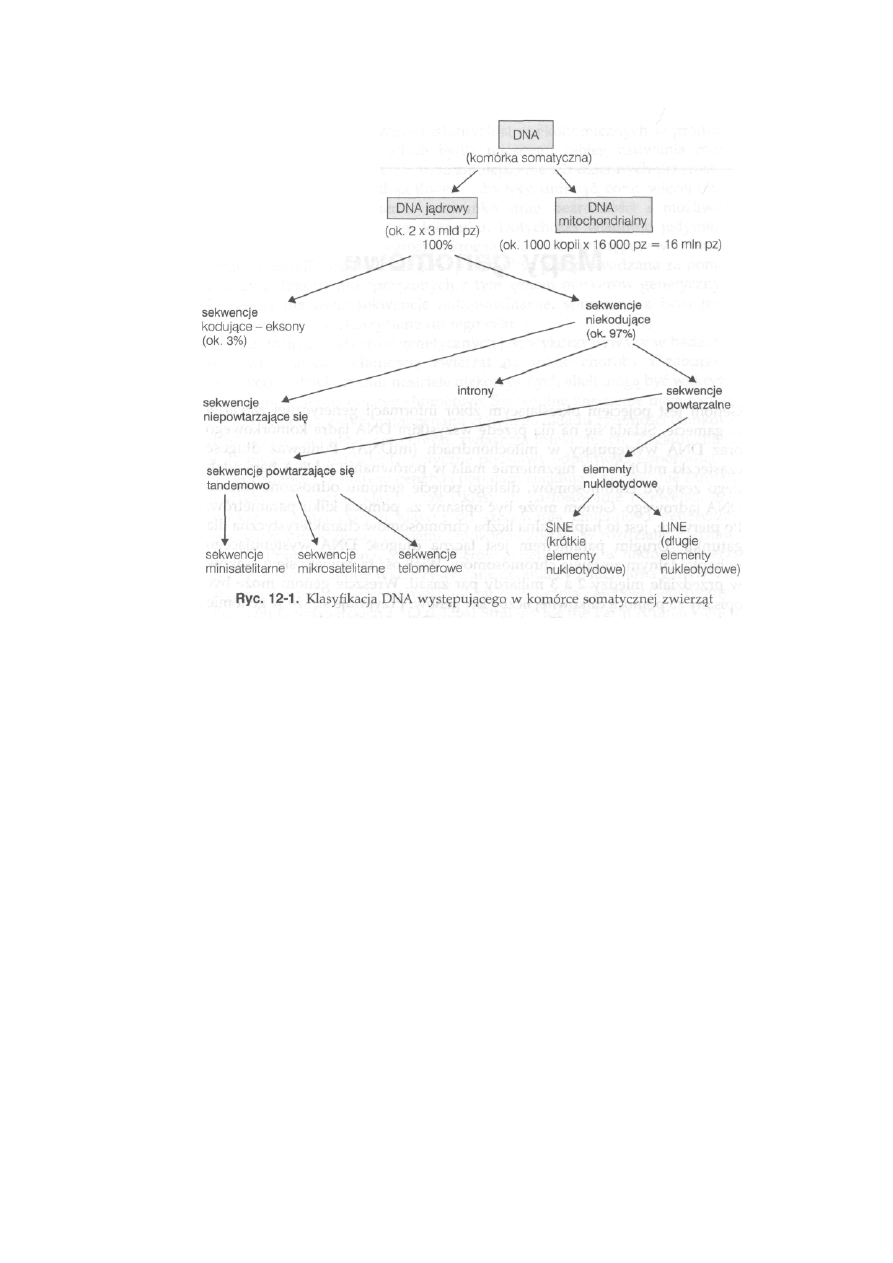
dzieli się na dwie podstawowe grupy: a) sekwencje powtarzające się
tandemowo i b) elementy nukleotydowe. Cechą charakterystyczną sekwencji
powtarzających się tandemowo jest to, że dana sekwencja nukleotydów (układ
podstawowy) występuje w wielu miejscach genomu, ale w każdym z tych
miejsc (loci) sekwencja ta powtarza się wielokrotnie w układzie tandemowym
(jeden za drugim). Zależnie od długości układu podstawowego sekwencje
powtarzalne dzielone są na mikrosatelitarne (do 10 nukleotydów) i minisateli-
tarne (powyżej 10 nukleotydów). W genomie występują duże skupienia
sekwencji niekodujących w obrębie heterochromatyny konstytutywnej, którą
uwidocznia się metodą barwienia prążkami C. Niezależnie od tego, sekwencje
powtarzalne są rozproszone również w obszarach euchromatynowych.
Sekwencje mikrosatelitarne okazały się bardzo bogatym źródłem wysoce
polimorficznych markerów genetycznych. Szacuje się, że w genomie ludzkim
sekwencje te pojawiają się przeciętnie co kilka tysięcy par zasad. Oznacza
to, że genom jest bardzo silnie nimi nasycony. Szczególnie cenne, z punktu
widzenia przydatności tych sekwencji do analizy genetycznej, jest to, że
liczba powtórzeń danego układu podstawowego wykazuje w obrębie
276

populacji silne zróżnicowanie. Inaczej mówiąc, dla danego locus funkcjonują
niejednokrotnie długie serie alleli wielokrotnych, które różnią się między
sobą liczbą powtórzeń podstawowego układu nukleotydów. Szczególnym
przykładem sekwencji mikrosatelitarnych ze względu na funkcję biologiczną,
jest sekwencja telomerowa (TTAGGG/AATCCC), która stanowi zakończenia
ramion chromosomowych. Funkcją biologiczną tych sekwencji jest ochrona
zakończeń chromosomów przed degradacją podczas kolejnych replikacji.
Skróceniu ulegają właśnie sekwencje telomerowe. Na chromosomach ludz-
kich każdy telomer zawiera od 250 do 1500 powtórzeń tej sekwencji.
W przypadku sekwencji minisatelitarnych stopień równomierności
rozproszenia w genomie jest mniejszy. Zazwyczaj są one zlokalizowane
w częściach dystalnych — tzn. w pobliżu końców ramion chromosomów.
Elementy nukleotydowe to takie sekwencje, które również są rozproszone
w wielu miejscach genomu, ale w danym miejscu występuje tylko jedna
kopia sekwencji. Podobnie jak poprzednio, sekwencje te dzieli się na dwie
grupy zależnie od ich długości: a) SINE (short interspersed nucleotide
element), których długość mieści się poniżej 500 pz oraz b) LINE (long
interspersed nucleotide element), których długość jest powyżej 500 pz i może
dochodzić do kilku tysięcy pz. Przykładem elementu nukleotydowego z gru-
py SINE jest rodzina sekwencji Alu, której nazwa pochodzi od enzymu
restrykcyjnego mającego miejsce cięcia cząsteczki DNA w obrębie tej sekwen-
cji. Sekwencje te bardzo często są zlokalizowane w okolicach centromerów.
Funkcja sekwencji powtarzalnych, z wyjątkiem sekwencji telomerowych,
jest do tej pory słabo rozpoznana. Przy okazji badań molekularnych
kompleksów synaptonemalnych okazało się, że sekwencje te, podobnie jak
elementy nukleotydowe, leżą w pobliżu białkowych struktur kompleksów.
Może to świadczyć o roli tych sekwencji w kotwiczeniu domen pędowych
DNA w strukturze białkowej kompleksu synaptonemalnego.
Organizacja chromatyny w jądrze interfazowym charakteryzuje się pew-
nymi szczególnymi cechami. Wiadomo, że utrzymywana jest w nim nukleo-
somowa organizacja nici chromatynowej. Poszczególne chromosomy zajmują
w jądrze wyodrębnione domeny, przy czym domeny chromosomów homolo-
gicznych zazwyczaj nie kontaktują się ze sobą. Z kolei części chromosomów
zbudowane z heterochromatyny konstytutywnej pozostają mocniej skonden-
sowane i niejednokrotnie wykazują tendencje do tworzenia chromocentrów,
w których asocjuje heterochromatyna z kilku chromosomów.
12.2. Organizacja DNA mitochondrialnego
Badania na zwierzętach i roślinach przyczyniły się do wykrycia obecności
informacji DNA poza jądrem komórkowym — w mitochondriach (u
roślin i zwierząt) i chloroplastach (u roślin).
277
Mitochondria należą do organelli komórkowych przekształcających ener-
gię. W nich odbywa się większość reakcji oddychania komórkowego. Niektóre
białka uczestniczące w oddychaniu nie mogą przechodzić przez błony
organelli, zatem muszą być syntetyzowane wewnątrz mitochondriów. Infor-
macja o składzie aminokwasowym tych białek jest zapisana w mitochondrial-
nym DNA. Wiadomo już, że mitochondrialny DNA (mtDNA) koduje białka
będące składnikami mitochondrialnego łańcucha oddechowego i systemu
fosforylacji oksydacyjnej (ang. oxidative phosphorylation — OXPHOS), poza
tym 2 rodzaje rRNA i 22 rodzaje tRNA. Całkowita liczba genów w ludzkim
mtDNA wynosi 37. Geny kodujące białka i rRNA są od siebie oddzielone
genami kodującymi tRNA. Białka będące podjednostkami enzymów łańcucha
oddechowego (np. ubichinon/koenzym Q, cytochrom c czy oksydoreduktaza)
należą do pięciu kompleksów oznaczonych cyframi rzymskimi I-V, przy czym
kompleks V jest to syntaza ATP. Większość białek wchodzących w skład tych
kompleksów jest kodowana przez DNA jądrowy, a tylko część przez mtDNA.
Porównanie liczby podjednostek białkowych kodowanych przez DNA mito-
chondrialny i jądrowy zawiera poniższe zestawienie.
Struktura mtDNA jest inna niż DNA jądrowego, mtDNA jest bowiem
kolisty. Jego długość wynosi około 16 tysięcy par zasad. Większość komórek
zawiera od tysiąca do 10 tyś. kopii mtDNA. Jedynie w oocytach II rzędu liczba
kopii jest wyraźnie większa i sięga około 100 tyś. Zwielokrotnienie liczby kopii
w oocycie jest częścią przygotowań tej komórki do podołania przez zygotę
wymaganiom energetycznym związanym z wczesnym rozwojem zarodka lub
zapewnieniem wystarczającej liczby kopii mtDNA dla pierwszych blastome-
rów zarodka, w których być może replikacja mtDNA nie zachodzi.
278

