
1
ZAKŁAD BIOCHEMII
ENZYMOLOGIA
Biochemia i Biotechnologia
UMCS
(KP/KR)
2012
KINETYKA REAKCJI ENZYMATYCZNYCH – II
Ćwicz. 3
Wyznaczanie stałych kinetycznych (V
max
i K
M
)
Celem badań kinetyki reakcji enzymatycznych jest uzyskanie informacji o ich
przebiegu w czasie, a więc o szybkości zaniku substratów i powstawania produktów.
Wyznaczone stałe kinetyczne pozwalają na wnioskowanie o liczbie i kolejności przyłączania
cząsteczek substratu do enzymu i odłączania cząsteczek produktów z kompleksu aktywnego,
o liczbie kompleksów pośrednich i izomeryzujących form enzymu oraz o efektach działania
inhibitorów lub aktywatorów.
Badania te prowadzą do stworzenia modelu kinetycznego, będącego zbiorem
wyrażeń matematycznych pozwalających przewidywać efekt działania enzymu w obecności
określonego stężenia substratu, z jednoczesnym uwzględnieniem ilościowego udziału
wszystkich etapów reakcji enzymatycznej.
Najstarszy model kinetyczny dla ilościowego opisu nieodwracalnej reakcji
enzymatycznej z jednym substratem, przebiegającej według schematu:
2
1
k
1
E
S
ES
E
P
przedstawili Leonor Michaelis i Maud Menten (1913).
Podstawy teorii Michaelisa–Menten są następujące:
1. Główne założenie:
Istnieje szybko ustalający się stan równowagi reakcji:
1
1
E
S
(ES)
który nie jest zaburzony przez dużo powolniejszy rozpad kompleksu (ES) na enzym
i produkt:
P
E
(ES)
2
k

2
2. Maksymalna szybkość reakcji: V
max
= k
+2
[E]
0
3. Stała K:
1
1
-
S
k
k
K
K
S
jest stałą dysocjacji kompleksu przejściowego (tzw. stała substratowa)
-1
2
2
M
S
1
1
k
k
k
K
K
k
k
K
M
jest stałą Michaelisa (wyprowadzoną przez Briggsa i Haldane’a), jej wartość liczbowa
odpowiada takiemu stężeniu substratu, przy którym początkowa szybkość reakcji równa jest
połowie szybkości maksymalnej.
4. Warunek równoważności K
M
K
S
Równoważność stałych K
M
i K
S
występuje bardzo często w reakcjach enzymatycznych,
ponieważ kompleks przejściowy tworzy się przy udziale słabych oddziaływań
drugorzędowych, natomiast rozpad kompleksu na produkty i enzym wymaga przegrupowań
atomowych i rozerwania lub syntezy wiązań kowalencyjnych.
Wtedy k
-1
> k
+2
5. Warunek nierównoważności K
M
K
S
Utworzenie kompleksu przejściowego wymaga utworzenia wiązań kowalencyjnych, wtedy
k
-1
≤ k
+2
Zależność początkowej szybkości reakcji enzymatycznej w stanie stacjonarnym
(warunek d[ES]/dt = 0) od początkowego stężenia substratu jest określona równaniem:
max
0
0
M
0
V
S
v
K
S
v
0
– początkowa szybkość reakcji
V
max
– maksymalna szybkość reakcji
[S]
0
– początkowe stężenie substratu
K
M
– stała Michaelisa

3
Występujące w podanym powyżej równaniu hiperbolicznej kinetyki stacjonarnej
stałe kinetyczne K
M
i V
max
można wyznaczyć przez graficzne dopasowanie hiperboli do
doświadczalnych wartości v
0
i [S]
0
, ale postępowanie takie obarczone jest dużym błędem.
Istnieje kilka przekształconych form równania Michaelisa-Menten dających prostoliniową
zależność obu zmiennych. Najczęściej stosowaną metodą linearyzaji krzywej hiperbolicznej
jest metoda Lineweavera-Burke’a, która określa zależność 1/v
0
od 1/[S]
0
zgodnie
z równaniem:
M
0
max
max
0
K
1
1
1
v
V
V
S
Wykonanie ćwiczenia
Przygotować probówki zawierające po 3 ml następujących roztworów sacharozy
w 0,1 M buforze octanowym o pH 4,7:
0,2 M;
0,1 M;
0,05 M;
0,025 M
Do każdej probówki dodać po 3 ml roztworu rozcieńczonego preparatu enzymatycznego,
zawierającego inwertazę (o optymalnym stężeniu, wyznaczonym na poprzednich ćwiczeniach
– ćwicz. 2).
Natychmiast po wymieszaniu (próby odczynnikowe), a następnie dokładnie po 5, 10,
15 i 30 min. pobierać po 1 ml mieszaniny inkubacyjnej do odpowiednio oznakowanych
probówek zawierających po 4 ml 0,1 M buforu glicynowego o pH 10 (w celu zatrzymania
reakcji enzymatycznej).
Dalszy tok postępowania jest zgodny z procedurą podaną przy wyznaczaniu
optymalnego stężenia preparatu enzymatycznego (ćwicz. 2).
Z krzywej kalibracyjnej odczytać stężenie (μg/ml) monosacharydów uwolnionych
podczas enzymatycznej hydrolizy sacharozy, a następnie obliczyć ilość powstałych
produktów uwzględniając rozcieńczenie mieszaniny inkubacyjnej (30
):
Ilość produktu (μg) = A
520
× f (
μg/ml) × 1 ml × 30
Dla każdego stężenia substratu sporządzić krzywą progresji - wykres zależności
ilości produktu (mg) od czasu reakcji (min.) i wyznaczyć szybkość początkową reakcji.
