5. Enzymy
Wiadomości wstępne
Podstawy katalizy enzymatycznej
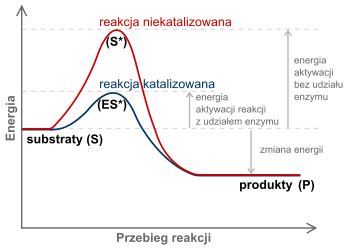
Enzymami nazywamy białka o specyficznych właściwościach katalitycznych, tj. przyspieszających przebieg reakcji chemicznych w komórce. Przyspieszanie reakcji następuje w wyniku obniżania przez enzym wielkości tzw. energii aktywacji, czyli energii, jaką muszą dysponować cząsteczki związków, aby mogły z sobą reagować. Obniżenie energii aktywacji powoduje bowiem, że większy procent cząsteczek posiada wystarczająco dużą energię kinetyczną i dzięki temu może wziąć udział w reakcji, przez co reakcja ta przebiega wielokrotnie szybciej (Rys. 5.1) . Enzymy nie mogą natomiast zmieniać kierunku reakcji, a więc przyspieszają przebieg reakcji tylko wówczas, gdy prowadzi ona w kierunku stanu równowagi.
Rys. 5.1. Udział enzymu w obniżeniu energii aktywacji cząsteczek substratu w reakcji chemicznej
Budowa enzymu i jego działanie
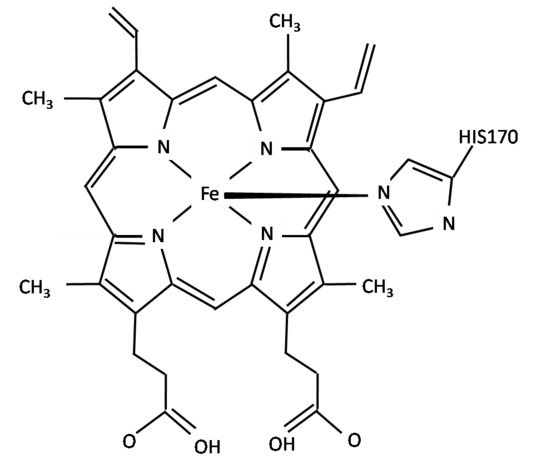
Część enzymu odpowiedzialną za przyłączanie substratu oraz prowadzenie katalizy nazywamy centrum katalitycznym (aktywnym). Centrum katalityczne enzymu może być zbudowane wyłącznie z reszt aminokwasowych, lub też może je tworzyć część niebiałkowa, stanowiąca tzw. grupę prostetyczną lub koenzym. W tym drugim przypadku część białkowa cząsteczki enzymu nazywa się apoenzymem. Grupa prostetyczna jest trwale związana z apoenzymem, natomiast koenzym może funkcjonować samodzielnie. Rolą koenzymu jest przenoszenie jakiejś grupy związków, atomów lub elektronów. Zarówno koenzym jak i grupa prostetyczna czy jony metali łączące się chelatowo z atomami apoenzymu nazywane są kofaktorami. Rys. 5.2. przedstawia strukturę peroksydazy z jonem żelaza w centrum jako kofaktorem. Enzymy wykazują silną specyficzność w stosunku do substratu, stąd też czasem porównuje się pasowanie enzymu do substratu jak klucza do zamka.
Rys. 5.2. Struktura peroksydazy z atomem żelaza w centrum katalitycznym jako kofaktorem
Cząsteczka enzymu wchodzi w reakcję z cząsteczką substratu. Zmienia się przy tym zarówno struktura enzymu, jak i substratu. Dzięki zmianie struktury substrat może ulec reakcji chemicznej, natomiast zmiana struktury enzymu jakiej weszła do niej. Reakcję katalizowaną enzymatycznie można zatem przedstawić następującym schematem:
E + S → ES → E + P
gdzie: E - enzym, S - substrat, ES - kompleks enzym-substrat, P - produkt
Teoria aktywnego kompleksu zakłada, że reakcja katalizowana enzymatycznie zachodzi poprzez wytworzenie kompleksu pomiędzy centrum katalitycznym a cząsteczką substratu. Wytworzenie tego kompleksu jest koniecznym warunkiem zajścia reakcji enzymatycznej. Efektowność własności katalitycznych określonego enzymu definiuje tzw. „liczbę obrotów”, czyli liczba pełnych cykli jednostkowej reakcji (wytworzenie kompleksu ES, reakcja chemiczna przeprowadzona na cząsteczce substratu i rozpad kompleksu enzym - produkt) w czasie jednej sekundy. Jeśli wszystkie cząsteczki enzymu występują w formie kompleksu z substratem, wówczas stan ten określamy jako maksymalną szybkość procesu enzymatycznego (Vmax).