DNA występujący w mitochondriach nie jest powiązany z białkami,
w odróżnieniu od DNA jądrowego. Prawie cały mtDNA zawiera sekwen-
cje kodujące, a sekwencje powtarzające się tandemowo są reprezentowane
bardzo nielicznie. Kolejną cechą charakterystyczną mtDNA jest to, że geny
mitochondrialne nie zawierają intronów, co więcej, transkrypcji podlegają
obie nici kolistego DNA, a niektóre geny nawet nakładają się na siebie.
Łańcuchy komplementarne, wchodzące w skład cząsteczki mtDNA, różnią
się udziałem nukleotydów i dlatego wyodrębnia się łańcuch ciężki (H),
który ma większy udział nukleotydów guanylowych — G i tymidylo-
wych - T niż łańcuch lekki (L). Na łańcuchu ciężkim występuje 12
genów kodujących białka oraz 14 genów kodujących cząsteczki tRNA
i 2 geny kodujące rRNA, a na łańcuchu lekkim umiejscowiony jest tylko
jeden gen kodujący białko i 8 genów kodujących cząsteczki tRNA.
W strukturze łańcucha ciężkiego wyróżnia się również pętlę D, w której
obrębie występuje potrójna struktura DNA, uformowana przez krótki
fragment nowego łańcucha H, pozostający w asocjacji z cząsteczką
macierzystą. W mtDNA występuje jedno miejsce inicjujące transkrypcję,
która przebiega równocześnie, ale w przeciwnych kierunkach, na obu
łańcuchach. Rola i zachowanie się mtDNA w dziedziczeniu pozajądrowym
opisane są w rozdziale: Podstawowe mechanizmy dziedziczenia cech.
12.3. Markerowa mapa genomu
Wiedza o lokalizacji genów oraz sekwencji niekodujących w DNA jądrowym
jest bardzo słabo rozwinięta w porównaniu z wiedzą o mapie mtDNA.
Dlatego w dalszej części tego rozdziału będą omówione zagadnienia
związane z poznawaniem (mapowaniem) DNA jądrowego, który stanowi
dominującą część genomu.
Markerowa mapa genomu jest zbiorem informacji o:
a) położeniu loci sekwencji kodujących, tzn. genów (markery klasy
I),
i niekodujących, czyli tandemowo powtarzających się sekwencji anonimo
wych (markery klasy II) w chromosomach,
b) stopniu polimorfizmu w obrębie znanych loci (wykaz alleli),
c) odległościach genetycznych, wyrażonych w cM, pomiędzy sprzężo
nymi loci oraz ich uszeregowaniu.
Mapa, która wskazuje położenia loci w chromosomach, nazywa się
cytogenetyczną lub fizyczną mapą genomu. Z kolei mapa opisująca sprzę-
żenia, uszeregowanie i odległości genetyczne pomiędzy takimi loci określana
jest terminem genetycznej lub sprzężeniowej mapy genomu. Ponieważ
mapy genomowe budowane są przede wszystkim na bazie polimorficznych
markerów genetycznych, dlatego niejednokrotnie mówi się o markerowej
mapie genomu.
279
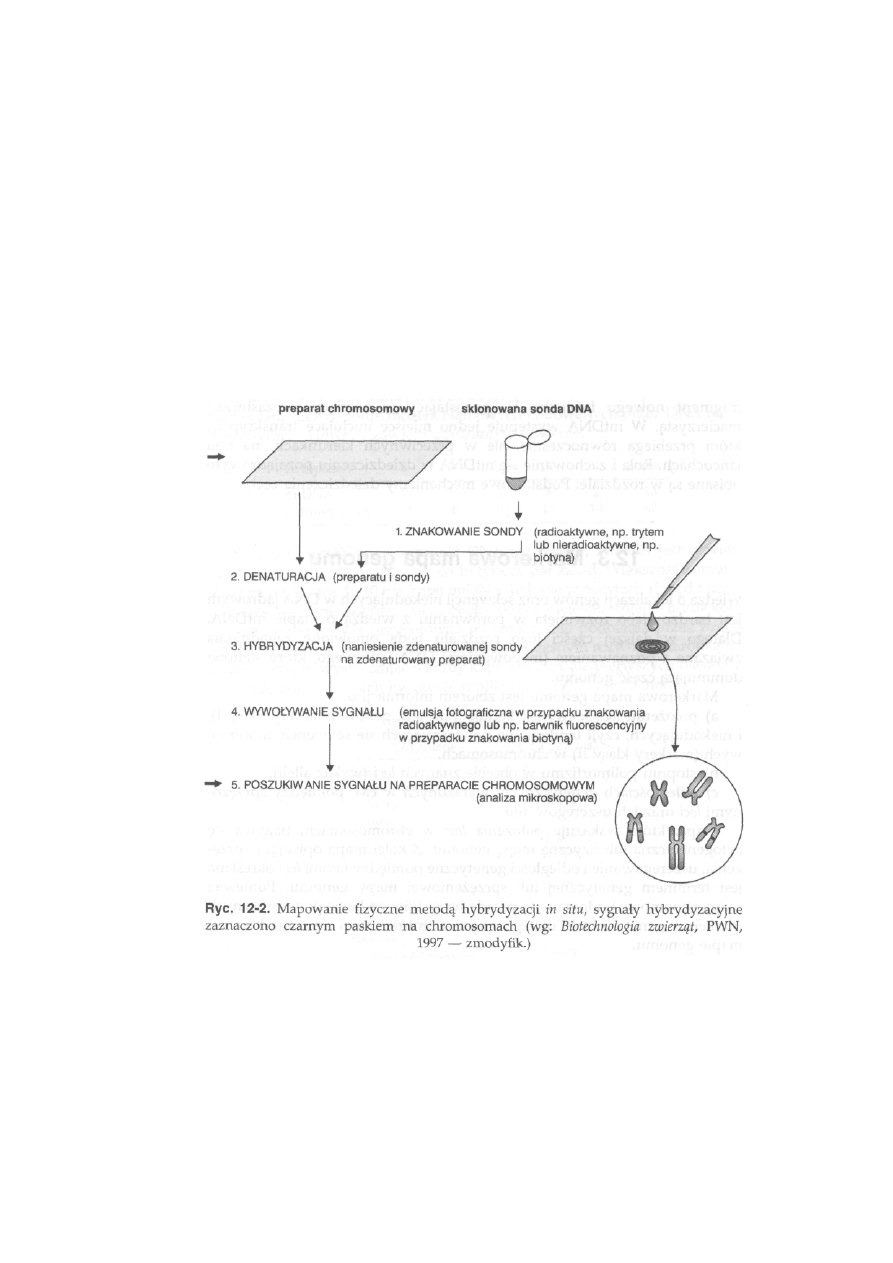
12.3.1. Fizyczna mapa genomu
Podstawową, ale nie jedyną, metodą mapowania fizycznego jest hybrydyzacja
in situ pomiędzy zdenaturowaną i wyznakowaną sondą DNA a zdenaturowa-
nymi chromosomami metafazowymi utrwalonymi na preparacie mikroskopo-
wym. Sonda jest to sklonowana molekularnie sekwencja nukleotydowa,
reprezentująca gen (lub jego część) bądź fragment DNA obejmujący niekodują-
cą sekwencję, na przykład mikrosatelitę — marker klasy II. Często sondę
stanowi tzw. cDNA (komplementarny DNA) genu. Jest to taka sekwencja
nukleotydowa, która powstała na drodze syntezy DNA z udziałem enzymu
odwrotnej transkryptazy na bazie wyizolowanego mRNA. Tak utworzony
cDNA jest wbudowywany do wektorów i klonowany. Różnica między DNA
genu a jego cDNA polega na tym, że w cDNA nie występują introny, ponieważ
w mRNA, na bazie którego syntetyzowano cDNA, sekwencje intronowe
zostały już wcześniej wycięte podczas obróbki potranskrypcyjnej (ang.
280

Ryć. 12-3. Lokalizacja markera mikrosatelitarnego ZuBeCa4 metodą fluorescencyjnej
hybrydyzacji in situ (ang. FISH) na chromosomie 3 psa; (a) chromosomy barwione
metodą prążków Q; (b) ta sama metafaza po hybrydyzacji; jasne punkty wskazują
miejsce, gdzie sonda hybrydyzowała z DNA chromosomu
splicing). Schemat mapowania genów metodą hybrydyzacji in situ jest
przedstawiony na rycinie 12-2. Trzeba podkreślić, że do identyfikacji chromo-
somu, zgodnie z przyjętym wzorcem kariotypu, wykorzystuje się techniki
prążkowego barwienia (G, Q lub R), które stosuje się albo przed hybrydyzacją,
albo po hybrydyzacji. Miejsce, w którym sonda połączy się z DNA chromo-
somu, jest rozpoznawane przez obserwację mikroskopową, dzięki wyzna-
kowaniu sondy pierwiastkiem promieniotwórczym lub analogiem jednego
z nukleotydów, związanego z barwnikiem fluorescencyjnym (ryć. 12-3).
Hybrydyzacją in situ ma wiele odmian, które w ostatnim czasie są szeroko
wykorzystywane do opisu genomu zwierząt. Warto wspomnieć o dwóch:
tzw. malowaniu chromosomów (ang. chromosome painting) i mapowaniu
na chromatynie jąder interfazowych.
Malowanie chromosomów polega na zastosowaniu sondy, która zawiera
unikatowe sekwencje DNA wybranego chromosomu. Pierwszym etapem
prowadzącym do uzyskania takiej sondy jest pozyskanie jednorodnej frakcji
wybranego chromosomu na drodze ich sortowania w cytometrze prze-
pływowym lub mikrodysekcji przeprowadzonej za pomocą mikromanipu-
latora na preparacie mikroskopowym. Kolejnym etapem jest amplifikacja
(metoda PCR) lub klonowanie molekularne fragmentów DNA tego chromo-
somu. W trakcie amplifikacji blokowane są sekwencje powtarzalne i dzięki
temu następuje zwielokrotnienie unikatowych sekwencji nukleotydowych
danego chromosomu. Przeprowadzenie hybrydyzacji in situ z użyciem
takiej sondy daje efekt w postaci sygnału (np. świecenie barwnika flurescen-
cyjnego związanego z sondą) na obszarze całego chromosomu (ryć. 12-4
wkładka kolorowa). Stosowanie techniki malowania chromosomów umoż-
liwia detekcję mutacji genomowych (głównie aneuploidii) i chromosomo-
wych. Podejście takie jest alternatywne w stosunku do klasycznej procedury
opartej na identyfikacji chromosomów za pomocą barwień prążkowych.
Ponadto, technika ta wykorzystywana jest do identyfikacji homologii między
chromosomami różnych gatunków. Analizę taką określa się terminem
porównawczego malowania chromosomów (ang. comparative chromosome
281
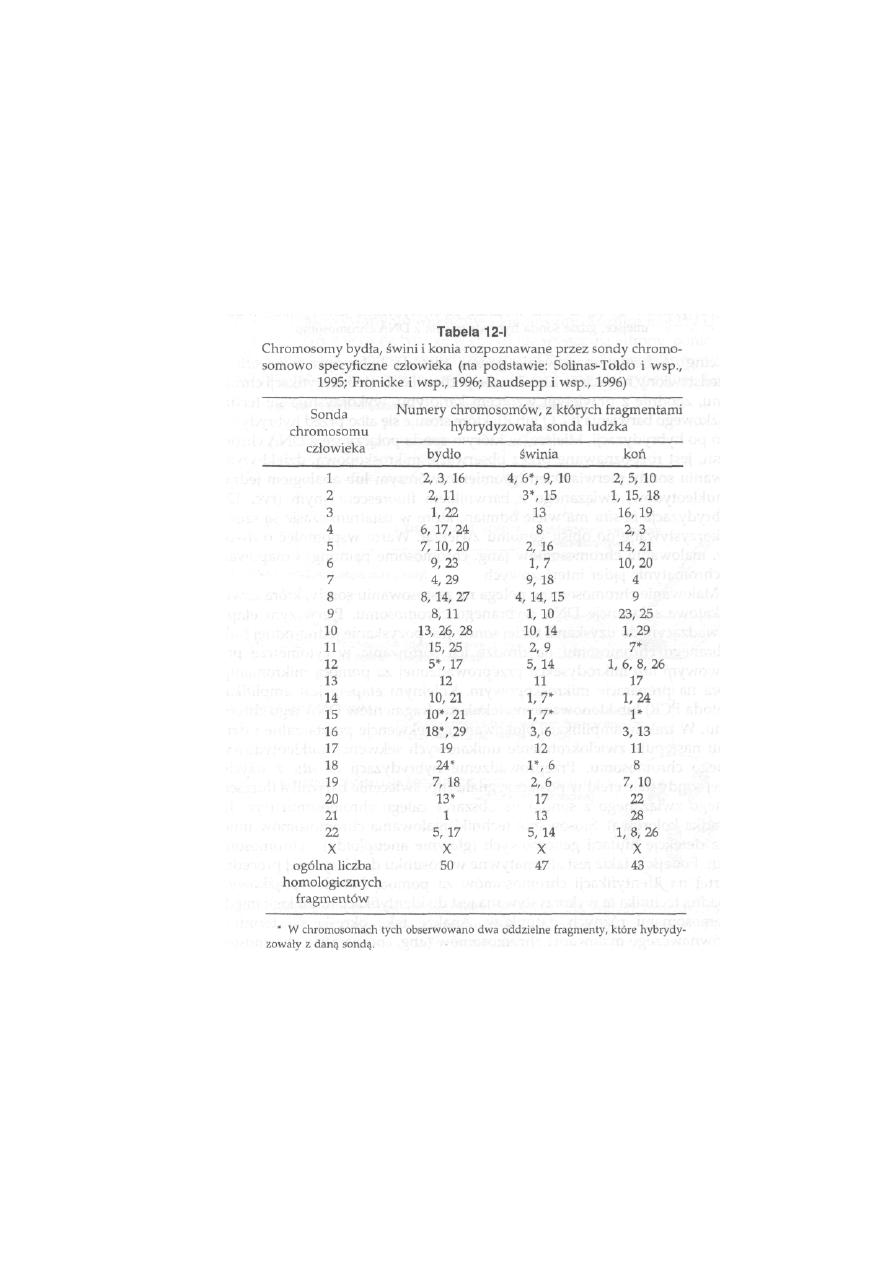
painting) i polega ona na tym, że sondę chromosomowo specyficzną,
utworzoną dla jednego gatunku (np. człowieka), stosuje się do hybrydyzacji
na preparatach chromosomowych pochodzących od innego gatunku (np.
bydła). W ten sposób można wskazać, które chromosomy bydła zawierają
fragmenty homologiczne względem konkretnego chromosomu ludzkiego.
Przeprowadzone eksperymenty ujawniły, że w genomach bydła, konia czy
świni można zidentyfikować ponad czterdzieści fragmentów chromosomo-
wych, które są „malowane" sondami reprezentującymi wszystkie chromo-
somy człowieka (tab. 12-1). Dowodzi to, że zmiany na poziomie chromo-
somowym, jakie zaszły podczas ewolucji, są bardzo złożone. Należy jednak
282

