Analiza chemicznych zanieczyszczeń środowiska - WYKŁADY
Wykład 1 02.04.2009
Spektrochemiczne metody oznaczania
Oddziaływanie promieniowania elektromagnetycznego z materią.
Odkrywcy analizy spektralnej
Gustaw Robert Kirchhoff
Robert Wilhelm Bunsen
Spektralna analiza jakościowa - odkrycie nowych pierwiastków. W latach 1860-1864 odkryto cez, rubid, bal, ind, gal. Pierwszy spektrometr absorpcji atomowej - sir Alan Walsh.
Zakresy widma elektromagnetycznego
Gamma - X - ultrafiolet - zakres widzialny - podczerwień - mikrofale - fale radiowe.
Oddziaływanie promieniowania elektromagnetycznego z materią
Odbicie |
→
|
Spektroskopia |
Rozproszenie |
|
Ramana (rozpraszania) |
Absorpcja |
|
Absorpcyjna |
Emisja |
|
Emisyjna |
Atom |
→
|
Atomowa |
Cząsteczka |
|
Molekularna |
Jądro |
|
Jądrowa NMR |
Elektron |
|
Elektronów (XPS) |
Jon |
|
Jonowa(SIMS, ISS) |
|
|
Masowa (stosunek masy jonu do ładunku) |
Spektrometria atomowa:
- fotometria płomieniowa,
- spektrografia i spektrometria emisyjna,
- atomowa spektrometria absorpcyjna (ASA)
- atomowa spektrometria fluorescencyjna (FSA)
- spektrometria rentgenowska,
- spektrometria elektronowego rezonansu jądrowego (EPR),
Spektrometria cząsteczkowa:
- spektrometria absorpcyjna,
- spektrofluorymetria
- Ramana (rozproszenia)
Spektrometria cząsteczkowa
- przejścia elektronowe (możliwe dwa przejścia: *z niewiążącego na pi, *z pi na pi)
- powstawanie widma cząsteczkowego
![]()
Transmitancja - informuje o stosunku natężenia promieniowania przechodzącego i padającego.
![]()
T = 1, kiedy wszystko przejdzie przez próbkę
T = 2, kiedy połowa przejdzie przez próbkę.
Prawo Beera (absorbancja zależy od ε, b, c.)
Absorbancja - ujemny logarytm z transmitancji
![]()
Elementy rozszczepiające (pryzmat, siatka dyfrakcyjna)
Lampy: Deuterowe 18 - 380 nm,
Wolframowo - halogenowe > 380 nm
Ksenonowe
Lasery
Detektory: Fotokomórka
Fotopowielacz
Fotoopornik
Matryca diodowa
Spektrometr jednowiązkowy
Źródło → szczelina wejściowa → element rozszczepiający → szczelina wyjściowa → próbka → pomiar (detektor).
Wady: nie mierzy się ślepej próby i próbki w tych samych warunkach (pomiary w innych warunkach), nie dokładne pomiary.
DAD (detektor diodowy) - drugi typ spektrometru jednowiązkowego
Źródło → próbka → szczelina wejściowa → pomiar (detektor).
Następuje tu zarejestrowanie całego widma jednocześnie.
Spektrometr dwuwiązkowy
Źródło światła → szczelina wejściowa → rozszczepienie → szczelina wyjściowa → odbicie od lustra → rozdzielenie → 2 próbki → 2 detektory.
Wady: spada czułość pomiaru (próbka rozdzielana na dwa strumienie), niejednakowa praca detektorów.
Inny typ detektora dwuwiązkowego
- zawiera czoper ( zwierciadło obracające), jeden detektor. Ten typ jest lepszym rozwiązaniem.
Rodzaje kuwet pomiarowych:
- kwarcowe (pomiar od ultrafioletu) - ważna jest grubość kuwety, gdyż od tego zależy absorbancja.
Wpływ rozproszenia promieniowania
Szerokość szczeliny i rozdzielczość
- od szczeliny zależy rozdzielczość (zdolność do rozdzielania pików)
Nakładanie pasm absorpcyjnych
- próbki rozdzielają się (absorbują) przy tych samych pasmach.
![]()
błąd 10 % gdy dobrze rozdzielone
![]()
błąd 100 % gdy źle rozdzielone
Spektrometria pochodna (gdy widma źle się rozdzielają (nakładają się) należy zróżniczkować.
A = εbc
![]()
![]()
![]()
Walidacja metod spektrofotometrycznych
Wykresy wzorcowe - dlaczego wykres się zagina?
- cząsteczki asocjują
- problemy aparaturowe
Jak zapewnić dobrą liniowość - przy pomiarze piku wybiera się zawsze maksimum piku.
Jak zapewnić dobra dokładność - przy pomiarze wybiera się tam gdzie jest względnie stała absorbancja.
Jak zapewnić dobrą czułość pomiarów - w maksimum piku.
Jak wybrać warunki zapewniające dobrą selektywność - tam gdzie absorbancja jednego widma jest najmniejsza.
Analiza jakościowa
Rodzaj chromoforu, otoczenia grup chromoforowych, rozpuszczalnik, temperatura, pH.
Zastosowanie:
- analiza jakościowa,
- analiza ilościowa: jonów metali (barwne kompleksy chelatowe ze związków organicznych, kompleksy z ligandami nieorganicznymi), anionów nieorganicznych, związków organicznych (po połączeniu z HPLC),
- badanie równowagi reakcji chemicznych: wyznaczanie stałych dysocjacji kwasów i zasad, ustalanie składu i stałych trwałości związków kompleksowych.
Metody polegające na oddziaływaniu promieniowania z atomem to:
Spektrometria atomowa
Zasada działania spektrometru AAS:
Spektralne źródło → Io atomizer (próbka) It → monochromator (zespół zwierciadeł, pryzmatów, siatek dyfrakcyjnych) → detektor
![]()
Stosowane są lampy, które mają promieniowanie monochromatyczne (o określonej długości fali).
