Stanowiskowa instrukcja BHP
INSTYTUT TECHNOLOGII NIEORGANICZNEJ I NAWOZÓW MINERALNYCH
POLITECHNIKI WROCŁAWSKIEJ
TECHNOLOGIA CHEMICZNA kurs Technologia
Ćwiczenie nr 5.
Oczyszczanie gazów metodami absorpcyjnymi:
usuwanie tlenków azotu roztworami alkalicznymi
Wrocław 2012
Zawartość
Załączniki
Załącznik 1 - Formularz sprawozdania (w przygotowaniu)
Załącznik 2 - DTR regulatora TCH -CR92C
Załącznik 3 - Dobór nastaw PID regulatora wg procedury Zieglera-Nicholsa
Wstęp
Źródłem emisji NOx jest przemysł chemiczny - technologia wytwarzania kwasu azotowego, azotynu amonu czy też procesy nitrowania - a także transport oraz energetyka, w których tlenki azotu powstają podczas procesów spalania. W związku ze szkodliwym działaniem NOx na środowisko naturalne (kwaśne deszcze, niszczenie dziury ozonowej) konieczne jest ich wiązanie i usuwanie przed wypuszczeniem do atmosfery.
Pośród metod usuwania tlenków azotu z gazów odlotowych wyróżnia się:
selektywną redukcję katalityczną (SCR);
nieselektywną redukcję katalityczną (NSCR);
adsorpcję;
absorpcję.
W metodach absorpcyjnych wykorzystywana jest rozpuszczalność niektórych tlenków azotu w wodnych roztworach kwaśnych (roztwory kwasu azotowego i/lub siarkowego) a także zdolność do tworzenia azotanów (III) i (V) w roztworach o charakterze alkalicznym (wodorotlenki sodu/potasu/wapnia lub węglany w tym węglan amonowy). W związku z tym rozróżnia się absorpcję kwaśną oraz absorpcję alkaliczną przebiegające wg odmiennego mechanizmu chemicznego.
Mechanizm procesu absorpcji w roztworze alkalicznym.
W przypadku absorpcji w roztworze alkalicznym mają miejsce następujące reakcje:
w fazie gazowej
![]()
(1)
![]()
(2)
![]()
(3)
![]()
(4)
![]()
(5)
oraz w fazie ciekłej
![]()
(6)
![]()
(7)
![]()
(8)
![]()
(9)
![]()
(10)
W gazie poddawanym absorpcji znajduje się głównie słabo rozpuszczalny w wodzie tlenek azotu NO oraz powstający w wolnej reakcji (1) dobrze rozpuszczalny NO2, o ile w strumieniu jest dostępny wolny tlen. Całkowite przereagowanie NO jest osiągane w czasie dziesiątek lub setek sekund w zależności od temperatury i ciśnienia. Jako miarę ilości dwutlenku azotu w gazie stosuje się często parametr „stopień utlenienia tlenku azotu w gazie Su” definiowany jako ![]()
, gdzie yi są ułamkami molowymi. W laboratorium stosuje się te tlenki przechowywane w butlach w postaci czystej: tlenek azotu NO jako gaz pod ciśnieniem do 50at (stopień utlenienia Su=0) oraz NO2 w postaci cieczy (a właściwie jego dimer N2O4 - reakcja 3) pod ciśnieniem własnych par ok. 1at w temperaturze otoczenia (Su=1). Korzystając z tego źródła można przygotować mieszaninę tlenków o dowolnym stopniu utlenienia Su. Oprócz stopnia utlenienia Su zasadniczą miarą zanieczyszczenia gazu tlenkami azotu, określaną jako NOx, jest całkowita ich zawartość w przeliczeniu na czysty tlenek azotu NO. Przeliczenie stężenia NO2 na stężenie NO dokonuje się na podstawie reakcji (1).
