
Kinetyka reakcji enzymatycznych
Teoria Michaelisa-Menten i Briggsa-
Haldane’a
Kinetyka reakcji z jednym i z dwoma
substratami
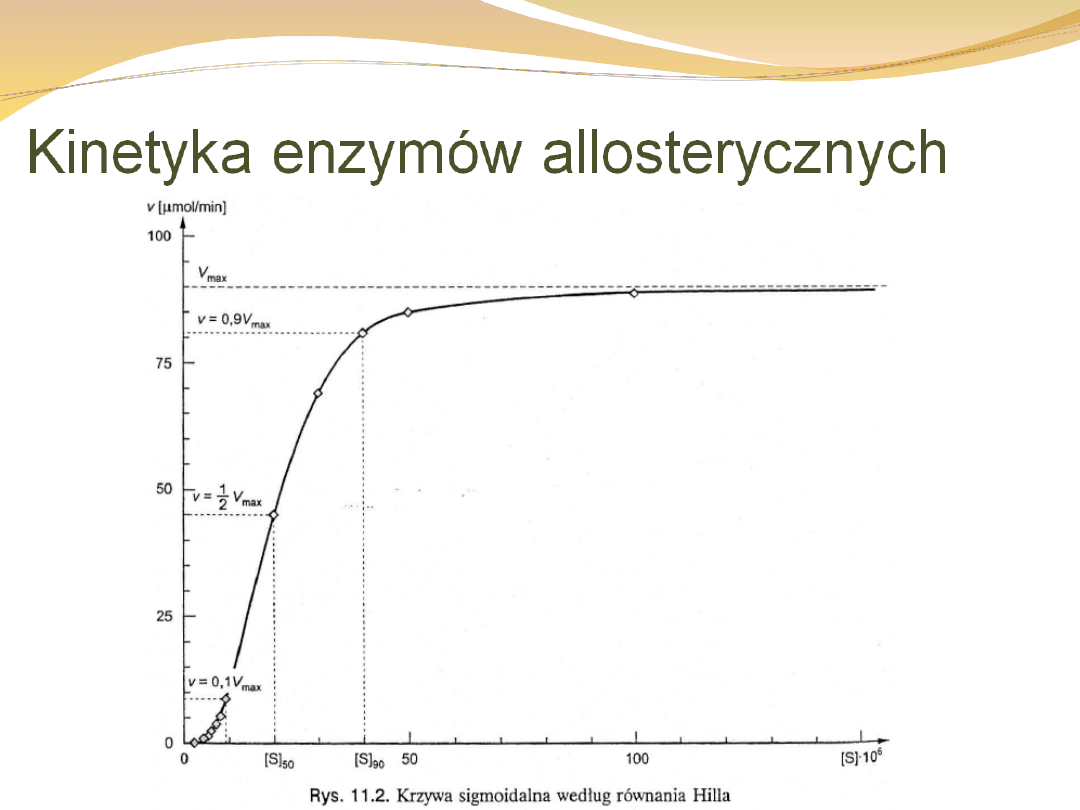
Kinetyka enzymów allosterycznych
Stałe kinetyczne reakcji
Stała wydajności (specyficzności)
katalitycznej
Inhibicja enzymów i jej kinetyka
Kinetyka enzymów immobilizowanych

