
Genetyka
Część II

Zmienność
Zmiennością nazywamy występowanie
dziedzicznych lub niedziedzicznych różnic:
między komórkami (zmienność wewnątrzosobnicza),
między osobnikami należącymi do tej samej
populacji (zmienność osobnicza)
pomiędzy populacjami (zmienność grupowa).
Zmienność fenotypowa może mieć charakter:
ciągły (zmienność fluktuacyjna), dający się określić
w jednostkach miary (wzrost, masa ciała, IQ, liczba
erytrocytów i leukocytów)
charakter nieciągły (grup krwi Rh (+) i Rh(-).

Zmienność
Czynniki środowiskowe mają duży wpływ na
przejawianie się cech dziedzicznych.
Ten sam zespół genów w różnych warunkach
może dać różne efekty fenotypowe.
Może istnieć również sytuacja odwrotna.
Różne zespoły genów mogą dawać podobny
fenotyp. Sytuację tę określamy jako fenokopia.
Rozwój danego organizmu zależy więc z jednej
strony od genotypu, a z drugiej od czynników
środowiskowych.

Zmienność
Stąd też choroby w zależności od udziału
czynników genetycznych i środowiskowych
podzielono na trzy grupy:
- choroby zależne tylko od genotypu organizmu (np.
hemofilia A, albinizm);
- choroby, w których przebiegu dużą rolę odgrywa
zarówno genotyp jak i warunki środowiska (np.
cukrzyca, choroba wieńcowa);
- choroby warunkowana tylko czynnikami
środowiskowymi (choroby zakaźne i inwazyjne).
Zmienność możemy podzielić na niedziedziczną
(zmienność modyfikacyjna) i dziedziczną
(zmienność rekombinacyjna i mutacyjna).

ZMIENNOŚĆ
MODYFIKACYJNA
Ten rodzaj zmienności zachodzi pod wpływem
środowiska. Może mieć zarówno charakter zmienności
fluktuacyjnej jak i alternatywnej.
Jest ona możliwa dzięki „plastyczności” organizmów,
czyli możliwością zmian morfofizjologicznych pod
wpływem środowiska.
Służy to jak najlepszemu przystosowaniu danego
osobnika do określonych warunków środowiska.
Organizm nie może zmieniać się bez ograniczeń.
Istnieje tzw. norma reakcji, czyli dopuszczalny zakres
zmian organizmu pod wpływem środowiska.
Po przekroczeniu tej granicy mogą rozpocząć się
procesy patologiczne, które niekiedy prowadzą do
śmierci.

ZMIENNOŚĆ
MODYFIKACYJNA
W zależności od cechy ulegającej zmianie, zakres tych
zmian może być różny.
Należy jednak pamiętać, że nie wszystkie cechy mogą
ulegać modyfikacji.
W zależności od warunków środowiska można stwierdzić
jego wpływ np. na wzrost lub masę organizmu.
Z kolei np. w przypadku daltonizmu wpływ środowiska
nie odgrywa żadnej roli.
Ze względu na niedziedziczny charakter, zmienność
modyfikacyjna nie ma żadnego wpływu na procesy
ewolucyjne. Jest natomiast bardzo ważna dla danego
osobnika.

ZMIENNOŚĆ
REKOMBINACYJNA
Pod pojęciem rekombinacji genetycznej rozumiemy
każdy proces, w wyniku którego kontakt dwóch
organizmów lub komórek, różniących się pod względem
jednej lub więcej par genów, prowadzi do powstania
osobników, w których allele połączone są inaczej niż u
rodziców.
Rekombinanty mogą się pojawiać w wyniku mejozy,
mitozy lub koniugacji.
U organizmów eukariotycznych rekombinacja może
nastąpić w wyniku jednego z wymienionych procesów:
- losowej segregacji chromosomów w spermatogenezie lub
oogenezie;
- losowego łączenia się gamet;
- losowego doboru rodziców lub koniugantów;
- rekombinacji wewnątrzchromosomowej (crossing-over).

Mutacja
Mutacją nazywamy nagłą, skokową zmianę fenotypu,
spowodowaną zmianą genotypu.
Ze względu na sposób powstania mutacji dzieli się je na:
spontaniczne (samorzutne)
indukowane, które są efektem działania tzw. mutagenów
(np. promieniowanie UV, niektóre substancje chemiczne).
Powstała mutacja może w różny sposób wpływać na
danego osobnika.
Te, które powodują śmierć danego osobnika określamy
jako mutacje letalne (np. zespół Retta w przypadku
osobników męskich).
Mutacje subletalne w znacznym stopniu upośledzają
danego osobnika, ale w odpowiednich warunkach może
on przeżyć (np. niektóre hemoglobinopatie, lub
hemofilia).

Mutacja
Istnieją także mutacje, które nie mają żadnego wpływu na
przeżywanie danej osoby (mutacje obojętne). Przykładem
takiej mutacji może być np. ruda barwa włosów.
Mutacje można także podzielić ze względu na poziom, na
jakim zachodzą. Wyróżniamy w tym przypadku mutacje
genowe i chromosomowe.
MUTACJE GENOWE
Mutacją genową (punktową) nazywamy każdą zmianę
sekwencji nukleotydów w obrębie genu inną od sekwencji
nukleotydów genu wyjściowego.
Mutacje punktowe dotyczą zarówno genów kodujących
białka enzymatyczne jak i strukturalne.
Do mutacji na poziomie DNA zalicza się:
substytucje (tranzycja i transwersja), delecje i insercje.

Mutacja. Tranzycja
Tranzycja polega na zamianie jednej zasady purynowej na
inną purynową lub zasady pirymidynowej na inną
pirymidynową.
W prawidłowym DNA tymina jest połączona z adeniną
trzema wiązaniami wodorowymi.
Na przykład adenina w formie tautomerycznej łączy się z
cytozyna zamiast z tyminą.
Natomiast cytozyna wbudowana uprzednio do
komplementarnej nici DNA połączy się z guaniną.
Powstaną więc dwa rodzaje cząsteczek DNA.
Jedna cząsteczka identyczna z macierzystym DNA i druga, w
której doszło do substytucji pary zasad AT na CG.
Te dwa rodzaje cząsteczek rozdzielą się w wyniku mitozy do
dwóch komórek potomnych, które utworzą dwa różne klony
komórkowe.
W wyniku takiej mutacji może powstać kodon dla innego
aminokwasu, co w sumie może spowodować syntezę
niewłaściwego peptydu.

Mutacja. Transwersja
Transwersja polega na zamianie zasady purynowej na
pirymidynową lub pirymidynowej na purynową.
Jedną z przyczyn powstania tego typu mutacji jest
występowanie dimerów C-C, C-T a zwłaszcza dimerów T-T.
Dimery pirymidynowe powstają w wyniku wytwarzania
wiązań kowalencyjnych pomiędzy leżącymi obok siebie
pirymidynami w łańcuchu DNA.
Powstałe dimery zaburzają replikację DNA.
Należy jednak zaznaczyć, że obecność dimerów nie jest
mutacją, a tylko uszkodzeniem DNA.
Natomiast mutacje mogą powstać podczas procesów
usuwania dimerów.
Najbardziej znanym efektem transwersji u ludzi jest
anemia sierpowata.

INSERCJA (ADDYCJA) I DELECJA
Addycja polega na dodaniu jednej lub więcej par
nukleotydów (ale nie trzech i wielokrotności trzech) w
łańcuchu DNA. Natomiast delecją oznacza ubytek jednej
lub więcej par nukleotydów (nie trzech i wielokrotności
trzech) z łańcucha DNA.
O mutacji punktowej mówimy wówczas, gdy wystąpi
substytucja.
W tym przypadku mogą być dwa efekty: mutacja zmiany
sensu i mutacja typu nonsens.
Mutacja zmiany sensu wystąpi, gdy jeden kodon
oznaczający aminokwas zostanie zamieniony na drugi
kodon, również oznaczający aminokwas. Na przykład w
wyniku zmiany tripletu TTC na CTC zamiast lizyny
wbudowany zostanie kwas glutaminowy.