Jeżeli w gazie zawierającym NO pojawia dwutlenek azotu NO2 (wprowadzony z zewnątrz lub w wyniku reakcji 1), tworzą się pozostałe tlenki w tym N2O3. Szybkość jego tworzenia jest duża i stan równowagi wynikający z reakcji (2) - ![]()
jest osiągany w ciągu pojedynczych sekund a jego stężenie może być stosunkowo duże ok. 10-3 tak, że można go nawet wydzielić z układu w czystej postaci - błękitna ciecz z temperaturą wrzenia ok. 3,5oC. Jednak gdy równocześnie biegną reakcje absorpcji tlenków w roztworze alkalicznym wg mechanizmu (6-10), to stężenie równowagowe N2O3 nigdy nie jest osiągane, ponieważ szybkość reakcji absorpcji jest bardzo szybka - usuwanie N2O3 z fazy gazowej zachodzi w ułamkach sekund. Zatem w fazie gazowej ustalić się może jedynie pewne niewielkie kwazi-równowagowe (nie mylić ze stężeniem stacjonarnym) stężenie N2O3
ok. 10-5 a reakcja (2) efektywnie go wytwarza zużywając w równym stopniu niereaktywny NO jak i reaktywny NO2, ponieważ reakcja ta jest daleko od stanu równowagi.
W rezultacie proces ten prowadzi do otrzymywania roztworu głównie azotanu(III) (azotynu) z niewielkim dodatkiem azotanu(V). Tlenek NO2 zużywany jest głównie do tworzenia N2O3 a N2O4 pozostając na nieznacznym poziomie 10-7, nie jest w stanie wytworzyć dużych ilości kwasu azotowego w wyniku przebiegu reakcji (5). Tę właściwość układu wykorzystuje się np. w technologii produkcji azotynu amonowego dla potrzeb przemysłu tworzyw sztucznych (melamina) gdzie konieczne jest minimalizowanie zawartości azotanu amonowego, który jako substancja termicznie niestabilna przy zatężaniu w roztworach technologicznych może prowadzić do zagrożenia wybuchem.
Na podstawie mechanizmu reakcji (1-5) i (6-10), uwzględniając ponadto szybkość procesów przenoszenia masy między fazą gazową i ciekłą, która wynika z parametrów stosowanej kolumny absorbcyjnej (średnica, wysokość, rodzaj wypełnienia, strumienie zasilające) oraz właściwości fizyko-chemicznych układu (gęstość, lepkość, współczynniki dyfuzji, prężność par), wykonuje się obliczenia bilansowe prowadzące do określenia wszystkich parametrów procesu, w szczególności zmian stężeń poszczególnych składników wzdłuż kolumny absorpcyjnej. Szczegóły tych obliczeń leżą poza zakresem informacji wymaganych w niniejszym ćwiczeniu a na rys. 1-4 przedstawiono jedynie wybrane wyniki dla absorpcji 1,1% obj. NO. Należy przeanalizować podane na rysunkach zależności w celu przygotowania się do odpowiedzi na pytania w teście kontrolnym na początku zajęć, dotyczących znajomości przedstawionego wyżej mechanizmu absorpcji.
Rys. 1 Zmiana stężenia tlenku azotu NO w gazie podczas absorpcji w roztworze alkalicznym (górna część wykresu - lewa oś OY) oraz stężenia dwutlenku azotu NO2 (dolna część wykresu - prawa oś OY) dla różnych stopni utlenienia tlenków azotu w gazie wlotowym do absorbera.
Rys. 2 Stopień absorpcji (konwersji) tlenków azotu w zależności od stopnia utlenienia w gazie wlotowym o zawartości początkowej NO 1,1%obj..
Rys. 3 Zawartość tlenków azotu na wylocie absorbera w zależności od stopnia utlenienia w gazie wlotowym do absorbera.
Rys. 4. Stopień absorpcji (konwersji) tlenków azotu NOx oraz związana z nim zawartość jonu azotanowego NO3- , w zależności od stopnia utlenienia w gazie wlotowym o zawartości początkowej NO 1,1%obj.
