1. Zasada działania baterii słonecznych
2. Efekt fotowoltaiczny
3. Otrzymywanie, właściwości i zastosowanie powłok niklowych
Na pytanie 3, jeżeli się pojawi, odpowiadać bez względu na to, jakie padnie pytanie z galwanotechniki.
4. Klasyfikacja polimerów + oznaczenia (skróty)
5. *Kompozyty - charakterystyka (+budowa) i podział
6. *Nanoproszki - charakterystyka i zastosowanie
7. Biomateriały - charakterystyka; bioceramiczne i *węglowe
8. Biodegradacja - charakterystyka
9. Recykling - charakterystyka i rodzaje.
10. *Reakcje metali z wodą, kwasami i zasadami
11. Proces wielkopiecowy (reagenty, reakcje, zanieczyszczenia)
12. Proces otrzymywania glinu metodą Halla - Heroulta (elektroliza stopionego Al2O3 w kriolicie Na3AlF6)
13. Otrzymywanie wysokiej czystości tytanu w procesie van Arkela
14. Topienie strefowe (na czym polega).
15.* CVD - Chemiczna Depozycja Par
16. CIEKŁE KRYSZTAŁY - pytania z wykładu
Przypuszczam, że pytania będą mniej więcej takie:
a) Podział faz ciekłokrystalicznych (wraz podziałami podrzędnymi)
b) Charakterystyka faz termotropowych
c) Budowa ciekłych kryształów
d) *Cechy decydujące o właściwościach ciekłych kryształów (uporządkowanie, energia swobodna, anizotropia współczynnika załamania, anizotropia elektryczna, tekstury)
UWAGA!! PKT. 16 TO TYLKO MOJE PRZYPUSZCZENIA!
17. *Co to są kompozyty (patrz pkt. 5)
18. Budowa polimerów amorficznych i krystalicznych.
19. *Podział włókien i różnice między nimi (np. między termo- a duroplastem)
20. *Mer - monomer - polimer - makrocząsteczka <- różnice
21. Budowa, podział, cechy, zastosowanie światłowodów.
22. Metoda dwutyglowa produkcji światłowodów.
1. Zasada działania baterii słonecznych
Zasada działania kolektora słonecznego
Zadaniem kolektora słonecznego jest pobieranie energii z promieniowania bezpośredniego, rozproszonego i odbitego, a następnie przekazywanie jej do instalacji grzewczej.:
Słońce ogrzewa umieszczony w kolektorze absorber, który pochłania promieniowanie słoneczne i zamienia je w ciepło. Skuteczność pochłaniania zależy od rodzaju absorbera. Zwykły, czarny absorber dużą część promieniowania odbija. Skuteczniejszy jest tzw. absorber selektywny - pochłania on 95% padającego na niego promieniowania.
Od absorbera ogrzewa się czynnik grzewczy (może to być woda lub płyn niezamarzający), który przepływa przez kolektor.
Ogrzany płyn przepływa do zasobnika.
Tam oddaje ciepło ogrzewanej wodzie użytkowej, znajdującej się w zasobniku, i ochłodzony wpływa z powrotem do kolektora.
Fotoogniwo jest zbudowane z półprzewodnika i tworzy złącze p-n, na które pada światło. Padające na złącze fotony o energii większej od szerokości przerwy energetycznej półprzewodnika powodują powstanie par elektron-dziura. Pole elektryczne wewnątrz półprzewodnika, związane z obecnością złącza p-n, przesuwa nośniki różnych rodzajów w różne strony. Elektrony trafiają do obszaru n, dziury do obszaru p. Rozdzielenie nośników ładunku w złączu powoduje powstanie na nim zewnętrznego napięcia elektrycznego. Ponieważ rozdzielone nośniki są nośnikami nadmiarowymi (mają nieskończony czas życia), a napięcie na złączu p-n jest stałe, oświetlone złącze działa jako ogniwo elektryczne.
2. Efekt fotowoltaiczny
Zjawisko fotowoltaiczne (efekt fotowoltaiczny) - zjawisko polegające na powstaniu siły elektromotorycznej w ciele stałym pod wpływem promieniowania świetlnego. W związku z tym należy do zjawisk fotoelektrycznych wewnętrznych. Zjawisko fotowoltaiczne jako pierwszy zauważył w roku 1839 Aleksander Edmund Becquerel.
Zjawisko to jest wykorzystywane w ogniwach fotowoltaicznych, które coraz częściej zastępują inne rodzaje źródeł energii.
W fotoogniwach wykorzystuje się zjawisko fotoelektryczne wewnętrzne, które polega na uwalnianiu elektronów walencyjnych z wiązań atomowych
w kryształach półprzewodnikowych. Oswobodzone elektrony pozostają wewnątrz kryształu i mogą poruszać się w nim swobodnie. Miejsce po uwolnionym elektronie może zająć elektron z wiązania sąsiedniego. Wtedy brak elektronu w wiązaniu, czyli tzw. dziura przenosi się do wiązania sąsiedniego. Zatem zarówno elektrony jak i dziury mogą przemieszczać się w krysztale, a tym samym przewodzić prąd.
Efektem wewnętrznego zjawiska fotoelektrycznego jest więc zwiększone przewodnictwo elektryczne kryształu. Elektrony walencyjne mają energię zawartą jedynie w pewnym przedziale wartości energii zwanym pasmem walencyjnym. Podobnie elektrony przewodnictwa przyjmują wartości energii tylko z pasma przewodnictwa (leżącego powyżej pasma walencyjnego).
W półprzewodniku pasmo przewodnictwa i pasmo walencyjne oddzielone są wzbronionym dla elektronów przedziałem energii zwanym pasmem wzbronionym lub przerwą energetyczną. Szerokość tej przerwy Eg równa jest energii wiązania elektronów walencyjnych. Wartość Eg określa minimalną częstość vg światła, które zdolne jest przenieść elektron z pasma walencyjnego do pasma przewodnictwa. Warunkiem uwolnienia elektronu jest, aby energia fotonu, Ef = hvf, była większa od Eg.
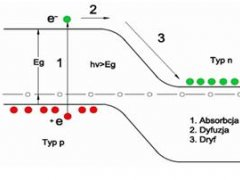
Rysunek. Zjawisko fotowoltaiczne w złączu p-n
Jeżeli elektron przewodnictwa i dziura znajdą się w tym samym miejscu
w półprzewodniku wówczas elektron zajmie wolne miejsce w wiązaniu
i obydwa nośniki prądu zanikają. Proces taki nazywamy rekombinacją. Liczba rekombinacji w jednostce czasu jest zależna od liczby nośników prądu i gdy w miarę upływu czasu oświetlania powierzchni półprzewodnika nośników przybywa, wzrasta również liczba rekombinacji. Po pewnym czasie ustala się równowaga dynamiczna, w której dodatkowa liczba nośników zależy od liczby generowanych par elektron-dziura w jednostce czasu, czyli od natężenia oświetlenia. światło dociera tylko do warstw przypowierzchniowych półprzewodnika i aby zwiększyć wpływ oświetlenia, fotoczuły materiał powinien mieć jak najmniejszą grubość i jak największą powierzchnię. Zjawisko fotoelektryczne wewnętrzne w warstwach podwójnych, z których każda charakteryzuje się innym typem przewodnictwa, przejawia się w postaci efektu fotowoltaicznego. Polega ono na tym, że oświetlone warstwy podwójne, stają się źródłem prądu elektrycznego. światłoczułe warstwy podwójne nazywamy fotoogniwami.