Spektrometr jednowiązkowy i dwuwiązkowy
Źródła promieniowania:
- lampy z katodą wnękową - HCl (katoda - z pierwiastka, który chcemy oznaczać, anoda - z innego pierwiastka)
- lampy wielopierwiastkowe
- ultra lampy
Promieniowanie ciągłe
- lampa deuterowa - D2
- ksenonowa
Atomizacja
- płomieniowa FAAS
- bezpłomieniowa ETAAS (V, Mo, Ta), GFAAS (c).
Sposoby wprowadzenia próbki
- aerozol FAAS
- ciecz ETAAS
- generowanie lotnych wodorków (HG - hydrie generation) lub lotnych form analitu (VS - volatile species) FAAS, GFAAS,
- zawiesina (slurry) ETAAS,
- stała próbka (soild sampling) ETAAS
Technika płomieniowa - procesy zachodzące w płomieniu
M+ + A- (roztwór)
Nebulizacja M+ + A- (aerozol)
odparowanie rozpuszczalnika MA (suchy aerozol)
topnienie MA (ciecz)
odparowanie MA (gaz)
atomizacja M + A (wolne atomy)
wzbudzenie M+ (gaz)
jonizacja M+ + e- (gaz)
Technika płomieniowa FAAS
- powietrze C2H2/2125 - 2400 (˚C): Cu, Pb, K, Na
- (powietrze) N2O/2600 - 2800 (˚C): Al, Si, W
- oba rodzaje: As, Ca, Cr, Mg, Cs, Se, Sr,
- gaz palny C2H2*N2O
- utleniacz - powietrze w płomieniu powietrze (C2H2*N2O w N2O/ C2H2)
Płomień bogaty w paliwo = redukujący
Płomień ubogi w paliwo = utleniający
Wykład 2 07.04.2009
Atomizacja bezpłomieniowa
Gaz - argon
Musi być wysoka temperatura
Wstrzyknięcie próbki → suszenie → spopielanie (rozkład związków organicznych do prostych związków nieorganicznych) → atomizacja (przeprowadzenie do wolnych atomów) → czyszczenie → chłodzenie
Zastosowanie alternatywnego gazu w etapie rozkładu termicznego, np. oznaczanie selenu we krwi.
Technika dozowania zawiesiny (koloid fazy stałej w fazie ciekłej)
- próbka (może być problem z powtarzalnością pomiarów, więc należałoby dodać stabilizatora, aby zawiesina była w tej samej postaci).
Analiza próbek stałych (solid sampling) - problem z dokładnością.
Porównanie technik atomizacji:
FAAS |
GFAAS |
Próbka jest wprowadzana do atomizera w sposób ciągły |
Próbka jest wprowadzana do atomizera w sposób dyskretny |
Duża objętość próbki do analizy (1-2 ml) |
Mała objętość próbki do analizy (10-50 µl) |
Duża precyzja |
Mniejsza precyzja |
Duża szybkość analizy |
Mniejsza szybkość analizy |
Brak aparaturowych możliwości kontrolowania interferencji |
Aparaturowe możliwości kontrolowania interferencji, ale duże wpływy matrycowe |
Mniejsza czułość (0,0X - X,00 µg/ml) |
Duża czułość (0,0X - X,0 ng/ml) |
|
Możliwości analizowania zawiesin i próbek stałych |
Np. FAAS µg/ml → Ca stężenie analitu jest 1000 razy większe niż w technice płomieniowej
GFAAS ng/ml → Pb
Generowanie lotnych par
generowanie lotnych wodorków: As, Bi, Pb, Se, Sn, Sb, Te, Ge
BH4- + 3 H2O + H2 = B(OH)3 + 6 H + H2
Men+ + 3nH = MeHn + nH2
Ważne parametry:
- stężenie reduktora i kwasu,
- transport do atomizera,
- wpływ matrycy,
- kinetyka redukcji zależna od formy występowania analitu Se(IV)/Se(VI).
Bardzo czuła metoda oznaczania pierwiastków, uwalniany analit z matrycy, wolny od interferencji znalazła zastosowanie w analizie specjacyjnej, dlatego że w reakcji nie wszystkie formy utleniacza biorą udział.
Wprowadzenie wodorków do atomizera:
Atomizer - kuweta, rurka kwarcowa
FAAS - umieszczenie w płomieniu, wodorek przepływa do kuwety i jest w płomieniu zatrzymywany dłużej.
GFAAS - transportowany do normalnej kuwety grafitowej.
Metoda zimnych par - oznaczanie rtęci CV AAS
2 Hg(I) + Sn2+ + SCl- = 2 Hg(0) + SnCl62- (najczęściej stosowana, reakcja zachodzi w temperaturze pokojowej)
BH4- + 3 H2O + H+ = B(OH)3 + 6 H + H2
Men+ + 3nH = MeHn + nH2
LOD = 0,5 - 1 ng/ml
Interferencja Ni, Fe, Pb, Cu, Ag, Pt, Pd.
Np. oznaczanie rtęci w pojedynczym włosie - włos wkłada się do kubeczka z próbką → ogrzewanie → transport do pułapki → rtęć ulega rozpuszczeniu → ogrzewanie pułapki z rtęcią → odparowanie rtęci (po ogrzaniu)
Interferencje:
Spektralne
- rozproszenie promieniowania
- absorpcja cząsteczkowa (analitu więcej niż jest w rzeczywistości - błąd dodatni)
- absorpcja atomowa
- emisja płomieniowa lub grafitu
Niespektralne (związane ze składem próbki)
- chemiczne (wtedy gdy analit tworzy trudno lotne związki)
- fizyczne (np. wzorzec H2O, próbka w roztworze gliceryny - większa gęstość, lepkość, zmniejszy się wydajność transportu - błędy ujemne)
Jonizacja
Matrycowe
Specyficzne (dla naszego analitu, związane z naturą danego pierwiastka) i niespecyficzne (związane z rodzajem próbki, matrycy)
Modyfikatory chemiczne (stosuje się je aby pozbyć się interferencji)
- mieszane (przeprowadzenie analitu w związki trudno lotne
Pd + Mg(NO3)2
Cd + Pd
- długotrwałe Pd, Rh, Ru (permanent) : redukcja termiczna i redukcja elektrolityczna.