Opis układu instalacji doświadczalnej dla absorpcji tlenków azotu w środowisku alkalicznym
Absorpcję tlenków azotu prowadzi się w warunkach bezciśnieniowych w układzie dwukolumnowym (rys.1), z których pierwsza pełni rolę absorbera natomiast druga jest pomocniczą kolumną myjącą, usuwającą niezaabsorbowane tlenki azotu z odgazów z absorpcji przed wypuszczeniem ich do atmosfery. Ponadto przez śledzenie zmian składu roztworu absorpcyjnego w kolumnie myjącej w wyniku pochłaniania tlenków, poprzez kontrolę zmiany pH i potencjału utleniająco/redukcyjnego (redox), możliwe jest bilansowanie efektywności procesu w kolumnie absorpcyjnej. Absorbentem w kolumnie myjącej jest zawsze roztwór kwasu azotowego z dodatkiem nadtlenku wodoru do utleniania kwasu azotawego do azotowego w roztworze, niezależnie od rodzaju stosowanego absorbenta w kolumnie absorpcyjnej.
Jako źródło tlenków azotu o zadanym stopniu utlenienia stosuje się zamiennie:
- mieszaninę czystego tlenku azotu z powietrzem sporządzaną w butli stalowej przy ciśnieniu całkowitym nie przekraczającym 20atm, z uwagi na zjawisko dimeryzacji utworzonego dwutlenku azotu
- strumień czystego NO z butli dodawanego do strumienia powietrza, doprowadzany do absorbera rurociągiem o zadanej długości i wynikającego z niej czasu reakcji utleniania NO
- strumień czystych NO i NO2 z butli w odpowiednich proporcjach
- reaktor katalitycznego utleniania amoniaku wytwarzający gaz nitrozowy (mieszanina NO i NO2) o zawartości ok. 7%obj. rozcieńczany azotem lub powietrzem do wymaganego w ćwiczeniu stężenia tlenków - patrz Dodatek A.
Absorber składa się z dwóch segmentów wypełnionych pierścieniami o rozmiarze 12x12x0,4mm ze stali kwasoodpornej, z których każdy posiada własny dystrybutor cieczy absorpcyjnej, zapewniając właściwe rozprowadzenie cieczy na całej długości wypełnienia. W zależności od rodzaju pomiaru, przede wszystkim zawartości tlenków azotu w fazie gazowej, wykorzystuje się jeden lub obydwa segmenty wypełnione, tak aby zapewnić na wylocie absorbera odpowiednio dużą siłę napędową procesu absorpcji.
Stosowane elementy aparatury kontrolno-pomiarowej zestawiono w tabeli 1.
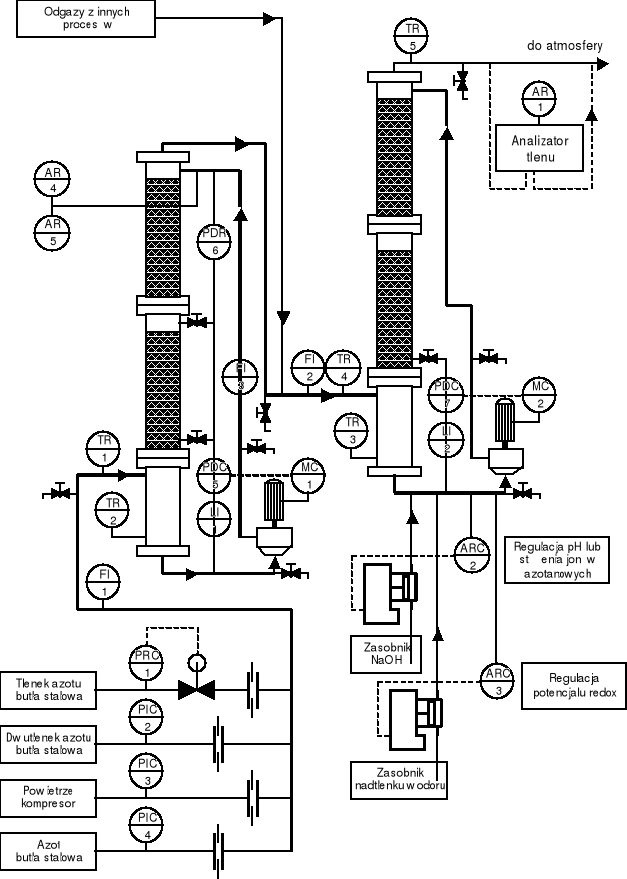
Rys. 5. Układ do badań absorpcji tlenków azotu o różnym stopniu utlenienia przy niskiej zawartości tlenków w fazie gazowej.
