
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
MINISTERSTWO EDUKACJI
NARODOWEJ
Alicja Królak
Wykonywanie towaroznawczych badań żywności
321[09].Z4.05
Poradnik dla ucznia
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy
Radom 2006

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
1
Recenzenci:
mgr inż. Teresa Kubiak
mgr inż. Aleksandra Ptak
Opracowanie redakcyjne:
mgr inż. Alicja Królak
Konsultacja:
mgr inż. Maria Majewska –
Korekta:
Poradnik stanowi obudowę dydaktyczną programu jednostki modułowej 321[09].Z4.05
Wykonywanie towaroznawczych badań żywności zawartego w modułowym programie nauczania dla
zawodu technik technologii żywności.
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy, Radom 2006

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
2
SPIS TREŚCI
1. Wprowadzenie
3
2. Wymagania wstępne
5
3. Cele kształcenia
6
4. Materiał nauczania
7
4.1. Badanie niektórych cech produktów żywnościowych
7
4.1.1. Materiał nauczania
7
4.1.2. Pytania sprawdzające
10
4.1.3. Ćwiczenia
10
4.1.4. Sprawdzian postępów
14
4.2. Chemiczne badanie produktów żywnościowych
15
4.2.1. Materiał nauczania
15
4.2.2. Pytania sprawdzające
22
4.2.3. Ćwiczenia
22
4.2.4. Sprawdzian postępów
26
4.3. Wykrywanie obcych substancji w żywności
27
4.3.1. Materiał nauczania
27
4.3.2. Pytania sprawdzające
27
4.3.3. Ćwiczenia
28
4.3.4. Sprawdzian postępów
33
4.4 Analiza sensoryczna i ocena organoleptyczna żywności
34
4.4.1. Materiał nauczania
34
4.4.2 Pytania sprawdzające
35
4.4.3. Ćwiczenia
35
4.4.4. Sprawdzian postępów
37
5. Sprawdzian osiągnięć
38
6. Literatura
43

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
3
1. WPROWADZENIE
Poradnik będzie Ci pomocny w przyswajaniu wiedzy dotyczącej stosowania różnych metod
oznaczania zawartości składników w produkowanej żywności. Program jednostki modułowej
obejmuje treści dotyczące wykonywania oznaczeń chemicznych, fizycznych, fizykochemicznych,
W poradniku zamieszczono:
−
wymagania wstępne, w których określono co powinieneś umieć przystępując do realizacji tej
jednostki modułowej,
−
cele kształcenia, które określają umiejętności jakie powinieneś opanować w wyniku procesu
kształcenia,
−
materiał nauczania, który pomoże Ci samodzielne przygotować się do wykonania ćwiczeń
i zaliczenia sprawdzianów. Wykorzystaj do poszerzenia wiedzy wskazaną literaturę oraz inne
źródła informacji. Obejmuje on również ćwiczenia zawierające polecenie, sposób wykonania
oraz wyposażenie stanowiska pracy.
−
sprawdzian postępów, który umożliwi Ci sprawdzenie poziomu wiedzy po wykonaniu ćwiczeń,
−
wykaz literatury,
−
sprawdzian osiągnięć opracowany jest w formie testu zawierającego:
−
instrukcję,
−
zestaw zadań testowych,
−
punktację zadań,
−
kartę odpowiedzi.
Bezpieczeństwo i higiena pracy
Przebywając w laboratorium analizy żywności musisz przestrzegać regulaminu pracowni,
przepisów bezpieczeństwa i higieny pracy oraz przepisów przeciwpożarowych. Przy wykonywaniu
ćwiczeń zachowaj ostrożność podczas ogrzewania roztworów. Szyjkę kolby lub probówki trzymaj
otworem od siebie. Postępuj ostrożnie z roztworami kwasów i zasad szczególnie stężonych. Kwasy
do pipety naciągaj za pomocą pompki a nie ustami.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
4
Schemat układu jednostek modułowych
321[09].04
Analiza żywności w przetwórstwie spożywczym
321[09].04.01
Wykonywanie wagowej analizy żywności
321[09].04.02
Wykonywanie objętościowej analizy żywności
321[09].04.05
Wykonywanie towaroznawczych badań żywności
321[09].04.03
Wykonywanie
instrumentalnej analizy
żywności
321[09].04.04
Wykonywanie
mikrobiologicznych
badań żywności

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
5
2. WYMAGANIA WSTĘPNE
Przystępując do realizacji programu jednostki modułowej powinieneś umieć:
−
korzystać z różnych źródeł informacji,
−
posługiwać się normami,
−
zastosować odczynniki w wykonywaniu analiz,
−
łączyć w zestawy sprzęt laboratoryjny,
−
przygotowywać naważki analityczne,
−
korzystać z wag analitycznych,
−
przygotowywać i przechowywać próbki do badań laboratoryjnych,
−
przeliczać stężenia molowe na procentowe i odwrotnie,
−
przestrzegać zasady Dobrej Praktyki Laboratoryjnej,
−
obsługiwać aparaturę stosowaną do wykonywania towaroznawczych analiz żywności,
−
przygotowywać potrzebne do badań odczynniki i wskaźniki,
−
opracowywać i interpretować wyniki badań,
−
przeprowadzić ocenę organoleptyczną i sensoryczną,
−
wykazać różnice między oceną sensoryczną a organoleptyczną.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
6
3. CELE KSZTAŁCENIA
W wyniku realizacji programu jednostki modułowej powinieneś umieć:
−
oceniać organoleptycznie surowce i produkty produkowane z zastosowaniem różnych
technologii przetwórstwa żywności,
−
oznaczyć cechy fizyczne i chemiczne surowców i produktów spożywczych,
−
określić zawartość wody, suchej masy, kwasowości i składników odżywczych w surowcach
i wyrobach przetwórstwa spożywczego,
−
wykryć i oznaczyć zawartość dodatków w żywności,
−
wykryć i oznaczyć zawartość pozostałości substancji ochrony roślin w surowcach przetwórstwa
spożywczego,
−
wykryć drobnoustroje w surowcach i produktach przetwórstwa spożywczego,
−
wykryć i oznaczyć pozostałości środków higieny w produktach przetwórstwa spożywczego,
−
opracować wyniki badań laboratoryjnych z wykorzystaniem techniki komputerowej,
−
zinterpretować wyniki analiz laboratoryjnych i zarejestrować w dzienniku laboratoryjnym,
−
wykonać laboratoryjne analizy żywności zgodnie z zasadami Dobrej Praktyki Laboratoryjnej,
−
wykonać analizy laboratoryjne (HACCP) w zakładach przetwórstwa spożywczego
monitorujących procesy produkcyjne w celu otrzymywania bezpiecznej żywności,
−
ustalić krytyczne punkty kontroli (CCP) w procesach produkcji artykułów spożywczych
poprzez wykonywanie monitorujących analiz żywności,
−
skorzystać z dokumentacji technicznej i technologicznej przy wykonywaniu oznaczeń
mikrobiologicznych, fizycznych, chemicznych i fizykochemicznych żywności,
−
zastosować przepisy bezpieczeństwa i higieny pracy, ochrony przeciwpożarowej, ochrony
środowiska, oraz wymagania ergonomii na stanowisku pracy,
−
skorzystać z różnych źródeł informacji.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
7
4. MATERIAŁ NAUCZANIA
4.1. Badanie niektórych cech produktów żywnościowych
4.1.1. Materiał nauczania
Oznaczanie temperatury krzepnięcia i topnienia
Oznaczanie temperatury topnienia pozwala na określenie składu chemicznego tłuszczów.
Temperaturą topnienia nazywa się temperaturę, w której substancja przechodzi ze stanu stałego
w stan ciekły. W praktyce za temperaturę topnienia przyjmuje się taką temperaturę, w której badany
tłuszcz osiąga całkowitą przezroczystość. Zasada oznaczania temperatury topnienia tłuszczów
polega na określeniu temperatury, w której w wyniku topnienia następuje unoszenie się do góry
słupka tłuszczu znajdującego się w otwartej z obu stron kapilarze. Wykonując oznaczanie
temperatury topnienia tłuszczów za wynik końcowy przyjmuje się średnią arytmetyczną dwóch
równoległych pomiarów różniących się nie więcej niż 0,5°C. Uzyskany wynik należy porównać
z wymaganiami normy przedmiotowej smalec 28 ÷ 40°C, łój topiony 42 ÷ 48°C.
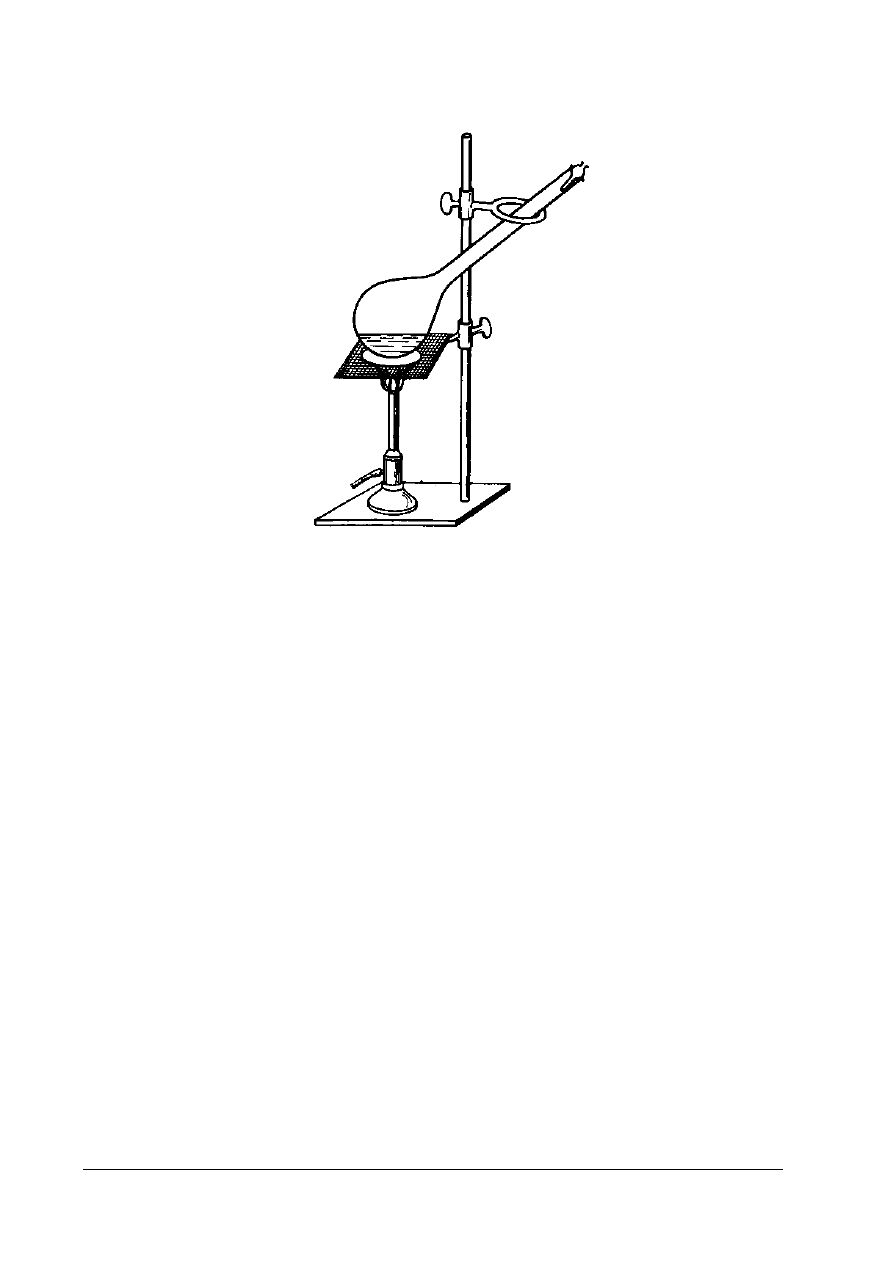
Rys. 1. Schemat aparatu Thielego do oznaczania temperatury topnienia tłuszczu [3, s. 178]
Charakter mieszaniny stałych tłuszczów spożywczych, zachodzące zjawiska uboczne w czasie
topnienia lub krzepnięcia sprawiają, że tłuszcze posiadają temperaturę całkowitego sklarowania
(temp. topnienia) różną od temperatury krzepnięcia. Należy dodać, że temperaturę krzepnięcia
można często oznaczyć dokładniej niż temperaturę topnienia. Wygląd tłuszczu w czasie krzepnięcia
nie jest na tyle charakterystyczny, aby określić temperaturę odpowiadającą temu procesowi, dlatego
w metodzie oznaczania temperatury krzepnięcia wykorzystano zjawisko fizyczne. Polega ono na
tym, że temperatura substancji krzepnącej, wolno chłodzonej pozostaje w ciągu pewnego czasu bez
zmiany, albo po pewnym przechłodzeniu wzrasta (substancja sama się ogrzewa). Za temperaturę
krzepnięcia przyjmuje się temperaturę, która przez pewien czas jest stała w czasie krzepnięcia
substancji (pomimo jej chłodzenia) lub tę temperaturę maksymalną, którą substancja osiągnie po
wyjściu ze stanu pewnego przechłodzenia. Oznaczanie temperatury krzepnięcia tłuszczów
przeprowadza się metodą termostatową lub uproszczoną metodą statyczna. Obie metody oparte są
na tej samej podanej zasadzie, różnią się nieco odmiennym wykonaniem oznaczenia. Metodę
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
8
termostatową
stosuje
się
do
badania
tłuszczów
roślinnych,
jadalnych
o temperaturze topnienia poniżej 30ºC. Metodę statyczną stosuje się do tłuszczów roślinnych
jadalnych o temperaturze topnienia 30ºC i powyżej 30ºC i stosuje się inną aparaturę niż w metodzie
termostatowej.
Do oznaczania temperatury krzepnięcia tłuszczu może służyć aparat przedstawiony na rysunku 2.
Dla mieszanin takich jak tłuszcze (mieszanina glicerydów) temperatura topnienia różni się od
temperatury krzepnięcia, ponieważ w czasie topnienia lub krzepnięcia zachodzą zjawiska uboczne,
np. zmiana rozpuszczalności ze zmianą stanu skupienia. Temperatury te wahają się w pewnych
granicach charakterystycznych dla tłuszczów naturalnych.