INSERCJA (ADDYCJA) I DELECJA
Mutacje typu nonsens polegają na zmianie tripletu
kodującego jakiś aminokwas, na jeden z trzech kodonów
nonsensownych (terminalnych).
Wówczas zamiast polipeptydu kodowanego przez dany
gen powstaje krótszy, w zależności od miejsca mutacji,
fragment polipeptydu.
Jako przykład może służyć triplet TTC kodujący lizynę,
który w wyniku substytucji ATC staje się kodonem stop.
Na skutek delecji lub insercji trzech lub wielokrotności
trzech par nukleotydów następuje ubytek lub włączenie
nowych aminokwasów cło syntetyzowanego łańcucha
polipeptydowego.
Mutacje nonsensowne i zmiany fazy odczytu prowadzą
zwykle do całkowitej utraty aktywności enzymu

MUTACJE
CHROMOSOMOWE
Zmiany genetyczne w komórce mogą dotyczyć
nie tylko pojedynczego genu, ale także grupy
genów (odcinka chromosomu), jak również
całego chromosomu lub kilku chromosomów.
Anomalie chromosomowe nazywane są także
aberracjami chromosomowymi.
Dzielimy je na strukturalne i liczbowe.
Do najlepiej poznanych czynników
powodujących mutacje chromosomowe należy
promieniowanie jonizujące oraz wiele
substancji chemicznych, zwłaszcza tych, które
wywierają działanie cytostatyczne.

MUTACJE CHROMOSOMOWE
STRUKTURALNE
Przyczyną powstania aberracji strukturalnych jest
najczęściej przerwanie ciągłości jednego lub kilku
chromosomów, nazywane także złamaniem
chromosomu.
Ułamany fragment, nie posiadając centromeru,
może być wyeliminowany z komórki w czasie jej
podziału. Możliwe jest także połączenie tego
fragmentu z chromosomem macierzystym lub z
innym.
Wyróżnia się następujące anomalie strukturalne:
delecja, duplikacja, inwersja, translokacja,
chromosom kolisty, izochromosom.

Delecja
Delecją nazywamy utratę odcinka chromosomu.
Gdy utrata dotyczy części dystalnej chromosomu
określa się ją jako delecję terminalną, a gdy fragment
środkowy — delecją interstycjalną.
Ułamany fragment chromosomu zostaje z reguły
utracony w czasie podziału komórki, ponieważ brak
centromeru uniemożliwia przyczepienia się do niego
włókna wrzeciona podziałowego podczas metafazy.
Skutki genetyczne delecji wynikają z utraty informacji
genetycznej oraz ze zmian ilościowych w genomie.
Osobniki z drobnymi delecjami mogą być żywotne,
podczas gdy z utratą dużych fragmentów chromosomów
giną wcześnie, nawet jeżeli są heterozygotami.

Duplikcja
Ta anomalia strukturalna polega na powtórzeniu tych
samych odcinków chromosomów leżących obok siebie
lub w innym miejscu chromosomu.
Najczęstszą przyczyną duplikacji są translokacje
fragmentów jednego chromosomu na drugi,
homologiczny.
Może to nastąpić podczas nieprawidłowego przebiegu
crossing-over.
Duplikacje prawdopodobnie odegrały istotną rolę w
procesach ewolucyjnych.
Przykładem mogą być geny białek hemoglobiny. Nowe
rodzaje łańcuchów hemoglobinowych człowieka
powstały na skutek duplikacji pierwotnych genów.
W wyniku licznych mutacji stały się one zupełnie
nowymi, kodującymi odrębne białka hemoglobinowe.

Inwersja
Jest efektem dwóch pęknięć jednego
chromosomu i połączenia jego wolnych
końców w odwrotnym kierunku.
Mimo, że w tym chromosomie znajduje
się ten sam materiał genetyczny
(aberracja zrównoważona), efekt
fenotypowy może być inny, na skutek
innej lokalizacji genów (tzw. efekt
pozycji).

Translokacja
Jest rodzajem aberracji polegającym na przemieszczeniu
fragmentu chromosomu w inne miejsce tego samego
chromosomu (translokacja intrachromosomalna lub
wewnętrzna), innego chromosomu (translokacja
interchromosomalna, transpozycja), wzajemnej
wymianie odcinków między chromosomami
niehomologicznymi (translokacja wzajemna lub
wymienna), jak również połączeniu się dwóch
chromosomów.
Translokacja może zachodzić także między
chromosomami homologicznymi (translokacja
siostrzana). Prowadzi to do intrachromosomalnych
duplikacji w jednym z chromosomów homologicznych
przy jednoczesnej delecji translokowanego odcinka w
drugim.

- Chromosom kolisty
Jest efektem połączenia nowych
zakończeń po uprzedniej delecji obu
końców chromosomu.
Przyczyną objawów chorobowych u
człowieka nie jest w tym przypadku
nieprawidłowy kształt chromosomu,
szczególnie, że u niektórych gatunków
chromosomy koliste są regułą.
Zespoły chorobowe są efektem utraty
fragmentów chromosomów.

Izochromosom
Powstaje w wyniku nieprawidłowego,
poprzecznego podziału centromeru w
chromosomie.
Izochromosom składa się z połączonych
ramion krótkich lub długich.
Powoduje to brak genów zawartych w
utraconych ramionach oraz podwójną ilość w
ramionach, które zostały podwojone.
Izochromosomy spotyka się w zespołach
chorobowych powstających w następstwie
mutacji autosomów oraz heterochromosomów.

MUTACJE CHROMOSOMOWE
LICZBOWE (GENOMOWE)
Przyczyną tego typu mutacji jest brak rozdziału
chromosomów (non-disjunction) podczas podziału
komórki.
W zależności od liczby chromosomów wyróżnia się
euploidię i aneuploidię.
- Euploidia. W tej aberracji występuje zwielokrotniony
cały podstawowy genom (np. 3n - triploid, 4n tetraploid
itd.). Wyróżnia się dwie kategorie poliploidów:
autopoliploidy i allopoliploidy (amfiploidy).
U autopoliploidów zespół chromosomów jest
zwielokrotniony o ten sam zestaw chromosomów
homologicznych.
Natomiast allopoliploidy mają dwa lub więcej
niehomologicznych zespołów chromosomów.

MUTACJE CHROMOSOMOWE
LICZBOWE (GENOMOWE)
Poliploidia (w formie triploidii) we
wszystkich komórkach organizmu
człowieka była stwierdzana tylko u
spontanicznie poronionych płodów.
Triploidie są przyczyną poronień
samoistnych (około 20%).
Dotychczas nie opisano u człowieka
monoploidii (1n).
Natomiast u wielu roślin, szczególnie
uprawnych, poliploidia jest zjawiskiem
bardzo częstym.

Aneuploidia
W tym przypadku liczba chromosomów u danego
osobnika nie jest wielokrotnością „n”.
Aneuploidia może polegać na braku bądź
obecności dodatkowego chromosomu
(chromosomów).
Organizm, u którego jest brak jednego
chromosomu jest nazywany monosomikiem.
Obecność jednego dodatkowego chromosomu
określa się jako trisomię.
Jeśli występują dwa dodatkowe chromosomy
homologiczne, jest to tetrasomia
Aneuploidia może dotyczyć zarówno autosomów
jak i heterochromosomów.

RODZICIELSKA DISOMIA
Jest to sytuacja, gdy dany osobnik
posiada dwa chromosomy jednej
pary pochodzące tylko od jednego
rodzica, a nie po jednym
chromosomie od każdego z
rodziców.

PIĘTNO GENOMOWE
W przedstawionych prawach i prawidłowościach
dotyczących dziedziczenia, materiał genetyczny
każdego z rodziców był „równocenny”.
Badania wykazały, że w niektórych przypadkach
materiał genetyczny wykazuje zróżnicowanie
ekspresji w zależności od pochodzenia
(matczynego lub ojcowskiego).
Prawdopodobną przyczyną tego stanu jest różna
metylacja DNA fragmentów regulacyjnych
podczas spermatogenezy i oogenezy.

