
Właściwości koligatywne roztworów

Właściwości koligatywne roztworów
Obniżenie prężności par
Rozważamy roztwór złożony z rozpuszczalnika 1 i nielotnej
substancji 2. Ponieważ roztwór jest doskonałym roztworem
rozcieńczonym, rozpuszczalnik spełnia prawo Raoulta:
ponieważ x
1
= 1 – x
2
, czyli:
Stąd
ΔP jest obniżeniem prężności par roztworu w porównaniu z
czystym rozpuszczalnikiem.
1
1 1
*
P
x P
=
(
)
*
1
2
1
1
P
x P
= -
1
1
2 1
*
*
P
PΔP x P
-
=
=
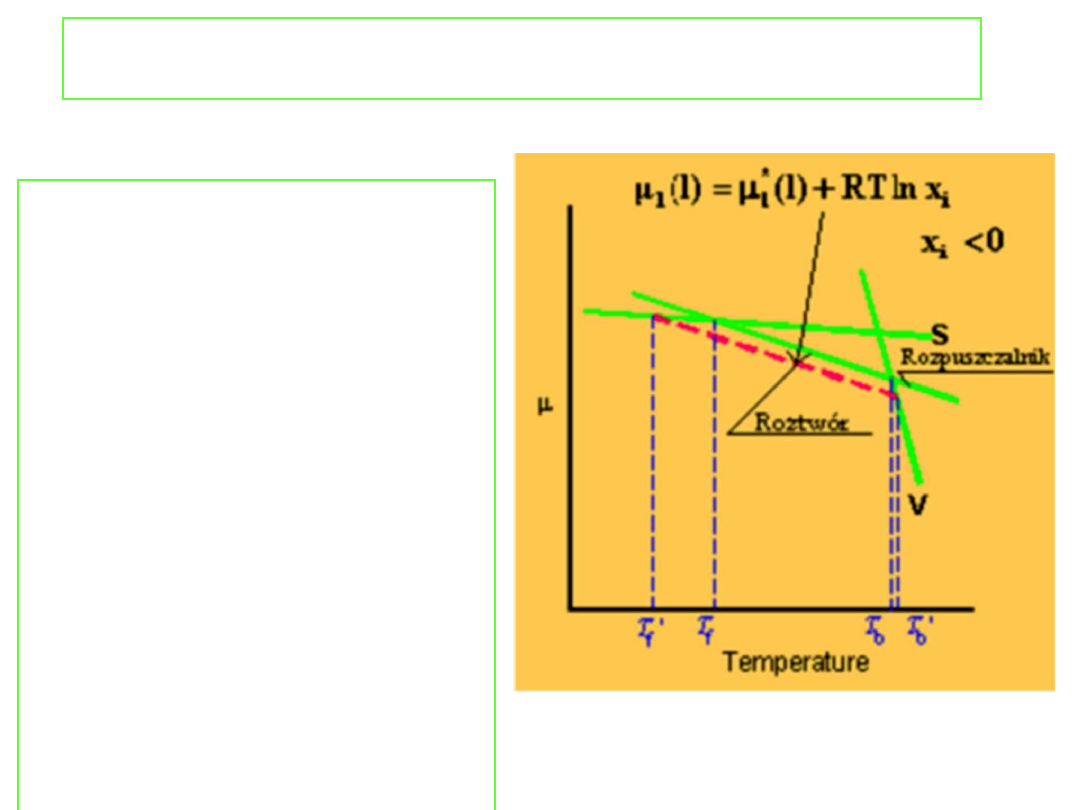
Podwyższenie temperatury
wrzenia
Punkt wrzenia jest
określony przez
temperaturę, w której
prężność par wrzącej
cieczy jest równa ciśnieniu
zewnętrznemu. Można
więc oczekiwać, że skoro
dodatek nielotnej
substancji rozpuszczonej
obniża prężność par
roztworów to również
temperatura wrzenia tego
roztworu musi ulec
podwyższeniu. Jakościowo
efekt ten przedstawia
rysunek:
Właściwości koligatywne roztworów

Właściwości koligatywne roztworów
Właściwości koligatywne roztworów
Dlatego
Na podstawie Gibbsa – Helmholtza
możemy zapisać
gdzie to molowa entalpia parowania
rozpuszczalnika z roztworu. Ponieważ roztwór jest
rozcieńczony możemy przyjąć, że wielkość ta jest równa
molowej entalpii parowania czystego rozpuszczalnika.
Z poprzedniego równania:
*
1
1
1
ln
par
(g)
(c)
Δ G
μ
μ
R x
T
T
-
=
=
2
G
d
H
T
dT
T
D
� �
� �
D
� �=-
1
2
ln
par
par
Δ G
d
T
Δ H
d( x )
R
dt
T
dT
�
�
�
�
�
� -
�
�=
=
par
H
D
1
2
ln
par
Δ H
d x
dT
RT
-
=

Po całkowaniu w granicach między x
1
= 1 czysty rozpuszczalnik,
któremu odpowiada T
w
a x
1
, ułamek molowy rozpuszczalnika w
roztworze, czemu odpowiada temperatura wrzenia roztworu T
w
’:
Właściwości koligatywne roztworów
dT
RT
H
Δ
x
d
x
'
T
T
par
w
w
ln
1
1
ln
ln
2
1
czyli
2
1
1
1
ln
w
par
w
w
w
w
par
w
w
par
T
ΔT
R
H
Δ
' T
T
T
'
T
R
H
Δ
T
'
T
R
H
Δ
x
przy założeniach, że
(1)
H
Δ
par
jest niezależna od temperatury
(2)
w
w
T
'
T
czyli
2
w
w
w
T
' T
T

Chcemy znaleźć zależność ΔT od x
2
ułamka molowego substancji rozpuszczonej
2
2
1
1
ln
ln
w
par
T
ΔT
R
H
Δ
x
x
Dla
2
,
0
2
x
...
3
2
1
ln
3
2
2
2
2
x
x
x
x
2
x
(dla x
2
« 1)
Stąd
2
2
x
H
Δ
RT
ΔT
par
w
Zmieniamy ułamek molowy na molalność (m
2
)
1
1
2
1
2
2
1
2
2
M
W
n
n
n
n
n
n
x
W
1
- masa rozpuszczalnika w kg
M
1
– masa molowa rozpuszczalnika
2
1
2
m
W
n
stąd x
2
= m
2
M
1

Właściwości koligatywne roztworów
A w ięc
2
1
2
m
H
Δ
M
RT
ΔT
par
w
Przyjm ując
H
Δ
M
RT
K
par
w
e
1
2
stała ebulioskopow a
otrzym ujem y
2
m
K
ΔT
e

