
7.91 / 7.36 / BE.490
Lecture #1
Feb. 24, 2004
Genome Sequencing
&
DNA Sequence Analysis
Chris Burge
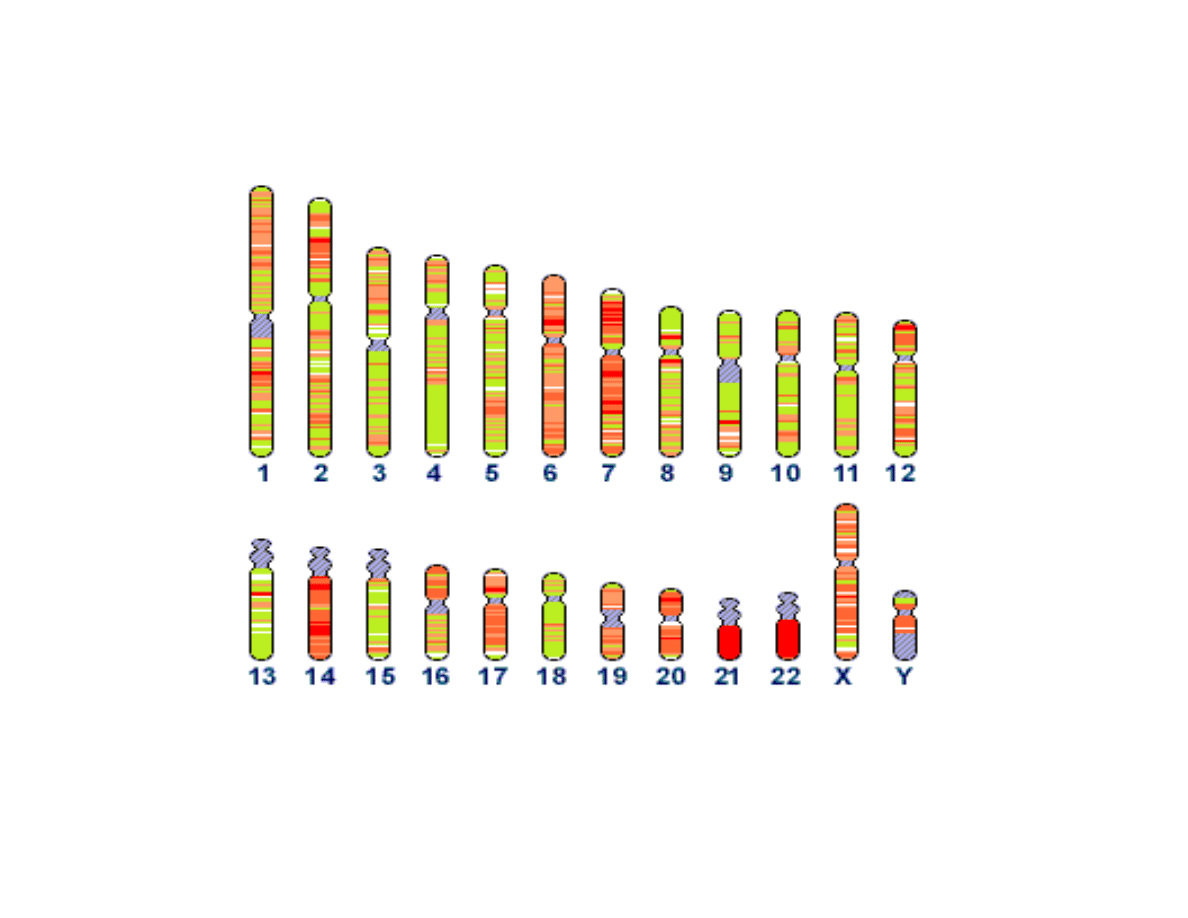
What is a Genome?
A genome is NOT a bag of proteins

What’s in the Human Genome?