Problemy: |
Rozwiązania: |
Wpływ interferencji na wynik analiz → błędy oznaczeń |
Efektywna korekcja tła Modyfikatory matrycy Wydzielanie analitu z matrycy: - pułapki - generowanie par HG, VG - metody chemiczne LLE, SPE |
Zbyt wysoka granica wykrywalności : |
Zwiększenie efektywności nebulizacji Wzbogacanie analitu - rozwiązanie aparaturowe - chemiczne metody wzbogacania |
Pierwiastki ważne w biologii i medycynie
- istotne fizjologicznie Na, Mg, K, Ca, Sr, Ba
- toksyczne Al., Cd, Hg, TL, Pb, Bi
- istotne w farmakologii i diagnostyce
Techniki emisyjne (wielopierwiastkowe)
- wykorzystują emisję promieniowania, można oznaczyć kilka, kilkanaście pierwiastków.
Spektrometr emisji atomowej:
- brak lampy (spektralnego źródła promieniowania)
- jest źródło wzbudzania:
*klasyczne: iskra, łuk elektryczny (detektorem była klisza fotograficzna)
*DPC (plazma prądu stałego) - układ trójelektrodowy, jedna z nich jest próbka, stosuje się do badania jakości produkcja,
*płomień (np. fotometria płomieniowa)
Monochromatory: Czerny - Turnera, Eberta - Fastie'go, Littrowa.
Są dwa typy spektrometrów:
- spektrometr sekwencyjny z monochromatorem Czerny - Turnera (jedno źródło wzbudzania, jeden detektor, zmienia się długość fali)
*polichromator - analiza jednoczesna (wiele detektorów, każda długość fali dociera do innego detektora), analiza krótsza, droższa,
- spektrometr z polichromatorem opartym na kole Rowlanda
Detektor DDA - detekcja na matrycy diodowej
Detektor CCD - analiza trójwymiarowa.
Plazma indukcyjnie sprzężona (ICP)
Plazma - gaz posiadający właściwości magnetyczne, w którym występują atomy w stanie zjonizowanym. Gaz, w którym jest powyżej 1 % elektronów. Plazma niskotemperaturowa (ok. 6000 - 10000 ˚C [K] występuje w atomowej spektrometrii absorpcyjnej i spektrometrii mas.
Wprowadzenie próbki do płomienia i plazmy indukcyjnie sprzężonej następuje w postaci aerozolu bądź w postaci zawiesiny.
Interferencje:
- przyczyny nieliniowości wykresów wzorcowych:
*samo absorpcja,
*samo odwrócenie linii - linie rezonansowe mogą ulec poszerzeniu, a nawet obniżać się w maksimum (efekt większy dla płomienia niż plazmy)
- jonizacja atomów
- addytywne - ślepej próby
- spektralne: *emisja innych atomów, *pasma cząsteczkowe,
- fizyczne,
- chemiczne: *bufory uwalniające, *bufory ochronne, *bufory jonizacyjne.
Analizowanie pierwiastków ICP-OES: C, P, Na, Mg, Li, lantanowce, aktynowce.
Wykład 3 21.04.2009
Zasada powstawania widma mas (ICP-MS)
Droga jonów polu elektrycznym i magnetycznym: rezultat działających sił
- siła pola magnetycznego (Fm)
- siła odśrodkowa (Fo)
- energia kinetyczna w polu elektrycznym (E)
Fm = Bev , gdzie B - natężenie pola magnetycznego, v - prędkość cząsteczki, e - ładunek jonu
Fo = mv2/r
Ek = eU = ½ mv2
Fm = Fo → Bev = mv2/r → v = Ber/m
![]()
![]()
Masy atomowe - izotopy (technika ta pozwala na analizę izotopów)
Schemat blokowy spektrometru mas
1) Układ wprowadzania próbki |
Jonizacja strumieniem Jonizacja z desorpcją jonów |
2) Komora jonizacyjna |
Jonizacja chemiczna
Jonizacja chemiczna pod cisnieniem atmosferycznym |
|
Jonizacja w plazmie sprzężonej indukcyjnie (ICP-LA) |
3) Analizator jonów |
Kwadrupolowy (najprostszy i najtańszy) Pułapka jonowa (T) Czas przelotu ToF Magnetyczny (wysokorozdzielczy) Cyklotronowy ICR |
4) Detektor jonów |
|
5) Analiza danych |
|
Pojęcie rozdzielczości widma mas
- z próbki wytwarzane jest wiele jonów,
- im droższy aparat tym lepsza rozdzielczość,
- im lepsza rozdzielczość tym gorsza granica wykrywalności
Spektrometr mas ICP-MS
Musi być wysoka próżnia, która jest redukowana w układzie wielopolowym.
Sector-field ICP-MS
- droższe, dużo większe od kwadrupolowych,
- lepsza rozdzielczość, gorsza granica wykrywalności.
Układ wprowadzania próbki
- stosowana jest nebulizacja - próbka wprowadzana w postaci cieczy. Nebulizatory mogą być pneumatyczne lub ultradźwiękowe. Najmniej aerozolu tworzy się w pneumatycznym, w ultradźwiękowym 30 %, 40 % w termospreju.
- plazma,
- próbka może być wprowadzana w postaci ciała stałego, czyli za pomocą kuwety grafitowej, próbkę wstrzykuje się i ogrzewa, a następnie transportuje do plazmy.
- ablacja laserowa - próbki stałe (można prowadzić analizę stałych próbek). Na stałą próbkę kierowana jest wiązka z lasera o określonej długości fali, część atomów ulega wyparowaniu (ablacji), strumień gazu kieruje to co powstało do plazmy.
ICP - plazma indukcyjnie sprzężona
Impuls elektryczny powoduje jonizację (zapoczątkowuje reakcję jonizacji), a pole elektryczne następnie utrzymuje reakcję. Temperatura 10000 K - to jonizacja zachodzi z prawie 100 % efektywnością.
Interferencje:
- spektralne izobaryczne i wieloatomowe: *jony wieloatomowe, *jony pierwiastkowe, *jony naładowane, *jony termicznie trwałych tlenków,
Przykłady: m/z = 40 , interferują Ca, Ar, K
m/z = 58 , interferują 58Ni i 58Fe
m/z = 48 , z 48Ti interferują 32S 16O-, 31P16O1H+, z 58Fe+ interferuje 49Ar16O+.
