
1
TECHNOLOGIA CHEMICZNA – WYKŁAD – CZĘŚĆ ORGANICZNA
1. Podstawowe surowce naturalne (ropa naftowa, węgiel, gaz ziemny)
Ropa naftowa jest złożoną mieszaniną związków chemicznych, podstawową masę ropy (80-95%)
stanowią ciekłe oraz rozpuszczone w nich stałe węglowodory parafinowe, naftenowe i aromatyczne.
Skład elementarny rop naftowych:
węgiel 83-87%
wodór 12-14%
siarka 0,01-8%
azot 0,01-1,2%
tlen 0,05-4%
Procesy technologiczne pozyskiwania paliw i surowców do przemysłu chemicznego:
- procesy pierwotne – przygotowanie ropy do przeróbki (osuszenie, odsalanie) i destylacja
atmosferyczna, próżniowa (refrakcjonowanie)
- przeróbka wtórna (procesy wtórne) – reforming, hydroodsiarczanie (hydrotreating – traktowanie
wodorem)
- procesy przeróbki pogłębionej – krakin katalityczny i termiczny, hydrokraking, hydroodsiarczanie
gudronu, koksowanie
- procesy pozyskiwania komponentów benzyn – alkilacja, izomeryzacja
A. klasyfikacja rop naftowych ze względu na:
gęstość:
- konwencjonalne ropy naftowe
- ciężkie ropy naftowe
- piaski bitumiczne
zawartość siarki (skład frakcyjny):
- ciężka ropa (3,7% siarki) więcej metali: nikiel, wanad (wartości w ppm)
- lekka ropa (0,5% siarki) mniej metali
zawartość składników grupowych:
- nierozpuszczalne: asfalteny (najcięższe)
- rozpuszczalne: składniki nasycone, węglowodory aromatyczne, frakcje: żywice 1, żywice 2.
Podstawowe właściwości fizykochemiczne rop naftowych:
- liczba kwasowa – ilość w mg KOH przypadająca na 100g próbki charakteryzuje składniki ropy naftowej.
Wraz ze wzrostem liczby kwasowej zwiększa się charakter aromatyczny oraz zawartość heteroatomów
(N, S, O).
Składniki nasycone
Węglowodory aromatyczne wzrost liczby kwasowej
Żywice 1
Żywice 2
Asfalteny
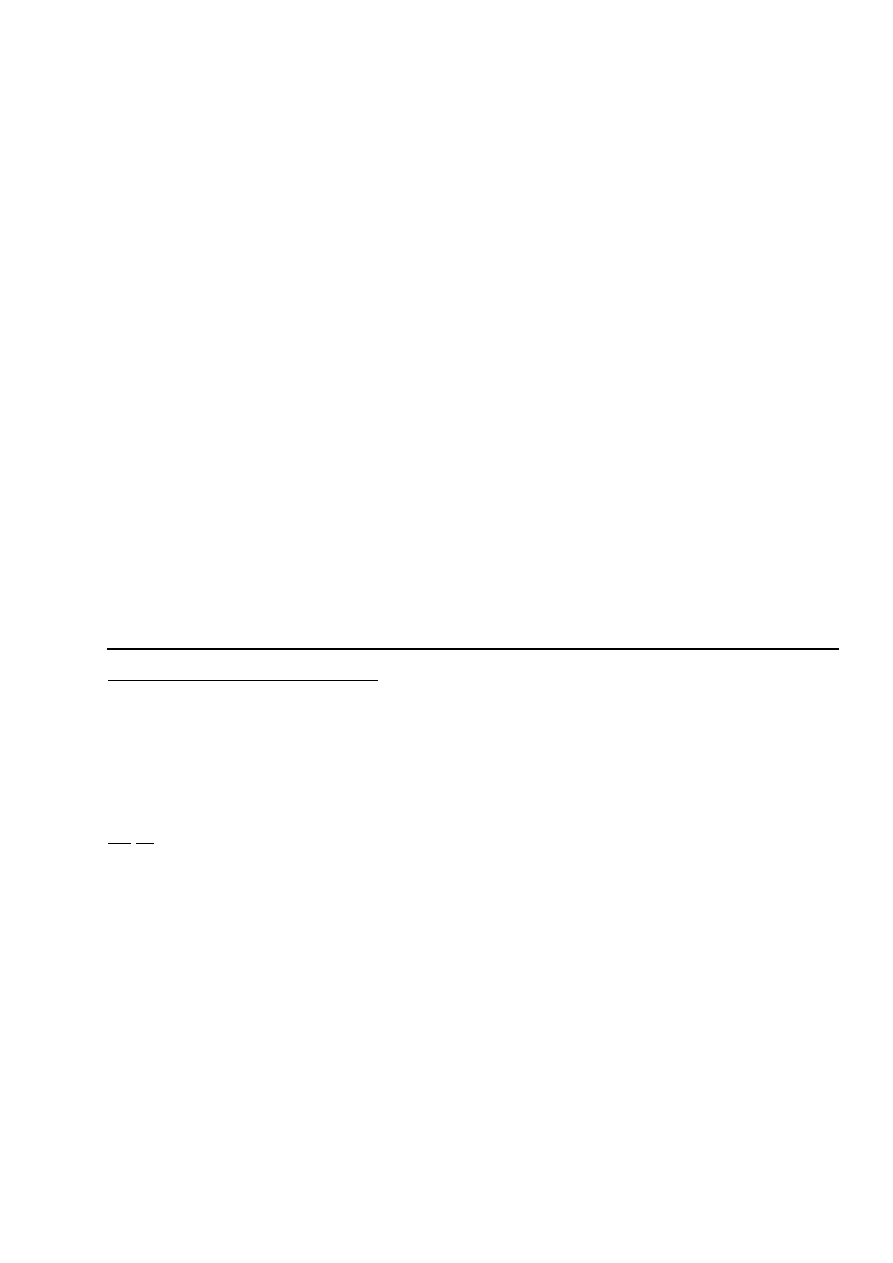
2
Zawartość w ropie naftowej:
a) siarki:
nisko siarkowe ropy <0,5%
średnio siarkowe ropy 0,5-1%
siarkowe ropy 1-3%
wysoko siarkowe ropy >3%
b) parafin (świecie, parafiny farmaceutyczne)
nisko parafinowe ropy 0-5%
średnio parafinowe ropy 5-10%
wysoko parafinowe ropy <10%
c) żywic
nisko żywiczne ropy 0-5%
średnio żywiczne ropy 5-10%
wysoko żywiczne ropy <10%
d) asfaltenów
nisko asfaltenowe ropy <1%
średnio asfaltenowe ropy 1-3%
wysoko asfaltenowe ropy >3%
B. Systematyka technologiczna węgli (typy węgli, składniki węgli, asortyment węgli). Podstawowe
właściwości fizykochemiczne węgli.
Typy węgli, w zależności od zawartości węgla (rosnąco):
torf (ok. 60%C)
Właściwości torfu uzależnione są od składu florystycznego zbiorowisk torfotwórczych oraz
stosunków wodnych i temperaturowych (klimatycznych).
węgiel brunatny (62-75%C)
Węgiel brunatny jest nieodnawialnym źródłem energii. Jego wartość opałowa waha się od 7,5 do 21
MJ/kg
węgiel kamienny (75-97%C)
Ma czarną barwę, matowy połysk, czarną rysę. Wartość opałowa czystego pierwiastka węgla wynosi
ok. 33,2 MJ/kg. Węgiel kamienny jest nieodnawialnym źródłem energii.
Asortyment węgli aktywnych:
granulowane (formowane) węgle aktywne cylindryczne granule wytwarzane są z pyłu węgla
kamiennego i substancji lepiszczowej.
ziarniste (łamane) – nieregularne ziarna. Wytwarzane są z wyreparowanych form ziarnistych węgli
kamiennych podczas ich karbonizacji, a następnie następuje aktywacja parą wodną.
węgle aktywne pyliste – wytwarzane SA na drodze rozdrabniania węgli aktywnych ziarnistych oraz
partii nieudanych węgli formowanych.
Węgiel aktywny wytwarzany z surowców:
kopalnych (węgiel kamienny, brunatny i torf),
roślinnych(drewno drzew liściastych, pestki owoców i skorupy orzechów kokosowych).
3
C. Klasyfikacja gazów ziemnych ze względu na skład, składniki węglowodorowe i nie
węglowodorowe gazów ziemnych. Podstawowe właściwości fizykochemiczne gazów ziemnych.
Podział gazu ziemnego:
gaz suchy metanowy, 97,8 % CH
4
, 1% N
2
, wartość opałowa nie mniejsza niż 31 MJ/m
3
(warunki normalne)
gaz suchy zaazotowany,(typ Lw) 79% CH
4
+ 19,5 % N
2
, wartość opałowa nie mniejsza niż 27
MJ/m
3
gaz suchy zaazotowane,(typ Ls) 71% CH
4
+ 27% N
2
,wartość opałowa nie mniejsza niż 27
MJ/m
3
gaz ziemny azotowy, ponad 90% N
2
Zależnie od pochodzenia gaz ziemny stanowi mieszaninę homologów (od metanu do pentanu),
związków siarki, CO
2
, O
2
, H
2
, N
2
, He. W zależności od składu rozróżnia się 2 gazy:
- suchy, zawierający łącznie 95% metanu i etanu
- mokry, w którym obok metanu i etanu występują także cięższe węglowodory tego szeregu w
ilościach dochodzących do 30%;
Gaz ziemny towarzyszący ropie naftowej jest gazem mokrym.
Oprócz węglowodorów gaz ziemny może zawierać także azot, dwutlenek węgla i
siarkowodór(niekiedy do 30% N3,do 40% CO2 i do 50% H2S) oraz niewielkie ilości helu(najbogatsze,
wydobywane w USA gazy zawierają do 1,35% He).
Właściwości chemiczne gazu ziemnego:
Gaz ziemny w stanie naturalnym jest bezbarwny, bezwonny, palny (w powietrzu spala się on
jasnoniebieskim płomieniem, przy czym wydzielają się duże ilości ciepła), lżejszy od powietrza.
Liczba Wobbego –– jest to stosunek wartości kalorycznej odniesionej do jednostki objętości gazu,
do pierwiastka kwadratowego jego gęstości względnej, w tych samych warunkach odniesienia. Jeśli przez Q
oznaczymy wartość kaloryczną, a przez d gęstość względną, wtedy liczbę Wobbego W można przedstawić
następująco
Wyróżnia się:
- dolną Liczbę Wobbego – gdy za wartość kaloryczną przyjmuje się jego wartość opałową;
- górną Liczbę Wobbego – gdy za wartość kaloryczną przyjmuje się jego ciepło spalania.
Wartość liczby Wobbego jest podstawą do podziału paliw gazowych na podgrupy.
Jednostki: MJ/m³, MJ/mol, MJ/kg. Liczba Wobbego – wskaźnik oceny stałości właściwości użytkowych
gazu.
2. Biogaz, pozyskiwanie biogazu.
Biogaz – paliwo gazowe produkowane z biomasy i/lub ulegającej biodegradacji części odpadów, które
można oczyścić do jakości naturalnego gazu i używać jako biopaliwo.
Skład:
- metan (40-70%)
- CO
2
(40-50%)
- inne składniki (azot, siarkowodór, tlenek węgla, amoniak, tlen)
Do produkcji energii cieplnej lub elektrycznej może być wykorzystywany biogaz zawierający powyżej 40%
metanu.