Outline of Unit II:
DNA/RNA Sequence Analysis
Reading*
2/24
2/26
3/2
M Ch. 4
3/4
M Ch. 4
3/9
DNA Sequence Evolution
3/11
RNA Structure Prediction & Applications
M Ch. 5
3/16
Literature Discussion
TBA
Genome Sequencing & DNA Sequence Analysis
M Ch. 3
DNA Sequence Comparison & Alignment
M Ch. 7
DNA Motif Modeling & Discovery
Markov and Hidden Markov Models for DNA
M Ch. 6
* M = Mount, “Bioinformatics: Sequence and Genome Analysis”

Feedback to Instructor
Examples from past years:
• Comic font looks stupid
• Burge uses too much genomics jargon
• Better synergy between Yaffe/Burge sections
• Asks questions to the class, student answers,
but I didn’t hear/understand the answer…

DNA vs Protein Sequence Analysis
Protein Sequence Analysis
DNA Sequence Analysis
- emphasis on chemistry
- emphasis on regulation
- protein structure
- RNA structure
- selection is everywhere
- signal vs noise (
statistics
)
- multiple alignment
- motif finding
- comparative proteomics
- comparative genomics
- data:
O(10^8) aa
- data:
O(10^10) nt
Read your probability/statistics primer!

Genome Sequencing
& DNA Sequence Analysis
•
The Language of Genomics
•
Shotgun Sequencing
•
DNA Sequence Alignment I
•
Comparative Genomics Examples
- Progress: genomes, transcriptomes, etc.
- How to choose a mismatch penalty
- PipMaker, Phylogenetic Shadowing
Recent Media Attention

Genomespeak
Bork, Peer, and Richard Copley. " Genome Speak."
Nature
409 (15 February 2001): 815.
Learn to speak genomic

In the following article, note the use of the following genomic terms:
euchromatic, whole-genome shotgun sequencing, sequence reads,
5.11-fold coverage, plasmid clones, whole-genome assembly,
regional chromosome assembly.
Venter, JC, MD Adams, EW Myers, PW Li, RJ Mural, GG Sutton, HO Smith, … "The
Sequence of The Human Genome." Science 291, no. 5507 (16 February 2001): 1304-51.