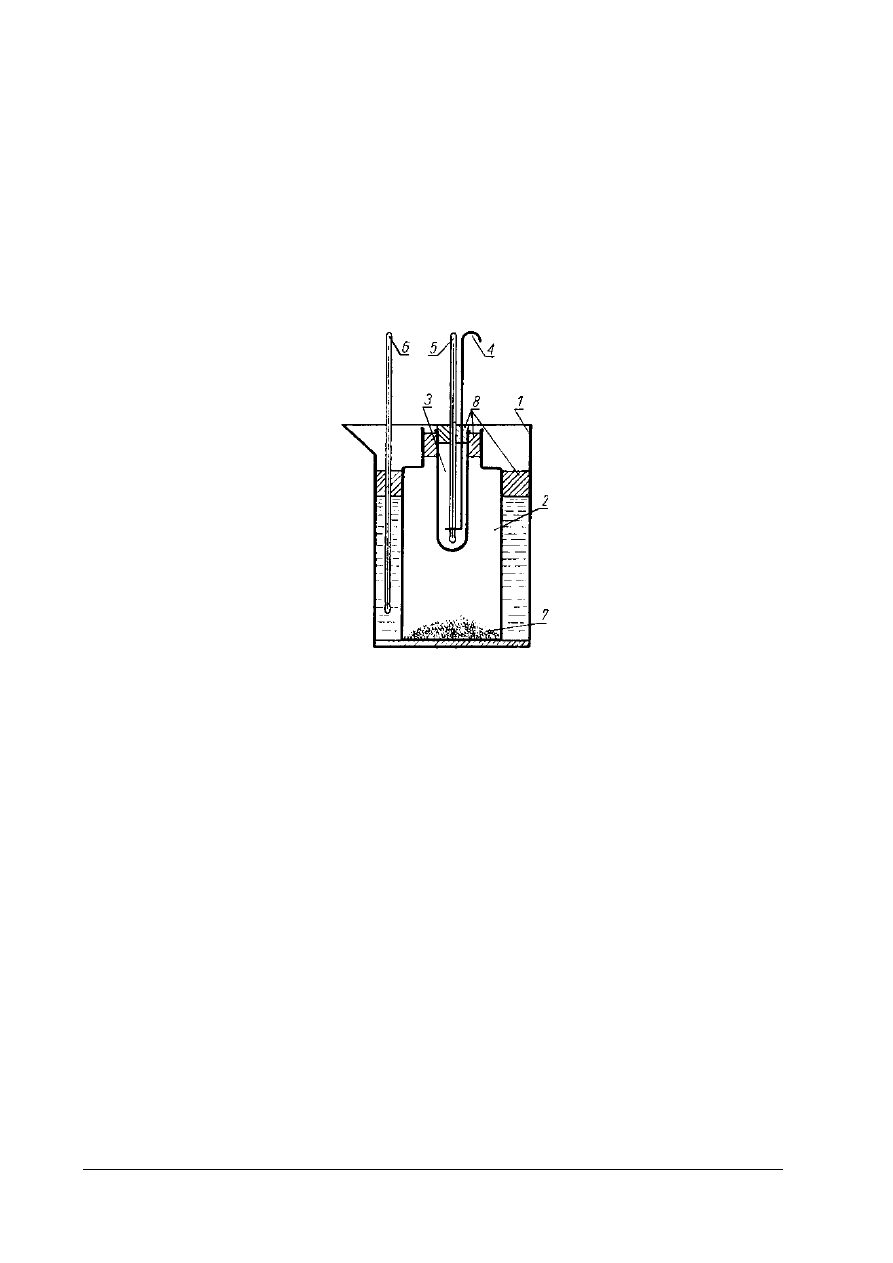
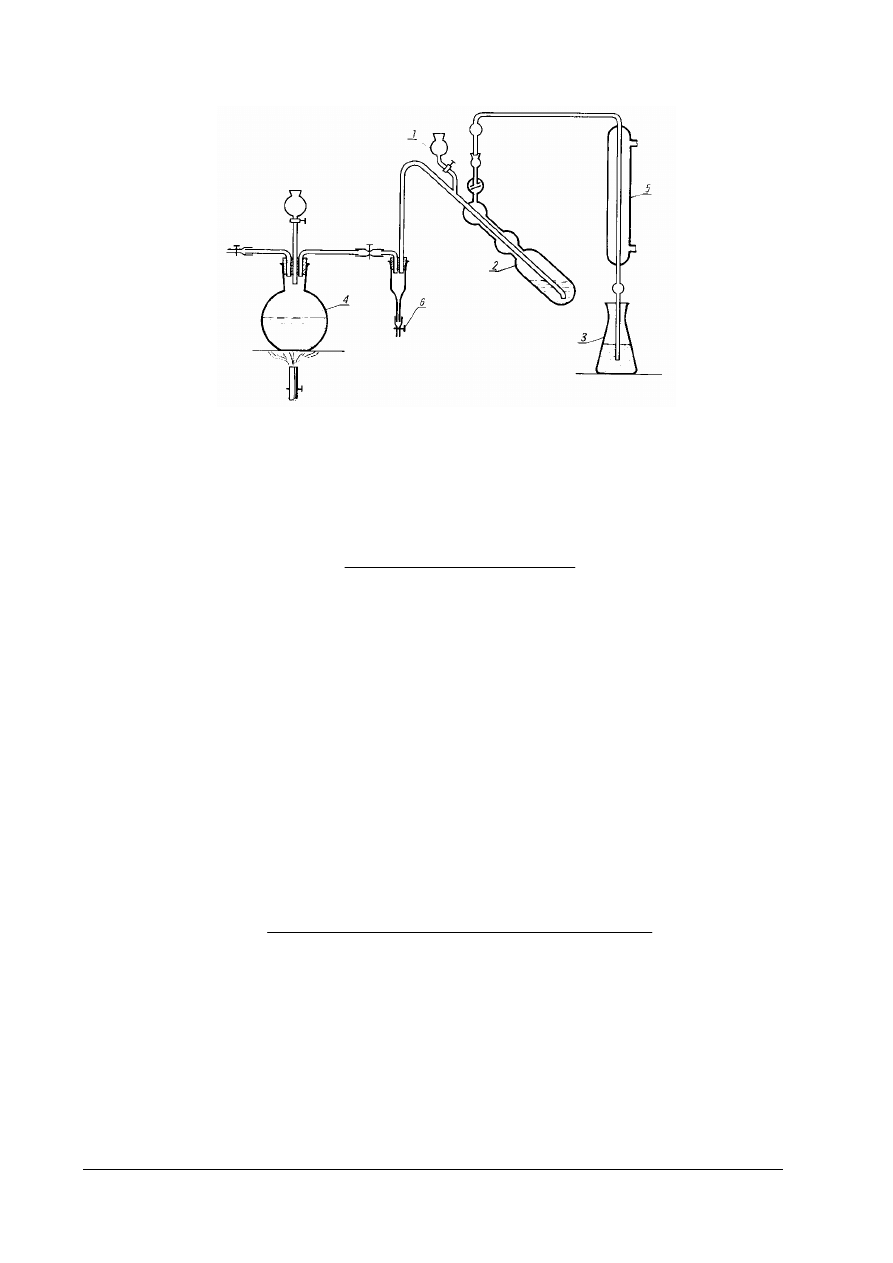
Rys. 2. Schemat aparatu do oznaczania temperatury krzepnięcia tłuszczu 1 – zlewka, 2 – słoik, 3 – probówka,
4 - mieszadło, 5 i 6 – termometr laboratoryjny, 7 – obciążenie (śrut), 8 – korek [ 1, s. 179]
Oznaczanie kwasowości
Kwasowość surowców i produktów przemysłu spożywczego jest uwarunkowana
występowaniem w nich wielu kwasów np. jabłkowego, winowego, cytrynowego, szczawiowego.
Występują w nich także kwasy tłuszczowe jak masłowy, stearynowy, palmitynowy, linolowy
linolenowy, oleinowy, stearynowy. Oprócz wymienionych kwasy mogą powstawać w wyniku
fermentacji cukrów np. octowy, propionowy, mlekowy. Z punktu widzenia chemicznego można
wyróżnić dwa rodzaje kwasowości: potencjalną (bierną) i aktywną, uwarunkowaną stężeniem lub
aktywnością jonów wodorowych (H
+
) lub hydroniowych (H
3
O
+
).
Kwasowość lub zasadowość produktu może być wyrażona ilością zasady lub kwasu
potrzebnego do zobojętnienia zawartych w nim związków reagujących kwaśno (lub zasadowo)
wobec wskaźnika. Jest to tzw. kwasowość miareczkowa lub potencjalna. Miareczkowy sposób
wyrażania kwasowości nie jest bardzo dokładny, ale nie wymaga stosowania specjalnej aparatury jak
w przypadku mierzenia pH. Istnieje wiele metod oznaczania kwasowości, których wyniki mogą się
między sobą różnić. Dlatego należy przy podawaniu wyników zaznaczyć jaką metodą zostały
otrzymane.
W analizie surowców i produktów przemysłu spożywczego wyróżnia się oprócz kwasowości
potencjalnej i aktywnej kwasowość lotną, kwasowość związaną (estry, sole) oraz kwasowość
całkowitą, która jest sumą kwasowości potencjalnej i związanej.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
9
Kwasowość potencjalna
Kwasowość potencjalną oznacza się procesie miareczkowania mianowanymi roztworami zasad
w obecności wskaźników, które pozwalają na uchwycenie końcowego punktu miareczkowania.
Procesy miareczkowania kwasów zasadami i odwrotnie można prowadzić śledząc zmiany wartości
liczbowych pH na potencjometrze lub pehametrze. Podstawą oznaczeń kwasowości potencjalnej są
reakcje zobojętniania zgodnie z reakcją:
H
+
+ R
-
+ M
+
+ OH
-
→
H
2
O + M
+
+ R
-
kwas zasada woda sól
Reakcje zachodzą podczas miareczkowania mianowanymi roztworami zasad lub odwrotnie przy
czym w miarę zobojętniania jonów wodorowych jonami wodorotlenowymi zmieniają się wartości
liczbowe pH, które charakteryzują odczyn środowiska. Nie zawsze zmiany wartości liczbowych pH
są proporcjonalne do stopnia zobojętnienia kwasu zasadą bądź odwrotnie. W przypadku
miareczkowania
roztworów
słabych
kwasów
mocnymi
zasadami
lub
odwrotnie
i mocnych kwasów słabymi zasadami powstają roztwory buforowe, które mają zdolność stabilizacji pH.
Oznaczanie kwasowości aktywnej
Kwasowość aktywną można oznaczyć orientacyjnie metodą kolorymetryczną lub dokładnie
metodą elektrometryczną za pomocą potencjometrów kompensacyjnych lub lampowych.
Metoda kolorymetryczna polega na dodaniu do roztworu odpowiedniego wskaźnika
i porównaniu intensywności powstałego zabarwienia ze skalą wzorców przygotowanych na
roztworach o znanych wartościach pH. Można też stosować paski bibuły nasycone odpowiednimi
wskaźnikami, które po zanurzeniu w badanym roztworze przyjmują określone zabarwienie,
porównywane z odpowiednimi wzorcami.
Metody elektrometryczne polegają na bezpośrednim lub pośrednim pomiarze siły
elektromotorycznej ogniw złożonych z dwóch elektrod, tj. porównawczej (odniesienia) i badanej
(wskaźnikowej). Elektrodami są płytki metalowe zanurzone w roztworach elektrolitów. Metal
zanurzony w roztworze soli ładuje się dodatnio lub ujemnie w zależności od tego czy przeważa
energia krystaliczna siatki czy energia hydratacji. W przypadku roztworu rozcieńczonego kationy
metalu przechodzą z płytki do roztworu, przy jednoczesnym pozostawianiu jonów ujemnych na
powierzchni płytki. Jeżeli nie ma ciągłego odpływu ładunków ujemnych z płytki, wówczas przy jej
powierzchni gromadzą się jony dodatnie i powstaje podwójna warstwa elektryczna przypominająca
płaski kondensator. W roztworach stężonych procesy te przebiegają inaczej. Jony dodatnie
wydzielają się na powierzchni płytki metalowej, która przyciąga ładunki ujemne z roztworu
i podobnie jak poprzednio tworzy się podwójna warstwa elektryczna. W obydwu przypadkach
dochodzi do ustalenia się stanów równowagi pomiędzy przechodzeniem jonów do roztworu i ich
wydzielaniem się na powierzchni płytek metalowych czemu odpowiadają pewne skoki potencjałów
na granicy faz: metal-roztwór. Wielkości skoku potencjału nie można mierzyć bezpośrednio
doświadczalnie, dlatego do roztworu elektrolitu zanurza się dwie płytki z różnych metali, które łączy
się ze sobą przewodem zewnętrznym i mierzy różnicę potencjałów odpowiadającą różnicy
poszczególnych
skoków
potencjałów
na
powierzchniach
rozdziału:
metal-elektrolit
i elektrolit-metal. Układ tego typu odpowiada ogniwu galwanicznemu.
Do pomiaru kolorymetrycznego stosowane są spektrofotometry. Szczegółowe uwagi o obsłudze
kolorymetrów zawarte są w instrukcjach załączonych przez producentów. Nie należy posługiwać się
aparatem w sposób nie przewidziany przez instrukcję, gdyż można go trwale uszkodzić.
Oznaczanie kwasowości lotnej
Wśród kwasów występujących w produktach spożywczych występują kwasy lotne i nielotne.
Do lotnych zaliczane są niskocząsteczkowe kwasy jednokarboksylowe: mrówkowy, octowy,
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
10
propionowy i masłowy, a także siarkawy i węglowy. Do oznaczania kwasów lotnych stosuje się
aparaturę przedstawioną na rysunku 3.
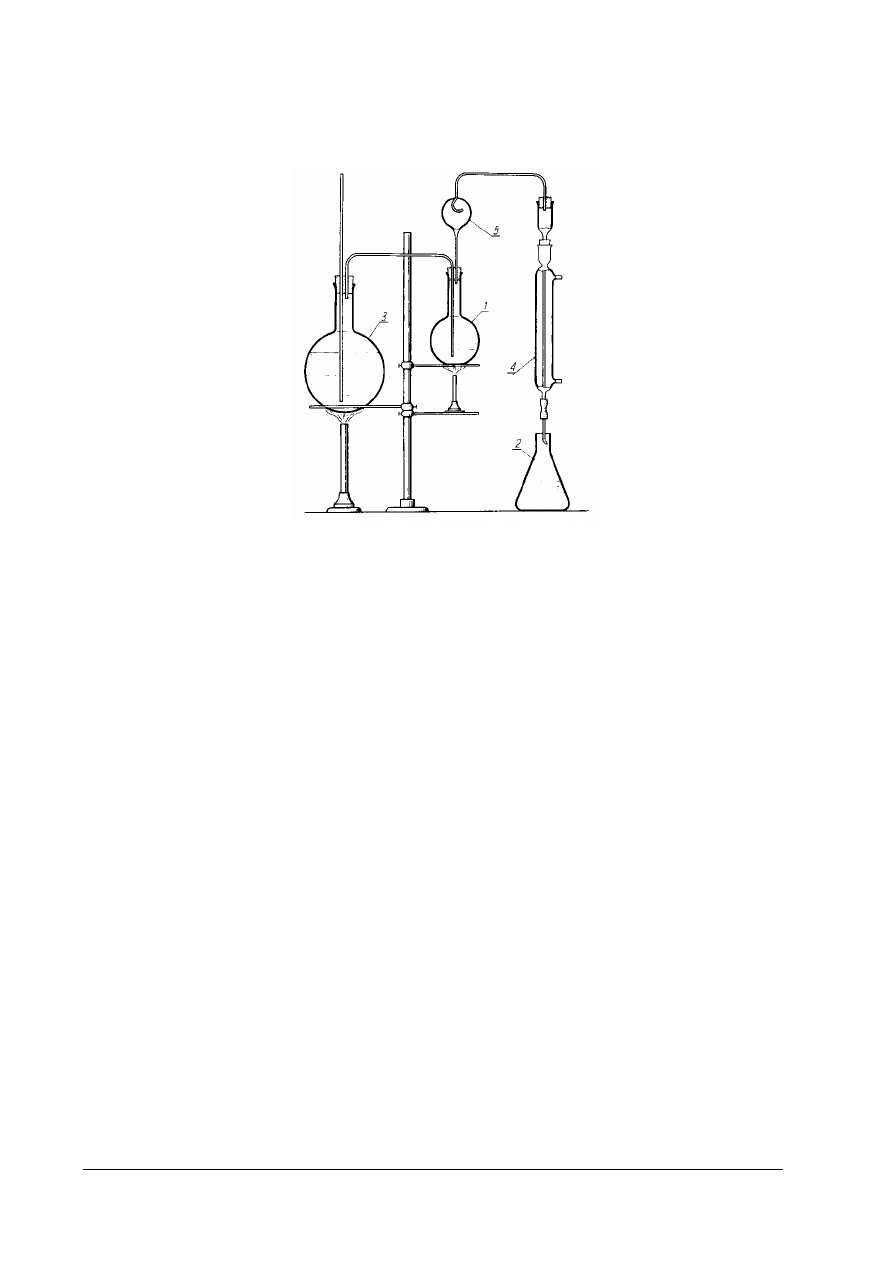
Rys. 3. Zestaw do destylacji kwasów lotnych z parą wodną 1 – kolba destylacyjna o pojemności 500cm
3
, 2 – kolba
stożkowa o pojemności 250cm
3
, 3 – kolba destylacyjna o pojemności 1000cm
3
, (wytwornica pary), 4 – chłodnica,
5 – deflegmator [ 1,s. 179]
4.1.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1. Jak wyznaczyć temperaturę topnienia i krzepnięcia tłuszczów?