ANTYCYPACJA
Zjawisko polegające na
wcześniejszym ujawnianiu się i
cięższej postaci choroby w
kolejnych pokoleniach.
W wielu przypadkach związane jest
to z mutacjami dynamicznymi.

Genetyka człowieka
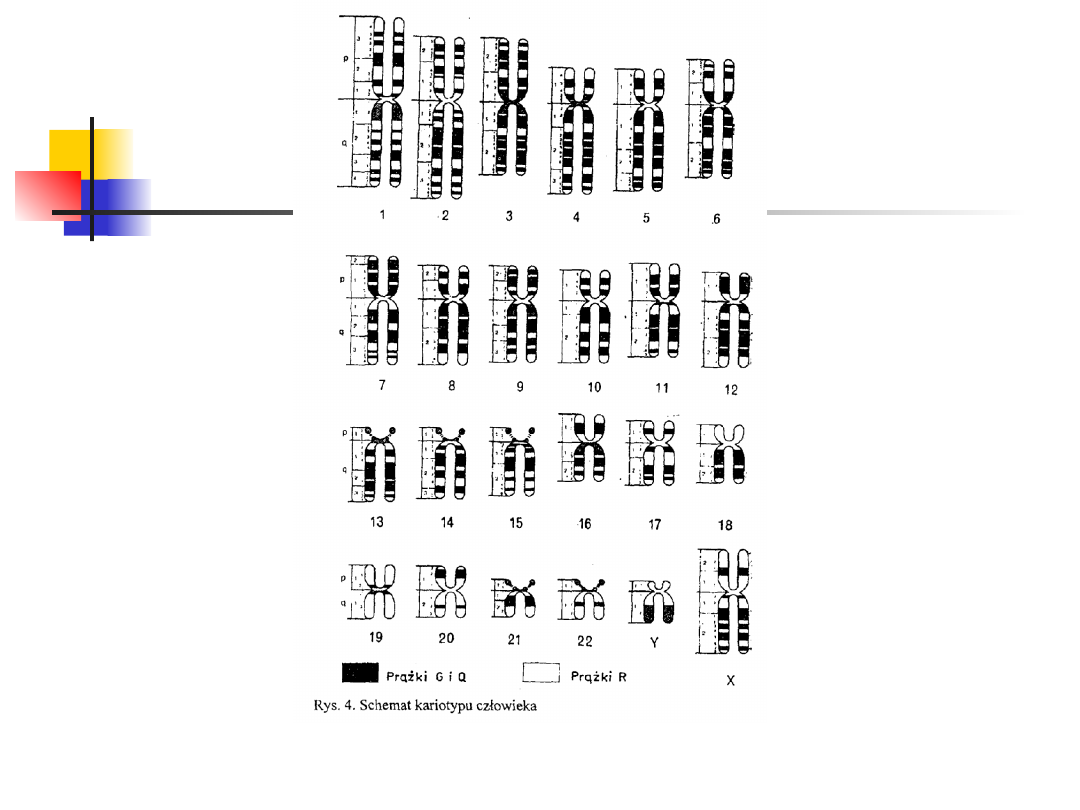
Kariotyp jest to zestaw chromosomów typowy dla danego
osobnika, grupy osobników lub gatunku, natomiast
kariogram jest obrazem zespołu chromosomów jednej
komórki uszeregowanych systematycznie według ich
długości i położenia centromeru.
Pod względem położenia centromeru wyróżnia się
chromosomy:
metacentryczne (centromer znajduje się w środku
chromosomu),
submetacentryczne (centromer jest przesunięty w kierunku
jednego końca),
akrocentryczne (wyraźne przesunięcie centromeru; wyróżnia
się ramiona długie oznaczone literą „q” oraz krótkie,
oznaczone literą „p”)
telocentryczne (centromer znajduje się na końcu
chromosomu; brak jednych ramion)
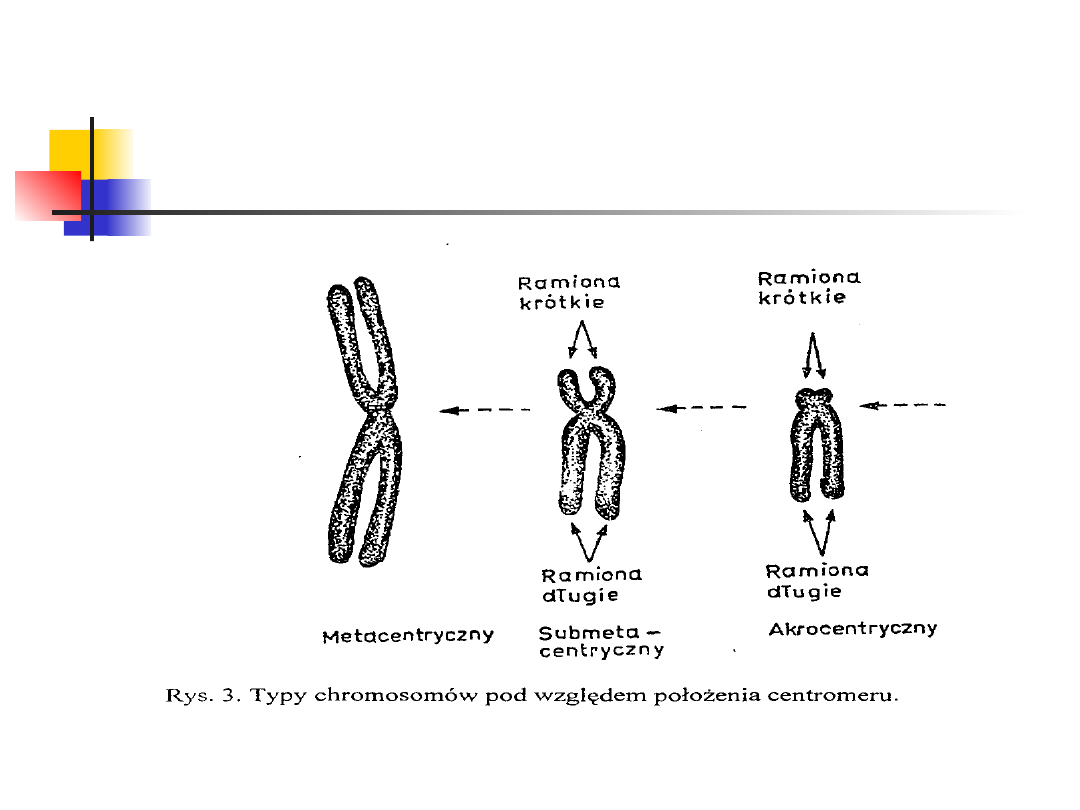
Typy chromosomów

Typy chromosomów
U człowieka występują tylko trzy pierwsze typy
chromosomów. Dla ujednolicenia oceny badań
cytogenetycznych przyjęto jednolity system klasyfikacji
chromosomów.
Jako kryteria klasyfikacji przyjęto:
- wielkość chromosomów w stosunku do innych
chromosomów tej samej komórki określoną na podstawie
wartości względnej. Względną długość chromosomu określa
się przyjmując za 1000 sumę długości chromosomów
jednego całego zespołu haploidalnego;
- położenie centromeru;
- rozmieszczenie prążków w chromosomach.
Na początku lat siedemdziesiątych opracowano metody
wyodrębniania w chromosomach prążków pozytywnych
(barwiących się intensywnie) i negatywnych (barwiących się
słabiej).

Typy chromosomów
W oparciu o przedstawione wyżej kryteria, u człowieka
wyodrębniono pary autosomów od 1 do 22.
Chromosomy płciowe wyodrębniono jako osobną parę.
Autosomy podzielono na siedem grup od A do G (I - VII),
zaś chromosomy płciowe oznaczono przez X i Y .
Grupa I. Należą do niej duże chromosomy
metacentryczne. Są to pary 1, 2, 3. Ich względna
długość waha się w granicach 90 — 72.
Grupa II. Należą do niej chromosomy
submetacentryczne pary 4 i 5. Względna długość 64 —
60.
Grupa III. W tej grupie występują chromosomy
submetacentryczne średniej wielkości. Są nimi:
chromosom X oraz autosomy par od 6 do 12. Względna
długość 59—43.