Inhibicja
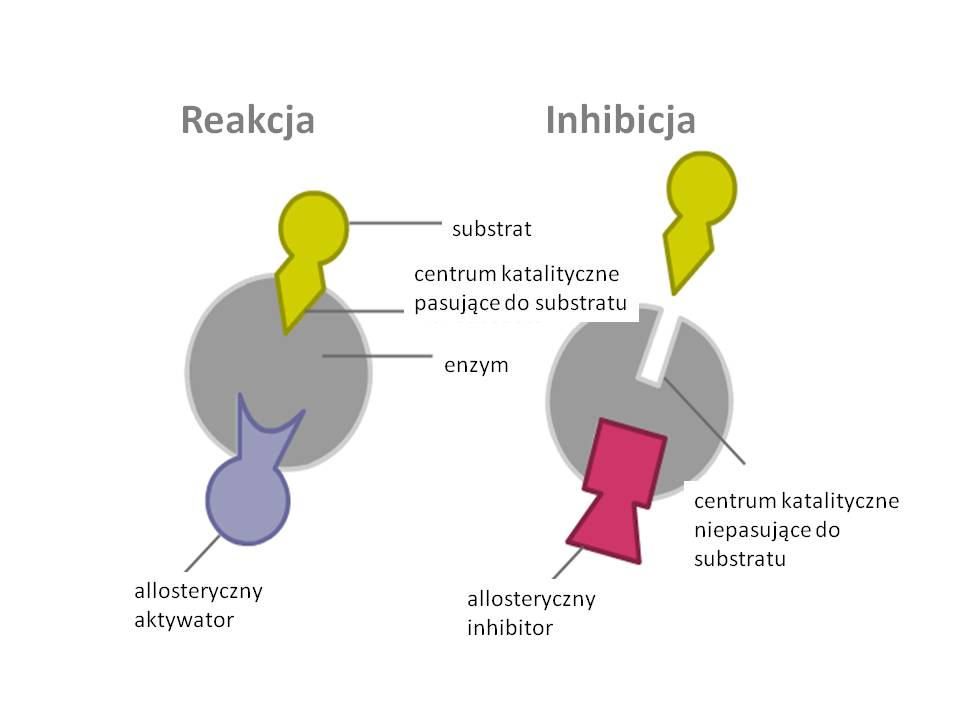
Jeśli cząsteczka nie będąca substratem ma budowę dostatecznie podobną do naturalnego substratu, staje się zdolna do wytworzenia kompleksu z enzymem, chociaż nie jest możliwe zajście procesu enzymatycznego. W takiej sytuacji powstały kompleks nie może się rozpaść i dana cząsteczka enzymu pozostaje zablokowana. O substancji mającej opisane właściwości mówimy, że jest inhibitorem kompetycyjnym, czyli konkurującym z naturalnym substratem o centrum katalityczne enzymu na zasadzie podobieństwa strukturalnego. Inhibicja kompetycyjna jest odwracalna, ponieważ w obecności nadmiaru substratu inhibitor może zostać wyparty przez substrat z kompleksu. W przypadku, gdy substancja wiąże się z centrum katalitycznym, chociaż nie wykazuje strukturalnego podobieństwa do substratu, mówimy o niekompetycyjnym hamowaniu enzymu. Inhibicja niekompetycyjna jest nieodwracalna, ponieważ tego rodzaju inhibitor zmienia strukturalnie centrum katalityczne.