2. Jakiej aparatury należy użyć do przeprowadzenia oznaczenia temperatury topnienia
i krzepnięcia tłuszczów?
3. W jakim celu prowadzi się oznaczanie kwasowości w analizie towaroznawczej?
4. Jakie znasz metody oznaczania kwasowości?
5. Jakie rodzaje kwasowości można wyróżnić z chemicznego punktu widzenia?
6. Na czym polega sposób oznaczania kwasowości potencjalnejaktywnej?
4.1.3. Ćwiczenia
Ćwiczenie 1
Oznacz temperaturę topnienia smalcu.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbkę do badania zgodnie z obowiązującą normą,
2) ogrzać smalec do temperatury wyższej o 8 ÷ 10°C , niż przypuszczalna temperatura topnienia
i przesączyć,
3) zanurzyć dwie kapilary tak, aby słupek tłuszczu w nich był na wysokości ok. 1cm,
4) wytrzeć zewnętrzne ścianki kapilary skrawkiem bibuły,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
11
5) wstawić kapilary do lodówki o temperaturze 0°C i po skrzepnięciu tłuszczu przymocować do
pierścienia gumowego do termometru tak, aby koniec kapilary z tłuszczem był na wysokości dna
zbiorniczka z rtęcią termometru,
6) utrzymywać termometr z kapilarami w temperaturze 15 ÷ 17°C,
7) napełnić aparat Theliego wodą destylowaną odpowietrzoną przez gotowanie i ochłodzoną do
temperatury 15 ÷ 17°C,
8) umieścić termometr z kapilarami dokładnie w centrum aparatu,
9) ogrzewać boczne odgałęzienie aparatu małym płomieniem palnika tak, aby wzrost temperatury
następował z prędkością 1÷ 2°C/min,
10) zanotować temperaturę, w której słupek zaczyna się podnosić,
11) odczytać i zinterpretować wynik. Za wynik końcowy przyjmij średnia arytmetyczna dwóch
równoległych pomiarów różniących się nie więcej niż 0,5ºC. Wynik podaj z dokładnością do
0,1ºC. Uzyskany wynik porównaj z następującymi wymaganiami normy przedmiotowej (PN-
84/A-8502):
– smalec - 28÷40ºC; łój topiony - 42÷ 48ºC.
Wyposażenie stanowiska pracy:
−
próbka smalcu,
−
rurki szklane, kapilarne otwarte na obu końcach, o średnicy wewnętrznej 1,1 ÷ 1,3mm, średnicy
zewnętrznej 1,4 ÷ 1,7mm i długości 50÷ 60mm,
−
termometr o zakresie skali 0 ÷ 50°C i działce elementarnej co 0,1 lub 0,2 stopnia,
−
aparat Theliego z instrukcją obsługi,
−
termostat,
−
siarczan sodowy,
−
chłodziarka,
−
norma przedmiotowa.
Ćwiczenie 2
Oznacz temperaturę krzepnięcia tłuszczu roślinnego metodą termostatową.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) zmontować aparaturę do oznaczania temperatury krzepnięcia tłuszczów roślinnych (rys.2),
2) napełnić zlewkę wodą tak, aby jej powierzchnia znalazła się o 10mm nad przewidywaną
powierzchnią tłuszczów w probówce,
3) ustalić
temperaturę
w
zlewce
na
15÷20°C
poniżej
przewidywanej
temperatury
krzepnięcia tłuszczu,
4) próbkę tłuszczu sklarowanego i bezwodnego, ogrzać do temperatury o 15°C powyżej jego
temperatury mięknienia i wlać do probówki, umieścić termometr w probówce w pierścieniu
mieszadełka w jednakowej odległości od ścianek tak, aby zakończenie znajdowało się 10mm
powyżej dna,
5) rozpocząć mieszanie po obniżeniu się temperatury tłuszczu do temperatury krzepnięcia ruchem
posuwistym z góry na dół z prędkością 100 ruchów na minutę,
6) przerwać mieszanie, gdy temperatura zatrzyma się na tym samym poziomie w ciągu 30 sekund
lub zacznie wzrastać,
7) podnieść mieszadełko na powierzchnię tłuszczu,
8) notować temperaturę w odstępach 1-minutowych do momentu, gdy słupek rtęci zatrzyma się
przez pewien czas na jednym poziomie lub osiągnie maksymalną wartość przy wznoszeniu,
9) podać wynik ostateczny przyjmując średnią dwóch pomiarów, których różnica nie przekracza 1°C.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
12
Wyposażenie stanowiska pracy:
−
próbka tłuszczu roślinnego,
−
zestaw aparatury do oznaczania tłuszczów roślinnych metodą termostatową,
Ćwiczenie 3
Oznacz kwasowość miareczkową (ogólną, potencjalną) mleka.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) popłukać pipetę o pojemności 50cm
3
niewielką ilością badanej próbki mleka,
2) odmierzyć pipetą 50cm
3
próbki do kolby stożkowej o pojemności 250cm
3
,
3) dodać do próbki 2% roztworu fenoloftaleiny jako wskaźnika,
4) miareczkować z biurety mianowanym roztworem NaOH o stężeniu 0,25 mol/dm
3
do chwili
uzyskania jasnoróżowej barwy, utrzymującej się co najmniej 30s,
5) zapisać wynik miareczkowania,
6) wykonać drugą identyczną próbę,
7) przyjąć wartość średniej arytmetycznej zużycia roztworu NaOH do obliczenia kwasowości mleka
wyrażając ją w stopniach Soxleta-Henkla,
8) wykonać obliczenia. Stopnie Soxleta-Henkla wyrażają liczbę cm
3
ściśle 0,25-molowego
roztworu NaOH zużytych do miareczkowania 100cm
3
mleka. Średni wynik miareczkowania
próbek mleka, o objętości 50cm
3
każda, należy pomnożyć prze 2, a następnie sprowadzić
stężenie roztworu zasady do ściśle 0,25mol/dm
3
i skorygować obliczoną objętość. W tym celu
wynik
poprzedniego
mnożenia
należy
przemnożyć
przez
faktyczne
stężenie
zasady(ok.0,25mol/dm
3
) i podzielić przez 0,2500, pamiętając że dwa ostatnie działania
sprowadzają się do mnożenia objętości przez współczynnik przeliczeniowy (W), wyrażający
stosunek stężenia faktycznego do żądanego ( w tym przypadku
2500
,
0
/
25
,
0
3
dm
mola
).
Wyposażenie stanowiska pracy:
−
próbka mleka,
−
biureta o pojemności 50 cm
3
ze ściskaczem na statywie,
−
pipeta jednomiarowa o pojemności 50cm
3
,
−
kolba stożkowa o pojemności 250cm
3
,
−
pipeta wielomiarowa o pojemności 5cm
3
,
−
wodorotlenek sodu, roztwór o stężeniu ok. 0,25 mol/dm
3
, ściśle mianowany,
−
fenoloftaleina, roztwór alkoholowy 2%.
Ćwiczenie 4
Oznacz kwasowość miareczkową (ogólną, potencjalną) jabłek.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbki (wyciąg z jabłek) do badania:
2) odważyć na wadze technicznej 20 g homogenizowanej próbki jabłek
3) dodać 60 cm
3
wody destylowanej,
4) ogrzać do wrzenia, utrzymać w tym stanie około 3 minuty, ochłodzić,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
13
5) przenieść ilościowo za pomocą lejka i bagietki do kolby miarowej o pojemności 100cm
3
,
6) uzupełnić zawartość kolby wodą destylowaną do kreski,
7) zamknąć kolbę doszlifowanym korkiem i wymieszać, sączyć przez sączek z bibuły filtracyjnej,
8) odrzucić pierwsze kilka cm
3
przesączu, a resztę wykorzystać do miareczkowania w następnym
ćwiczeniu,
9) wykonać oznaczenie:
10) odmierzyć do zlewki o pojemności 150cm
3
pipetą 20cm
3
wyciągu z jabłek,
11) dodać cylindrem 20cm
3
świeżo przegotowanej i ostudzonej wody destylowanej,
12) dodać z biurety mianowany roztwór NaOH o stężeniu 0,25 mol/ dm
3
13) miareczkować w obecności papierka lakmusowego jako wskaźnika (pobrać bagietką na
papierek
lakmusowy
krople
miareczkowanego
płynu
obserwować
zmianę
barwy
z różowej na jasnoniebieską),
14) pobierać w pierwszej fazie miareczkowania próbki kontrolne po dodaniu każdego następnego
1 cm
3
roztworu NaOH, następnie po 0,25 cm
3
i w końcu po każdych 2 ÷ 3 kroplach,
15) nanosić krople kontrolne miareczkowanego za każdym razem na nowe paski papierka
lakmusowego,
16) zapisać wynik miareczkowania z dokładnością do 0,1cm
3
,
17) wykonać drugą identyczną próbę z tym, że od razu dodać roztwór NaOH w ilości ok. 1cm
3
mniejszej niż w próbie wstępnej,
18) miareczkować z biurety tym samym roztworem ługu płyn w zlewce,
19) prowadzić kontrolę odczynu środowiska po dodaniu każdych 2 kropli roztworu NaOH,
20) zapisać wynik miareczkowania,
21) przyjąć wynik miareczkowania do obliczenia kwasowości miareczkowej,
22) wykonać obliczenia. Kwasowość jabłek wyraża się umownie w przeliczeniu na kwas jabłkowy.
Szukane stężenie molowe kwasu w wyciągu z jabłek można obliczyć ze wzoru:
2
1
⋅
⋅
⋅
=
k
z
z
k
V
V
Cm
Cm
Podstawiając dane do powyższego równania: Cm
z
– ok.0,25mol/dm
3
, V
z
– liczba cm
3
zasady
ustalona w procesie miareczkowania, V
k
– 20cm
3
, obliczyć stężenie kwasu jabłkowego w wyciągu
z jabłek, a następnie pomnożyć je przez masę molowa kwasu jabłkowego (134g), otrzymując w ten
sposób liczbę gramów kwasu jabłkowego w przeliczeniu na 1000cm
3
wyciągu. Wiedząc, że z 20g
homogenizowanej próbki jabłek uzupełniono do objętości 100cm
3
, łatwo obliczyć, że objętości
1000cm
3
wyciągu odpowiada 200g jabłek. Aby wyrazić wynik w % należy podzielić go przez 2.
Wyposażenie stanowiska pracy:
−
próbka jabłek homogenizowanych,
−
zlewki o pojemności 100 i 150cm
3
,
−
kolba miarowa o pojemności100cm
3
,
−
łyżeczka porcelanowa,
−
waga techniczna z kompletem odważników,
−
lejek szklany,
−
bagietka szklana,
−
trójnóg,
−
siatka,
−
palnik gazowy,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
14
−
sączek z bibuły filtracyjnej fałdowany,
−
kolba stożkowa o pojemności 250cm
3
,
−
pipeta jednomiarowa o pojemności 20cm
3
,
−
biureta o pojemności 50cm
3
ze ściskaczem na statywie,
−
cylinder miarowy o pojemności 100cm
3
,
−
wodorotlenek sodu, roztwór o stężeniu ok. 25mol/dm
3,
−
papierki lakmusowe czerwone,
−
woda destylowana, świeżo przegotowana i ochłodzona do temperatury pokojowej,
−
norma jakościowa.
4.1.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) zdefiniować pojęcia temperatury topnienia i krzepnięcia?
2) oznaczyć temperaturę topnienia i krzepnięcia tłuszczów?
3) określić pojęcie kwasowości miareczkowej (potencjalnej)?
4) dobrać sprzęt i aparaturę do oznaczania kwasowości ogólnej i lotnej?
5) określić pojęcie kwasowości aktywnej?
6) oznaczyć kwasowość produktów żywnościowych?
7) wykonać obliczenia i interpretować wyniki po wykonaniu oznaczenia?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
15
4.2. Chemiczne badanie produktów żywnościowych
4.2.1. Materiał nauczania
Oznaczanie zawartości suchej masy (wody)
Pod pojęciem wody w produkcie żywnościowym rozumie się ogólnie taką ilość wody, którą
można w nim oznaczyć jakąkolwiek z dostępnych dla danego produktu metod. Z pojęciem wody
nieodłącznie wiąże się pojęcie suchej substancji (suchej masy).
Pod pojęciem suchej masy rozumie się pozostałość po odparowaniu wody. Przyjmuje się, że
wszystko to co ulega odparowaniu w czasie 1÷3 godzinnego suszenia kilkugramowej masy
w temperaturze ok. 100ºC stanowi wodę, a pozostałość po suszeniu suchą masę.
Woda w produktach żywnościowych występuje w różnych postaciach. Rozróżnia się wodę
wolną i związaną. Woda wolna występuje w cieczach ustrojowych jako rozpuszczalnik substancji
organicznych i mineralnych, wypełnia wolne przestrzenie, ale nie podlega zjawiskom kapilarnym.
Woda związana może wiązać się w produkcie w różny sposób. Dlatego rozróżnia się wodę
higroskopijną powlekającą cienką warstwą powierzchnie wolne produktu, wodę kapilarną
występującą w naczyńkach włoskowatych, wodę krystaliczną i wodę związaną chemicznie.
Podstawowymi składnikami suchej masy w przypadku surowców i produktów przemysłu
spożywczego są cukrowce, białka, tłuszcze, związki mineralne, kwasy organiczne, barwniki, garbniki
i witaminy. Składniki suchej masy można podzielić na dwa rodzaje : rozpuszczalne
w wodzie i nierozpuszczalne w wodzie (błonnik, oraz część białek, pektyn itp.). Przeważającą część
suchej masy stanowią składniki ekstraktowe takie jak cukry i częściowo kwasy organiczne.
Oznaczanie suchej masy nie jest skomplikowane, ale jest pracochłonne, dlatego do
badań wykorzystuje się refraktometr lub aerometr. Do oznaczania suchej masy stosowane są
metody:
−
suszenia,
−
destylacji mieszanin azeotropowych
−
chemiczne,
−
oparte na gęstości wodnych roztworów,
−
refraktometryczne.