Types of Nucleotides
• ribonucleotides
• deoxyribonucleotides
• dideoxyribonucleotides
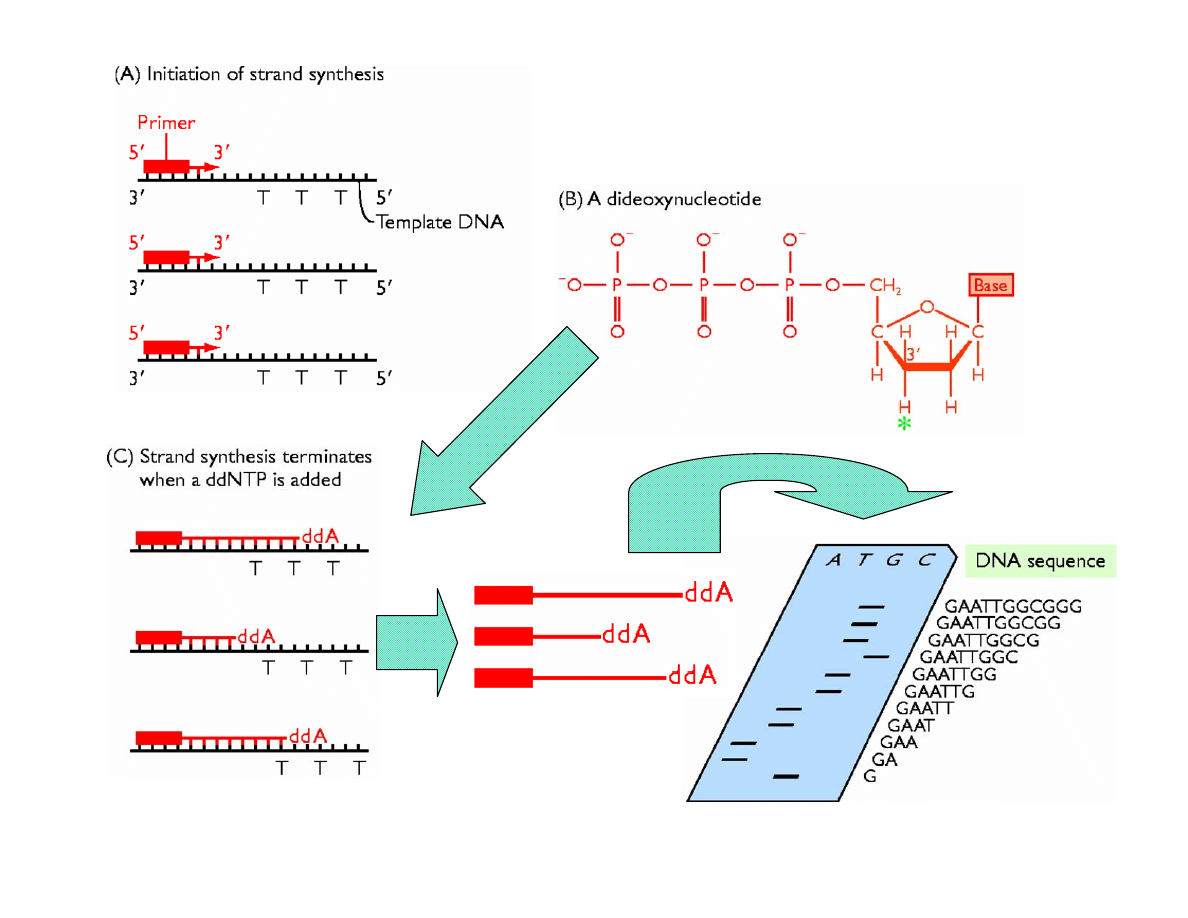
DNA Sequencing
Adapted from Fig. 4.2 of “Genomes” by T. A. Brown, John Wiley & Sons, NY, 1999
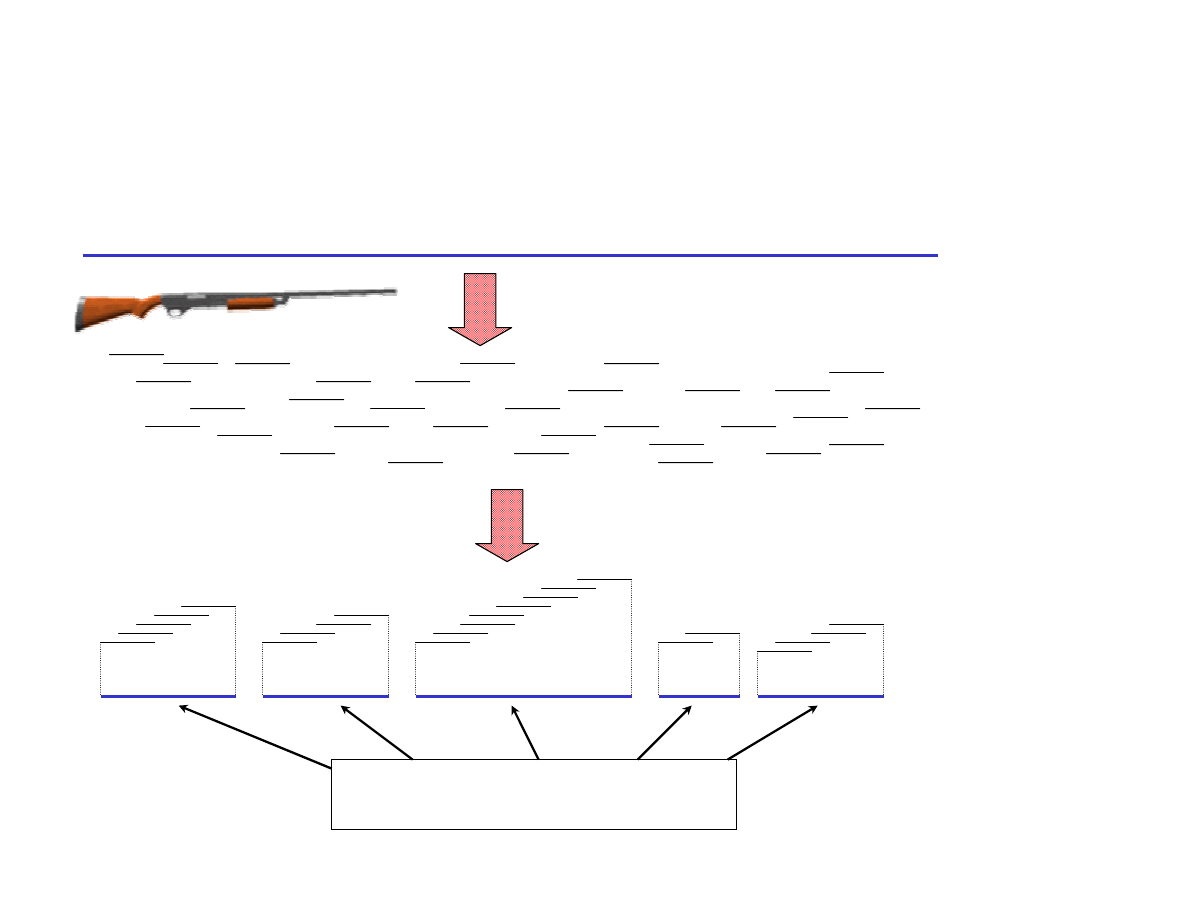
Shotgun Sequencing a BAC or a Genome
200 kb (NIH)
3 Gb (Celera)
Sequence, Assemble
Sonicate, Subclone
Subclones
Shotgun Contigs
What
would
cause
problems
with
assembly?