- matrycowe - oddzielenie matrycy i zmiana sposobu wprowadzania próbki do plazmy: membrany desolwatacyjne, komory zderzeniowe.
Zastosowanie:
- granica wykrywalności ppt (ng/l = pg/l = ppt = 10-12 g/ml)
- bardzo czuła technika,
- technika wielopierwiastkowa,
- najczęściej sekwencyjna.
Zakres pomiarowy technik spektralnych:
cena ↓ |
|
|
Ilość próbki |
Granica wykrywalności |
|
FAAS |
ppm |
ml |
10 ppb |
|
CP-AAS |
ppb |
µl |
0,1 ppb |
|
ICP-OES |
Wielopierwiastkowa, zależy od pierwiastka |
ml |
2 ppb |
|
ICP-MS |
Szeroki zakres, wielopierwiastkowa |
ppb-ppt |
|
Przykłady:
100 próbek do analizy, oznaczyć różne pierwiastki - zastosujemy technikę emisyjną
oznaczenie Pb we krwi dziecka (100 próbek) - zastosujemy technikę elektrotermiczną
oznaczenie pierwiastków w wodzie morskiej (100 próbek) - zastosujemy technikę wielopierwiastkową
Klasyfikacja technik chromatograficznych
rodzaj fazy ruchomej i stacjonarnej
- gazowa,
- cieczowa,
-fluidalna (w stanie nadkrytycznym - brak granicy faz, ani ciecz ani gaz)
konstrukcja
- kolumnowa (kapilarna)
- planarna (bibułowa)
rodzaj oddziaływań pomiędzy próbką a fazą stacjonarną
- adsorpcyjna (siły Van del Waalsa)
- podziałowa,
- wymiana jonowa, jonowymienna (IEC), jonowa (IC)
- wykluczania (SEC), żelowa, sitowa, sączenie molekularne, powinowactwa
Pik chromatograficzny
- czas retencji - powinien być jak najkrótszy
- szerokość piku - mierzona w połowie wysokości piku
- czas martwy
Wielkości charakteryzujące proces podziału
- stosunek podziału D = Cs/Cm
Jak szybko migruje substancja kiedy D jest małe? → szybko migruje
Jakie powinny być różnice w D, żeby szybko rozdzielić substancje? → różnice muszą być nie duże
- objętość retencji: Vr = Vm + DVs , gdzie Vr - objętość retencji, Vm, Vs - objętość faz ruchomej i stacjonarnej.
- czas retencji tr = tm (1+k) , gdzie tr - czas retencji, k - współczynnik retencji, tm - czas martwy.
Jaki powinien być współczynnik retencji, aby proces chromatograficzny przebiegał szybko? → musi być mały.
- względny czas retencji tr (próbka), tr' (wz)
- współczynnik rozdzielenia
Α = ((tr)B - tm)/((tr)A - tm) = kB/kA
Charakterystyka kolumny:
1) sprawność
- zależy od ilości półek teoretycznych, od długości kolumny, od czasu w którym kolumna osiąga równowagę
- sprawność musi być jak najlepsza
2) rozdzielczość
- piki muszą być dobrze rozdzielone, nie mogą nachodzić na siebie
R>1,5 rozdzielczość
Α (1-10) selektywność
Analiza jakościowa i ilościowa
- porównanie parametrów retencji
- dodawanie znanych substancji do próbek
- sprzęganie chromatografu ze specyficznym detektorem, np. MS
Pik analitu musi zostać zidentyfikowany i oddzielony od innych pików. Muszą być dostępne wzorce o znanej częstości. Należy używać uznanej procedury kalibrowania: *wykres wzorcowy, *dodatek wzorca wewnętrznego (substancja podobna do identyfikowanej ale nie identyczna), *metoda dodatku wzorca.
Wykład 4 28.04.2009.
Chromatografia planarna - cienkowarstwowa TLC
Faza ruchoma porusza się dzięki silom kapilarnym.
Fazy stacjonarne: żel krzemionkowy, proszek celulozowy, żywice jonowymienne, materiały o ograniczonych rozmiarach jonów, selektory chiralne.
Fazy ruchome: rozpuszczalniki o różnej porowatości i ich mieszaniny.
Detekcja w TLC - mineralizacja chemiczna
- spryskiwanie płytki odczynnikiem chromogennym, np. kwasem fosforomolibdenowym, ninhydryną (aminokwasy i aminy), rodamina (lipidy),
- oglądanie płytki w świetle UV,
- spryskiwanie płytki stężonym kwasem siarkowym, azotowym co powoduje zwęglanie matrycy organicznej,
- densytometryczne - pomiar promieniowania odbitego od powierzchni.
Zastosowanie TLC:
- rozdzielanie związków organicznych
*analiza jakościowa próbek biochemicznych,
*farmaceutycznych (steroidy)
*klinicznych,
*analiza sądowa (narkotyki),
- do sprawdzania czystości związków,
- w procesach produkcyjnych do monitorowania przebiegu reakcji.
Schemat chromatografu HPLC
Układ pobierania rozpuszczalników → układ mieszania rozpuszczalników → pompa (strzykawkowa, tłokowa) → degazer (do odgazowywania próbek, aby nie powstawały pęcherzyki powietrza, odparowywanie ultradźwiękami, odgazowywanie przez membranę) → autosampler (pobiera i wstrzykuje próbkę na kolumnę; od 10 µl do 100-200 µl) → prekolumna (krótsza od kolumny, 10 - 15 cm, służy do wstępnego oczyszczania próbki → kolumna → detektor → system zbierania danych.
Elucja
- izokratyczna - od początku do końca procesu chromatograficznego stosuje się ten sam rozpuszczalnik,
- gradientowa - zmienia się wzajemny stosunek (objętościowy, masowy) jednego rozpuszczalnika do drugiego.
