Postępy Biochemii 58 (3) 2012
273
Joanna Drozdowska
*
Warszawski Uniwersytet Medyczny, Wydział
Nauki o Zdrowiu, Zakład Biochemii Ogólnej i
Żywności, Warszawa
*
Warszawski
Uniwersytet
Medyczny,
Wydział Nauki o Zdrowiu, Zakład Biochemii
Ogólnej i Żywności, ul. Żwirki i Wigury 61,
02-091 Warszawa; tel.: (22) 599 18 35, e-mail:
jdrozdowska@wum.edu.pl
Artykuł otrzymano 13 lutego 2012 r.
Artykuł zaakceptowano 10 maja 2012 r.
Słowa kluczowe: czynnik tkankowy, TF, asTF,
latentny TF, śródbłonek, HUVEC
Wykaz skrótów: asTF (ang. alternatively spli-
ced Tissue Factor) — rozpuszczalna izoforma
czynnika tkankowego; flTF (ang. full length
Tissue Factor) — błonowy czynnik tkankowy;
HUVEC — komórki izolowane z ludzkiej żyły
pępowinowej (ang. Human Umbilical Vein En-
dothelial Cells); IL-1β (ang. Interleukin-1 β) — in-
terleukina pierwsza beta; LPS (ang. lipopolysac-
charide) — lipopolisacharyd; PDI (ang. Protein
Disulfide Isomerase) — izomeraza disulfidowa;
PS — fosfatydyloseryna; TF (ang. Tissue Fac-
tor) — czynnik tkankowy; TNFα (ang. Tumour
Necrosis Factor α) — czynnik martwicy nowo-
tworów alfa; VEGF (ang. Vascular Endothelial
Growth Factor) — czynnik wzrostu komórek
śródbłonka naczyniowego
Czynnik tkankowy w komórkach śródbłonka —
budowa i funkcja w świetle wyników najnowszych badań
STRESZCZENIE
C
zynnik tkankowy (TF) jest glikozylowanym białkiem błonowym wytwarzanym przez śród-
błonek i wiele innych komórek. Najlepiej poznaną jego rolą jest inicjacja procesu tworze-
nia skrzepu po uszkodzeniu naczynia, następująca po połączeniu z VII czynnikiem krzepnięcia
krwi, ale równie ważny jest udział TF w procesach zapalnych. W artykule przedstawiono aktu-
alny stan wiedzy na temat regulacji syntezy TF w komórkach śródbłonka. Przedstawiono też
poglądy dotyczące nieaktywnej formy TF oraz mechanizmów prowadzących do jej aktywacji.
WPROWADZENIE
Czynnik tkankowy (TF, ang. Tissue Factor) zwany inaczej tromboplastyną, czynni-
kiem III krzepnięcia krwi (F3, ang. Factor 3) lub CD 142 (ang. Cluster of Differentiation
142), jest 47 kDa glikoproteiną zakotwiczoną w błonie komórkowej. Pełni on istotną
rolę w procesach krzepnięcia i jego obecność w błonie komórkowej jest ściśle kontro-
lowana. TF uzyskuje kontakt z krwią między innymi, gdy dochodzi do uszkodzenia
naczynia krwionośnego i ekspozycji fibroblastów i miocytów mięśniówki gładkiej
(subendotelium). Jednak w przeciwieństwie do wspomnianych powyżej komórek
subendotelium konstytutywnie wytwarzających TF [1], nieaktywowane komórki
śródbłonka i monocyty nie wytwarzają TF. W komórkach tych pojawia się on w
formie aktywnej dopiero po ich stymulacji np. TNFα, IL-1β, LPS (odpowiednio, ang.
Tumour Necrosis Factor α, Interleukin-1 β, lipopolysaccharide).
