
Appl Microbiol Biotechnol (2005) 68: 774
–778
DOI 10.1007/s00253-005-1948-8
A P P L I E D G E N E T I C S A N D M O L E C U L A R B I O T E C H N O L O G Y
Yingfeng An . Jianfei Ji . Wenfang Wu . Anguo Lv .
Ribo Huang . Yutuo Wei
A rapid and efficient method for multiple-site mutagenesis
with a modified overlap extension PCR
Received: 3 February 2005 / Revised: 20 February 2005 / Accepted: 22 February 2005 / Published online: 14 June 2005
# Springer-Verlag 2005
Abstract A rapid and efficient method to perform site-
directed mutagenesis based on an improved version of
overlap extension by polymerase chain reaction (OE-PCR)
is demonstrated in this paper. For this method, which we
name modified (M)OE-PCR, there are five steps: (1)
synthesis of individual DNA fragments of interest (with
average 20-bp overlap between adjacent fragments) by
PCR with high-fidelity pfu DNA polymerase, (2) double-
mixing (every two adjacent fragments are mixed to im-
plement OE-PCR without primers), (3) pre-extension (the
teams above are mixed to obtain full-length reassembled
DNA by OE-PCR without primers), (4) synthesis of the
entire DNA of interest by PCR with outermost primers and
template DNA from step 3, (5) post-extension (ten cycles
of PCR at 72°C for annealing and extension are im-
plemented). The method is rapid, simple and error-free. It
provides an efficient choice, especially for multiple-site
mutagenesis of DNAs; and it can theoretically be applied to
the modification of any DNA fragment. Using the MOE-
PCR method, we have successfully obtained a modified
sam1 gene with eight rare codons optimized simultaneously.
Introduction
Site-specific mutagenesis of DNA is a very important tool
in genetic engineering. Changing the DNA sequence can
facilitate the study of the structure
–function relationships
of DNA, RNA, or protein coded by the DNA sequence. A
variety of methods have been applied for the introduction
of specific bases changes at predetermined sites in DNA
sequences (Higuchi et al.
; Warrens et al.
; Chiu
et al.
; Allemandou et al.
; Rabhi et al.
;
Tyagi et al.
; Li et al.
; Kirsch and Joly
;
Kegler-Ebo et al.
; Zoller and Smith
), the most
powerful among which is overlap extension by polymerase
chain reaction (OE-PCR), described by Higuchi et al.
(
). In some cases, it is desirable to introduce multiple
different substitutions at a particular position or at several
positions in a gene and to determine the consequences
of these changes on protein function or to optimize the
expression. However, traditional OE-PCR can introduce
mutations at only one site at a time; and efficiencies drop
drastically when a few sites are targeted simultaneously.
We have developed a rapid, efficient and high-fidelity
modification (M) of OE-PCR (i.e. MOE-PCR) which al-
lows one to generate multiple site-directed mutations in a
given DNA fragment synchronously. The method has been
successfully implemented to optimize eight rare codons in
the sam1 gene (ID 850877); and no unexpected mutagen-
esis was detected.
Materials and methods
Materials
Chemicals, high-fidelity pfu DNA polymerase, T4 DNA
ligase, DL2000 DNA marker and restriction endonucleases
Electronic Supplementary Material Supplementary material
is available for this article at http://dx.doi.org/10.1007/s00253-005-
1948-8.
Y. An . W. Wu (
*) . A. Lv
Institute of Applied Ecology,
Chinese Academy of Sciences,
Shenyang, China
e-mail: wshr100@sina.com
Y. An
Graduate School of Chinese Academy of Sciences,
Beijing, China
J. Ji
Beckman Research Institute,
City of Hope National Medical Center,
Duarte, USA
R. Huang . Y. Wei
Laboratory of Protein Engineering,
College of Life Science & Technology,
Guangxi University,
Guangxi, China

were purchased from Takara Co. (Dalian, China). Vector
pYES2, host strain Escherichia coli JM109 and Saccha-
romyces cerevisiae INVScI (products of Invitrogen Corp.)
were presented by the Laboratory of Protein Engineering,
Guangxi University. Oligonucleotide primers were synthe-
sized and purified by Sangon Co. (Shanghai, China). The
PCR purification kit and gel extraction kit were purchased
from Watson Biotechnologies (Shanghai, China); and the
DNA sequencing was performed by Takara Co. (Dalian,
China).
The five steps in the reaction process were as follows.