Enzymy allosterycznezny
Niektóre enzymy, oprócz centrum katalitycznego, posiadają również specyficzny fragment cząsteczki, którego stan decyduje o aktywności centrum katalitycznego, mimo iż przestrzennie mogą być znacznie od siebie oddalone. Jest to tzw. allosteryczne centrum regulatorowe, a substancje, które poprzez to centrum wpływają na aktywność enzymu, nazywają się inhibitorami lub aktywatorami allosterycznymi (Rys. 5.3).
Rys. 5.3. Wpływ aktywatora i inhibitora na przebieg reakcji z udziałem enzymu allosterycznego
Klasy enzymów
Stosownie do rodzaju katalizowanej reakcji rozróżniane są następujące klasy enzymów:
1) oksydoreduktazy - katalizujące reakcje utleniania i redukcji,
2) transferazy - katalizujące reakcje przenoszenia grup atomów, np. grup funkcyjnych,
3) hydrolazy - katalizujące reakcje hydrolizy, czyli rozpadu różnych wiązań z udziałem wody,
4) liazy - katalizujące niehydrolityczne rozrywanie różnych wiązań,
5) izomerazy - katalizujące reakcje izomeryzacji, czyli przegrupowania atomów lub grup atomów wewnątrz cząsteczek substratu,
6) ligazy - katalizujące tworzenie nowych wiązań połączone z hydrolizą ATP.
W poszczególnych klasach wyróżnia się kolejno ponumerowane podklasy i pod-podklasy, a każdy enzym wewnątrz pod-podklasy otrzymuje numer kolejny, zależny od tego, kiedy enzym ten został odkryty. Dzięki temu każdy enzym jest opisany przez zespół 4 liczb (numer klasy, podklasy, pod-podklasy i numer kolejny w pod-podklasie). Ten czteroczłonowy numer jednoznacznie identyfikuje każdy znany enzym i umieszcza go w międzynarodowym katalogu klasyfikacji enzymów (E.C. - Enzyme Commission). Na przykład trypsyna ma numer EC 3.4.21.4. Klasyfikacja enzymów, czyli przyporządkowanie każdemu znanemu enzymowi specyficznego zestawu liczb opisujących jego właściwości, dokonuje się poprzez określenie typu enzymu, rodzaju substratu poddawanego reakcji enzymatycznej i np. rodzaju przenoszonej grupy atomów, koenzymu uczestniczącego w procesie itp. Niektóre enzymy występują w postaci wolnych cząsteczek, inne, zwłaszcza takie, które katalizują reakcje wchodzące w skład np. zespołu większej liczby reakcji (np. łańcuch oddechowy), zajmują ściśle określone miejsce w organellach komórki i łączą w sobie dwie funkcje, tj. enzymatyczną i strukturalną.