3. Otrzymywanie, właściwości i zastosowanie powłok niklowych
Ciężar atomowy niklu wynosi 58,69, wartościowość 2,3 i 8, ciężar właściwy 8,88 G/cm3 ciepło właściwe 0,110 cal/g, współczynnik rozszerzalności liniowej 13,1 · 10-6, przewodnictwo cieplne 0,208 cal/cm·s°C, temperatura topnienia 1453°C, temperatura wrzenia 2800°C, potęcjał normalny - 0,25V, równoważnik elektrochemiczny k=1,095 G/Ah.
Mikrotwardość galwanicznie osadzonych pokryć niklowych zależy od składu zastosowanej kąpieli oraz stosowanych parametrów pracy i może wachać się od 200÷300 kG/mm2 do 700÷800 kG/mm2. Powłoki niklowe osadzone chemicznie, zawierające fosfor, po obróbce cieplnej osiągają twardość do 900÷1000 kG/mm2 zbliżając się do twardośći błyszczących powłok chromowych.
Po raz pierwszy nikiel został wyodrębniony w 1860 r. przez Adamsa i w kilka lat później zaczęto stosować powłoki niklowe przemysłowe.
Nikiel na powietrzu w normalnej temperaturze prawie się nie zmienia. W kwasie siarkowym i solnym rozpuszcza się bardzo powoli, a w rozcieńczonym kwasie azotowym dosyć intensywnie. Gorące tłuszcze i kwas octowy pozostawiają na powierzchni niklu ciemne plamy. Pod działaniem wysokiej temperatury, powierzchnia pokrycia niklowego pokrywa się elastyczną, twardą warstewką tlenków o odcieniu żółtym lub fioletowym w zależności od temperatury i czasu ogrzewania.
Powlekanie galwanicznie niklem jest obecnie jednym z najbardziej popularnych i rozpowszechnionych procesów elektrolitycznego powlekania metali. Do niklowania zużywa się prawie 10% światowego wydobycia tego metalu. Tak szerokie zastosowanie niklu w galwanotechnice można tłumaczyć przede wszystkim fizykochemicznymi właściwościami powłok osadzonych metodą elektrolityczną. Poza tym wpłynęły na to takie czynniki, jak uzyskiwany ładny biały kolor powłoki, odpowiednia twardość, a oprócz tego, jak już wspomniałem, wysoka odporność na wpływy atmosferyczne przewyższające w pewnych przypadkach odporność takiego metalu jak srebro. Na przykład w atmosferze zawierającej siarkowodór nikiel i jego powłoki galwaniczne prawie nie zmieniają się lub po bardzo długim czasie nieco ciemnieją, w przeciwieństwie do srebra lub jego powłoki, które w bardzo krótkim czasie czernieją. Powłoki niklowe służą obecnie nie tylko jako powłoki dekoracyjne, w mniejszym stopniu ochronne przed rdzewieniem, lecz również jako powłoki utwardzające stosunkowo miękki podkład czcionek i płyt drukarskich. Znalazły one zastosowanie w różnych przemysłach takich, jak samochodowy, sanitarno-chirurgiczny, poligraficzny, wyrobów użytku domowego oraz galanteryjno-dekoracyjny. Tzw. czarny nikiel jest stosowany w przemyśle optycznym i precyzyjnym oraz aparatury naukowej.
Galwaniczne elektrolity niklowe są bardzo wrażliwe na kwasowość; nieprzestrzeganie odpowiedniej kwasowości powoduje złą przyczepność powłoki do podłoża, rozwarstwianie się powłok i często jej odpryskiwanie. Nie mniejsze znaczenie przy niklowaniu odgrywa także temperatura kąpieli niklowej; zbyt wysoka temperatura powoduje polaryzacje metali grupy żelaza, a ponadto wpływa ujemnie na strukturę i równomierność powłoki niklowej. Natężenie prądu podczas niklowania, ma tak samo duży wpływ nie tylko na strukturę powłoki niklowej, ale i na jej własności mechaniczne. Jak widać warunki kąpieli podczas niklowania odgrywają dużą rolę; trzeba więc zawsze pamiętać, że powodzenie przeprowadzanego procesu zależy w dużym stopniu od utrzymania właściwej kwasowości kąpieli(pH), temperatury oraz gęstości prądu. Należy jeszcze zaznaczyć, że dostatecznie dobrze i trwale można poniklować miedź i jej stopy oraz żelazo i stal, a cynk i glin tylko w specjalnych kąpielach - bezpośrednio. Cynk i glin oraz bardzo często wyroby żelazne nikluje się po uprzednim pomiedziowaniu (w kąpielach cjankowych) w celu otrzymania dobrej powłoki nie tylko z powodu dekoracyjnego, ale głównie ochronnego (przed korozją).
W układzie stal-nikiel, nikiel jest bardziej elektrododatni i jest pokryciem katodowym nie mogącym ochraniać elektrochemicznie stali przed korozją. Odpowiednią odporność korozyjną można zapewnić na drodze szczelności pokrycia oraz odpowiedniej jego grubości. Powłoki galwaniczne obdarzone są zawsze pewną porowatością, a dla otrzymania powłok nieporowatych stosuje się wielowarstwowe powłoki. Istnieje bardzo małe prawdopodobieństwo, żeby w wielowarstwowych powłokach pory powtarzały się w tych samych miejscach. Oprócz tego wielowarstwowe powłoki znacznie obniżają ich cenę, ponieważ miedź stosowana najczęściej jako podwarstwa pod nikiel, jest wielokrotnie tańsza od niklu. Grubość stosowanej podwarstwy miedzi najczęściej wynosi połowę całkowitej grubości powłoki dekoracyjno-ochronnej.
4. Klasyfikacja polimerów + oznaczenia (skróty)
Klasyfikacje nie są jednoznaczne. Carothers(1929) wprowadził podział względem budowy:
Polimery
Kondensacyjne addycyjne
Inny podział, np. wg Flory'ego(1953) uwzględnia mechanizm polimeryzacji
Polimery otrzymywane w reakcji
Stopniowej łańcuchowej
Polimery kondensacyjne
Otrzymywane z są z wielofunkcyjnych monomerów w reakcjach kondensacji znanych z chemii organicznej. Powstaje przy tym małocząsteczkowy produkt uboczny, zwykle woda.
*Polimery, w których budowa jednostki konstytucyjnej (meru) różni się od budowy monomeru tym fragmentem, który tworzy produkt uboczny (Carothers, 1929).
*A zatem celuloza, skrobia (polisacharydy), wełna, jedwab (poliaminokwasy) są polimerami kondensacyjnymi
Polimery addycyjne
*Wg Carothers'a-polimery otrzymywane z monomerów bez utraty przez nie małej cząsteczki.