Najczęściej stosowane są metody suszenia termicznego w suszarkach wyposażonych
w termoregulatory pozwalające na utrzymanie stałej temperatury podczas suszenia. Suszenie ma
miejsce wówczas gdy ciśnienie pary wodnej w materiale suszonym jest wyższe od ciśnienia
panującego w otoczeniu (komorze, suszarni). Suszenie przebiega tym szybciej, im większa jest
różnica ciśnień. Przy suszeniu materiałów z dużą ilością wody np. warzywa suszenie należy
prowadzić dwustopniowo. Suszenie dwustopniowe polega na tym, że naważkę badanego surowca
poddaje się wstępnemu suszeniu na płytce Petriego na wrzącej łaźni wodnej w ciągu 45 minut,
a potem dosusza w suszarce o temperaturze 95 ÷ 98°C w czasie dalszych 45 minut. Bardzo ważny
jest dobór parametrów suszenia do charakteru i właściwości suszonych próbek. Przy ustalaniu
parametrów należy uwzględnić, że są one względem siebie odwrotnie proporcjonalne (czas
i temperatura) tzn. przy podwyższaniu temperatury suszenia skraca się czas trwania tego procesu
i odwrotnie. Za najbardziej optymalne warunki suszenia uważa się suszenie próżniowe, czyli suszenie
pod zredukowanym ciśnieniem. Jest ono przydatne w końcowej fazie suszenia, gdy usuwa się resztki
wody.
Metody destylacji azeotropowej polegają na tym, że woda zawarta w badanych próbkach
tworzy z rozpuszczalnikami organicznymi (ksylen, toluen) mieszaniny azeotropowe, czyli takie,
które mają stałą temperaturę wrzenia. W destylacji azeotropowej musi być spełniony warunek, że
rozpuszczalniki muszą mieć wyższe od wody temperatury wrzenia (ksylen 135 ÷ 140°C toluen
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
16
111°C), a jednocześnie nie mogą mieszać się z wodą. Metoda stosowana jest do oznaczania wody
w surowcach i produktach zawierających ją w niewielkich ilościach jak zboża i produkty ich
przemiału. Do destylacji stosowane są aparaty (rys. 4) zaopatrzone w wyskalowane biuretki,
w których odczytywane są objętości destylowanej wody. Opary rozpuszczalnika i wody ulegają
skropleniu w chłodnicy i zbierają się w wyskalowanej biuretce. Woda posiada gęstość inną niż
rozpuszczalnik i zbiera się w dolnej części biuretki, a nadmiar rozpuszczalnika jest zawracany do
kolby destylacyjnej. Destylację prowadzi się ok. 30 ÷ 60 minut, po czym odczytuje się objętość wody
w cm
3
. Przyjmując, że w 1 cm
3
wody jest równy 1 g oblicza się procentową zawartość wody
w badanym materiale.
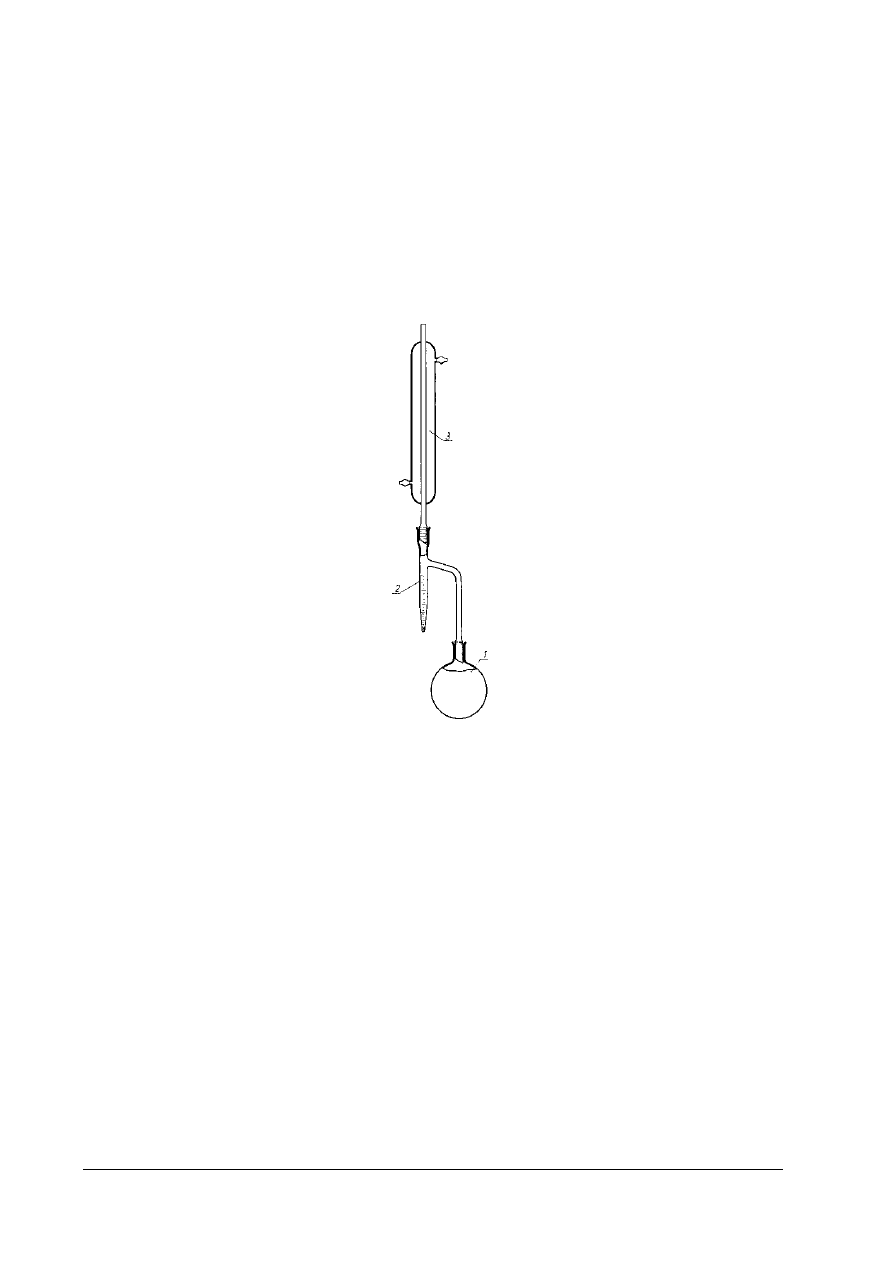
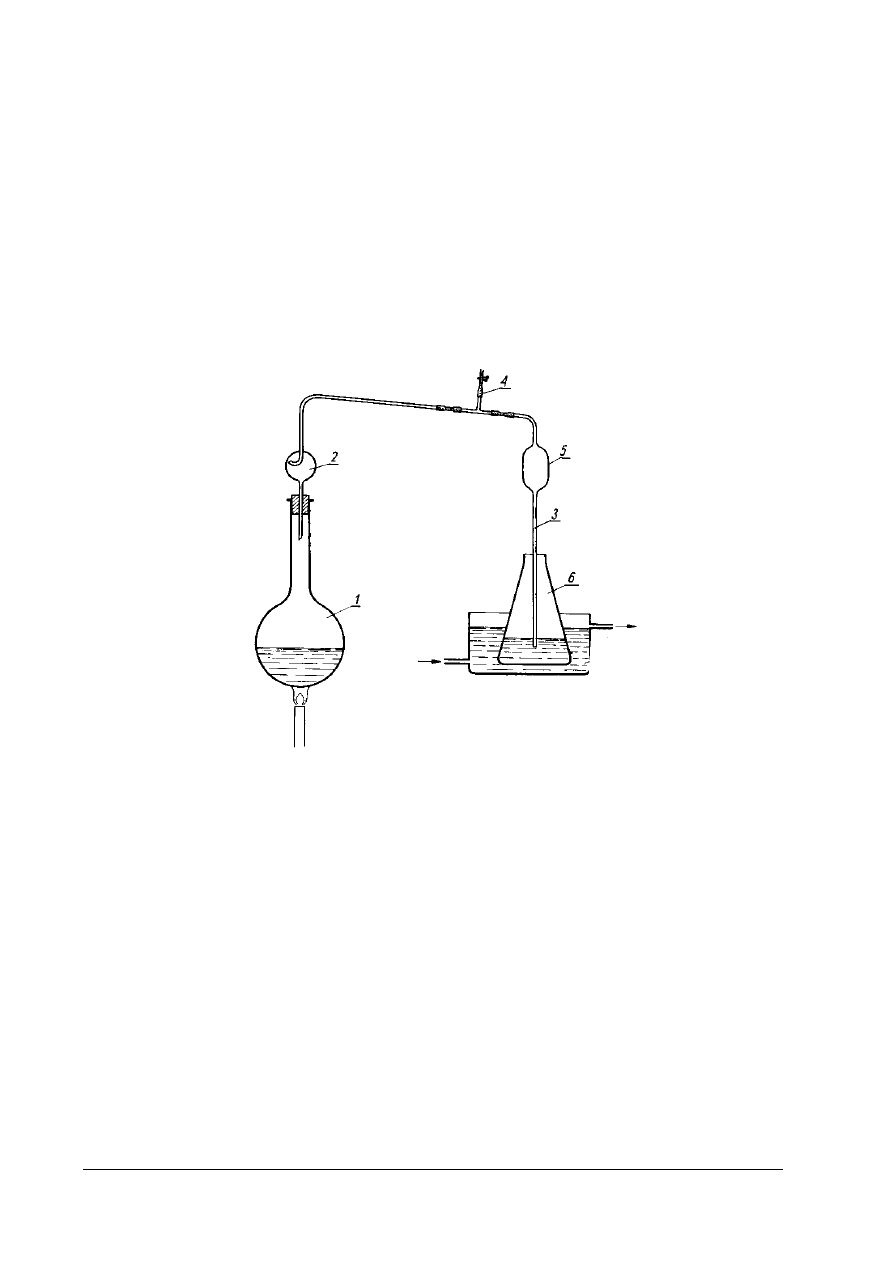
Rys. 4. Aparat do oznaczania zawartości wody metodą destylacyjną 1- kolba destylacyjna o pojemności 750 cm
3
,
2 – wyskalowana biureta, 3- chłodnica [4, s. 306]
W metodach chemicznych wykorzystuje się reakcje chemiczne zachodzące między wodą
zawartą w badanych próbkach, a niektórymi substancjami celowo do niej dodawanymi np. karbid,
wodorek wapnia lub odczynnik K. Fischera. Powstałe produkty oblicza się ilościowo i na tej
podstawie oraz w oparciu o równania reakcji oblicza się procentową zawartość wody w badanych
surowcach lub produktach.
Oznaczanie zawartości białek
Białka należą do związków azotowych. Zawierają w swoim składzie azot, którego przeciętna
ilość wynosi 16%. Ogólną zawartość białek oznacza się najczęściej klasyczną metoda Kjeldahla,
która polega na mineralizacji substancji organicznej w stężonym kwasie siarkowym z dodatkiem
siarczanu potasu i katalizatorów w temperaturze wrzenia tj. ok. 300°C. Związki organiczne ulegają
utlenieniu do dwutlenku węgla i wody, a zawarty w nich azot wydziela się w postaci amoniaku.
Amoniak w środowisku kwasu siarkowego przechodzi w siarczan amonu, ten po zalkalizowaniu
wydziela amoniak, który oddestylowuje się do kwasu.
Spalanie (mineralizację próbek do oznaczeń zawartości białka metodą Kjeldahla prowadzi się
w specjalnych kolbach Kjeldahla o jajowatym kształcie i pojemnościach 250, 500 lub 750cm
3
pod
wyciągiem ze względu na żrący charakter wydzielających się gazów (rysunek 5)
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
17
Rys. 5. Spalanie w kolbie Kjeldahla – oznaczanie zawartości azotu [3, s. 48]
Wielkość próbki do mineralizacji uzależnia się od zawartości białka w badanym produkcie. Jako
optymalną przyjmuje się próbkę zawierającą 10 ÷ 30mg azotu co odpowiada 60 ÷ 200mg czystego
białka. Zwykle jest to próbka 0,5 ÷ 2g w przypadku materiałów stałych i kilka, a nawet
kilkadziesiąt cm
3
w przypadku materiałów płynnych. Przed rozpoczęciem mineralizacji dodaje się
katalizatory np. rtęciowe (Hg, HgO), miedziowe (Cu, CuO, CuSO
4
5 H
2
O) lub selenowe
(CuSeO · 2 H
2
O). Dodaje się także substancję podnoszącą temperaturę wrzenia (K
2
SO
4
).
Do destylacji amoniaku stosuje się aparat Parnasa-Wagnera rysunek 6. Zawartość kolby
stożkowej, do której oddestylowano amoniak miareczkuje się mianowanym roztworem HCl
W przypadku stosowania kwasu borowego lub mianowanym roztworem NaOH
odmiareczkowuje się nadmiar kwasu solnego (siarkowego) pobranego przed destylacją.
Miareczkowanie mianowanym roztworem HCl w obecności wskaźnika Tashiro prowadzi się do
chwili przejścia barwy zielonej w fioletową, a w obecności wskaźnika Ma Zuazaga do chwili
przejścia barwy zielonej w różową. W ten sam sposób postępuje się z próbą ślepą, odczynnikową,
którą mineralizowano przy użyciu tych samych odczynników i w tych samych ilościach jak
w przypadku próby właściwej.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
18
Rys. 6. Aparat Parnasa-Wagnera do destylacji amoniaku w metodzie Kjeldahla oznaczania zawartości azotu (białka)
1 – lejek z kranem, 2- kolba destylacyjna, 3- kolba stożkowa o pojemności 300 cm
3
, 4 – kolba destylacyjna
o pojemności 1500 cm
3
(wytwornica pary), 5 – chłodnica, 6 – odprowadzenie próbki do kanału po zakończeniu
destylacji amoniaku. [4, s. 354]
Na podstawie wyników miareczkowania prób właściwej i odczynnikowej oblicza się zawartość azotu
w badanym materiale:
(
)
m
Cm
V
Cm
V
x
k
k
100
01401
,
0
1
⋅
⋅
−
⋅
=
gdzie:
x – iczba gramów azotu odpowiadająca 100g (cm
3
) badanego produktu,
V – liczba cm
3
mianowanego roztworu HCl o stężeniu ok. 0,1 mol/dm
3
, zużyta do miareczkowania
w próbie właściwej,
V
1
– liczba cm
3
mianowanego roztworu HCl o stężeniu ok. 0,1 mol/dm
3
,zużyta do miareczkowania
w próbie ślepej,
Cm
k
– dokładne stężenie molowe roztworu HCl, użytego do miareczkowania (ok. 0,1 mol/dm
3
),
m – liczba gramów (cm
3
) badanego produktu pobrana do mineralizacji, a następnie destylacji,
0,01401 – liczba gramów azotu odpowiadająca 1 milimolowi HCl lub 1cm
3
roztworu HCl
o stężeniu ściśle 1mol/dm
3
.
W przypadku destylacji amoniaku do określonej objętości mianowanego roztworu HCl, a następnie
odmiareczkowywania nadmiaru tego kwasu za pomocą mianowanego roztworu NaOH, ilość azotu
oblicza się według wzoru:
(
) (
)
m
Cm
V
Cm
V
Cm
V
Cm
V
x
z
k
z
k
100
01401
,
0
2
1
⋅
⋅
−
⋅
−
⋅
−
⋅
=
gdzie:
x – liczba gramów azotu odpowiadająca 100g (cm
3
) badanego produktu (% masowy),
V – liczba cm
3
mianowanego roztworu HCl o stężeniu ok. 0,1 mol/dm
3
, pobrana do odbieralnika
przed destylacją amoniaku,
V
1
– liczba cm
3
mianowanego roztworu NaOH o stężeniu ok. 0,1mol/dm
3
, zużyta do
odmiamiareczkowania nadmiaru HCl w próbie właściwej,
V
2
– liczba cm
3
mianowanego roztworu NaOH o stężeniu ok. 0,1mol/dm
3
, zużyta do
odmiareczkowania nadmiaru HCl w próbie ślepej,
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
19
Cm
k
– dokładne stężenie molowe roztworu HCl pobranego do odbieralnika przed destylacją
amoniaku (ok. 0,1 mol/dm
3
),
Cm
z
– dokładne stężenie molowe roztworu NaOH użytego do odmiareczkowania nadmiaru HCl (ok.