Cele badań kinetyki reakcji
enzymatycznych:
uzyskanie informacji o przebiegu reakcji w czasie,
a więc o:
szybkości zaniku substratów i powstawania
produktów
liczby i kolejności przyłączania cząsteczek
substratów do enzymu i odłączania cząsteczek
produktów z kompleksu aktywnego
liczby kompleksów pośrednich i izomeryzujących
form enzymu
o efektach czynników modyfikujących przebieg
reakcji – inhibitorów i aktywatorów
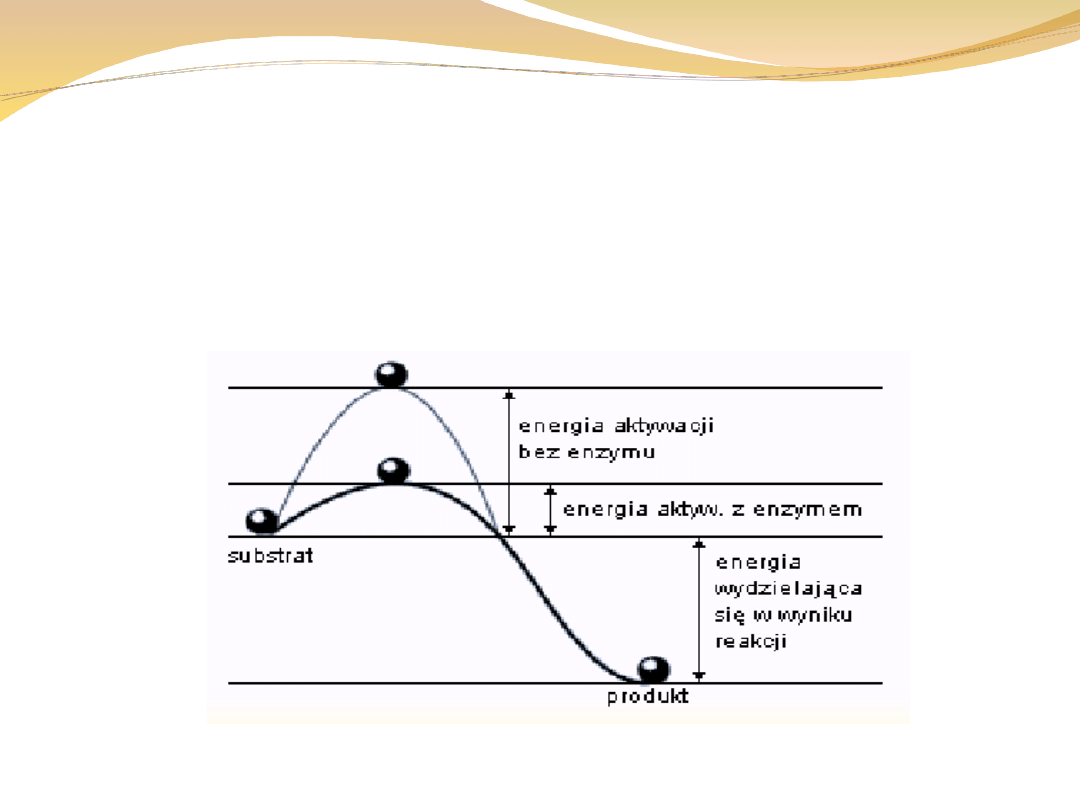
Energia aktywacji
Jest to energia, którą muszą mieć cząsteczki (jony, atomy), aby były zdolne do
określonej reakcji chemicznej. Energię aktywacji wyraża się zwykle w kJ/mol
reagujących cząsteczek. Im mniejsza jest energia aktywacji, tym reakcja zachodzi
szybciej. (Energia aktywacji, najmniejsza
, jaką muszą posiadać cząsteczki
substratów, by wskutek zderzenia tych cząsteczek, mogła zajść
.;
różnica energii swobodnej między stanem przejściowym a substratem. )

Szybkość reakcji jest miara ilości zużywanych substratów lub
powstających produktów w jednostce czasu. Opisujemy ją wzorem:
Gdzie: - współczynniki stechiometryczne reagentów
A- substraty
B- produkty
Doświadczalnie wyznaczone równanie tego typu nosi nazwę równania
kinetycznego, przyjmuje ono postać:
Współczynnik k nosi nazwę stałej szybkości reakcji, jest niezależna od
stężeń reagentów, a zależna m.in. od temperatury. Wykładniki
potęgowe nie wynikają z równania sumarycznego, a jedynie z
mechanizmu reakcji. Wykładniki te mogą mieć zarówno wartości
całkowite jak i ułamkowe.
dt
v
dc
dt
v
dc
r
B
B
A
A
B
A
v
v ,
...
b
a
B
A
k
v

Jednostki enzymatyczne:
Najpowszechniejsza- podawana jako wartość
w czasie zerowym- czyli jako początkową
szybkość reakcji Vo, jednostka: µmol/min
2 standardowe jednostki aktywności
enzymatycznej:
Jednostka enzymatyczna (U)
Katal (kat)

Jednostka enzymatyczna-czyli taka ilość
enzymu, która katalizuje przemianę 1 μmola
substratu w ciągi 1 minuty w temperaturze
30°C i określonym pH
katal (kat), czyli ilość enzymu katalizująca
przemianę 1 mola substratu w ciągu 1
sekundy.
1 µmol/min=1U=16.67 nanokat