4
Biogaz powstaje w wyniku:
- fermentacji odpadów organicznych na składowiskach odpadów (gaz wysypiskowy) – Polska: 18 instalacji
1 tona odpadów = 200 m
3
biogazu
- przetwarzania odchodów zwierząt hodowlanych
1 m
3
płynnych odpadów = 20 m
3
biogazu
1 m
3
obornika = 30 m
3
biogazu
- fermentacja: roślin uprawnych (kukurydza, rzepak), słoma, trawa, łodygi, odpady rolnicze (obierki,
wysłodki)
- osady ściekowe (oczyszczalnie ścieków)
1 m
3
osadu = 10-20m
3
biogazu
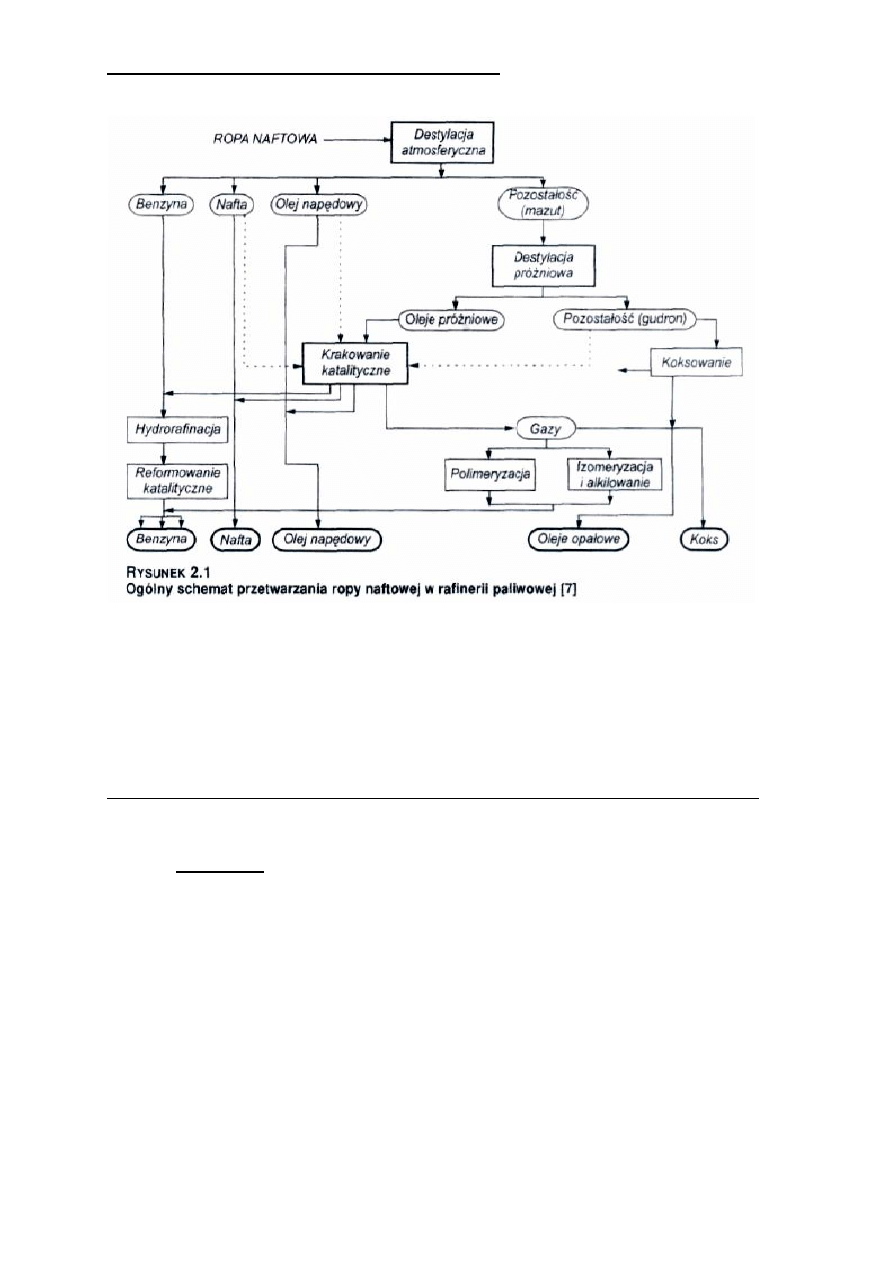
3. Przerób ropy naftowej; destylacja, procesy pogłębionej przeróbki ropy naftowej.
Pierwotna przeróbka ropy naftowej:
- przygotowanie ropy do przeróbki (osuszanie i odsalanie)
Ropa wydobyta ze złoża zawiera zawsze zanieczyszczenia mechaniczne oraz pewną ilość wody
z rozpuszczonymi w niej solami (głównie chlorkami). Woda (solanka) występuje w ropie w postaci emulsji,
w której stanowi fazę rozproszoną, która utrudnia destylację. Głębokie odsolenie ropy jest konieczne, gdyż
zmniejsza korozję aparatury w instalacjach destylacji ropy i w instalacji dalszej przeróbki różnych frakcji
naftenowych.
- destylacja rurowo-wieżowa (DRW)
W instalacjach destylacji ropy naftowej głównymi aparatami są piece rurowe i kolumny rektyfikacyjne
zwane wieżami stąd nazwa tej destylacji (DRW). Nowoczesne instalacje DRW pracują w sposób ciągły
i zazwyczaj są dwustopniowe. Pierwszy stopień stanowi destylacja pod ciśnieniem atmosferycznym, drugi
pod ciśnieniem zmniejszonym (próżniowa)
Frakcje otrzymywanie w procesie destylacji ropy:
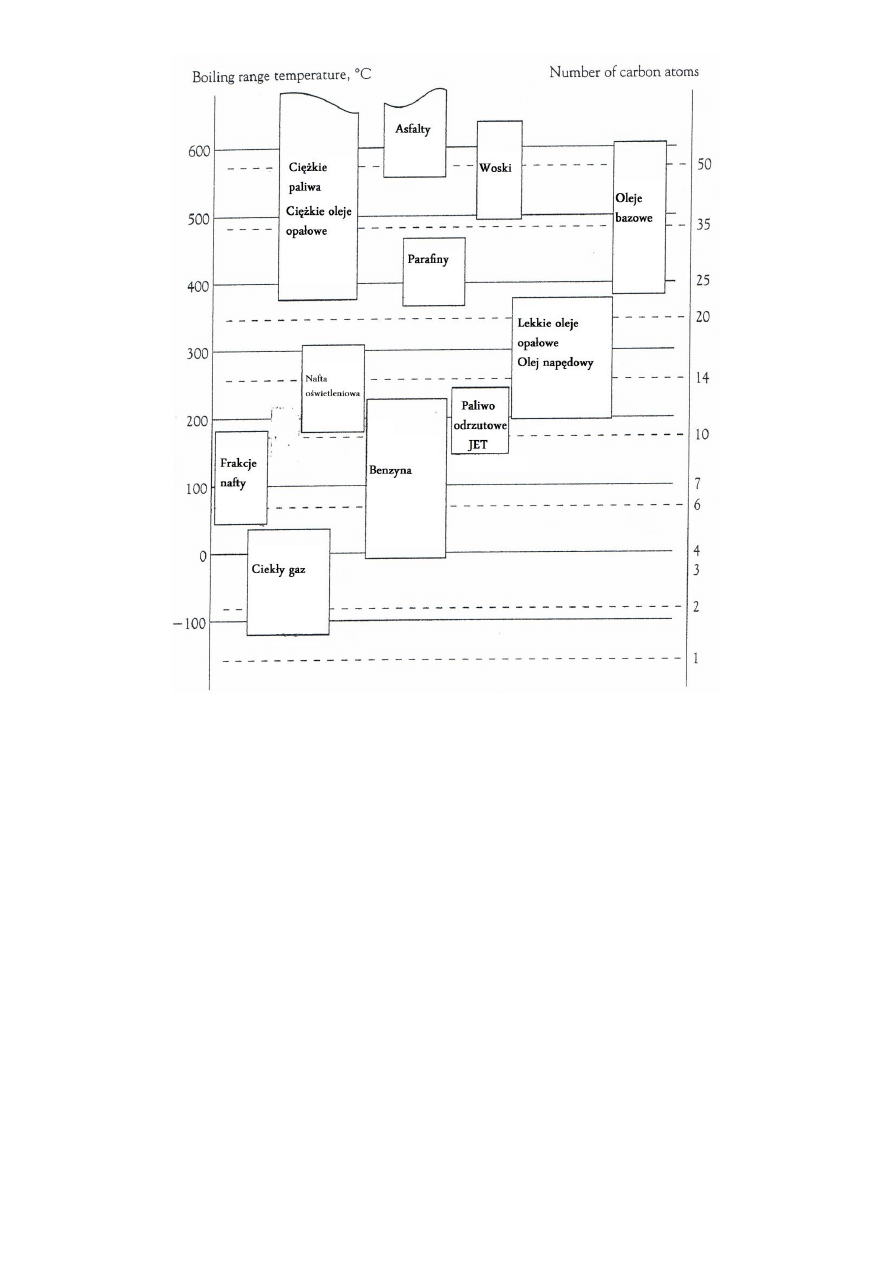
5
6
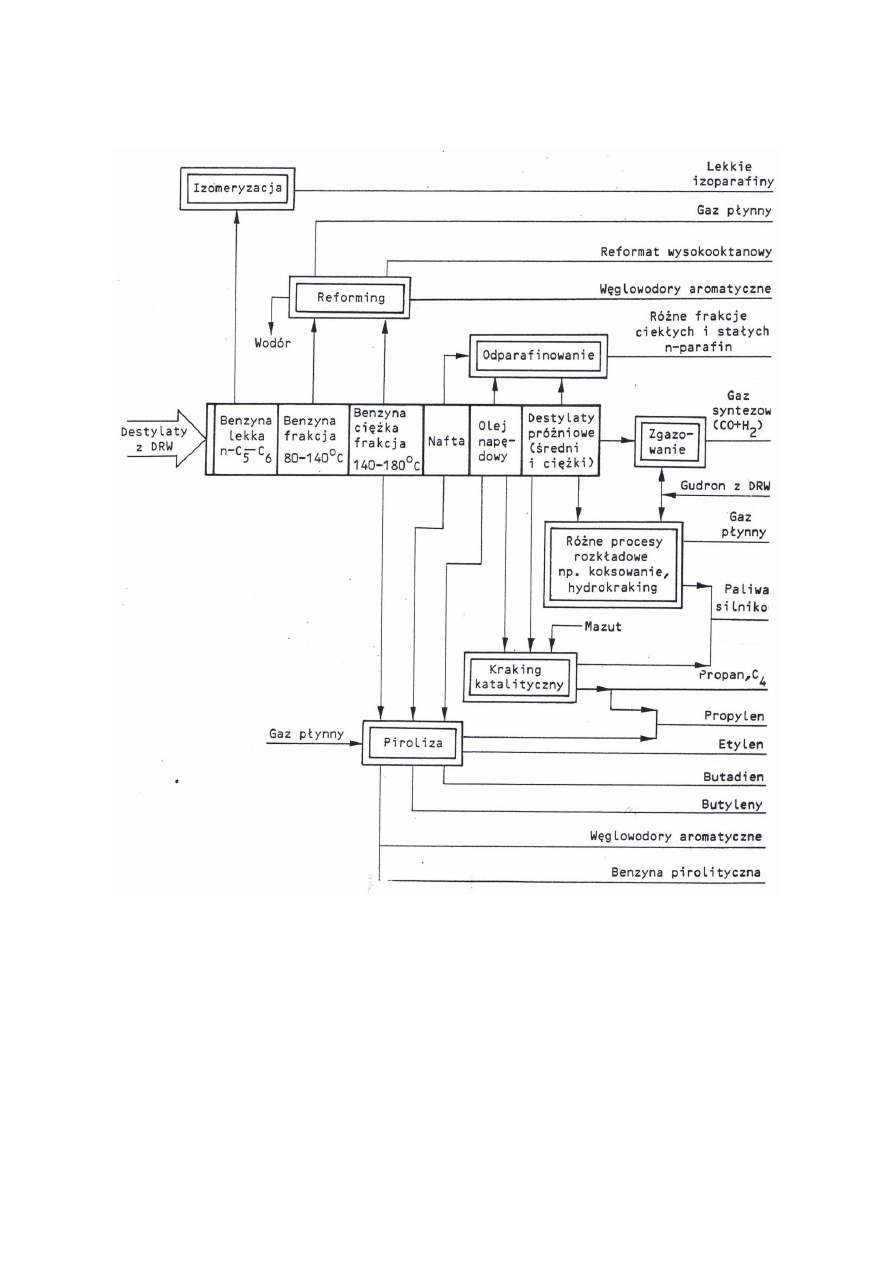
- kierunki dalszej przeróbki produktów DRW
W procesach przeróbki ropy naftowej i frakcji węglowodorowych z niej otrzymywanych wytwarza się:
- gaz płynny
- paliwa silnikowe (benzyny, oleje napędowe, paliwa odrzutowe JET)
- oleje smarowe i smary
- oleje opałowe
- stałe węglowodory naftowe (parafiny, cerezyny, wazeliny)
- asfalty drogowe i przemysłowe
- surowce węglowodorowe do syntez organicznych
7
4. Procesy przemysłu rafineryjnego dla pozyskiwania paliw: hydrorafinacja, reforming, hydrokraking,
kraking katalityczny
Kraking katalityczny
a) Kraking termiczny, kraking katalityczny: typowe surowce, parametry procesu, produkty
Proces
Typowe surowce
Parametry procesu
Produkty
Kraking termiczny
(proces wysokociśnieniowy)
pozostałość atm.,
destylaty próżniowe
550ºC, 2-5MPa
benzyna
olej napędowy
lekkie oleje opałowe
Kraking termiczny
(proces niskociśnieniowy)
lekki olej próżniowy
destylaty próżniowe
> 500ºC, < 0,7MPa
< 500ºC, 2,8-7MPa
benzyna
olej napędowy
olej opałowy
frakcje gazowe
Kraking katalityczny
lekki olej próżniowy
destylaty próżniowe
470-525ºC, 0,7-1,4MPa
benzyna. olej napędowy
olej opałowy,
frakcje gazowe
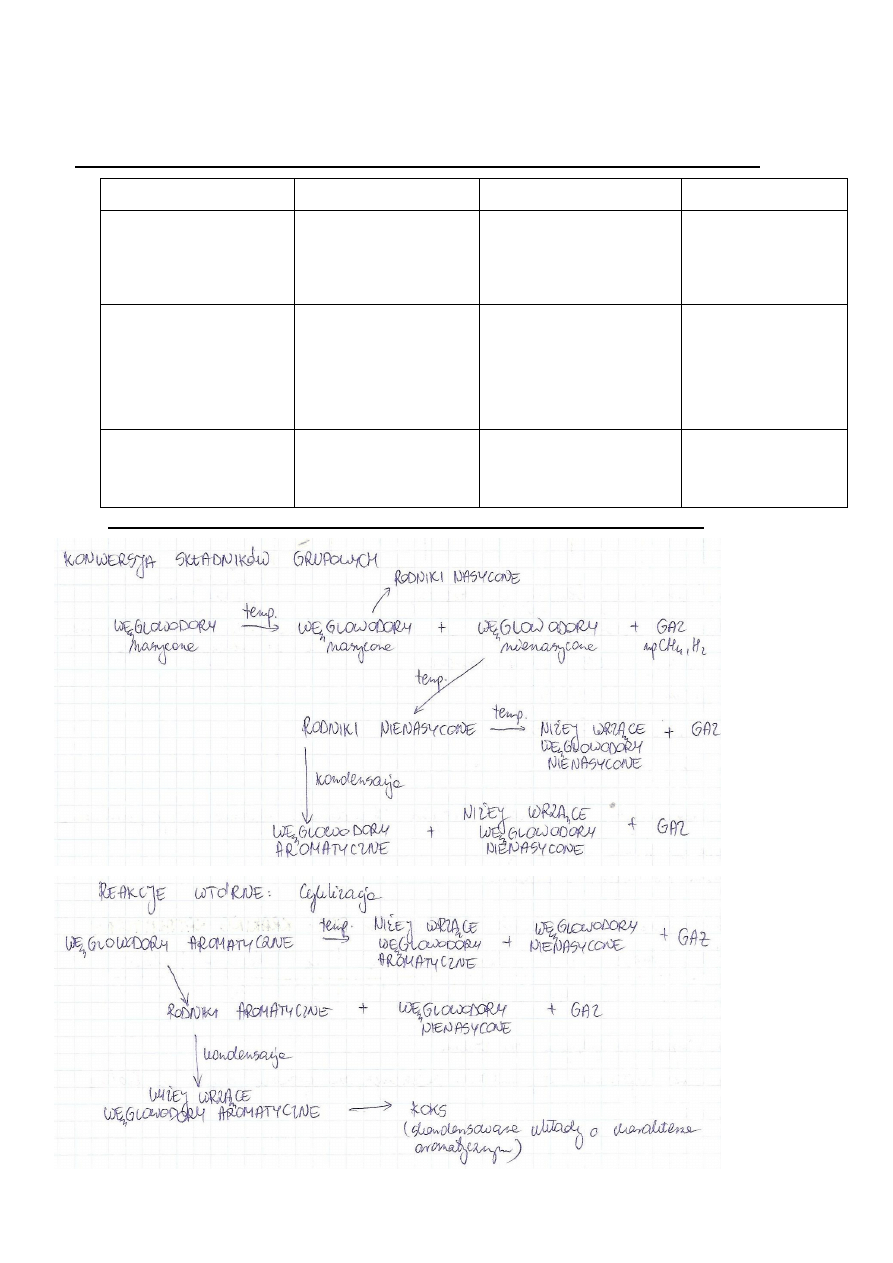
b) Konwersja składników grupowych frakcji ropy naftowej w procesie krakingu
8
c) Przebieg procesu przemysłowego. Wpływ rozwiązań aparaturowych reaktora na wydajność
produktów procesu krakingu katalitycznego
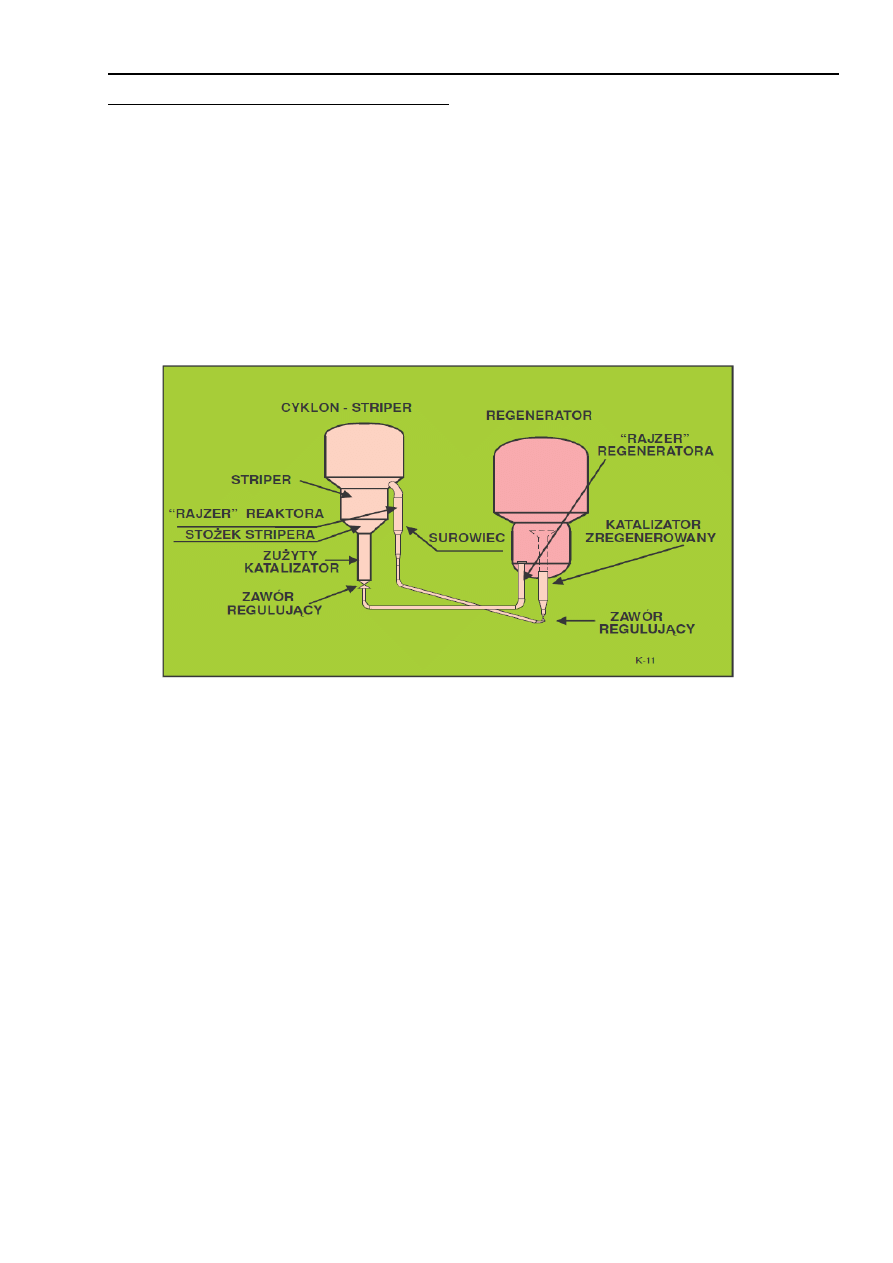
Proces krakingu katalitycznego zachodzi w temperaturach 470-525°C przy ciśnieniu bliskim
atmosferycznemu (maksymalnie 1.5 bar nadciśnienia). W obecnie stosowanych procesach reakcja krakingu
nie zachodzi w złożu fluidalnym - katalizator jest transportowany pneumatycznie w strumieniu par surowca
i właśnie wówczas zachodzą reakcje krakingu. Niemniej jednak nadal utrzymuje się nazwa FCC (procesy
fluidalne) chociaż naprawdę w złożu fluidalnym prowadzona jest tylko regeneracja katalizatora.
Nowoczesne instalacje są wyposażane w podnośnik pneumatyczny (ang. riser). Nazwa ta - "raizer" -
przyjęła się w międzynarodowej gwarze naftowej. W typowej instalacji jest pionowa rura o średnicy około 1
m i wysokości do ok. 30 m. Stanowi ona właściwy reaktor, czas przebywania surowca i katalizatora w
podnośniku wynosi około 1 sek. W tych warunkach ważna jest budowa zakończenia "raizera". Występują
tam duże siły i erozja związane ze zmianą kierunku ruchu. Przykładem współczesnych rozwiązań może być
Flexicraking IIIR opracowany przez firmę Exxon, jest ono zbliżone do modelu IV.
Przed wejściem do "raizera", surowiec poprzez specjalny system wstrzykujący jest kontaktowany
z katalizatorem transportowanym strumieniem pary wodnej. Górne zakończenie "raizera" stanowią
sprzężone cyklony oddzielające katalizator od produktu, są dwa stopnie oddzielania pyłu. Katalizator opada
poprzez zasuwę dławiącą do regeneratora. Po drodze jest przeparowywany aby usunąć zaadsorbowane
węglowodory i zmniejszyć ilość koksu w katalizatorze. Para wodna odcina przestrzeń z palnymi parami
węglowodorów od części gdzie jest podawane powietrze do regeneracji. Na wypadek niebezpieczeństwa są
mechaniczne zawory odcinające. Regenerator stanowi jedno naczynie z urządzeniami zapewniającymi
ciągłość długotrwałego ruchu. Można prowadzić wypalanie całkowite lub częściowe. Z regeneratora można
otrzymywać gaz opałowy. Jest on odpylany w cyklonach i myty w skruberach. W instalacji K-13 można
przetwarzać destylaty próżniowe, ekstrakty z olejów smarowych, oleje odasfaltowane i (lub) pozostałości.
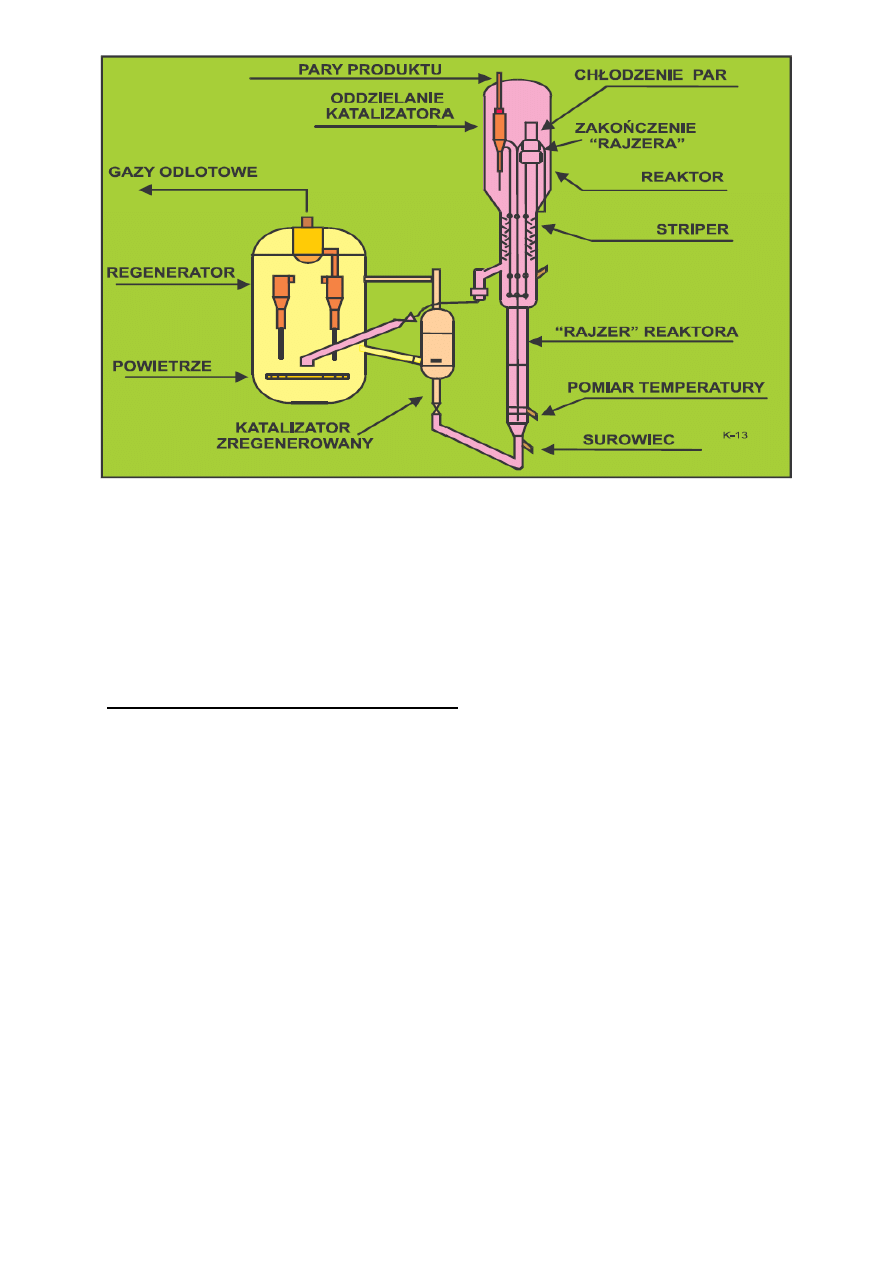
9
Instalacje krakingu katalitycznego należą w rafinerii do tych, które w największym stopniu obciążają
środowisko. W trakcie regeneracji katalizatora z siarki zawartej w osadach koksu powstają tlenki siarki.
Dodatkowo, tworzą się znaczne ilości tlenków azotu. W zależności od lokalnych uwarunkować, niezbędnym
może być odsiarczanie (deSOx) i odazotowanie (deNOx) spalin z regeneratora. Oprócz tego, w zależności
od skuteczności pracy cyklonów, różne ilości pyłu katalizatora (zawierającego metale ciężkie) również
mogą być emitowane. Siarka zawarta w surowcu trafia do produktów ciekłych reakcji, tworzy H
2
S
zanieczyszczający lekkie gazy krakingowe oraz osadza się z koksem na katalizatorze.
Reforming
a) Surowiec i jego przygotowanie do reformingu
Surowcem do procesu jest benzyna o granicach wrzenia od 60-80°C do 160-190°C. W wyniku
reformowania temperatura końca wrzenia wzrasta o około 15°C ponieważ węglowodory aromatyczne mają
wyższe temperatury wrzenia. W wielu przypadkach stosowane są węższe frakcje, zwłaszcza gdy celem jest
wytwarzanie węglowodorów aromatycznych a nie benzyny wysokooktanowej. Maksymalna dopuszczalna
temperatura końca wrzenia surowca wynosi 204°C. Obecnie, w związku z ograniczaniem zawartości
benzenu w benzynach, z wsadu na reforming usuwa się prekursory benzenu, zwłaszcza nafteny C6. Dlatego
początek temperatury wrzenia wsadu jest obecnie powyżej 85oC – dla usunięcia wszystkich prekursorów
należałoby stosować surowiec o początku wrzenia 104-110oC.
Lekka benzyna, wrząca poniżej tej temperatury jest zwykle kierowana do procesu hydroizomeryzacji,
jednak możliwość ta jest ograniczona ze względu dopuszczalną zawartością benzenu i heptanu we wsadzie
na ten proces. Ilość benzenu powstającego w procesie reformowania zależy od temperatury i ciśnienia
procesu. Surowiec do reformowania nie może zawierać olefin (starsze procesy prowadzone przy wyższym
ciśnieniu dopuszczały zawartość olefin do 2 %). Siarka (i jej związki) są trucizną dla katalizatora
reformingowego, dlatego surowce muszą być głęboko rafinowane. Kiedyś dopuszczano zawartość siarki do
10 ppm. Współczesne instalacje wymagają zawartości siarki poniżej 0,5 ppm a nawet poniżej 0,2 ppm,
zawartość azotu poniżej 0,5 ppm. Surowce zawierające dużo węglowodorów parafinowych są mniej
przydatne dla reformowania, wymagają wyższych temperatur pracy i częstszych regeneracji, dają mniejszą
wydajność produktu o określonej liczbie oktanowej. Zawartość węglowodorów aromatycznych w surowcu
wynosi około 20 % lub zwykle mniej. Dla określenia jakości surowca do reformowania przyjmuje się sumę
N+2A gdzie N oznacza zawartość naftenów (% vol.) a A- węglowodorów aromatycznych; pożądane są
surowce, które mają wartość N+2A powyżej 60. Główny składnik większości katalizatorów reformingu –
10
platyna – jest wrażliwy na zatrucie siarką (siarkowodorem), amoniakiem oraz organicznymi związkami
siarki i azotu. Dlatego surowiec reformingu musi być wcześniej poddany hydrorafinacji.
b) Przebieg procesu przemysłowego. Instalacje z ciągłą cyrkulacją i regeneracją katalizatora, inst
alacje z cykliczną regeneracją katalizatora
Reformowanie prowadzi się w temp.480-525 st.C i pod ciśnieniem 0,7-3 MPa. Bardzo ważne jest
utrzymanie intensywności cyrkulacji gazu wodorowego dobranej odpowiednio masowego natężenia
dopływu surowca (benzyny). Zapobiega to zakoksowaniu i dezaktywacji katalizatora. W praktyce
przemysłowej ostrość procesu reformingu ocenia się na podstawie dwóch parametrów: molowego stosunku
wodoru do surowca (węglowodorów) i szybkości objętościowej.
Ogólnie biorąc, ze względu na sposób przeprowadzania regeneracji można wyróżnić trzy rodzaje procesów:
1. semiregeneratywne
2. cykliczne
3. z ruchomym złożem
Instalacje semiregeneratywne są okresowo zatrzymywane dla przeprowadzenia regeneracji katalizatora we
wszystkich reaktorach. Takimi były pierwsze instalacje platformingu. Okresy między regeneracjami
wynosiły od 6 miesięcy do roku. Pracowały one przy stosunkowo wysokim ciśnieniu do 3 a nawet 3,5 MPa.
W Polsce były to stare reformingi w
rafinerii w Płocku. Uzyskiwane liczby oktanowe w zasadzie były poniżej 100.
Instalacje cykliczne
Instalacje te oprócz reaktorów aktualnie wykorzystywanych w procesie, miały dodatkowy reaktor, tzw.
swing reaktor, który mógł być włączany zamiast każdego z 3 – 4 reaktorów normalnych, gdy był on
wyłączany na czas regeneracji.
Jednostki te pracowały przy niższym ciśnieniu 0,7-1,5 MPa dzięki czemu można było uzyskać wyższe
liczby oktanowe, ponad 100. Ale niskie ciśnienie powodowało znacznie szybsze zakoksowanie i regenerację
trzeba było prowadzić co 5 do 14 dni. Częściej regenerowano reaktor ostatni niż pierwszy ponieważ w nim
była wyższa średnia temperatura. W instalacjach cyklicznych wydajności wodoru i produktu ciekłego były
bardziej stabilne. Katalizator wytrzymywał do 600 regeneracji. Instalacją tego rodzaju był stary reforming w
rafinerii w Gdańsku.
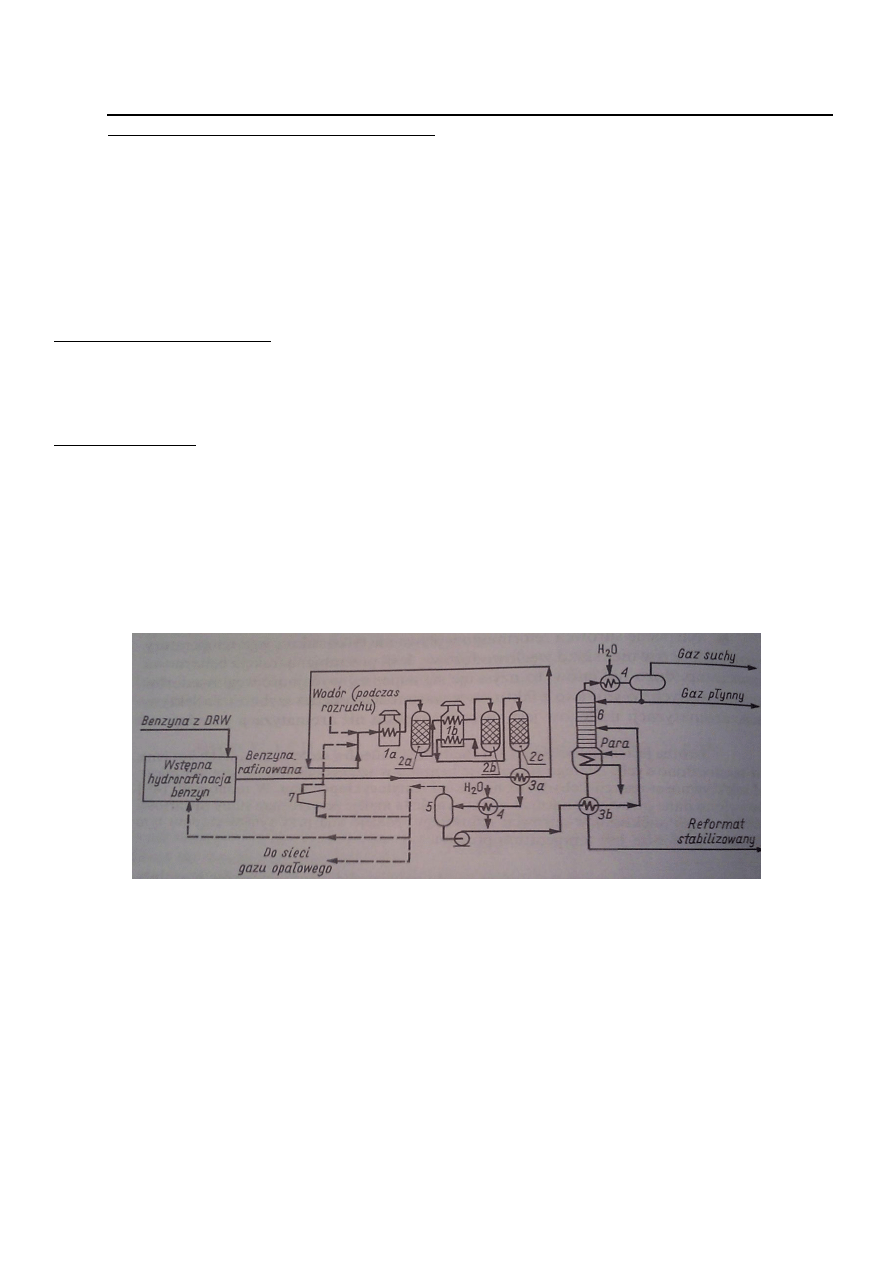
Benzynę po wstępnej hydrorafinacji ogrzewa się w wymienniku 3a. Po zmieszaniu z obiegowym gazem
wodorowym (ze sprężarki 7) wprowadza się ją do pieca rurowego 1a. Mieszanina jest ogrzewana w tym
piecu do temp.480st.C i kierowana następnie do pierwszego reaktora reformingu. Aby zdążyły zajść
wszystkie pożądane reakcje, niezbędne jest przeprowadzenie reagującej mieszaniny przez trzy szeregowo
pracujące reaktory (2a,2b,2c). Pomiędzy reaktorami jest umieszczony piec 1b. Takie między stopniowe
podgrzewanie jest konieczne ze względu na endotermiczność reakcji zachodzących w dwóch pierwszych
reaktorach. W pierwszym reaktorze 2a przeważają reakcje odwodornienia naftenów do węglowodorów
aromatycznych, w drugim 2b reakcja dehydrocyklizacji parafin, a w trzecim reakcje ich izomeryzacji.
Hydrorafinacja
a) Podstawowe reakcje zachodzące w tym procesie (konwersja heterozwiązków, konwersja węglo
wodorów aromatycznych)