Czynniki wpływające na aktywność enzymu
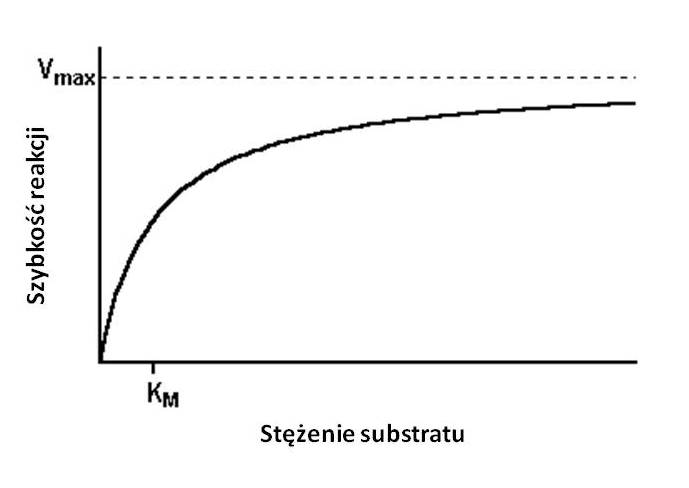
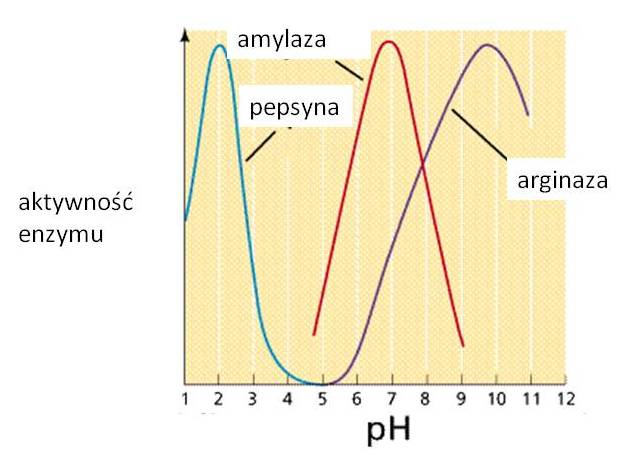
Ważną cechą enzymu jest szybkość katalizowanej przez niego reakcji. Szybkość ta jest wprawdzie cechą charakterystyczną enzymu, jednak mają na nią wpływ liczne czynniki takie jak: temperatura, pH, stężenie cząsteczek enzymu, obecność i stężenie aktywatorów, inhibitorów i jonów aktywujących (lub jonów metali ciężkich dezaktywujących enzym) i wreszcie siła jonowa roztworu, czyli sumaryczne stężenie wszystkich jonów. Ponadto generalnie aktywność enzymów jest hamowana produktami reakcji, a stymulowana zwiększonym stężeniem substratów (Rys. 5.4). Optimum temperaturowe wynosi zazwyczaj 30-45°C, natomiast większość enzymów ulega dezaktywacji w temperaturze powyżej 60°C (Rys. 5.5). Każdy enzym posiada pewne optymalne pH dla swojej aktywności (Rys. 5.6).
Rys. 5.4. Wpływ stężenia substratu na szybkość reakcji enzymatycznej (Vmax - maksymalna szybkość reakcji enzymatycznej, KM - stała Michaelis'a
Rys. 5.5. Wpływ temperatury na aktywność enzymów
Rys. 5.6. Wpływ pH na aktywność różnych enzymów: amylazy, pepsyny i arginazy
ĆWICZENIA
5.1. Oznaczanie aktywności kwaśnej fosfatazy (fosfomonoesterazy)
Zasada:
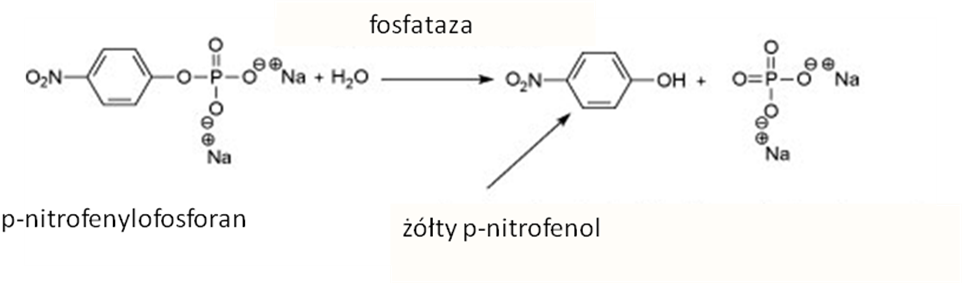
Fosfatazy są enzymami hydrolizującymi estry fosforanowe różnych metabolitów. Specyficzność fosfataz jest bardzo zróżnicowana. Jednym ze standardowych substratów dla pomiaru aktywności fosfatazy jest p-nitrofenylofosforan (PNP): O2N-C6H4-O-PO3H2. Związek ten jest hydrolizowany przez fosfatazę, która uwalnia z niego cząsteczkę kwasu fosforowego pozostawiając p-nitrofenol, związek o intensywnie żółtym zabarwieniu, uwidaczniający się zwłaszcza w alkalicznym środowisku (Rys. 5.7). Umożliwia to spektrofotometryczny pomiar ilości uwolnionego p-nitrofenolu przy = 400 nm. Przebieg analizy jest następujący: w probówce miesza się enzym i PNP jako substrat w odpowiednim buforze, a następnie przetrzymuje się mieszaninę przez określony czas w stałej temperaturze, po czym zatrzymuje się reakcję poprzez nagłą alkalizację mieszaniny inkubacyjnej i wykonuje pomiar spektrofotometryczny.