Shotgun Coverage
(Poisson distribution)
Sequence
N
reads, 500 bp each, from a 200kb BAC
Coverage/read
p
= 500/200,000 = 0.0025
Total coverage
C
=
Np
Y
= no. of reads covering the point
x
P(Y=k) = (N!/(N-k)!k!) p
k
(1-p)
N-k
≈ e
-c
c
k
/ k!
.
x
P(Y=0)= e
-c
Examples:
e
-2
≈ 0.14
e
-4
≈ 0.02
What could cause reality to differ from theory?

Clickable Genomes
Eukaryotes
Protists
Eubacteria
S. cerevisiae
Plasmodium
E. coli
S. pombe
Giardia
B. subtilis
C. elegans
…
S. aureus
Drosophila
(several)
…
Anopheles
(>100)
Ciona
Archaea
Arabidopsis
Methanococcus
Human
Sulfolobus
Phages/Viruses
Mouse
…
Lots
Tetraodon
(total of ~16)
Fugu
Organelles
Zebrafish
Lots
Neurospora
Aspergillus
…

Large-scale Transcript Sequencing
Please see the following example article that uses large-scale
transcript sequencing.
Nature
420
, no. 6915 (5 December 2002): 563-73.
Okazaki, Y, M Furuno, T Kasukawa, J Adachi, H Bono, S Kondo, … "Analysis of The
M
ouse Transcriptome Based On Functional Annotation of 60,770 Full-length cDNAs."