Aktywność:
Całkowita- odnosi się do całkowitej liczby
jednostek enzymatycznych w próbce
Specyficzna- liczba jednostek enzymatycznych
na miligram białka, miara czystości enzymu

Model Michaelisa-Menten:
model oparty na założeniu o nieodwracalnym
mechanizmie reakcji

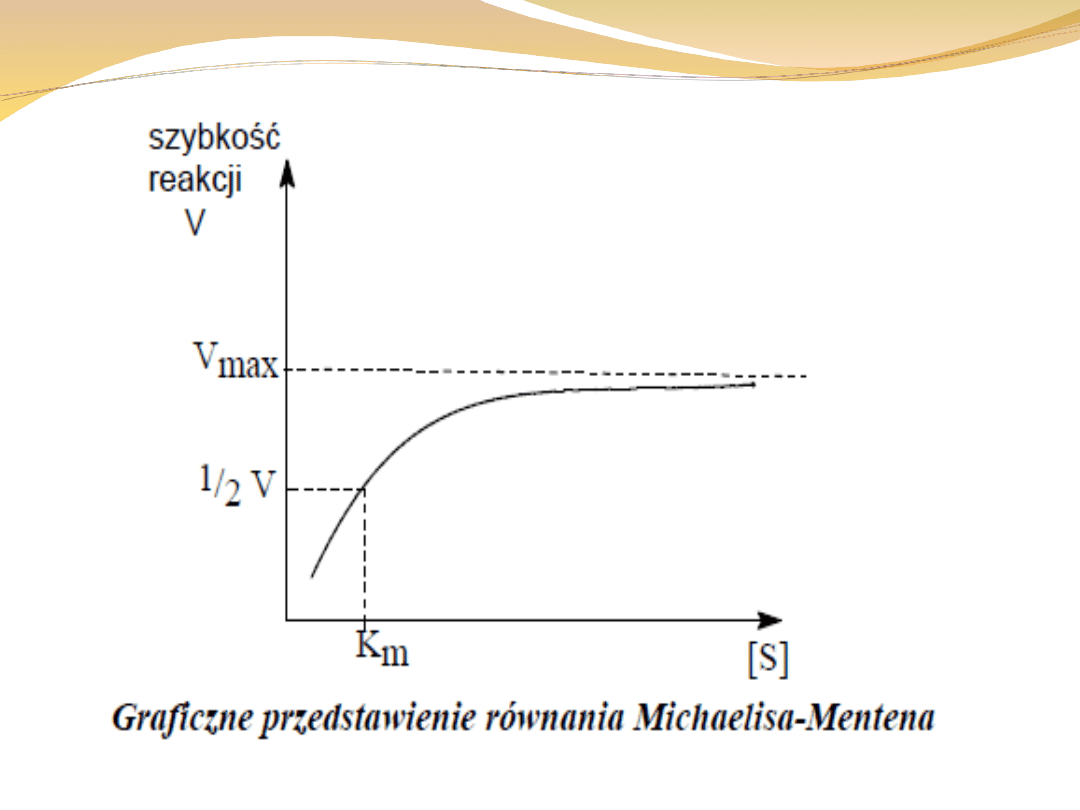

Stała Michaelisa:
jest wyrazem powinowactwa enzymu do substratu
enzymy o niskiej wartości stałej Michaelisa działają
znacznie sprawniej niż enzymy o wysokiej jej wartości
szybkość tworzenia produktu zależy od [ES], a więc im
wartość [ES] jest większa, a tym samym stała
Michaelisa mniejsza, tym szybsze powstawanie
produktu.
dla większości enzymów stała Michaelisa mieści się w
przedziale od 10
-1
do 10
-7
M
zależy ona od substratu, a także warunków
zewnętrznych takich jak: temperatura i siła jonowa
oznacza ona takie stężenie substratu, w którym połowa
miejsc aktywnych jest obsadzona
kiedy znamy jej wartość możemy obliczyć ułamek
obsadzonych miejsc dla każdego stężenia substratu

Model Briggsa Haldane
’
a
Model oparty o założenie obecności stanu
stacjonarnego w układzie nieodwracalnym