Właściwości koligatywne roztworów
Obniżenie temperatury krzepnięcia
W identyczny, jak poprzednio sposób możemy wyprowadzić wzór na obniżenie
temperatury krzepnięcia roztworu
'
t
T
w stosunku do czystego rozpuszczalnika T
t
2
m
K
T
T
ΔT
k
'
t
t
k
K
stała krioskopowa
gdzie
k
K
stała krioskopowa dana jest wyrażeniem
H
Δ
M
RT
K
top
t
k
1
2
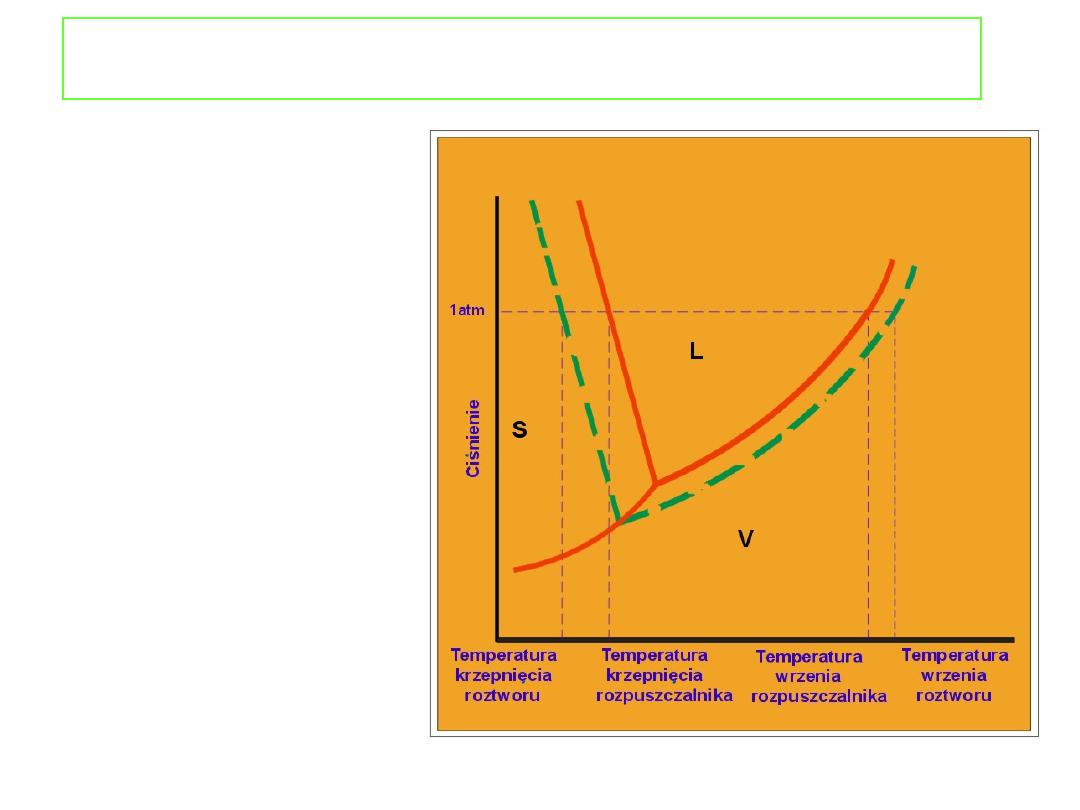
Właściwości koligatywne roztworów
Podwyższenie
temperatury wrzenia i
obniżenie temperatury
krzepnięcia może być
także przedstawione na
wykresie fazowym (np.
wody).
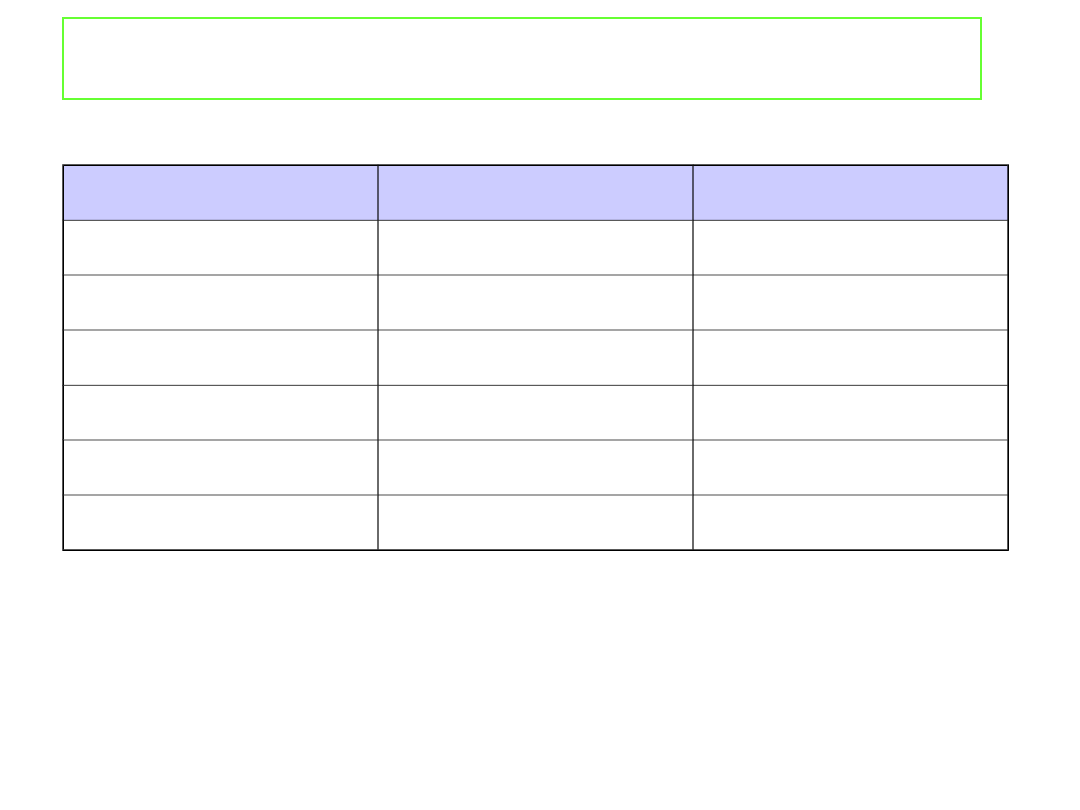
Właściwości koligatywne roztworów
Rozpuszczalnik
K
b
/K mol
-1
·kg
K
f
/K mol
-1
kg
H
2
O
0.51
1.86
C
2
H
5
OH
1.22
-
C
6
H
6
2.53
5.12
CHCl
3
3.63
-
CH
3
COOH
2.93
3.90
CCl
4
5.03
-
Właściwości koligatywne roztworów
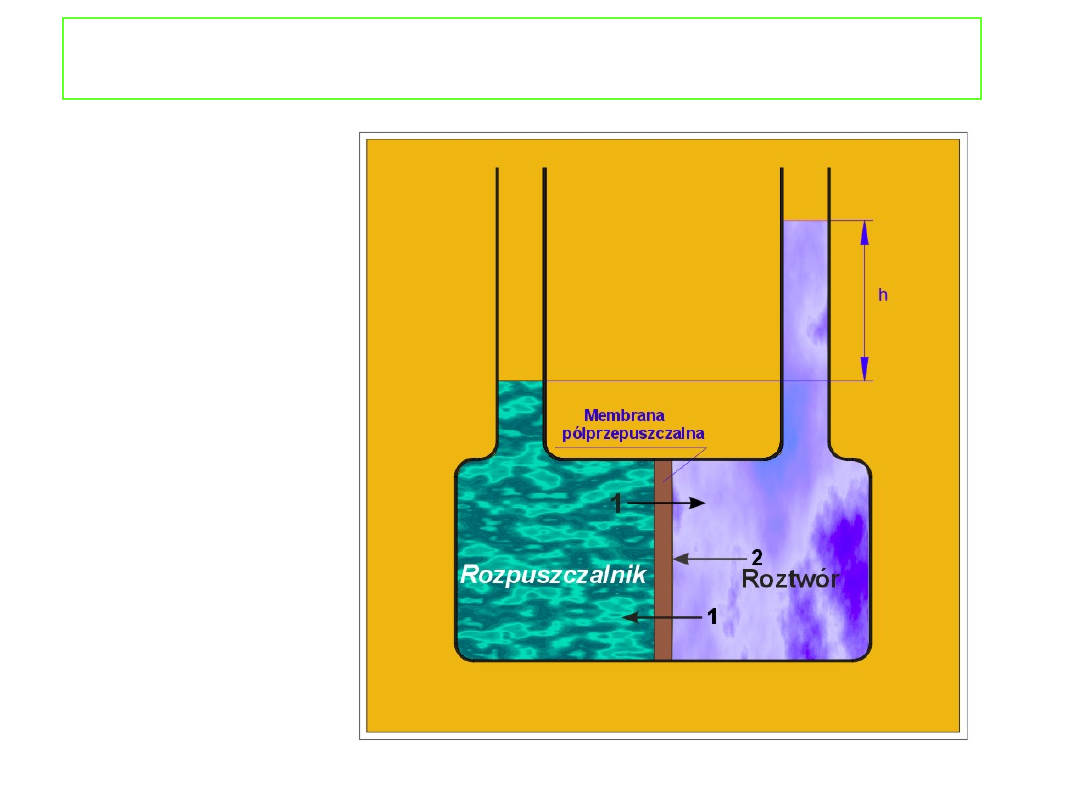
Ciśnienie osmotyczne