Proces tworzenia skrzepu przebiega przy udziale kilkunastu czynników krzep-
nięcia, które są proteazami serynowymi aktywowanymi po proteolizie fragmentów
swoich cząsteczek. Pierwszy etap kaskady krzepnięcia następuje po związaniu TF
ze stale obecnym we krwi czynnikiem VII. W wyniku tej reakcji powstaje aktywny
czynnik VII (VIIa). Kompleks VIIa/TF aktywuje czynnik X, który w kolejnych reak-
cjach z udziałem V czynnika krzepnięcia indukuje przekształcenie protrombiny w
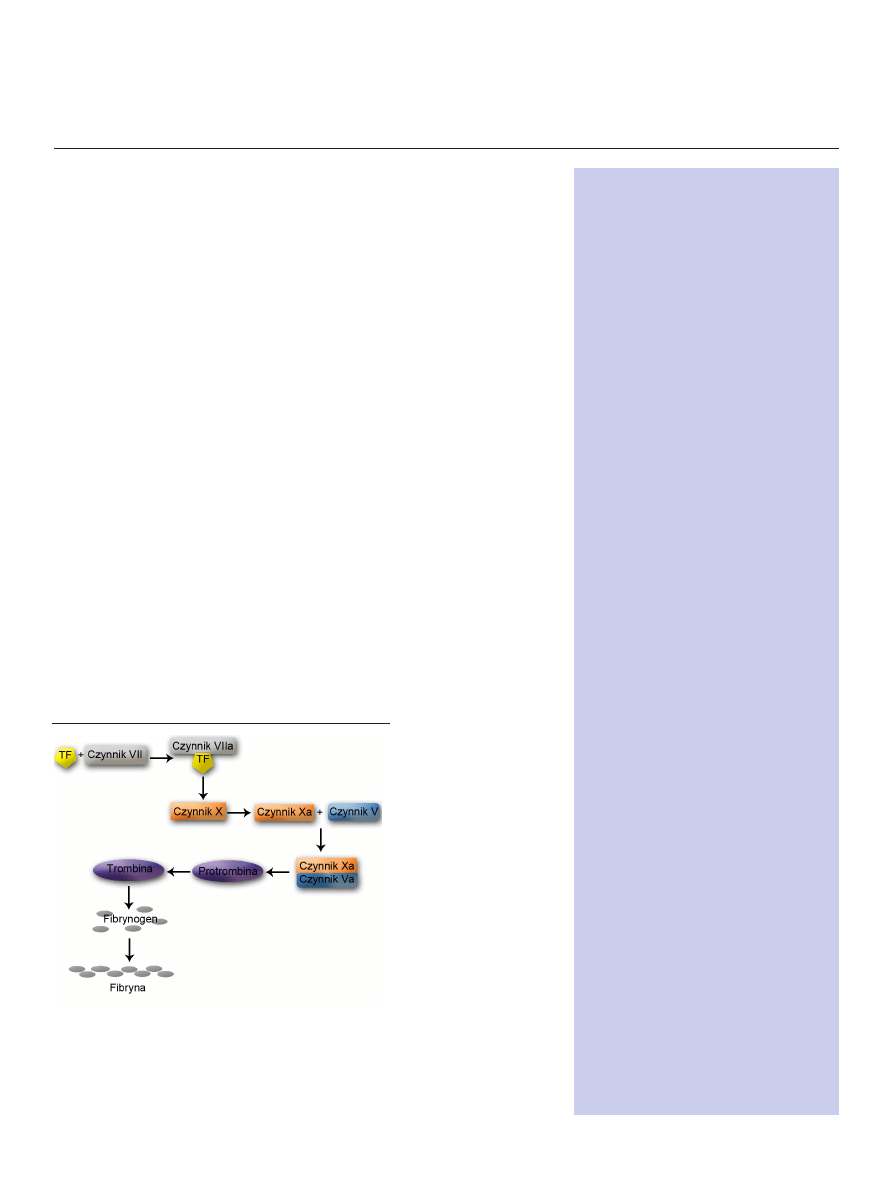
trombinę. Trombina następnie aktywuje fibrynę i tworzy się skrzep (Ryc. 1).
Opisana powyżej rola TF w krzepnięciu krwi jest dobrze poznana i udoku-
mentowana [2,3]. W ostatnich latach coraz więcej uwagi zwraca się jednak na
inną, ważną funkcję tego białka. Bierze ono udział w szlakach przekazywania
sygnału angażujących
białka PAR (ang. Pro-
tease-Activated Recep-
tors) i regulujących
m.in. odpowiedź za-
palną,
angiogenezę
oraz tworzenie prze-
rzutów nowotworo-
wych [4-10]. Rola TF
w szlakach przekazy-
wania sygnału nie bę-
dzie omawiana w tym
artykule.
Oprócz TF obecne-
go na powierzchni ko-
mórek, jego obecność
stwierdza się także we
krwi obwodowej. Na
frakcję rozpuszczal-
nego, osoczowego TF
składa się uwalniane
z komórek rozpusz-
Rycina 1. Kaskada aktywacji czynników krzepnięcia krwi prowadzą-
ca do polimeryzacji fibryny. TF oddziałuje z czynnikiem VII tworząc
kompleks zdolny do proteolizy czynnika X, który następnie aktywuje
czynnik V. Protrombina przekształcana jest w trombinę dzięki proteoli-
tycznym właściwościom kompleksu utworzonego przez czynnik X oraz
czynnik V. Trombina aktywuje fibrynogen, który polimeryzuje i staje się
elementem skrzepu, fibryną.

274
www.postepybiochemii.pl
czalne białko pozbawione domeny transbłonowej (asTF,
ang. alternatively spliced Tissue Factor) oraz TF związany z
błoną komórkową mikrocząstek (MPs, ang. microparticles,
microvesicles) czyli bardzo drobnych fragmentów błon ko-
mórkowych uwalnianych przez różne typy komórek i płyt-
ki krwi. Funkcja i pochodzenie osoczowego TF nadal budzi
wiele kontrowersji [2,11-14]. Pojawiły się także wątpliwości
dotyczące mechanizmów aktywacji procesów krzepnięcia
z udziałem TF. W świetle bieżących doniesień wydaje się,
że samo pojawienie się TF na powierzchni komórek śród-
błonka nie warunkuje zapoczątkowania procesów tworze-
nia skrzepu, jak zakłada to klasyczny model [3,15-17]. Nie
została również ustalona rola rozpuszczalnej izoformy TF
(asTF). Wydaje się, że może ona pełnić rolę czynnika angio-
gennego i sprzyjać metastazie komórek nowotworowych.
Zjawiska te wymagają jednak dalszych badań.