Step 1
Six parallel PCR reactions were performed to amplify each
DNA fragment, with 5 units of pfu DNA polymerase and
1× buffer in the presence of 200
μM dNTP, 1 mmol of each
primer (Table
) and 10 ng of S. cerevisiae genomic DNA
in a final volume of 100
μl. The PCR reaction programs
were optimized respectively as follows: 94°C for 2 min,
then 30 cycles of 94°C for 30 s for denaturation, 59.6°C for
30 s and 72° for 55 s, followed by 72°C for 10 min for
fragment 1; and the reaction conditions for the other frag-
ments were similar, except for the annealing temperature
and extension time. The annealing temperatures used for
fragments 2
–6 were 62.5, 58.8, 59.5, 55.1 and 53.3°C,
respectively; and the extension times used for fragments
2
–6 were 1 min 25 s, 55 s, 1 min 20 s, 50 s and 55 s,
respectively. Amplified products were loaded on a 1% aga-
rose electrophoresis gel and purified by a gel extraction kit.
Step 2
The following reactions were divided into five teams and,
in each team, equimolar aliquots of every two adjacent
DNA fragments (10 ng for each) above were mixed in the
presence of 1 unit of pfu DNA polymerase, 1× buffer,
200
μmol of each dNTP in a final volume of 20 μl. The
reaction programs include 15 cycles of PCR without prim-
ers. Denaturation was at 94°C for 20 s, with various an-
nealing and extension conditions: 59.6°C for 1 min 25 s for
fragments 1, 2, 58.8°C for 1 min 25 s for fragments 2, 3,
58.8°C for 1 min 20 s for fragments 3, 4, 55.1°C for 1 min
20 s for fragments 4, 5 and 53.3°C for 55 s for fragments
5, 6.
Step 3
Next, 20
μl of every product from step 2 were mixed
together to perform 20 cycles of 94°C for 20 s, 72°C for
30 s without primers.
Step 4
To add both outer primers (40 pmol for each) to the reaction
system, PCR was implemented as follows: 94°C for 20 s,
then 30 cycles of 94°C for 20 s, 53.3°C for 30 s and 72°C
for 2 min.
Step 5
Ten additional cycles of 94°C for 30 s and 72°C for 1 min
were implemented.
Comparisons omitting steps 2,3, 5
As comparisons, processes without steps 2, 3 or 5 were
implemented, respectively. The PCR product was subjected
Frag-
ments
Primers
Sequences for primers (listed 5’to3’)
Bases
substitute
F
0
ATTGGATCCCAACGATGGCACTAGACATA
-------
1
R1
AGCGGTTTCACAAGCAACTTTGGAGTG
C to A
F1
GCTTGTGAAACCGCTGCAAAGACTGGTAT
G to T
2
R2
AGCTAAAGAGCCATCTCTTCTAGCGTCAGCCATG
C to A
F2
GCTAGAAGAGATGGCTCTTTAGCTTGGTTGAGACC
G to T
3
R3
AGCTCTTAAGTCCTCGGTAGTGATTTCGTCAGC