0,1 mol/dm
3
),
m – liczba gramów azotu odpowiadająca 1 milimolowi HCl (lub 1cm
3
roztworu HCl o stężeniu
ściśle 1mol/dm
3
).
Obliczoną ilość azotu przelicza się na białko mnożąc przez współczynnik przeliczeniowy 6,25.
Współczynnik ten jest typowy dla białek mięsa i większości produktów spożywczych zawierających
niewielkie ilości białek.
Rys. 7. Zestaw do destylacji amoniaku 1 – kolba destylacyjna, 2- łapacz kropli, 3 – chłodnica powietrzna, 4- rura
bocznikowa ze ściskaczem do sprawdzenia końca procesu destylacji, 5 – rozszerzenie chłodnicy zabezpieczającej
przed wyrzucaniem cieczy, 6 – odbieralnik, 7 – naczynie z zimną wodą do chłodzenia odbieralnika [3, s. 51]
Oznaczanie tłuszczu
Do metod oznaczania zawartości tłuszczu w surowcach i produktach żywnościowych należą:
−
metody ekstrakcyjno-wagowe,
−
metody objętościowe (butyrometryczne),
−
metody ekstrakcyjno-refraktometryczne.
W przypadku stosowania wszystkich wymienionych metod należy wykonywać te same czynności
podczas oznaczania tłuszczu w próbkach, mianowicie:
−
rozpuszczenie lub rozkład substancji wiążących tłuszcz w badanym produkcie
−
wydzielenie tłuszczu w procesie ekstrakcji lub mechanicznie,
−
otrzymanie wolnego tłuszczu surowego.
Do ekstrakcji tłuszczu używa się eteru naftowego, eteru etylowego lub ich mieszaniny. Można
używać także benzenu, chloroformu, trichloroetylenu. Rozpuszczalniki stosowane do ekstrakcji nie
powinny zawierać wody, ponieważ może ona rozpuszczać niektóre składniki badanego produktu,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
20
które po przejściu do wyciągu eterowego i odparowaniu eteru zwiększają masę suchej pozostałości
przyjmowanej jako tłuszcz surowy.
Do metody ciągłej ekstrakcji tłuszczu stosuje się specjalne aparaty. Jednym z nich jest aparat
Soxhleta przedstawiony na rysunku 8. Metoda Soxhleta polega na umieszczeniu wysuszonego
produktu w gilzie z materiału porowatego lub w bibule filtracyjnej, a następnie w ekstraktorze
aparatu Soxhleta. Ogrzewany eter paruje i po skropleniu w chłodnicy spływa do ekstraktora, styka
się z próbką badanego produktu rozpuszczając stopniowo tłuszcz. W miarę skraplania nowych porcji
eteru, po zrównaniu się poziomu eteru z górnym poziomem rurki przelotowej eter wraz
z rozpuszczonym w nim tłuszczem spływa do kolby destylacyjnej. Operacje powtarza się z tym, że
tłuszcz pozostaje w kolbie, a paruje eter. W ten sposób nowe partie tłuszczu spływają do kolby.
Proces trwa 4 ÷ 8 godzin, powinno być 30 przelewów. Po zakończeniu ekstrakcji eter odparowuje
się na łaźni wodnej, a pozostałość suszy ok. godzinę w temperaturze 105°C, po czym kolbę wraz
z
tłuszczem
chłodzi
w
eksykatorze,
waży
i
na
podstawie
różnicy
mas
kolby
z tłuszczem i kolby pustej oblicza się zawartość tłuszczu.
Metody ekstrakcyjno-refraktometryczne oznaczania zawartości tłuszczu polegają na pomiarze
współczynnika załamania światła roztworu tłuszczu w odpowiednim rozpuszczalniku. W celu
oznaczenia zawartości tłuszczu metodą refraktometryczną naważkę badanego produktu traktuje się
określoną objętością rozpuszczalnika i po rozpuszczeniu tłuszczu mierzy wartość współczynnika
refrakcji roztworu oraz oznacza gęstość otrzymanego tłuszczu. Procentową zawartość tłuszczu
w badanym produkcie oblicza się na podstawie wzoru:
(
)
(
)
g
n
n
V
d
n
n
x
t
r
t
r
⋅
−
⋅
⋅
−
=
100
gdzie:
x – zawartość tłuszczu w %,
n – współczynnik załamania światła roztworu tłuszczu,
n
r
– współczynnik załamania światła rozpuszczalnika,
n
t
– współczynnik załamania światła tłuszczu,
d
t
– gęstość tłuszczu w g/cm
3
,
V
r
– objętość użytego rozpuszczalnika w cm
3
,
g – naważka badanego produktu w g.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
21
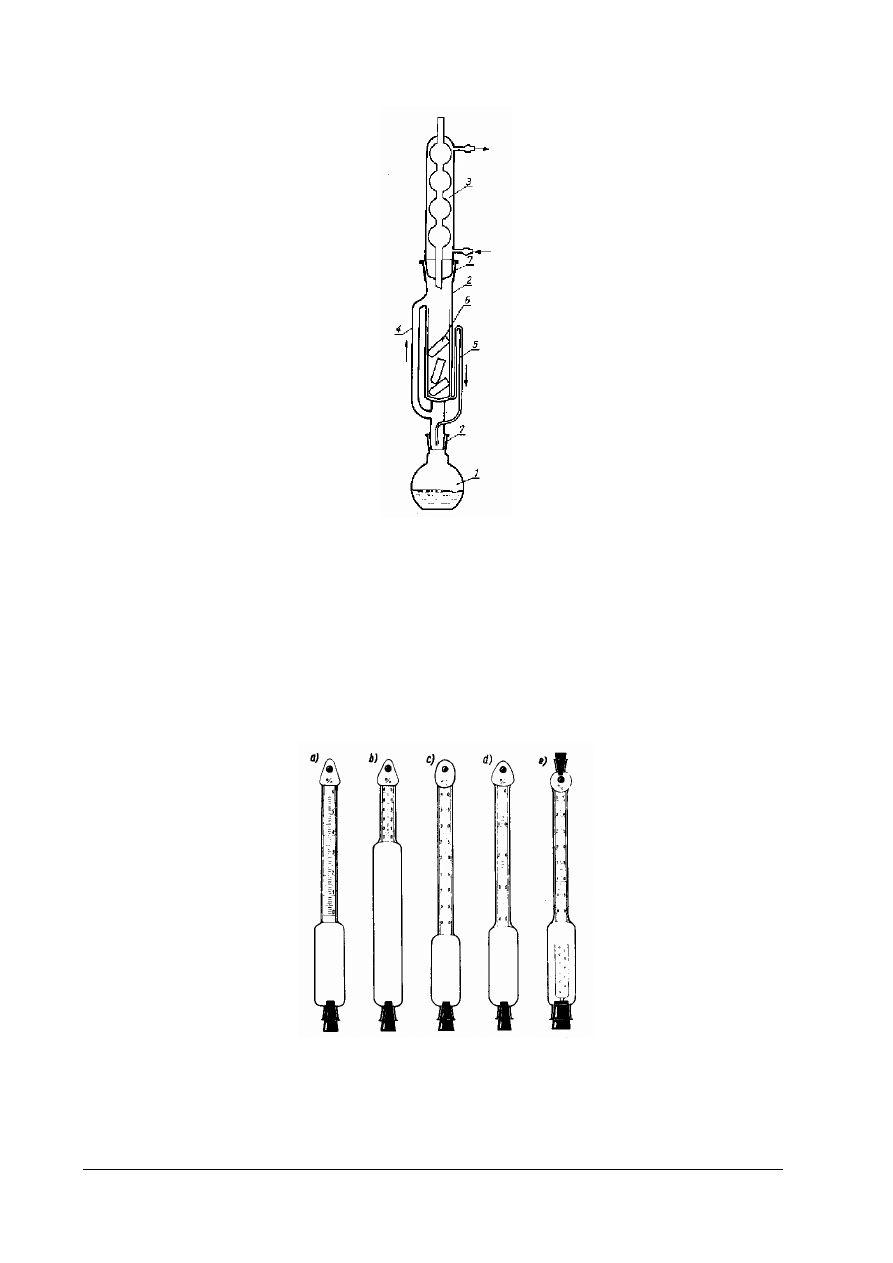
Rys. 8. Aparat Soxhleta do oznaczania zawartości tłuszczu
1 – kolba destylacyjna, 2 – ekstraktor, 3 – chłodnica, 4 – rurka odprowadzająca opary rozpuszczalnika, 5 – rurka
przelewowa (syfon), 6 – gilza z badaną próbką, 7 – połączenia na szlify [4, s. 363]
Metody objętościowe reprezentowane są przez metody butyrometryczne. Zasada oznaczania polega
na wydzieleniu tłuszczu z badanej próbki w oddzielną fazę i odczytaniu jego objętości na
wyskalowanej części butyrometru. Do wydzielenia tłuszczu niezbędne jest jego uwolnienie
z otoczek białkowych przez potraktowanie kwasem siarkowym i alkoholem izoamylowym, który
zmniejsza przyczepność tłuszczu do szkła i ułatwia zbicie go w jednolity słupek. Znanych jest kilka
typów butyrometrów (przedstawione na rysunku 9)
Rys.9. Różne typy butyrometrów (tłuszczomierzy: a) do mleka pełnego, b – do mleka odtłuszczonego (podwójny), c)
do mleka w proszku, d) do śmietany, e) do serów [4, s. 365]

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
22
4.2.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń
1. Jak określić pojęcie suchej masy?
2. Pod jakimi postaciami występuje woda w surowcach i produktach roślinnych i zwierzęcych?
3. Jaki jest podział metod oznaczania suchej masy (wody)?
4. Jakie czynności należy wykonać oznaczając tłuszcze niezależnie od metody?
5. Jak wyjaśnić pojęcie ekstrakcji tłuszczów?
6. Do czego służą butyrometry?
7. Jakie etapy wyróżnia się podczas oznaczania ogólnej zawartości białek?
4.2.3. Ćwiczenia
Ćwiczenie 1
Oznacz zawartość suchej masy i wody w mące metodą suszenia.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) wysuszyć naczyńko wagowe w suszarce w temperaturze 130°C w czasie 15 minut,
2) ochłodzić naczyńko w eksykatorze i zważyć na wadze technicznej z dokładnością do
10 mg,
3) zapisać wynik ważenia (a),
4) przenieść do naczyńka ok. 10g wymieszanej próbki badanej mąki,
5) zważyć naczyńko z próbką mąki na wadze technicznej,
6) zapisać wynik ważenia (b),
7) wstawić naczyńko do suszarki o temperaturze 130°C i suszyć przez 150 minut,
8) przenieść do eksykatora na 30 minut,
9) zważyć po ochłodzeniu na wadze technicznej, zapisując wynik ważenia (c),
10) powtórzyć suszenie w tej samej temperaturze przez 30 minut, ochłodzić i zważyć,
11) suszyć ponownie w czasie 30 minut, jeśli wynik ważenia różni się od pierwszego więcej niż
10 mg,
12) wykonać obliczenia według wzoru:
(
)
a
b
a
c
x
−
⋅
−
=
100
gdzie: x – zawartość suchej masy w badanej mące w %,
a – masa pustego naczyńka wagowego w g,
b – masa naczyńka wagowego z naważką mąki ( przed suszeniem) w g,
c – masa naczyńka wagowego z suchą pozostałością (po suszeniu) w g.
Wyposażenie stanowiska pracy:
−
naczyńko wagowe o pojemności 50 ÷ 100cm
3,
−
waga techniczna z kompletem odważników,
−
suszarka elektryczna z termoregulacją,
−
eksykator z bezwodnym CaCl.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
23
Ćwiczenie 2
Oznacz zawartości glutenu w mące.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować ciasto: odważyć 25g mąki pszennej (oprócz razowej i Graham), wsypać do
moździerza i mieszając tłuczkiem dodać 12,5cm
3
wody o temperaturze 15÷20°C,
przygotowanie ciasta jest zakończone, gdy utrzymuje się ono w całości na tłuczku,
2) zdjąć ciasto z tłuczka, uformować palcami kulkę i umieścić w miseczce zalewając
całkowicie wodą,
3) pozostawić na 20 minut,
4) wyjąć kulkę ciasta z wody, umieścić w cylindrze płuczki glutownika, uruchomić wirnik
i doprowadzić do lejka na wirniku cienki strumień wody,
5) wymywać gluten dopóki nie uzyska ujemnej reakcji na skrobię: pobrać w tym celu do zlewki
kilka cm
3
wody przeciekającej przez sito i dodać kroplę płynu Lugola, jeżeli próbka wody nie
zmieni zabarwienia wymywanie glutenu można uznać za zakończone,
6) zebrać gluten ze ścianek płuczki, dna płuczki i łopatek, połączyć w całość i wymywać ręcznie
pod cienkim strumieniem wody,
7) wymywać ręcznie nad sitem pod strumieniem wody i zakończyć, gdy woda spod sita nie daje
dodatniej reakcji na skrobię,
8) wycisnąć wodę z glutenu ściskając go w dłoniach,
9) wałkować gluten między dwiema szklanymi płytkami, aż zacznie się lepić w palcach,
10) zważyć wyciśnięty gluten z dokładnością do 0,01g,
11) wykonać obliczenie. Masę glutenu mokrego (X) obliczyć w % według wzoru: x = m
1
· 4,
w którym: m
1
– masa wymytego glutenu. Za wynik przyjmuje się średnia arytmetyczną co
najmniej dwóch oznaczeń, nie różniących się między sobą więcej niż 2%. Zawartość mokrego
glutenu w mąkach pszennych powinna wynosić: w mące krupczatce, luksusowej co najmniej
25%; w mące razowej co najmniej 22%; w mące tortowej co najmniej 18%; w mące graham co
najmniej 24%.
Wyposażenie stanowiska pracy:
−
próbka mąki,
−
glutownik do mechanicznego
wymywania glutenu lub naczynie o pojemności 2dm
3
do
wymywania ręcznego,
−
sito opięte gazą nr 180 lub 160 oraz 230,
−
moździerz porcelanowy, miseczka porcelanowa o średnicy 12 ÷ 16cm,
−
kolba miarowa o pojemności 100 cm
3
, kroplomierz,
−
2 płytki szklane o wymiarach 10 x 15cm o grubości ok. 0,8cm,
−
płyn Lugola.