Rys. 5.7. Schemat reakcji katalizowanej przez fosfatazę
Wynik aktywności enzymu, tj. masa przetworzonego substratu zależy między innymi od stężenia substratu, ilości enzymu oraz czasu reakcji. Przedłużenie czasu reakcji prowadzi do zużycia substratu, w wyniku czego szybkość reakcji maleje asymptotycznie do zera (zbliża się do tej wartości, ale nigdy jej nie osiąga). Zwiększenie stężenia substratu przy zachowaniu stałości pozostałych parametrów prowadzi początkowo do wzrostu szybkości reakcji aż do uzyskania maksymalnej prędkości, przy której wszystkie cząsteczki enzymu są maksymalnie aktywne i dalsze zwiększanie stężenia substratu nie przyspiesza reakcji. Osiągnięta została tzw. maksymalna aktywność enzymu.
Wykonanie:
a) Otrzymywanie ekstraktów z ziemniaka
Sporządzenie ekstraktu w buforze: Zetrzeć na tarce 10 g ziemniaka, dodać ok. 15 cm3 0,1 M buforu octanowego o pH = 5,5, wycisnąć sok przez warstwę gazy, po czym odwirować przez 15 min przy ok. 9 300 x g.
Sporządzenie ekstraktu wodnego: Przygotowanie jak wyżej, ale zamiast buforu octanowego użyć wody (ekstrakt wodny będzie potrzbny do ćwiczenia 5.1 d).
b) Dobranie czasu inkubacji
Do trzech probówek wlać po 0,1 cm3 ekstraktu enzymatycznego w buforze, dodać po 0,9 cm3 0,1 M buforu octanowego o pH = 5,5 i po 1 cm3 0,4% roztworu PNP w tym samym buforze octanowym. Zamieszać, odstawić na czas inkubacji reakcji enzymatycznej (odpowiednio: jedną probówką na 3, drugą na 6 a trzecią na 9 minut w temperaturze pokojowej), po czym zastopować reakcję 1 cm3 0,3 M NaOH i zmierzyć absorbancję próbek przy λ = 400 nm względem ślepej próby (bufor octanowy o pH=5,5). Wynik wyrazić jako liczbę optycznych jednostek aktywności enzymu (rozcieńczenie razy absorbancja). Za optymalny uznać należy czas inkubacji, po którym absorbancja osiągnie wartość pomiędzy 1 a 1,5. Jeśli żaden z czasów inkubacji nie okaże się odpowiedni, należy dokonać interpolacji liniowej.
Jednostką optyczną nazywamy taką ilość danej substancji, która rozpuszczona w 1 cm3 roztworu wykazuje absorbancję równą 1,0 na drodze optycznej 1 cm (tj. w naczynku pomiarowym o grubości 1cm).
Przykładowe obliczenia
Dodając do siebie objętości wszystkich roztworów: 0,1 cm3 ekstraktu, 0,9 cm3 buforu, 1 cm3 PNP oraz 1 cm3 NaOH, razem otrzymujemy 3 cm3. Absorbancja roztworu była mierzona w kuwetce spektrofotometrycznej o grubości 1 cm, zatem liczbę jednostek optycznych uzyskujemy mnożąc uzyskaną absorbancję przez 3.