Tabela 1. Zestawienie punktów pomiarowych w instalacji wg rys. 5
Oznaczenie |
Nazwa urządzenia |
Przeznaczenie |
PRC1 |
Regulator automatyczny przepływu |
Zawór elektromagnetyczny ze stali kwasoodpornej sterowany regulatorem PWM w zależności od wartości ciśnienia mierzonego manometrem rezystancyjnym; ze stałej wartości ciśnienia wynika stały strumień gazu w kapilarze o zadanej średnicy i długości. |
PIC2 |
Regulator nastawny (ręczny) przepływu |
Reduktor ciśnienia nastawny ustalający ciśnienie na wejściu do kapilary regulatora; zestaw ze stali kwasoodpornej |
PIC3,4 |
Regulator nastawny przepływu |
Jak wyżej w wykonaniu zwykłym |
PDC5, LI1 |
Regulator poziomu |
Manometr różnicowy o zakresie 9,6kPa mierzący wysokość słupa cieczy w absorberze; wersja pneumatyczna z szeroką membraną |
PDR6 |
Pomiar spadku ciśnienia na wypełnieniu absorbera |
Manometr różnicowy precyzyjny o zakresie 1kPa do pomiaru spadku ciśnienia na wypełnieniu |
PDC7, LI2 |
Regulator poziomu |
Jak dla PDC5 |
TR1-5 |
Pomiar temperatury |
Termometr rezystancyjny Pt100 w wykonaniu kwasoodpornym o małej pojemności cieplnej |
FI1-3 |
Pomiar strumienia |
Rotametry szklane o różnych zakresach i parametrach cechowania |
MC1, 2 |
Regulator PID mocy |
Inwerter częstotliwości (falownik) zasilający trójfazowy silnik pompy obiegowej absorbenta, utrzymujący stały poziom cieczy w kolumnie lub stały strumień cieczy |
AR1 |
Analizator |
Analizator tlenu Magnos6G |
AR2 |
Analizator |
Regulator pH złożony ze sterownika, ciśnieniowej elektrody szklanej do pomiaru pH oraz elektromagnetycznej pompy dozującej utrzymujący zadane pH kolumny myjącej; impulsy sterujące pompą podawane przez regulator, zliczane przez komputerowy układ rejestrujący, są miarą całkowitej zawartości niezaabsorbowanych tlenków azotu w gazie po kolumnie absorpcyjnej |
AR3 |
|
Regulator potencjału redoks złożony ze sterownika, ciśnieniowej elektrody redoks oraz elektromagnetycznej pompy dozującej utrzymujący zadany potencjał w roztworze kolumny myjącej; impulsy z regulatora zliczane przez sterownik są miarą ilości czynnika utleniającego koniecznego do utlenienia jonu azotynowego w roztworze kolumny myjącej, tym samym miarą stopnia utlenienia gazu po kolumnie absorpcyjnej |
AR4-5 |
|
Pomiar pH i potencjału redoks roztworu absorpcyjnego zrealizowany przez odpowiednią elektrodę i jej przetwornik; sygnał wyjściowy 4-20mA podawany do zewnętrznego układu rejestrującego |
Wykonanie doświadczenia
- uzyskać od prowadzącego informację o rodzaju i stężeniu absorbenta (wodorotlenek potasowy/sodowy, węglan potasowy, węglan amonowy). Sporządzić roztwór absorpcyjny o zadanym składzie i sprawdzić jego skład wg Dodatku D
- ustalić źródło tlenków azotu i stosowany zakres stężeń tlenków w gazie wlotowym do absorpcji; dokonać niezbędnych przeliczeń wskazań rotametrów
- wybrać punkt pracy (obciążenia gazem i cieczą) kolumny absorpcyjnej wykonując obliczenia zgodnie z dodatkiem B
- dla wybranego punktu pracy wykonać pomiar szybkości absorpcji poprzez pobranie próbek fazy ciekłej i gazowej i wykonanie oznaczeń zawartości kluczowych składników wg Dodatku C
- sporządzić bilans masowy azotu zawartego w tlenkach przenoszonego z fazy gazowej do ciekłej
- zestawić uzyskane wyniki w sprawozdaniu wg załączonego w załączniku 1. formularza
Dodatek A.