11
Termin hydrorafinacja (HR) stosuje się do grupy procesów uwodornienia (HYD), hydroodsiarczania
(HDS), hydroodazotowania (HDN) i hydrodemetalizacji (HDM), którym w rafinerii poddaje się
różne strumienie naftowe.
b) Wpływ temperaturowego zakresu wrzenia frakcji na parametry procesu hydrorafinacji
W miarę wzrostu średniej temperatury wrzenia surowca, jego hydrorafinacja wymaga coraz
ostrzejszych warunków: wyższej temperatury i ciśnienia, dłuższego czasu pobytu (czasu reakcji)
oraz większego nadmiaru wodoru.
12
5. Przykładowa struktura produkcji paliw w rafinerii
Produkt:
Udział w produkcji (%)
Gaz płynny (LPG)
2,6
Benzyny
30,8, w tym 7 % Super Plus 98
Paliwo lotnicze
4
Oleje napędowe
37,1
Ekoterm plus
17,1
Olej opałowy ciężki
8,4
6. Przykładowe procesy przemysłu rafineryjnego dla pozyskiwania surowców do syntez
a) Wytwarzanie węglowodorów aromatycznych: reforming i ekstrakcyjne wydzielanie węglowodorów
aromatycznych
Podczas reformingu ropy naftowej otrzymuje się związki aromatyczne, na drodze procesu, w
którym alkany są przepuszczane nad katalizatorem (platyna) pod temperaturą około 500°C, pod
zwiększonym ciśnieniem. Głównym produktem jest benzen, jest on jednym z najważniejszych
surowców w przemyśle organicznym; stosowany m.in. do produkcji tworzyw sztucznych, włókien
syntetycznych, barwników, detergentów, pestycydów, otrzymywania fenolu, aniliny, bezwodnika
maleinowego, także jako rozpuszczalnik.
13
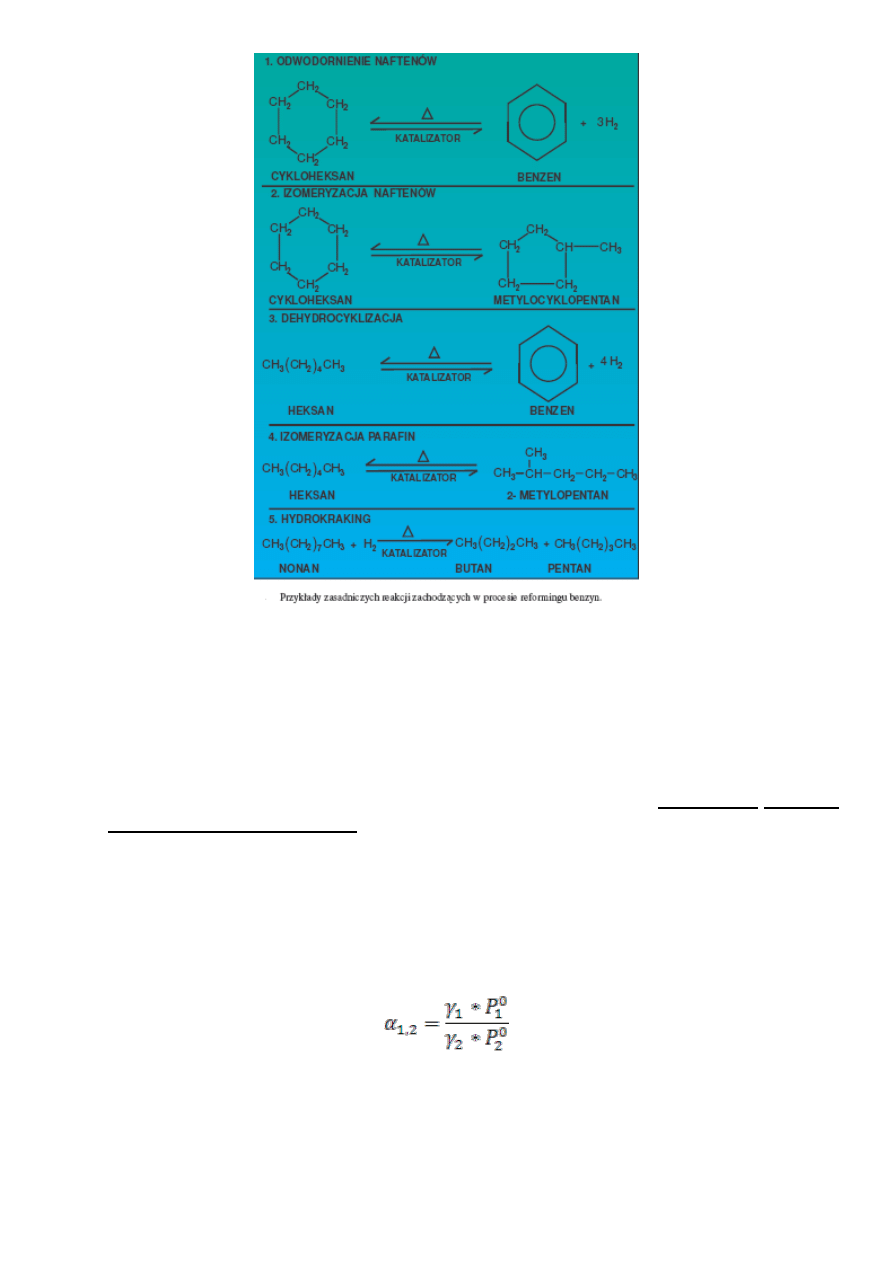
Na schemacie przedstawiono przykłady głównych reakcji zachodzących podczas reformingu katalitycznego.
Zasadnicze reakcje to:
- odwodornienie cykloparafin do węglowodorów aromatycznych,
- izomeryzacja cykloparafin o pierścieniach pięcio- lub sześcioczłonowych,
- dehydrocyklizacja parafin łańcuchowych do węglowodorów aromatycznych,
- izomeryzacja parafin, hydrokraking parafin i cykloparafin.
Powszechnie stosowaną techniką wydzielania węglowodorów aromatycznych z mieszanin
zawierających węglowodory aromatyczne i niearomatyczne jest destylacja ekstrakcyjna z czystym
selektywnym rozpuszczalnikiem lub mieszaniną rozpuszczalników. Układ destylacji ekstrakcyjnej
składa się z dwóch kolumn destylacyjnych. Do górnej części pierwszej kolumny (kolumny destylacji
ekstrakcyjnej) wprowadzany jest cyrkulujący rozpuszczalnik, a do środkowej części tej kolumny
mieszanina węglowodorów aromatycznych i niearomatycznych. Rozpuszczalniki to mieszaniny N-
metylopirolidon-woda.
Sposoby oparte na destylacji ekstrakcyjnej zapewniają uzyskanie najwyższego stopnia oczyszczenia
węglowodorów aromatycznych. Wielkością pozwalającą ocenić skuteczność rozdziału mieszaniny dwóch
składników 1 i 2 przez destylację jest lotność względna a zdefiniowana jako:
gdzie: α — lotność względna składnika 1 względem składnika 2, 1 γ1 i γ2 — współczynniki
aktywności składników 1 i 2 w fazie ciekłej, P°i i P°2 — prężność par nasyconych składników
czystych.
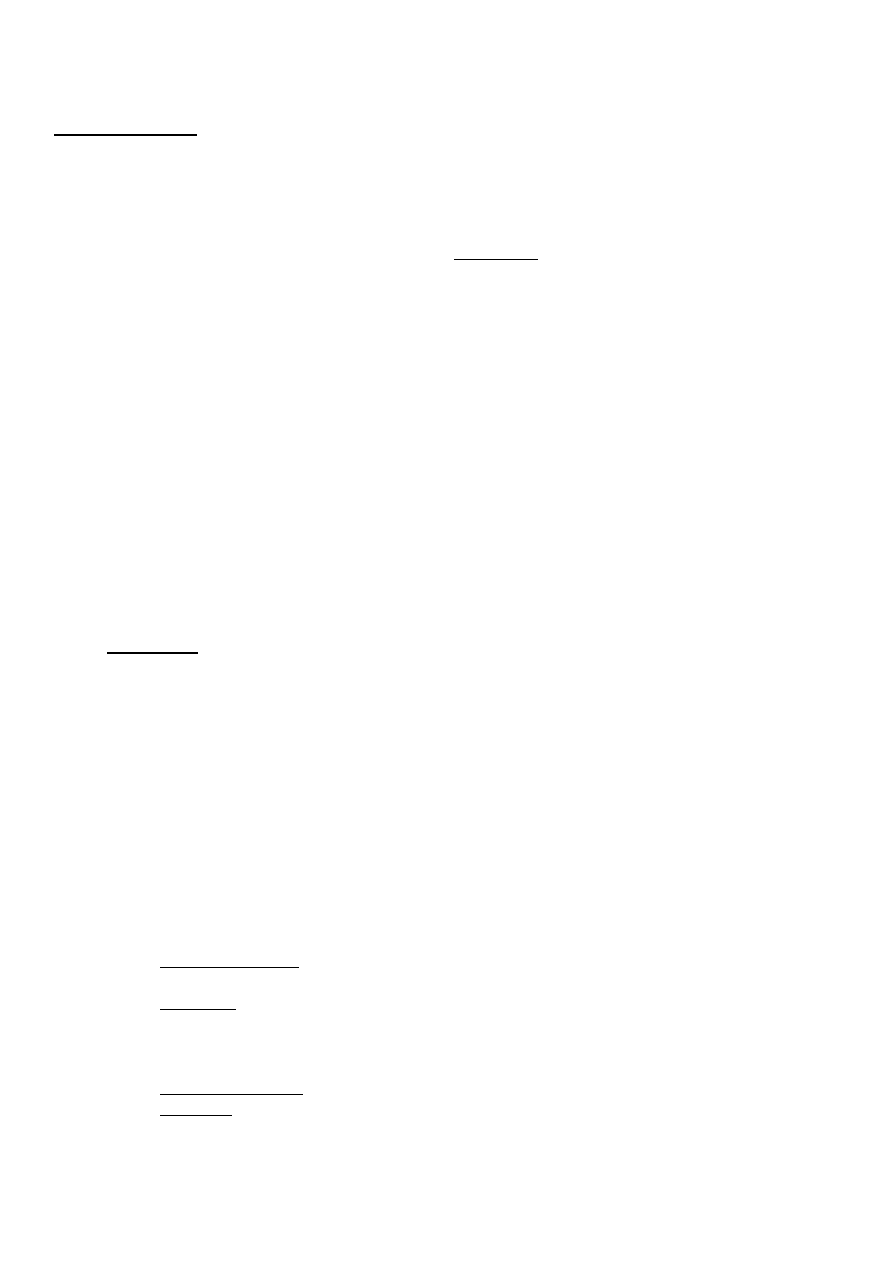
14
b) Procesy wytwarzania węglowodorów olefinowych (piroliza olefinowa), surowce i warunki procesu,
względny udział węglowodorów olefinowych.
Piroliza olefinowa stanowi proces polegający na degradacji cząsteczek wyższych węglowodorów pod
wpływem temperatury 780-900oC i ciśnieniem 0,1 do 0,3 MPa. Proces pirolizy przeprowadza się w celu
otrzymania nienasyconych węglowodorów alifatycznych (etylenu, propylenu, butenów i butadienu) cennych
surowców w syntezie organicznej. Substrat wyjściowy w procesie pirolizy stanowić mogą różne produkty
naftowe, najczęściej jednak są to ciężki benzyny i frakcje zbliżone do niej. Uzyskuje się głównie benzynę
pirolityczna w wydajności ok. 25%, etylen ok. 28%, propylen ok.17%.W procesie pirolizy, przebiegającej w
temperaturze znacznie wyższej aniżeli typowy proces krakowania dochodzi do bardzo głębokich przemian
surowca. Zwiększenie temperatury prowadzi do zwiększenia wydajności etylenu, natomiast nieznaczne
obniżenie temperatury procesu prowadzi do zwiększenia wydajności propylenu lub wyższych olefin. Górny
pułap zakresu temperatury określany jest przez temperaturę prowadzącą do całkowitego rozkładu
węglowodorów do składników prostych, jak metan, wodór i węgiel. Dla uniknięcia daleko posuniętych
przemian, przejawiających się poprzez nadmierne wydzielanie koksu, stosuje się dodatek pary przegrzanej.
Zabezpiecza ona przed wydzielaniem pierwiastkowego węgla już w chwili jego powstawania i w postaci
gazowej jest odprowadzany poza środowisko reakcyjne. Dodatek pary wodnej wpływa również na korzystne
ustalenie stanu równowagi reakcyjnej powstawania olefin w warunkach podwyższonego ciśnienia
panującego w czasie ogrzewania surowca. Z punktu widzenia wydajności powstawania propylenu lub
etylenu z odpowiednich parafin ogrzewanie surowca pod zwiększonym ciśnieniem jest niekorzystne.
Prowadzi, bowiem do zwiększenia objętości mieszaniny reakcyjnej ulegającej rozpadowi termicznemu i
odwodornieniu. Dlatego w praktyce stosuje się obniżone ciśnienie dla reakcji odwodornienia i rozdział
poreakcyjnych gazów, a następnie ich skroplenie celem sprawniejszego transportu i magazynowania.
Instalacje służące do otrzymywania olefin zbudowane są zazwyczaj jako ogrzewane reaktory, najczęściej w
postaci pieców rurowych umożliwiających bardzo szybkie ogrzanie surowca do bardzo wysokiej
temperatury. Czas pobytu mieszaniny w środowisku powinien być możliwie najkrótszy, tak, aby nie
doprowadzić substratów do elementarnego rozkładu. W rzeczywistości wynosi około jednej sekundy.
Opuszczające rektor gazy poreakcyjne są natychmiast chłodzone w celu uniknięcia rekcji wtórnych.
7. Gaz ziemny
a) Instalacje kompleksowego przygotowania gazu
Separacja – wstępne rozdzielanie gazu
Usunięcie pyłu, kropli cieczy (woda, kondensat węglowodorów C
3
)
Osuszanie – woda przyspiesza korozję (rozpuszczanie się kwaśnych składników (H
2
S, CO
2
), powoduje
tworzenie stałych hydratów (związek metanu z dużą ilością cząsteczek wody). Zawartość wody w
gazie kierowanym do transportu wymaga osuszenia do temperatury (-15 °C) – (-25°C)
- absorber:
absorpcja pary wodnej za pomocą glikolu etylenowego (TEG, DEG) przeciwprądowy
przepływ gazu (od dołu absorbera) i glikolu ( z góry absorbera) temp. 30°C.
DEG – glikol dietylenowy,
TEG - glikol trietylenowy.
-desorber:
I etap: roztwór glikolu
Wymiennik ciepła (glikol – glikol); roztwór glikolu nagrzewa się do 90°C i przepływa do
separatora.
Separator (0,12 MPa) – wydzielenie lekkich węglowodorów (gaz resztkowy – gaz
opałowy)
II etap: regeneracja glikolu
Wymiennik ciepła (glikol-glikol) : roztwór glikolu nagrzewa się do wrzenia
Desorber:
15
Dolna część desorbera (ogrzewana parą wodną) – roztwór nagrzewa się do temp. 190-
204 °C. Zregenerowany glikol (odbierany z dołu kolumny) zawiera do 97% TEG lub
DEG.
Środkowa część desorbera temp. 140°C (0,1 – 0,2 MPa)
Górna część desorbera (ok.100°C) Strumień pary wodnej kierowany do chłodnicy
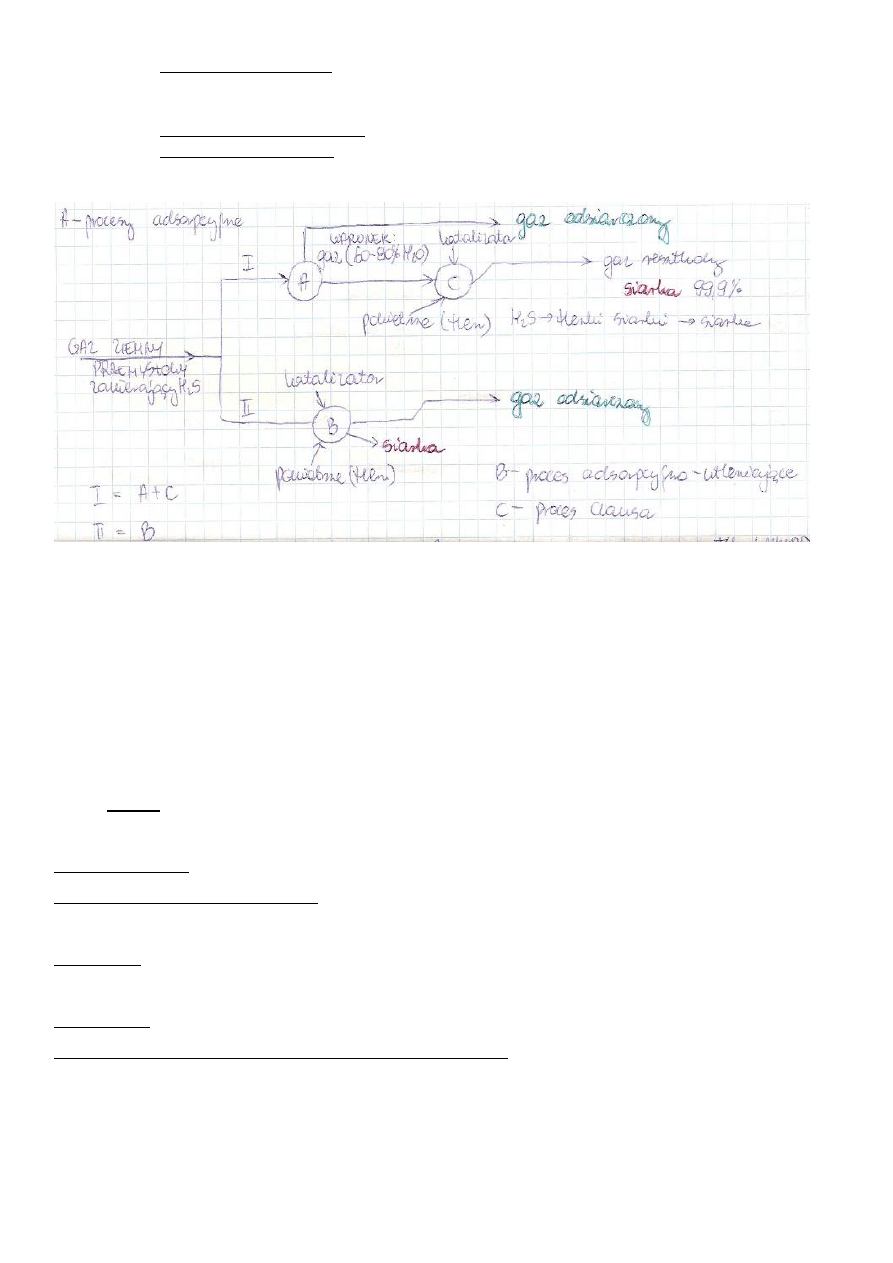
b) Warianty technologiczne odsiarczania gazu
c) Przetwarzanie gazu z uwzględnieniem jego składu/typu
Gaz ziemny metanowy (niekiedy bogaty w etan)
GAZ (etan)→ piroliza → etylen
→ paliwo (przemysł energetyczny gospodarka komunalna, transport)
→ bezpośrednio do syntez chem. (np. chlorometany, acetylen, sadza)
→ konwersja z parą wodną → gaz syntezowy→metanol,amoniak,alkohol
Gaz ziemny zaazotowany
GAZ (odazotowanie) → azot do atmosfery
→ paliwo (przemysł, transport itp.)
8. Węgiel
a) Procesy chemicznej przeróbki węgla.
Zgazowanie węgla
Uwodornienie węgla (upłynnianie): to destruktywne oddziaływanie na węgiel pod wpływem wysokiego
ciśnienia i w obecności wodoru.
Sortowanie: ma na celu podział węgla na klasy według wielkości ziaren i odbywa się ono na sitach
o różnych wymiarach oczek. Liczba sortymentów zależy od potrzeb i właściwości węgla.
Zagazowanie
Odgazowanie lub sucha destylacja( pirogenizacja, piroliza): polega na ogrzaniu węgla do temperatury
powyżej 5000C, w której węgiel rozkłada się na pozostałość stałą tj: półkoks, koks oraz na części lotne,
z których wydzielają się frakcje ciekłe (smoła , benzole) i gazy palne. Mają tu miejsce następujące procesy:
- wytlewanie, czyli proces odgazowania odbywający się w niskiej temperaturze. Powstaje półkoks, który jest