Ćwiczenie 3
Oznacz zawartość tłuszczu w mleku metodą butyrometryczną.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
24
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) odmierzyć do butyrometru ze specjalnego automatu 10cm
3
kwasu siarkowego,
2) dodać ostrożnie po ściance, z pipety 11cm
3
badanej próbki mleka i 1cm
3
alkoholu izoamylowego
unikając zwilżenia nim szyjki,
3) zakorkować butyrometr gumowym korkiem i trzymając go w lnianej ściereczce mieszać do
chwili całkowitego rozpuszczenia białka,
4) wstawić butyrometr korkiem w dół do łaźni wodnej o temperaturze 65°C i odczekać 5 minut,
5) wyjąć butyrometr z łaźni wodnej,
6) ustawić słupek tłuszczu na podziałce zerowej skali i wstawić do wirownicy Gerbera (parami na
krzyż) częścią wyskalowaną do środka bębna,
7) wirować przez 5 minut, przy prędkości obrotowej ok. 1000 obr./min.,
8) wyjąć butyrometr z wirownicy korkiem w dół i wstawić do łaźni wodnej na 5 minut, tak aby
wyskalowana część butyrometru była pod wodą,
9) wyjąć butyrometr i trzymając go pionowo na wysokości oczu (okulary ochronne) odczytać
procentową zawartość tłuszczu w mleku z dokładnością do 0,5%
Wyposażenie stanowiska pracy:
−
próbka mleka,
−
butyrometr do mleka,
−
pipeta jednomiarowa o pojemności 10cm
3
,
−
automat do odmierzania kwasu siarkowego (10cm
3
),
−
automat do odmierzania alkoholu izoamylowego (1cm
3
),
−
wirownica Gerbera,
−
łaźnia wodna
−
kwas siarkowy – 1000 cm
3
stężonego H
2
SO
4
o d= 1,84 g/cm
3
, wlać ostrożnie do 100 cm
3
wody
destylowanej (nigdy odwrotnie),
−
alkohol izoamylowy (d= 0,815 g/ cm
3
.
Ćwiczenie 4
Oznacz zawartość azotu metodą Kjeldahla i przelicz na białko. Przeprowadź mineralizację
próbki.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować
próbkę
przetworów
mięsnych
do
badania:
zmielić
dwukrotnie
w maszynce do mięsa z siatką o średnicy otworów 2mm,
2) przygotować ślepą próbę z 0,5g sacharozy zamiast próbki mięsa, (dalej postępować jak
z próbką mięsa)
3) odważyć w naczyńku wykonanym z małej probówki z dokładnością do 0,0001g 1,5 g próbki,
4) przenieść odważkę z naczyńkiem do kolby Kjeldahla,
5) dodać 0,31g CuSO
4
· 5 H
2
O; 7,5 g K
2
SO
4
oraz 15cm
3
H
2
SO
4
, (dla mięsa o zawartości tłuszczu
powyżej15% ilości odczynników są większe i wynoszą : 0,35 g CuSO
4
· 5 H
2
O; 8,5 g K
2
SO
4
oraz 18 cm
3
H
2
SO
4
),
6) wymieszać ostrożnie zawartość kolby,
7) umieścić pod wyciągiem w zestawie do spalań pod kątem 30 ÷ 40°,
8) dodać perełki szklane lub kawałki porcelanki,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
25
9) zwracać uwagę, by powstająca w czasie spalania piana nie przedostała się do szyjki kolby,
10) zwiększyć płomień, gdy płyn przestanie się pienić,
11) prowadzić ogrzewanie jeszcze 60 minut od momentu, gdy ciecz w kolbie stanie się
przezroczysta o jasnozielonkawym zabarwieniu,
12) ostudzić kolbę do temperatury pokojowej.
Wyposażenie stanowiska pracy:
−
próbka mięsa,
−
maszynka do mięsa z siatką o średnicy oczek 2mm,
−
kolba Kjeldahla poj. 250 ÷ 750 cm,
−
chłodniczka do kolby Kjeldahla,
−
perełki szklane,
−
zestaw do spalań: elektryczny lub gazowy,
−
waga analityczna,
−
siarczan miedzi (CuSO
4
· 5 H
2
O) cz.d.a.,
−
kwas siarkowy H
2
SO
4
(d=1,83 ÷ 1,84 g/ cm
3
),
−
siarczan potasu (K
2
SO
4
) cz.d.a.,
−
sacharoza cz. d. a.(do ślepej próby).
Ćwiczenie 5
Oznacz zawartość azotu w zmineralizowanej próbce badanego produktu oraz przelicz na białko.
(Ćwiczenie jest kontynuacją ćwiczenia 4).
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przeprowadzić destylację amoniaku w aparacie Parnasa-Wagnera
2) rozpuścić zawartość kolby Kjeldahla w ok. 200cm
3
wody destylowanej,
3) przenieść całość do kolby destylacyjnej,
4) przemyć trzykrotnie kolbę Kjeldahla niewielkimi porcjami wody destylowanej i przenieść do
kolby destylacyjnej,
5) wlać do odbieralnika 25cm
3
roztworu kwasu borowego i 8 kropli wskaźnika Tashiro,
6) podłączyć zestaw destylacyjny,
7) zanurzyć wylot rurki chłodnicy w roztworze kwasu borowego,
8) wlać przez lejek 50cm
3
roztworu NaOH oraz 3 ÷ 5 kropli oleju parafinowego do kolby
destylacyjnej,
9) prowadzić proces destylacji do zmniejszenia objętości cieczy w kolbie do około
1/3 pierwotnej zawartości,
10) sprawdzić papierkiem lakmusowym czy destylacja jest zakończona (kropla destylatu
spływającego z chłodnicy na papierek lakmusowy nie barwi go na niebiesko),
11) miareczkować zawartość odbieralnika kwasem solnym o stężeniu 0,1 mol/ dm
3
, aż do zmiany
barwy z zielonej w różowofioletową,
12) postąpić tak samo z próbą ślepą, którą mineralizowano używając tych samych odczynników,
13) obliczyć procentową zawartość azotu wg wzoru:
(
)
(
)
m
n
b
a
m
n
b
a
x
14
1000
100
14
⋅
⋅
−
=
⋅
⋅
⋅
⋅
−
=

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
26
gdzie:
a – objętość mianowanego HCl zużytego do miareczkowania w próbce właściwej , [ cm
3
],
b – objętość mianowanego roztworu HCl zużytego do miareczkowania w ślepej próbie, [cm
3
],
n – stężenie roztworu HCl określone z dokładnością do czwartego miejsca po przecinku,
m – masa badanej próbki, [g],
14 – ilość azotu, której odpowiada 1 cm
3
kwasu solnego o stężeniu 1 mol/dm
3
, [mg],
Procentową zawartość białka oblicz mnożąc uzyskany wynik przez 6,25 (współczynnik
przeliczeniowy)
14) zinterpretować wyniki, porównać dane z wymaganiami norm.
Wyposażenie stanowiska pracy:
−
aparat Parnasa-Wagnera do destylacji amoniaku,
−
zestaw do miareczkowania,
−
tryskawka zwodą destylowaną,
−
wodorotlenek sodu roztwór ok. 33 %,
−
kwas borowy, roztwór ok. 4%
−
kwas solny, roztwór o stężeniu 0,1 mol/dm
3
ściśle mianowany,
−
wskaźnik Tashiro,
−
papierki lakmusowe,
−
olej parafinowy,
−
kolba Kjeldahla z próbką otrzymaną w ćwiczeniu 4,
−
normy jakościowe.
4.2.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) oznaczyć zawartość tłuszczu w produkcie metodą butyrometryczną?
2) rozróżnić metody oznaczania tłuszczów?
3) wykonać oznaczenie ogólnej zawartości białek metodą Kjeldahla?
4) wykonać oznaczenie glutenu w mące?
5) wykonać obliczenia i zinterpretować wyniki na podstawie norm?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
27
4.3. Wykrywanie substancji obcych w żywności
4.3.1. Materiał nauczania
Niektóre środki utrwalające są dozwolone do konserwowania pewnych surowców,
półproduktów i produktów żywnościowych na podstawie odpowiednich przepisów. W mięsie
i przetworach mięsnych dopuszczone są do użytku następujące środki konserwujące: azotyn sodowy
oraz azotan sodowy i potasowy. W sokach, przetworach owocowych i warzywnych bezwodnik
kwasu siarkawego, kwas benzoesowy i benzoesan sodowy, w sokach surowych kwas mrówkowy.
W konserwach rybnych, przetworach owocowych i warzywnych, w margarynie, serach topionych,
masie jajowej można stosować ester propylowy i etylowy kwasu p-hydroksybenzoesowego.
W produktach mącznych dopuszcza się stosowanie kwasu propionowego. Maksymalne ilości
dozwolonych środków konserwujących są ściśle określone i normowane przepisami. W celu
kontrolowania ilości dozwolonych środków utrwalających i badania obecności niedozwolonych
istnieje konieczność wykrywania ich i ilościowego oznaczania.
Do wykrywania zafałszowań przetworów mięsnych dodatkami zawierającymi skrobię np.
dodatkiem mączki ziemniaczanej, mąki pszennej stosowana jest metoda jakościowa. Metoda
ilościowa stosowana jest do oznaczania zawartości skrobi w przetworach mięsnych z dodatkiem
tylko skrobi lub w przetworach mięsnych z dodatkiem różnych cukrowców. Zasada metody
jakościowej polega na reakcji barwnej płynu Lugola ze skrobią. Metoda ilościowa polega na
hydrolizie skrobi z kwasem solnym, a następnie oznaczaniu zawartości glukozy na podstawie
jodometrycznego ustalania ilości miedzi zredukowanej prze cukier prosty zawarty w badanej próbce
(wg Luffa-Schoorla).
W przemyśle mięsnym ważną rolę odgrywa oznaczanie azotanów i azotynów w mięsie i jego
przetworach. Zgodnie z obowiązującymi przepisami ustawodawstwa sanitarnego maksymalna ilość
azotanów i azotynów może wynosić: azotanu potasu(sodu) – 0,07g na 100g produktu; azotynu sodu
– 0,0125g na 100g produktu. Większa ilość tych związków w produkcie jest szkodliwa dla zdrowia.
Oznaczanie zawartości azotanów i azotynów prowadzi się metodami kolorymetrycznymi.
Środki spożywcze zawierają barwniki naturalne i barwienie tymi barwnikami bywa dozwolone.
W niektórych przypadkach dopuszcza się barwniki sztuczne, jeśli nie są szkodliwe dla zdrowia
ludzkiego. Nie mogą być dopuszczone barwniki zawierające arsen, antymon, cynę, rtęć, ołów, kadm,
chrom, cynk, bar, uran, barwniki zawierające niektóre kwasy organiczne. Listy dopuszczonych
barwników do barwienia niektórych środków żywności są podawane w Dziennikach Urzędowych.
Metody wykrywania polegają najczęściej na wykrywaniu odnośnych metali (kationów), a barwników
organicznych na zasadzie charakterystycznych reakcji. Barwniki wykrywa się w wyciągach wodnych
artykułów spożywczych lub w wyciągach innych rozpuszczalników, zależnie od rodzaju próbki
i barwnika. Przykładem może być wykrywanie obcych barwników w maśle. Masło może być
barwione barwnikami naturalnymi np. wyciągiem z marchwi lub szafranem. Niedopuszczalne są
barwniki smołowcowe jak żółcień anilinowy, żółcień masłowy itp.
4.3.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń
1. Jakie środki stosowane są do konserwowania żywności?
2. Jaki jest cel wykrywania substancji obcych w żywności?
3. Na czym polegają metody wykrywania substancji obcych w żywności?
4. Jak ustalić krytyczne punkty kontroli w procesach produkcji artykułów spożywczych
wykorzystując metody analizy żywności?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
28
4.3.3. Ćwiczenia
Ćwiczenie 1
Wykryj zafałszowanie masła margaryną – wykryj obecność skrobi.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) odważyć do zlewki ok. 10g badanego masła, dodać 20cm
3
wrzącej wody i wymiesza
2) przenieść do rozdzielacza, wstrząsać przez ok. 30 sekund, pozostawić do rozwarstwienia
zawartości i zlać warstwę wodną do probówki,
3) ogrzewać zawartość probówki do wrzenia na łaźni wodnej, ostudzić pod strumieniem
wody,
4) dodać 4 ÷ 5 kropel płynu Lugola i obserwować zabarwienie,
5) zinterpretować wyniki.
Wyposażenie stanowiska pracy:
−
próbka masła,
−
rozdzielacz o pojemności 50 ÷ 100cm
3
,
−
zlewka o pojemności 50cm
3,
−
płyn Lugola,
−
łaźnia wodna.
Ćwiczenie 2
Wykryj barwnik sztuczny w wodzie gazowanej.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) wlać do zlewki o pojemności 50cm
3
około 20cm
3
badanej próbki napoju gazowanego,
2) zakwasić 10% roztworem wodorosiarczanu potasowego,
3) wrzucić kilka nitek odtłuszczonej wełny (odtłuszczanie: pasmo wełny zanurzyć w 10%
roztworze sody na 24 godziny, wypłukać i wysuszyć),
4) przykryć zlewkę szkiełkiem zegarkowym ,
5) ogrzewać 10 ÷ 15 minut na łaźni wodnej,
6) wyjąć zabarwioną wełnę i wypłukać dokładnie pod strumieniem bieżącej wody (jeśli barwnik
został wypłukany świadczy to o jego nieobecności w badanym napoju),
7) przenieść zabarwioną wełnę do drugiej zlewki, dodać 20cm
3
, 1 % roztworu amoniaku,
8) ogrzewać na łaźni wodnej pod przykryciem przez 30 minut,
9) zakwasić zabarwiony płyn 10% roztworem wodorosiarczanu potasu, wrzucić nowe nitki
w mniejszej ilości niż w pierwszym barwieniu, aby uzyskać wyraźne zabarwienie nitek
świadczące o obecności sztucznego barwnika w badanym napoju,
10) zinterpretować wynik biorąc pod uwagę etykietę.
Wyposażenie stanowiska pracy:
−
próbka wody gazowanej z etykietą,
−
zlewki o pojemności 50cm
3
,
−
szkiełko zegarkowe,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
29
−
łaźnia wodna,
−
pasmo bezbarwnej wełny,
−
10% roztwór sody,
−
wodorosiarczan potasowy roztwór 10%,
−
1% roztwór amoniaku.
Ćwiczenie 3
Oznacz zawartość azotynów w mięsie i przetworach mięsnych oraz w solankach.
Sposób wykonania ćwiczenia
Uczeń powinien:
przygotować próbkę do badania:
1) zmielić mięso w maszynce do mięsa z siatką o średnicy otworów 4mm dokładnie wymieszać.
Po uzyskaniu jednolitej masy, pobrać 200g próbki i umieścić w szczelnie zamkniętym naczyniu
o pojemności 200cm
3
, upychając próbkę tak, aby nie było wolnych przestrzeni z powietrzem.