Właściwości koligatywne roztworów
O z n a c z a m y
rozp
μ
1
i
R-r
μ
1
- t o p o t e n c j a ł y c h e m i c z n e r o z p u s z c z a l n i k a w c z y s t y m
r o z p u s z c z a l n i k u i w r o z t w o r z e .
P r z e d u s t a l e n i e m s i ę r ó w n o w a g i :
*
rozp
μ
μ
1
1
( x
1
= 1 )
i
1
1
1
ln x
RT
μ
μ
*
R-r
( x
1
< 1 )
S t ą d :
1
1
1
1
1
ln x
RT
μ
μ
μ
μ
*
R-r
*
rozp
N i e r ó w n o ś ć t a w s k a z u j e , ż e w i ę c e j c z ą s t e c z e k p r z e j d z i e z o b s z a r u , w k t ó r y m z n a j d u j e s i ę
c z y s t y r o z p u s z c z a l n i k d o o b s z a r u , g d z i e z n a j d u j e s i ę r o z t w ó r .


Ciśnienie osmotyczne
jest to ciśnienie jakie musi
być przyłożone do roztworu, aby potencjał
chemiczny rozpuszczalnika w roztworze wzrósł do
wartości potencjału chemicznego czystego
rozpuszczalnika pozostającego pod ciśnieniem
atmosferycznym.
W stanie równowagi:
V
π
x
RT
μ
μ
μ
*
R-r
rozp
1
1
1
1
ln
Ponieważ
*
rozp
μ
μ
1
1
więc
1
lnx
RT
V
π

Z a k ł a d a m y , ż e
2
2
1
1
ln
ln
x
x
x
1
2
x
i ż e
1
2
2
1
2
2
n
n
n
n
n
x
2
1
n
n
W ó w c z a s
1
2
2
n
n
RT
RTx
V
π
1
n
V
V
1
2
1
n
n
RT
n
V
π
c z y l i
RT
n
πV
2
i
RT
c
RT
V
n
π
M
2
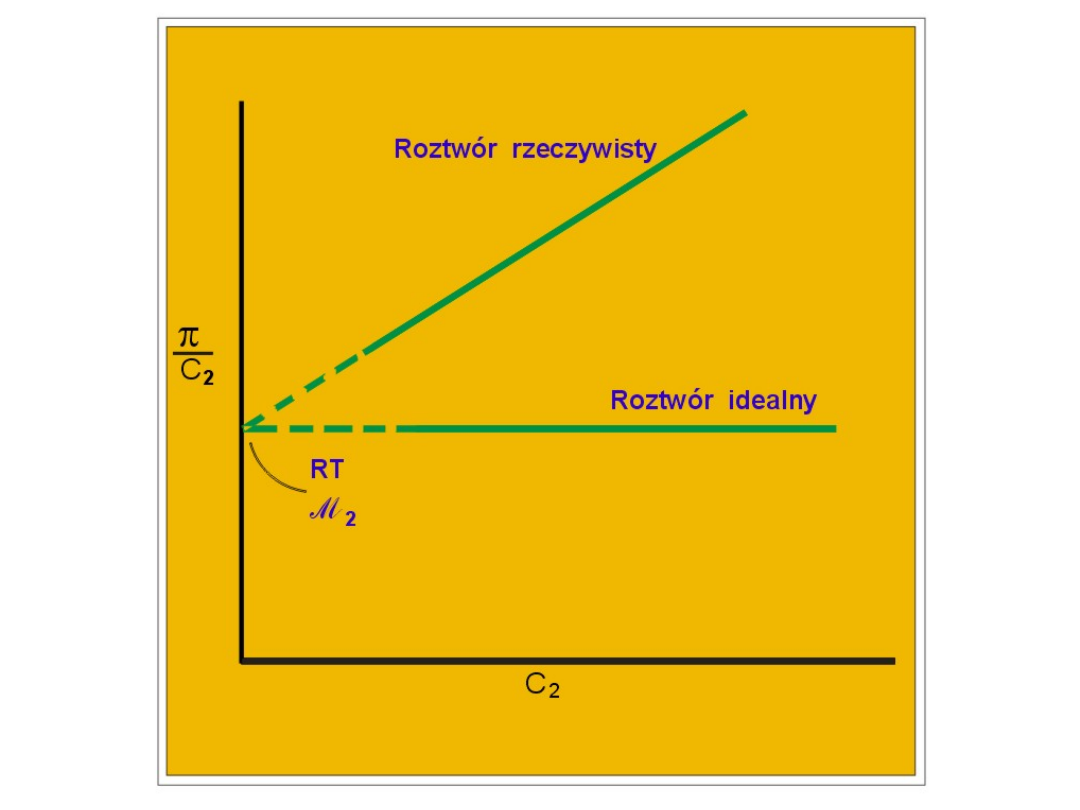
g d z i e c
M
to m o l a rn o ś ć ro z tw o ru .