Uruchomienie reaktora katalitycznego spalania amoniaku do wytwarzania tlenków azotu
Rotametrem ustalić strumień powietrza podawanego do reaktora przez pogrzewacz mieszaniny amoniakalno-powietrznej
Włączyć zasilanie chłodnicy gazów nitrozowych wodą chłodzącą
Włączyć ogrzewanie podgrzewacza przez wybranie wartości zadanej jego regulatora (150oC - temperatura za podgrzewaczem) i nagrzewać reaktor gorącym strumieniem powietrza aż do uzyskania temperatury zadanej także za reaktorem; dokumentacja techniczno-ruchowa stosowanego regulatora w załączniku 2.
Sprawdzić poprawność doboru nastaw PID regulatora zmniejszając i zwiększając strumień powietrza, notując zmiany temperatury za podgrzewaczem w arkuszu kalkulacyjnym; jeżeli zachodzi konieczność skorygować parametry PID metodą Zieglera-Nicholsa - opis metody w załączniku 3
Po osiągnięciu temperatury zadanej za reaktorem ustalić na rotametrze strumień amoniaku podawany do podgrzewacza tak, aby stężenie amoniaku było poniżej dolnej granicy wybuchowości w zakresie 7-10%obj. Sprawdzić poprawność nastaw rotametru przez pobranie próbki gazu przed podgrzewaczem i wykonanie oznaczenia zawartości amoniaku zgodnie z p. 2
Przy braku zapłonu na katalizatorze w przeciągu 2min. Operując zaworem amoniaku zwiększyć impulsowo strumień amoniaku na ok. 2 sekundy tak, aby osiągnąć stężenie 14% amoniaku w powietrzu; jeżeli trzy próby nie przyniosą pożądanego efektu (zapłonu mieszaniny na katalizatorze) zwiększyć temperaturę za podgrzewaczem o 10oC i ponowić próbę zapalenia przez impuls wzbogaconej w amoniak mieszanki
Po zapaleniu reaktora i upływie ok. 2min. pobrać próbkę gazu nitrozowego za chłodnicą i oznaczyć zawartość nieprzereagowanego amoniaku zgodnie z p. 3
2. Analiza amoniaku gazowego w mieszaninie amoniakalno-powietrznej podawanej do reaktora utleniania.
Próbkę amoniaku pobrać do suchej pipety gazowej o objętości 0,5dm3 po czym wprowadzić strzykawką wodę destylowaną w ilości ok. 20ml. Wytrząsać aż do całkowitego zaabsorbowania amoniaku i następnie przenieść ilościowo do zlewki, przepłukując pipetę małymi porcjami wody destylowanej. Miareczkować potencjometrycznie 0,2 M roztworem kwasu solnego sporządzając krzywą miareczkowania w arkuszu kalkulacyjnym. Obliczyć stężenie amoniaku w próbce gazu.
3. Oznaczenie zawartości nieprzereagowanego amoniaku w mieszaninie gazów po reaktorze utleniania (w gazie nitrozowym)
Próbkę do analizy pobiera się do pipety gazowej o objętości 0,5dm3 i absorbuje się w ok. 20ml wody. Otrzymany roztwór zobojętnia się 0,1 M roztworem NaOH - jeżeli roztwór ma odczyn kwaśny, albo 0,2 M roztworem kwasu siarkowego(VI) - jeżeli ma odczyn zasadowy. Następnie zobojętnia się 10 ml 10% roztworu formaliny wobec papierka lakmusowego stosując rozcieńczony roztwór NaOH i dodaje do roztworu badanego. W procesie mieszania zachodzi reakcja:
![]()
Powstały jon wodorowy zneutralizować dodatkiem mianowanego roztworu NaOH a jego nadmiar odmiareczkować 0,2n HCl potencjometrycznie stosując elektrodę do pomiaru pH. Obliczyć zawartość amoniaku w pobranej próbce gazu.