zaznaczyć, że metoda malowania chromosomów ma ograniczoną dokładność,
przyjmuje się bowiem, że sonda chromosomowo specyficzna da widoczny
sygnał, jeżeli obejmuje segment długości co najmniej kilku milionów par zasad.
Dużą rolę w postępie mapowania genomów odegrała metoda hyb-
rydyzacji komórek somatycznych. Polega ona na tym, że w warunkach
in vitro doprowadza się do połączenia komórek pochodzących od dwóch
różnych gatunków, np. mysich komórek nowotworowych, utrzymywanych
w hodowli, z fibroblastami pochodzącymi z mięśni czy skóry bydła. Komórki
takie po połączeniu powinny mieć liczbę chromosomów odpowiadającą
sumie liczb diploidalnych u obu gatunków, czyli 40 (mysz) plus 60 (bydło)
daje 100 chromosomów. Okazuje się jednak, że jeżeli takie komórki są
hodowane w warunkach in vitro, to stopniowo „gubione" są chromosomy,
które nie pochodzą z komórek nowotworowych. W tym przypadku są to
chromosomy bydła. W rezultacie można wyprowadzić klony (komórki
będące potomstwem jednej wyjściowej komórki), które będą się między
sobą różniły liczbą zachowanych chromosomów bydła. Liczba zachowanych
chromosomów zazwyczaj jest rzędu kilku, ale czasami może się zdarzyć tak,
że tylko pojedyncze chromosomy bydła zostaną zachowane w danym
klonie komórkowym. Tak wyprowadzone klony poddawane są analizie
elektroforetycznej, której celem jest identyfikacja białek lub markerów DNA
typowych dla komórek gatunku, którego genom jest badany — w tym
przykładzie są to komórki bydlęce. Objęcie badaniami dużej liczby klonów
stwarza możliwość wykrycia w komórkach hybrydowych loci, które zawsze
pojawiają się razem. Sytuacja taka świadczy o tym, że grupa tych loci jest
położona w tym samym chromosomie. Loci takie nazywa się syntenicznymi.
0 ich umiejscowieniu wiadomo jedynie tyle, że są położone w jednym
chromosomie. Nie można natomiast wskazać, który jest to chromosom
1 w jakim regionie mieszczą się badane loci, nieznana pozostaje również
wzajemna odległość między nimi. Lokalizacja chromosomowa grup syn-
tenicznych wymaga przeprowadzenia badań cytogenetycznych, których
celem jest identyfikacja chromosomu (-ów) gatunku mapowanego w danym
klonie hybrydowym.
Do techniki uzyskiwania hybrydowych linii komórek somatycznych
została ostatnio wprowadzona istotna modyfikacja. Polega ona na pod-
dawaniu komórek gatunku mapowanego, przed hybrydyzowaniem z komór-
kami nośnikowymi (np. komórki nowotworowe myszy), naświetlaniu
promieniami jonizującymi. Efektem naświetlania jest powstanie licznych
pęknięć cząsteczek DNA, a zatem fragmentacja chromosomów. Komórki
z pofragmentowanymi chromosomami poddawane są hybrydyzacji, podczas
której dochodzi do fuzji fragmentów chromosomowych z chromosomami
komórek nośnikowych. W ten sposób w komórkach hybrydowych za-
chowane są fragmenty chromosomów gatunku mapowanego. Na przykład,
naświetlanie fibroblastów psa promieniowaniem o dawce 5000 radów dało
fragmenty o średniej wielkości około 16,6 milionów par zasad, a w każdej
283

z uzyskanych linii hybrydowych zachowane fragmenty chromosomowe
stanowiły około 20% genomu psa. Tak zmodyfikowaną technikę hybrydyzacji
komórek somatycznych określa się terminem mapowania radiacyjnego.
Zaletą tej techniki mapowania jest możliwość identyfikacji loci markerowych
położonych bardzo blisko siebie, na tyle blisko, że klasyczne mapowanie
sprzężeniowe nie pozwala na określenie odległości genetycznej, ze względu
na zbyt małe prawdopodobieństwo zajścia crossing over. Dotyczy to
szczególnie loci o niskim stopniu polimorfizmu, w takim przypadku bowiem
analiza segregacji alleli (mapowanie genetyczne) jest nieefektywna. Dawki
promieniowania jonizującego, wykorzystywane w mapowaniu radiacyjnym,
wynoszą zazwyczaj 5 tyś., 7 tyś. lub 12 tyś. radów. Wielkość uzyskiwanych
fragmentów chromosomowych zależy od dawki promieniowania jonizują-
cego — czym większa dawka, tym mniejsze fragmenty. Uzyskiwanie
małych fragmentów chromosomowych zwiększa rozdzielczość mapowania,
przez którą rozumie się najmniejszą odległość między loci sąsiednich
markerów możliwą do wykrycia.
12.3.2. Genetyczna mapa genomu
Analiza segregacji alleli w loci markerowych, u potomstwa osobników
o znanych genotypach, służy tworzeniu genetycznej mapy genomu. Kolej-
ność postępowania jest następująca. Najpierw poszukuje się sprzężeń
pomiędzy loci. Dalej, na podstawie liczby zidentyfikowanych rekombinantów
szacuje się odległości genetyczne (cM) między sprzężonymi loci, które
odzwierciedlają częstość, z jaką zachodziło crossing over między nimi
podczas gametogenezy u rodziców. Poznanie wzajemnych odległości między
sprzężonymi loci stwarza warunki do poznania ich uszeregowania, w czym
pomocne może być przeanalizowanie rodzin z równoczesnym uwzględ-
nieniem trzech loci (tzw. krzyżówka trójpunktowa). Jednostką odległości
genetycznej jest l cM. Odległość taka oznacza, że crossing over między
dwoma sprzężonymi loci zachodzi jeden raz na sto podziałów mejotycznych.
Do wykrywania sprzężeń jest stosowana powszechnie funkcja zwana
logarytmem ilorazu wiarogodności (ang. loci).
L - - funkcja wiarogodności, obliczana jako prawdopodobieństwo wy-
stąpienia określonych genotypów, przy założonym współczynniku
rekombinacji (r), względem hipotezy alternatywnej, przyjmującej
niezależne dziedziczenie (r = 0,5)
r - - współczynnik rekombinacji, zawierający się w przedziale (0; 0,5)
Badanie sprzężenia dokonuje się poprzez analizę segregacji genów
w dwóch loci. W tym celu korzysta się z wiedzy o genotypach osobników
284
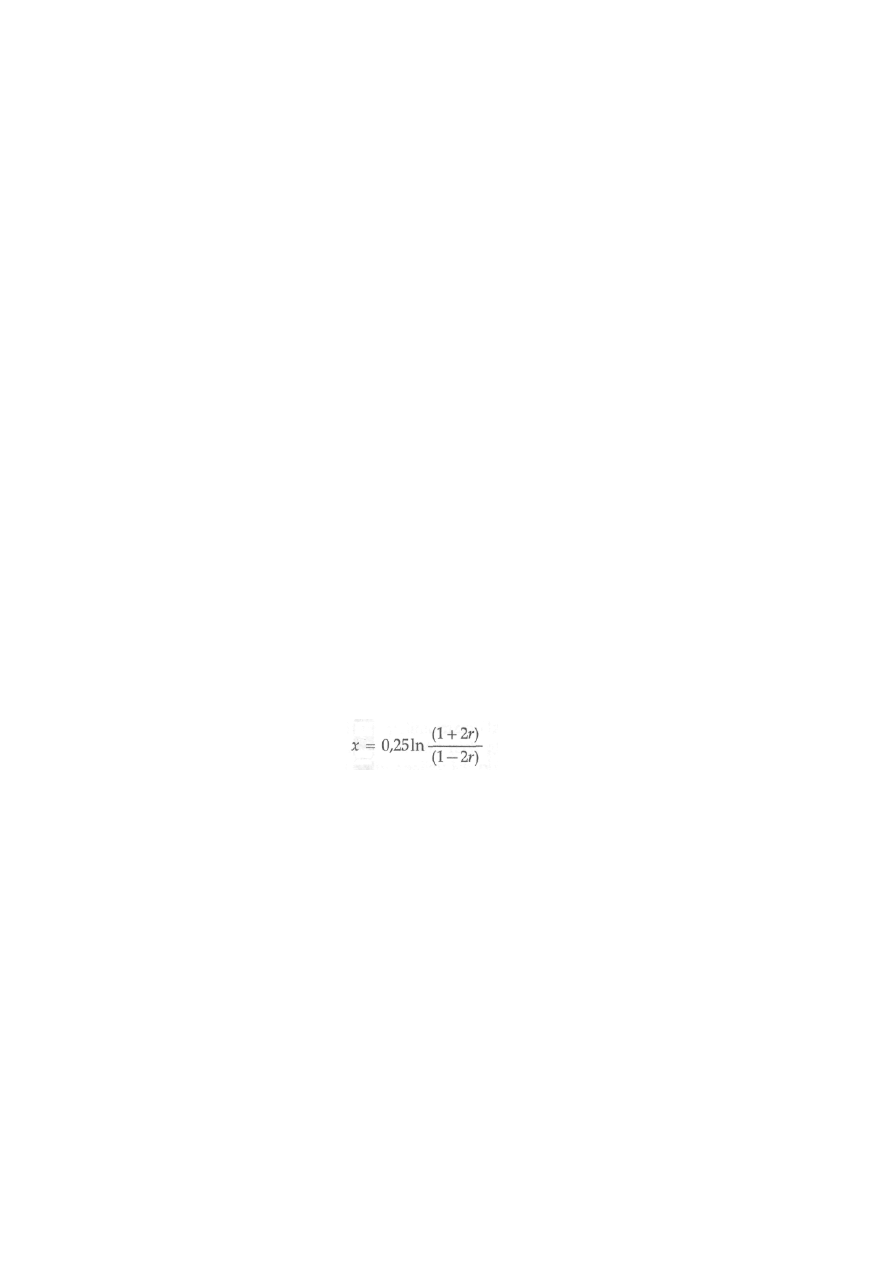
należących do rodzin co najmniej dwupokoleniowych. Jednak nie wszystkie
kojarzenia są informatywne. Jeżeli oboje rodzice są homozygotami w obu
loci, to oczywiście identyfikacja segregantów jest niemożliwa. Z kolei, jeżeli
oboje rodzice są heterozygotami o identycznych allelach, to niemożliwe jest
ustalenie fazy (cis lub trans), w jakiej allele analizowanych loci występują
u rodziców. Tego typu kojarzenie też nie jest informatywne. Wynika z tego,
że informatywne są kojarzenia typu podwójna heterozygota z podwójną
homozygotą lub kojarzenie między heterozygotami, u których w badanych
loci występują różne ałlele. Obliczenie wartości loci przeprowadza się
dla z góry założonych wartości współczynnika rekombinacji (r), mniejszych
niż 0,5. Jeżeli wartość loci, dla niektórych wielkości współczynnika
rekombinacji, będzie większa niż 3,0, to oznacza, że analizowane loci są ze
sobą sprzężone. Największą wartość loci przewyższającą 3,0 uznaje się za
oszacowanie współczynnika r, czyli częstości, z jaką dochodzi do
rekombinacji między analizowanymi loci. Wartość loci mniejsza niż —2
oznacza, że dla testowanej wartości współczynnika r nie stwierdza się
sprzężenia między analizowanymi loci. Natomiast wartości z przedziału ( — 2;
3) nie pozwalają na wyciągnięcie jednoznacznego wniosku dotyczącego
sprzężenia, czyli analizowany materiał rodzinowy był zbyt mały lub loci były
ze sobą na tyle słabo sprzężone, że zachowują się tak jakby się dziedziczyły
niezależnie. Badanie sprzężenia polega na wyliczaniu wartości loci dla
kolejnych wielkości współczynnika r (np. 0,1; 0,2; 0,3; 0,4). Jeśli dla
wszystkich testowanych wielkości współczynnika r wartość loci jest mniejsza
niż —2,0, to można przyjąć, że badane geny nie są ze sobą sprzężone.
Pojawienie się wartości loci powyżej 3,0 dowodzi, że geny są ze sobą
sprzężone i równocześnie poznajemy współczynnik r. Jeśli wartość loci
mieści się między —2,0 i 3,0, to należy zwiększyć liczbę badanych rodzin, tak
aby dojść do wiążących wniosków. Po ustaleniu, że loci są ze sobą
sprzężone, można przystąpić do oszacowania odległości genetycznej między
nimi. W tym celu wykorzystywana jest tzw. funkcja Kosambiego (x), która
wyraża odległości genetyczne między sprzężonymi loci w centymorganach
(cM).
r - - oszacowana częstość rekombinacji między analizowanymi loci
Funkcja ta uwzględnia dwie okoliczności wpływające na częstość
identyfikowanych zjawisk crossing over. Po pierwsze, bierze pod uwagę to,
że identyfikacja rekombinantów w potomstwie pozwala na wykrycie tylko
nieparzystych przypadków crossing over, parzysta ich liczba bowiem nie
zmienia układu alleli w chromatydzie i tym samym nie pojawiają się
w potomstwie osobniki o zrekombinowanym układzie alleli. Po drugie,
brane jest pod uwagę zjawisko interferencji, polegające na tym, że zajście
crossing over w danym miejscu wpływa ograniczająco na możliwość
285
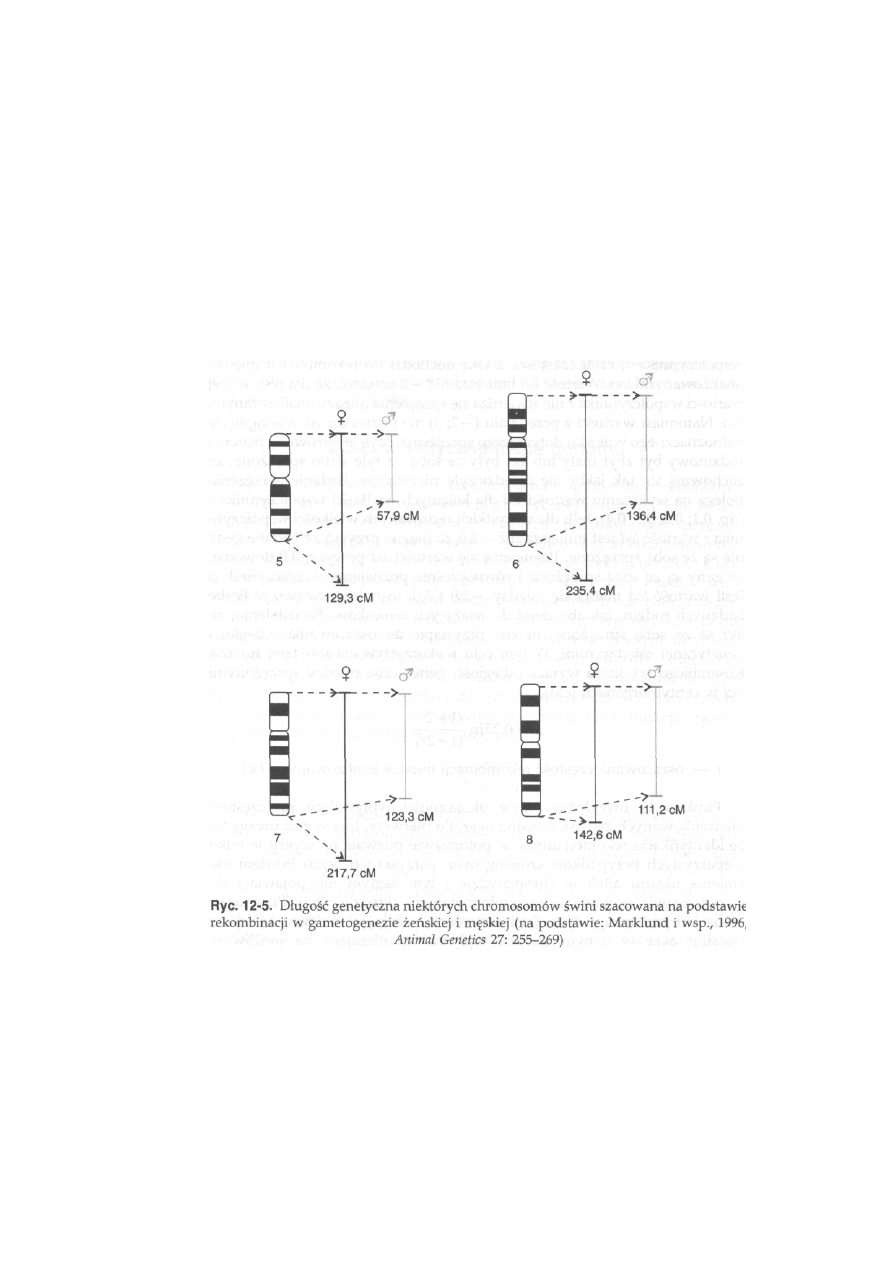
powtórnego wystąpienia crossing over w pobliżu. Należy podkreślić, że do
obliczania funkcji loci oraz funkcji Kosambiego opracowano programy
komputerowe.
Wyniki badań nad częstością crossing over ujawniły ciekawą zależność
polegającą na tym, że długość genetyczna chromosomu, szacowana na
podstawie rekombinacji zachodzącej w gametogenezie żeńskiej, jest za-
zwyczaj większa, aniżeli w przypadku gdy podstawą takiego oszacowania
były rekombinacje zachodzące podczas spermatogenezy (ryć. 12-5). Można
zatem sądzić, że w oogenezie crossing over zachodzi częściej aniżeli
w spermatogenezie. Brak jest jednoznacznej odpowiedzi na pytanie, dlaczego
tak jest. Być może prawidłowość ta wynika ze specyfiki spermatogenezy,
podczas której jest wytwarzanych nieporównanie więcej gamet niż podczas
oogenezy i dlatego żeńska rekombinacja wewnątrzchromatydowa (crossing
286