P
rzyjm
u
jąc, że c
2
to stężen
ie roztw
oru
w
g/ dm
-3
, M
2
to m
asa m
olow
a substan
cji
rozpu
szczonej m
ożem
y zapisać:
RT
M
c
π
2
2
i
2
2
M
RT
c
π
R
ów
n
an
ie to pozw
ala n
a w
yzn
aczen
ie m
asy m
olow
ej su
bstan
cji rozpu
szczon
ej.

Równowaga chemiczna w stanie gazowym -
gazy doskonałe
Równowaga chemiczna w stanie gazowym - gazy
doskonałe
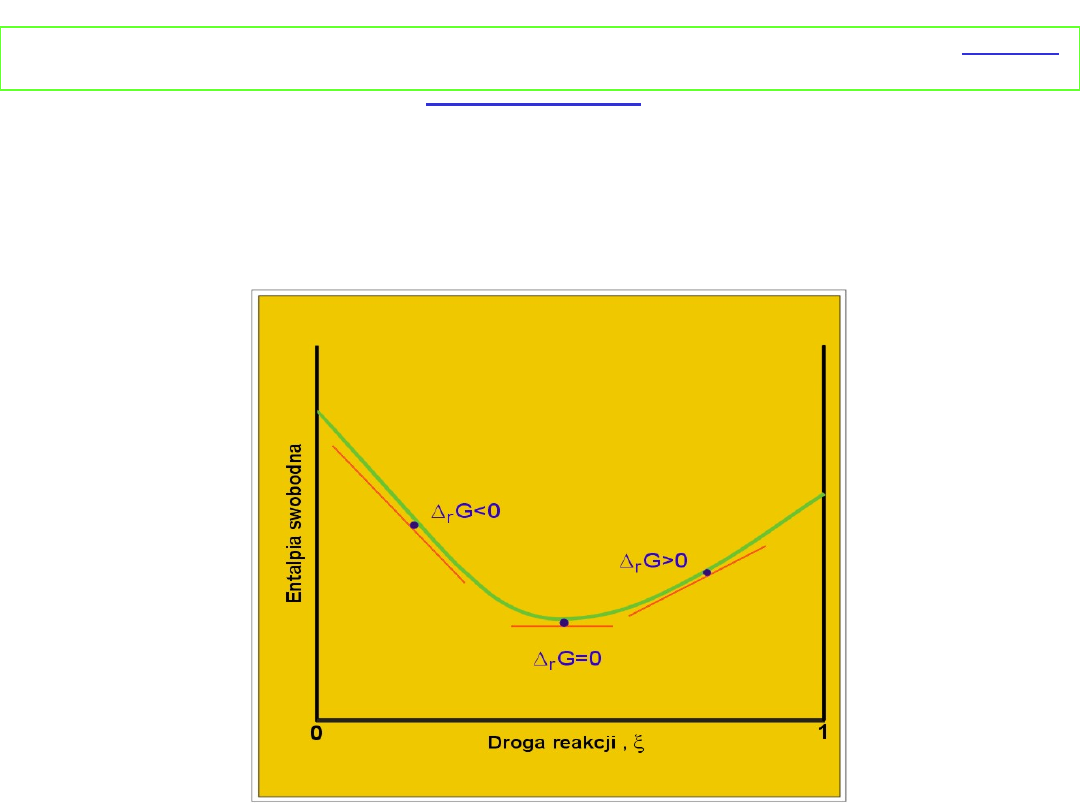
Reakcja przebiega w takim kierunku, aby następowało
zmniejszanie entalpii swobodnej, tzn. (∂G/ ∂ζ)
T,P
< 0.
Stąd reakcja A → B jest samorzutna gdy μ
B
> μ
A
, a reakcja B
→A jest samorzutna, gdy μ
B
< μ
A
. W stanie równowagi μ
A
= μ
B
i
(∂G/∂ξ)
T,P
= 0.

Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Δ
r
G < 0 reakcja A → B jest
samorzutna
Δ
r
G > 0 reakcja B → A jest
samorzutna
Δ
r
G = 0 układ jest w równowadze

Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Rozpatrzmy przypadek:
gdzie a i b to współczynniki stechiometryczne.
W roztworach doskonałych potencjał chemiczny i-tego
składnika opisuje równanie:
P
i
– ciśnienie cząstkowe składnika i
- standardowy potencjał chemiczny składnika i
P˚ = 1 bar
Stąd
(g)
(g)
bB
aA
P
P
RT
μ
μ
i
ln
1
1
1
μ
P
P
RT
μ
μ
A
A
A
ln
P
P
RT
μ
μ
rs
B
B
ln
Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Zmiana entalpii swobodnej reakcji A → B jest równa
więc
Ponieważ
- standardowa zmiana entalpii swobodnej reakcji
więc
A
B
aμ
bμ
P
P
a RT
P
P
b RT lu
aμ
bμ
G
Δ
A
B
A
B
r
ln
A
B
r
aμ
bμ
G
Δ
G
Δ
r
a
A
b
o
B
r
r
P
P
P
P
RT lu
G
Δ
G
Δ

Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Zdefiniujmy ciśnieniową stałą równowagi:
W stanie równowagi , więc:
i
Równanie to jest jednym z najważniejszych i użytecznych
równań w termodynamice chemicznej. Wiąże ono w bardzo
prosty sposób (ciśnieniową) stałą równowagi i zmianę
standardowej entalpii swobodnej reakcji Δ
r
G
o
. Wartość Δ
r
G
o
zależy od temperatury oraz rodzaju substratów i produktów.
b
a
o
a
A
b
B
a
o
A
b
o
B
p
P
P
P
P
P
P
P
K
0
G
Δ
r
p
o
r
K
RT
G
Δ
ln
0
p
o
r
K
RT
G
Δ
ln
Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Dyskusja równania Δ
r
G
o
= - RT ln K
p
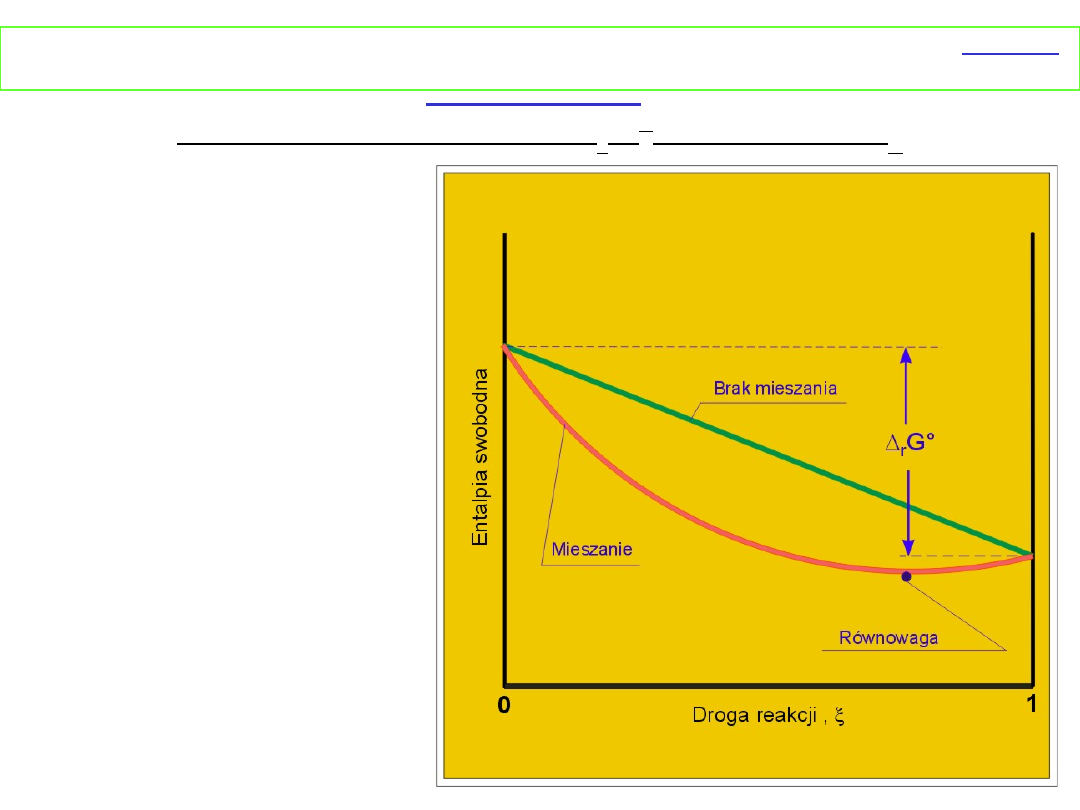
I. Zmiana
entalpii swobodnej
podczas reakcji
w
zależności od
postępu reakcji przy
założeniu,
że Δ
r
G
o
< 0
przedstawia rysunek
obok.
)
(
)
(
g
g
bB
aA

Równowaga chemiczna w stanie gazowym - gazy
doskonałe
Jeżeli reagenty nie mieszają się z produktami,
entalpia swobodna maleje liniowo
w miarę postępu reakcji i w rezultacie substraty zostaną
zamienione w produkty. Jak wyprowadziliśmy wcześniej
Δ
miesz
G jest zawsze ujemna, dlatego w przypadku mieszania
się substratów i produktów entalpia swobodna będzie
miała mniejsze wartości niż w poprzednim przypadku
(niemieszanie). Dlatego też stan równowagi zostanie
osiągnięty, gdy zrównają się entalpie swobodne dwóch
przeciwstawnych efektów przemiana substratów w produkt
i mieszanie produktów z substratami.
II. Znając Δ
r
G
o
możemy obliczyć K
p
. Ponieważ
sub
o
tw
prod
o
tw
o
r
G
Δ
G
Δ
G
Δ
III. Gdy:
Δ
r
G
o
< 0
K
p
> 1
Δ
r
G
o
> 0
K
p
< 1
Δ
r
G
o
= 0
K
p
= 1
Gdy Δ
r
G
o
> 0 nie oznacza to, że nie zachodzi reakcja. Np. gdy
Δ
r
G
o
= 10 kJ mol
-1
i T = 298 K, K
p
= 0,018 i pewna ilość produktów
powstanie, szczególnie gdy substraty są w dużych ilościach.

Równowaga chemiczna w stanie gazowym - gazy
doskonałe
IV. K
p
zależy wyłącznie od temperatury (ponieważ μ
o
zależy
wyłącznie od temperatury) i jest bezwymiarowa (dzielimy
ciśnienie przez P˚).
V.Dla dowolnego stanu w postępie reakcji możemy
obliczyć Δ
r
G
gdzie Q – iloraz stężeń produktów od substratów.
Pozwala nam to określić w jakim kierunku zachodzi
reakcja samorzutnie jeżeli znamy stężenia reagentów.
Q
RT
G
Δ
G
Δ
o
r
r
ln
Równowaga chemiczna w stanie gazowym - gazy
rzeczywiste
Zachowanie gazów
rzeczywistych opisano
poprzez równania
wirialne i van der Waalsa
(i mogą być inne).
Zastosowanie procedury
podobnej do użycia
aktywności w przypadku
roztworów ciekłych.
Ciśnienie cząstkowe
zastępuje się wielkością
zwaną lotnością (f) i dla
gazów rzeczywistych
otrzymujemy równanie:
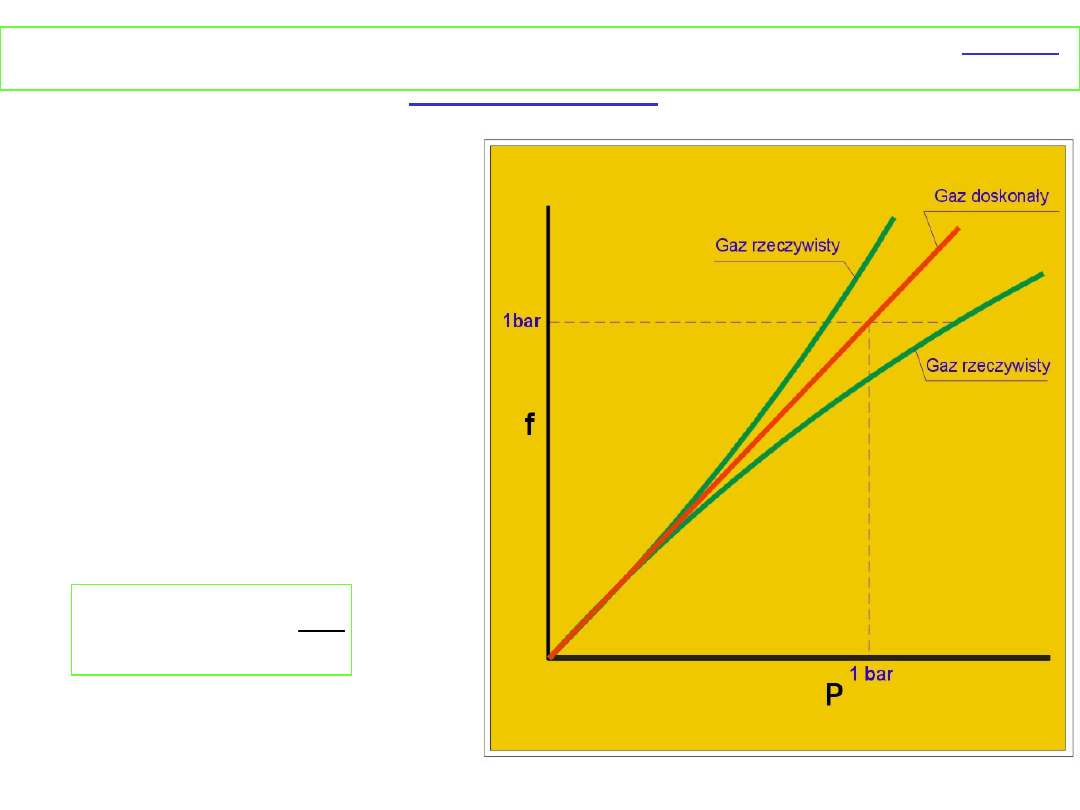
Zależność lotności od
ciśnienia przedstawia
rysunek
o
o
P
f
RT
μ
μ
ln