16
paliwem bezdymnym i łatwo palącym się, smoła wytlewna, zwana prasmołą mająca cechy ropy oraz gaz
wytlewny.
- gazownictwo, czyli proces którego celem jest uzyskanie dużej ilości gazu opałowego o wartości opałowej
co najmniej 5 MJ/m3. Oprócz gazu uzyskuje się koks gazowniczy o małej wytrzymałości mechanicznej,
smołę pogazową, benzole i wodę pogazową zawierającą amoniak. Aktualnie gazownictwo w swej
klasycznej postaci zanika.
- koksownictwo, czyli proces który ma na celu otrzymanie koksu
b)
Zgazowanie węgla i kierunki wykorzystania gazu syntezowego (punkt 16 analogiczny)
Zgazowanie węgla, czyli półspalanie z dodatkiem pary wodnej, przy nadmuchu powietrza prowadzi
do otrzymania gazu generatorowego, a przy zastosowaniu nadmuchu tlenowego – gazu syntezowego
(CO + H2). Gaz syntezowy może być wykorzystywany do syntezy amoniaku, metanolu oraz
węglowodorowych paliw płynnych (poprzez syntezę Fischera-Tropsch’a), syntetycznego gazu ziemnego.
Gaz syntezowy może być także wykorzystany w elektroenergetyce w turbinach gazowych i wtórnie
parowych, podnosząc sprawność przemiany węgla w energię elektryczną.
ZGAZOWANIE:
→ wydzielanie wodoru → wodór
→ synteza paliw → paliwa silnikowe
→ synteza metanolu → metanol
→ turbiny gazowe → energia elektryczna
Zgazowanie – nadaje się każdy rodzaj węgla, wysoka wartość opałowa powyżej 22MJ/kg
c) Produkty koksowania węgla (% wag.)
• koks - 70%-80%
• smoła - 2.5%-4.5%
• woda pogazowa - 3%-5%
• amoniak - 0.2%-0.4%
• benzol - 0.8%-1.4%
• gaz koksowniczy - 12%-18%
Koks – po rozsortowaniu jest głównym produktem zbytu.
Smoła oraz benzol są przerabiane w zakładach centralnych (obsługujących kilka koksowni) na wiele
produktów węglopochodnych, zwłaszcza aromatycznych, stanowiących cenne surowce przemysłu
organicznego.
Składniki wody pogazowej i amoniak służą bezpośrednio w koksowni do wyrobu soli amonowych siarczanu
lub fosforanu stosowanych jako nawozy sztuczne. Pozbawiona amoniaku woda pogazowa po odfelowaniu
trafia do ścieków. (NH
3
+ H
2
SO
4
→ (NH
4
)
2
SO
4
Surowy gaz koksowniczy uwolniony od smoły, amoniaku, benzolu i naftalenu może być kierowany
do ogrzewania pieców koksowniczych (ok. 50 %) reszta trafia do dyspozycji konsumentom komunalnym
(po uprzednim oczyszczeniu tj. odsiarczenie).