c) Wyznaczenie zależności aktywności kwaśnej fosfatazy od stężenia substratu
Zastosować uzyskany w poprzednim doświadczeniu a) ekstrakt z ziemniaka w buforze octanowym. Do probówki pobrać 4 cm3 roztworu substratu (PNP) o stężeniu 4% w buforze octanowym. Z tej probówki pobrać do kolejnej 2 cm3 roztworu substratu (PNP) a następnie dodać 2 cm3 samego buforu octanowego (pH 5,5), tym razem uzyskując tym samym stężenie 2% PNP. Uzyskany roztwór zamieszać i pobrać z niego 2 cm3 do kolejnej probówki, do której ponownie dodajemy 2 cm3 samego buforu octanowego (pH 5,5). Tym razem uzyskaliśmy stężenie 1% PNP. Procedurę powtórzyć jeszcze 7-krotnie, uzyskując w efekcie końcowym 10 probówek zawierających po 2 cm3 PNP o dalszych stężeniach: 0,5; 0,25; 0,125; 0,064; 0,032; 0,016 i 0,008% (w ostatniej probówce będą 4 cm3 roztworu). Następnie, do nowych 10 probówek pobrać po 1 cm3 przygotowanych roztworów PNP oraz po 0,9 cm3 buforu octanowego o pH 5,5. Reakcję enzymatyczną rozpocząć przez dodanie do każdej probówki po 0,1 cm3 ekstraktu z ziemniaka w buforze octanowym. Inkubację reakcji przeprowadzić w czasie dobranym w podpunkcie b) i w tej samej temperaturze. Reakcję zastopować NaOH (jak w punkcie a), po czym zmierzyć absorbancję przy λ=400 nm względem ślepej próby (bufor octanowy o pH=5,5).
d) Określanie zależności aktywności kwaśnej fosfatazy od pH
W 11 probówkach przygotować mieszaniny 0,4 M kwasu octowego i 0,4 M octanu sodu w proporcjach podanych w tabeli. Bufor (np. octanowy) składa się ze składnika kwaśnego (kwas octowy) i alkalicznego (octan sodu). Zmieszanie tych składników w różnych proporcjach pozwala uzyskać różne wartości pH buforu, w którym jest mierzona aktywność enzymu. Zmierzyć wartości pH każdego z buforów.
|
Nr probówki |
||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
octan sodu [cm3] |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
kwas octowy [cm3] |
10 |
9 |
8 |
7 |
6 |
5 |
4 |
3 |
2 |
1 |
0 |
pH buforu |
|
|
|
|
|
|
|
|
|
|
|
Do nowych 11 probówek pobrać po 0,9 cm3 poszczególnych sporządzonych buforów oraz po 1 cm3 0,4% wodnego roztworu PNP. Reakcję enzymatyczną rozpocząć przez dodanie po 0,1 cm3 wodnego ekstraktu z ziemniaka przygotowanego w zadaniu a). Następnie przeprowadzić inkubację w czasie i temperaturze jak w poprzednim doświadczeniu. Reakcję zastopować przez dodanie po 1 cm3 0,3 M NaOH. Zmierzyć absorbancję przy λ=400 nm względem ślepej próby (woda destylowana).
Sprawozdanie
a) Na podstawie otrzymanych wyników wykreślić funkcję zależności aktywności enzymu od stężenia substratu. Wykres zinterpretować.
b) Na podstawie otrzymanych wyników wykreślić wykres zależności aktywności enzymu od pH. Wykres zinterpretować.
5.2. Wykazanie aktywności oksydaz w ziemniaku
Zasada:
W tkankach występują różne enzymy powodujące utlenianie substratów, a wśród nich oksydazy (utleniające substraty przy udziale tlenu cząsteczkowego) i peroksydazy (wykorzystujące nadtlenek wodoru jako źródło tlenu dla utleniania substratów). Produkty utlenienia mają zwykle ciemne zabarwienie, stąd ciemnienie przekrojonego i poddanego działaniu powietrza jabłka lub ziemniaka. Zwilżenie powierzchni tkanki substratem dla działania enzymów utleniających (np. p-fenylenodiamina), jak i rozcieńczonym, ok. 0,1% roztworem nadtlenku wodoru przyspiesza proces ciemnienia tkanki. Oksydazy i peroksydazy są enzymami, w których istotną rolę odgrywają jony aktywujące. Stąd też zwilżenie powierzchni ok. 1% roztworem EDTA (kwasu etylenodiaminotetraoctowego) wiążącego w trwały kompleks różne dwuwartościowe kationy, opóźnia zjawisko ciemnienia powierzchni przekrojonej tkanki.