Równowaga chemiczna w stanie gazowym - gazy
rzeczywiste
Dla małych ciśnień gaz rzeczywisty zachowuje się jak gaz
doskonały i lotność jest równa ciśnieniu cząstkowemu, dla
wyższych ciśnień występują odstępstwa. Stan standardowy
lotności gazu rzeczywistego to stan, w którym lotność byłaby
równa 1 bar gdyby gaz zachował się doskonale
Ogólnie:
Współczynnik lotności α jest zdefiniowany jako:
i
Lotności nie można zmierzyć. Można ją jedynie obliczyć na
podstawie pomiarów ciśnienia.
P
f
P
0
lim
P
f
α
1
lim
0
α
P

Równowaga chemiczna w stanie gazowym - gazy
rzeczywiste
Dla dyskutowanej wcześniej reakcji
możemy zapisać wyrażenie na stałą równowagi K
f
(f-lotność)
gdzie K
f
to termodynamiczna stała równowagi
Ponieważ f = γP powyższe równanie można zapisać jako:
gdzie ,
a K
p
zdefiniowana była poprzednio – nazywana jest pozorną
stałą równowagi, ponieważ jej wartość dla danej temperatury
zależy od ciśnienia.
Dla gazu doskonałego K
f
= K
p
.
)
(
)
(
g
g
bB
aA
a
A
b
B
f
/
f
/
f
K
bar
1
bar
1
p
γ
a
A
b
B
a
A
b
B
f
K
K
/
P
/
P
γ
γ
K
1bar
1bar
a
A
b
B
γ
γ
γ
K
Równowaga chemiczna w stanie gazowym - gazy
rzeczywiste
Dla reakcji
stałe równowagi w temperaturze 450˚C przedstawia tablica:
(g)
(g)
(g)
NH
H
N
3
2
2
2
2
3
2
1
Ciśnienie
całkowite/bar
3
NH
P
2
N
P
2
N
P
p
K
γ
K
Kγ
K
K
p
f
10,2
0,204
2,30
7,67 0,0064
0,994
0,0064
30,3
1,76
6,68 21,9 0,0066
0,975
0,0064
50,6
4,65 10,7 35,2 0,0068
0,95
0,0065
101,0
16,6 19,4 65,0 0,0072
0,89
0,0064
302,8
108
42,8 152
0,0088
0,70
0,0062
606
326
56,5 223
0,0130
0,50
0,0065

Reakcje w roztworach ciekłych
Dla reakcji
zakładamy, że A i B są nieelektrolitami. Jeżeli roztwór
jest doskonały i stężenie wyrazimy jako molalność
otrzymujemy:
to stężenie
)
(
)
(
r
r
bB
aA
m
m
RT
μ
μ
A
o
A
A
ln
m
rozp
kg
mol
1
Reakcje w roztworach ciekłych
Stosując te same przekształcenia jak dla gazów doskonałych
otrzymujemy:
gdzie
Jeżeli stężenie wyrazimy jako molarność (mol dm
-3
) stała
równowagi przybiera postać:
stężeniowa stała
równowagi
Zarówno K
m
jak i K
c
są bezwymiarowe, ponieważ stężenia są
dzielone przez
standardowe (odpowiednio i
).
m
r
K
RT
G
Δ
ln
a
A
b
B
m
m
m
m
m
K
a
b
c
/
A
/
B
K
3
3
dm
mol
1
dm
mol
1
(rozp)
kg
mol
1
ru)
(r
dm
mol
1
3

Reakcje w roztworach ciekłych
W warunkach odbiegających od stanu równowagi entalpia
swobodna reakcji opisana jest równaniem
Q - iloczyn stężeń
W przypadku roztworów niedoskonałych stężenia zastępujemy
poprzez aktywności.
Potencjał chemiczny i-tego składnika wynosi więc
Wychodząc z wyrażenia opisującego potencjał chemiczny i-
tego składnika możemy otrzymać wyrażenie na
termodynamiczną stałą równowagi
a ponieważ
Q
RT
G
Δ
G
Δ
r
r
ln
i
o
i
i
a
RT
μ
μ
ln
a
A
b
B
a
a
a
K
m
γ
a

Reakcje w roztworach ciekłych
więc
m
γ
a
A
b
B
a
A
b
B
a
K
K
/m
m
/m
m
x
γ
γ
K

Równowaga w układzie heterogenicznym
Rozważmy rozkład termiczny CaCO
3
w układzie zamkniętym
CaCO
3(s)
CaO
(s)
+ CO
2(g)
Zwykle stężeń ciał stałych nie uwzględnia się w opisie stałej
równowagi.
Dlatego zapisując powyższe wyrażenie na stałą równowagi
jako:
i uwzględniając, ze stężenia CaCO
3
i CaO są stałe możemy
zapisać
3
2
CaCO
CO
CaO
'
c
K
2
3
CO
CaO
CaCO
'
c
K
2
CO
c
K