*Główna grupa to polimery otrzymane z monomerów nienasyconych
*Do tej grupy polimerów należą także polimery otrzymywane w tzw. polimeryzacji z otwarciem pierścienia
*Także w tym przypadku skład chemiczny jednostki monomerycznejjest taki sam, jak skład monomeru
Polimery kondensacyjne vs. addycyjne
*Polimery kondensacyjnemają budowę typu:
-R-Z-R-Z-R-Z-R-Z-R-
R -ugrupowanie alifatyczne lub aromatyczne,
Z -charakterystyczna grupa funkcyjna: -OC(O)-; -NHC(O)-; -OC(O)NH-;
-SO2-; -O-; itp.
*Polimery addycyjnezbudowane są zwykle z makrocząsteczek, posiadających wyłącznie atomy węgla w łańcuchu głównym, a ewentualne grupy funkcyjne stanowią podstawniki „boczne”.
*Przykładem niedoskonałości takiej klasyfikacji są żywice fenolowo‐formaldehydowe ,
a także produkt utleniającej dehydrogenacji p‐ksylenu
Klasyfikacja wg mechanizmu polimeryzacji
Polimery otrzymywane w reakcji polimeryzacji
stopniowej (step) łańcuchowej (chain)
Klasyfikacja została wprowadzona przez Flory'ego(1953). Być może lepszym tłumaczeniem angielskiego terminu step polymerizationjest polimeryzacja krokowa, ale literatura polska stosuje określenie stopniowa.
Podstawowe różnice to:
*sposób, w jaki reagują ze sobą cząsteczki,
*zależność średniej wielkości cząsteczek od stopnia przereagowania.
Polimeryzacja stopniowa
*Reagują grupy funkcyjne w monomerach, dimerach, trimerachitd. Schemat reakcji:
monomer + monomer->dimer
dimer + monomer ->trimer
dimer + dimer->tetramer
dimer + trimer->pentamer
trimer+ monomer ->tetramer
trimer+ dimer ->pentamer
trimer+ trimer->heksamer
itd.
*W przeważającej części przypadków otrzymujemy polimery należące do grupy polimerów kondensacyjnych.
Polimeryzacja łańcuchowa
*Do zapoczątkowania reakcji polimeryzacji potrzebny jest inicjator (układ inicjujący) lub katalizator.
*Inicjator, w stanie `wzbudzonym', reaguje z monomerem, tworząc centrum aktywne i zapoczątkowując reakcję polimeryzacji. Jest to proces inicjowania polimeryzacji.
*Kolejne cząsteczki monomeru przyłączają się do centrum aktywnego. Ten etap nosi nazwę wzrostu lub propagacji.
*Propagacja następuje aż do momentu terminacji, w wyniku której centrum aktywne ulega dezaktywacji
*Centrum aktywne stanowić może wolny rodnik, anion lub kation.
*Udział cząsteczek aktywnych jest zwykle mały -inicjator `wytwarza' coraz to nowe centra, które jednak ulegają dezaktywacji w reakcjach terminacji.
*Obok cząsteczek polimeru, które, bądź właśnie rosną, bądź zakończyły swój wzrost, w układzie pozostaje nieprzereagowany monomer i inicjator. W miarę ubywania monomeru ta sytuacja nie zmienia się zasadniczo.
*W wielu typach polimeryzacji łańcuchowej nie przebiega germinacja lub przebiega ona bardzo powoli. Jeżeli szybkość inicjowania jest duża, powstają centra, które 'konkurują' o cząsteczki monomeru. Powstaje tyle makrocząsteczek ile jest cząsteczek inicjatora. Przykładami takich reakcji są reakcje polimeryzacji z otwarciem pierścienia. Przykładami są polimeryzacja tlenku propylenu lub ε-kaprolaktamu
skróty dotyczące polimerów typu pp pe pet itp
poli(tereftalanetylenu)
polietylen dużej gęstości
poli(chlorek winylu)
polietylen małej gęstości
polipropylen
polistyren
5. Kompozyty - charakterystyka (+budowa) i podział
Kompozyt jest to materiał utworzony z co najmniej dwóch komponentów mający właściwości nowe (lepsze) w stosunku do komponentów.
Z czego składa się kompozyt
Kompozyt składa się z osnowy i umieszczonego w niej drugiego składnika (zbrojenia) o znacznie lepszych właściwościach mechanicznych
Osnowa pełni następujące funkcje:
*utrzymuje razem zbrojenie
*zapewnia wytrzymałość na ściskanie
*przenosi naprężenie zewnętrzne na zbrojenie,
*zatrzymuje rozprzestrzenianie się pęknięć,
*nadaje wyrobom żądany kształt.
najczęściej polimer (poliepoksyd, poliester)
*może być to metal (Ti, Ni, Fe, Al, Cu) lub ich stopy
*bądź ceramika (np.Al2O3,SiO2, SiC, TiO2)
Zbrojenie
Zadaniem zbrojenia jest wzmacnianie materiału, poprawianie jego właściwości mechanicznych.
Zbrojenie może mieć postać:
*Włókna ciągłego lub nieciągłego;
*Proszku
Podział kompozytów
Ze względu na wielką różnorodność materiałów kompozytowych dzieli się je ze względu na:
-właściwości
-rodzaj osowy
-rodzaj zbrojenia
Kompozyty
*cząsteczkowe
- duże cząsteczki
- z wydzieleniami
*wzmacniane włóknami
- ciągłe
- nieciągłe (uporządkowane i przypadkowe)
*strukturalne
- laminaty
- warstwowe
7. Biomateriały - charakterystyka; bioceramiczne i węglowe
Biomateriał (zwany też materiałem biomedycznym) - materiał, z którego można produkować urządzenia i elementy, mające bezpośredni kontakt z tkankami organizmu.
Z biomateriałów produkuje się implanty (np. protezy ortopedyczne, naczyniowe), a także pokrywa się nimi powierzchnie urządzeń wszczepianych do wnętrza organizmu (np. rozrusznik serca, sztuczne zastawki serca, elektrody endokawitarne, stenty), lub przeznaczonych do długotrwałego kontaktu z organizmem (np. rurki intubacyjne, cewniki, dreny, nici chirurgiczne).
Podstawową cechą biomateriałów jest ich biozgodność, czyli brak toksyczności oraz minimalne oddziaływanie na system immunologiczny. Biomateriały będące w styczności z krwią nie mogą wywoływać hemolizy.
Do najczęściej stosowanych biomateriałów zalicza się:
Jakie cechy powinien mieć biomateriał?
- Musi być biokompatybilny (nie może wywoływać reakcji obronnej tkanek);
- Może być przy tym neutralny dla organizmu (nie oddziałuje);
- Może być bioaktywny (oddziałuje z tkankami: następuje integracja materiału z tkanką).
- Może być biodegradowalny (rozkładać się w organizmie);
- Musi mieć odpowiednie właściwości. Np. implant kości nie może być ani słabszy, ani silniejszy niż kość.
Główne bio-ceramiki i szkło to fosforan wapnia (Ca3(PO4)2 - TCP), hydroksyapatyt (Ca10(PO4)6 x (OH)2 - HA) i szkła krzemianowe zawierające Ca i P. Wszystkie mają skład podobny do mineralnych składników kości. Na ich powierzchni, w organizmie powstaje
warstwa wiążąca się z tkanką. Mogą być ponadto źródłem jonów (Si) aktywujących różnicowanie się komórek kości.