Wykonanie:
Ćwiartkę jabłka lub ziemniaka zwilżyć roztworem p-fenylenodiaminy, drugą zwilżyć roztworem nadtlenku wodoru, trzecią zwilżyć roztworem EDTA i wreszcie ostatnią pozostawić jako kontrolną. Porównać szybkość ciemnienia poszczególnych połówek.
5.3. Wykazanie obecności katalazy w ziemniaku
Zasada:
Katalazy są enzymami rozkładającymi nadtlenek wodoru powstający jako produkt uboczny w niektórych procesach metabolicznych. Nadtlenek wodoru jest szkodliwy ze względu na jego utleniające właściwości. Przyjmuje się zatem, że katalazy pełnią w organizmie funkcję ochronną przed szkodliwym działaniem tego związku.
Wykonanie:
Do miazgi ziemniaczanej dodać kroplami wodę utlenioną. Następuje burzliwe wydzielanie się pęcherzyków tlenu świadczących o aktywności katalazy. Wydzielanie pęcherzyków tlenu następuje również podczas przemywania wodą utlenioną skaleczenia; oznacza to, że katalaza występuje nie tylko w tkankach roślinnych.
5.5. Spektrofotometryczne oznaczanie aktywności peroksydazy niespecyficznej
Zasada:
Peroksydazy są powszechnie występującymi enzymami utleniającymi różne substraty przy udziale nadtlenku wodoru H2O2. Nazwą peroksydaza niespecyficzna określa się całkowitą pulę peroksydaz cytoplazmatycznych i apoplastycznych. Najczęściej substratem dla peroksydaz są różne związki fenolowe oraz diaminy, których produkty utleniania charakteryzują się wyraźną barwą. W pomiarach aktywności peroksydaz wykorzystuje się reakcję utlenienia N,N'-difenylo-1,4-fenylenodiaminy (czyli p-fenylenodiaminy) do związku N,N'-difenylo-1,4-benzo-quinondiiminy.
Wykonanie:
Odważyć 200 mg świeżej masy liści. Ucierać w ciekłym azocie dodając stopniowo (3 x 1 cm3 = 3 cm3) bufor ekstrakcyjny (50 mM bufor fosforanowy pH 7,0 z 1mM EDTA). Następnie ekstrakt wirować przy 14000 × g przez 10 minut w temp. 4°C. Aktywność peroksydazy zmierzyć spektrofotometrycznie jako wzrost absorbancji przy długości fali 460 nm.
Do kuwety jednorazowej o pojemności 3 cm3 dodać: 2 cm3 buforu ekstrakcyjnego, 12 µl 0,5% roztworu p-fenylenodiaminy w buforze ekstrakcyjnym, 12 µl ekstraktu, a następnie „zerujemy” spektrofotometr. Natychmiast dodajemy 12 µl H2O2 w buforze ekstrakcyjnym (50 cm3 buforu ekstrakcyjnego + 0,15 cm3 30% H2O2). Należy odczytać początkową wartość absorbancji oraz po upływie 1 minuty. Aktywność enzymów wyrażamy w różny sposób: można np. podawać ilości (mg, μg, μmole, nmole) rozłożonego substratu lub powstającego produktu w reakcji w przeliczeniu na świeżą lub suchą masę, bądź też na mg białka w jednostce czasu. W tym przypadku aktywność peroksydazy niespecyficznej podajemy jako ΔABS/g świeżej masy/minutę.
Przykładowe obliczenia:
Jeżeli utarto 200 mg świeżej tkanki w 3 cm3 buforu, a potem do reakcji zostało z tego pobrane 12 µl ekstraktu, to z przeliczeń wynika, że w 12 µl ekstraktu było białko uzyskane z 0,8 mg świeżej masy. Przykładowo, obserwowano wzrost absorbancji (ΔABS) wynoszący 0,500 w ciągu minuty. Był on spowodowany aktywnością peroksydazy niespecyficznej zawartej w 0,8 mg tkanki, a zatem ilość tego enzymu zawartego w 1 g świeżej masy spowodowałaby wzrost ABS o 625.