over) zachodzi z większym nasileniem, aby dorównać ogromnej zmienności
genetycznej wśród plemników. Trzeba też pamiętać, że chiazmy utrzymują
strukturę biwalentu w oocycie przez znacznie dłuższy czas niż w sper-
ma tocy cię.
Należy również podkreślić, że odległości genetycznej (cM) między
sprzężonymi loci nie można w bezpośredni sposób przeliczać na odległości
fizyczne, mierzone np. liczbą par zasad, jaka je dzieli. Przyjmuje się
z dużym przybliżeniem, że odległość 1 cM odpowiada długości cząsteczki
DNA, liczącej około jednego miliona par zasad. Brak prostej zależności
między odległością względną i bezwględną wynika z tego, że losowość
zachodzenia crossing over jest ograniczona tym, iż w chromosomie są
obszary, gdzie zachodzi ono znamiennie częściej (prążki T), oraz takie, gdzie
zdarza się bardzo rzadko (heterochromatyna konstytutywna).
12.3.3. Strategia mapowania genomu
Markerowa mapa genomu powinna charakteryzować się kilkoma cechami,
które wyznaczają jej wartość poznawczą i aplikacyjną. Po pierwsze, markery
genetyczne powinny być możliwie wysoce polimorficzne, tzn. w puli
genowej populacji powinna występować możliwie długa seria alleli wielo-
krotnych. Wykrycie w drugiej połowie lat 80. wysoce polimorficznych
sekwencji anonimowych typu mini- i mikrosatelitarnego spowodowało, że
markery zostały podzielone na dwie klasy. Do klasy I zostały zaliczone
klasyczne markery genetyczne (sekwencje kodujące, czyli geny). Polimorfizm
tych markerów identyfikowany jest na poziomie produktu, za pomocą
metod serologicznych — układy grupowe krwi, antygeny głównego układu
zgodności tkankowej itd. lub elektroforezy — polimorficzne białka surowicy
krwi, mleka itd. Możliwe jest również wykrywanie polimorfizmu tych
markerów poprzez badanie polimorfizmu DNA tych genów metodami
opartymi na elektroforezie (RFLP i SSCP). W grupie markerów klasy II
znalazły się wszystkie rodzaje polimorfizmu związane z niekodującymi
sekwencjami nukleotydowymi, a wśród nich główną rolę odgrywają sek-
wencje mikrosatelitarne. Metody analizy ich polimorfizmu opisano w roz-
dziale: Markery genetyczne.
Ważną cechą mapy genomu jest równomierność rozproszenia loci
markerowych. Początkowo zakładano, że stopień równomierności powinien
być taki, aby maksymalna odległość genetyczna między dwoma dowolnymi
sąsiednimi loci nie przekraczała 20 cM. Spełnienie tego warunku stwarza
sytuację, w której dowolny locus genu znajduje się w odległości nie większej
niż 10 cM od jakiegoś markera. Odległość taka pozwala zidentyfikować
położenie dowolnego genu, gdy w badaniach rodzinowych analizowane loci
markerowe byłyby rozproszone równomiernie w całym genomie, a gen
będący obiektem zainteresowania byłby reprezentowany w analizowanej
287

rodzinie przez więcej niż jeden allel. Obecnie stopień nasycenia map
genomowych podstawowych gatunków zwierząt gospodarskich jest znacznie
większy i w przypadku bydła oraz świń kształtuje się poniżej poziomu 3 cM.
Kolejną istotną cechą mapy genomu jest informatywność jej mapy
fizycznej. Zakłada się, że na każdym ramieniu chromosomu powinny być
zlokalizowane co najmniej dwa markery, jeden w pobliżu centromeru
(marker proksymalny), a drugi w końcowej części ramienia (marker
dystalny). Spełnienie tego kryterium dowodzi, że na obszarze całego genomu
zostały zlokalizowane markery genetyczne. Ostatecznym celem programów
budowania map genomowych jest stworzenie zintegrowanej mapy, tzn.
takiej, w której wszystkie grupy genów sprzężonych oraz syntenicznych mają
znaną lokalizację chromosomową. Taka mapa może być przedmiotem
porównania z najbardziej rozwiniętymi mapami, do których zalicza się mapę
genomu człowieka i myszy. Porównanie to odnosi się przede wszystkim do
lokalizacji tzw. loci zakotwiczonych (ang. anchor loci). Należą one do
markerów klasy I, o których wiadomo, że są równomiernie rozproszone
w genomie człowieka, myszy, kota i bydła. Z przeprowadzonych badań
porównawczych wynika, że w genomach tych gatunków odległości między
dwoma dowolnymi loci zakotwiczonymi mieszczą się w granicach od 5 do 10
cM. Przyjmuje się, że ich lokalizacja odzwierciedla obszary konserwatywne
pod względem ewolucyjnym. Konserwatyzm genetyczny jest równie często
wykorzystywany w mapowaniu fizycznym markerów klasy I. Polega to na
tym, że sondy molekularne uzyskane na bazie genomu jednego gatunku są
przydatne do mapowania tegoż genu w genomie innego gatunku.
Spełnienie podstawowych warunków dotyczących nasycenia mapy
fizycznej i genetycznej genomu oznacza, że w przypadku genomu gatunków
ssaków, których wielkość wynosi około 2500 cM, minimalna liczba równo-
miernie rozproszonych (na poziomie 20 cM) polimorficznych markerów
genetycznych wynosi około 120. Oczywiście jest to rozważanie czysto
teoretyczne, ponieważ przystępując do mapowania kolejnych markerów
nie można przewidzieć, w którym miejscu mogą one mieć swoje loci. Może
się zdarzyć tak, że wiele takich markerów zostanie zlokalizowanych
w niewielu tylko chromosomach, lub wręcz w niektórych ich regionach.
Wynika z tego wniosek, że osiągnięcie stanu równomiernego nasycenia
mapy markerami genetycznymi wymaga zmapowania o wiele większej
liczby loci, niżby to wynikało z teoretycznego wyliczenia.
Wysiłkom skierowanym na tworzenie mapy markerowej towarzyszą
prace, których celem jest identyfikacja genów kontrolujących cechy użytkowe
zwierząt lub istotnie na nie wpływających. Osiągnięcie tego celu wymaga
skrupulatnych obserwacji zmienności fenotypowej interesującej cechy w tzw.
rodzinach referencyjnych. Są to zazwyczaj trzypokoleniowe rodziny, w któ-
rych rodzice wywodzą się z odległych genetycznie ras lub nawet blisko
spokrewnionych gatunków (np. świnia domowa i świnia dzika lub bydło
domowe i bydło zebu). Duży dystans genetyczny (patrz rozdział: Podstawy
288