Tematem niniejszej pracy jest regulacja wytwarzania
izoformy błonowej i rozpuszczalnej TF oraz jego aktywa-
cja w komórkach śródbłonka. Ze względu na ograniczenia
techniczne wiele doświadczeń dotyczących ekspresji genu
kodującego TF oraz uaktywnienia jego latentnej formy zo-
stało przeprowadzonych na innych niż śródbłonek komór-
kach. Zebrane dane zostały jednak tak przedstawione, aby
przybliżyć czytelnikom mechanizmy i zjawiska dotyczące
przede wszystkim komórek śródbłonka.
REGULACJA EKSPRESJI GENU TF
Gen kodujący TF u człowieka znajduje się na pierwszym
chromosomie i należy do grupy genów wczesnej odpowie-
dzi (IEG, ang. immediate early genes). Indukcja jego ekspresji
przebiega bezpośrednio w odpowiedzi na czynnik stymu-
lujący, bez wcześniejszej syntezy de novo innych białek. Sys-
tem regulacji wytwarzania TF w śródbłonku jest inny niż
w komórkach subendotelium. Różnica polega na tym, że
nieaktywowany śródbłonek nie wytwarza TF. Rozpoczęcie
ekspresji genu następuje dopiero w odpowiedzi na stymu-
lację, np. cytokinami. Natomiast w komórkach mięśni gład-
kich naczyń jest on wytwarzany stale [2]. Badania mające
na celu ustalenie mechanizmów regulacji transkrypcji genu
dla TF przeprowadzono na komórkach wykazujących jego
stałą, wysoką ekspresję. W dalszej części rozdziału opisa-
no, w jaki sposób dochodzi do indukcji syntezy mRNA tego
białka w śródbłonku.
Analiza struktury genu TF wykazała, że aktywną kontro-
lę jego transkrypcji umożliwiają znajdujące się w promoto-
rze liczne odcinki podatne na metylację uniemożliwiającą
przyłączenie białek kompleksu transkrypcyjnego. W pro-
motorze znajdują się również miejsca wiązania czynników
transkrypcyjnych (elementy regulatorowe cis), które od-
działują ze specyficznymi białkami umożliwiając utworze-
nie kompleksu transkrypcyjnego. W obszarze promotora
genu kodującego TF znajdują się dwa miejsca AP-1(-217 i
-204 ), jedno κB (-179) oraz trzy Sp1/Egr1.
Badania przeprowadzone na komórkach izolowanych z
ludzkiej żyły pępowinowej (HUVEC, ang. Human Umbili-
cal Vein Endothelial Cells) wykazały, że miejsca AP-1 są na
stałe związane z heterodimerami Fos-Jun [18]. Konstytu-
tywną obecność czynników trans w wymienionych miej-
scach potwierdziły także dane otrzymane z komórek śród-
błonka aorty świni (PAEC, ang. Pig Aortic Endothelial Cells)
[19]. Sprzeczne z nimi są wyniki badań przeprowadzonych
przez innych badaczy. Wskazują one indukcyjność wiąza-
nia hererodimerów Fos/JunD do jednego z miejsc AP-1 po
stymulacji TNFα. Analiza kompleksów czynników wiążą-
cych się z miejscami AP-1 ujawniła różny dla obu miejsc
skład oddziałujących białek [20].
Z kolei do miejsc κB, po stymulacji bakteryjną endotok-
syną (lipopolisacharydem, LPS) lub cytokinami, rekrutowane
są czynniki transkrypcyjne NF-κB składające się z białek
Rel. Ich nazwa wywodzi sie od homologicznej domeny o
takiej samej nazwie (RDH, ang. Rel homology domain). Struk-
tura motywu κB w promotorze genu TF zawiera w pozycji
pierwszej C zamiast G co sprawia, że nie może z nią oddzia-
ływać kompleks p50-p65, natomiast preferencyjnie wiąże
się c-Rel-p65 [18]. Warto zauważyć, że oba białka uprzy-
wilejowanego heterodimeru zawierają domenę TAD (ang.
Transcriptional Activation Domain) umożliwiającą aktywację
transkrypcji [21]. Przed pojawieniem się sygnału aktywują-
cego, białka Rel, znajdują się w cytoplazmie w formie nie-
aktywnej. Tworzą one kompleks z białkami hamującymi
IκBα, które przysłaniają fragment domeny Rel, zawierającej
informację o transporcie do jądra komórkowego (NLS, ang.
Nuclear Localization Signal). Ufosforylowanie białek inhibito-
rowych przez odpowiednią kinazę np. IκBK powoduje ich
dysocjację, odsłonięcie sekwencji sygnałowej i transport do
jądra komórkowego [21].
Doświadczenia wykazały, że białka przyłączane do
miejsc κB i AP-1 mogą ze sobą oddziaływać, tworząc frag-
ment kompleksu inicjującego transkrypcję genu TF w odpo-
wiedzi na LPS, TNFα i IL-1β [18-20]. Fragment promotora
zawierający dwa miejsca AP-1 i κB nazywany jest elemen-
tem LRE (ang. LPS Responce Element) [22]. Inne badania
dowiodły, że elementy kompleksu transkrypcyjnego NFκB
mogą także oddziaływać z Sp1[18].