17
9. Klasyfikacja procesów jednostkowych
W zależności od rodzaju dominującego zjawiska fizykochemicznego, procesy jednostkowe dzielimy na:
- mechaniczne (dynamiczne)takie jak np.: transport, magazynowanie, rozdrabnianie, sortowanie, flotacja,
sedymentacja, mieszanie, filtracja, wirowanie, odpylanie itp.
- cieplne takie jak np.: ogrzewanie, chłodzenie, skraplanie, odparowywanie, zatężanie itp.
- dyfuzyjne takie jak np.: destylacja, rektyfikacja, absorpcja, adsorpcja, ekstrakcja, ługowanie, suszenie,
nawilżanie, krystalizacja itp.
- reaktorowe (z reakcją chemiczną) takie jak np. utlenianie, estryfikacja, sulfonowanie itp.
10. Procesy jednostkowe w technologii chemicznej
Procesem jednostkowym nazywamy część procesu technologicznego, w którym dominuje jedno zjawisko
fizykochemiczne. Proces jednostkowy zazwyczaj zachodzi w jednym aparacie.
11. Najlepsze dostępne techniki, zapobieganie i minimalizacja zanieczyszczeń na przykładzie
procesów rafinacji frakcji ropy naftowej
Główne produkty końcowe zachowawczej przeróbki ropy naftowej
W wyniku zachowawczej przeróbki ropy w rafineriach uzyskuje się następujące główne rodzaje
produktów:
a) Paliwa do silników gaźnikowych (benzyny) uzyskiwane z destylatów z wieży atmosferycznej;
b) Paliwa do silników samo zapłonowych (oleje napędowe) uzyskiwane z destylatów ciężkich z wieży
atmosferycznej i destylatów lekkich z wieży próżniowej.
c) Oleje opałowe uzyskiwane z pozostałości podestylacyjnej z wieży atmosferycznej - mazutu ) i innych
wysokowrzących frakcji uzyskiwanych w procesach rafineryjnych.
d) Rozpuszczalniki(benzyny ekstrakcyjne, benzyny lakowe) uzyskiwane z lekkich i średnich destylatów
z wieży atmosferycznej.
e) Produkty parafinowe (parafina stała, petrolatum, cerezyna) uzyskiwane przede wszystkim przez
odparafinowanie destylatów z ropy typu parafinowego.
f) Oleje smarowe uzyskiwane z destylatów z wieży próżniowej. Ze względu na zastosowanie i
podobieństwo warunków pracy wyróżnia się następujące podstawowe rodzaje olejów:
oleje maszynowe, stosowane do smarowania łożysk, ruchomych części maszyn itp.
oleje silnikowe-samochodowe, traktorowe, dieslowskie i lotnicze,
oleje smarowe o specjalnym przeznaczeniu –do turbin parowych i wodnych, do sprężarek, chłodziarek
itp.
oleje cylindrowe do maszyn parowych,
oleje niesmarujące – np. olej transformatorowy.
g) Asfalty uzyskiwane z pozostałości podestylacyjnej z wieży próżniowej.
h) Gaz płynny (frakcja propanowo -butanowa) uzyskiwany z instalacji rozdziału gazów rafineryjnych.
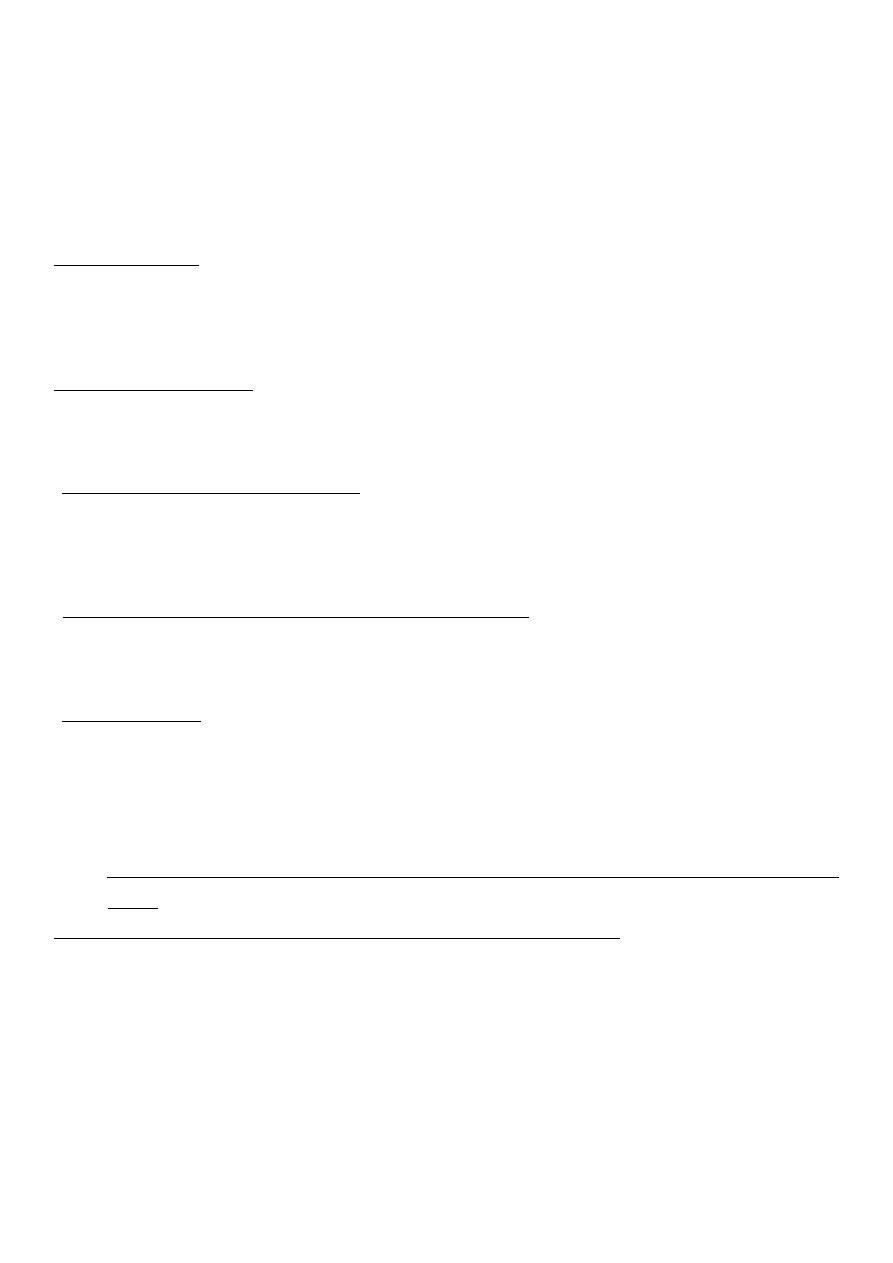
18
Rafinacja produktów naftowych
Celem rafinacji jest oczyszczenie surowych produktów przeróbki zachowawczej i rozkładowej. Podczas
rafinacji z surowych produktów przeróbki zachowawczej, która przebiegała w instalacji DRW i
rozkładowej, usuwa się przede wszystkim związki siarki, składniki kwaśne i zasadowe, żywice, asfalteny, a
w poszczególnych przypadkach także określone rodzaje węglowodorów, np. parafinę stałą.
Do rafinacji stosuje się zarówno metody fizyczne – np. ekstrakcję selektywnymi rozpuszczalnikami,
krystalizację lub sorpcję zanieczyszczeń na ziemiach aktywnych, jak i metody chemiczne – np.
zobojętnianie zanieczyszczeń kwaśnych i zasadowych, uwodornianie, utlenianie.
Ważniejsze metody rafinacji:
a) Rafinacja ługowa – ma na celu usunięcie z produktów naftowych zanieczyszczeń o charakterze
kwasowym takich jak np. siarkowodór, przez ich zobojętnianie wodnymi roztworami wodorotlenków.
Powstające w wyniku zobojętniania sole przechodzą do warstwy wodnej. Ługowaniu poddaje się najczęściej
surowe destylaty z wieży atmosferycznej, które nie tworzą z roztworami wodorotlenków emulsji trudnych
do rozdzielenia. Ługowanie jest najpospolitszą metodą odsiarczania benzyn, w których większość
zanieczyszczeń siarkowych stanowią związki o charakterze kwaśnym.
b) Rafinacja uwodorniająca – inaczej hydrorafinacja – jest skuteczną metodą usuwania z produktów
naftowych związków siarki, azotu i tlenu. Hydrorafinacja polega na działaniu wodorem (pod ciśnieniem 20-
70 at. I w temp. 300-500OC) wobec katalizatora kobaltowo – molibdenowego osadzonego na tlenku glinu
jako nośniku. Źródłem wodoru do hydrorafinacji w rafineriach są instalacje reformingu. Podczas
hydrorafinacji organiczne związki siarki ulegają rozkładowi na odpowiednie węglowodory i siarkowodór.
c) Odparafinowanie produktów naftowych – ma ono na celu usunięcie składników podwyższających
temperaturę krzepnięcia olejów napędowych i smarowych, tzn. przede wszystkim długołańcuchowych
alkanów, będących składnikami parafiny stałej. Do odparafinowania stosuje się krystalizację parafiny z
roztworów rafinowanych produktów w lotnych rozpuszczalnikach. Jako rozpuszczalnik wykorzystuje się
ciekły propan, benzynę, mieszaninę dwuchloroetanu z benzenem oraz mieszaniny benzenu i toluenu z
ketonami – acetonem lub ketonem metyloetynowym.
d) Rafinacja olejów smarowych selektywnymi rozpuszczalnikami – ma na celu polepszenie jakości produktu
przez usunięcie żywic, asfaltenów oraz węglowodorów aromatycznych i naftenowych o krótkich łańcuchach
bocznych. Jako rozpuszczalnik selektywny stosuje się najczęściej furfurol (rzadziej fenol). Pierwszym
stadium procesu jest ekstrakcja surowych olejów furfurolem, a po oddestylowaniu go z ekstraktu uzyskuje
się oleje rafinowane o zmniejszonej gęstości i o lepszym, zwiększonym wskaźniku lepkości.
e) Oksydacja asfaltu – ma na celu podwyższenie temperatury mięknienia asfaltu surowego, tj. pozostałości
podestylacyjnej z wieży próżniowej. Proces prowadzi się metodą okresową w zbiornikach mieszczących ok.
100 ton surowca w temperaturze 250-280
O
C. Po napełnieniu zbiornika surowcem o temp. 200
O
C tłoczy się
do niego powietrze pod ciśnieniem ok. 1,5 at. Oksydacja jest procesem egzotermicznym i dla zapobieżenia
nadmiernemu wzrostowi temperatury asfaltu (ponad wartość temperatury zapłonu, tj. ok. 280
O
C) w dalszych
stadiach procesu do zbiornika doprowadza się parę wodną.
12. Problemy środowiskowe na przykładzie wybranych procesów technologicznych (przetwórstwo
węgla)
Zanieczyszczenie środowiska w wyniku emisji spalin i popiołów do atmosfery;
Spalaniu węgla towarzyszy emisja pyłów i szkodliwych gazów. Największym zagrożeniem środowiska jest
emisja dwutlenku siarki i tlenków azotu powodująca kwaśne deszcze, które niszczą życie w akwenach,
dewastują olbrzymie obszary lasów i powodują korozję konstrukcji metalowych i niszczenie budynków.
Polska zajmuje czołowe miejsce w Europie pod względem wielkości emisji dwutlenku węgla do atmosfery.
Głównymi przyczynami tak dużej emisji dwutlenku węgla są:
· produkcja energii elektrycznej i cieplnej niemal wyłącznie z węgla, który charakteryzuje się znacznie
wyższymi współczynnikami emisji dwutlenku węgla w stosunku do gazu ziemnego i paliw ciekłych,
· nieefektywność systemu energetycznego i marnotrawstwo energii,
· struktura przemysłowa z dużym udziałem gałęzi najbardziej energochłonnych (przemysł wydobywczy i
hutnictwo),
· duży udział ogrzewania indywidualnego zużywającego węgiel w sektorze mieszkaniowym.