W opisanych warunkach łatwiej zmierzyć ciśnienie CO
2
więc
możemy zapisać
K
c
i K
p
to wielkości bezwymiarowe, bowiem stężenie CO
2
i
ciśnienie CO
2
są
podzielone przez odpowiednie wielkości standardowe (
i 1 bar).
Dla termodynamicznej stałej równowagi
Z definicji
i
Dla niezbyt wysokich ciśnień możemy założyć, że CO
2
zachowuje się jak gaz doskonały
2
CO
P
K
P
3
-
dm
mol
mol
1
3
2
CaCO
CO
CaO
a
a
a
K
a
1
CaO
CaCO
3
a
a
bar
1
2
CO
f
K
a
bar
1
2
CO
P
K
K
p
a
Równowaga w układzie heterogenicznym
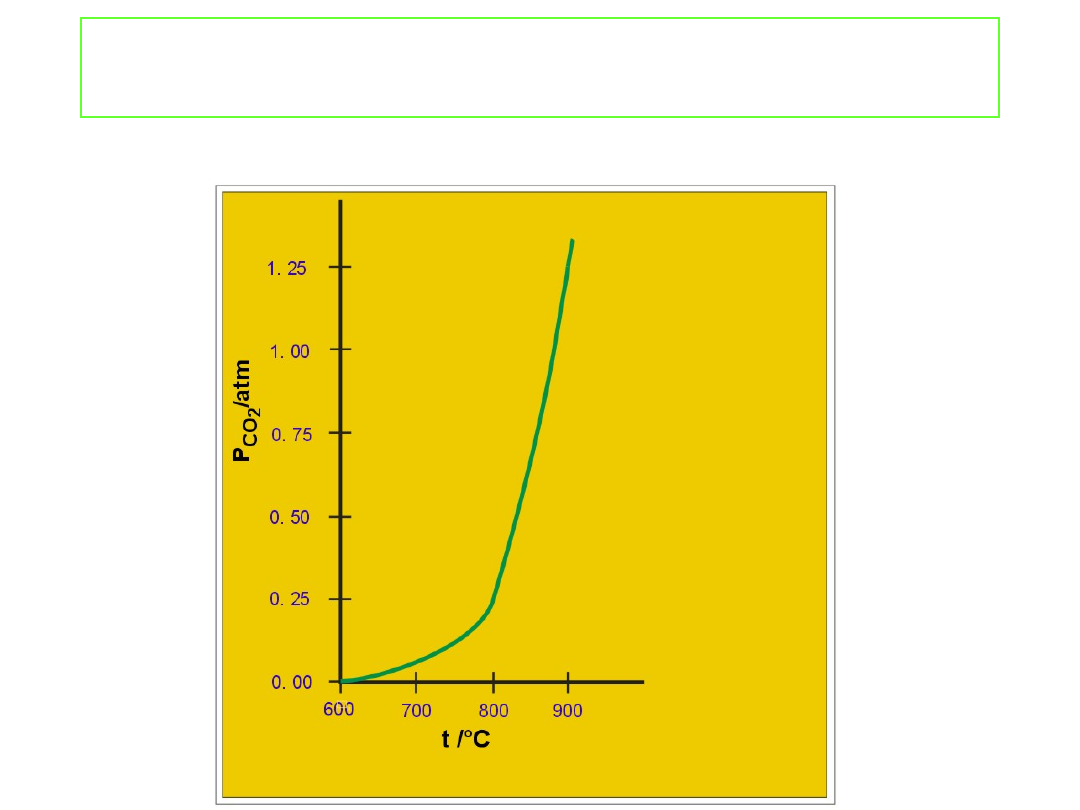
Równowaga w układzie heterogenicznym
Równowagowe ciśnienie CO
2
w kontakcie z CaCO
3
i CaO w
funkcji temperatury.
Wpływ temperatury na stałą równowagi.
Dla reakcji w stanie równowagi
z równania Gibbsa – Helmholtza
K
RT
G
Δ
r
ln
2
T
H
Δ
T
T
G
Δ
r
P
r
Otrzymuje
i
Jest to równanie van’t Hoff’a.
2
ln
T
H
Δ
T
T
K
RT
r
P
2
ln
RT
H
Δ
K
K
r
P
Wpływ temperatury na stałą równowagi.
Wpływ temperatury na stałą równowagi.
Zakładając, że Δ
r
H˚ nie zależy od temperatury można
scałkować powyższe równanie otrzymując:
Ponieważ
i
otrzymujemy
Jest to inna postać równania vant’a Hoffa.
2
1
1
2
2
1
1
2
1
1
ln
T
T
T
T
R
H
Δ
T
T
R
H
Δ
K
K
r
r
RT
G
Δ
K
r
ln
S
TΔ
H
Δ
G
Δ
r
r
r
R
S
Δ
RT
H
Δ
K
r
r
ln
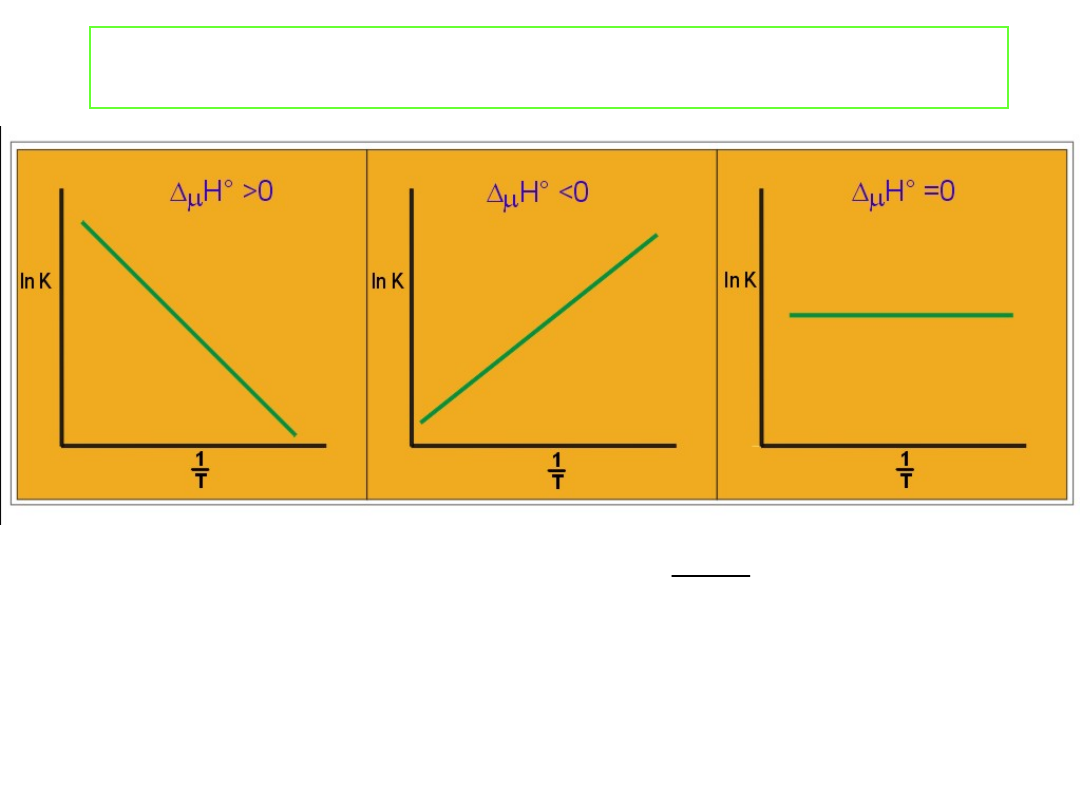
Wpływ temperatury na stałą równowagi.
Współczynnik kierunkowy Δ
r
H˚/R, a wyraz wolny
Linie proste otrzymamy jedynie wówczas gdy Δ
r
H˚ i Δ
r
S˚ są
niezależne od temperatury. Dla małych zakresów temperatur
około 50 K założenie to jest spełnione dość dobrze i metoda ta
pozwala w dogodny sposób wyznaczyć Δ
r
H˚
i Δ
r
S˚ dla danej reakcji.
R
S
Δ
r