8. Biodegradacja - charakterystyka
Biodegradacja (z greckiego "bios" - życie i łaciny "degradatio" - obniżenie), biochemiczny rozkład związków organicznych przez organizmy żywe (pierwotniaki, bakterie, promieniowce, grzyby, glony, robaki) na prostsze składniki chem.
Termin biodegradacja, w odróżnieniu od terminu mineralizacja, używany jest na ogół w odniesieniu do substancji szkodliwych, np. pestycydów. Rozkładowi ulegać może nawet 95% substancji organicznej. Biodegradację wykorzystuje się w biologicznych oczyszczalniach ścieków oraz w stawach biologicznych (służących do fermentacyjnego oczyszczania ścieków np. z cukrowni). Konieczna jest do tego odpowiednia temperatura oraz brak w ściekach substancji toksycznych dla mikroorganizmów (np. detergentów czy pestycydów).
Biodegradacja ma zastosowanie przy produkcji biogazu z odpadów i ścieków, biomasy paszowej ze ścieków, a także pestycydów w opakowaniach podatnych na biodegradację, rozpuszczalnych w wodzie (np. Tilt Premium 37,5 WP). Dużą biodegradacją charakteryzują się gleby biologicznie aktywne, zasobne w próchnicę.
9. Recykling - charakterystyka i rodzaje.
Recykling - w rozumieniu ustawy z dnia 27 kwietnia 2001 roku O odpadach to taki odzysk, który polega na powtórnym przetwarzaniu substancji lub materiałów zawartych w odpadach w procesie produkcyjnym w celu uzyskania substancji lub materiału o przeznaczeniu pierwotnym lub o innym przeznaczeniu, w tym też recykling organiczny, z wyjątkiem odzysku energii.
Funkcjonuje wiele różnych pojęć recyklingu. Oto niektóre z nich:
Recykling chemiczny - obejmuje procesy, w których zużyte materiały odpadowe, przetwarzane są do materiałów o innych właściwościach fizyko - chemicznych, np.:
wytwarzanie materiałów termoizolacyjnych ze stłuczki szklanej,
wytwarzanie olejów opałowych z tworzyw sztucznych,
wytwarzanie materiału termoizolacyjnego z makulatury,
wytwarzanie gazu energetycznego ze zużytych opon, itd.
Odzysk to szerokie pojęcie, które zgodnie z ustawą z dnia 27 kwietnia 2001 roku O odpadach obejmuje wszelkie działania nie stwarzające zagrożenia dla życia, zdrowia ludzi lub dla środowiska, polegające na wykorzystaniu odpadów w całości lub w części, lub prowadzące do odzysku z odpadów substancji, materiałów lub energii i ich wykorzystania.
Odzysk obejmuje również recykling energetyczny, zwany też odzyskiem energii. Jest to proces, w którym odzyskuje się w części energię zużytą na wytworzenie wyrobów i towarów, usuniętych po zużyciu na wysypisko, w tym także odpadów opakowaniowych.
Recykling energetyczny obejmuje nie tylko spalanie odpadów, lecz także wytwarzanie z odpadów paliw stałych, ciekłych i gazowych oraz przetwarzanie ich na materiały termoizolacyjne - czyli recykling chemiczny.
Przykładem może być makulatura, która w różny sposób uczestniczy w obiegu materiałowo-energetycznym. Spalanie makulatury - to recykling energetyczny. Z kolei materiał termoizolacyjny wytworzony z makulatury (patrz recykling chemiczny) można wykorzystać do ocieplania budynków i zaoszczędzić w związku z tym znaczne ilości energii - to również jest recykling energetyczny. Podobnie można zdefiniować energetyczny recykling stłuczki szklanej.
Recykling energetyczny można prowadzić dla każdej grupy materiałowej odpadów osobno. Wymaga on wtedy selektywnej zbiórki, segregacji i selekcji odpadów opakowaniowych. W zależności od rodzaju odpadów i użytej technologii można otrzymać paliwo stałe, ciekłe lub gazowe.
Współpraca Zakładów Małej Energetyki wytwarzających "energię środowiskową" w procesie recyklingu energetycznego odpadów (ERO) z lokalnymi zakładami energetycznymi, powinna być poszerzona o współpracę z lokalnymi źródłami surowców, jakimi są systemy selektywnej zbiórki w gminach.
Recykling surowcowy jest procesem przetwarzania materiałów i wyrobów odpadowych do postaci surowców, z których te materiały i wyroby zostały wytworzone.
Proces ten obejmuje wysokotemperaturowy rozkład organicznych materiałów odpadowych na proste związki chemiczne. Przykładem takiego recyklingu może być proces rozkładu tworzyw sztucznych do postaci gazu złożonego z tlenku węgla CO i wodoru H2, który jest z kolei surowcem wykorzystywanym do produkcji pierwotnych tworzyw sztucznych.
Istnieje również recykling organiczny, który rozumie się przez obróbkę tlenową, w tym kompostowanie, lub beztlenową odpadów, które ulegają rozkładowi biologicznemu w kontrolowanych warunkach przy wykorzystaniu mikroorganizmów, w wyniku której powstaje materia organiczna lub metan.
11. Proces wielkopiecowy (reagenty, reakcje, zanieczyszczenia)
Żelazo to metal ciężki, który jest niezwykle rozpowszechniony na ziemi. Ze względu na jego właściwości fizyczne, które można zmieniać w bardzo szerokich granicach poprzez małe dodatki innych pierwiastków, jest on często i chętnie wykorzystywany przez człowieka. Niestety, w stanie wolnym żelazo nie występuję w przyrodzie. Na szczęście, licznie można go spotkać w stanie związanym, w postaci rud. Do najważniejszych rod żelaza zaliczamy:
- magnetyt, inaczej nazywany żelaziakiem magnetycznym (Fe3O4)
Ruda ta jest niezwykle bogata w żelazo, zawiera go niemal 50 do 67 %, ponadto wykazuje właściwości magnetyczne.
- hematyt , inaczej nazywany żelaziakiem czerwonym (Fe2O3)
Hematyt zawiera mniej żelaza, niż magnetyt, ale także dużo od 30 do 64 %.
- limonit, inaczej nazywany żelaziakiem brunatnym (Fe2O3 ∙ n H2O)
Ruda ta złożona jest z uwodnionego tlenku metalu, w zależności od ilości wody, może zawierać nawet 40 % żelaza.
- syderyt (FeCO3)
Syderyt należy do bardziej ubogich w żelazo rud, zawiera go ok. 25 %.
- piryt (FeS2)
Do krajów posiadających duże zasoby tych minerałów zależą: Rosja, USA oraz Szwecja.
Polskę zalicza się do krajów ubogich w te rudy. Posiadamy jedynie rudy syderytu i limonitu.
Z powyżej wymienionych rud, na skale przemysłową otrzymuje się stopy żelaza, czyli stopione mieszaniny żelaza z innymi pierwiastkami. Odbywa się to najczęściej na drodze procesu wielkopiecowego.