5.6. Oznaczanie aktywności katalazy w ekstrakcie z roślin (EC: 1.11.1.6.)
Zasada:
Katalazy są enzymami rozkładającymi nadtlenek wodoru powstający jako produkt uboczny w niektórych procesach metabolicznych. Ponieważ nadtlenek wodoru jest silnym utleniaczem, podobnie jak inne reaktywne formy tlenu, stąd też katalazy pełnią w organizmie funkcję ochronną przed jego szkodliwym działaniem.
Wykonanie:
Odważyć 200 mg świeżej masy liści. Ucierać w ciekłym azocie stopniowo dodając (3 x 1cm3) 3 cm3 buforu ekstrakcyjnego (50 mM buforu fosforanowego pH 7,5 z 1mM EDTA). Następnie ekstrakt wirować przy 14000 × g przez 10 min w temp. 4°C.
Do kuwety spektrofotometrycznej (kwarcowej!) dodać kolejno: 2 cm3 H2O2 w buforze (50 cm3 buforu fosforanowego o pH 7,5 + 0,3 cm3 30% H2O2). Na nim „zerujemy” spektrofotometr, a następnie dodajemy bardzo szybko mieszając 200 µl ekstraktu. Odczytać wartość absorbancji przy długości fali 240 nm na początku pomiaru oraz po 1 minucie. Jeżeli dokonujemy pomiaru na spektrofotometrze mierzącym kinetykę enzymatyczną, odczytujemy tzw. slope (kąt nachylenia krzywej) i notujemy jego wartość. Spadek absorbancji o 0,0145 odpowiada rozkładowi 1µmola H2O2. Aktywność katalazy wyrażamy w µmolach H2O2/mg białka/min (w tym przypadku należy zmierzyć ilość białka w badanym materiale roślinnym, np. metodą Bradford) lub w przeliczeniu na g świeżej masy.
Przykładowe obliczenia:
Przy spadku absorbancji wynoszącej 0,5 na minutę z przeliczeń wynika, że odpowiada to rozkładowi 34,48 µmoli H2O2. Jeżeli tkanka o masie 200 mg została utarta w 3 cm3 buforu ekstrakcyjnego, a do pomiaru enzymatycznego zostało pobrane 200 μl, to w tych 200 μl znajdowało się białko pochodzące z 13,33 mg tkanki. Należy wyliczyć z proporcji: białko (w domyśle katalaza) z 13,33 mg tkanki rozłożyło 34,48 µmoli H2O2 , to z 1000 mg świeżej masy rozłoży x µmoli nadtlenku wodoru.
Piśmiennictwo:
Bradford M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-254.
73
Wyszukiwarka
Podobne podstrony:
Kwasy nukleinowe nowy skrypt, Studia, UR OŚ, semestr III, biochemia
zagadnienia kol I 2012-2013, Studia, UR OŚ, semestr III, biochemia
biochemia-osr2013-2014 PLAN CW, Studia, UR OŚ, semestr III, biochemia
zagadnienia kol I 2012-2013, Studia, UR OŚ, semestr III, biochemia
EGZAMIN Z GOSPODARKI WODNO, Studia, UR OŚ, semestr III, gospodarka wodno-ściekowa, egzamin
gosp.wodn, Studia, UR OŚ, semestr III, gospodarka wodno-ściekowa, egzamin
odpowiedzni moje gospo, Studia, UR OŚ, semestr III, gospodarka wodno-ściekowa, egzamin
agrometeo (2) - Kopia, Studia UR OŚ, semestr I, meteorologia
ZC dwa kolosy, Studia, UR OŚ, semestr IV, zagrożenia cywilizacyjne, ćwiczenia, kolosy
koloss 1, Studia, UR OŚ, semestr IV, zagrożenia cywilizacyjne, ćwiczenia, kolosy
kolokwia, Studia, UR OŚ, semestr IV, zagrożenia cywilizacyjne, ćwiczenia
Ekologia - egzamin(, Studia UR OŚ, semestr II, ekologia
Rozw. zad. 10, Studia, UR OŚ, semestr VII, prawo i ekonomia w ochronie środowiska, kolos 2
OCHRONA POWIETRZA 2012, Studia, UR OŚ, semestr VI, ochrona powietrza, ćwiczenia
więcej podobnych podstron