Detektory
Granice wykrywalności [ng]
UV-VIS |
0,1 - 1 |
Gradient |
|
Refraktometryczny |
100 - 1000 |
|
Niespecyficzny, bada rozpraszanie, tani, nie wie co wykrywa |
Elektrochemiczny |
0,01 - 1 |
|
Może zidentyfikować co się bada |
Fluorescencyjny |
0,001 - 0,01 |
Gradient |
Drogi, szeroko stosowany w analizie klinicznej, białka, lipidy |
Konduktometryczny |
0,5 - 1 |
|
Zmiana przewodnictwa, rejestruje zmiany, niespecyficzny |
Spektrometr mas |
0,1 - 1 |
Gradient |
Jeden z najlepszych, liczy masę cząsteczki danej substancji |
FTIR |
1000 |
Gradient |
Spektrometria w podczerwieni z transformacją Fouriera, specyficzny |
Chromatografia podziałowa
Cieczowa - fazą stacjonarna jest ciecz utrzymywana na nośniku w wyniku adsorpcji na powierzchni cząstek wypełnienia.
Z faza powiązaną - fazą stacjonarną jest substancja organiczna chemicznie związana z powierzchnią cząstek wypełnienia - C18 powszechniej stosowane nie wymagają regeneracji, ograniczona pojemność.
Chromatografia podziałowa:
- w normalnym układzie faz - faza stacjonarna bardziej polarna niż faza ruchoma, faza ruchoma - węglowodory, alkohole lub rozpuszczalniki chlorowane,
- w odwróconym układzie faz - faza stacjonarna mniej polarna niż faza ruchoma, najczęściej stosowana faza ruchoma: bufor - metanol, woda - acetonitryl, THF.
Jakie anality będą wymywane jako pierwsze? → polarne.
Jak na szybkość wymywania wpłynie zwiększenie polarności fazy ruchomej? → znacznie przyśpieszy szybkość wymywania.
- z tworzenia par jonowych do związków dysocjujących. Wybór rozpuszczalnika na podstawie szeregu eluotropowego.
Fazy stacjonarne
- polarne fazy stacjonarne:
*SH(CH3)2NH2 (aminoalkilowe, aminopropylowe)
*SH(CH3)2CN (cyjanoalkilowe)
*SH(CH3)2OCHOHCH2OH (diolowe)
- niepolarne fazy stacjonarne
- R-(CH2)3CH3 (akta decylowa, oktylowe, butylowe, etylowe)
- (CH2)2C6H11
Zastosowanie:
- farmaceutyki - antybiotyki, uspokajające, przeciwzapalne, stereoidy,
- biochemiczne - aminokwasy, białka,
- żywność,
- zanieczyszczenia,
- chemia kryminalistyczna,
- medycyna kliniczna.
Chromatografia adsorpcyjna
Faza stacjonarna: żel krzemionkowy, tlenek glinu,
Faza ruchoma: rozpuszczalnik organiczny lub mieszanina rozpuszczalników
Adsorpcja powierzchniowa na grupach silanolowych -Si-OH, wymywanie w kolejności wzrastającej polarności.
Zastosowanie:
Tabela: Adsorbenty stosowane w chromatografii kolumnowej oraz przykłady ich zastosowania.
Tabela: Rozpuszczalniki uszeregowane zgodnie z wzrastającą mocą elucyjną w chromatografii adsorpcyjnej na żelu krzemionkowym.
Chromatografia żelowa
Rozdział: wypełnienie polimeryczne (pory o różnej średnicy). Cząsteczki które są za małe lub za duże przechodzą przez pory, a inne są zatrzymywane. Wybiera się odpowiedni bufor do kolumny. Na samym końcu wychodzą te, które są zatrzymywane najdłużej.
Faza stacjonarna - porowaty żel o określonej wielkości porów hydrofilowy lub hydrofobowy.
Faza ruchoma - ciecz lub gaz, brak oddziaływań miedzy faza stałą a analitem.
Zastosowanie:
- cukry, białka, szybkie oznaczanie masy cząsteczkowej lub rozkładu mas cząsteczkowych polimerów, produktów naturalnych.
W porach zatrzymywane są jedynie małe cząsteczki analitu, duże cząsteczki są wykluczane.
Chromatografia powinowactwa
Faza stacjonarna: agarowa porowate kulki szklane z unieruchomionym ligandem powinowactwa.
- ligandy, przeciwciała, inhibitory enzymów,
- elucja po zmianie warunków fazy ruchomej,
- funkcje fazy ruchomej,
- wysoka specyficzność.
Chromatografia jonowymienna
Faza stacjonarna: zwykle żywica zawierająca związane kowalencyjnie grupy funkcyjne, anionowe (np. HSO-) lub kationowe (np. -N(CH3)3+)
Faza ruchoma: ciecz lub gaz.
Oddziaływanie elektrostatyczne między fazą stałą a analitem.
Budowa jonitu
- zbudowane z polimeru, który stanowi rdzeń wypełnienia cząsteczki, na nim znajdują się grupy funkcyjne odpowiedzialne za rozdzielanie kationowymienne (aminy III-rzędowe), anionowymienne.
Tabela: rodzaje jonitów.
Anionity:
- silnie zasadowe,
- średnio i słabo zasadowe,
Kationity:
- silnie kwasowe -SO3-
- średnio kwasowe -PO3-
- słano kwasowe -COO-
- bardzo słabo kwasowe -OH
Amfoteryczne
Chromatografia jonowa
- wysokosprawna chromatografia jonowa IC
- wysokosprawna chromatografia wykluczania jonowego HPICE
- chromatografia par jonowych MPIC
Aparatura
Dozowanie → pompa → zawór wstrzykowy → prekolumna → kolumna → degazer → supresor (dodatkowy element) → detektor → monitor
Wzrost powinowactwa
- ze wzrostem wartościowości, np. (3+) silniej zatrzymywany niż (1+)
- ze wzrostem promienia jonowego, np. Ca (większy) silniej zatrzymywany niż Mg (mniejszy)
- ze wzrostem stopnia jonizacji
Supresor
- podwyższa czułość oznaczeń
- poprawia wykrywalność
- wydłuża czas analizy, gdyż wymaga regeneracji
- oddziela wstępnie nadmiar kationów 1 wartościowych lub anionów biogennych aby nie przeszkadzały na chromatografie.