Badania powinno się rozpocząć bezpośrednio po przygotowaniu próbki.
przygotować przesącz:
2) odważyć 10 g próbki z dokładnością do 0,001g i przenieść ilościowo do kolby miarowej
o poj. 200 cm
3
używając 100 cm
3
gorącej wody, lejka i bagietki,
3) dodać 5 cm
3
nasyconego roztworu boraksu i wstrząsać do rozbicia grudek mięsa,
4) ogrzewać zawartość kolby na wrzącej łaźni wodnej przez 15 minut wstrząsając co 2 ÷ 3 minuty,
5) ochłodzić kolbę z zawartością do temperatury pokojowej,
6) dodać kolejno po 1cm
3
odczynników odbiałczających I i II, wymieszać zawartość kolby,
7) odstawić kolbę na 30 minut w temperaturze pokojowej, uzupełnić wodą do kreski
i wymieszać,
8) sączyć przez sączek karbowany do suchej kolby stożkowej,
9) użyć przesącz do oznaczania zawartości azotynów:
przygotować roztwory do krzywej kalibracji:
10) miareczkować 10cm
3
roztworu KMnO
4
o stężeniu 0,02 mol/dm
3
rozcieńczonego
10 cm
3
wody i zakwaszonego 2cm
3
H
2
SO
4,
roztworem azotynu sodu zawierającym 0,2g NaNO
2
w 100cm
3
,
11) przygotować roztwór A: rozpuścić w kolbie miarowej o pojemności 1000cm
3
1,0g NaNO
2
w wodzie destylowanej, rozcieńczyć wodą do kreski,
12) przygotować roztwór B: przenieść 5cm
3
roztworu A do kolby miarowej o pojemności 1000 cm
3
i uzupełnić wodą do kreski,
13) przygotować rozcieńczenia z roztworu B wprowadzając do kolb miarowych o pojemności
100 cm
3
kolejno pipetą: 5, 10 i 20cm
3
roztworu B i uzupełnić wodą destylowaną do kreski,
14) odmierzyć do 4 probówek po 10cm
3
wody oraz po 10cm
3
poprzednio przygotowanych
roztworów w kolbach miarowych po 100cm
3
,
15) dodać do każdej probówki po 10cm
3
odczynnika Griessa (za pomocą dozownika)
i wymieszać,
16) zmierzyć po 20 minutach ekstynkcję roztworów w kolorymetrze przy długości fali
λ
=520 nm
wobec ślepej próby zawierającej wodę,
wykreślić krzywą kalibracji przedstawiającą zależność ekstynkcji od stężenia azotynu sodu
wyrażonego w μg/cm
3
,
zmierzyć ekstynkcję,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
30
17) pobrać do probówki pipetą 10cm
3
przesączu przygotowanego w punkcie 3 tego ćwiczenia,
dodać 10cm
3
odczynnika Griessa, wymieszać i po 20 minutach zmierzyć ekstynkcję
w kolorymetrze o długości fali
λ
=520 nm wobec ślepej próby,
18) odczytać z krzywej kalibracji ilość azotynu sodu odpowiadającą ekstynkcji badanego roztworu,
19) obliczyć wyniki według wzoru:
a
m
c
x
⋅
⋅
=
2000
gdzie:
c – stężenie NaNO
2
odczytane z krzywej kalibracji, które odpowiada ekstynkcji roztworu próbki
[
µ
g/cm
3
],
m – masa próbki [g],
a – objętość roztworu pobranego do oznaczania kolorymetrycznego [cm
3
].
Środki dydaktyczne:
−
badane próbki,
−
waga analityczna,
−
łaźnia wodna,
−
kolorymetr,
−
kolby miarowe o pojemności 100, 200 i 100cm
3
,
−
pipety poj. 1, 5, 10 i 20cm
3
−
lejki szklane o średnicy 5cm,,
−
kolby stożkowe poj. 200cm
3
,
−
probówki szklane długości 15cm i średnicy 20 ÷ 25mm,
−
sączek,
−
bagietka,
−
KMnO
4
roztwór o stężeniu 0,02mol/dm
3
,
−
dozownik ustawiony na wylew 10cm
3
płynu
−
I – żelazocyjanek potasu,
−
II – octan cynku,
−
boraks,
−
azotyn sodu,
−
kwas siarkowy rozcieńczony w stosunku 1:4,
−
odczynnik Griessa
Ćwiczenie 4
Oznacz zawartość azotanów w mięsie i przetworach mięsnych oraz w solankach.
Sposób wykonania ćwiczenia
Uczeń powinien:
przygotować kadm:
1) umieścić 5 ÷ 7 lasek cynkowych w zlewce zawierającej 1000cm
3
roztworu siarczanu kadmu,
2) zbierać kadm tworzący się na laskach do innej zlewki wodą,
3) przemyć zebrany kadm wodą (2 razy po 1dm
3
wody),
4) przenieść kadm za pomocą 400cm
3
roztworu kwasu solnego o stężeniu 0,1 mol/dm
3
do
homogenizatora i rozdrabniać przez 10 sekund,
5) przenieść do zlewki zawartość homogenizatora, pozostawić pod działaniem kwasu mieszając od
czasu do czasu bagietką,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
31
6) zlać płyn znad kadmu na drugi dzień i przemyć go wodą (2 razy po 1dm
3
),
7) napełnić szklaną kolumnę kadmem, po uprzednim zatkaniu końca rurki watą szklaną,
8) przenieść kadm za pomocą wody formując złoże wysokości ok. 17cm,
9) odprowadzać wodę znad złoża kadmu, nie dopuścić, aby poziom wody opadł poniżej poziomu
złoża,
przygotować kolumnę kadmową do redukcji:
10) przemywać (kolejno wprowadzając przez lejek): 25cm
3
roztworu kwasu solnego o stężeniu ok.
0,1 mol/ dm
3
, 50cm
3
wody i 25cm
3
buforu amonowego rozcieńczonego wodą w stosunku 1:9,
sprawdzić zdolność redukcyjną kolumny kadmowej:
11) przygotować roztwór A rozpuszczając w kolbie miarowej o poj. 1000cm
3
1,465 g azotanu
potasu wysuszonego do stałej masy w temperaturze 105°C i uzupełnić wodą do kreski,
12) przygotować roztwór B przenosząc 5cm
3
roztworu A do kolby miarowej poj. 1000cm
3
i uzupełnić wodą do kreski,
przeprowadzić redukcje azotanów do azotynów:
13) wprowadzić do zbiornika znajdującego się na szczycie kolumny jednocześnie 20cm
3
roztworu B
i 5cm
3
buforu amonowego o pH 9,6,
14) przemyć zbiornik wodą 2 razy po 15cm
3
, opróżnić zbiornik i napełnić całkowicie wodą,
15) zbierać wyciek z kolumny do kolby miarowej o poj. 100cm
3
; po zebraniu
ok. 100cm
3
wycieku usunąć kolbę spod kolumny i uzupełnić wodą do kreski. Wyciek zawiera w
1cm
3
1,465μg KNO
2
,
16) pobrać pipetą do probówki 10cm
3
wycieku, dodać 10cm
3
odczynnika Griessa,
17) zmierzyć ekstynkcję (po 20 minutach) barwnego roztworu wobec próby odniesienia,
zredukować azotany do azotynów:
18) wprowadzić
do
zbiornika
na
szczycie
kolumny
20cm
3
przesączu
otrzymanego
w punkcie 3 poprzedniego ćwiczenia i 5cm
3
buforu amonowego o pH 9,6,
19) opróżnić zbiornik, przemyć ścianki dwukrotnie wodą po 15cm
3
oraz napełnić go wodą,
20) zbierać wyciek do kolb miarowych poj. 100cm
3
,
21) wykonać
próbę
odniesienia
dla
każdej
serii
oznaczeń
stosując
wodę
zamiast
przesączu,
wykonać oznaczenie kolorymetryczne:
22) pobrać pipetą do probówki 10 cm
3
wycieku, dodać 10cm
3
odczynnika Griessa,
23) zmierzyć ekstynkcję po 20 minutach przy długości fali 520nm,
24) odczytać wynik z krzywej kalibracji,
25) obliczyć i zinterpretować wyniki. Zawartość azotanów (x
1
) wyrażoną w mg KNO
3
na 1kg
obliczyć wg wzoru:
x
b
m
c
x
−
⋅
⋅
⋅
=
10000
465
,
1
1
gdzie:
c – stężenie azotynów odczytane z krzywej kalibracji wyrażone w [μg/cm
3
], które odpowiada
ekstynkcji roztworu próbki,
m – masa próbki [g],
b – objętość wycieku pobranego do pomiaru kolorymetrycznego [ cm
3
],
x – stężenie azotynów oznaczone w tej samej próbce [mg/kg].
Za wynik ostateczny oznaczania azotynów i azotanów przyjąć średnią arytmetyczną z dwóch
równoległych oznaczeń, które nie powinny różnić się więcej niż 10%. Otrzymane dane porównać
z wymaganiami normy. Wyciągnąć wnioski dotyczące badanego produktu.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
32
Środki dydaktyczne:
−
przesącz, przygotowany na poprzednich ćwiczeniach,
−
kolorymetr,
−
kolumna szklana z metalicznym kadmem do redukcji azotanów,
−
kolby miarowe o poj. 100, 200 i 1000cm
3
,
−
lejki szklane,
−
kolby stożkowe,
−
probówki,
−
homogenizator,
−
wata szklana,
−
zlewki,
−
bagietki,
−
cynk w laskach, siarczan kadmu,
−
kwas solny o stężeniu 0,1 mol/dm
3
,
−
bufor amonowy o pH 9,6,
−
azotan potasu,
−
odczynnik Griessa.
Ćwiczenie 5
Oznacz zawartość skrobi w lodach.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) przygotować próbkę lodów do badań chemicznych:
2) wstawić pobraną do słoika próbkę lodów do łaźni wodnej o temperaturze wyższej o około 3°C
od temperatury próbki i ogrzewać stopniowo podwyższając temperaturę łaźni wodnej, aż do
uzyskania 38 ÷40°C w próbce,
3) oziębić próbkę lodów do 18 ÷ 20°C, zamknąć słoik,
4) wymieszać zawartość słoika przed każdym pobraniem próbki do badań,
5) wlać kilka cm
3
badanych lodów do parownicy porcelanowej,
6) dodać równą ilość gorącej wody,
7) dodać kilka kropel płynu Lugola, wymieszać i obserwować zabarwienie (niebieskie lub
niebieskofioletowe zabarwienie świadczy o obecności skrobi w lodach),
8) zinterpretować wynik.
Wyposażenie stanowiska pracy:
−
naczynie do pobrania próbki lodów,
−
próbka lodów,
−
parownica porcelanowa,
−
płyn Lugola,
−
łaźnia wodna.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
33
4.3.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) uzasadnić oznaczanie substancji obcych w żywności?
2) określić metody wykrywania substancji obcych w żywności ?
3) wskazać substancje dozwolone do konserwowania żywności?
4) określić metody wykrywania barwników?
5) określić zagrożenie jakości zdrowotnej surowców i produktów?
6) interpretować uzyskane wyniki analiz?
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
34
4.4. Analiza sensoryczna i ocena organoleptyczna żywności
4.4.1. Materiał nauczania
Ocena organoleptyczna polega na najogólniej pojętej ocenie jakości produktów spożywczych za
pomocą zmysłów: smaku, zapachu, wzroku, węchu i dotyku. Zależy ona od uwrażliwienia osoby
oceniającej, jej stanu fizycznego i psychicznego. Ocena organoleptyczna zastępowana jest oceną
sensoryczną, która jest bardziej obiektywna polegającą na ocenianiu żywności w specjalnych
warunkach i przez osoby odpowiednio przygotowane do jej wykonywania. W analizie sensorycznej
produktów spożywczych wyróżnia się wiele specyficznych określeń np.: wrażliwość sensoryczna
czyli zdolność odczuwania wrażeń powstających w wyniku bodźców zewnętrznych; próg
wrażliwości tj. najmniejsze natężenie bodźca, wyczuwalne przez organ zmysłu, pozwalające na
zdefiniowanie np. rodzaju smaku lub zapachu; próg wyczuwalności, tj. minimalne natężenie bodźca
powodujące uchwytne, ale jeszcze jakościowo możliwe do zidentyfikowania; próg rozpoznania, tj
minimalne natężenie bodźca, wywołujące wrażenie dające się określić; próg różnicy, tj. minimalna
różnica natężenia dwóch bodźców dająca uchwytną różnicę intensywności wrażeń; pamięć
sensoryczna, tj. zdolność zapamiętywania i rozpoznawania rożnych bodźców i wrażeń zmysłowych;
minima sensoryczne, tj. minimalna wymagana wrażliwość i sprawność poszczególnych zmysłów
biorących udział w ocenie u osób wykonujących analizy sensoryczne; indywidualna powtarzalność
wyników, tj. zdolność uzyskiwania przez tę samą osobę jednakowych wyników analizy sensorycznej
produktu ściśle w tych samych warunkach, ale w różnym czasie; wyróżnik jakościowy, tj. cecha
będąca częścią składową jakości produktu jak np. smak, zapach; smakowitość, tj. kompleksowe
wrażenie smakowo-zapachowo-czuciowe, odczuwalne przez oceniającego przy rozprowadzaniu
próbki produktu spożywczego w jamie ustnej. Osoby oceniające powinny mieć kwalifikacje
w zakresie minimów sensorycznych oraz zdolności do uzyskiwania indywidualnej
i grupowej powtarzalności wyników analizy sensorycznej. Analizy sensoryczne należy
prowadzić komisyjnie, w zespołach 3 ÷ 6 osobowych, przy zachowaniu identycznych warunków
oceniania. Próbki powinny być podawane anonimowo w oznakowanych naczyniach. W ciągu
jednego dnia nie należy wykonywać zbyt dużej ilości próbek, a przy podawaniu trzeba zwracać
uwagę na odpowiednią kolejność próbek od najsłabszego do najsilniejszego natężenia cech
produktu.Pracownia analizy sensorycznej (właściwa) powinna mieć utrzymaną stałą temperaturę
w zakresie 18 ÷ 20°C i wilgotność względną powietrza ok. 75%. Wymiana powietrza powinna być
stała. Ważne jest dobre oświetlenie pracowni i usytuowanie stanowisk. W bezpośrednim sąsiedztwie
z pracownią właściwą powinno być pomieszczenie do przygotowywania próbek.
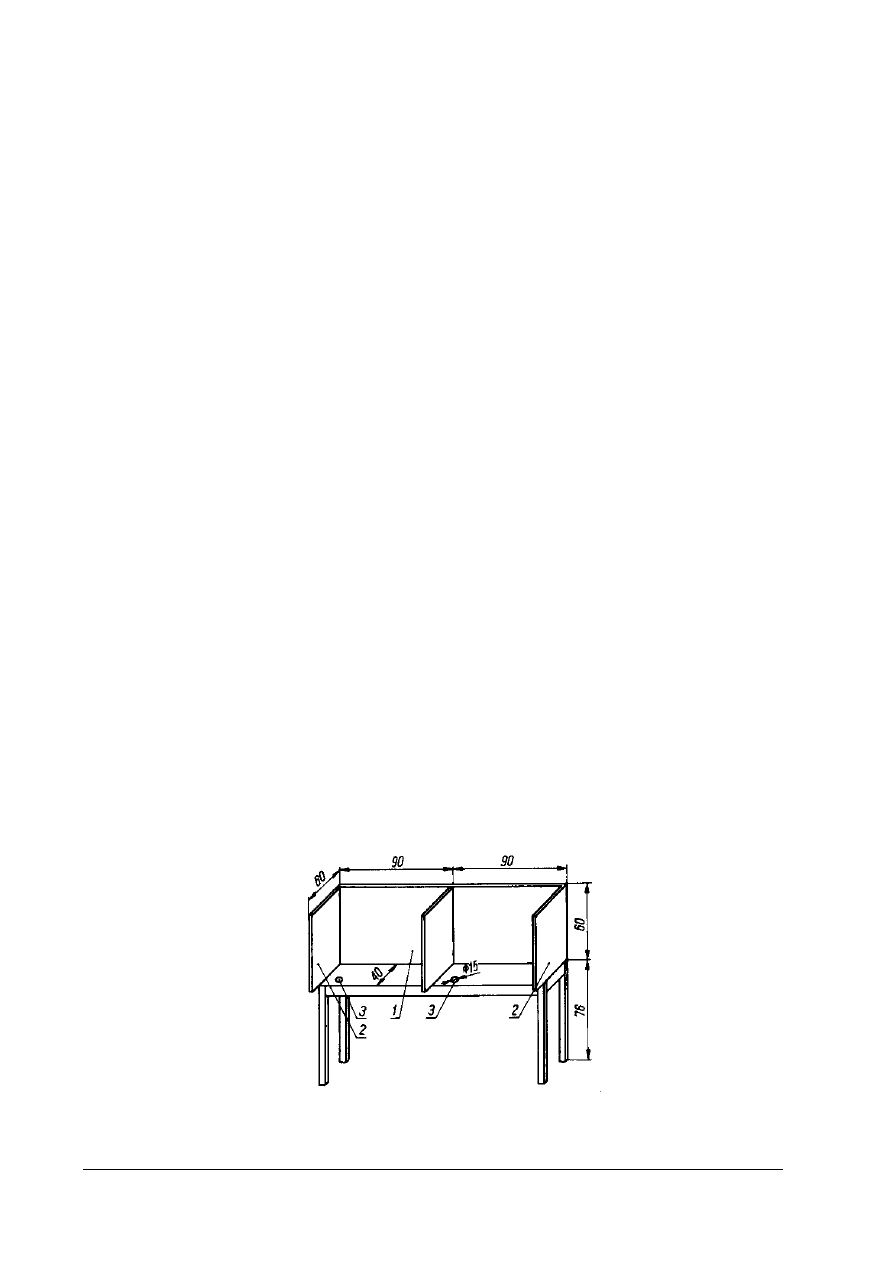
Rys. 10. Schemat podwójnego stołu do analizy sensorycznej 1 – osłona czołowa, 2 – osłony boczne, 3 – otwory lejków
spływowych] [4, s. 181]

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
35
Stanowiska dla osób przeprowadzających analizę sensoryczną powinny być tak usytuowane, by
zapewnić oceniającym optymalne warunki pracy, a jednocześnie uniemożliwić kontaktowanie się ze
sobą. Stanowisko takie składa się z krzesła i specjalnego stołu rys. 10, zaopatrzonego w osłony
boczne oraz ruchomą osłonę czołową, pozwalającą na przyjmowanie próbek. W bezpośrednim
sąsiedztwie z pracownią właściwą powinno znajdować się pomieszczenie do przygotowywania
próbek.