Porównanie modeli:
1
1
'
k
k
m
K
]
[
]
[
S
m
K
S
m
V
v
]
0
[
2
E
k
m
V
1
2
1
k
k
k
m
K
Michaelis-Menten
kiedy k
2
<< k-1,
1
2
1
'
k
k
k
m
K
m
K
]
[
'
]
[
S
m
K
S
m
V
v
]
0
[
2
E
k
m
V
Briggs-Haldane
d[ES]/dt ≈
0
]
[
1
]
][
[
1
ES
k
S
E
k
założenie:
równanie:
Prędkość
maksymalna:
Stała:

Kinetyka reakcji z wieloma
substratami
Reakcje z kilkoma substratami można podzielić na dwie
klasy:
Przeniesienie sekwencyjne
Przeniesienie podwójne (reakcje ping-pong)
Reakcje z kilkoma substratami można podzielić na dwie
klasy:
Przeniesienie sekwencyjne
Przeniesienie podwójne (reakcje ping-pong)
Q
P
B
A

Przeniesienie sekwencyjne
W tej grupie wszystkie substraty muszą się
związać z enzymem. Jeżeli są dwa substraty to
powstaje trójskładnikowy kompleks enzymu z
obydwoma Substra- tami. Kolejność wiązania
substratów z enzymem może być uporządkowana