Normy IC w analizie wody i ścieków.
Zalety chromatografii jonowej:
- możliwość jednoczesnego oznaczania kilkunastu jonów
- wykrywalność µg/l
- tania
- małe zużycie rozpuszczalnika
Przygotowanie próbek do analizy, gdy konieczne jest wzbogacanie.
Przygotowanie próbek gazowych
- absorpcja w r-rach HF, HCl, NOx,
- adsorpcja w wodzie z CO32-, o,1 M NaOH.
Przygotowanie próbek ciekłych
- filtracja,
- SPE do usuwania jonów Cl-, SO32-, metali
- deprywatyzacja analitów
- techniki membranowe
- zmiana pH
Chromatografia
Jonowa |
Jonowymienna |
Słabe eluenty mmol/l |
Silne eluenty mol/l |
Oznaczanie mieszanin |
Analiza pojedynczych składników |
Jonity o małej pojemności wymiennej |
Jonity o dużej pojemności wymiennej |
Kolumna supresyjna |
|
Duża szybkość analizy |
Niewielka szybkość analizy |
Różne metody detekcji |
Detekcja konduktometryczna |
Granica wykrywalności µg/l |
Granica wykrywalności mg/l |
Jak wybrać odpowiednią techniką?
- lotne, stałe techniczne
- małe masy cząsteczkowe
- tani sprzęt
- nielotne (chromatografia cieczowa)
- specyficzne (chromatografia powinowactwa)
- rozpuszczalne (zależy w czym)
*hydrofobowe
**rozmiar (żelowa)
**grupy funkcyjne (chromatografia adsorpcyjna)
**polarność (podziałowa w normalnym układzie faz)
*hydrofilowe
**rozmiar (wykluczania, żelowa)
**polarność (podziałowa, odwrócony układ faz)
- po dodaniu odpowiednich odczynników (odwrócony układ faz)
Wykład 5 05.05.2009
MeHg 1950 - pochodne organiczne
Katastrofa nad zatoką Mina mate, Japonia
- ścieki z fabryki tworzyw sztucznych i poliwinylowych
Specjacja
Występowanie i rozmieszczanie różnych chemicznych i fizycznych form danego pierwiastka w rzeczywistych próbkach.
Analiza specjacyjna
Analiza mająca na celu identyfikację i/lub pomiar zawartości jednej lub kilku form chemicznych pierwiastka w próbce.
Badania specjacji dotyczą:
- atmosfery
- hydrosfery
- litosfery
- biosfery:
*materiał biologiczny
*tkanki zwierzęce
*żywność
*tkanki ludzkie, płyny ustrojowe
Frakcjonowanie
Proces klasyfikacji analiz lub grupy analitów w danej próbce zgodnie z ich właściwościami:
- fizycznymi (rozmiar, rozpuszczalność)
- chemicznymi ( rodzaj wiązania, reaktywność)
Rozmieszczenie metali w glebach:
- łatwo rozpuszczalne: wodorowęglany
- wymienne: przy obniżonym pH
- sorbowane przez wodorotlenki i tlenki Fe, Mn, Al.
- związane z substancją organiczną - niedostępna dla roślin
- związane z glinokrzemianami
Ekstrakcja sekwencyjna Pb, porównanie trzech procedur:
- procedura Tessiera
Łatwo wymienne MgCl2
Węglany NaAc/HAc
Tlenki NH2OH*HCl / HAc
Związki organiczne H2O2 / HNO3 / NH4Ac (octan amonowy)
Pozostałość HF / HClO4
*procedura BCR (organizacja europejska)
*zmodyfikowana procedura Tessiera
Ekstrakcja sekwencyjna
- stosowanie różnych procedur nie pozwala porównać wyników ze sobą
- błędy mogą wystąpić z powodu redystrybucji, readsorpcji, wpływu różnych warunków reakcji.
Aspekty strukturalne badan specjacji:
- skład izotopowy
- stopień utlenienia
- związki i kompleksy nieorganiczne
- kompleksy organiczne (kompleks metalu i związku organicznego)
- związki metaloorganiczne (różnią się wiązaniem miedzy metalem a związkiem organicznym - koordynacyjne, kowalencyjne miedzy metalem a węglem)
- związki i kompleksy małocząsteczkowe
Aspekty toksykologiczne
- oznaczanie antropogenicznych zanieczyszczeń środowiska związkami metaloorganicznymi, np. alkilowymi pochodnymi Hg, Pb, Sn.
Me4Pb, Me3EtPb, Me2Et2Pb, MeEt3Pb, Et4Pb (dodawanie do benzyny ołowiu), Me2Pb2+, Et2Pb2+, Me3Pb+, Et3Pb+.
- oznaczanie pierwiastków na różnych stopniach utlenienia
Cr(III)/Cr(VI), As(III)/As(V)
As(III) jako As2O3
Aspekt kliniczny i biochemiczny
- kontrola homeostazy, metabolizmu, detoksykacji pierwiastków niezbędnych do prawidłowego funkcjonowania organizmów
- wyjaśnienie mechanizmu transformacji biochemicznej i prostych związków nieorganicznych w organizmach żywych
- oznaczanie związków o wysokich masach cząsteczkowych: *metaloporfiryny, *kompleksy z białkami: fitochelatynami i metalotioneinami, *metaloenzymy, *metalocukry, pochodne chlorofilu, kobalaminy, glikoproteiny.
Metody bezpośrednie badania specjacji:
- potencjometria bezpośrednia z elektrodami jonoselektywnymi do pomiaru stężenia wolnych jonów,
- woltamperometria do pomiaru stężeń labilnych form pierwiastków elektroaktywnych,
- jądrowy rezonans magnetyczny (Al w liściach herbaty).
Techniki rozdzielania:
Metody fizyczne: filtracja, ultrafiltracja, odwrócona osmoza, dializa.
Metody chemiczne: np. ekstrakcja, chromatografia: GC, LC, HPLC, SFC, SFE, elektroforeza kapilarna strefowa CZE, elektrochromatografia EC.