Dodatek B
Wybór parametrów pracy kolumny absorpcyjnej
W celu wyznaczenia parametrów pracy kolumny absorpcyjnej sporządzić charakterystykę wypełnienia mierząc dla danego obciążenia cieczą L spadek ciśnienia na wypełnieniu p przy wzrastającym obciążeniu gazem G. Jako punkt zalewania kolumny przyjąć obciążenie gazem G, przy którym wystąpi spadek ciśnienia na wypełnieniu równy 1kPa (w0,kr). Sporządzić wykres zależności spadku ciśnienia od G i L. Sprawdzić na ile jest spełniona korelacja Kafarowa-Dytnierskiego dla układu gaz-ciecz w postaci:
![]()
gdzie:
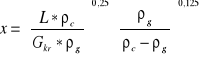
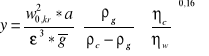
L,G - strumień cieczy i gazu, m3/h
ρ c, ρ g,- gęstość cieczy i gazu, kg/m3
η c, η w, - lepkość cieczy absorpcyjnej i wody, mPas
w0,kr - liniowa prędkość fazy gazowej w punkcie przeciążenia wypełnienia, m/s
a= 440 m2/m3, ε= 0.94 m3/m2 - parametry wypełnienia
Jako punkt pracy kolumny przyjąć obciążenie gazem L aby liniowa prędkość gazu liczona na pusty przekrój kolumny wynosiła, dla danego obciążenia cieczą L, 0,7 wartości krytycznej w0,kr. Obciążenie cieczą L wybrać na podstawie zmierzonej charakterystyki wypełnienia w uzgodnieniu z prowadzącym.
Uwagi:
- do rozwiązywania korelacji użyć narzędzia „Szukaj wyniku” arkusza kalkulacyjnego Excel lub dowolnego oprogramowania umożliwiającego rozwiązywanie równania z jedną niewiadomą metodą iteracyjną (Polymath, Matlab, MathCad etc.)
Dodatek C
Oznaczenie zawartości azotanów (III) i (V) w roztworze
Oznaczenia azotanów(III) oraz azotanów(V) w roztworze są wykonywane fotometrycznie w zakresie ultrafioletu metodą krzywej wzorcowej dla azotanów(III) oraz azotanów(V).
a) Oznaczania zawartości azotanów(III)
Przygotowanie krzywej wzorcowej:
w kolbach miarowych o obj. 100ml przygotować roztwory wzorcowe azotanu(III), o stężeniach: 2, 5, 10, 15, 25 mmol/dm3; w oparciu o roztwór sporządzony z naważki NaNO2 w kolbie miarowej o poj. 250ml w odpowiednim stężeniu, przez dodawanie określonych objętości do kolb miarowych wzorców.
Uruchomić aplikację Scan fotometru i wprowadzić ustawienia:
Zakładka Cary: zakres długości fal (X Mode): 200-400 nm; szybkość skanowania (Scan Controls): wolna (slow);
Zakładka Reports: należy załączyć wszystkie przebiegi oraz pary punktów;
Stosując przycisk Baseline: wykonać linię bazową dla aktualnie stosowanej wody redestylowanej w celu zastosowania korekcji linii bazowej (Baseline correction)
Wykonać pomiary spektrofotometryczne próbek wzorcowych;
Wybrać długość fali, dla której azotan(III) ma maksymalną absorbancję i wykreślić krzywą wzorcową z odczytanych dla tej fali absorbancji w zależności od stężenia azotanu(III) A = f(c).