Elementami trans wiążącym się z trzema miejscami
Sp1 jest konstytutywnie syntetyzowane białko Sp1 (ang.
Specificity protein 1) zawierające w swojej strukturze trzy
palce cynkowe, którymi wiąże się z DNA. Badania prze-
prowadzone na komórkach linii nowotworowej HeLa
wskazują, że przyłączenie dwóch Sp1 jest konieczne, aby
utrzymać aktywność promotora TF. Sp1 bierze udział w
inicjacji powstania kompleksu transkrypcyjnego w odpo-
wiedzi na czynniki wzrostu, a obszar na którym się znaj-
duje ( -111 do +14) nazwano SRR (ang. Serum Response
Region). Warto podkreślić, że w tym fragmencie zlokali-
zowane są również trzy miejsca, do których możliwe jest
przyłączenie kolejnego elementu trans Egr-1 (ang. Early
growth response protein 1). Odcinki te zachodzą na obsza-
ry wiązania Sp1 sześcioma parami zasad. Wydaje się, że
wymienione czynniki transkrypcyjne konkurują ze sobą
o miejsce wiązania [22]. Sekwencje nukleotydów w obrę-
bie miejsc regulatorowych dla genu TF nie są zachowane
w ewolucji. Jak wykazano, występuje zróżnicowanie ga-
tunkowe w składzie białkowych kompleksów rekrutowa-
nych do miejsc AP-1 w odpowiedzi na cytokiny [19,20].
Być może dotyczy to również innych elementów regula-
torowych w obrębie genu TF.

Postępy Biochemii 58 (3) 2012
275
ROZPUSZCZALNA IZOFORMA
CZYNNIKA TKANKOWEGO
Tradycyjnie TF jest opisywany jako transbłonowe białko
występujące na powierzchni śródbłonka i innych komórek, nie
jest to jednak jego jedyna forma. We krwi obecna jest frakcja
tego białka nie związana z komórkami ani płytkami krwi, na-
zwana rozpuszczalnym TF albo osoczowym TF. Tworzą go TF
związany z powierzchnią mikrocząstek oraz asTF (lub asHTF;
ang. alternatively spliced Tissue Factor), czyli forma TF, którego
transkrypt uległ alternatywnemu różnicowemu cięciu i skła-
daniu (ang. splicing), w wyniku czego nie jest kotwiczony w
błonie komórkowej. Badania przeprowadzone na komórkach
śródbłonka aktywowanych TNFα lub IL-6 ujawniły, że oprócz
klasycznego TF (flTF, ang. full length Tissue Factor) syntezują
one również jego rozpuszczalną izoformę [23].
Wprawdzie w komórkach śródbłonka nie analizowano
struktury transkryptu asTF, ale doświadczenia przeprowa-
dzone na komórkach linii HL-60 (mieloblastów ostrej bia-
łaczki promieloblastycznej) i monocytach CD 14
+
, ujawniły,
że jego alternatywna forma nie zawiera eksonu 5 odpowia-
dającego domenie transbłonowej i cytoplazmatycznej [4].
Ekson 4 jest dołączony do eksonu 6, który rozpoczyna się
„niekompletnym” kodonem. W wyniku przesunięcia ramki
odczytu powstaje specyficzny dla asHTF C koniec. Składa
się on z pierwszych 166 reszt aminokwasowych transbłono-
wej formy oraz ciągu 40 reszt aminokwasowych unikalnych
dla TF. Opisane powyżej modyfikacje sprawiają, że mRNA
asTF jest o 160 nukleotydów krótszy od pełnej formy.
Rola TF w procesach tworzenia skrzepu przy udziale ko-
mórek śródbłonka została dość dobrze poznana, ale funkcja
asTF nadal pozostaje zagadką. W literaturze pojawiają się
na ten temat różne hipotezy. Według niektórych badaczy
asTF ułatwia powstawanie rozległego skrzepu [24], inni
zaś w ogóle poddają w wątpliwość jego zdolności proko-
agulacyjne [25]. Wiadomo także, że u ludzi jego synteza nie
jest jednakowa we wszystkich narządach. W płucach sto-
sunek liczby kopii mRNA asTF do flTF jest podwyższony
w stosunku do nerek, serca czy mózgu. Wyjaśnienie tego
zjawiska może pomóc w zidentyfikowaniu mechanizmów
kontrolujących powstawanie rozpuszczalnej formy TF, jak
również w ustaleniu jego funkcji [4, 23].