C to A
F3
ACTACCGAGGACTTAAGAGCTCAACTAAAGTCC
G to T
4
R4
CAATGGTTCAGCAATACCGATGGC
C to A
F4
ATGCCATCGGTATTGCTGAACCATTGTCC
G to T
5
R5
GTCAGACTTGGTAGCAGTACCATAGGTGTCAAC
C to A
F5
ACCTATGGTACTGCTACCAAGTCTGACGAAG
G to T
6
R
0
GCAGAATTCGAGGTTGAAGGCAGAAAA
------
Table 1 The mutagenic primers
used for optimizing rare
codons in sam1 with MOE-PCR
The bases for substitutions were
shadowed. The restriction sites
for BamHI and EcoRI were
in boxes
775
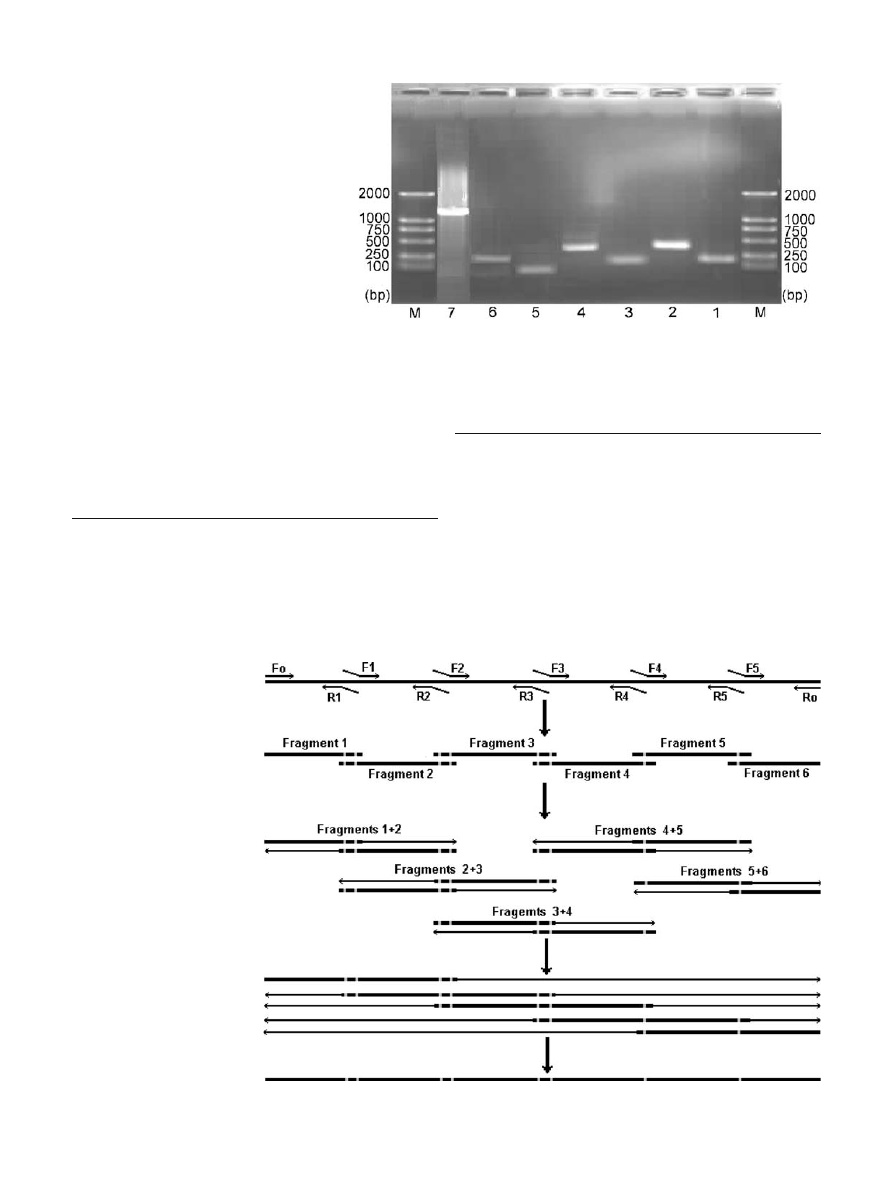
to agarose gel electrophoresis and the desired bound was
then excised and purified with the gel extraction kit. The
purified DNA was subjected to BamHI and EcoRI diges-
tion and cloned into similarly digested pYES2 vector, ac-
cording to standard procedures (Sambrook et al.
). The
recombinant plasmid was transformed into E. coli JM109;
and recombined vector was extracted and sequenced to
verify the accurate mutations.
Results
We successfully applied MOE-PCR to introduce eight
codon-substitutions into the DNA of sam1. After a round
of reassembly and extension, DNA of the desired size was
obtained (Fig.
). DNA was cloned into the vector and the
sequences analyzed; and all the rare codons were optimized
properly without additional mutations introduced (Supple-
mentary Material, Appendix 1).
Discussion
Compared with OE-PCR for site-directed mutagenesis,
MOE-PCR (Fig.
) has some apparent advantages, one of
which is that, when multiple-site mutations are implemen-
ted, relative fewer PCR cycles are required for MOE-PCR.
As for the present research, eight sites were mutated and a
total of 255 cycles of PCR reaction were implemented,
including 180 cycles for fragment synthesis (30 cycles for
each fragment), 15 cycles for double-mixing, 20 cycles
pre-extension, 30 cycles for PCR extension and 10 cycles
Fig. 1 One percent agarose
electrophoresis gel showing
eight site substitutions in sam1
by MOE-PCR. Lane M DNA
size marker DL2000, lanes 1
–6
fragments 1
–6 synthesized by
PCR, lane 7 extension of the
target DNA by PCR with out-
ermost primers and template
DNA from step 3
Fig. 2 The use of MOE-PCR
for eight site-substitutions in
sam1. a Synthesis of DNA
fragments by PCR, gray bars
represent substitution sites.
b Double-mixing: every two
adjacent fragments are mixed to
implement OE-PCR without
primers. c Pre-extension: the
teams above are mixed to obtain
full-length reassembled DNA by
OE-PCR without primers.