W celu określenia zawartości azotanu(III) w próbce roztworu absorpcyjnego należy wykonać pomiar absorbancji, a następnie, korzystając ze wzoru wykreślonej krzywej wzorcowej, wyliczyć stężenie azotanu(III) w roztworze. W przypadku, gdy absorbancja jest większa niż zakres krzywej wzorcowej należy roztwór rozcieńczyć.
Oznaczanie zawartości azotanów(V)
Podczas absorpcji w roztworze zawartość azotanu(V) wzrasta powoli przyjmując niewielkie wartości natomiast azotan(III) występuje w znacznych ilościach. Tradycyjne oznaczenie zawartości azotanu(III) oraz azotanu(V) podczas jednego pomiaru spektrofotometrycznego prowadzi do oznaczenia azotanu(V) z bardzo dużym błędem.. W celu precyzyjnego oznaczenia zawartości azotanów(V) konieczne jest uprzednie usunięcie z roztworu azotanów(III) za pomocą kwasu aminosulfonowego. W rzeczywistym roztworze absorpcyjnym znajduje się także węglan amonu, który również reaguje z kwasem aminosulfonowym.
Przygotowanie krzywej wzorcowej:
W kolbach miarowych o obj. 50ml sporządzić roztwory wzorcowe azotanu(V) o stężeniach: 0,2, 0,5, 0,1, 1,5, 2,5 mmol/dm3; w oparciu o wyjściowy roztwór KNO3 z naważki. Do każdego roztworu dodać azotanu(III) tak aby jego stężenie wynosiło 25mmol/dm3
Każdą z próbek przed pomiarem wymieszać w zlewce z roztworem kwasu aminosulfonowego o stężeniu 0,2mol/dm3 w stosunku 2:1 (np. 4 ml analizowanego roztworu i 2 ml H2NSO3H);
Wykonać pomiary spektrofotometryczne wszystkich próbek (ustawienia aplikacji Scan - patrz: oznaczenie zawartości azotanów(III));
Wybrać długość fali, dla której azotan(V) ma maksymalną absorbancję i wykreślić krzywą wzorcową A = f(c).
Oznaczenie azotanu(V) w roztworze absorpcyjnym:
do zlewki pobrać 4 ml badanego roztworu i zneutralizować porcjami 10 μl kwasu siarkowego(VI), badając jego pH za pomocą papierka wskaźnikowego, aż do osiągnięcia pH w przybliżeniu 4
dodać 2 ml roztworu kwasu amino sulfonowego i mieszać delikatnie aż do zaniku wydzielania pęcherzyków gazu;
wykonać pomiar spektrofotometryczny i za pomocą równania krzywej wzorcowej wyliczyć stężenie azotanu(V) w badanym roztworze.
Dodatek D
Oznaczenie zawartości węglanów w roztworze absorpcyjnym
(na podstawie: Z. S. Szmal, T. Lipiec, Chemia analityczna z elementami analizy instrumentalnej, Wydawnictwo Lekarskie PZWL, Warszawa 1996)
Oznaczanie węglanów i wodorowęglanów obok siebie wykonuje się metodą potencjometryczną stosując elektrodę do pomiaru pH. Próbkę roztworu pobrać pipetą miarową o objętości 2,0ml, rozcieńczyć w zlewce o obj. 150ml do 2/3 wysokości i mieszać mieszadłem magnetycznym. Elektrodę zanurzyć odpowiednio głęboko aby klucz elektrolityczny znalazł się pod powierzchnią roztworu, ale nie za głęboko aby nie uszkodzić końcówki elektrody szklanej wirującym mieszadłem. Miareczkować 0,2M roztworem kwasu solnego odczytując i notując w arkuszu kalkulacyjnym wartość pH. Kwas wprowadza się porcjami po 0,5 do 1,0ml a w pobliżu punktu równoważnikowego pojedynczymi kroplami aby uchwycić wyraźną skokową zmianę pH. Miareczkowanie należy zakończyć, gdy roztwór miareczkowany jest mocno kwaśny - pH poniżej 3. Z otrzymanych wyników należy sporządzić wykres w układzie: oś odciętych - objętość roztworu HCl (ml), oś rzędnych - pH. Wykresy powinny wykazać dwa punkty rónoważnikowe zgodnie z 2-stopniową reakcją węglanu z mocnym kwasem, z których pierwszy odpowiada zobojętnieniu węglanu do wodorowęglanu, a drugi - całkowitemu zobojętnieniu węglanu i wodorowęglanu. Na podstawie objętości zużytego kwasu oblicza się stężenia obu oznaczanych anionów wg zależności:
Załącznik 1. Formularz sprawozdania (w przygotowaniu)
Załącznik 2.