Typy chromosomów
Grupa IV. Należą do niej chromosomy akrocentryczne,
średniej wielkości. Należą tu autosomy par od 13 do 15.
Względna długość chromosomów te grupy wynosi 36 —
35. Chromosomy 13 pary mają wyraźne satelity.
Chromosomy pary 14 mają satelity znacznie mniejsze.
Grupa V. Należą tu małe chromosomy. Chromosomy 16
pary są metacentryczne, natomiast chromosomy 17 i 18
pary — submetacentryczne. Względna długość
chromosomów tej grupy wynosi 33 — 27.
Grupa VI. Należą do niej małe chromosomy
metacentryczne z par 19 i 20. Ich względna długość
wynosi 26 — 22.
Grupa VII. Należą do niej bardzo małe chromosomy
akrocentryczne par 21 i 22. Ich względna długość wynosi
20 — 12. Chromosom płciowy Y jest akrocentryczny i jego
względna długość wynosi 22— 11.

ZASADY ZAPISYWANIA
KARIOTYPÓW
Zgodnie z obowiązującym systemem zapisu wyników
cytogenetycznych zapisuje się całkowitą liczbę chromosomów
danego kariotypu, a następnie po przecinku, chromosomy
płciowe. Zapis prawidłowego kariotypu męskiego wygiąda
następująco: 46,XY. Natomiast żeńskiego 46,XX.
Jeśli nieprawidłowość dotyczy liczby chromosomów płciowych
wówczas dodaje się odpowiedni symbol w przypadku
chromosomów dodatkowych (np. 47,XXY; 47,XXX itp.) lub
opuszcza się w zapisie jego symbol w przypadku braku (np.
45,X). Mozaicyzm chromosomowy określa się symbolem „mos”
lub dzieli się poszczególne kariotypy ukośnymi kreskami (np.
46,XY/47,XXY).
Zmiany dotyczące liczby autosomów oznacza się znakiem (+)
lub (-) przed jego symbolem (np. 47,XX,+21 co oznacza płeć
żeńską, trisomia chromosomu 21). Zmiany strukturalne części
chromosomu określa się odpowiednim znakiem.

ZASADY ZAPISYWANIA
KARIOTYPÓW
Na przykład zapis 46,XX,del(5p) oznacza osobnika
żeńskiego u którego stwierdzono delecję ramienia
krótkiego 5 pary chromosomów.
Izochromosom oznacza się literą „i”, chromosom kolisty
(pierścieniowy) — literą „r”.
Zapis 46, XY, t(Bp-,Dq+) oznacza, że fragment krótkich
ramion chromosomu z grupy B przemieścił się na
ramiona długie chromosomu z grupy D.
Natomiast jeśli w wyniku translokacji powstaje nowy
chromosom, np. przez połączenie chromosomów z
grupy D i 0, zapis wygląda następująco:
45,XY,-D,-G,+t(Dq,Gq). Oznacza to, że w zestawie brak
jest po jednym chromosomie z grupy D i G, natomiast
powstał nowy chromosom DqGq (przykład translokacji
robertsonowskiej).

Choroby genetyczne
Choroby genetyczne definiuje się jako
upośledzające sprawność życiową odchylenia od
stanu prawidłowego (statystycznej normy),
które są
przekazywane jako cecha dziedziczna z
pokolenia na pokolenie,
albo powstają de novo na skutek zmian i
zaburzeń w mechanizmach przekazywania cech
dziedzicznych.
Powstałe de novo zmiany mogą być
przekazywane jako cecha (choroba) dziedziczna.

Choroby genetyczne
Podział chorób genetycznych opiera się na
podstawowych prawach dziedziczenia. Wyróżnia się:
1) choroby monogenowe (jednogenowe), które
można podzielić na autosomowe i sprzężone z płcią
warunkowane allelem recesywnym lub
dominującym,
2) wielogenowe, które nie są przekazywane zgodnie
z prostym dziedziczeniem według schematu Mendla.
Bardzo często objawy tych chorób uzależnione są od
czynników środowiska, stąd często choroby te określa się
jako wieloczynnikowe,
3) choroby chromosomalne.

ZABURZENIA
MONOGENOWE
Ten typ zaburzeń jest efektem mutacji w jednym
lub obydwu allelach genu na chromosomie
autosomowym, płciowym lub genu
mitochondrialnego.
Choroby te dziedziczą się w sposób podobny do
opisanego przez Mendla dziedziczenia barwy
groszku.
Dotychczas opisano u człowieka ponad 6 000
chorób będących efektem zaburzenia
monogenowego.
Choroby monogenowe mogą być uwarunkowane
allelem recesywnym lub dominującym.

ZABURZENIA
AUTOSOMOWE
RECESYWNE
Cechą charakterystyczną tego typu dziedziczenia
jest taka sama częstość występowania tej choroby u
kobiet i u mężczyzn.
Choroba ujawni się tylko u homozygot recesywnych.
W rodowodach przypadki choroby mają układ
„poziomy”, czyli ujawniają się wśród dzieci rodziców
będących nosicielami.
Jeśli rodzice są nosicielami zmutowanego genu
(heterozygoty), wówczas prawdopodobieństwo
wystąpienia choroby u dzieci wynosi 25%.
Natomiast u 75% potomstwa choroba nie ujawni
się.

ZABURZENIA
AUTOSOMOWE
RECESYWNE
Jednak tylko 25% będzie homozygotami
dominującymi, a 50% heterozygotami
(czyli nosicielami nieprawidłowego allelu).
Osoby heterozygotyczne mogą
oczywiście przekazać nieprawidłowy allel
swoim dzieciom (prawdopodobieństwo
wynosi w tym przypadku 50%).
Opisano wiele chorób dziedziczonych jako
autosomowe recesywne.

Zaburzenia przemiany
aminokwasów
- Fenyloketonuria.
Gen warunkujący tę chorobę jest zlokalizowany na
chromosomie l2q. Występuje z częstością I na 10 000
urodzeń.
Dzieci rodzą się bez szczególnych zaburzeń, jednak
w ciągu pierwszych sześciu miesięcy życia zaznacza
się wyraźne zahamowanie rozwoju, a w wieku 4 — 5
lat większość chorych jest głęboko upośledzona
umysłowo.
Chorzy wykazują charakterystyczne zaburzenia
barwnikowe. Skóra jest jasna z tendencją do
wyprysku, włosy jasnoblond z odcieniem szarawym.
Mocz i pot mają „mysi” zapach (kwas fenylooctowy).

Zaburzenia przemiany
aminokwasów
Fenyloketonuria
Stwierdza się także wzrost napięcia
mięśniowego, wygórowanie odruchów, a
także często napady padaczkowe.
Zaburzenie charakteryzuje się podwyższoną
zawartością fenyloalaniny we krwi oraz
wydalaniem fenyloalaniny i niektórych jej
pochodnych z moczem.
Defekt metaboliczny w fenyloketonurii
spowodowany jest syntezą nieprawidłowego
enzymu, hydroksylazy fenyloalaninowej ,
przekształcającego fenyloalaninę w tyrozynę.

Zaburzenia przemiany
aminokwasów
Fenyloketonuria
Pierwotnym skutkiem defektu jest zwiększenie
zawartości fenyloalaniny w organizmie i
zmniejszenie zawartości tyrozyny. Badania
biochemiczne pozwalają na wykrycie
fenyloketonurii u noworodków.
Wykluczenie mleka z diety noworodka jako
źródła fenyloalaniny i podawanie hydrolizatów
białkowych o małej zawartości tego
aminokwasu, zapobiega występowaniu
objawów chorobowych

Zaburzenia przemiany
aminokwasów
Albinizm.
Częstość występowania I na 40 000.
Jest wynikiem braku enzymu tyrozynazy, czego
efektem jest zaburzenie syntezy melaniny.
Skóra jest różowoczerwona i nie opala się po
wystawieniu na światło UV.
Włosy są białe, tęczówki niebieskie lub różowe
z zaznaczonym czerwonym połyskiem.
Ostrość wzroku jest zmniejszona.