Osoby, które przewidziane są do wykonywania analiz sensorycznych w zakresie smaku i zapachu
poddawane są testowi, w którym uwzględnia się oznaczenia:
−
próbę na daltonizm smakowy,
−
próbę na progi wrażliwości smakowej,
−
próbę na wartości smakowych progów różnicy: parzystą i trójkątowi,
−
próbę na pamięć smakową,
−
próbę na rozpoznawanie i definiowanie zapachów,
−
próbę na węchowe progi różnicy.
4.4.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń
1. Co oznaczają pojęcia: ocena organoleptyczna i analiza sensoryczna?
2. Jakim warunki musi spełniać pracownia analizy sensorycznej?
3. Jakim testom poddawane są osoby przewidziane do wykonywania analiz sensorycznych
w zakresie zmysłów smaku i zapachu?
4.4.3.Ćwiczenia
Ćwiczenie 1
Przeprowadź próbę na daltonizm smakowy.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) nalać na łyżkę roztwór z 1butelki, a następnie spróbować
2) ustawić butelkę na kartonie w odpowiednim polu,
3) przemyć łyżkę wodą destylowaną i wytrzeć ją do sucha,
4) przepłukać jamę ustną wodą destylowaną,
5) powtórzyć czynności z pozostałymi butelkami,
6) wypełnić formularz wpisując w odpowiednie miejsca numery roztworów,
7) zinterpretować wyniki. Prawidłowa identyfikacja smaków roztworów zawartych w 9 butelkach
jest uważana za minimum sensoryczne w zakresie próby na daltonizm smakowy. W przypadku
błędnego określenia smaku, nawet jednego z roztworów, próbę należy powtórzyć po upływie 30
minut lub następnego dnia. Osoby, które uzyskały wynik pozytywny kwalifikują się do dalszych
prób.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
36
Tabela.1
Stężenia roztworów do próby na daltonizm smakowy
Rodzaj smaku
Substancja rozpuszczona(bodźcowa)
Stężenie roztworu g/cm
3
słodki
sacharoza
0,8
słony
chlorek sodu
0,25
kwaśny
kwas winowy
kwas cytrynowy
0,018
0,03
gorzki
kofeina
chlorowodorek chininy
0,0004
0,0002
Wyposażenie stanowiska pracy:
−
roztwory rozcieńczone o czterech smakach i stężeniach podanych w tabeli, w buteleczkach
100 cm
3
oznakowane numerami 1 ÷ 9, po dwie buteleczki z roztworami do trzech smaków
i 3 buteleczki z roztworem o czwartym smaku,
−
karton o wymiarach 25x25 cm, podzielony na 4 równe części z zaznaczonymi smakami,
−
łyżka stołowa ze stali kwasoodpornej.
−
formularz,
−
ręczniki papierowe,
−
łyżki.
Ćwiczenie 2
Wykonaj badanie organoleptyczne olejów roślinnych jadalnych.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) sprawdzić zapach oleju w probówce w temperaturze 20°C (powinien być nikły, swoisty, bez
obcych zapachów),
2) zbadać smak (powinien być lekko wyczuwalny, swoisty, przyjemny bez obcego posmaku),
3) określić konsystencję w temperaturze pokojowej (powinna być płynna),
4) określić klarowność, po przechowywaniu próbki w termostacie w temperaturze 35°C
w szklanym naczyniu przez 24 godziny (powinien być przejrzysty, klarowny i bez osadu)
5) zinterpretować wynik, porównać z normą.
Wyposażenie stanowiska pracy:
−
próbki oleju,
−
probówki,
−
termostat,
−
norma jakościowa.
Ćwiczenie 3
Wykonaj ocenę organoleptyczną pieczywa.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) zastosuj pięciopunktowy system oceniania pieczywa. W systemie tym wszystkie wyróżniki
jakościowe oceniane są w zakresie od 1 do 5 punktów. Wszystkim punktom odpowiadają
zróżnicowane poziomy jakości: poziom jakości bardzo dobrej - 5 pkt, jakości dobrej – 4 pkt,
jakości dostatecznej – 3 pkt, jakości niedostatecznej – 2 pkt, jakości złej – 1 pkt.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
37
2) określić kształt pieczywa stanowiącego próbkę (okrągły, owalny, podłużny),
3) zbadać skórkę pieczywa biorąc pod uwagę wygląd powierzchni, elastyczność, chrupkość oraz
grubość,
4) zbadać barwę skórki (od jasnobrązowej do ciemnobrązowej),
5) określić grubość skórki pieczywa - nie powinna być mniejsza od 2 mm,
6) zbadać miękisz poprzez określenie barwy, elastyczności, porowatości, wilgotności i lepkości
wyczuwalnej w dotyku
7) określić barwę i porowatość badanego pieczywa po obejrzeniu miękiszu na przekroju (przekrój
przez środek)
8) zbadać elastyczność miękiszu oraz jego cechy fizyczne według wymagań norm,
9) określić zapach przez wąchanie natychmiast po przekrojeniu pieczywa przez środek sztuki,
10) określić smak przez powolne przeżuwanie próbki pobranej ze środka rozkrojonego pieczywa
(właściwy dla danego pieczywa).
11) zinterpretować wyniki.
Wyposażenie stanowiska pracy:
−
próbki pieczywa,
−
nóż,
−
norma
Ćwiczenie 4
Oceń organoleptycznie solankę.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie powinieneś:
1) określić przez oględziny w świetle dziennym rozproszonym barwę i wygląd zewnętrzny solanki,
2) ocenić przez wąchanie próbki o temperaturze 15 ÷ 20°C,
3) sprawdzić smak przez pobranie do ust niewielkiej ilości solanki,
4) zinterpretować wyniki, określić jakość i przydatność technologiczną solanki.
Wyposażenie stanowiska pracy:
−
próbki solanki,
−
zlewki o pojemności 250cm
3
,
−
łyżka stołowa ze stali nierdzewnej.
4.4.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) ocenić przydatność osób do wykonywania analiz sensorycznych?
2) zdefiniować pojęcie wrażliwości sensorycznej?
3) określić warunki wykonywania analiz sensorycznych?
4) przeprowadzić próbę na daltonizm smakowy?
5) oceniać organoleptycznie produkty przetwórstwa spożywczego?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
38
5. SPRAWDZIAN OSIĄGNIĘĆ
INSTRUKCJA DLA UCZNIA
1. Przeczytaj uważnie instrukcję.
2. Zapoznaj się z zestawem zadań testowych.
3. Udzielaj odpowiedzi tylko na załączonej karcie odpowiedzi.
4. Podpisz imieniem i nazwiskiem kartę odpowiedzi.
5. Pracuj samodzielnie, pamiętaj, że w przyszłej pracy nie możesz liczyć na podpowiedzi kolegów.
6. Na udzielenie odpowiedzi masz 45 minut.
Powodzenia !

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
39
ZESTAW ZADAŃ TESTOWYCH
1. Podstawowymi składnikami suchej masy są
a) węglowodany, białka i witaminy,
b) tłuszcze i kwasy organiczne,
c) związki mineralne, barwniki i garbniki,
d) wszystkie wymienione składniki.
2. Liczba pobieranych próbek pierwotnych do analizy zależy od
a) kształtu surowców,
b) wielkości badanej partii,
c) miejsca pobrania,
d) ilości składników w próbce.
3. Kwasowość aktywna uwarunkowana jest stężeniem jonów:
a) wodorowych i hydroniowych,
b) tlenowych i wodorotlenowych,
c) wodorowych i węglowych,
d) tlenowych i wodorowych.
4. Metoda destylacji mieszanin azeotropowych stosowana jest do oznaczania zawartości
a) błonnika,
b) cukrowców,
c) suchej masy,
d) skrobi.
5. Do pobierania sypkich mas próbek stosuje się
a) zgłębniki,
b) zlewki,
c) łyżki,
d) cylindry.
6. W analizie surowców i produktów przemysłu spożywczego wyróżnia się kwasowość
a) lotną,
b) związaną,
c) całkowitą,
d) wszystkie wymienione.
7. Do suszenia próbek pod normalnym ciśnieniem stosuje się temperatury
a) 60 ÷ 80°C,
b) 60 ÷ 90°C,
c) 60 ÷ 100°C,
d) 60 ÷ 130°C.
8. Woda związana występuje w produktach w postaci wody
a) konstytucyjnej i kapilarnej,
b) krystalizacyjnej,
c) higroskopijnej,
d) wymienionej we wszystkich punktach powyżej.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
40
9. Kolba Kjeldahla służy do oznaczania
a) witamin,
b) tłuszczów,
c) białek,
d) cukrów.
10. Stosowane do ekstrakcji tłuszczu rozpuszczalniki nie mogą zawierać
a) wody,
b) białek,
c) soli mineralnych,
d) błonnika.
11. Oznaczanie temperatury topnienia tłuszczu pozwala określić
a) zawartość azotynów,
b) skład chemiczny,
c) zawartość wody,
d) liczbę kwasową.
12. Aparat Parnasa-Wagnera stosowany jest do
a) destylacji,
b) mineralizacji,
c) mierzenia gęstości,
d) oznaczania popiołu.
13. Oznaczanie skrobi w przetwórstwie mięsnym służy do
a) określania jakości produktu,
b) wykrywania zafałszowań,
c) określania ilości składników,
d) wykrywania środków higieny.
14. Metody butyrometryczne oznaczania tłuszczów należą do metod
a) ekstrakcyjnych,
b) wagowych,
c) objętościowych,
d) refraktometrycznych.
15. Do oznaczania tłuszczów wykorzystuje się ich zdolność do
a) topnienia,
b) rozpuszczania w eterze,
c) krzepnięcia,
d) ekstrakcji.
16. Zafałszowanie masła margaryną przeprowadza się wykrywając obecność
a) skrobi,
b) soli mineralnych,
c) laktozy,
d) kazeiny.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
41
17. Metoda Gerbera znajduje zastosowanie do oznaczania
a) kwasowości w jabłkach,
b) białek w mięsie,
c) tłuszczu w mleku,
d) cukru w lodach.
18. Oznaczanie temperatury topnienia tłuszczów można prowadzić w aparacie
a) Gerbera,
b) Parnasa-Wagnera,
c) Thielego,
d) Kjeldahla.
19. Próbki do analizy pobierane są
a) losowo,
b) przypadkiem,
c) zgłębnikiem,
d) specjalnym przyrządem.
20. Ilość i jakość glutenu stanowi o właściwościach
a) ziarna zbóż,
b) wypiekowych mąki,
c) zanieczyszczeń w mące,
d) zanieczyszczeń w ziarnie.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
42
KARTA ODPOWIEDZI
Imię i nazwisko..........................................................................................
Wykonywanie towaroznawczych badań żywności
Zakreśl poprawną odpowiedź.
Nr
zadania
Odpowiedź
Punkty
1.
a
b
c
d
2.
a
b
c
d
3.
a
b
c
d
4.
a
b
c
d
5.
a
b
c
d
6.
a
b
c
d
7.
a
b
c
d
8.
a
b
c
d
9.
a
b
c
d
10.
a
b
c
d
11.
a
b
c
d
12.
a
b
c
d
13.
a
b
c
d
14.
a
b
c
d
15.
a
b
c
d
16.
a
b
c
d
17.
a
b
c
d
18.
a
b
c
d
19.
a
b
c
d
20.
a
b
c
d
Razem:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
43
6. LITERATURA
1. Budsławski J., Drabant Z.: Metody analizy żywności. WNT, Warszawa 1999
2. Drewniak T., Drewniak E.: Mikrobiologia żywności. WSiP, Warszawa 1997
3. Drewniak T.: Analiza techniczna w przemyśle mięsnym. WSiP Warszawa1993
4. Drzazga B.: Analiza techniczna w przemyśle spożywczym. WSiP, Warszawa 1992
5. Drzazga B.: Analiza techniczna w przemyśle owocowo-warzywnym, WSiP, Warszawa 1992,
6. Gaweł J., Molska M.: Analiza techniczna w przemyśle mleczarskim, WSiP, Warszawa 1990
7. Harber T., Horubałowa A.: Analiza techniczna w przetwórstwie zbóż, WSiP, Warszawa 1992
8. Horubałowa A., Harber T.: Analiza techniczna w piekarstwie. WSiP, Warszawa 1989
9. Klepaczko-Filipiak B., Łonin J.: Pracownia chemiczna. Analiza techniczna. WSiP, Warszawa
2001
Wyszukiwarka
Podobne podstrony:
36 Wykonywanie towaroznawczych badań żywności
35 Wykonywanie mikrobiologicznych badań żywności
35 Wykonywanie mikrobiologicznych badań żywności
32 Wykonywanie wagowej analizy żywności
33 Wykonywanie objętościowej analizy żywności
34 Wykonywanie instrumentalnej analizy żywności
32 Wykonywanie wagowej analizy żywności
36 Wykonywanie powłok malarskich w technice kazeinowej
32 Wykonywanie wagowej analizy żywności
34 Wykonywanie instrumentalnej analizy żywności
14 Wykonywanie szczegółowych badań strabologicznych
Towaroznawstwo żywności - zagadnienia na egzamin, Nauka, Towaroznawstwo żywności, Zagadnienia
dr hab RG II cz swoboda towarowa ograniczenia ilościowe art 34 36 TFUE
05 Wykonywanie badań biochemicznych
więcej podobnych podstron