Dokumentacja techniczno-ruchowa (DTR) regulatora podgrzewacza mieszaniny amoniakalno-powietrznej reaktora katalitycznego utleniania amoniaku do tlenków azotu - typ TCH CR92C
Pobrać ze strony WWW.itn.pwr.wroc.pl plik z DTR regulatora i zapoznać się z następującymi zagadnieniami:
- znaczenie wszystkich kontrolek na panelu regulatora
- ustawianie wartości zadanej
- przełączanie regulatora do trybu ręcznego nastawiania wartości sygnału regulowanego; rozpoczęcie rozruchu reaktora odbywa się w trybie ręcznym, podobnie zatrzymanie jego pracy; w trybie automatycznym jest duże ryzyko przekroczenia temperatury samozapłonu stosowanej mieszaniny amoniaku z powietrzem
- odczyt i zmiana aktualnych parametrów PID regulatora poprzez jego panel sterujący oraz programem obsługi poprzez łącze komunikacyjne RS-485
- obsługa i definiowanie dostępnych stanów alarmowych w regulatorze
Załącznik 3.
Reguła Zieglera-Nicholsa - wytyczne stosowania
Regulatory typu P, PI, PID są regulatorami uniwersalnymi do regulacji dowolnych obiektów pod warunkiem prawidłowego doboru nastaw. Stosowane zakresy nastaw wynoszą najczęściej:
współczynnik wzmocnienia kp=0.25..25,
czas zdwojenia Ti=3s..30 min,
czas wyprzedzenia Td=0..15 min.
Powiększanie czasu Ti powoduje późniejsze włączanie do pracy członu całkującego, a powiększanie czasu Td zwiększa wpływ elementu różniczkującego. Należy pamiętać, że w regulatorach mikroprocesorowych współczynnik wzmocnienia (najczęściej podawany w DTR jako P) jest odwrotnością kp, tzn. gdy przyjmuje wartość równą zeru wtedy wzmocnienie jest największe.
Nastawy P, I, oraz D dobiera się na podstawie znajomości charakterystyk dynamicznych obiektu. W przypadku, gdy charakterystyka jest nieznana, nastawy dobiera się wg reguły Zieglera-Nicholsa. W regulatorze włącza się tylko człon proporcjonalny P. Człon całkujący zamyka się całkowicie (Ti = max), człon różniczkujący całkowicie otwiera (Td = 0). Zwiększa się współczynnik wzmocnienia kp tak, aby układ doprowadzić do granicy stabilności (uzyskać oscylacje o stałej ampitudzie). Z wyznaczonego przebiegu wielkości regulowanej określić okres drgań niegasnących Tkryt. Znając kp,kryt i Tkryt obliczyć nastawy regulatorów wg wzorów:
dla regulatora P: kp = 0,5 kp kryt;
dla regulatora PI: kp = 0,45 kp,kryt, Ti = 0,75 Tkryt;
dla regulatora PID: kp = 0,6 kp,kryt, Ti = 0,5 Tkryt, Td = 012 Tkryt.
Metody tej nie można stosować gdy względy technologiczne nie pozwalają na doprowadzenie układu do granicy stabilności (ciągła oscylacja wartości regulowanej), jednak zastosowanie okresu oscylacji gasnących jako Tkryt (przyjmując aktualną wartość kp jako kp,kryt) prowadzi również do poprawy skuteczności regulacji. Zaleca się stosowanie tego przybliżenia podczas prowadzenia doświadczeń jako wystarczającego.
![]()
Wyszukiwarka