19
Ze względu na to, że każde z zanieczyszczeń atmosfery ma określone źródło emisji i specyficzne metody
jego zwalczania, to możemy je podzielić na:
· Pyłu;
· Dwutlenku siarki;
· Tlenków azotu (NO, NO2);
· Tlenków węgla (CO, CO2);
· Lotnych związków organicznych;
· Metali ciężkich.
Zanieczyszczenia te powstają w czasie procesów produkcyjnych, głównie przez spalanie węgla. Wydostając
się z kominów fabrycznych, elektrowni, kotłowni lokalnych i pojedynczych budynków mieszkalnych
rozchodzą się na zewnątrz w promieniu kilku a nawet kilkudziesięciu kilometrów w zależności od
warunków terenowych.
Udział poszczególnych czynników w emisji szkodliwych gazów
Pyły powstają wszędzie tam, gdzie występuje tarcie, np. podczas ścierania jezdni i opon pojazdów
mechanicznych. Powstawanie pyłów wiąże się z różnymi procesami produkcyjnymi jak: spalanie węgla
kamiennego i brunatnego, produkcja cementu czy stali i metali nieżelaznych. Najpoważniejszym źródłem
emisji pyłowych jest niewątpliwie przemysł paliwowo-energetyczny (ponad 50%), a zwłaszcza przemysł
elektroenergetyczny i ciepłowniczy (46% wszystkich pyłów) oraz przemysł metalurgii żelaza i stali (9%).
Negatywna działalność pyłów polega między innymi na:
· zamykaniu szparek oddechowych u roślin;
· ograniczaniu procesu fotosyntezy;
· powodowaniu licznych chorób układu oddechowego u człowieka i zwierząt, np. pylicy;
· wpływaniu na chemizm wody i gleb;
· toksycznym działaniu metali ciężkich na organizmy żywe.
W powietrzu miast jest wiele sadzy. Są to bardzo drobne cząstki węgla, które mają bardzo wielką
powierzchnię w stosunku do masy. Pociąga to za sobą wielką aktywność i absorpcję różnych substancji ze
środowiska. Cząsteczki sadzy niosą na sobie między innymi ciężkie węglowodory, wśród których nie brak
rakotwórczych.
Działanie pyłów jest zróżnicowane, gdyż z jednej strony rozpraszają światło słoneczne skierowane do Ziemi
zwiększając albedo, co powoduje ochłodzenie, a z drugiej strony absorpcja i reemisja promieniowania
podczerwonego działa odwrotnie, powodując wzrost temperatury powierzchni kuli ziemskiej.
Wpływ pyłów na bilans cieplny Ziemi i na jej klimat jest tym większy, im większe jest ich stężenie w
powietrzu atmosferycznym.
Cząsteczki pyłów są ośrodkami skraplania się pary wodnej. Pyły mogą także wpływać na reakcje chemiczne
odbywające się w atmosferze.
Toksyczność pyłów zależy od średnicy ziaren, od składu chemicznego, a zwłaszcza od zawartości metali
toksycznych, takich jak ołów, kadm, nikiel, miedź, cynk, chrom. Bardzo szkodliwy dla zdrowia jest tzw. pył
zawiesinowy ze związkami Pb, Cd, Zn i węglowodorami wielopierścieniowymi.
W wyniku całkowitego spalenia paliwa mineralnego w komorze paleniskowej kotła energetycznego
powstają spaliny zawierające: dwutlenek węgla (CO
2
), parę wodną (H
2
O), azot (N
2
), dwutlenek siarki
(SO
2
), trójtlenek siarki (SO
3
) oraz popiół. Ze składników tych toksycznymi są: SO
2
, SO
3
oraz częściowo
popiół (pył) ze względu na zawartość w nim takich pierwiastków jak kadm, ołów, arsen.
Przy wysokiej temperaturze w jądrze płomienia komory paleniskowej elektrowni zachodzi częściowe
utlenienie azotu z powietrza i azotu z paliwa, a w jego wyniku tworzenie się tlenków azotu. Tlenki azotu
(NOx) nawet w minimalnych stężeniach w powietrzu działają drażniąco na organy układu oddechowego,
niszczą urządzenia i materiały, przyczyniają się do powstawania smogów, pogarszają widoczność i
ograniczają nasłonecznienie powierzchni Ziemi. Są one szkodliwe dla organizmów żywych, co stawia je
zaraz za dwutlenkiem siarki SO
2
, jako najgroźniejsze zanieczyszczenie powietrza atmosferycznego.
W przypadku niezupełnego spalania w komorze paleniskowej, powstaje: tlenek węgla CO, sadza oraz
rakotwórczy benzo-a-piren.
Związki chemiczne takie jak tlenki siarki i azotu, uwolnione w wyniku spalania paliw kopalnych i drewna,
reagują z parą wodną zawartą w powietrzu. W wyniku tej reakcji powstają kwasy siarkowy i azotowy, które
z wodą deszczową powracają na powierzchnię Ziemi - są to tzw. kwaśne deszcze. Chmury mogą przenieść