Szybkość początkowa reakcji v
0
to tg α, nachylenie stycznej do krzywej progresji
w punkcie odpowiadającym stężeniu produktu w czasie t = 0 min., przechodzącej zawsze
przez początek układu współrzędnych.
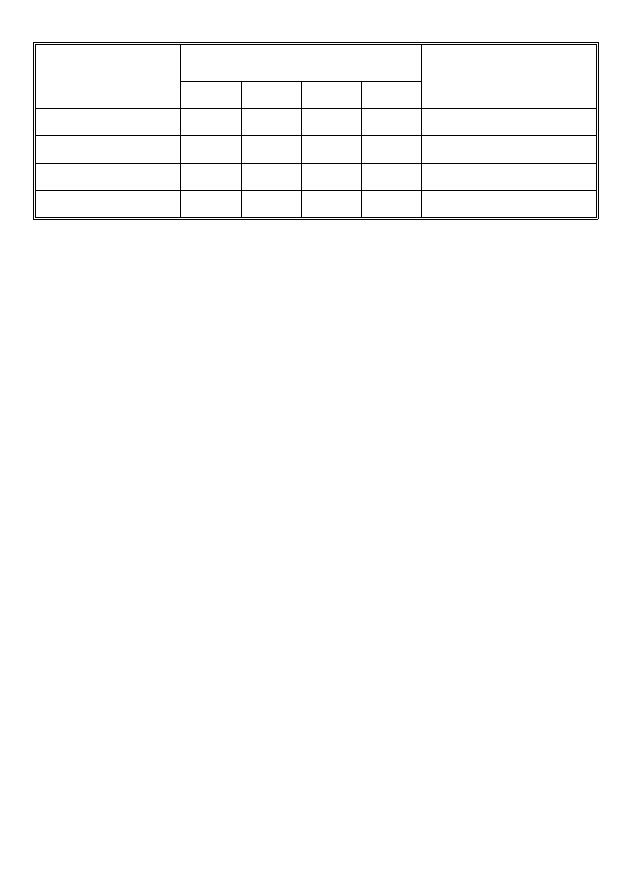
4
Stężenie początkowe
substratu [S]
0
Ilość produktu (mg)
w zależności od czasu
Szybkość początkowa
(mg/min)
v
0
= tg α
5 min
10 min
15 min
30 min
0,2 M
0,1 M
0,05 M
0,025 M
Na podstawie szybkości początkowych wyznaczonych dla każdego stężenia
substratu wykreślić zależność tych szybkości od stężenia początkowego substratu (krzywą
Michaelisa-Menten).
Sporządzić wykres dla równania Lineweavera-Burke’a, wyznaczyć stałą Michaelisa
(K
M
) i szybkość maksymalną reakcji (V
max
).
Odczynniki
1. 0,1 M bufor octanowy (octan sodu – kwas octowy) o pH 4,7
2. 0,2 M roztwór sacharozy w buforze octanowym o pH 4,7
3. 0,1 M bufor glicynowy (glicyna – NaOH) o pH 10
4. Odczynnik Somogyi I
288 g Na
2
SO
4
(bezw.) rozpuścić w 1l H
2
O dest. Dodać 24 g soli Rochelle’a
(winian sodowo potasowy), 48 g Na
2
CO
3
, 32 g NaHCO
3
. Roztwór rozcieńczyć
do 1600 ml gotowaną H
2
O dest. i przechowywać w temp. 27
O
C.
5. Odczynnik Somogyi II
72 g Na
2
SO
4
(bezw.) rozpuścić w 300 ml wrzącej H
2
O dest. Dodać 8 g
CuSO
4
×H
2
O. Roztwór rozcieńczyć do 400 ml gotowaną H
2
O dest. i
przechowywać w temp. 27
O
C.
6. Mieszaninę odczynników Somogyi I i Somogyi II (stosunku 4:1) przygotowują
studenci bezpośrednio przed użyciem.
7. Odczynnik Nelsona
100 g molibdenianu amonu rozpuścić w 1,8 l H
2
O dest. Dodać 84 ml stęż.
H
2
SO
4
i roztwór arsenianu sodu (12 g Na
2
H-arsenian w 100 ml H
2
O).
Odczynnik przechowywać w ciemnej butelce szklanej przez 24-48 godz.
w temp. 37
O
C, potem w pokojowej.
Sprzęt
1. łaźnia wodna (100
o
C)
2. micro-shaker (wytrząsarka)
3. spektrofotometr
4. stoper
5. probówki ze szczelnymi korkami
6. probówki krótkie
7. pipety automatyczne
8. cylindry miarowe, zlewki, kolbki
(poj. 50, 100, 250 ml)
Wyszukiwarka
Podobne podstrony:
Enzymologia Skrypt III id 16216 Nieznany
Kolos ekonimika zloz II 2 id 24 Nieznany
cyw1 skrypt zobowiazania id 126 Nieznany
BIOCHEMIA skrypt 2010 id 86508 Nieznany
kolokwium organiczna II id 2408 Nieznany
ASW CANTIUS II id 71219 Nieznany (2)
MGLab Formularz II 5 id 297630 Nieznany
Chemia polimerow II id 113148 Nieznany
Podstawy psychiatrii II id 3681 Nieznany
Nowy skrypt z OPC (1) id 324288 Nieznany
MGLab Formularz II 4 id 297629 Nieznany
m kawinski cz ii id 274819 Nieznany
Anatomia Skrypt Godowicz id 628 Nieznany
Fizyka Ciala Stalego II id 1766 Nieznany
historyczna kolo II id 204904 Nieznany
Bliski wschod II id 90148 Nieznany
Modul II id 305650 Nieznany
Fuzzy Logic II id 182423 Nieznany
Grupa II id 196511 Nieznany
więcej podobnych podstron