Zaburzenia przemiany
aminokwasów
- Ketoacyduria (choroba syropu klonowego,
leucynoza).
Częstość występowania wynosi 0,004 na 1 000 żywo
urodzonych dzieci.
Przyczyną choroby jest brak aktywności enzymu
dekarboksylazy leucyny, odpowiedzialnego za
dekarboksylację takich aminokwasów jak walina,
leucyna i izoleucyna.
Skutkiem tego komórki nie metabolizują tych
aminokwasów.
Objawy choroby ustępują po podaniu dużych dawek
tiaminy, która spełnia funkcję koenzymu
niezbędnego do aktywacji enzymu.

Zaburzenia przemiany
aminokwasów
- Cytrulinemia.
Przyczyną tej choroby jest brak
aktywności enzymu syntetazy
arginino-bursztynianowej.
Jest możliwa do wykrycia w badaniach
prenatalnych za pomocą biopsji
trofoblastu.

Zaburzenia przemiany
węglowodanów
- Galaktozemia.
Gen zlokalizowany jest na chromosomie 9p.
Częstość występowania galaktozemii wynosi 0,025 na 1
000 urodzeń.
Objawami choroby jest spadek wagi ciała u noworodków,
powiększenie wątroby, żółtaczka, wrażliwość na
zakażenia.
Może prowadzić do upośledzenia umysłowego.
Przyczyną zaburzeń jest brak aktywności jednego z
enzymów niezbędnych w przemianie galaktozy:
urydililotransferazy heksozo-1 -fosforanowej
Możliwa jest diagnostyka prenatalna tego zaburzenia, a
zastosowanie odpowiedniej diety przez matkę umożliwia
urodzenie zdrowego dziecka.

Zaburzenia przemiany
lipidów
Najbardziej charakterystycznymi objawami tych
schorzeń są zaburzenia ośrodkowego układu
nerwowego.
- Choroba Taya i Sachsa.
Gen jest zlokalizowany na chromosomie 15.
Choroba często występująca u Żydów
Aszkenazyjskich (1 na 3 600 urodzeń).
Jej przyczyną jest brak enzymu 3-heksozaminidazy
A.
Powoduje to gromadzenie się gangliozydów w
komórkach nerwowych. Objawami są postępujące
nieprawidłowości neurologiczne od późnego
niemowlęctwa. Śmierć następuje w wieku 3 - 4 lat.

Zaburzenia przemiany
lipidów
Choroba Niemanna I Picka.
Przyczyną tej choroby jest brak aktywności enzymu
sfingomielinazy.
Efektem bloku jest odkładanie sfingomielin w różnych
narządach.
normalna długość życia.
- Choroba Gauchera.
Gen jest zlokalizowany na chromosomie 1.
Choroba często występuje u Żydów Aszkenazyjskich.
Przyczyną defektu jest brak aktywności
enzymatycznej 3-D-glukozydazy.

Zaburzenia przemiany
lipidów
Choroba Gauchera (c.d.)
Skutkiem tego następuje odkładanie
glukocerebrozydów w komórkach układu
siateczkowo-śródbłonkowego i układu
nerwowego.
Postać dziecięca ostra jest postępującą chorobą
neurologiczną z powiększeniem wątroby i
śledziony.
Śmierć następuje w wieku 1 — 2 lat.
Postać przewlekła typu dorosłego powoduje bóle
kostne i powiększenie śledziony. W tej postaci
choroby, przy leczeniu podtrzymującym

Inne choroby dziedziczone w
sposób autosomalny, recesywny
- Mukowiscydoza (fibrosis cistica CF, zwyrodnienie
torbielowate trzustki).
Lokalizacja genu na chromosomie 7q.
Częstość występowania 0,5 na 1 000 urodzeń.
Jest to jedna z najczęściej spotykanych chorób
metabolicznych rasy białej.
Cechą charakterystyczną tej choroby jest dysfunkcja
gruczołów zewnątrzwydzielniczych, co powoduje
uszkodzenie płuc, trzustki i niektórych narządów jamy
brzusznej oraz gruczołów potowych.
Objawia się w różnych postaciach i nie jest do końca
poznany czynnik (czynniki) odpowiedzialny za zmiany
chorobowe.

Inne choroby dziedziczone w
sposób autosomalny, recesywny
- Zwyrodnienie wątrobowo-soczewkowe.
Częstość choroby I na 20 000 urodzeń.
Początek choroby może ujawniać się w wieku
młodzieńczym lub później.
Zaburzenie jest spowodowane defektem przemiany
miedzi ze zmniejszonym stężeniem ceruloplazminy w
surowicy.
Powoduje to podwyższenie zawartości miedzi w wątrobie
oraz jej odkładanie w mózgu.
Choroba nie leczona prowadzi do przewlekłego zapalenia
wątroby i następstw neurologicznych, natomiast u osób
leczonych — normalna długość życia.
Wyłączenie miedzi z diety lub wzmożone jej usuwanie z
organizmu hamuje proces chorobowy.

Dziedziczenie autosomalne,
dominujące
W tym typie dziedziczenia choroba
ujawnia się zarówno u homozygoty
dominującej jak i heterozygoty.
Podobnie jak w przypadku dziedziczenia
autosomowego recesywnego, stwierdza
się taką samą częstość choroby i
ciężkość jej przebiegu u osób obu płci.
Obserwuje się bardzo zmienną ekspresję
genu, nawet z możliwością
„przeskoczenia” jednego pokolenia.

Dziedziczenie autosomalne,
dominujące
- Achondroplazja.
Występuje z częstością 1 na 26 000 urodzeń.
Charakteryzuje się pełną penetracją i małą
zmiennością ekspresji.
Objawami tej choroby są krótkie kończyny,
szczególnie odcinki proksymalne, natomiast
tułów jest normalnej długości z lordozą
lędźwiową.
Przeciętny wzrost dorosłego mężczyzny wynosi
132 cm, kobiety 123 cm.
Współczynnik inteligencji i długość życia w
normie.

Dziedziczenie autosomalne,
dominujące
Pląsawica Huntingtona
. Częstość występowania waha się od 1 na
333 000 w Japonii do 1 na 5 740 w Tasmanii.
Ekspresja genu zależy od wieku.
Początek choroby najczęściej jest
obserwowany w czwartej dekadzie życia.
Objawia się zaburzeniami psychicznymi,
postępującą pląsawicą i otępieniem.
Prowadzi to do postępującej inwalidyzacji.
Śmierć następuje w 10—12 lat od początku
choroby.

Dziedziczenie autosomalne,
dominujące
- Hipercholesterolemia rodzinna.
Częstość występowania choroby 1 na 500.
Prawdopodobnie objawy chorobowe są skutkiem
braku lub defektów receptorów wiążących
lipoproteiny o małej gęstości (LDL).
Pierwsze oznaki defektu pojawiają się na początku
trzeciej lub czwartej dekady życia, m.in. w postaci
niedokrwiennej choroby serca.
Homozygoty dominujące mają bardzo wysokie
stężenie cholesterolu, a objawy wieńcowe mogą
pojawiać się już w wieku dziecięcym.
Jeśli choroba nie jest odpowiednio wcześnie
rozpoznana i leczona, prawie połowa chorych umiera
przedwcześnie z powodu choroby niedokrwiennej
serca (przed 60 rokiem życia).

Dziedziczenie autosomalne,
dominujące
- Zespól Marfana.
Częstość występowania choroby wynosi 1 na
10 000, z czego 25% stanowią nowe mutacje.
Objawia się arachnodaktylią, długimi
kończynami, zmniejszonym stosunkiem
długości tułowia do kończyn dolnych oraz
skłonnościami do powikłań.
W następstwie tych zmian pojawia się m.in.
skolioza oraz tętniak aorty.
Średni wiek przeżycia wynosi 40 — 50 lat.