20
taki deszcz daleko od miejsca jego powstania.
Kwaśne deszcze powodują:
· obumieranie wielkich połaci lasów (szczególnie wrażliwe są lasy iglaste - a zwłaszcza jodła) - najbardziej
dotknięte tą klęską są lasy Europy Wschodniej
· wymarcie ryb w jeziorach (w Skandynawii deszcze znad Wielkiej Brytanii zabiły ryby w 80% tamtejszych
jezior)
· niszczenie zabytkowych posągów i budowli kamiennych
· rdzewienie metalowych konstrukcji, mostów, ogrodzeń
· zatruwanie wody pitnej
· spadek wydajności rolnictwa
· choroby płuc osób mieszkających w okolicy, gdzie często padają kwaśne deszcze.
Procesowi spalania węgla towarzyszą również związki organiczne emitowane w gazach spalinowych, są one
szczególnie uciążliwe z uwagi na ich toksyczność miedzy innymi kancerogenność i skutki mutagenne.
Wśród tych związków występują wysokowrzące wielopierścieniowe węglowodory aromatyczne (WWA)
jest benzo(a)piren. WWA występują w postaci zaadsorbowanej na cząsteczkach pyłów oraz w formie
gazowej tworzą również sadzę w czasie spalania paliw kopalnianych do atmosfery są uwalniane również
nitro-WWA.
Zanieczyszczeniami pierwotnymi wywołanymi spalaniem węgla są węglowodory, tlenki azotu które są
rakotwórcze, tlenek węgla i dwutlenek siarki są trujące. Dwutlenek siarki, tlenki azotu (głównie NO2)i
węglowodory gromadzą się w atmosferze wchodzą w reakcję pod wpływem promieniowania
słonecznego(powstaje smog). Oprócz ww związków toksycznych w wyniku spalania paliw powstaje CO2 ,
który nie jest związkiem toksycznym, ale powoduje efekt cieplarniany, na skutek, którego podnosi się
temperatura Ziemi i następują zmiany klimatyczne. Gazem cieplarnianym jest także podtlenek azotu ( N2O),
który długi czas zalega w atmosferze.
Efektem pierwotnym zanieczyszczeń są zanieczyszczenia wtórne czyli smog, który jest efektem utleniania
spalonych węglowodorów, zainicjowane przez wolne rodniki hydroksylowe. Kolejne to kwaśne deszcze
(SO
2
, NO
x
) oczyszczają atmosferę ziemska z tych toksycznych związków. Natomiast niszczą lasy i uprawy
roślin. Dowiedziono natomiast, że wypłukują z gleby magnez, niszczą budowle z kamienia wapiennego oraz
skał. Kolejne to wzrost temperatury ziemi i dziura ozonowa, która przyczynia się do obecności silniejszego
promieniowania ultrafioletowego napowierzchni ziemi.

21
13. Procesy alkilowania
a) Alkilowanie i-butanu olefinami C
3
-C
4
, wykorzystanie produktu jako komponenta benzyn
b) Alkilowanie benzenu α-olefinami (alkilowanie benzenu propylenem) propylobenzen (kumen)
Kumenowa metoda produkcji fenolu; utlenienie kumenu → rozkład wodoronadlenku kumenu do
fenolu i acetonu.
c) Alkilowanie benzenu α-olefinami (alkilowanie benzenu etylenem); etylobenzen
Wykorzystanie etylobenzenu (Odwodornienie etylobenzenu → styren )
d) O-alkilowanie izobutylenu metanolem (zastosowanie MTBE)

22

23
14. Wykorzystanie etylenu do syntez.
a) Otrzymywanie etanolu. Proces pośredniej i bezpośredniej hydratacji etylenu. Porównanie
procesów. (Inna metoda otrzymywania etanolu).
Etanol do celów spożywczych (→ napój alkoholowy) otrzymuje się w wyniku fermentacji alkoholowej.
Podstawowymi surowcami do produkcji etanolu są: buraki cukrowe, trzcina cukrowa, ziemniaki, i wiele
innych roślin.
Etanol do celów przemysłowych jest też często produkowany poprzez bezpośrednią syntezę z gazu
syntezowego (mieszaniny CO, H
2
O), która jest tańsza od fermentacji i prowadzi do czystszego
chemicznie etanolu, jednak w wielu krajach świata użycie etanolu syntetycznego do celów spożywczych jest
zabronione. Jest to podyktowane nie tyle względami zdrowotnymi, ile chęcią utrzymania tradycyjnego
przemysłu spirytusowego i związanych z nim miejsc pracy
.
W Polsce przed laty dostępna była w sprzedaży wódka "Vistula" produkowana na drodze chemicznej, a nie
poprzez fermentację.
Etanol jest tanim w produkcji surowcem i jest jednocześnie stosunkowo niegroźny dla środowiska, dlatego
jest dość powszechnie wykorzystywany jako rozpuszczalnik w przemyśle chemicznym.
Cena prostych napojów alkoholowych (czystych wódek i spirytusu spożywczego) w wielu krajach wynika z
podatków nakładanych na te produkty, a nie z kosztów ich produkcji. Rzeczywisty koszt wytworzenia 1 l
spirytusu to ok. 0,50 Euro, gdy jest on otrzymywany przez fermentację i jeszcze taniej jeśli pochodzi z
syntezy bezpośredniej.
Stosowany także jako paliwo napędowe np. w Brazylii 86% sprzedawanych nowych aut jest
przystosowanych do spalania etanolu.

24
b) Chlorek winylu. Trzy koncepcje syntezy chlorku winylu: oksychlorowanie etylenu,
bezpośrednie chlorowanie etylenu, z użyciem etylenu i acetylenu)
Chlorek winylu (chloroetylen, chloroeten, CH
2
=CHCl)
A) Oksychlorowanie etylenu
W procesie wykorzystuje się chlorowodór z instalacji krakingu DCE. W procesie tym stosuje się katalizator
Deacona, którego aktywnym składnikiem jest CuCl
2
naniesiony na ilość ok. 10% mas. na Al2O3 o silnie
rozwiniętej powierzchni. Stosuje się często dodatek soli innych metali np.: KCl, NaCl co zwiększa
selektywność. Katalizator dla procesów w warstwie fluidalnej przygotowuje się z Al2O3 o średnicy ziaren
wynoszącej kilkadziesiąt mikrometrów. Powinien on wykazywać duża odporność na ścieranie aby nie
powstał pył.
Cu
2
Cl
2
+ 2 HCl + 0,5 O
2
= 2 CuCl
2
+ H2O
Przereagowanie chlorowodoru jest całkowite. Otrzymuje się DCE- 1,2-dichloroetan z wydajnością około
96% w odniesieniu do przereagowanego etylenu i około 98% w stosunku do chlorowodoru. Reszta
substratów przereagowuje na produkty uboczne, które są oddzielane od DCE przed jego krakingiem
Przebieg procesu w instalacji przemysłowej – temp: 230-260 st.C, ciśnienie ok. 0,3 MPa. Warstwę fluidalną
tworzy katalizator unoszony przez gazowe reagenty: etylen, techniczny tlen i chlorowodór.
B) Bezpośrednie chlorowanie etylenu
W fazie ciekłej, pod ciśnieniem 0,5MPa
Reakcja przebiega w środowisku produktu reakcji, gdyż substraty zarówno chlor, jaki i etylen są dość
dobrze rozpuszczalne w dichloroetanie
W środowisku reakcji rozpuszczony jest również katalizator (FeCl3)
Proces prowadzi się z kilkuprocentowym nadmiarem etylenu, co ogranicza uboczne reakcje podstawiania.
Chlor w reakcji przereagowuje całkowicie
C) Produkcja chlorku winylu z etylenu i acetylenu
Zbilansowane chlorowanie mieszaniny etylenu i acetylenu
25
Zbilansowanie polega na tym, że w chlorku winylu odbiera się całą ilość zużytego w procesie chloru
Odchlorowodorowaniu 1,2-dichloroetanu, w temp. ok. 500ºC, pod ciśnieniem 2,5-3,5MPa
Proces w fazie ciekłej uznaje się za korzystniejszy ze względu na lepszą kontrolę nad nim, lepsze wydobycie
ciepła i zwiększenie bezpieczeństwa pracy.
D) Produkcja chlorku winylu z acetylenu
Wady:
- wysoka cena acetylenu
- wybuchowość
- toksyczność HgCl2 stosowanego jako katalizator
- stosowanie bardzo suchych substratów bo katalizator przyśpiesza hydratację acetylenu do aldehydu
octowego
Proces prowadzi się w fazie gazowej w reaktorze rurowym. Rurki SA wypełnione katalizatorem
(10% HgCl2 w suchej masie węgla aktywnego). W przestrzeni międzyrurowej cyrkuluje czynnik chłodzący
– wrząca woda lub organiczny nośnik ciepła. Reakcja jest silnie egzotermiczna. Wewnątrz rur utrzymuje się
temperatura około 170 st. C, która w miarę zmniejszania się aktywności katalizatora zwiększa się stopniowo
do 220 st. C. Katalizator przyspiesza selektywnie tylko chlorowodorowanie acetylenu do chlorku winylu.
Stopień przereagowania acetylenu przekracza 98%.
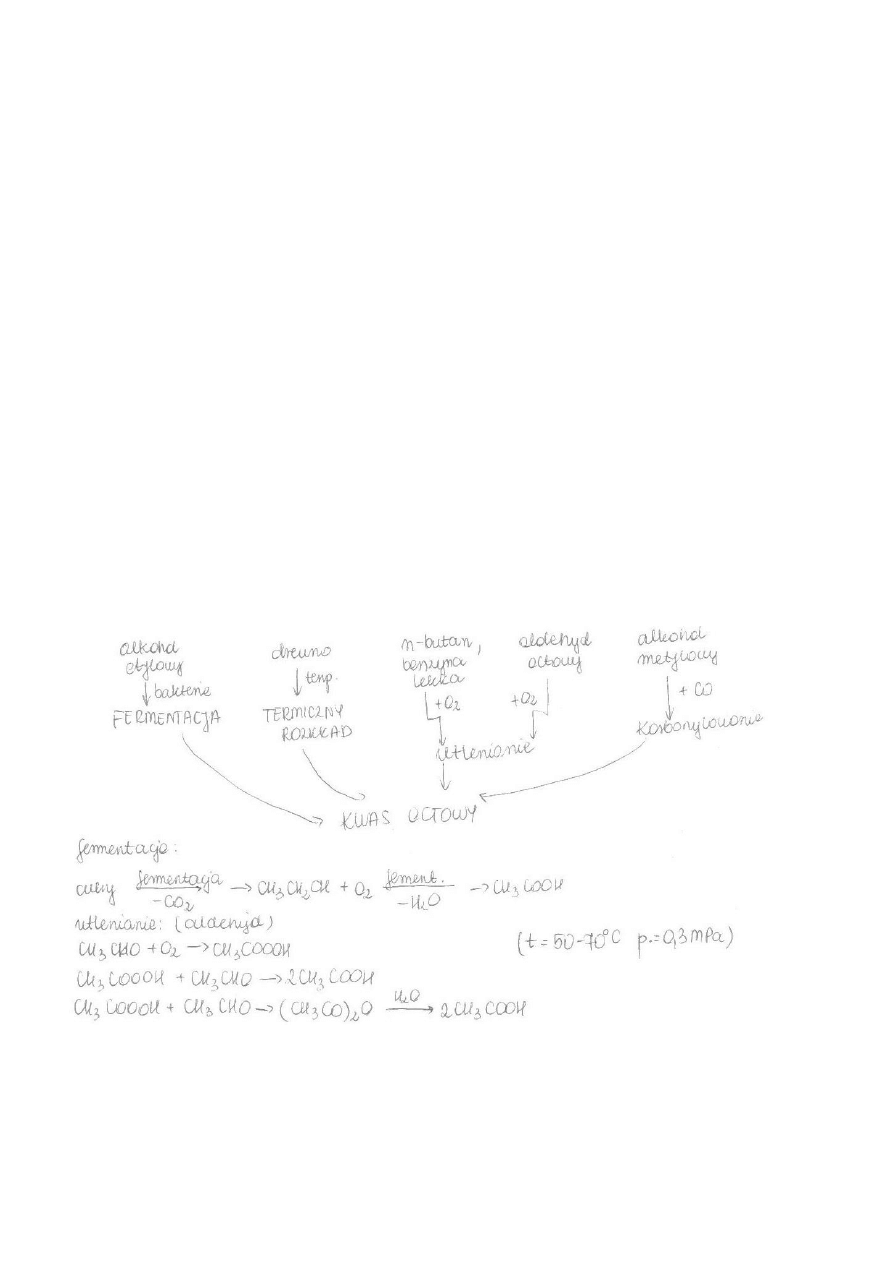
c) Otrzymywanie kwasu octowego i bezwodnika octowego. Najważniejsze zastosowania.
CHCl
CH
HCL
CH
CH
HgCl
2
2
CHCl
CH
Cl
CH
CH
CH
CH
e
sumaryczni
CHCl
CH
HCl
CH
CH
b
HCl
CHCl
CH
Cl
ClCH
CH
Cl
ClCH
CH
Cl
CH
CH
a
C
2
2
2
2
2
2
500
2
2
2
2
2
2
2
2
:
)
)
0

26
Najważniejsze zastosowania:
KWAS OCTOWY:
- do produkcji sztucznego jedwabiu, leków (aspiryna), niepalnej taśmy filmowej i esencji octowej, kwasu
chlorooctowego, octanów, karboksymetylocelulozy, octany celulozy, w technice grzewczej do usuwania
kamienia kotłowego, w postaci kilkuprocentowego roztworu (produkt fermentacji octowej) jako ocet
spożywczy do konserwacji żywności, jako składnik roztworów buforowych i wiele innych. Bardzo duże
ilości kwasu octowego używane są jako rozpuszczalnik w rafinacji kwasu tereftalowego, używanego do
wielkotonażowej produkcji poli(tereftalanu etylenu) (butelki PET). Jest stosowany również jako konserwant.
Jego numer jako dodatku do żywności to E260.
BEZWODNIK OCTOWY:
- Stosowany jako środek acetylujący, np. do otrzymywania kwasu acetylosalicylowego (aspiryny), octanu
celulozy, włókien sztucznych (poli(octan winylu), błony filmowe, taśmy magnetofonowe), barwników,
materiałów wybuchowych, ale także heroiny. Z uwagi na te dwa ostatnie zastosowania bezwodnik octowy
znajduje się na liście substancji objętych obrotem kontrolowanym.