Aby otrzymać żelazo z rud należy go zredukować. Najczęściej używanymi reduktorami są węgiel oraz tlenek węgla (II). Substancje te wyjątkowo łatwo reagują z tlenkami żelaza, zwłaszcza w wysokiej temperaturze.
Cały proces przeprowadza się w wielkich, szybowych piecach, o wysokości ok. 30 metrów i pojemności nawet 2000 [m3]. Zasada działania pieca oparta jest o zjawisko przeciwprądów. Od góry, warstwami, do pieca wprowadza się za pomocą urządzeń zasypowych surowce. Surowcami są tutaj:
- ruda żelaza
- koks, czyli produkt odgazowania węgli kopalnych, najczęściej węgla kamiennego, zawiera ok. 80 % czystego węgla
- topniki, czyli substancje, które obniżają temperaturę topnienia rud oraz ułatwiającą oddzielenie metalu od innych domieszek, zawartych w rudzie. Te bezużyteczne domieszki zostają związane w postaci żużla. Najczęściej używanymi topnikami są piasek, wapień lub dolomit.
Natomiast od dołu pieca wdmuchuje się rozgrzane powietrze.
W górnej części pieca zachodzą reakcje utleniania węgla, które prowadzą do powstania tlenku węgla (II):
C + O2 → CO2
CO2 + C → 2 CO
Tlenek ten łatwo reaguje ze stopioną, w wysokiej temperaturze, pod wpływem topników rudą żelaza.
Zachodzą następujące reakcje:
3 Fe2O3 + CO → 2 Fe3O4 + CO2
Fe3O4 + CO → 3 FeO + CO2
FeO + CO → Fe + CO2
Jest to tzw. redukcja częściowa, redukcja właściwa, czyli redukcja węglem zachodzi w dolnej strefie wielkiego pieca, w której panuje najwyższa temperatura, dochodząca do ok. 1200 - 1500 ºC.
FeO + C → Fe + CO
Jednocześnie z reakcja pomiędzy węglem , a tlenkami żelaza, odbywa się reakcja z topnikami oraz innymi zanieczyszczeniami obecnymi w mieszaninie. W efekcie powstaje ciekły żużel, czyli produkt uboczny zawierający stopione substancje mineralne. Żużel jest lżejszy do surówki, więc oddziela się od niej i wypływa na jej powierzchnię.
Surówka, czyli pierwszy produkt procesu wielkopiecowego zawiera ok. 4 % węgla oraz niewielkie ilości krzemu, manganu, fosforu oraz siarki. Pod wpływem powolnego oziębiania surówki część węgla wykrystalizowuje się w postaci grafitu.
Powstające w dużych ilościach gazy: tlenki węgla, są ponownie wykorzystywane do ogrzewania pieca. Proces wielkopiecowy jest przeprowadzany nieprzerwanie.
12. Proces otrzymywania glinu metodą Halla - Heroulta (elektroliza stopionego Al2O3 w kriolicie Na3AlF6)
Proces Halla-Héroulta - podstawowy proces używany do elektrolitycznego otrzymywania aluminium. Opracowany w 1886 roku niezależnie przez C. M. Halla (1863-1914) i P. L. Héroulta (1863-1914).
W procesie tym tlenek aluminium (Al2O3), uprzednio otrzymany z boksytu w procesie Bayera, jest rozkładany elektrolitycznie na metaliczny glin oraz gazowy tlen. Proces ten zachodzący w sposób ciągły (w wypadku konieczności przerwania procesu, ciekły metal ulega zakrzepnięciu w wannie elektrolitycznej, co powoduje konieczność kosztownej odbudowy aparatury), rozpoczyna się od etapu rozpuszczenia tlenku aluminium w kriolicie wypełniającym wannę elektrolityczną. Uzyskiwany elektrolit wykazuje wysoką rezystancję powodującą wydzielenie dużych ilości ciepła podczas przepływu prądu, dzięki czemu elektrolit może być utrzymywany w stanie ciekłym bez dodatkowego ogrzewania.
Temperatura utrzymywana jest na poziomie 920-980 °C. Aluminium powstające w procesie elektrolizy jest oddzielane od elektrolitu oraz sukcesywnie usuwane z komory elektrolizera. Elektrolizery połączone są na ogół w baterie połączone szeregowo (prąd płynie w kierunku od węglowej anody poprzez roztwór tlenku aluminium w kriolicie do węglowej katody).
W trakcie elektrolizy, aluminium, którego gęstość jest minimalnie większa od elektrolitu, osiada na dnie wanny elektrolitycznej. Wydzielający się tlen reaguje z grafitową okładziną anody tworząc dwutlenek węgla (CO2). W wyniku tego procesu następuje ubytek masy anody, która musi być w regularnych odstępach czasu wymieniana. Energiczny proces wydzielania się dwutlenku węgla na anodzie pozwala na lepsze mieszanie się tlenku aluminium (Al2O3) w elektrolicie, jednocześnie jednak dwutlenek węgla jest nośnikiem dla szkodliwych lotnych substancji ubocznych (np. fluorowodoru (HF), dwutlenku siarki (SO2), fluoropochodnych węglowodorów (PFC, np. CF4 i C2F6), smoły i wielopierścieniowych węglowodorów aromatycznych (WWA) itd.), a czasami stałych (np. pył).
W trakcie postępu procesu elektrolizy stężenie tlenku aluminium w elektrolicie spada i jest on uzupełniany przez podajnik tak, aby zachować stężenie na poziomie 2-5%. Jeśli stężenie tlenku aluminium spadnie do około 1,5 - 2%, mogą zacząć występować niekorzystne zjawiska na anodzie. Elektrolit nie zwilża powierzchni anody, która otoczona jest poprzez film utworzony z gazu. Warstwa gazu zwiększa rezystancję, a to powoduje z kolei (zgodnie z prawem Ohma) zwiększenie napięcia panującego na elektrodach o około 10-15 razy powyżej normalnego poziomu 4-5 V.
Elektrolit
Roztopiony elektrolit składa się głównie z kriolitu (fluoroglinianu sodu, Na3AlF6) z dodatkiem fluorku glinu (AlF3), 6-10% wagowych fluorytu (CaF2) oraz 2-5% tlenku glinu (Al2O3). Właściwa kontrola składu elektrolitu ma istotne znaczenie dla procesu elektrolizy. W celu obniżenia temperatury topnienia (czysty kriolit topi się w 1009 °C) stosuje się topniki (fluorek wapnia lub glinu), które powodują obniżenie temperatury do poziomu 920-980 °C. Zmiana temperatury zwiększa sprawność procesu elektrolizy.