Dziedziczenie autosomalne,
dominujące
Glejak siatkówki (retinoblastoma).
Częstość występowania wynosi 1 na 18 000.
Pierwsze objawy pojawiają się w ciągu dwóch
pierwszych lat życia m.in. w postaci białego refleksu
źrenicznego lub zeza (w 20 — 30% obustronnego).
Jeżeli guz jest mały i jednostronny, to w 90%
przypadków choroba jest uleczalna.
W pozostałych przypadkach rozwija się kolejny
pierwotny guz, szczególnie mięsak kościopochodny w
dzieciństwie, czerniak złośliwy skóry lub raki
pęcherza, płuc lub trzustki u dorosłych pacjentów.

DZIEDZICZENIE SPRZĘŻONE Z
CHROMOSOMEM X, RECESYWNE
Dystrofia mięśniowa Duchenne’a.
Częstość występowania u chłopców 1 na 3 000.
Lokalizacja genu na chromosomie Xp22.l.
Przyczyną choroby jest delecja w genie DMD.
Choroba ujawnia się we wczesnym dzieciństwie
postępującym osłabieniem mięśni
proksymalnych i pozornym przerostem łydek.
W dalszym przebiegu prowadzi do lekkiego
upośledzenia umysłowego, inwalidztwa około
10 roku życia i śmierci około 20 roku życia.

DZIEDZICZENIE SPRZĘŻONE Z
CHROMOSOMEM X, RECESYWNE
- Dystrofia mięsniowa Beckera.
Lokalizacja genu na chromosomie Xp22.l.
Objawy podobne jak w przypadku dystrofii
mięśniowej Duchenne”a, ale rozpoczynają
się w późnym dzieciństwie.
Często inwalidztwo około 25 lat od
rozpoczęcia choroby, natomiast długość
życia może być normalna.

DZIEDZICZENIE SPRZĘŻONE Z
CHROMOSOMEM X, RECESYWNE
Zespól jąder feminizujących (zespół
niewrażliwości na androgeny).
Częstość wśród osobników z chterochromosomami
XY - 1 na 62 400.
Lokalizacja genu na chromosomie Xp11-q11.
Skutkiem mutacji jest defekt komórek wiążących
testosteron i dihydrotestosteron.
Objawia się fenotypem kobiecym z normalnie
rozwiniętymi piersiami, ale z pierwotnym brakiem
miesiączki, słabym owłosieniem wzgórka łonowego,
ślepą pochwą jądrami wewnątrz brzucha.
Kariotyp 46,XY.
Osoby te są bezpłodne, natomiast długość życia i
inteligencja normalne.

DZIEDZICZENIE SPRZĘŻONE Z
CHROMOSOMEM X, RECESYWNE
- Hemofilia A.
Gen jest zlokalizowany na chromosomie Xq28.
Skutkiem mutacji jest brak czynnika VIII, niezbędnego w
procesie krzepnięcia krwi.
Powoduje to nawracające krwotoki pooperacyjne i
samoistne do tkanek miękkich i stawów.
Przy leczeniu polegającym na podawaniu czynnika VIII
Niemal normalna długość życia.
- Hemofilia B.
Występuje z częstością 1 na 30 000 urodzonych
chłopców. Lokalizacja genu na chromosomie Xq27.1-
q27.2.
Mutacja powoduje w tym przypadku brak czynnika IX.
Podobnie jak w poprzednim typie hemofilii, leczenie
warunkuje prawie normalną długość życia.

DZIEDZICZENIE SPRZĘŻONE Z
CHROMOSOMEM X, RECESYWNE
- Ślepota na barwy (daltonizm).
Częstość występowania 80 na 1 000
urodzonych chłopców.
Lokalizacja genu na chromosomie
Xq28.
Skutkiem defektu genetycznego jest
synteza nieprawidłowego barwnika w
czopkach, co powoduje nie rozróżnianie
barwy czerwonej i zielonej.

Dziedziczenie mitochondrialne
W tym typie dziedziczenia informacja genetyczna jest
przekazywana wyłącznie od matki.
Stąd określane jest także jako tzw. dziedziczenie
matczyne.
Podstawową cechą dziedziczenia mitochondrialnego jest
specyficzność tkankowa i nasilanie objawów choroby wraz
z wiekiem.
Częstość powstawania mutacji w mtDNA jest
prawdopodobnie kilkanaście razy większa niż w DNA
jądrowym.
Przyczyną tego może być brak systemu naprawczego DNA
oraz istnienie tzw. wolnych rodników, które mogą
indukować mutacje.
W mtDNA stwierdza się występowanie wszystkich znanych
typów mutacji, ale najczęściej spotykane są delecje.

Dziedziczenie mitochondrialne
W komórce znajduje się kilkaset mitochondriów i
kilka tysięcy cząsteczek mtDNA.
Objawy kliniczne niektórych chorób ujawniają się
dopiero, gdy około 85% cząsteczek mtDNA ulegnie
mutacji.
Mutacje w genach mitochondrialnego DNA są
przyczyną wielu zwyrodnieniowych chorób układu
nerwowego, mięśniowego, nerek, gruczołów
wydzielania wewnętrznego.
Przykładami chorób warunkowanych tego typu
mutacjami mogą być:
padaczka miokloniczna, dziedziczny zanik nerwu
wzrokowego typu Lebera, zespół Pearsona, choroba
Leigh.

Dziedziczenie wieloczynnikowe
Ten typ dziedziczenia jest uwarunkowany przez wiele genów,
których efekt sumuje się (poligeny, geny kumulatywne,
addytywne). Każdy z tych genów z osobna ma mały wpływ
na fenotyp.
Na ujawnienie się wady wpływają w tym przypadku zarówno
geny, jak i środowisko.
Dlatego mówi się o dziedziczeniu wieloczynnikowym.
Sama skłonność (czyli genotyp) nie decyduje o wystąpieniu
choroby lub wady.
Dopiero współdziałanie określonych czynników
środowiskowych z wysoką predyspozycją genetyczną może
przejawić się wystąpieniem objawów choroby lub wadą.
Ryzyko wystąpienia choroby jest tym większe, im więcej
dany osobnik ma nieprawidłowych alleli oraz im więcej
działa na niego zewnątrzpochodnych czynników
szkodliwych.

Dziedziczenie wieloczynnikowe
Przykładami chorób dziedziczonych w ten
sposób mogą być:
rozszczep wargi i/lub podniebienia,
wrodzone zwichnięcie stawów biodrowych,
schizofenia,
cukrzyca typu I,
wrodzone wady serca, stopy końsko-szpotawe,
choroba Hirschsprunga,
wady ośrodkowego układu nerwowego
(bezczaszkowie, przepukliny rdzeniowe),
wrodzone zwężenie odźwiernika,
padaczka.

CHOROBY
CHROMOSOMALNE
Przyczyną tych chorób mogą być:
aberracje strukturalne
chromosomów,
dotyczące liczby chromosomów.
nieprawidłowości mogą dotyczyć
autosomów i heterochromosomów.

ZABURZENIA CHROMOSOMOWE
STRUKTURALNE
- Zespól Pradera-Williego.
Częstość występowania 1 na 10 000 urodzeń.
Przyczyną jest mikrodelecja w chromosomie
15 pochodzenia ojcowskiego [46,XX,del(15)(q
11 .2-q13) lub 46,XY,del( 15)(q 11 .2-q 13)].
U noworodków może występować obniżone
napięcie mięśniowe, twarz jest płaska, waga
górna uniesiona, hypogenitalizm.
W późniejszym wieku hipotonia ustępuje, ale
pojawia się otyłość i upośledzenie umysłowe.