– czyli substraty wiążą się w ściśle określonej
kolejności lub przypadkowa.
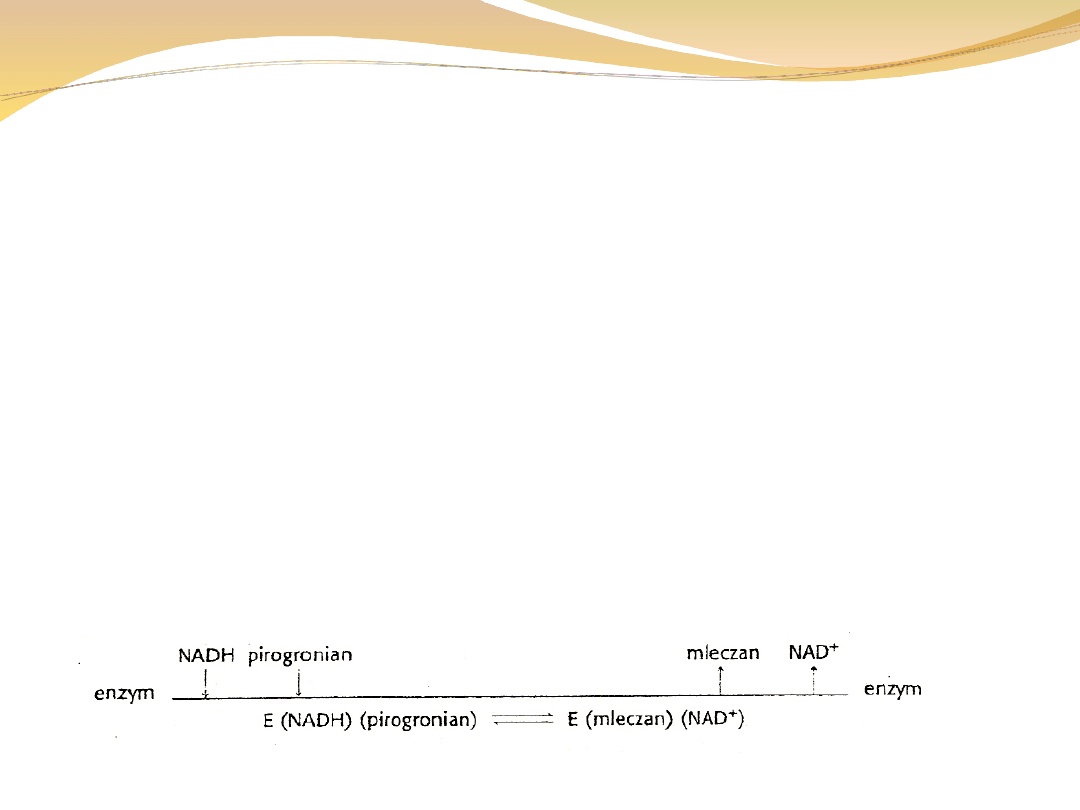
Przykładem reakcji uporządkowanej jest przekształcenie
pirogronianu w mleczan
Pirogronian + NADH + H+ ↔ mleczan + NAD+
W uporządkowanym mechanizmie sekwencyjnym koenzym
zawsze wiąże
się jako pierwszy i dysocjuje od enzymu jako ostatni.
Diagram Clelanda:
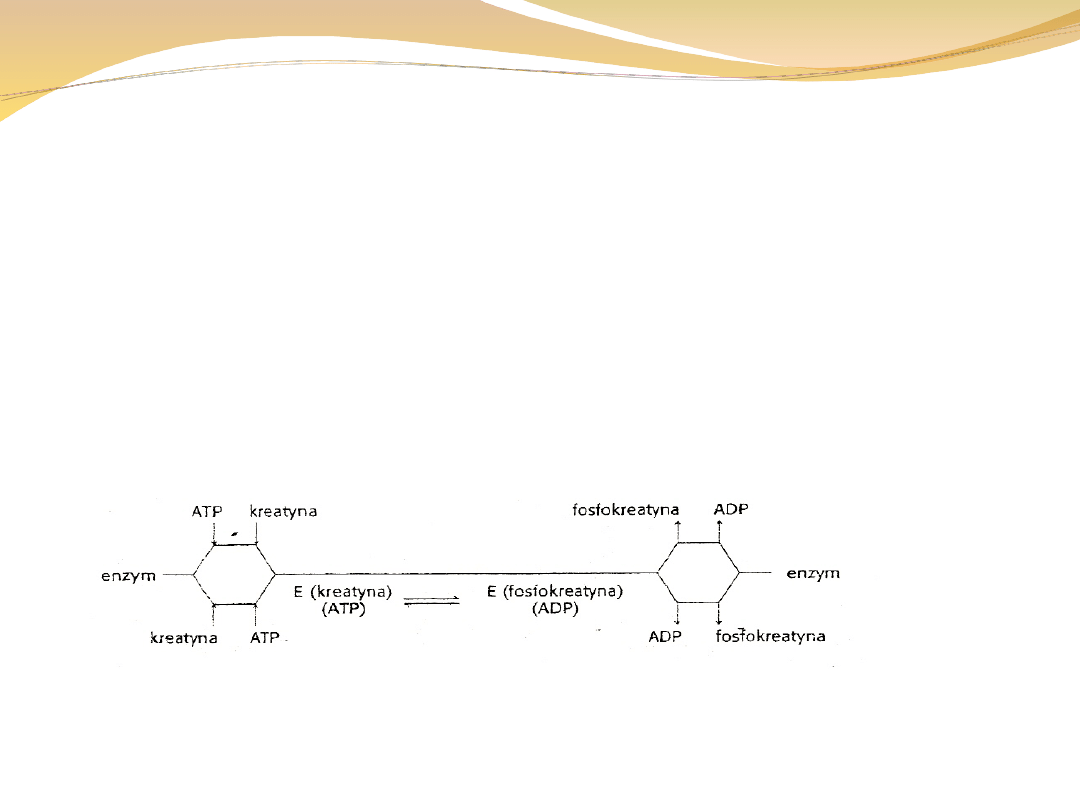
W mechanizmie przypadkowym kolejność jest
dowolna:
Kreatyna + ATP ↔ fosfokreatyna + ADP
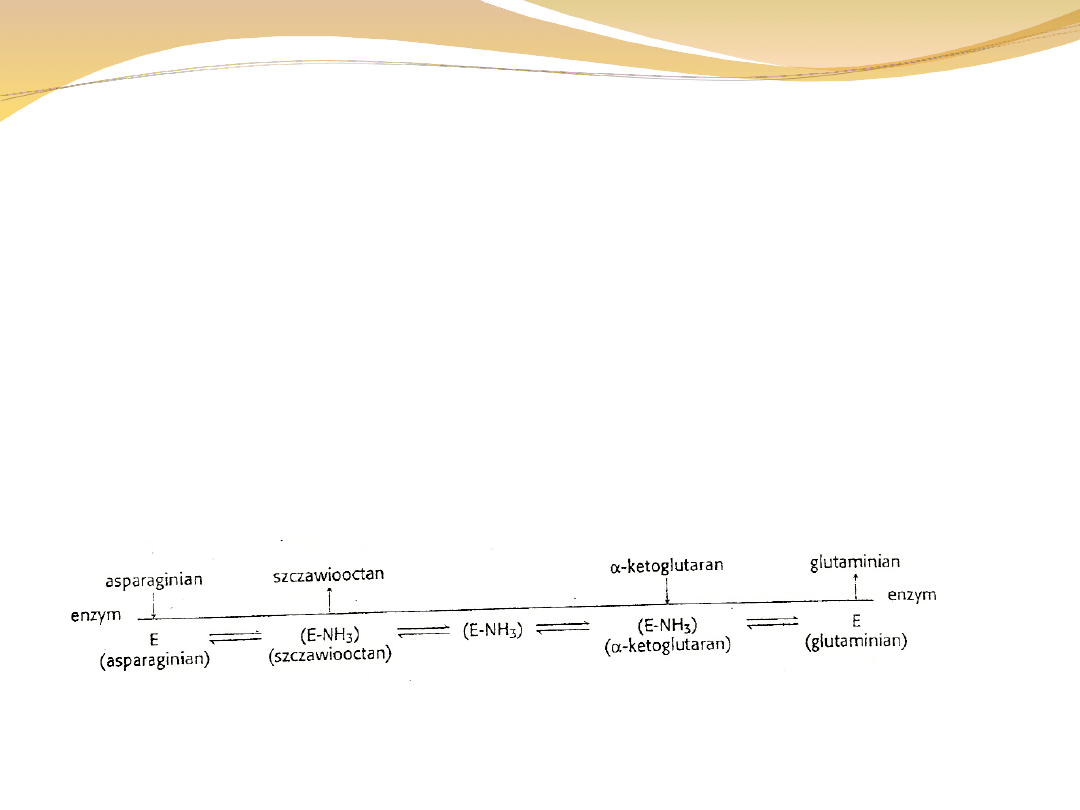
W reakcjach podwójnego przeniesienia jeden z
substratów jest uwalniany przed związaniem wszystkich
substratów przez enzym.
Cechą charakterystyczną jest związanie przenoszonej grupy
przez enzym i późniejsze przeniesienie jej na drugi substrat
Asparaginian + ketoglutaran ↔ glutaminian + szczawiooctan
Sekwencje zdarzeń opisuje diagram:

Enzymy allosteryczne- zbudowane z kilku podjednostek,
mające najczęściej dwa rodzaje centrów:
• Centra aktywne- decydujące o właściwościach
katalitycznych
• Centra regulatorowe (miejsca allosteryczne, efektorowe)-
zostają w nich związane związki niskocząsteczkowe zwane
efektorami lub ligandami allosterycznymi.

Przyłączenie efektora powoduje modyfikację działania enzymu.
Jest to tzw. kooperacja, będąca jedną z głównych cech
zjawiska allosterii.
Kooperacja dodatnia powoduje, że związanie jednego
reagenta zwiększa siłę wiązania następnego i w rezultacie
otrzymujemy krzywą sigmoidalną a nie hiperboliczną.
Właściwości katalityczne enzymów allosterycznych, w których
występują efekty kooperacyjne, opisuje się za pomocą dwóch
podstawowych modeli:
• sprzężonego czyli jednoprzejściowego
• sekwencyjnego

Model sprzężony- zakłada, że enzym istnieje w dwóch
stanach znajdujących się w równowadze:
• w stanie rozluźnionym R (ang. relaxed)- o silnym
powinowactwie do reagentów
• w stanie napiętym T (ang. tense)- o słabym
powinowactwie
Przejście konformacyjne pomiędzy stanami T i R zachodzi
jednocześnie we wszystkich protomerach
danej
cząsteczki enzymu i ma charakter sprzężony. Ligandy
przesuwają stan równowagi enzymu w jedną lub drugą stronę
i konformacja podjednostek zmienia się w tym samym czasie
symetrycznie- nie ma stanów hybrydowych.

Powinowactwo miejsca wiążącego dany ligand zależy od
konformacji protomeru. Pewne ligandy wiążą się
preferencyjnie z jedną konformacją oligomeru, podczas,
gdy inne ligandy- z drugą.
Związanie ligandu z jedną poszczególną konformacją
powoduje przesunięcie równowagi na korzyść konformacji,
z którą związany jest dany ligand.
W formie wolnej przeważa stan T. Równowaga R
0
T
0
jest
więc przesunięta w prawo w kierunku formy o słabym
powinowactwie do ligandu.