genetyki populacji) wskazuje, że pule genowe tych ras są inne zarówno ze
względu na występowanie w populacjach różnych alleli dla danego locus,
jak i różnic we frekwencji tych samych alleli. Zróżnicowanie to dotyczy tak
loci markerowych, jak i loci genów wpływających na kształtowanie się cech
będących obiektem zinteresowania hodowcy. Skrzyżowanie tak dobranych
rodziców daje wysoce heterozygotyczne pokolenie F
l
, które jest kojarzone
między sobą w celu uzyskania pokolenia F
2
. W pokoleniu F
2
analizuje się
odległości genetyczne między markerami oraz asocjacje między markerami
i cechami użytkowymi, będącymi obiektem zainteresowania hodowców.
Zbudowanie markerowej mapy genomowej, która mogłaby być użytecz-
nym narzędziem służącym do identyfikacji genów kontrolujących ważne
cechy użytkowe, jest zadaniem wymagającym współpracy specjalistów
z wielu dziedzin, takich jak: genetyka molekularna, immunogenetyka,
cytogenetyka czy biometria. Przekracza to zatem możliwości pojedynczych
zespołów badawczych. Na początku lat 90. zostały utworzone między-
narodowe programy badawcze. Jako pierwszy powstał program Mapowania
Genomu Świni - - PiGMaP. W przedsięwzięcie to zostało włączonych
kilkanaście laboratoriów, wywodzących się z państw Wspólnoty Europejskiej.
Rok później, na podobnych zasadach, powołano Program Mapowania
Genomu Bydła — BovMap. W tym programie zarejestrowano udział ponad
dwudziestu laboratoriów. Oprócz tych dwóch wiodących programów
rozwijały się inne międzynarodowe przedsięwzięcia, zmierzające do stwo-
rzenie mapy genomu kury, konia, owcy, kozy czy psa (DogMap). W nie-
których programach biorą udział również polskie laboratoria (mapa genomu
konia i psa, a także świni). Ponadto, powstało wiele przedsięwzięć realizo-
wanych przez laboratoria jednego kraju. Na przykład, w Polsce prowadzono
badania w ramach Polskiego Projektu Mapowania Genomu Świni, który był
realizowany w ramach współpracy między trzema zespołami badawczymi.
Głównym celem tego projektu było poszukiwanie genów głównych, od-
działujących istotnie na kształtowanie cech związanych z tuczem i użyt-
kowością rzeźną.
12.3.4. Aktualny stan map genomowych zwierząt
gospodarskich
Dynamiczny rozwój badań nad mapami genomowymi utrudnia prezentację
aktualnego stanu wiedzy z tego zakresu. Nieomal każdy dzień przynosi
nowe informacje wzbogacające mapy różnych gatunków. Dane pochodzące
z drugiej połowy 1996 roku wykazały, że w genomie bydła opisano już
ponad 1600 loci, a w genomie świni — ponad 1300 loci. Rok później liczba
ta wzrosła do 3300 u bydła i 1600 u świni. Wśród loci markerowych
zdecydowanie przeważają markery klasy II, a bardzo ważne jest to, że są
one równomiernie rozproszone w genomach obu gatunków. Oczywiście są
289
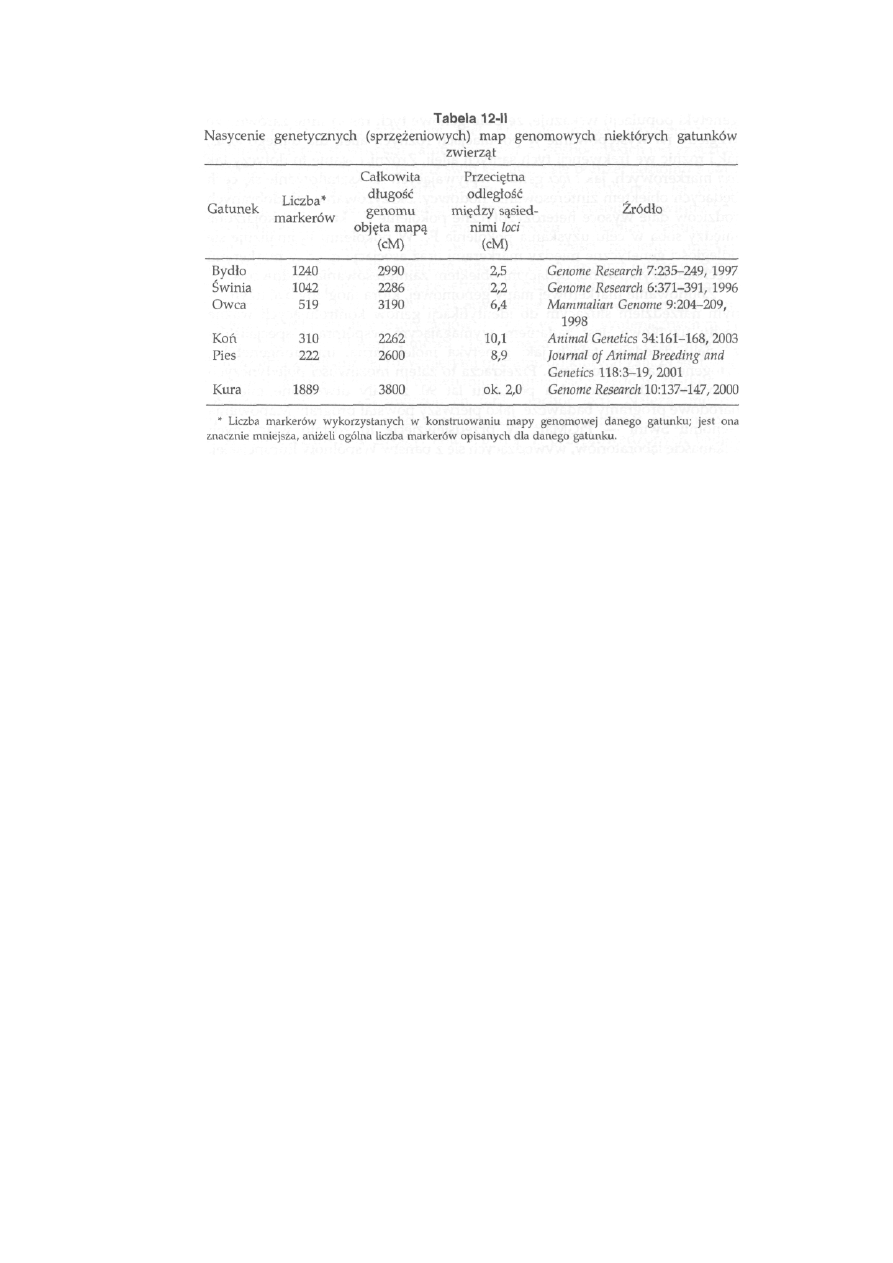
regiony silniej nasycone markerami, gdzie odległość między sąsiednimi loci
wynosi poniżej l cM, ale są również i takie, gdzie odległość ta sięga 20 cM.
Można jednak stwierdzić, że zakładany na początku lat 90. podstawowy cel,
polegający na zbudowaniu mapy, w której odległość między dowolnymi
sąsiednimi loci nie będzie większa niż 20 cM, został w odniesieniu do genomu
bydła i świni już osiągnięty (tab. 12-11). Opracowana w 1997 roku mapa
genetyczna genomu bydła, oparta na analizie segregacji 1240 markerów,
charakteryzuje się znacznym nasyceniem loci markerowych. Średnia odległość
między dwoma dowolnymi sąsiednimi loci wyniosła 2,5 cM. Ogólną długość
genomu, będącą sumą cząstkowych odległości między kolejnymi loci w chro-
mosomach, oszacowano na poziomie 2990 cM. W przypadku świni mapę
skonstruowano na podstawie analizy segregacji 1042 loci. Średnia odległość
między sąsiednimi loci wyniosła 2,2 cM, a ogólna długość genomu — 2286 cM.
Prawie wszystkie badane loci należały do kategorii anonimowych sekwencji
mikrosatelitarnych. Na uwagę zasługuje bardzo rozwinięta mapa genomu
kury (tab. 12-11). Zauważyć trzeba nie tylko dużą liczbę markerów wykorzysta-
nych do budowy mapy sprzężeniowej, ale także i to, że całkowita długość
mapy (3800 cM) jest znacznie większa niż w przypadku ssaków.
Szerokie zastosowanie analizy molekularnej hybrydowych linii komór-
kowych, powstałych na bazie komórek poddanych napromieniowaniu (tzw.
mapowanie radiacyjne) przyczyniło się do gwałtownego zwiększenia nasy-
cenia map genomowycn loci markerowymi. Obecnie coraz częściej pub-
likowane są mapy poszczególnych chromosomów, a nie całych genomów.
Na mapach takich uwzględnione są dane pochodzące z mapowania
fizycznego (informacje pochodzące z zastosowania techniki hybrydyzacji in
290
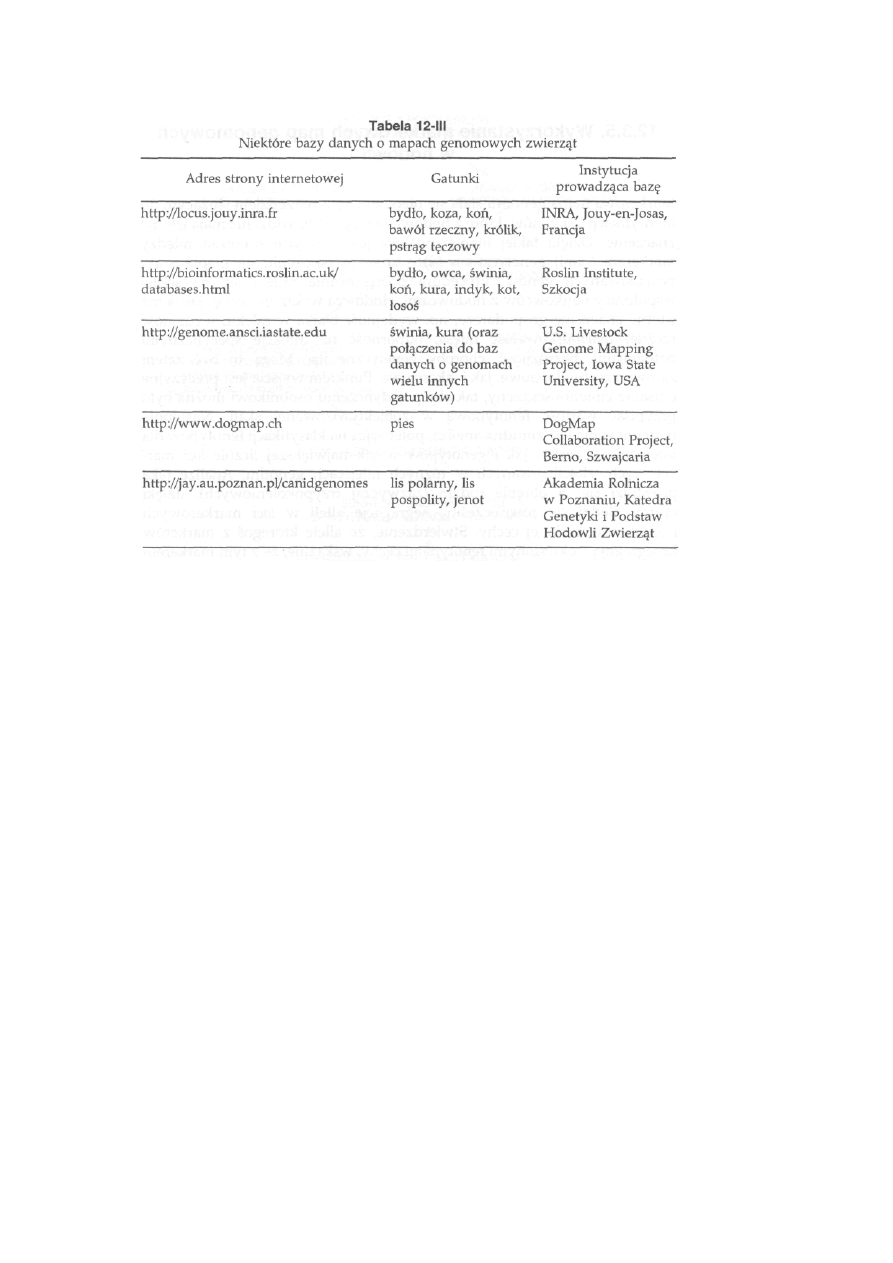
situ, analizy hybrydowych komórek somatycznych, a w tym także mapowa-
nia radiacyjnego) i sprzężeniowego. Tak opracowane mapy określane są
terminem map zintegrowanych (ang. comprehensive map).
Szczegółowe dane dotyczące map poszczególnych genomów, a w tym
i konkretnych chromosomów, są zamieszczone w internetowych bazach
danych (tab. 12-111). Warto zwrócić uwagę, że również mapy genomów
innych gatunków zwierząt gospodarskich, obok wymienionych w tabeli
12-11, są coraz lepiej poznane. Coraz więcej informacji pojawia się o mapach:
kota, królika, bawoła, a także łososia i pstrąga tęczowego. Do grupy
gatunków objętych mapowaniem dołączyły ostatnio również zwierzęta
futerkowe, takie jak: lis pospolity, lis polarny oraz jenot.
W ślad za programem sekwencjonowania genomu człowieka podjęto
analogiczne przedsięwzięcia dotyczące gatunków zwierząt laboratoryjnych
(sekwencję genomu myszy opublikowano w grudniu 2002 r.) i domowych.
Wśród tych ostatnich pierwszym genomem, dla którego ustalono sekwencję
nukleotydów, jest genom psa. Opublikowano ją we wrześniu 2003 r.
Przewiduje się, że do 2005 r. zostanie ustalona sekwencja genomu bydła,
świni i kury. Powiązanie ze sobą markerowych map genomowych z sek-
wencją DNA genomu danego gatunku umożliwi jeszcze efektywniejsze
poszukiwanie genów o dużym znaczeniu w hodowli zwierząt.
291

12.3.5. Wykorzystanie markerowych map genomowych
w hodowli
Markerowa mapa genomu stała się nieodzownym narzędziem służącym do
identyfikacji loci genów, które z hodowlanego punktu widzenia mają istotne
znaczenie. Dzięki takiej mapie możliwe jest wykrycie sprzężeń między
markerem (-ami) genetycznym (-i) o znanym położeniu na mapie a loci
poszukiwanych genów. Procedura postępowania opiera się na ścisłej
współpracy naukowców z hodowcami. Hodowca wskazuje cechę, która ma
istotne znaczenie gospodarcze, np. wydajność białka w mleku, wydajność
rzeźna, marmurkowatość mięsa, odporność na infekcje specyficznymi
patogenami, bezrożność, choroby genetyczne itp. Mogą to być zatem
zarówno cechy ilościowe, jak i jakościowe. Punktem wyjścia jest precyzyjne
opisanie zmienności cechy, tak aby pojedynczemu osobnikowi można było
przypisać wartość fenotypową w zobiektywizowanej skali. Następnie
podejmowana jest żmudna analiza, polegająca na klasyfikacji fenotypów dla
interesującej cechy, jak i genotypów w jak największej liczbie loci mar-
kerowych, zlokalizowanych w różnych miejscach genomu. Analizę taką
prowadzi się w obrębie rodzin (zazwyczaj trzypokoleniowych), dzięki
czemu śledzi się równocześnie segregację alleli w loci
markerowych i fenotypów badanej cechy. Stwierdzenie, że allele któregoś
z markerów kosegregują z określonymi fenotypami cechy, wskazuje, że z
tym markerem jest sprzężony gen kontrolujący kształtowanie się badanej
cechy lub istotnie na nią wpływający (tzw. gen główny w przypadku cech
ilościowych). Wykrycie sprzężenia jest pierwszym krokiem na drodze do
molekularnego scharakteryzowania poszukiwanego genu, które polega na
opisie struktury genu, a w tym przede wszystkim ustaleniu sekwencji
nukleotydów i wskazaniu miejsc zmutowanych, odpowiedzialnych za
występowanie różnych alleli. Jednocześnie poznawany jest produkt tego
genu oraz jego szczegółowa funkcja biologiczna. Osiągnięcie takiego
poziomu wiedzy o genie zazwyczaj pozwala na opracowanie testu
molekularnego (najczęściej typu PCR-RFLP), który znajduje zastosowanie
w identyfikacji genotypu osobnika na podstawie bezpośredniej analizy
DNA. Ogólny schemat postępowania, którego celem jest identyfikacja
nieznanego genu, jest pokazany na rycinie 12-6.
Klasycznym przykładem wykorzystania badań markerów genetycznych
do identyfikacji genu wpływającego na ważną cechę produkcyjną jest
historia 20-letnich poszukiwań genu odpowiedzialnego za podatność świń
na stres i związanej z tym zmniejszonej wartości technologicznej mięsa
(tzw. mięso blade, miękkie i wodniste, ang. pale, soft, exudative — PSE).
Najpierw ustalono, że cecha ta warunkowana jest monogenowo, a konkretnie
przez allel recesywny. Następnie opracowano test (inhalacja halotanem) do
identyfikacji zwierząt podatnych na stres, tzn. homozygot recesywnych
292