NaAlO2 + 6HF + Na2CO3 → Na3AlF6 + 3H2O + CO2
Elektrody
Reakcje elektrodowe:
Katoda
Węglowe okładziny, którymi wyłożona jest wanna elektrolityczna, są wstępnie spiekane w odrębnym procesie produkcyjnym. Okładziny te są umieszczane wewnątrz metalowej obudowy wanny. Szczeliny między płytami wypełnia się uszczelniającą pastą grafitową. Izolacja termiczna wykonana z materiałów ogniotrwałych wypełnia przestrzeń między węglową okładziną a stalową obudową wanny. Do katody przymocowane są grube (natężenia prądu w granicach 180-350 kA) stalowe pręty będącymi kolektorami dostarczającymi prąd, oraz służące łączeniu wanien w baterie. Okładzina węglowa tworząca katodę wystarcza w normalnej eksploatacji na okres od 4 do 6 lat. Czasami jednak może wydarzyć się penetracja płynnego aluminium do warstwy metalowych kolektorów, które ulegają roztwarzaniu. Nagłe zwiększenie zawartości żelaza w aluminium świadczy o konieczności wykonania naprawy (wymiany okładziny) .
Anoda
W procesie Halla-Héroulta główną rolę odgrywają anody węglowe. Do wyprodukowania każdej tony aluminium zużywane jest ok pół tony węgla. Wykorzystywane są głównie dwa typy anod: wstępnie spieczona oraz Söderberga; obydwa typy są wykonane z tego samego materiału oraz podlegają identycznym reakcjom zachodzącym w procesie elektrolizy. Mieszanka koksu oraz smoły po uformowaniu ogrzewane są do wysokiej temperatury powodując spiekanie cząstek węgla. Anoda „wstępnie spieczona” jest spiekana w zewnętrznym piecu i w gotowym stanie dodawana jest do wanny elektrolitycznej. Anoda Söderberga formowana jest z pasty grafitowej bezpośrednio w wannie elektrolitycznej, tam też ulega procesowi spieczenia.
14. Topienie strefowe (na czym polega)
Topienie strefowe, metoda otrzymywania czystych materiałów ze stopów. Stop poddawany topieniu strefowemu odlany jest w kształcie pręta, wzdłuż niego przesuwa się pierścieniowy grzejnik, powodując lokalne topienie stopu, a następnie jego krzepnięcie.
Kryształy wydzielające się w strefie krzepnięcia wykazują wyższą czystość aniżeli wyjściowy stop, tymczasem zanieczyszczenia przesuwają się wraz ze strefą topioną do jednego z końców pręta. Wielokrotne powtarzanie tej operacji, z zachowaniem kierunku przesuwania grzejnika pozwala na uzyskanie materiału o wysokiej czystości.
Topienie strefowe jest stosowane do otrzymywania materiałów półprzewodnikowych (półprzewodniki).
16. CIEKŁE KRYSZTAŁY
a) Podział faz ciekłokrystalicznych (wraz podziałami podrzędnymi)
Liotropowe i termotropowe
Faza ciekłokrystaliczna może być generowana na dwa sposoby:
poprzez ogrzewanie stałych kryształów - jest nazywana wtedy mezofazą termotropową. Kryształy, zamiast od razu topić się w zwykłą ciecz, przechodzą w pewnej określonej temperaturze w stan mezofazy, a dopiero w wyższej temperaturze następuje izotropizacja mezofazy, czyli zamiana ciekłych kryształów w ciecz izotropową. W zależności od rodzaju ciekłego kryształu może on przechodzić przez różne mezofazy, w miarę obniżania temperatury.
poprzez rozpuszczanie cząsteczek mających tendencję do tworzenia mezofazy w odpowiednim rozpuszczalniku - faza jest nazywana wtedy mezofazą liotropową. W układzie takim rozpuszczone pręto- lub dyskopodobne cząsteczki tworzą mezofazę "zmuszając" niejako cząsteczki rozpuszczalnika do uczestniczenia w tej fazie.
Fazy termotropowe
Mezofazy termotropowe dzieli się na trzy główne klasy:
fazę cholesterolową, w której uporządkowanie jest związane ze skręceniem o określony kąt przy przechodzeniu między warstwami, czyli ich direktor nie jest linią prostą lecz układa się w kształt helisy. Faza cholesterolowa jest w istocie podtypem fazy nematycznej. Fazę cholesterolową nazywa się też często fazą nematyczną skręconą i oznacza symbolem N*
Fazy smektyczne
Fazy smektyczne podzielić można dalej na dwa sposoby. Względem stopnia uporządkowania cząstek:
smektyki cieczopodobne - brak uporządkowania molekuł w warstwach (poza uporządkowaniem środków ciężkości) np.: SmA, SmC
smektyki kryształopodobne - wykazujące pozycyjne ułożenie molekuł w warstwie np.: SmB (ułożenie heksagonalne), SmH, SmG, SmE.
Podział ten jednak jest nieco sztuczny, gdyż w istocie stopień uporządkowania molekuł w fazach smektycznych (których jest łącznie kilkadziesiąt) zmienia się płynnie od układów bardziej cieczopodobnych do bardziej kryształopodobnych.
Drugi podział, wynika z orientacji warstw smektycznych, względem direktora:
smektyki ortogonalne: direktor jest prostopadły do powierzchni warstw (SmA, SmB, SmE).
smektyki pochylone (nieortogonalne) direktor przecina powierzchnie warstw pod kątem innym niż 90° (SmC, SmG, SmH). Nazwa pochodzi od tego, że w fazach tych, prętopodobne cząsteczki są "pochylone" w warstwach, w stosunku do płaszczyzny warstw.
Fazy kolumnowe
Fazy kolumnowe tworzą się poprzez układanie się cząsteczek o kształcie dysków w długie kolumny, przypominające trochę stosy monet. Kolumny te mogą być dodatkowo zorganizowane w rodzaj siatki, której rodzaj jest podstawą podziału faz kolumnowych:
faza kolumnowa rombowa (Drh), w której komórka elementarna siatki kolumn przybiera kształt rombu,
faza kolumnowa kwadratowa (Dsq), najrzadziej spotykana, w której komórka elementarna przybiera kształt kwadratu.
Oprócz tego cząsteczki dyskopodobne mogą też generować mezofazę nematyczną i niektóre mezofazy smektyczne.
Fazy skręcone (chiralne)
Smektyki pochylone mogą dodatkowo posiadać, podobnie jak to jest w przypadku fazy nematycznej, swoje wersje "skręcone", w których direktor nie jest linią prostą lecz helisą. Analogicznie do fazy nematycznej skręconej (cholesterolowej) - fazy smektyczne skręcone oznacza się symbolem wyjściowej fazy z dodaną gwiazdką (SmC*, SmH* itd). Smektyki ortogonalne z natury rzeczy nie posiadają swoich odpowiedników skręconych, gdyż łatwo można dowieść, że jest to niemożliwe geometrycznie. Z faz kolumnowych jedynie faza rombowa może występować w formie skręconej.
Fazy skręcone tworzą cząsteczki chiralne, zaś fazy nie skręcone tworzą cząsteczki niechiralne. Podobnie jak wszystkie cząsteczki chiralne, również chiralne fazy ciekłokrystaliczne skręcają światło spolaryzowane, przy czym kąt skręcenia jest tutaj dużo bardziej zależny od temperatury niż w przypadku związków nie generujących mezofaz.