Trwałość specjacji zależy od:
- formy i właściwości analitu
- poziomu stężeń analitów
- matrycy próbki,
- rodzaju materiału pojemnika do przechowywania/przygotowania próbki i jego historii (np. procedury mycia, np. na powierzchni)
Techniki połączone w analizie specjacji
LC, EC Absorption, QF, QP MASS, ICP, EI
Chromatografia + Spektrometria
Super… GC Emisyjne fluororescencyjne
Chromatografia gazowa
- lotne związki lub … przeprowadza w lotne pochodne
*tworzenie wodorków
*etylowanie
*stabilne termicznie
*dobra wydajność wprowadzania substancji gazowych do źródła wzbudzania lub źródła jonów
Ładunek cząsteczek
HPLC - jonowymienne (układy redoks, organiczne związki Se, organiczne związki As, polipeptydy, kompleks metabolitów z białkami), faz odwróconych i par jonowych(układy redoks, polipeptydy, związek selenowy), wykluczeniowa(wielocukry, metaloproteiny, fitochelatyny, metaloenzymy, białka)
Identyfikacja
- proste związki metaloorganiczne
*porównanie czasów retencji,
*metoda dodatku wzorca
*analiza certyfikowanych materiałów odniesienia
Chromatografia wielowymiarowa
Klucz do sukcesu
- zachowanie oryginalnych form analitów w próbce
- prowadzenie pomiarów w oryginalnej niezmienionej próbce w jej naturalnym środowisku(analiza In situ) w czasie rzeczywistym
- właściwa kalibracja, poprawne obliczenia
- walidacja metody
Proteonika - porównanie budowy cząsteczek z jej właściwościami.
Wykład 6 12.05.2009
Elementy chemometrii
Chemometria - dziedzina chemii wykorzystującej matematykę, rachunek prawdopodobieństwa, statystykę, informatykę oraz teorię podejmowania decyzji do optymalizacji procedur eksperymentalnych w celu uzyskania maksymalnej ilości użytecznej informacji o obiekcie badan na podstawie analizy danych.
Specyfika danych chemometrycznych
x11… … x1j … … x1m
………………………. n<<m
xn1 … … xnj … … xnm
Etapy rozwiązywania problemów
- sformuowanie problemu
- planowanie problemu
- wykonanie problemu
- przechowywanie i kontrola wyników: eliminacja błędów grubych za pomocą np. analizy wizualnej
- wykonanie oparte na testach statystycznych
Obszary zastosowań technik chemometrycznych
Przedstawienie danych kolumny reprezentują zmienne
Wiersze - obiekty (próbki)
Analiza chemometryczna
- analiza czynników,
- konstruowanie modelu zależności
- analiza podobieństw
- analiza trendów czasowych
Planowanie doświadczeń
- na etapie planowania eksperymentu musimy wiedzieć jaką technika chemometryczną będziemy się posługiwać,
- do analizy podobieństw wystarczy jedna zmienna
- analiza czynnikowa wymaga n ≥ 3 oraz ilość obiektów/cech =3:1
- określenie optymalnej próby pomiarów: liczba pomiarów powinna być większa niż minimalna → 4 lub 5 < n < 10
Liczebność zbioru testowego około 10 % zbioru liczącego, ale nie mniej niż 5 obiektów
- wybór rozmieszczenia punktów pomiarowych
* kryterium zupełności
* kryterium proporcjonalności (częstość występowania obiektu w próbie generalnej)
Plan optymalny - to taki który przy danej liczbie punktów pomiarowych n zapewni największą wiarygodność uzyskanego rozwiązania.
Archiwizacja i kontrola danych:
Cel kontroli danych:
- wykrycie ewentualnych „błędów grubych”
- zmniejszenie ryzyka popełnienia nowych błędów na kolejnych etapach analizy chemometrycznej
- wykrycie pewnych relacji tkwiących w danych
- wskazanie na potrzebę wykonania dodatkowych operacji na danych, np. transformacji zmiennych.
Dokumentacja danych i wyników:
Brakujące dane:
- puste miejsca w tabeli
- w tabeli wpisujemy zero tylko wtedy, gdy jest to wartość rzeczywista mierzonej cechy.
Przyczyny braku danych:
- brak analizy zaplanowanej próbki,
- wykonanie oznaczeń tylko niektórych analitów (jeśli zmienna wykazuje 10 % braków to należy je usunąć)
- stężenie analitu poniżej granicy oznaczalności stosowanej techniki analitycznej
Kontrola danych:
- analiza pojedynczych zmiennych
- analiza relacji pomiędzy zmiennymi
- wieloparametrowa analiza rozkładu badanych obiektów
Kontrola danych:
- eliminowanie błędów grubych
- statystyczne wykrywanie obiektów różniące się istotnie od innych
- przetestowanie rozkładu zmiennych w celu uzyskania przesłanek do ewentualnej transformacji
- określenie jednorodności zbioru danych (potwierdzenie, że uzyskane dane należą do jednej populacji)
Analiza pojedynczej zmiennej
- wartość najmniejsza (min)
- wartość największa (max)
- stosunek min/max
- rozstęp rozkładu r = max - min
- środek rozkładu d = (max + min)/2
- wartość średnia
- mediana
- odchylenie standardowe (s)
- normalność rozkładu i indeks skośności rozkładu (q)
Badanie charakteru rozkładu zmiennej
- tworzenie histogramów
- liczba przedziałów histogramu k: n/4 ≥ k
- przedziały muszą mieć jednakową szerokość
- krańce przedziałów powinny być liczbami „okrągłymi”, np. 1,5 a nie 1,48
Relacje między zmiennymi:
- współczynnik korelacji liniowej (r)
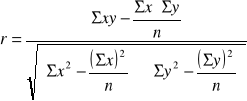
Gdy r jest bliskie 0 to brak związku pomiędzy zmiennymi.
- współczynnik determinacji (r2) - jaki % jednej zmiennej jest określany przez drugą zmienną.
Transformacja zmiennych
Celem jest umożliwienie lub ułatwienie przeprowadzenia dalszych etapów analizy chemometrycznej. Gdy chcemy lub musimy doprowadzić rozkład zmiennej do rozkładu zbliżonego do rozkładu normalnego. Gdy dzięki transformacji chcemy uzyskać liniową zależność pomiędzy zmienną zależną i objaśniającą.