L=T
0
/R
0
stała równowagi
sprzężonej
Powinowactwo reagenta A do
protomerów w stanie R i T określone
jest przez dwie stałe dysocjacji K
R
i K
T
:
[R
0
]
[A]
[R
1
]
K
R
=
[T
0
]
[A]
K
T
=
[T
1
]

K
R
/K
T
= c współczynnik
wiązania
Kooperatywność
wiązania
substratu
zależy od wartości L i c. Krzywa
szybkości reakcji staje się bardziej
sigmoidalna, gdy wzrasta L, tj. gdy
równowaga przesunięta jest na korzyść
T i kiedy zmniejsza się wartość c,
czyli wtedy, gdy powinowactwo formy T
do substratu zmniejsza się w stosunku
do formy R.

Model
sekwencyjny-
zakłada,
że
związanie substratu (lub innego ligandu)
z jedną z podjednostek indukuje
powstawanie zmian konformacyjnych w
tej
podjednostce,
które
zmieniają
powinowactwo do substratu innych
wolnych miejsc wiążących. W efekcie
tego,
każda
związana
cząsteczka
substratu ułatwia wiązanie następnej.

Na
podstawie
właściwości
wiązania
substratów,
które
uwzględnia
model
sekwencyjny można wyprowadzić równanie:
v
0
V
max
[S]
n
K’ + [S]
n
=
Równanie to znane jest jako równanie Hilla,
gdzie:
n- liczba miejsc wiążących substrat w
cząsteczce enzymu, czyli tzw. współczynnik
interakcji Hilla;
K’-
stała
uwzględniająca
czynniki
współdziałania i stałą dysocjacji K
s

Wartość K’ jest w pewnym sensie
odpowiednikiem
Km,
ponieważ
również odnosi się ona do [S], przy
którym v = Vmax/2, ale nie
jest równa temu stężeniu (w
przeciwieństwie do Km).


k
A
= k
kat
/K
m
Stała katalityczna
enzymu k
3
, liczba obrotów
enzymu
Stała wydajności (specyficzności)-
sprawność katalityczna
Porównanie stałych specyficzności enzymu dla dwóch
różnych substratów jest miarą selektywności enzymu
wobec jednego substratu w stosunku do innego
substratu.
Dla reakcji gdzie [S]>>K
m
, k
3
jest stałą reakcji I rz.
Dla reakcji gdzie [S]<<K
m
, k
A
jest stałą reakcji II rz.
V
max
= k
3
[E
T
]

„Biochemia” J.M. Berg; J.L. Tymoczko; L. Stryer Wyd.
Naukowe PWN W-wa 2005
„Biochemia Tom I Wprowadzające wiadomości z
chemii ogólnej, składniki chemiczne ustrojów
metabolizmu (enzymy)” B. Filipowski; W.Więchowski
PWN wyd. V W-wa – Łódź 1983
„Biochemia ilustrowany przewodnik” J. Koolman;
K.H.Rochm Wyd.Lekarskie PZWL W-wa 2005
„ Chemia bioorganiczna”, P. Kafarski, B. Lejczak.
„Chemia fizyczna dla biologów. Termodynamika i
kinetyka”- G. Bartosz, Wydawnictwo Uniwersytetu
Rzeszowskiego, Rzeszów 2005
„Obliczenia biochemiczne”- A. Zgirski, R. Gondko,
PWN W-wa 1998
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
Wyszukiwarka
Podobne podstrony:
06 Kinetyka reakcji enzymatycznych
kinetyka reakcji enzymatycznych I
Wyznaczanie parametrów kinetyki reakcji enzymatycznej za pomocą metod polarymetrycznych 5x
KINETYKA REAKCJI ENZYMATYCZNYCH
Kinetyka reakcji enzymatycznej m poteraj
Kinetyka reakcji enzymatycznych Nieznany
cwiczenie 4 inwertaza kinetyka reakcji enzymatycznych 05 05 2014
5 Kinetyka reakcji enzymatycznych
Kinetyka Reakcji Enzymatycznych
trusek hołownia, procesy membranowe, KINETYKA REAKCJI ENZYMATYCZNYCH
ENZYMY KINETYKA REAKCJI ENZYMATYCZNYCH
Kinetyka Reakcji Enzymatycznych
ĆWICZENIE 4 kinetyka reakcji enzymatycznej
Kinetyka reakcji enzymatycznych
I Wyznaczanie parametrow kinetyki reakcji enzymatycznej polarymetr
kinetyka reakcji enzymatycznych
rybiak,biologia i ekologia, Kinetyka reakcji enzymatycznych
06 Kinetyka reakcji enzymatycznych
więcej podobnych podstron