Ryć. 12-6. Ogólny schemat postępowania zmierzającego do identyfikacji nieznanego
genu o dużym znaczeniu hodowlanym, np. genu głównego wpływającego na kształ-
towanie poligenicznych cech produkcyjnych
(nn). Na podstawie tego testu wprowadzono symbol HAL na oznaczenie
genu. Kilka lat później zidentyfikowano pierwsze markery genetyczne
(m.in. locus układu grupowego krwi H oraz loci polimorficznych białek krwi
- GP7, PGD i A1BG), które okazały się ściśle sprzężone z locus HAL. Loci
tych markerów umiejscowiono, za pomocą hybrydyzacji in situ, w chromo-
somie 6 świni (ryć. 12-7a). Wreszcie w 1991 roku zidentyfikowano gen
kandydujący (ang. candidate gene). Był nim gen receptora rianodyny
(RYR1), którego produkt jest zaangażowany w transport jonów wapniowych
w mięśniach szkieletowych. Badania molekularne tego genu u osobników
wrażliwych na stres wykazały, że jedna z kilkunastu zidentyfikowanych
mutacji punktowych prowadzi do zmiany w łańcuchu polipeptydowym,
która polega na zamianie argininy na cysteinę w pozycji 615. Zamiana ta
jest skutkiem tranzycji C
→T w pozycji 1843 cDNA genu RYR1. Odkrycie to
pozwoliło na wskazanie enzymu restrykcyjnego HhaI, który nie rozpoznaje
293

Ryć. 12-7. Lokalizacja niektórych genów głównych o znanej już budowie molekularnej:
(a) KYR1, (b) MSTN, (c) BMPR-IB, (d) PRKAG3 oraz regionów chromosomowych,
w których prawdopodobnie występują geny główne kontrolujące: (e) otłuszczenie tuszy
i tempo wzrostu/ (f) otłuszczenie tuszy i zawartość mięsa w tuszy
sekwencji zawierającej mutację. W ten sposób opracowano test PCR-RFLP
(patrz rozdział: Zmienność cech), który umożliwia wykrycie allelu recesyw-
nego w genotypie homozygot recesywnych i heterozygot.
Od połowy lat 80. wiadomo było, że hipertrofia mięśniowa (tzw.
podwójne umięśnienie) u belgijskiego bydła błękitnego zależy od re-
cesywnego genu mh (ang. muscular hyperthrophy). Poszukiwanie markerów
genetycznych sprzężonych z locus mh pozwoliło ustalić, że gen ten jest
położony w części przycentromerowej ramienia długiego chromosomu
2 bydła. Porównanie tego regionu z homologicznym segmentem chro-
mosomu ludzkiego doprowadziło do wskazania w 1997 roku na gen
miostatyny (MSTN), jako gen kandydujący do miana odpowiedzialnego
za hipertrofię mięśniową (ryć. 12-7b). Analiza molekularna tego genu
u zwierząt z hipertrofią mięśniową, wywodzących się z rasy belgijskiej
294

błękitnej i asturiana, wskazała na obecność w ich genotypie dwóch
zmutowanych allełi (homozygota recesywna). Okazało się, że mutacja
polegała na braku (delecji) 11 nukleotydów. Skutkiem tej mutacji jest
powstanie skróconego łańcucha polipeptydowego (przedwczesne po-
jawienie się kodonu „stop"), który jest biologicznie nieaktywny, co
fenotypowo objawia się hipertrofią mięśniową. Badania molekularne
genu MSTN w innych europejskich rasach mięsnych doprowadziły
do identyfikacji kolejnych czterech, różnych mutacji uszkadzających
funkcję białka miostatyny. Osiągnięcie to pozwala na ustalanie genotypu
zwierząt metodami genetyki molekularnej (patrz rozdział: Zmienność
cech).
Rozwijające się dynamicznie badania nad mapami genomów zwierząt
gospodarskich umożliwiły identyfikację markerów genetycznych sprzężo-
nych z różnymi genami głównymi, które na poziomie molekularnym nie
zostały dotychczas jeszcze rozpoznane. Co więcej, nie udało się jeszcze
wskazać genów kandydujących. Przykładem może być gen Fec
B
, wpływający
na plenność owiec rasy merynos booroola, a konkretnie kontrolujący liczbę
owulujących oocytów podczas rui. Locus tego genu jest sprzężony z mar-
kerami mikrosatelitarnymi BM1329 i OarAE101, które umiejscowione są
w chromosomie 6 (ryć. 12-7c). W 2001 r. ustalono, że gen Fec
B
to zmutowana
postać genu BMPR-IB. Substytucja A > G w tym genie odpowiada za
wbudowanie w pozycji 249 łańcucha polipeptydowego argininy zamiast
glutaminy. Powoduje to zmianę siły wiązania ligandów przez receptor
kodowany przez gen BMPR-IB. Innym przykładem jest gen RN, który
odpowiada za zawartość glikogenu w mięśniach. Dominujący gen RN~
powoduje większą, a allel recesywny rn
+
mniejszą zawartość glikogenu.
Efektem zwiększonej zawartości glikogenu jest uzyskanie po uboju tzw.
kwaśnego mięsa, które ma mniejszą wartość technologiczną. Locus genu RN
umiejscowiono w chromosomie 15, dzięki identyfikacji sprzężonych mar-
kerów mikrosatelitarnych Sw120 i Sw936 (ryć. 12-7d). W 2000 r. zakończyły
się wieloletnie poszukiwania genu RN. Okazało się, że jest nim zmutowany
allel genu PRKAG3, który koduje podjednostkę y kinazy białkowej ak-
tywowanej przez AMP. Mutacja polegała na zmianie sekwencji nukleotydów
G > A w kodonie 200 tego genu.
Badania zmierzające do identyfikacji genów głównych, które w znaczący
sposób wpływają na cechy użytkowe, są prowadzone w licznych ośrodkach
naukowych. W początkowym stadium badań zakłada się, że zmienność
wybranej cechy produkcyjnej może zależeć od jakiegoś genu z dużym
efektem działania, czyli genu głównego. Analiza segregacji licznych mar-
kerów genetycznych, pokrywających w miarę równomiernie większość
genomu, oraz zmienności cechy produkcyjnej w rodzinie referencyjnej
(najczęściej 3-pokoleniowej) może doprowadzić do wskazania regionu
chromosomowego, w którym prawdopodobnie występuje locus genu głów-
nego. W 1994 roku ukazała się praca dowodząca, że w chromosomie 4 świni,
295

między markerami S0001 i S0067, występuje prawdopodobnie locus (loci?)
genu głównego wpływającego na tempo wzrostu oraz otłuszczenie tuszy
(ryć. 12-7e). Sugestia ta została potwierdzona także w innych badaniach. Do
tej pory jednak nie udało się zaproponować genu (-ów?) kandydującego,
który odpowiadałby za kształtowanie tych cech ilościowych. Podobne
badania przeprowadzone w ramach Polskiego Projektu Mapowania Genomu
Świni sugerują, że w chromosomie 12, między markerami mikrosatelitarnymi
S0083 i S0090, występuje locus (loci?) genu głównego wpływającego na
odkładanie tłuszczu brzusznego i zawartość mięsa w tuszy (ryć. 12-7f).
Z kolei w październiku 2003 r. doniesiono, że mutacja w intronie 3 genu
IGF2 (insulinopodobnego czynnika wzrostu 2) wywołuje bardzo duży
wpływ na mięsność świń. Prosta substytucja G > A prawdopodobnie zakłóca
interakcję DNA z nieznanym białkiem represorowym, czego efektem jest
3-krotny wzrost ekspresji genu IGF2. Trzeba zaznaczyć, że gen ten podlega
piętnowaniu gametycznemu. W przypadku genu IGF2 ekspresji podlega
jedynie allel pochodzący od ojca, czyli duży efekt fenotypowy pojawia się
tylko wtedy, gdy zmutowany allel jest odziedziczony od ojca. Odnotowano
też znaczące osiągnięcie dotyczące bydła. Z badań prowadzonych w rodzi-
nach referencyjnych wynikało, że w chromosomie 14 zlokalizowany jest gen
wpływający w znaczący sposób na wydajność i skład mleka. W 2001 r.
wykazano, że mutacja typu podstawienia dwunukleotydowego AA > GC
w genie DGTA1 (acylo-CoA: diacylogliceroloacylotransferaza) wywołuje
zmianę lizyny na alaninę, a to upośledza funkcję kodowanego enzymu.
W efekcie dochodzi do znacznego zwiększenia wydajności tłuszczu w mleku.
Mapy genomowe są powszechnie wykorzystywane do identyfikacji
genów odpowiedzialnych za monogenowe choroby genetyczne lub marke-
rów genetycznych z nimi sprzężonych. Jednym z przykładów może być
letalna wada rozwojowa — achondroplazja, określana symbolem bd (ang.
bulldog disease), ze względu na wywoływane zmiany m.in. w budowie
czaszki. Nosicielstwo genu recesywnego bd stwierdzono w 1999 roku
w genotypie francuskiego buhaja, charakteryzującego się wybitną wartością
hodowlaną. W początkowym okresie jego użytkowania stwierdzono, że około l
% potomków był obarczony letalnymi zniekształceniami czaszki i kończyn.
Oznaczało to, że również wiele krów było nosicielkami zmutowanego genu
bd. Badacze francuscy wytypowali 75 rodzin (buhaj nosiciel, krowa nosicielka,
chore cielę — homozygota recesywna), w których przeprowadzono analizę
dziedziczenia markerów genetycznych rozproszonych równomiernie po
całym genomie. Wkrótce potem udało się zidentyfikować marker genetyczny
silnie sprzężony z genem bd. Osiągnięcie to pozwalało na identyfikację
potomków buhaja nosiciela, którzy również byli nosicielami tego niepożąda-
nego genu. Dzięki temu udało się zapobiec rozprzestrzenianiu niepożądane-
go genu, w wyniku wykorzystywania do inseminacji nasienia buhaja
nosiciela. Można się spodziewać, że wkrótce zostanie również zidentyfikowa-
ny gen odpowiedzialny za powstanie tej wady wrodzonej.
296

W tym samym czasie w populacji duńskiej bydła holsztyńsko-fryzyjskiego
zidentyfikowano inną letalną wadę rozwojową, polegającą na zniekształceniu
kręgów (ang. CVM — central vertebral malformation). Również dla tego
genu udało się zidentyfikować sprzężone markery genetyczne, a ostatnio
scharakteryzowano gen oraz mutację w nim występującą. Niestety badań
tych nie opublikowano, a opracowany test molekularny skomercjalizowano.
Szczególne osiągnięcia dotyczące identyfikacji genów odpowiedzialnych
za rozwój monogenowych chorób dziedzicznych odnotowano u psów.
U gatunku tego znanych jest ponad 300 jednostek chorobowych uwarun-
kowanych przez mutacje pojedynczych genów. Wiele z tych mutacji
występuje w ściśle określonych rasach. Dotychczas opisano na poziomie
molekularnym trzydzieści genów i ich mutacji odpowiedzialnych za rozwój
choroby dziedzicznej. Wśród nich są geny wywołujące takie schorzenia, jak:
dziedziczna dystrofia siatkówki u owczarków francuskich (briardów),
narkolepsja u dobermanów i labradorów, ciężki złożony niedobór odporności
psów rasy welsh corgi czy fukozydoza u angielskich springer spanieli.
W latach 2001-2003 podjęto udane próby terapii genowej niektórych chorób
dziedzicznych psa (np. dziedziczny zanik siatkówki u owczarków fran-
cuskich).
Przedstawione powyżej przykłady wykorzystania markerowych map
genomowych do rozwiązywania konkretnych problemów hodowlanych
pokazują, że genetyka molekularna stała się kluczowym narzędziem służą-
cym postępowi w hodowli zwierząt.