ZABURZENIA CHROMOSOMOWE
STRUKTURALNE
- Zespół „cri du chat” (zespół ‚„miauczenia
kota”).
Częstość występowania 1 na 50 000
urodzeń.
Przyczyną jest delecja ramion krótkich
chromosomu 5 pochodzenia ojcowskiego.
Objawami klinicznymi są:
niedorozwój umysłowy, małomózgowie,
hipotonia, niskie osadzenie uszu, mała żuchwa
oraz wiele innych nieprawidłowości.

ZABURZENIA CHROMOSOMOWE
LICZBOWE
Zaburzenia liczby chromosomów mogą
polegać na zwielokrotnieniu liczby haploidalnej
podstawowego, diploidalnego garnituru
(np. 3n, 4n itd.) — tzw. poliploidia,
w przypadku aneuploidii, liczba chromosomów
nie stanowi prostej wielokrotności liczby
haploidalnej (np. 2n + 1, 2n — 1 itp.).
1. POLIPLOIDIA
Ten typ aberracji jest u człowieka zazwyczaj
cechą letalną.

TRIPLOIDIA
Triploidia (69,XXY lub 69, XXX) jest
przyczyną poronień przed ósmym
tygodniem życia płodowego.
Ten typ aberracji powstaje zazwyczaj w
wyniku zapłodnienia oocytu przez dwa
plemniki lub przez połączenie dwóch
gamet, z których jedna jest diploidalna.
Opisywano przypadki urodzenia żywych
osobników z mozaiką 69,XXY/46,XY.
We wszystkich przypadkach stwierdzano
niedorozwój fizyczny i umysłowy.

Tetraploidia. Aneuploidia
TETRAPLOIDIA
Tetraploidia (92,XXYY lub 92,XXXX) jest wynikiem braku
pierwszego podziału zygoty.
Płody z tetraploidią są jeszcze bardziej upośledzone niż
z triploidią.
Nawet jeśli urodzą się żywe, to tylko wyjątkowo
przeżywają pierwsze miesiące życia.
ANEUPLOIDIA
Aneuploidia należy do najczęściej występujących u
człowieka aberracji chromosomowych (1 na 200
noworodków).
Przyczyną tej aberracji może być nieprawidłowa gameta
żeńska lub męska
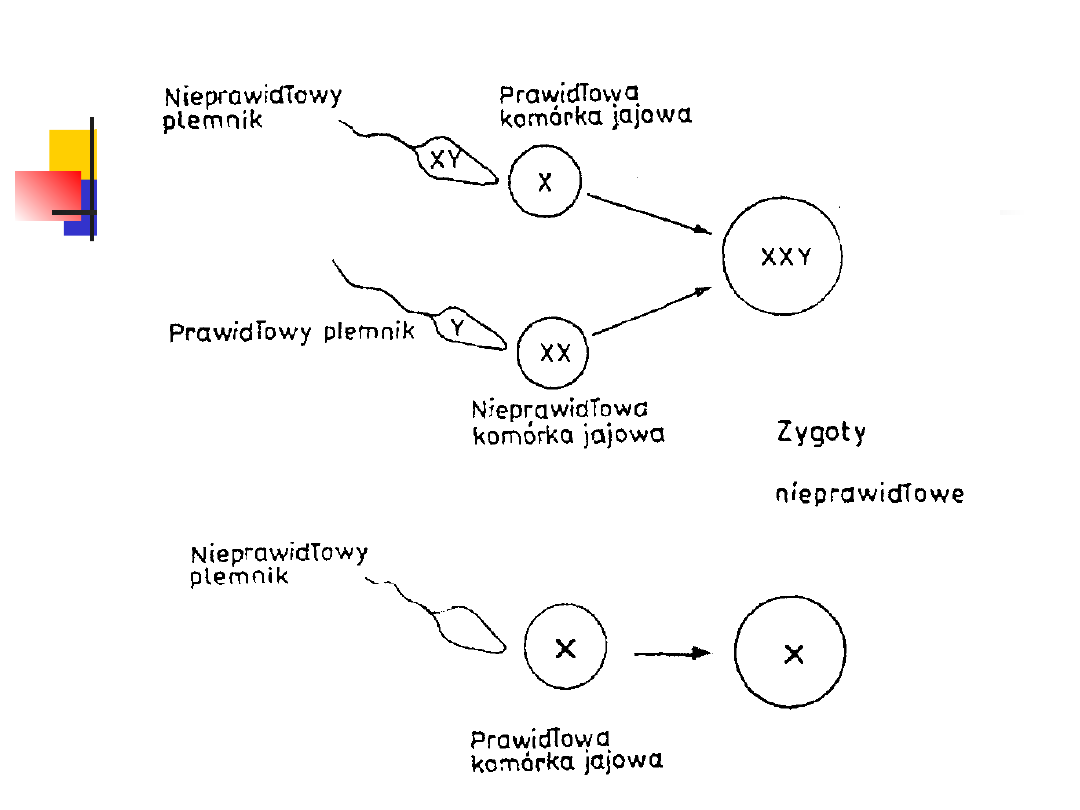

MONOSOMIA
Brak jednego chromosomu (2n — 1) w
garniturze chromosomowym może
dotyczyć zarówno autosomów jak i
heterochromosomów.
Jednak monosomie autosomów (45,XY
lub 45,XX) mają charakter letalny.
Jedyny przypadek osoby z monosomią,
która przeżywa, dotyczy
heterochromosomów.

MONOSOMIA
- Zespół Turnera.
Kariotyp takiej osoby jest 45,X.
Charakteryzuje się fenotypem żeńskim.
Objawami tego zespołu są: niski wzrost,
dysgenezja gonad, infantylność narządów
płciowych, często pierwotny brak miesiączki,
zwykłe bezpłodność.
Innymi wadami są: płetwistość szyi,
koślawość łokci, wady wrodzone układu
moczowo-płciowego, krążenia i inne.

TRISOMIE AUTOSOMALNE
- Zespół Patau.
Kariotyp 47,XX,+13 lub 47,XY,+13.
Występuje 1 na 10 000 żywych urodzeń.
Częstymi zaburzeniami jest niedorozwój mózgu (np.
holoprosencephalia, czyli brak płatów czołowych), wady
czaszki, rozszczep podniebienia, polidaktylia. Ponadto
stwierdza się wady wrodzone serca, niedorozwój
umysłowy, wady oczu, nisko osadzone i zdeformowane
uszy, głuchota, anomalie w układzie moczowo-
płciowym, przepuklina pępkowa.
Około 50% dzieci z tym zespołem umiera w pierwszym
miesiącu, a 90% - w pierwszym roku życia.

TRISOMIE AUTOSOMALNE
Zespół Edwardsa.
Kariotyp 47,XX,±18 lub 47,XY,+18.
Występuje z częstością 1 na 6 000 żywo urodzonych.
Objawami klinicznymi są: niska masa po urodzeniu,
zahamowanie rozwoju fizycznego, hipertonia mięśniowa,
opóźnienie rozwoju psychoruchowego, nieprawidłowa
budowa czaszki, nisko osadzone i zdeformowane uszy,
mała żuchwa.
Chorzy z tym zespołem żyją krótko. Zwykle umierają w
okresie niemowlęcym, około 30% umiera w pierwszym
miesiącu życia, a jedynie 10% dożywa I roku.
Sporadycznie chorzy ci mogą dożyć 15 lat.

TRISOMIE AUTOSOMALNE
- Zespól Downa. Kariotyp 47,XX,+21 lub 47,XY,+21.
Częstość występowania 1 na 800 urodzeń.
Objawami klinicznymi są:
zahamowanie wzrostu, niedorozwój umysłowy, dysmorfia
twarzy (krótkogłowie, płaska twarz i potylica, skośnogórne
szpary powiekowe, zmarszczka nakątna, małe uszy).
Ponadto występuje opóźnienie w rozwoju
psychomotorycznym jak również mogą wystąpić wrodzone
wady serca.
Innymi objawami, rzadziej występującymi, są: zaćma,
padaczka, niedoczynność tarczycy, ostra białaczka
Do śmierci w okresie niemowlęcym dochodzi wówczas, gdy
występują ciężkie wady rozwojowe serca.
W innych przypadkach długość życia może być tylko
nieznacznie skrócona.