Fazy liotropowe
Fazy liotropowe, są specyficznym rodzajem emulsji, w której cząsteczki rozpuszczalnika koordynują się względem wstępnie uporządkowanych cząsteczek mezogenu, albo na odwrót, siłą napędową tworzenia się fazy ciekłokrystalicznej jest wymuszanie przez rozpuszczalnik określonego uporządkowania rozpuszczonych w nim mezogenów. Przyjmuje się, że przy wyższych stężeniach mezogenów zachodzi pierwsze zjawisko, a przy niższych drugie. Fazy tworzące się w drugim przypadku opatruje się często mianem faz "odwróconych".
Przejścia od jednych faz do drugich można tu generować zarówno przez zmianę temperatury całego układu jak i poprzez zmiany stężenia związku generującego.
Cząsteczki zdolne do tworzenia faz liotropowych mają zazwyczaj własności amfifilowe tzn. posiadają długie łańcuchy, które na jednym końcu są hydrofilowe (a ogólniej liofilowe) a na drugim hydrofobowe (a ogólniej liofobowe). Powoduje to, że w roztworze cząsteczki takie mają skłonność do mikroseperacji i tworzenia złożonych, uporządkowanych układów micelarnych, która jest główną siłą napędową tworzenia się tych faz. Istnieją jednak też przypadki gdy fazy liotropowe są tworzone przez związki nie posiadające własności amfifilowych.
Ze względu na to, że tworzenie się faz liotropowych zależne jest od bardzo subtelnych oddziaływań międzycząsteczkowych, które niezwykle łatwo zaburzyć, badanie własności tych faz jest znacznie trudniejsze od faz termotropowych. Na przykład nawet delikatne wstrząśnięcie naczyniem z roztworem lub zmiana temperatury o 1 °C potrafi zaburzyć strukturę fazy liotropowej, a po ponownym osiągnięciu równowagi układ może przejść do innej fazy.
Fazy liotropowe występują powszechnie w układach biologicznych. Na przykład błona komórkowa jest w gruncie rzeczy bardzo cienką, lamelarną fazą liotropową, tworzoną przez lipidy, która powstaje w naturalny sposób na granicy cytoplazmy z otoczeniem.
b) Charakterystyka faz termotropowych
Faza ciekłokrystaliczna może być generowana na dwa sposoby:
poprzez ogrzewanie stałych kryształów - jest nazywana wtedy mezofazą termotropową. Kryształy, zamiast od razu topić się w zwykłą ciecz, przechodzą w pewnej określonej temperaturze w stan mezofazy, a dopiero w wyższej temperaturze następuje izotropizacja mezofazy, czyli zamiana ciekłych kryształów w ciecz izotropową. W zależności od rodzaju ciekłego kryształu może on przechodzić przez różne mezofazy, w miarę obniżania temperatury.
Fazy termotropowe
Mezofazy termotropowe dzieli się na trzy główne klasy:
fazę cholesterolową, w której uporządkowanie jest związane ze skręceniem o określony kąt przy przechodzeniu między warstwami, czyli ich direktor nie jest linią prostą lecz układa się w kształt helisy. Faza cholesterolowa jest w istocie podtypem fazy nematycznej. Fazę cholesterolową nazywa się też często fazą nematyczną skręconą i oznacza symbolem N*
c) Budowa ciekłych kryształów
Nazwą ciekłe kryształy określa się substancje (zazwyczaj organiczne) znajdujące się w tzw. stanie ciekłokrystalicznym. Stan ten wyróżnia unikalne wprost połączenie typowej cechy cieczy, jaką jest płynność, ze swego rodzaju uporządkowaniem dalekiego zasięgu — typowym dla struktur krystalicznych, pociągającym za sobą anizotropię niektórych właściwości fizycznych (dielektrycznych, optycznych i innych). Stan ciekłokrystaliczny występuje w charakterystycznym dla danej substancji zakresie temperatur (typowy jest przedział — 5... + 55°C), gdy temperatura jest niższa, substancja zestala się, przechodząc w stan krystaliczny, gdy temperatura zaś jest wyższa, przechodzi w stan ciekły (normalny), czyli staje się cieczą (bezpostaciową, izotropową). Cząsteczki substancji ciekłokrystalicznych mają wydłużony kształt, ich rozmieszczenie przestrzenne, decydujące o właściwościach fizycznych, jest kryterium podziału ciekłych kryształów na trzy zasadnicze typy: nematyczny, smektyczny, cholesteryczny.
Najważniejsze rodzaje struktury ciekłych kryształów: a) nematyczna, b) smektyczna; c) cholesteryczna
Ciekłe kryształy najczęściej stosuje się w postaci cienkiej warstwy umieszczonej między dwiema płaszczyznami (elektrodami). Oddziaływania powierzchniowe między cząsteczkami ciekłego kryształu a materiałem elektrod powodują powstanie określonej tekstury molekularnej w warstwie ciekłokrystalicznej. Spośród wielu znanych tekstur molekularnych ciekłych kryształów najważniejsze są: tekstura planarna, charakteryzująca się równoległym ułożeniem cząstek ciekłego kryształu w stosunku do płaszczyzny elektrod i tekstura homeotropowa o prostopadłym układzie cząsteczek. Bardzo ważną odmianą tekstury planarnej jest konfiguracja TN (ang. twisted nematics) tzw. skręconego nematyka, która charakteryzuje się skręceniem o kąt 90° lub 45° osi cząsteczek równolegle ułożonych przy obu powierzchniach (rys. c). Wewnątrz warstwy uzyskuje się ciągłą deformację śrubową ułożenia cząsteczek, nadającą jej zdolność skręcania płaszczyzny polaryzacji światła odpowiednio o 90° lub 45°.
Podstawowe typy tekstury warstwy ciekłokrystalicznej: a) planarna; b) homeopolarna; c) skręconego nematyka (TN 90)
Struktura przestrzenna cząsteczek ciekłego kryształu związanych niewielkimi siłami jest bardzo podatna na wpływ oddziaływań zewnętrznych w postaci temperatury, oświetlenia, naprężeń oraz pól elektrycznych i magnetycznych. Na przykład działanie pola elektrycznego, wywołując zmianę konfiguracji przestrzennej cząsteczek ciekłego kryształu powoduje zmianę właściwości przepuszczania światła. Po ustaniu działania pola oddziaływania powierzchniowe przywracają pierwotny układ przestrzenny cząsteczkom ciekłego kryształu (warstwy ciekłokrystalicznej).
Ułożenie cząsteczek w warstwie ciekłokrystalicznej w przypadku: a) braku polaryzacji (E = 0) — warstwa przepuszcza światło; b) przy polaryzacji (E ≠ 0) — warstwa nie przepuszcza światła, lecz je rozprasza
18. Budowa polimerów amorficznych i krystalicznych
Polimery amorficzne zwane również bezpostaciowymi zgodnie z zasadą termodynamiki są w stanie cieczy przechłodzonej. Makrocząsteczki przyjmują postać kłębka tworząc struktury nieuporządkowane skłębione, o słabych oddziaływaniach energetycznych. W tej strukturze jest możliwe jedynie uporządkowanie bliskiego zasięgu do ok. l nm.