Literatura uzupełniająca
Podręczniki
Biochemia (1997), L. Stryer (tłum. z j. ang. pod red. J. Augustyniaka i J. Michejdy),
Wydawnictwo Naukowe PWN. Biologia molekularna w medycynie. Elementy genetyki
klinicznej (2001), pod red. J. Bala,
Wydawnictwo Naukowe PWN. Biotechnologia zwierząt (1997), pod red. L.
Zwierzchowskiego, K. Jaszczaka i J. Modlińskiego,
Wydawnictwo Naukowe PWN.
Cytobiochemia (1995), L. Kłyszejko-Stefanowicz, Wydawnictwo Naukowe PWN. Genetyka
człowieka. Rozwiązywanie problemów medycznych (2003), B.R. Korf (przekład pod
red. A. Pawlaka), Wydawnictwo Naukowe PWN.
Genetyka molekularna (1995), pod red. P. Węgleńskiego, Wydawnictwo Naukowe PWN.
Genom człowieka. Największe wyzwanie współczesnej genetyki i medycyny molekularnej (1997),
pod red. W. Krzyżosiaka, Wydawnictwo Naukowe PWN. Genomy (2001), T.A.
Brown (przekład pod red. P. Węgleńskiego), Wydawnictwo
Naukowe PWN. Immunologia (1996), I. Roitt, J. Brostoff, D. Małe (tłum. z j. ang. pod
red. J. Żeromskiego),
Wydawnictwo Medyczne Słotwiński Yerlag, Brema. Jeżyk genów. Poznawanie zasad
dziedziczenia (1997), P. Berg i M. Singer (tłum. z j. ang.
M. Waśniewska-Brzeska i J. Brzeski), Prószyński i S-ka, Warszawa. Karty z Dziejów
Zootechniki Polskiej. Cz. II— lata 1972^-1997 (1997), pod red. A. Filistowicza,
J. Juszczaka i S. Wężyka, Wydawnictwo Polskiego Towarzystwa Zootechnicznego,
Warszawa.
Leksykon biologiczny (1992), pod red. Cz. Jury i H. Krzanowskiej, Wiedza Powszechna.
Leksykon rozrodu zwierząt (1996), pod red. K. Rosłanowskiego, Wydawnictwo Akademii
Rolniczej w Poznaniu. Leksykon terminów z zakresu genetyki i hodowli zwierząt
(1994), B. Nowicki i wsp.,
Wydawnictwo Polskiego Towarzystwa Zootechnicznego, Warszawa. Słownik
terminów genetycznych (2002), R.C. King, W.D. Stansfield (przekład zbiorowy pod
red. W. Prus-Głowackiego), Ośrodek Wydawnictw Naukowych PAN, Poznań. The
Major Histocompatibility Complex Region of Domestic Animal Species (1996), pod red.
L.B. Schook i S.J. Lamont, CRC Press Inc., USA.
298

Artykuły przeglądowe
Barsh G. (1996). The genetics of pigmentation: from fancy genes to complex traits. Trends
in Genetics 12 (8): 299-305. Bartnik E. (1997). Ludzki genom mitochondrialny —
mutacje, polimorfizmy i choroby.
Postępy Biologii Komórki 24 (8): 69-75. Charon K.M. (1998). Wykorzystanie metod
molekularnej analizy genomu zwierząt
w pracy hodowlanej. Postępy Nauk Rolniczych 7 (nr 5): 87-97. Charon K.M. (1998).
Znaczenie głównego układu zgodności tkankowej w hodowli
zwierząt. Medycyna Weterynaryjna 54 (2): 92-96. Cotinot C. i wsp. (2002). Molecular
genetics of sex determination. Seminars in Reproductwe
Medicine 20: 157-167. De Gortari M.J., Freking B.A., Cuthbertson R.P., Kappes S.M.,
Keele J.W., Stone R.T.,
Leymaster K.A., Dodds K.G., Crawford A.M., Beattie C.W. (1998). A second-generation
linkage map of the sheep genome. Mammalian Genome 9: 204-209. Ellis S.A. (1994).
Minireview — MHC studies in domestic animals. European Journal of
Immunogenetics 21: 209-215.
Friend S.H., Stoughton R.B. (2002). Magiczne mikromacierze. Świat Nauki 4 (128): 36-43.
Grzybowski G. (1997). Wybrane praktyczne aspekty technik molekularnych — PCR-RFLP
i automatycznego sekwencjonowania DNA — w badaniach genomu zwierząt gos-
podarskich. Prace i Materiały Zootechniczne 50: 69-78. Grzybowski G., Dymnicki E.
(1997). Metody oraz znaczenie sterowania proporcją płci
u bydła. Biotechnologia 3 (38): 38-50. International Human Genome Seąuencing
Consortium (2001). Initial seąuencing and
analysis of the human genome. Naturę 409: 860-921. Kamiński S. (1996). Bovine
kappa-casein (CASK) gene — molecular naturę and application
in dairy cattle breeding. Journal of Applied Genetics 37: 179-196. Kamiński S. (2002).
DNA microarrays — a methodological breakthrough in genetics.
Journal of Applied Genetics 43: 123-130. Kappes S.M., Keele J.W., Stone R.T.,
McGraw R.A., Sonstegard T.S., Smith T.P.L.,
Lopez-Corrales N.L., Beattie C.W. (1997). A second-generation linkage map of bovine
genome. Genome Research 7: 235-249. Kirkness E.F. i wsp. (2003). The dog
genome: survey seąuencing and comparative
analysis. Science 301: 1898-1903. Krzanowska H. (1999). Długa historia krótkiego
chromosomu — jak poznano geny
płodności sprzężone z chromosomem Y u ssaków. Postępy Biologii Komórki 26 (supl.
12): 53-58. Kurył J. (1998). Geny oddziałujące na jakość tuszy i mięsa świń.
Prace i Materialy
Zootechniczne. Zeszyt specjalny 8: 9-18. Lechniak D. (1996). Anomalie
chromosomowe przyczyną zamierania gamet i zarodków
bydlęcych. Medycyna Weterynaryjna 52 (2): 89-93. Long S.E. (1997). Chromosome
abnormalities in domestic sheep (Ovis aries). Journal of
Applied Genetics 38: 65-76. Pareek Ch.S., Kamiński S. (1996). Bovine leukocyte
adhesion deficiency (BŁĄD) and its
worldwide prevalence. Journal of Applied Genetics 37 (3): 299-312. Parham P., Ohta T.
(1996). Population biology of antigen presentation by MHC class
I molecules. Science 272: 67-74. Pierzchała M. (1996). Mapa genomu świni (Sus
scrofa domestica). Prace i Materialy
Zootechniczne 49: 7-28. Ramikssoon Y., Goodfellow P. (1996). Early steps in
mammalian sex determination.
Current Opinion in Genetics & Development 6: 316-321.
299

Rejduch B., Świtoński M., Słota E., Danielak B., Kozubska-Sobocińska A. (1994). Aberracje
chromosomowe u bydła o użytkowości mięsnej. Medycyna Weterynaryjna 50 (8):
379-382. Rohrer G.A., Alexander L.J., Hu Z., Smith T.P.L., Keele J.W., Beattie
C.W. (1996).
A comprehensive map of the porcine genome. Genome Research 6: 371-391. Ruvinsky
A. (1999). Basics of gametic imprinting. Journal of Dairy Science 82 (suppl.
2):288-237. Słomski R., Napierała D., Kwiatkowska J. (1998). Reakcja łańcuchowa
polimerazy (PCR)
w badaniach naukowych i diagnostyce molekularnej. Postępy Biologii Komórki 25 (supl.
10): 195-217.
Stachurska A. (1997). Inheritance of colours in horses. Journal of Applied Genetics 38:195-204.
Sysa P.S. (1997). Krajowy system kontroli cytogenetycznej buhajów. Medycyna Weterynaryjna
53 (10): 581-584. Szatkowska L, Zych S., Kulig H. (2003). Determinacja płci u ssaków.
Medycyna Weterynaryjna
59: 847-852. Szwaczkowski T. (1993). Identyfication of animal major genes in field
collected data by
use of statystical methods. Journal of Animal and Feed Sciences 2/3: 91-103. Szydłowski
M. (1994). Wybrane aspekty analizy loci markerowych w doskonaleniu
ilościowych cech zwierząt. Prace i Materiały Zootechniczne 46: 29-40. Szymański M.,
Barciszewska M.Z., Żywiecki M., Barciszewski J. (2003). Noncoding RNA
transcripts. Journal of Applied Genetics 44: 1-20. Świtoński M. (1998). Markerowe
mapy genomowe i ich wykorzystanie w hodowli
zwierząt. Postępy Biologii Komórki 25 (supl. 10): 113-122. Świtoński M., Kurył J. (1998).
Poszukiwanie genów kontrolujących cechy tuczne, opasowe
i rzeźne — najnowsze osiągnięcia. Prace i Materiały Zootechniczne 52: 37-42. Świtoński
M., Stranzinger G. (1998). Synaptonemal complexes in domestic mammals —
a review. Journal of Heredity 89 (6): 473-480. Walawski K. (1999). Genetic aspects of
mastitis resistance in cattle. Journal of Applied
Genetics 40 (2): 117-128. Waterston R.H. i wsp. (2002). Initial seąuencing and
comparative analysis of the mouse
genome. Naturę 420: 520-562. Węgrzyn J., Skiba E. (1997). Antigenic markers of
protein genes and their nomenclature
in cattle. Journal of Applied Genetics 38 (3): 319-328. Wierzbicki H. (1998).
Genetyczne uwarunkowania odmian barwnych lisa pospolitego
(Vulpes vulpes) i lisa polarnego (Alopex lagopus). Prace i Materiały Zootechniczne 53: 35-48.
Yenter C. i wsp. — Celera Genomics (2001). The seąuence of the human genome. Science
291: 1304-1351. Zwierzchowski L., Rosochacki S.J. (1998). Transgeneza —
możliwości i perspektywy
zastosowania w produkcji zwierzęcej. Prace i Materiały Zootechniczne 50: 11-39.
Zwierzchowski L. (1998). Biotechnologiczne wykorzystanie gruczołu mlecznego -
perspektywy manipulacji genetycznych białkami mleka. Biotechnologia 2 (41): 33-56.

Skorowidz
Opracowali:
Krystyna M. Charon, Marek Świtoński
Numery stron, na których znajduje się główny opis, są pogrubione.
Gwiazdką oznaczono numery stron, na których hasła znajdują się na rysunku
lub w jego podpisie.






