d Synthesis of the entire DNA
of interest by PCR with outer-
most primers and template DNA
from step3 (c). e Post-extension:
ten cycles of PCR with 72°C for
annealing and extension are
implemented
776

post-extension. If OE-PCR was used, not less than 540
cycles would be needed for an eight-codon optimization
(within six fragments), because three fragments (including
a full-length DNA) need to be synthesized by PCR within
each round of the reaction; and six rounds for the whole
process (30 cycles for every round) are necessary tradi-
tionally. Additionally, only seven fragments need be pu-
rified in MOE-PCR, because no purification is needed after
steps 2, 3 and 4, while if OE-PCR was implemented,
purification would be necessary after every step and 18
fragments as a whole would be purified, respectively. Ad-
ditionally, to ensure efficient and error-free extension, every
program for PCR must be optimized, so for OE-PCR, 21
program-optimizations are needed, while MOE-PCR re-
quires only nine (pre-extension, post-extension programs
fixed for 94°C denaturation, 72°C annealing, extension). To
sum up, it is obviously more efficient and time-saving for
the application of MOE-PCR.
Through comparison, the necessity of pre-extension was
verified since, without it, only the dispersed band was ob-
tained through electrophoresis. Through alignment anal-
ysis between DNA fragments synthesized in step 1 with
Vector NT software, the results show that there were ob-
vious identity (average 40%) between every two of the
fragments (Supplementary Material, Appendix 2). As a re-
sult, when all the fragments are mixed together directly, the
multiple fragments interrupt each other for correct overlap.
To avoid this trouble, double-mixing was implemented.
When only two fragments were mixed and overlap-ex-
tended, the reaction component was relatively simple and
the interruption was minimized. More importantly, the gen-
eration of longer identical ends between adjacent fragments
in this step facilitated the overlap extension in the following
step.
In the pre-extension, owing to the long overlap regions
generated between newly extended fragments, a higher
temperature (72°C) was applied for the annealing and ex-
tension process, which was the optimized temperature for
pfu DNA polymerase. At the same time, a higher tem-
perature also decreases the possibility of any mismatch.
We also tested the necessity of PCR without primers,
after which reassembly was possible, while it was un-
available when primers were added directly to the mixture
of fragments (i.e. without steps 2, 3). This result affords
some thought, because theoretically the fragment overlap-
extensions and PCR with primers can occur simultaneously
in a reaction system and there seems to be no substantial
difference between the two treatments, except for that some
full-length templates are generated after PCR without
primers. Therefore, the prepared full-length template may
be essential for PCR in addition to overlapped fragments.
Although uncertain, the disturbance of template generation
by primers may be crucial for this result. Because the ratio
of primer to template is 10
6
in the PCR system, which
means that every molecular DNA fragment was enveloped
with 10
6
primers, the spatial obstruction by primers could
encumber the annealing process between the fragment ends
and thus restrain the generation of full-length templates.
This was especially the case when multiple-step overlaps
and extension with multiple DNA fragments were im-
plemented. Additionally, after steps 2 and 3, the full-length
template was ready for the exponential extension of the
desired DNA, while the template needed to be reassembled
and accumulated for preliminary cycles in the absence of
PCR without primers, which could be another factor to be
mentioned.
The necessary of post-extension was also tested for ob-
taining the reassembled DNA; and only a disperse band
was obtained without post-extension (data not shown).
After reassembly by PCR, there existed both remnant and
incompletely extended DNA fragments; and such frag-
ments had to be minimized, because the annealing of large
amounts of these fragments to full-length single-strand-
ed DNA (generated during denaturation) would give less
chance for the generation of the target double-stranded
DNA (dsDNA). As a result, distinct dsDNA cannot be de-
tected, although it is available in theory; and this was the
case especially when multiple original fragments were used
for reassembly. In the post-extension process, 72°C was
used for annealing and extension, which was higher than
the annealing temperatures of both primers. At 72°C, the
DNA recombining by overlap and extension of fragments
should induce full-length DNAs to be generated, accom-
panied by a decrease in DNA fragments, but the synthesis
of new DNA fragments by primers is avoided. The con-
ditions for the above steps were optimized and all the re-
action processes were repeated to attest the recurrence.
In summary, the MOE-PCR method we have demon-
strated here allows the rapid and efficient introduction of
multiple mutations into DNA. Although multiple-site mu-
tagenesis is also available using older methods, those pro-
cesses are tedious and time-consuming, while the method
presented here minimizes the process of multiple-site mu-
tagenesis with error-free results, which is an important im-
provement; and it can be widely applied wherever gene
substitutions, deletions or insertions in different sites are
needed, including the protein pharmaceutical industry, ag-
riculture, chemical industry, biotechnology, etc., especially
for enzyme engineering.