Na całkowitą pulę osoczowego TF składa się antygen odpo-
wiadający skróconej formie TF (10–30%) oraz TF o pełnej dłu-
gości związany z błoną mikrocząstek. Wydaje się jednak, że w
miejscu działania cytokin asHTF pełni nieco odmienną rolę niż
ten zlokalizowany w błonie komórkowej. Badania przeprowa-
dzone na komórkach śródbłonka izolowanych z ludzkiej żyły
pępowinowej ujawniły zróżnicowaną dynamikę wytwarzania
obu form białka. Komórki te odpowiadają szybką i intensyw-
ną syntezą asHTF (45-krotny wzrost) po upływie zaledwie 10
minut od stymulacji TNFα. Następnie białko to jest uwalniane
z komórki. Najwyższy poziom ekspresji genu kodującego bło-
nowy TF stwierdzano dopiero po 1 godzinie stymulacji cytoki-
ną [23]. Powstała więc hipoteza, według której pod wpływem
czynnika prozapalnego dochodzi najpierw do uwolnienia
asHTF z komórki, natomiast później aktywność prozakrzepo-
wa uzależniona jest od flTF występującego na mikrocząstkach
[23]. W świetle badań dotyczących właściwości latentnego TF
(patrz poniżej) wydaje się ona jednak nieprawdziwa. Autorzy
opisanych badań sugerują, że wczesne uwolnienie asHTF z ko-
mórki może być związane z przygotowaniem warunków do
szybszego stworzenia skrzepu. Po pojawieniu się w środowi-
sku cytokiny zmniejsza się wytwarzanie inhibitora TF (TFPI,
ang. Tissue Factor Pathway Inhibitor), jednak pewna jego ilość
nadal jest obecna we krwi. Skrócona forma TF mogła by two-
rzyć kompleks z czynnikiem VII i wiązać TFPI, przez co cała
pula TF związanego z błoną brałaby udział w procesie krzep-
nięcia. Hipoteza ta ma potwierdzenie w dynamice wytwarza-
nia obu form mRNA dla białka, czyli następującą w czasie re-
dukcją liczby wytwarzanych transkryptów dla asTF oraz ich
podwyższoną produkcją dla flTF [23]. W opisanych badaniach
do wyrażenia właściwości prozakrzepowych asTF wymagała
obecności fosfatydylocholiny i fosfatydyloseryny [23]. Donie-
sienia te potwierdzają wcześniejsze obserwacje wskazujące,
że prozakrzepowe właściwości kompleksu TF/czynnik VII
wymagają obecności ujemnie naładowanych fosfolipidów, o
czym będzie mowa w następnym rozdziale.
Kolejnym dowodem świadczącym o właściwościach pro-
koagulacyjnych asTF jest jego obecność w wytworzonym i w
tworzącym się ex vivo skrzepie. Zaproponowano więc nowy
model jego powstawania, w którym rozpuszczalna forma
TF pełni ważną funkcję w jego propagacji. TF związany z
błoną komórkową śródbłonka, w miarę przyłączania czyn-
ników krzepnięcia krwi (VII, IV, X) oraz płytek krwi, ulega
fizycznemu odseparowaniu od krwi przez co nie może peł-
nić swojej protrombotycznej funkcji. Uwolniony do światła
naczynia rozpuszczalny asHTF łączy się z czynnikiem VII
tworząc aktywny kompleks, a dzięki temu, że nie jest zwią-
zany z błoną plazmatyczną, oddziałuje z płytkami krwi i re-
krutuje w to miejsce kolejne cząsteczki fibryny [24].
Niektórzy badacze zakwestionowali jednak rolę asHTF
jako czynnika krzepnięcia, gdyż jego potencjał do genero-
wania aktywnego czynnika X jest mniejszy niż ten prezen-
towany przez flTF [4,23]. Nie jest ponadto jasne, jak pozba-
wiona domeny transbłonowej izoforma TF miałaby oddzia-
ływać ze związanym z błoną komórkową czynnikiem X.
Przeprowadzone badania sugerują, że białko to pełni raczej
rolę inhibitorową i zmniejsza pulę czynnika VII, z którym
flTF tworzy aktywny kompleks. Istnieje ogromna różnica
pomiędzy stężeniami asHTF i czynnika VII obecnymi we
krwi a stężeniami niezbędnymi do wywołania ich niewiel-
kiej proteolitycznej aktywności [4,26].
Inne doświadczenia przeprowadzone na komórkach
śródbłonka ujawniły, że asTF może pełnić odmienną niż
flTF funkcję. Komórki te w obecności podwyższonego stę-
żenia rozpuszczalnej formy TF intensywniej migrowały i
wykazywały szereg innych cech wskazujących na aktyw-
ność angiogenną asTF. Efekt ten był niezależny od czynnika
VII [27]. Skrócona forma TF łączy się z integrynami obecny-
mi na powierzchni błony komórkowej, aktywując migrację
i tworzenie kapilar. Co ciekawe, oddziaływanie TF z różny-
mi integrynami wywołuje odmienną odpowiedź komórko-
wą. Migracja następuje w skutek oddziaływania asTF z inte-
gryną αvβ3 i fosforylacji białek przekaźnikowych p38 MAP
i PI3 kinazy. Tworzenie kapilar jest natomiast zależne od
połączenia asTF z integrynami α6β1, a następnie aktywacji
kinazy p42/p44 MAP i kinazy Akt. Klasyczna, pełna forma

276
www.postepybiochemii.pl
TF również może łączyć się z integrynami, jednak reakcja ta
przebiega z zaangażowaniem receptora PAR2 [28].
Uwalnianie asTF wykryto w wielu typach komórek no-
wotworowych, co wzmacnia hipotezę o jego udziale w an-
giogenezie [27-29]. W modelu doświadczalnym wykorzy-
stującym komórki nowotworu trzustki transfekowane asTF
wykazano, że białko to znacząco nasilało powstawanie wo-
kół nich sieci naczyń krwionośnych oraz rozrost guza [29].