Dyskusja równania van’t Hoffa
Jeżeli proces A → B jest endotermiczny, wówczas Δ
r
H˚ jest
dodatnie.
Gdy T
2
> T
1
prawa strona równania
jest dodatnia, a więc
Jeżeli proces A → B jest egzotermiczny Δ
r
H˚ jest ujemne to
lewa strona powyższego równania jest również ujemna i
Wzrost temperatury przesuwa równowagę z lewa na prawo -
dla procesu endotermicznego, a dla procesu egzotermicznego,
równowaga wraz ze wzrostem temperatury przesuwa się z
prawa na lewo. Jest to zgodne z zasadą Le Chatelier’a (regułą
przekory).
Reguła przekory (reguła Le Chateliera i Brauna)
określa
kierunek przesunięcia równowagi chemicznej. Zgodnie z tą
regułą, w przypadku zmiany parametrów reakcji chemicznej
(temperatury, ciśnienia, stężenia) równowaga układu
przesuwa się w takim kierunku, aby przeciwdziałać tej
zmianie.
1
2
1
2
1
2
ln
T
T
T
T
R
H
Δ
K
K
r
1
2
K
K
1
2
K
K

Dyskusja równania van’t Hoffa
Reakcja syntezy amoniaku:
N
2(g)
+ 3H
2
(g)
2NH
3
(g)
ΔH˚ = – 46,19 kJ mol
-1
ΔH˚ = „-” reakcja łączenia azotu z wodorem jest
egzotermiczna - przebiega z wydzieleniem ciepła, procesowi
temu będzie sprzyjało obniżenie temperatury. Wzrost
temperatury spowoduje natomiast przesunięcie równowagi w
stronę azotu i wodoru (rozpad amoniaku).
Reakcja zachodzi w fazie gazowej: z wartości współczynników
stechiometrycznych wynika, że w przypadku stałej objętości
układu przebieg reakcji w prawą stronę prowadzi do
zmniejszenia ciśnienia – z 4 objętości (N
2
+ 3H
2
) powstają 2
objętości (2NH
3
). Zatem wzrost ciśnienia zewnętrznego
powoduje przesunięcie równowagi w stronę prawą, a obniżenie
ciśnienia w lewą.
Dodanie do układu będącego w równowadze jakiejkolwiek
substancji biorącej udział
w reakcjach spowoduje przesunięcie równowagi w stronę
przeciwną niż ta, po której znajduje się dana substancja. Tak
więc wzrost stężenia wodoru i azotu przesuwa równowagę
rozpatrywanej reakcji w prawo, a wzrost stężenia amoniaku w
lewo.
Wpływ ciśnienia na stałą równowagi
Δ
r
G˚ = – RT ln K
p
Δ
r
G˚ nie zależy od ciśnienia, ponieważ jako wartość
standardowa jest zdefiniowana dla określonego ciśnienia 1
bar.
Pozorna stała równowagi K
p
jest niezależna od ciśnienia tylko
wówczas, gdy reagują gazy doskonałe. Dla gazów
rzeczywistych K
p
zależy od ciśnienia. Termodynamiczna stała
równowagi K
f
jest niezależna od stężenia zarówno dla gazów
doskonałych jak i rzeczywistych.
Dla reakcji, w których uczestniczą gazy doskonałe K
f
= K
p
i dla
stałej temperatury możemy zapisać
0
P
K
T
p
Wpływ ciśnienia na stałą równowagi
Reakcja między doskonałymi gazami, będącą w stanie
równowagi:
n - liczba moli substancji A, przed reakcją,
α – ułamek mola substancji A, który przereagował wówczas w
stanie równowagi
liczba moli substancji A n(1-α)
liczba moli substancji B 2nα
Sumaryczna liczba moli w stanie równowagi:
n - nα + 2nα = n + nα = n (1 + 2)
a ułamki molowe substancji A i B wynoszą:
(g)
(g)
2B
A
α
α
α
n
α
n
x
A
1
1
1
1
α
α
α
n
nα
x
B
1
2
1
2
Ciśnienia cząstkowe A i B
P – całkowite ciśnienie panujące w układzie
Stałą równowagi opisuje zależność:
P
α
α
P
A
1
1
P
α
α
P
B
1
2
P
α
α
P
α
α
P
α
α
P
P
K
A
B
p
2
2
2
2
2
1
4
1
1
1
2
Wpływ ciśnienia na stałą równowagi

Wpływ ciśnienia na stałą równowagi
K
p
jest wielkością stałą, więc wartość α zależy wyłącznie od P.
Jeżeli P jest duże
α jest małe i odwrotnie, jeżeli P jest małe α jest duże. Jest to
zgodna z zasadą
Le Clateliera. Jeżeli zwiększymy ciśnienie, równowaga
przeniesie się w stronę, która daje mniej cząsteczek (od
prawej do lewej w rozpatrywanym przypadku)
i α maleje. Gdy obniżymy ciśnienie, zachodzą przeciwne efekty.
P
K
K
α
p
p
4
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
Wyszukiwarka
Podobne podstrony:
Chemia fizyczna wykład 11
chemia fizyczna wykłady, sprawozdania, opracowane zagadnienia do egzaminu Sprawozdanie ćw 7 zależ
chemia fizyczna I wykład(1)
chf wykład 6, Studia, Chemia, fizyczna, wykłady
Chemia fizyczna wykład 10
Chemia fizyczna wykład 4
chf wykład 3, Studia, Chemia, fizyczna, wykłady
2015 pyt tren do wykł VII, Technologia żywnosci i Żywienie człowieka, 2 semestr, chemia fizyczna, ch
Chemia fizyczna wykład 12
chf wykład 8, Studia, Chemia, fizyczna, wykłady
Chemia fizyczna wykład 9
Chemia fizyczna wykład 1
tech.chem. PGd chemia fizyczna wyklady, Technologia chemiczna, Chemia fizyczna, Wykłady, wykłady na
Chemia fizyczna wyklad, Studia, Mibm, semestr II, Chemia Fizyczna, Chemia fizyczna
Chemia fizyczna (wyklad 3), Studia, Mibm, semestr II, Chemia Fizyczna, Chemia fizyczna
Chemia fizyczna (wyklad 4), Studia, Mibm, semestr II, Chemia Fizyczna, Chemia fizyczna
chf wykład 1, Studia, Chemia, fizyczna, wykłady
Chemia fizyczna wykład 7 i 8
pytania treningowe wykład II 2015, Technologia żywnosci i Żywienie człowieka, 2 semestr, chemia fizy
więcej podobnych podstron