TRISOMIE
HETEROCHROMOSOMALNE
Zespół Klinefeltera.
Kariotyp 47,XXY. Częstość występowania 1 na 1 000
urodzonych chłopców.
Zespół ten jest najczęstszą przyczyną hipogonadyzmu i
bezpłodności u mężczyzn.
Jądra, które z reguły są małe, wytwarzają zbyt mało
testosteronu. Powoduje to słaby rozwój wtórnych cech
płciowych oraz ginekomastię.
Kończyny są długie, a stosunek długości górnej części ciała
do dolnej jest zbyt mały.
Bardzo rzadko może wystąpić także skrzywienie boczne
kręgosłupa, rozedma płuc, cukrzyca, osteoporoza i rak
sutka.
Również rzadkim objawem jest upośledzenie umysłowe
znacznego stopnia.
Terapia testosteronem od wczesnego wieku młodzieńczego
poprawia rozwój wtórnych cech płciowych, jednak ludzie ci
w dalszym ciągu pozostają bezpłodni.

TRISOMIE
HETEROCHROMOSOMALNE
Zespół 47,XYY.
Występuje z częstością 1 na 1 000 urodzonych
chłopców.
To zaburzenie jest zazwyczaj bezobjawowe.
Czasami stwierdza się obniżony iloraz
inteligencji (o 10 — 15 punktów w porównaniu
ze zdrowym rodzeństwem) oraz tendencje do
zaburzeń zachowania (frustracja i agresja).
Najczęściej ludzi ci są wysocy, z prawidłowymi
proporcjami ciała, bez innych objawów
klinicznych.

TRISOMIE
HETEROCHROMOSOMALNE
- Zespół 47,XXX.
Występuje z częstością 1 na 1000 urodzonych
dziewczynek.
Najczęściej nie stwierdza się objawów
klinicznych. Rzadko może wystąpić lekkie
upośledzenie umysłowe.
Na częstość wystąpienia zaburzeń liczby
chromosomów w niektórych przypadkach ma
wiek rodziców, szczególnie matki.
Wraz z wiekiem matki wzrasta ryzyko
wystąpienia u dzieci zespołów: Downa, Patau,
Edwardsa, Klinefeltera, 47,XXX.

TRISOMIE
HETEROCHROMOSOMALNE
Istotne jest także, że w poszczególnych zespołach,
oprócz aneuploidii, stwierdza się również mozaikowość.
Na przykład w zespole Turnera, oprócz klasycznej
monosomii, obserwuje się czasami inne
nieprawidłowości: 45,X/46,XX; 45,X!46,XY; 45 „X!
47,XXX.
Obecność prawidłowej linii komórkowej (46,XX) łagodzi
objawy kliniczne.
W przypadku zespołu Klinefełtera, oprócz opisanej
trisomii, bardzo rzadko zdarzają się chorzy, których
kariotyp jest 48,XXXY lub nawet 49,XXXXY.
Ale w tym przypadku, każdy dodatkowy chromosom X
powoduje pogłębienie upośledzenia umysłowego.

PORADNICTWO GENETYCZNE
Poradnictwo genetyczne ma na celu przekazanie
informacji o chorobach dziedzicznych osobom z taką
chorobą lub zwiększonym ryzykiem wystąpienia tej
choroby w rodzinie.
Jest podstawowa metodą profilaktyki chorób
genetycznych.
Istnieją sytuacje, w których wskazany jest kontakt
rodziny z poradnią genetyczną.
Należą do nich:
urodzenie się dziecka z izolowaną wadą wrodzoną,
rozpoznanie u dziecka choroby o nieznanej etiologii,
w rodzinie występowała określona choroba genetyczna,
między małżonkami istnieje pokrewieństwo
podczas ciąży kobieta była narażona na działanie
czynników teratogennych.

PORADNICTWO GENETYCZNE
Proces poradnictwa genetycznego obejmuje m.in.
zebranie wywiadu, konstrukcję rodowodu, badanie
kliniczne, ustalenie rozpoznania oraz udzielenie porady.
Na tej podstawie osoba konsultowana powinna uzyskać
informacje o chorobie (rozpoznanie, powikłania,
rokowania itp.) oraz zrozumieć genetyczny charakter i
ryzyko jej wystąpienia u członków rodziny.
Osoba udzielająca porady genetycznej powinna
przedstawić różne możliwości (np. sztuczne
zapłodnienie lub diagnostyka prenatalna w następnej
ciąży) oraz zasugerować wybór najlepszej możliwości w
przypadku ryzyka nawrotu choroby.
Bardzo ważnym aspektem poradnictwa genetycznego
jest pomoc w przystosowaniu chorego do życia w
środowisku.

PORADNICTWO GENETYCZNE
Prawidłowe rozpoznanie choroby jest istotne dla
poradnictwa genetycznego, dlatego ten etap nie
powinien wyprzedzać żadnego z wcześniej
wymienionych.
Porada powinna być udzielona obojgu rodzicom.
Podczas udzielania porady należy omówić cechy
kliniczne choroby jej przebieg, powikłania,
rokowania i ewentualne leczenie.
Można przedstawić także zasady opieki nad chorym.
Należy natomiast przedstawić porównanie wielkości
przekazanego ryzyka wystąpienia choroby u dzieci z
wartością tego ryzyka w populacji ogólnej.

PORADNICTWO GENETYCZNE
Przyjęto w tym przypadku zasadę, że jeżeli
ryzyko jest ponad 10% - jest ono wysokie, gdy
poniżej 5% - niskie.
Należy także przedyskutować z osobami
zgłaszającymi się po poradę problemy
dotyczące posiadania i planowania potomstwa.
W poradnictwie genetycznym istotne jest, że
osoba udzielająca porady nie może rodzicom
niczego nakazywać, ani zakazywać.
Jej rola musi ograniczyć się tylko do
przedstawienia rzetelnych informacji.

BADANIA PRENATALNE
Podczas porady genetycznej należy
poinformować rodzinę, jeśli są ku temu
wskazania, że istnieje możliwość wykonania
badań prenatalnych.
Istnieje wiele technik badawczych stosowanych
w diagnostyce prenatalnej chorób genetycznych.
Jednak decyzję o wykonaniu tych badań
podejmują wyłącznie rodziny, a nigdy lekarz
udzielający porady.
Niektóre sytuacje są wskazaniem do
przeprowadzenia badań prenatalnych.

BADANIA PRENATALNE
Należą do nich m.in.:
wiek matki powyżej 35 lat,
kobieta urodziła już dziecko z aberracją chromosomów
jedno z przyszłych rodziców jest nosicielem
udokumentowanej aberracji chromosomowej,
w rodzinie istnieje wysokie ryzyko urodzenia dziecka z
chorobą metaboliczną itp.
Należy przy tym pamiętać, aby w poradnictwie
genetycznym odróżniać wady warunkowane
genetycznie od wad uwarunkowanych czynnikami
środowiskowymi (np. działanie czynników
teratogennych, zakażenia niektórymi wirusami lub
pasożytami, zażywanie podczas ciąży niektórych leków
itp.).

Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
Wyszukiwarka
Podobne podstrony:
Fizjologia cz II skr
Analiza śladów genetycznych jako dowód w procesie karnym – cz II
Genetyka cz I skr
Praktyczne zastosowanie genetyki w hodowli ryb akwariowych cz II
socjologia cz II
BADANIA DODATKOWE CZ II
Wykład 5 An wsk cz II
AUTOPREZENTACJA cz II Jak w
Podstawy Pedagogiki Specjalnej cz II oligo B
J Poreda Ewangelia zdrowia, cz II
mmgg, Studia PŁ, Ochrona Środowiska, Chemia, fizyczna, laborki, wszy, chemia fizyczna cz II sprawka
!Spis, ☆☆♠ Nauka dla Wszystkich Prawdziwych ∑ ξ ζ ω ∏ √¼½¾haslo nauka, hacking, Hack war, cz II
więcej podobnych podstron