Stopień krystalizacji
• Struktura krystaliczna polimeru nigdy nie jest w 100% uporządkowana. Istnieją w polimerze obszary o grubości rzędu 10 nm uporządkowane, na przemian z nieuporządkowanymi
Stopień krystalizacji zależy od wielu czynników:
- Ilość rozgałęzień (im więcej tym mniejsza tendencja do
krystalizacji);
- Wielkość i asymetria grup bocznych
- Długość łańcucha
- Szybkość chłodzenia
- Odkształcenie łańcuchów i inne sposoby wymuszania
kierunku w czasie chłodzenia
21. Budowa, podział, cechy, zastosowanie światłowodów
Pod względem szybkości i jakości przepływu informacji światłowody stanęły wysoko ponad wszelką konkurencją. Transmisja światła jest niewrażliwa na zakłócające pola elektromagnetyczne, co jest szczególnie istotne środowisku przemysłowym. Innym powodem stosowania optycznej transmisji sygnału jest możliwość wykorzystania bardzo szerokiego pasma, dlatego nadaje się on szczególnie do telefonii, transmisji danych i sygnałów telewizyjnych w formie cyfrowej. W światłowodzie do transmisji danych, zamiast prądu elektrycznego, wykorzystywana jest odpowiednio modulowana wiązka światła. Rozwiązanie takie zapewnia większe pasmo przenoszenia - nawet do 3 Tb/s - , oraz większe odległości na jakie sygnał może być transmitowany bez potrzeby dodatkowego wzmacniania.
Światłowód zbudowany jest ze specjalnego rodzaju szkła kwarcowego. Główną jego częścią jest rdzeń, który okrywa płaszcz i warstwa ochronna. Czasami rdzeń składa się z wielu włókien.Zasada działania światłowodu polega na użyciu dwóch materiałów przewodzących światło o różnych współczynnikach załamania. Współczynnik załamania w rdzeniu jest nieco wyższy niż w płaszczu. Promień świetlny przemieszcza się cały czas w rdzeniu, ponieważ następuje całkowite wewnętrzne odbicie promień odbija się od płaszczyzny przejścia rdzenia do płaszcza. Wokół płaszcza znajduje się izolacja ochronna. Światłowody wykonuje się zasadniczo jako jednomodowe i wielomodowe. Światłowody wielomodowe, można podzielić na dwa typy: o współczynniku skokowym i gradientowym najczęściej spotykane są światłowody o płynnej zmianie współczynnika załamania pomiędzy rdzeniem a płaszczem, czyli gradientowe.
Transmisja światłowodowa polega na przekazaniu wiązki światła, którego źródłem może być laser lub dioda LED. Po drugiej stronie światłowodu jest ona odbierana przez element światłoczuły np. fotodiodę. Aby zapewnić prawidłową i szybką transmisję, wiązka światła jest modulowana. Zapobiega to mogącym pojawiać się zniekształceniom sygnału.
Światłowody dzieli się na jedno i wielo modowe oraz na wewnętrzne i zewnętrzne. Pierwszy podział wynika z ilości przesyłanych modów (fal).
W światłowodzie jednomodowym, przenosi się tylko jeden mod. Oznacza to, że wszystkie promienie odbijane są pod tym samym kątem do powierzchni płaszcza. Wszystkie promienie mają wiec jednakową drogę do przebycia i zajmuje to taki sam czas. Oznacza to, że nie powstaje dyspersja. Światłowody jednomodowe są lepsze. Umożliwiają transmisję danych bez ich wzmacniania na odległość do 100km. Prędkość transmisji sięga 3Tb/s.
Źródłem światła jest tu laser. Główną ich wadą, hamującą ich powszechne stosowanie, jest wysoki koszt interfejsów przyłączeniowych
W wielomodowym światłowodzie, jest możliwość występowania różnych kątów odbicia i w związku z tym następuje rozmycie krawędzi przesyłanego sygnału, czyli dyspersja
Światłowody wielomodowe przesyłają wiele modów o różnej długości co powoduje zniekształcenia impulsu wyjściowego a co za tym idzie, zmniejszenie prędkości i odległości transmisji. W tym rodzaju światłowodu, źródła światła jest dioda LED.
Czymś pośrednim miedzy światłowodem o pojedynczym modzie i kablami światłowodowymi o współczynniku skokowym, jest kabel światłowodowy gradientowy. W kablu takim współczynnik załamania zmniejsza się sukcesywnie od środka rdzenia na zewnątrz. Promień świetlny, który ukośnie chce wydostać się z centrum kabla jest uginany w sposób ciągły i kierowany z powrotem w stronę środka kabla. Rdzeń w światłowodzie gradientowym jest tak gruby, że jednocześnie może on przenosić wiele modów światła.
W światłowodzie wielomodowym, rdzeń jest dosyć gruby, ma ok. 50 mikrometrów, czyli jego średnica jest wielokrotnie większa niż długość fali przenoszonego światła. Promień światła może składać się z wielu składowych, z wielu modów, które mogą być przenoszone jednocześnie. Jeżeli zmniejszymy rdzeń dostatecznie (do ok. 5-10 mikrometrów, dla długości fali światła 1,3 mikrometra), to światłowód może przewodzić jedynie jeden mod. Będzie to światłowód typu jednomodowego. Ze względu na bardzo dobre własności częstotliwościowe posiada on możliwość gęstego upakowania informacji - posiada dużą pojemność kanału przenoszenia. Wadą takiego rozwiązania jest cienki rdzeń, co utrudnia łączenie światłowodów ze sobą.
22. Metoda dwutyglowa produkcji światłowodów.
metoda dwutyglowa (podwójnej dyszy) polegająca na jednoczesnym wyciąganiu niskotopliwej masy szklanej rdzenia i płaszcza z dwóch współosiowo umieszczonych tygli. Wymagane jest wcześniejsze oczyszczenie składników z jonów OH, homogenizacji masy szklanej i uformowania prętów szklanych (średnica 3 - 10 mm, długość 1 - 2 m) zasilających tygiel rdzeniowy i płaszczowy;
Wyszukiwarka
Podobne podstrony:
28fizyczna, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyc
FIZYCZNE ZAGROŻENIA ŚRODOWISKA, Inżynieria Środowiska (PWR), semestr 3, FZŚ - (A. Szczurek)
sekuła, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyczna,
zawiejski, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizycz
sprawko 13, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyc
22fizyczna, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyc
landolt, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyczna
calosc, Inżynieria Środowiska (PWR), semestr 3, FZŚ - (A. Szczurek)
Chemia materialow, Inżynieria materiałowa PWr, semestr I, Chemia materiałów
18. SEM ogniw, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fi
28fizyczna, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyc
Biofizyka pytania z kola, Biotechnologia PWR, Semestr 5, Biofizyka - Wykład, Biofizyka - materiały
Opracowanie - materialy, Technologia INZ PWR, Semestr 1, Materiałoznastwo, Materiały - opracowania
BIOFIZYKA- rozwiązania, Biotechnologia PWR, Semestr 5, Biofizyka - Wykład, Biofizyka - materiały
Materiałoznawstwo, Inżynieria środowiska, inż, Semestr II, Materiałoznawstwo ogólne i instalacyjne
ŚCIĄGA - MATERIALY BUD, Budownictwo PWr, SEMESTR 3, Materiały Budowlane, Laborki (O.Mierzejewska)
więcej podobnych podstron