EST Sequencing
dbEST release 022004
No. of public entries: 20,039,613
Summary by Organism - as of February 20, 2004
Homo sapiens
(human)
5,472,005
Mus musculus + domesticus
(mouse)
4,055,481
Rattus sp. (rat)
583,841
Triticum aestivum
(wheat)
549,926
Ciona intestinalis
492,511
Gallus gallus
(chicken)
460,385
Danio rerio
(zebrafish)
450,652
Zea mays
(maize)
391,417
Xenopus laevis
(African clawed frog)
359,901
Hordeum vulgare + subsp. vulgare
(barley)
352,924
Source: NCBI - http://ncbi.nlm.nih.gov
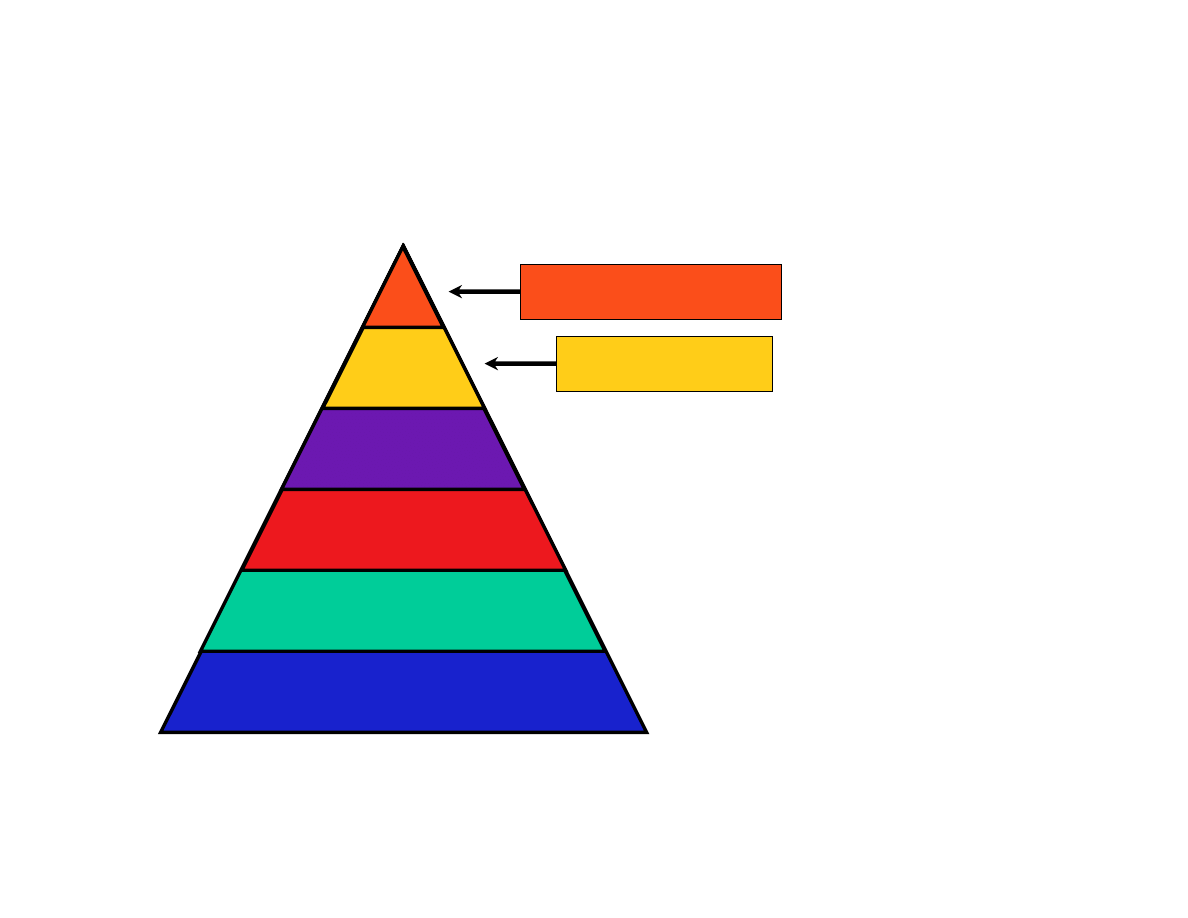
*
-omes and -omics
Proteome
Variome
Transcriptome
Genome
Mass spec, Y2H, ?
SNPs, haplotypes
ESTs, cDNAs, microarrays
Genome sequences
Ribonome?
Glycome ???
*Warning: some of the words on this slide may not be in Webster’s dictionary

DNA Sequence Alignment I
How does DNA alignment differ from protein alignment?
S
ubject:
Use BLASTN instead of BLASTP
1
ttgacctagatgagatgtcgttcacttttactgagctacagaaaa 45
|||| |||||||||||| | |||||||||||||||||||||||||
403 ttgatctagatgagatgccattcacttttactgagctacagaaaa 447
Query:

Nucleotide-
nucleotide
BLAST Web
Server
(BLASTN)

DNA Sequence Alignment II
Translating searches:
translate in all possible reading frames
search peptides against protein database (BLASTP)
ttgacctagatgagatgtcgttcactttactgagctacagaaaa
ttg|acc|tag|atg|aga|tgt|cgt|tca|ctt|tta|ctg|agc|tac|aga|aaa
L T x M R C R S L L L S Y R K
t|tga|cct|aga|tga|gat|gtc|gtt|cac|ttt|tac|tga|gct|aca|gaa|aa
x P R x D V V H F Y x S T E
tt|gac|cta|gat|gag|atg|tcg|ttc|act|ttt|act|gag|cta|cag|aaa|a
D L D E M S F T F T E L Q K
Also consider reading frames on complementary DNA strand

DNA Sequence Alignment III
Common flavors of BLAST:
Program
Query
Database
BLASTP
aa
aa
BLASTN
nt
nt
BLASTX
nt
(
⇒
aa
)
aa
TBLASTN
aa
nt
(
⇒
aa
)
TBLASTX
nt
(
⇒
aa
)
nt
(
⇒
aa
)
PsiBLAST
aa
(
aa
msa)
aa
Which would be best for searching ESTs against a genome?