Jego angiogenny potencjał potwierdzają również doświad-
czenia z użyciem matriżelu zawierającego zrekombinowa-
ny asTF, który był wszczepiony myszom. Z badań wynika,
że intensywność angiogenezy indukowanej przez asTF była
porównywalna do odpowiedzi wywołanej przez VEGF
(ang. Vascular Endothelial Growth Factor) [27]. Autorzy tych
badań sugerują także, że asTF może wpływać na utratę in-
tegralności blaszki miażdżycowej poprzez udział w tworze-
niu unaczynienia własnego dużych naczyń krwionośnych,
vasa vasorum. Przypuszczenia takie powstały po wykryciu
jego obecności w płytce miażdżycowej. Wyjaśnienie tego
zjawiska wymaga jednak dalszych badań [27].
Istnieją jednak prace dowodzące, że asTF nie wpływa bez-
pośrednio na procesy angiogenezy, a pośrednio. Wykazano,
że nadproducja asTF w linii mysich kardiomiocytów (HL-1),
indukuje syntezę czynników angiogennych i nasilających mi-
grację komórek, FGF2 (ang. Fibroblast Growth Factor-2), Cyr61
(ang. Cysteine-rich 61) i VEGF. Wytwarzanie tych białek pod-
lega regulacji przez ERK 1/2 i p38 MAP w odpowiedzi na
aktywację PAR1 i PAR2 [30]. Doniesienia świadczące o wtór-
nym działaniu asTF nie przeczą wynikom doświadczeń z
użyciem matriżelu z asTF, gdyż były one przeprowadzane
na modelu mysim, a nie in vitro. Nie można zatem wykluczyć
wpływu FGF2, Cyr61 i VEGF na tworzenie naczyń krwiono-
śnych oraz proliferację śródbłonka.
Nie jest do końca wyjaśnione, jak powstaje alternatyw-
na forma TF. Wydaje się jednak, że proces ten w śródbłon-
ku i w innych komórkach powinien przebiegać podobnie.
Cząsteczka pre-mRNA TF zawiera motywy sekwencyjne
i strukturalne, które biorą udział w przebiegu różnicowe-
go cięcia i składania mRNA. Fragmenty transkryptu okre-
ślane jako SRE (ang. Splicing Regulatory Elements) regulują
jego obróbkę oddziałując z białkami hnRNP (ang. hetero-
geneous nuclear Ribonucleoproteins) i SR (ang. Serine- and
arginine-Rich proteins) [31]. W eksonie 5 pre-mRNA dla
TF znajduje się pięć sekwencji ESE (ang. Exon Splicing En-
hanser), z którymi mogą się wiązać ufosforylowane białka
SF2/ASF (ang. Splicing Factor 2/Alternative Splicing Factor)
oraz jedno miejsce dla białek SR -SRp75 (ang. Serine and
arginine Rich protein 75) [32,33] lub/i SRp55 [27,34]. Do-
wiedziono, że wymienione czynniki transkrypcyjne biorą
udział w regulacji obróbki transkryptów dla dwóch izo-
form TF w komórkach śródbłonka. Doświadczenia polega-
ły na stymulowaniu TNFα komórek HUVEC i wyciszaniu
transkryptów poszczególnych SR. Badania wykazały, że
obecność ufosforylowanych SR75 i SF2/ASF wpływa na
zwiększenie liczby transkryptów dla flTF [32]. Doniesie-
nia te są w pewnym stopniu zgodne z wynikami badań
przeprowadzonych na monocytach stymulowanych LPS.
W komórkach tych wycinanie eksonu 5 z transkryptu
TF zależy od konkurujących ze sobą o miejsce wiązania
SRp40/SC35 – SC55 oraz SC35 - SF2/ASF. Białkami biorą-
cymi udział w tworzeniu mRNA dla asTF są zatem SRp40
i SC35, natomiast dla flTF SF2/ASF i SR55. Autorzy suge-
rują, że SRp75, którego rolę odkryto przy regulacji różni-
cowego cięcia i składania mRNA w komórkach HUVEC,
pełni raczej funkcje pomocnicze niż kluczowe przy two-
rzeniu spiceosomu [34].
Proces alternatywnego różnicowego cięcia i składania
mRNA nie tylko zależy od przyłączenia odpowiednich bia-
łek regulatorowych, ale również, a może przede wszystkim
od ich aktywacji przez odpowiednie kinazy. Kontrolowa-
nie procesowania pre–mRNA dla TF przez kinazy jest zło-
żone, hierarchiczne i zależne od wielu czynników. Badania
ujawniły, że białkami biorącymi udział w uaktywnianiu
elementów spiceosomu są prawdopodobnie Clk (ang. Cdc2-
-like kinase), DNA topoizomeraza I (DNA Topo I), p38 MAP
i białka szlaku przekazywania sygnału PI3/Akt [23,32,33].