27
d) Otrzymywanie glikolu etylenowego z:
- gazu syntezowego (CO + H
₂)
Parametry procesu: temperatura: 220-280°C, ciśnienie > 150MPa, proces nieselektywny
- szczawianu butylu
Parametry procesu: temperatura: 70-90°C, niskie ciśnienie, proces perspektywiczny
- uwodornienia odpowiedniego estru
- tlenku etylenu
Parametry proces: temperatura: 180-200°C, ciśnienie 1,8-2 MPa, proces dominujący
Integracja produkcji tlenku etylenu i glikolu etylenowego. (schemat pytanie 18)
Od 50 do nawet 70% wytwarzanego tlenku etylenu przerabia się na glikol etylenowy, chociażby ze względu
na właściwości (tlenek etylenu jest substancją skrajnie łatwo palną, toksyczną), dlatego też transport glikolu
jest znacznie bezpieczniejszy. Między innymi dlatego wytwarzanie tych dwóch produktów bardzo często
realizuje się w zintegrowanym kompleksie instalacji. Instalacja taka pozwala na oszczędność energii
wynikająca z możliwości wykorzystania ciepła utleniania etylenu podczas produkcji tlenku etylenu
w instalacji wytwarzającej glikol. Zdolności produkcyjne instalacji zintegrowanych w sposób przedstawiony
na schemacie mogą być tak dobrane, ze instalacja tlenku etylenu wytwarza jedynie glikol, ale możliwy jest
oczywiście wariant z nadmiarem zdolności produkcyjnej tlenku i z równoległym wytwarzaniem
obu produktów (EG, EO).
OH
CH
OH
CH
H
CO
2
2
2
3
2

28
15. Wykorzystanie propylenu do syntez
a) Chlorek allilu - epichlorohydryna – glicerol
b) Utlenianie propylenu do akroleiny. Produkcja kwasu akrylowego
16. Wykorzystanie węgla.
a) Energetyczne wykorzystanie węgla:
- gaz
- woda
- współspalanie biomasy
- OZE (odnawialne zasoby energii)
- węgiel brunatny
- węgiel kamienny
b) Chemiczna przeróbka węgla; koksowanie (wykorzystanie produktów), upłynnianie, zgazowanie
(rozwiązania technologiczne procesu zgazowania, kierunki wykorzystania gazu procesowego)
29
Koksowanie – produkty:
- 75-80% koks
- 20-25% surowy gaz koksowniczy: gaz koksowniczy (12-18%), benzol koksowniczy (1-1,5%),
amoniak (0,3%), smoła węglowa (3-4%).
Podstawową metodą przerobu smoły jest jej destylacja (atmosferyczna, atmosferyczno-próżniowa).
Produkuje się pak elektrodowy oraz oleje karbochemiczne.
Ważniejsze produkty otrzymywanie z paku (60%) to:
- koks palny (produkcja elektrod, mas anodowych, wykładzin pieców)
- smoły preparowane (smoły dachowe, drogowe, hutnicze)
ZGAZOWANIE:
→ wydzielanie wodoru → wodór
→ synteza paliw → paliwa silnikowe
→ synteza metanolu → metanol
→ turbiny gazowe → energia elektryczna
Zgazowanie – nadaje się każdy rodzaj węgla, wysoka wartość opałowa powyżej 22MJ/kg.
Kierunki wykorzystania gazu syntezowego:
Proces Fischera – Tropscha
180 – 240 ºC
300 – 350 ºC
0,5 – 5 MPa
Frakcje benzynowe, olej
napędowy, woski
Proces Mobil
(gaz syntezowy metanol)
360 – 415 ºC
2 MPa
Frakcje benzynowe
PROCES
PARAMETRY
PRODUKTY
ODGAZOWANIE
- koksowanie
- wytlewanie
- szybkie wytlewanie (piroliza)
Temp °C
900-1100
500-600
600
Ciśnienie
MPa
normalne
normalne
normalne
- koks,smoła węglowa,benzol,
g.koksowniczy
- półkoks, smoła wytlewna,
g.wytlewny
- koksik, smoła
EKSTRAKCJA
Metoda Pott-Broche’a
380-450
10-15
Ekstrakt węglowy (przeznaczony
do uwodorniania lub produkcji
koksu elektrodowego)
UPŁYNNIANIE
Uwodornianie metodą Bergiusa
Uwodornianie metodą H-Coal
300-350
420-450
6-25
15-18
Ropa węglowa
ZGAZOWANIE
Metoda Lurgi (złoże stałe)
Metoda Koppers-Totzka
Metoda Winklera
800-900
1500-1900
800-1000
3
normalne
normalne
Gazy nisko- lub średniokaloryczne
Gazy syntezowe

30
Synteza Fischera-Tropscha (w skrócie synteza/reakcja F-T lub FTS) to katalityczna reakcja chemiczna
tworzenia węglowodorów z mieszaniny tlenku węgla i wodoru, czyli tak zwanego gazu syntezowego. Celem
syntezy F-T jest produkcja paliw płynnych. Jedną z zalet syntezy F-T jest możliwość wytwarzania paliwa
wolnego od związków siarki i azotu a przez to czystszego dla środowiska naturalnego.
17. Gaz ziemny - procesy oczyszczania i wykorzystania
a) Odgazolinowanie gazu ziemnego. Cel i schemat technologiczny:
Operacja ta polega na usunięciu z gazów ziemnych mokrych wyższych alkanów (węglowodory C3+).
Obecnośd tych składników może byd kłopotliwa w czasie transportu rurociągowego lub w trakcie
niektórych operacji niskotemperaturowych. W warunkach podwyższonego ciśnienia i niskiej temperatury
może zachodzid wykroplenie alkanów co może prowadzid do trudności eksploatacyjnych. Węglowodory
usuwane w tej operacji są bardzo wygodnymi paliwami, a przede wszystkim cennymi surowcami
chemicznymi. Należy zatem traktowad operację odgazolinowania gazów ziemnych mokrych jako formę
uzyskiwania gazu ziemnego suchego (wygodnego do transportu) oraz wyodrębnienia cennych
węglowodorów wyższych zawartych w gazie.
Operację odgazolinowania prowadzi się zwykle jedną z trzech metod:
1. METODA ABSORPCYJNA - polega na wykorzystaniu różnicy rozpuszczalności węglowodorowych
składników gazu ziemnego w odpowiednio dobranych absorbentach. Najczęściej jako czynniki
pochłaniające stosuje się ciekłe frakcje ropy naftowej( np. benzyna, nafta). W odpowiednich warunkach
(podwyższone ciśnienie, niska temperatura) wskutek większej absorpcji wyżej wrzących składników gazu
ziemnego następuje usunięcie tych składników. Operacja nie jest selektywna i nie wszystkie C3+ ulegają
pochłonięciu, poza tym na skutek pewnej rozpuszczalności częśd metanu i etanu przechodzi do strumienia
gazoliny. W separatorze usuwa się rozpuszczone częściowo alkany C1-C2 i dołącza do strumienia gazu
suchego lub przerabia oddzielnie. W tej metodzie stopieo rozdzielenia składników nie jest doskonały,
dlatego dla poprawy selektywności absorpcji stosuje się czasem metodę niskotemperaturowej absorpcji
(NTA) - obniżona temperatura w kolumnie absorpcyjnej.
2. METODA NISKOTEMPERATUROWEJ KONDENSACJI - polega na wykropleniu cięższych składników gazu
ziemnego po osiągnięciu punktu rosy (warunków, w których prężnośd par danego węglowodoru osiąga
wartośd niższą od równowagowej ciecz - pary węglowodoru). W praktyce stosuje się temperaturę znacznie
31
niższą od odpowiadającej punktowi rosy oraz wykorzystuje możliwości głębokiego wykroplenia przez
zastosowanie podwyższonego ciśnienia (kilka MPa). Obniżenie temp. i wzrost ciśnienia podwyższa stopieo
odgazolinowania ale wiąże się z wyższymi kosztami.
3.METODA ADSORPCYJNA - stosowany rzadko i tylko w sytuacji, gdy konieczne jest usuwanie śladowych
ilości ciężkich alkanów. Operację przeprowadza się wykorzystując cykliczne pracujące adsorbery, zwykle
wypełnione węglem aktywnym jako masa pochłaniającą.
b) Osuszanie gazu ziemnego, cel i główne metody oraz ich efekty
Gaz ziemny wydobywany ze złoża zawiera parę wodną. Jej obecnośd jest przyczyną poważnych trudności
eksploatacyjnych w instalacjach niskotemperaturowej przeróbki gazu oraz podczas transportu gazociągami.
Występuje również intensywna korozja (zwłaszcza gdy gaz zawiera siarkowodór i CO2). Osuszenie nie musi
oznaczad całkowitego usunięcia pary wodnej, wystarczy usunąd tę cześd, która w określonej temperaturze
mogłaby wydzielid się w postaci skroplin. Gaz powinien byd osuszony aby jego temperatura punktu rosy
była 3-10 st C niższa od najniższej temperatury, w jakiej może się on znaleźd podczas przeróbki lub
transportu gazociągiem.
1. Absorbcyjne -do tej metody stosuje się glikol dietylenowy i trietylenowy. Etap absorpcji prowadzi
się w typowym absorberze półkowym, w którym w przeciwprądzie gaz kontaktuje się w temp. 20-
30oC z absorbentem. Zawilgocony glikol regeneruje się w desorberze w temperaturze uzależnionej
od rodzaju użytego absorbenta.
2. Metody adsorpcyjne - stosuje się do głębokiego osuszania gazu(np. przed operacjami
niskotemperaturowymi). Jako masy adsorpcyjne wykorzystuje się najczęściej żel krzemionkowy,
różne formy Al2O3 oraz sita molekularne(te ostatnie nabierają ostatnio bardzo dużego znaczenia i
obecnie dominują w technikach adsorpcyjnego osuszania gazów)
c) Osuszanie
gazu
w
instalacjach
kolumnowych za pomocą glikoli

32
d) Osuszanie gazu metodami adsorpcyjnymi:
Metody adsorpcyjne - Stosuje się do głębokiego osuszania gazu(np. przed operacjami
niskotemperaturowymi). Jako masy adsorpcyjne wykorzystuje się najczęściej żel krzemionkowy ,różne
formy Al2O3 oraz sita molekularne(te ostatnie nabierają ostatnio bardzo dużego znaczenia i obecnie
dominują w technikach adsorpcyjnego osuszania gazów).
e) Metody odsiarczania gazu ziemnego (wymienić):
- metody absorpcyjne (mokre)
- metody adsorpcyjne (suche)
f) Metody absorpcyjne odsiarczania gazu ziemnego (etanoloaminy):
Metoda wykorzystuje możliwośd absorpcji (pochłaniania) kwaśnych składników gazu ziemnego(H2S,
CO2) w odpowiednio dobranych absorbentach.
Wyróżniamy 2 rodzaje absorpcji:
- absorpcja połączona z reakcją chemiczną (zwykle reakcje typu kwas - zasada)
absorbentami mogą byd np. aminy, w tym szczególnie etanoloaminy, roztwory alkaliczne, K2CO3
Absorpcja - podwyższone ciśnienie, obniżona temperatura
Desorpcja - obniżone ciśnienie, zwiększona temperatura
WADY - duże zużycie ciepła, zwłaszcza przy oczyszczaniu gazów o dużej zawartości CO2 (tu
znaczenie ma absorpcja fizyczna - tu regeneracja absorbenta bez zużycia ciepła)
- absorpcja fizyczna - selektywna rozpuszczalnośd kwaśnych składników w odpowiednio dobranych
rozpuszczalnikach (metanol, sulfolan...)
WADY - wysoka cena rozpuszczalników i skłonnośd do absorpcji niektórych wyższych
węglowodorów zawartych w gazach ziemnych (utrudnia to przeróbkę H2S w instalacji Clausa)
g) Wytwarzanie gazu syntezowego z gazu ziemnego
Gaz syntezowy (gaz wodny) - palny gaz powstający podczas reakcji węgla lub gazu ziemnego z parą
wodną w obecności odpowiednich katalizatorów. Jest to mieszanina tlenku węgla (CO) i wodoru
(H2), może zawierad znaczne ilości azotu.
CH
4
+ H
2
O
⇌ CO + 3H
2
H
2
O + C
⇌ CO + H
2
Gaz ziemny użyty do wytworzenia gazu syntezowego nie może zawierad innych substancji
(związków chemicznych) które wykazują silne powinowactwo do powierzchni aktywnej kontaktu i
mają zdolnośd „zatruwania” katalizatorów w dalszych etapach procesu.
Katalizatory niklowe reformingu parowego są bardzo wrażliwe na śladowe ilości niektórych
związków, które mogą byd obecne w reagentach np. związki siarki, arsenu i halogenki. Z tego
powodu gaz ziemny przed skierowaniem do reform era musi byd odsiarczony do zawartości siarki
mniejszej niż 5 ppm.
18. Podać przykłady procesów zintegrowanych (ze względu na wytwarzane produkty uboczne,
ze względu na surowce, produkt)

33
produkt uboczny
Wyszukiwarka
Podobne podstrony:
pytania z mojego rocznika, Studia (Geologia,GZMIW UAM), I rok, Chemia, Egzamin, chemijap
Opracowanie mojego rozdziału Elwi
PROJEKT NR 1 - Opracowanie danych z roczników hydrologicznych, Hydrologia i Hydraulika
opracowanie wspólne rocznik 12
ROCZNIKIi, Polonistyka, staropolka, lektury - opracowania, Średniowiecze, renesans
Opracowane pytania na koło 3 7 11 15, Budownictwo UTP, III rok, DUL stare roczniki, GEODEZJA, geodez
pytania- opracowania, Budownictwo UTP, III rok, DUL stare roczniki, nawierzchnie
ROCZNIKIi, Polonistyka, staropolka, lektury - opracowania, Średniowiecze, renesans
Wokol tajemnicy mojego poczecia
Opracowanka, warunkowanie
OPRACOWANIE FORMALNE ZBIORÓW W BIBLIOTECE (książka,
postepowanie w sprawach chorob zawodowych opracowanie zg znp
opracowanie 7T#2
opracowanie testu
Opracowanie FINAL miniaturka id Nieznany
Opracowanie dokumentacji powypadkowej BHP w firmie
przetworniki II opracowane
więcej podobnych podstron