DNA Sequence Alignment IV
Which alignments are significant?
Q:
1
S:
Identify high scoring segments whose score
S
exceeds
a cutoff
x
using dynamic programming.
Scores follow an extreme value distribution:
P(S > x) = 1 - exp[-Kmn e
-
λx
]
For sequences of length
m, n
where
K,
λ
depend on the score
matrix and the composition of the sequences being compared
(Same theory as for protein sequence alignments)
ttgacctagatgagatgtcgttcacttttactgagctacagaaaa 45
|||| |||||||||||| | |||||||||||||||||||||||||
403 ttgatctagatgagatgccattcacttttactgagctacagaaaa 447

Notes (cont)
From M. Yaffe
Lecture #2
• The random sequence alignment scores
would give rise to an “extreme value”
distribution – like a skewed gaussian.
• Called Gumbel extreme value
distribution
For a normal distribution with a mean m and a variance
σ, the height of the
curve is described by Y=1/(
σ√2π) exp[-(x-m)
2
/2
σ
2
]
For an extreme value distribution, the height of the curve is described by
Y=exp[-x-e
-x
] …and P(S>x) = 1-exp[-e
-
λ(x-u)]
where u=(ln Kmn)/
λ
Can show that mean extreme score is ~ log
2
(nm), and the probability of
getting a score that exceeds some number of “standard deviations” x is:
P(S>x)~ Kmne
-
λx.
***K and
λ are tabulated for different matrices ****
-
λS
For the less statistically inclined: E~ Kmne
-2 -1
0.2
Yev
0.4
-4
4
0.4
B.
Yn
Probability values for the extreme value distribution (A) and the
normal distribution (B). The area under each curve is 1.
0
1
2
X
X
A.
3
4
5

i
DNA Sequence Alignment V
How is
λ
related to the score matrix?
λ
is the unique positive solution to the equation*:
∑
p p
j
e
λ
s
ij
= 1
i
i,j
p
= frequency of nt i,
s
ij
= score for aligning an i,j pair
What kind of an equation is this?
(transcendental)
What would happen to
λ
if we doubled all the scores?
(reduced by half)
What does this tell us about the nature of
λ
?
(scaling factor)
*Karlin & Altschul, 1990

DNA Sequence Alignment VI
What scoring matrix to use for DNA?
Usually use simple match-mismatch matrices:
i
j: A
C
G
T
A
1
m
m
m
C
m
1
m
m
s
i,j
:
G
T
m
m
m
m
1
m
m
1
m =
“mismatch penalty” (must be negative)

DNA Sequence Alignment VII
How to choose the mismatch penalty?
Use theory of High Scoring Segment composition*
High scoring alignments will have composition:
q
ij
= p
i
p
j
e
λs
ij
where
q
ij
=
frequency of
i,j
pairs (“target frequencies”)
p , p =
freq of
i, j
bases in sequences being compared
i
j
What would happen to the target frequencies if we
doubled all of the scores?
*Karlin & Altschul, 1990
Wyszukiwarka
Podobne podstrony:
Genome sequencing & DNA sequence analysis(1)
Biological Sequence Analysis
DNA sequencing methods(1)
DNA sequencing4(1)
DNA Sequencing3
Patterns of damage in genomic DNA sequences from a Neandertal
DNA sequencing(1)
05 DFC 4 1 Sequence and Interation of Key QMS Processes Rev 3 1 03
SHSBC135 THE OVERT MOTIVATOR SEQUENCE
Einschalt Sequenzer
SequenceDiagram 1
Causes and control of filamentous growth in aerobic granular sludge sequencing batch reactors
8 Wire?M V20 cut sequencing
9 INSTALLATION SEQUENCE
więcej podobnych podstron