Normalizacja - procedura prowadząca do uzyskania rozkładu zmiennej zgodnego z rozkładem normalnym.
Autoskalowanie (standaryzacja) prowadzi do takiego przekształcenia danych, gdzie średnia równa jest 0, a odchylenie = 1, pozwala na analizę zmiennych o różnej naturze i różnym zakresie.
Centrowanie - wszystkie wartości średnie wszystkich zmiennych pokrywają się z początkiem układu współrzędnych.
Budowanie modeli
Ma na celu ustalenie zależności pomiędzy jedną zmienną zależną y ( lub wieloma zmiennymi y) i bardzo licznym zbiorem zmiennych objaśniających Xi.
Typy modeli:
- model w pełni określony, gdy znamy postać funkcji i wartości wszystkich występujących w niej parametrów
- model półempiryczny, gdy znana jest postać funkcji lecz dla danego obiektu brakuje informacji o jego parametrach
- model empiryczny, gdy nie znamy ani postaci funkcji ani jej parametrów.
Analiza podobieństw
- technika chemometryczna bazująca na pojęciu odległości obiektów lub zmiennych w przestrzeni wielowymiarowej
- obiekty uznajemy za podobne, jeśli są położone blisko siebie w wielowymiarowej przestrzeni zmiennych objaśniających.
Rodzaje podobieństw:
- podobieństwa obiektów ze względu na wartość cech
- podobieństwo cech przy opisie obiektów
Analiza taka może stanowić:
- niezbędny etap wstępnej obróbki danych
- źródło danych do poprawnej redukcji liczby zmiennych objaśniających
- podstawę optymalnej metody graficznej prezentacji danych
- samodzielny cel analizy chemometrycznej
Wizualizacja w składzie obiekty - cechy
Dane jednorodności zbioru obiektów
Analiza podobieństw
Opracowanie reguł przypisania obiektów do określonych grup
Techniki aglomeracji: algorytm odległości (odległość tangenowa, wektorowa,)
Analiza wiązkowa
Dendryty
Metoda głównych składowych (PCA)
Chemometryczna strategia przeznaczona do analizowania wielowymiarowych zbiorów wyników,
- prezentacja graficzna zależności wielowymiarowych
- redukcja wielowymiarowości problemu, przekształcenie J skorelowanych zmiennych wyjściowych w A głównych składowych (A<J)
- umożliwienie merytorycznej interpretacji uzyskanych wyników i zależności
Liczbę istotnych składowych można określić stosując następujące kryteria:
- kryterium poglądowości
- kryterium zasobu zmienności
- kryterium spadku wartości własnych
Przykłady zastosowań chemometrii
- identyfikacja źródeł pochodzenia amfetaminy (PCA)
- wieloparametrowa charakterystyka szczepów arylobakterii
- analiza win
- optymalizacja procedur
Wykład 7 26.05.2009
Sensor chemiczny
Urządzenie przetwarzające informację chemiczną począwszy od stężenia określonego składnika próbki, po ogólny skład matrycy na sygnał analityczny:
Sygnał analityczny ← przetwornik ← część receptorowa ← analit
Historycznie
- szklana elektroda pH (lata 30-te XX w.)
- elektrody jonoselektywne (lata 60-te), pomiary potencjometryczne
- gazowa elektroda tlenowa Clarka - pomiary wolt amperometryczne
- elektroda jonoselektywna modyfikowana enzymem - pierwszy biosensor 1977 r.
Rodzaje oddziaływań
- oddziaływania powierzchniowe substancji zaabsorbowanej na powierzchni
- oddziaływania wewnątrzfazowe, efekt podziału analitu miedzy dwie fazy próbki i warstwy aktywnej sensora
- na selektywność mogą wpływać:
* rozmiar i kształt
* udział entropii w wiązaniu
* kinetyka procesów
Cechy dobrych czujników:
- mocne i stabilne w działaniu
- szybka i odtwarzalna odpowiedź na analit
- dobra selektywność
- szeroki zakres pracy
- niezależność od warunków pracy, temperatury, ciśnienia, itp.
Rodzaje czujników:
- elektrochemiczne
- optyczne
- elektryczne
- termiczne
- masowe
- biochemiczne
Wg typu zastosowanego przetwornika odwracalne (ISE) i nieodwracalne (najczęściej enzymatyczne)
Sensory elektrochemiczne
Sygnaly: potencjometryczne, amperometryczne, wolt amperometryczne, kulometryczne, konduktometryczne, powstają dzięki przebiegowi reakcji elektrochemicznych analitu lub dzięki obecności określonych cząstek z nim związanych.
Sensory optyczne
- badana wielkość podlega przetwarzaniu w warstwie receptorowej na skorelowany w stężeniu sygnał optyczny. W warstwie przetwornikowej sygnał zamieniany jest na końcowy sygnał elektryczny,
- odpowiadają za absorpcję lub fluororescencyjna emisję promieniowania przez anality, wskaźniki lub kompleksy analit-receptor.
Optrody: spektrofotometryczne, luminescencyjne, optometryczne.
Światłowody jednomodowe, światłowody wielodomowe.
Sensory światłowodowe i wykorzystujące wiązkę światłowodów.
Zastosowanie
- procesowa kontrola analityczna - czyste technologie,
- chemia kliniczna,
- ochrona środowiska
- przemysł motoryzacyjny
Chromatografia
Gazowa
Cieczowa
gaz - ciecz
gaz - ciało stałe
ciecz - ciecz
ciecz - ciało stałe
Chromatografia
Gazowa
Cieczowa
Nadkrytyczna
Adsorpcyjna
Podziałowa
Jonowa
Żelowa
Adsorpcyjna
Adsorpcyjna
Podziałowa
Podziałowa
Eksperyment terenowy lub laboratoryjny
Gromadzenie danych eksperymentalnych, wyniki analiz, obserwacje, szczegółowy opis eksperymentu, archiwizacja parametrów eksperymentu
Zmienne
Dane surowe
Obiekty
Wyszukiwarka