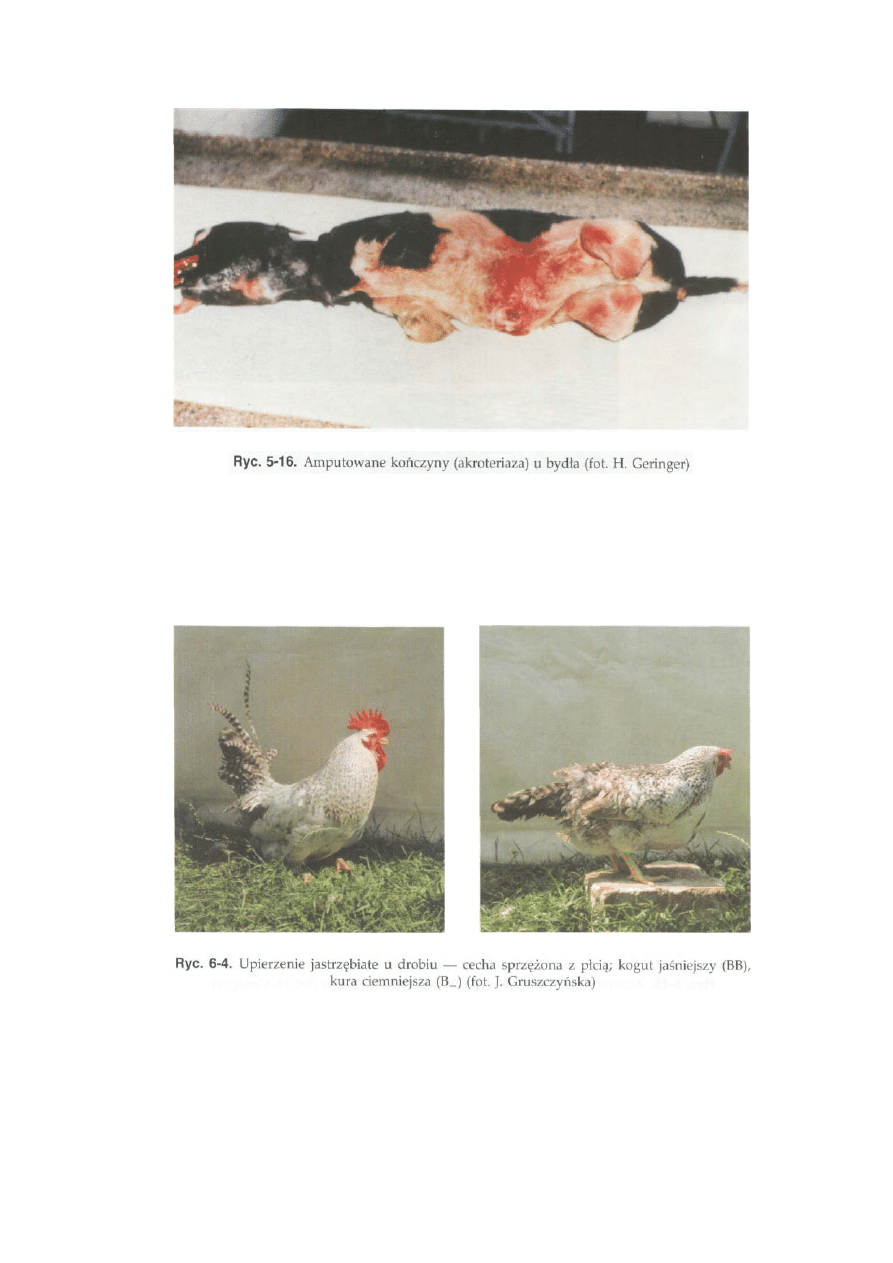




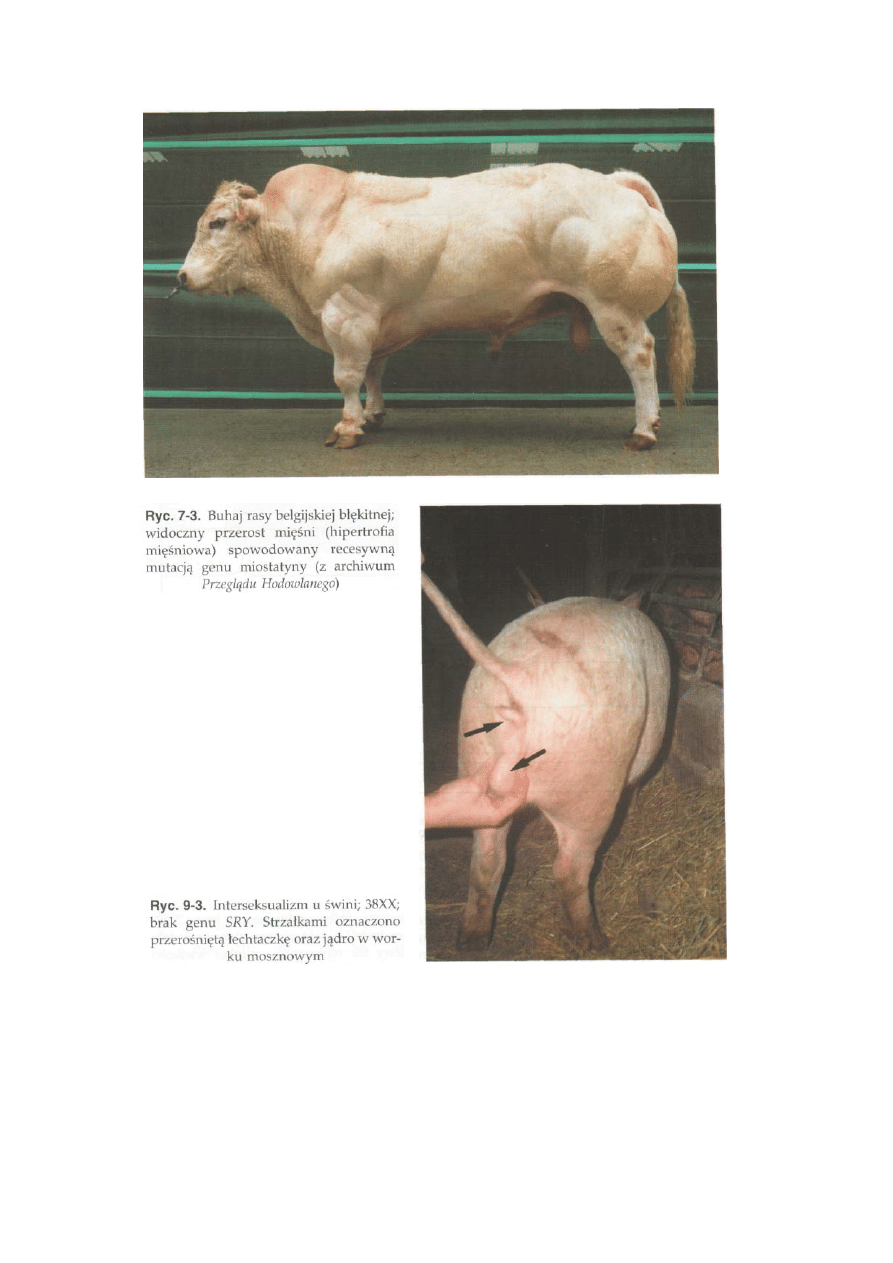
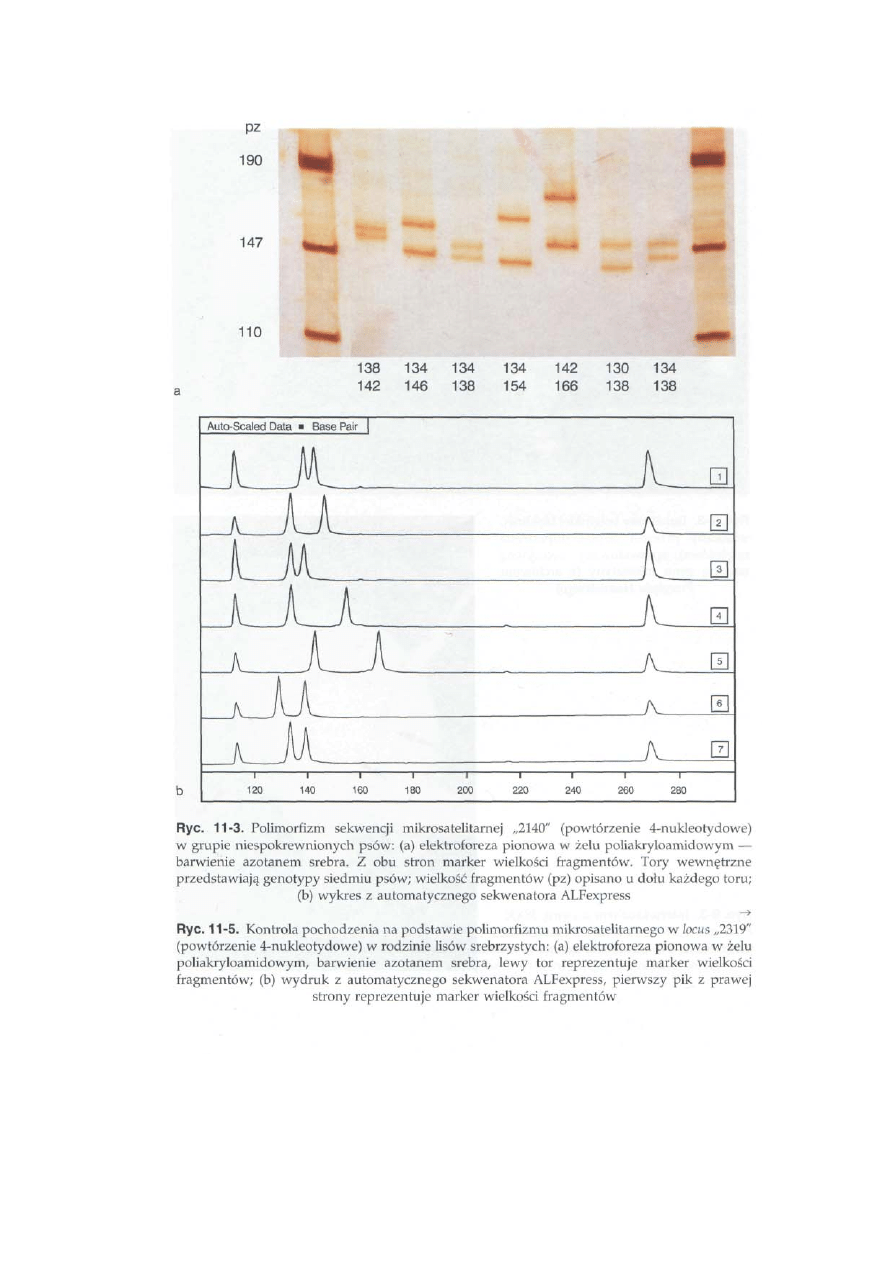
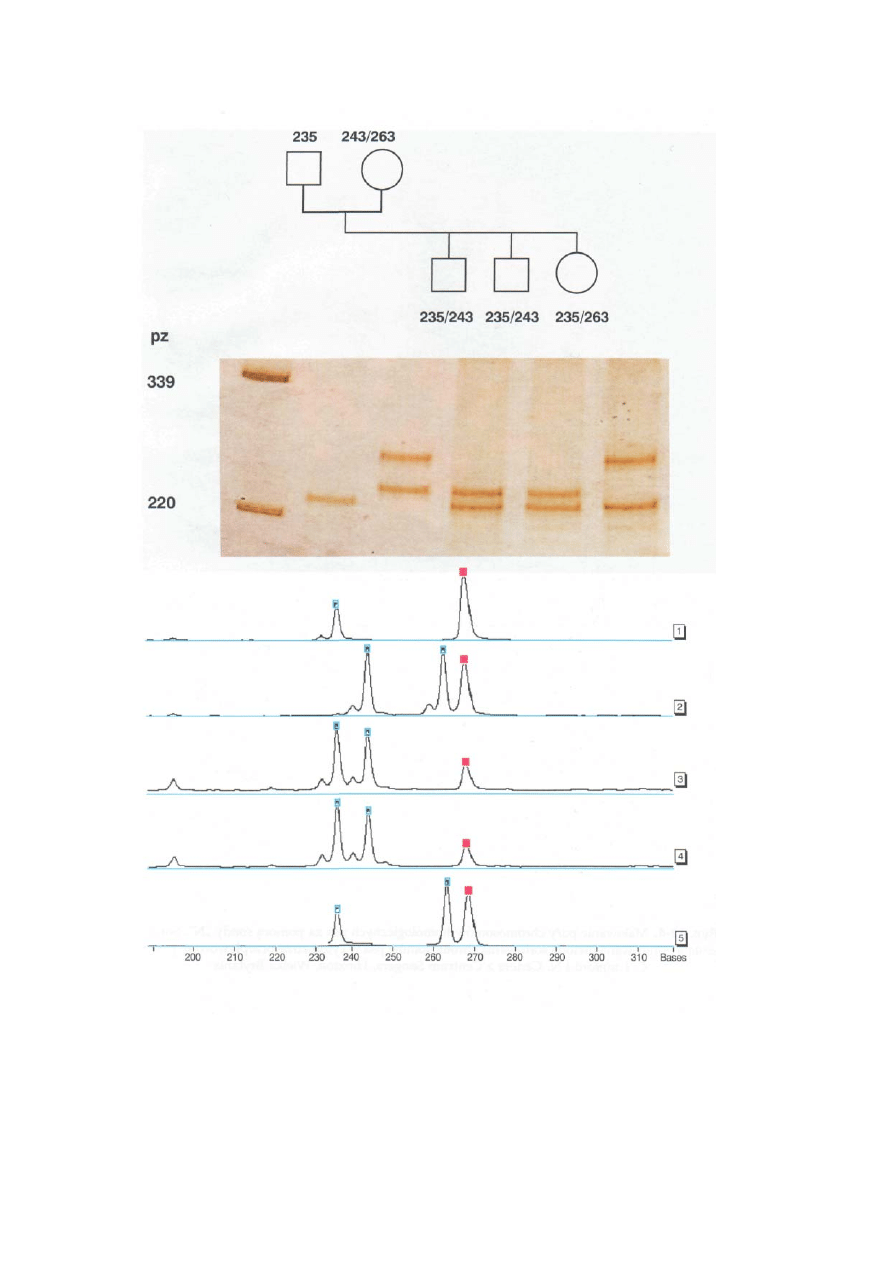
Document Outline
- Okładka
- Przedmowa
- Spis Treści
- Rozdział 1
- Rozdział 2
- Rozdział 3
- Rozdział 4
- 4.1. Endonukleazy restrykcyjne
- 4.2. Wektory
- 4.3. Rekombinacja molekularna i klonowanie
- 4.4. Amplifikacja DNA za pomocą łańcuchowejreakcji polimerazy (PCR)
- 4.5. Techniki hybrydyzacji i sondy molekularne
- 4.6. Polimorfizm fragmentów restrykcyjnychDNA (RFLP)
- 4.7. Biblioteki genomowe i genowe
- 4.8. Sekwencjonowanie DNA
- 4.9. Badanie ekspresji genów
- 4.10. Transgeneza
- Rozdział 5
- 5.1. Mutacje genomowe
- 5.2. Mutacje chromosomowe
- 5.3. Mutacje genomowe i chromosomowezidentyfikowane u zwierząt hodowanych w Polsce
- 5.4. Mutacje genowe
- 5.5. Mutacje w mitochondrialnym DNA
- Rozdział 6
- Rozdział 7
- Rozdział 8
- 8.1. Prawo równowagi genetycznej
- 8.2. Frekwencja genów i genotypów wprzypadku dominowania
- 8.3. Frekwencja genów i genotypów w przypadkucech uwarunkowanych szeregiem (serią)alleli wielokrotnych
- 8.4. Frekwencja genów i genotypów w przypadkucech sprzężonych z płcią
- 8.5. Czynniki naruszające równowagę genetyczną
- 8.6. Wykorzystanie frekwencji alleli do szacowaniazmienności genetycznej wewnątrz i międzypopulacjami
- Rozdział 9
- Rozdział 10
- 10.1. Genetyczne podłoże różnorodnościprzeciwciał
- 10.2. Genetyczne podłoże różnorodnościreceptorów limfocytów J
- 10.3. Główny układ zgodności tkankowej — MHC
- 10.4. Zarys przebiegu reakcji odpornościowej
- 10.5. Genetyczna oporność na choroby
- Rozdział 11
- Rozdział 12
- Literatura uzupełniająca
- Skorowidz
- Zdjęcia
Wyszukiwarka
Podobne podstrony:
genetyka zwierzątx
Genetyka zwierząt
Genetyka zwierzat agroturystyka Cw3
wyklady -ochrona zasobow genetycznych zwierzat gospodarskich2, polityka w ochronie srodowiska, polit
Choroby genetyczne zwierząt fakultet
metody, Makrokierunek, genetyka zwierząt i metody hodowlane
dziedziczenie dwóch cech, Makrokierunek, genetyka zwierząt i metody hodowlane
Genetyka zwierzat agroturystyka Cw3
Choroby genetyczne zwierząt
Fizjologia zwierząt wszystkie opracowania, chemia organiczna, biologia ewolucyjna-wykłady, genetyka,
Metody inzynierii genetycznej w hodowli zwierzat wyklady(calosc1)
Metody
więcej podobnych podstron