Acknowledgements
The authors thank Frances H. Arnold for data
and Han Siqin, Xu Mei and Ni Jianfeng for helpful discussion and
technical assistances. We are grateful to the Laboratory of Protein
Engineering, Guangxi University for technical support.
References
Allemandou F, Nussberger J, Brunner HR, Brakch N (2003) Rapid
site-directed mutagenesis using two-PCR-generated DNA frag-
ments reproducing the plasmid template. J Biomed Biotechnol
2003:202
–207
Chiu J, March PE, Lee R, Tillett D (2004) Site-directed, ligase-
independent mutagenesis (SLIM): a single-tube methodology
approaching 100% efficiency in 4 h. Nucleic Acids Res 32:
e174
Higuchi R, Krummel B, Saiki RK (1988) A general method of in
vitro preparation and specific mutagenesis of DNA fragments:
study of protein and DNA interactions. Nucleic Acids Res 16:
7351
–7367
777

Kegler-Ebo DM, Docktor CM, DiMaio D (1994) Codon cassette
mutagenesis: a general method to insert or replace individual
codons by using universal mutagenic cassettes. Nucleic Acids
Res 22:1593
–1599
Kirsch RD, Joly E (1998) An improved PCR-mutagenesis strategy
for two-site mutagenesis or sequence swapping between related
genes. Nucleic Acids Res 26:1848
–1850
Li CH, Fang HY, Deng XY, Xia K, Zheng D, Xia JH (2004)
Construction of middle fragment deletion mutant with im-
proved gene splicing by overlap extension. Zhonghua Yixue Yi
Chuan Xue Zazhi 21:52
–55
Rabhi I, Guedel N, Chouk I, Zerria K, Barbouche MR, Dellagi K,
Fathallah DM (2004) A novel simple and rapid PCR-based site-
directed mutagenesis method. Mol Biotechnol 26:27
–34
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a
laboratory manual. Cold Spring Harbor Laboratory, Cold
Spring Harbor, N.Y.
Tyagi R, Lai R, Duggleby RG (2004) A new approach to
‘megaprimer’ polymerase chain reaction mutagenesis without
an intermediate gel purification step. BMC Biotechnol 4:2
Urban A, Neukirchen S, Jaeger KE (1997) A rapid and efficient
method for site-directed mutagenesis using one-step overlap
extension PCR. Nucleic Acids Res 25:2227
–2228
Warrens AN, Jones MD, Lechler RI (1997) Splicing by overlap
extension by PCR using asymmetric amplification: an im-
proved technique for the generation of hybrid proteins of
immunological interest. Gene 186:29
–35
Zoller MJ, Smith M (1982) Oligonucleotide-directed mutagenesis
using M13-derived vectors: an efficient and general procedure
for the production of point mutations in any fragment of DNA.
Nucleic Acids Res 10:6487
–6500
778
Document Outline
Wyszukiwarka
Podobne podstrony:
Rapid and efficient purification and refolding of a (His) tagged recombinant protein produced in E c
A Simple and Effective Method for the Reduction of Acyl
Free Energy Bedini Device And Method For Pulse Charging A Battery Patent Info 2004
Improvements in Fan Performance Rating Methods for Air and Sound
Numerical Methods for Engineers and Scientists, 2nd Edition
Programming (ebook PDF) Efficient Algorithms For Sorting and Synchronization
Bangia, Diebold, Schuermann And Stroughair Modeling Liquidity Risk, With Implications For Traditiona
System and method for detecting malicious executable code
Suggestions for Those With Restrictive Eating Patterns and or Anorexia
Mellin Transform Method for Integral Evaluation [Intro and Appln for Electromagnetics] G Fikioris (
Szewczyk, Rafał i inni Rapid method for Mycobacterium tuberculosis identification using electrospra
Bedini Device and method for pulse charging a battery (patent info) (2004)
Energy performance and efficiency of two sugar crops for the biofuel
Improvements in Fan Performance Rating Methods for Air and Sound
Old McDonnalds song for kids with lyrics and pictures
Handbook for Working with Defendants and Offenders with Mental Disorders Third Edition
04 Matsumoto K i inni Fatigue life prolonging methods for welded flange attachment joint with a gap
AMACOM, A Survival Guide for Working With Bad Bosses Dealing With Bullies, Idiots, Back stabbers, A
Vatican lists conditions for ties with China Conflict Resolutions and World Security Solutions wor
więcej podobnych podstron