Aktualne dane wskazują, że kinaza Akt 2 może oddziały-
wać na białka SR bezpośrednio, tak jak się to dzieje się w
przypadku czynnika SF2/ASF [33,35] lub pośrednio, po-
przez aktywację innej kinazy, Clk, która fosforyluje SRp75,
SRp55 [35] i SF2/ASF [32]. Farmakologiczne zahamowanie
aktywności Clk powoduje spadek liczby transkryptów dla
obu izoform TF i hypofosforylację SF2/ASF, SRp55, SRp75
[32]. W innych badaniach ten sam autor pokazał, że po ak-
tywacji komórek śródbłonka przez TNFα, ufosforylowa-
nie SF2/ASF, SRp75 i SRp55 jest regulowane przez białka
szlaku PI3K/Akt. Ich hamowanie jednak nie prowadzi do
zmniejszenia liczby transkryptów dla flTF [33].
W proces regulacji alternatywnej obróbki transkryptu
dla TF może być zaangażowana również kinaza p38 MAP,
która bierze udział w organizacji przestrzennej czynników
uczestniczących w procesowaniu mRNA. Białka hnRNP A1
i SF2/ASF są antagonistami i konkurują ze sobą o miejsce
wiązania. Badania przeprowadzone na stymulowanych
TNFα komórkach śródbłonka ujawniły, że kinaza p38 MAP
fosforyluje hnRNP A1, w wyniku czego następuje eksport
tej nukleoproteiny z jądra do cytoplazmy [23]. Dzięki opisa-
nej translokacji białko SF2/ASF może swobodnie oddziały-
wać z białkami splicesomu, pre-mRNA i regulować powsta-
nie alternatywnego transkryptu.
Nie ma jeszcze żadnych badań potwierdzających, że ki-
nazy ścieżek PI3/Akt i p38 MAP oddziałują ze sobą podczas
procesu obróbki transkryptu dla TF. Przedstawione dane
sugerują jednak nadrzędną role ścieżki PI3K/Akt w regu-
lacji całego alternatywnego różnicowego cięcia i składania
mRNA TF. Zależności te wydają się jednak skomplikowane,
a wymieniony mechanizm może regulować powstawanie
zarówno asTF, jak i flTF.
W proces alternatywnego różnicowego cięcia i składa-
nia mRNA TF zaangażowana jest również DNA Topo I.
Hamowanie tego białka pociąga za sobą obniżenie liczby
aktywnych form SF2/ASF i SRp55 oraz spadek liczby trans-
kryptów flTF, a wzrost asTF. Powstały przypuszczenia, że
DNA Topo I reguluje fosforylację SF2/ASF i SRp55 [32]. Nie
wiadomo, czy aktywność tego białka w procesie alterna-
tywnego różnicowego cięcia i składania mRNA TF jest re-
gulowana przez kinazy.
Postępy Biochemii 58 (3) 2012
277
Regulacja alternatywnego różnicowego cięcia i składania
mRNA TF jest złożona i prawdopodobnie hierarchiczna. Na-
dal jednak mało o niej wiemy. Zebrane dane są niewystarcza-
jące, by móc stwierdzić, jak przebiega proces tworzenia trans-
kryptów obu form TF. Konieczne są zatem dalsze badania.
UTAJONY TF
Do niedawna sądzono, że synteza TF w komórkach
śródbłonka możliwa jest dopiero po ich aktywacji. Jednak
w świetle ostatnich badań ten klasyczny model wydaje się
być błędny [15,17]. Stwierdzono bowiem, że w błonie ko-
mórkowej niestymulowanych komórek występuje TF, który
jest wykrywany przez przeciwciała, lecz nie wykazuje ak-
tywności prozakrzepowej (nieaktywny, latentny, utajony,
uśpiony, ang. encripted, cryptic TF). Wiąże się on z czynni-
kiem VII/VIIa, jednak wytworzony kompleks nie ma zdol-
ności do przyłączenia i oddziaływania ani z czynnikiem X
ani z TFPI. Obecność uśpionego TF potwierdzono także w
innych niż śródbłonek komórkach, np. w HL-60 (ang. Hu-
man promyelocytic Leukemia cells) [3], monocytach [3,16] oraz
fibroblastach [36]. Okazało się, że nieaktywny TF występuje
także, gdy jego transkrypt zostanie wprowadzony do komó-
rek nie wytwarzających go de novo [37]. Dane te dowodzą,
że występowanie latentnej formy białka prawdopodobnie
jest właściwą jemu cechą.
Proces uaktywnienia TF w komórkach śródbłonka nastę-
puje prawdopodobnie po ich stymulacji, nie jest jednak na-
dal jasne, jaki jest mechanizm tego zjawiska. Wiadomo jed-
nak, że latentny TF związany z błoną komórkową zyskuje
proteolityczne właściwości w komórkach ulegających pro-
cesom apoptotycznym. Naukowcom udało się opracować
sposób sztucznego wzbudzania aktywności proteolitycznej
TF poprzez traktowanie komórek jonoforami (np. wapnio-
wym) oraz silnymi utleniaczami np. chlorkiem rtęci (II) lub
cykloheksymidem. Zastosowanie tych związków powoduje
wzrost liczby cząsteczek TF wyrażających właściwości pro-
zakrzepowe bez wpływu na jego syntezę czy obecność anty-
genu na powierzchni błony komórkowej [37,38]. Nadal nie
zostało ostatecznie rozstrzygnięte, jaki mechanizm reguluje
przemianę latentnego TF w aktywny i odwrotnie. Wiadomo
natomiast, że pewna liczba tej formy TF jest zawsze obecna
w błonie komórkowej. Wymienia się szereg mechanizmów,
które mogłyby odpowiadać za zjawisko uśpienia TF. Ogól-
nie można je podzielić na związane ze strukturą samego
białka oraz umiejscowieniem go w obrębie domen błony
komórkowej. Należy dodać, że mechanizmy te nie wyklu-
czają się wzajemnie.
WPŁYW STRUKTURY TF NA JEGO AKTYWACJĘ
TF jest przedstawicielem superrodziny receptorów cy-
tokin. Cechą charakterystyczną tej grupy białek jest zdol-
ność do tworzenia homodimerów i przyłączania swoistych
ligandów [3]. W przeciwieństwie jednak do pozostałych
członków swojej rodziny, dimeryzacja podjednostek TF ha-
muje jego aktywność. Analiza czwartorzędowej struktury
kompleksu TF/czynnik VII wykazała, że homodimer TF ma
zdolność do przyłączania czynnika VII, ale nie czynnika X.
Miejsce wiązania czynnika X w tym kompleksie jest przy-
słonięte, co oznacza zablokowanie jego funkcji prozakrze-
powych. Powyższe dane sugerują, że jedynie monomerycz-
na forma TF jest zdolna do udziału w procesie wytwarza-
nia skrzepu [3]. TF, podobnie jak inni członkowie rodziny
receptorów klasy II cytokin, łączy się z ligandem, którym
jest czynnik VII (Ryc. 1). Badania strukturalne ujawniły, że
wiązanie disiarczkowe w cząsteczce TF, występujące po-
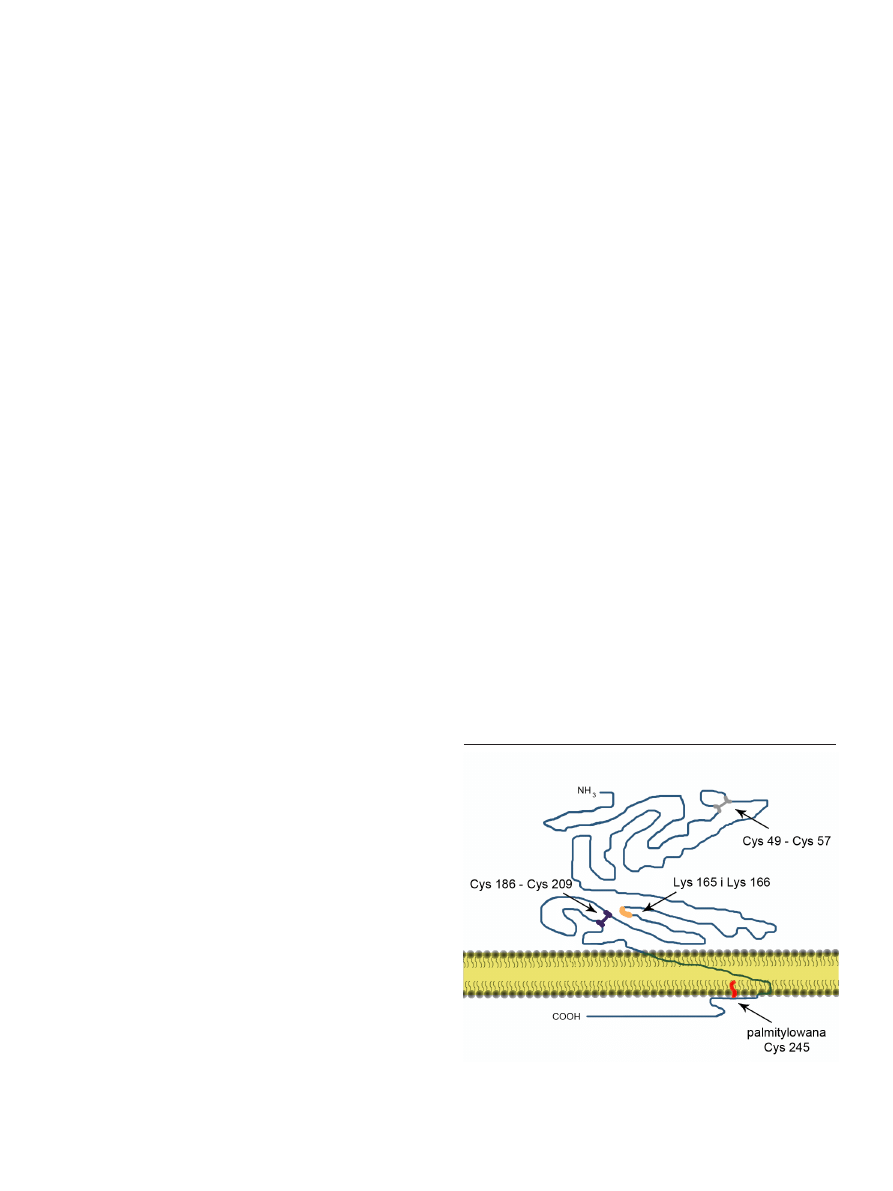
między resztami Cys 186 i Cys 209 (Ryc. 2), jest niezbędne
do prawidłowego wiązania czynnika VII oraz wyrażenia
aktywności prokoagulacyjnej kompleksu [39]. Aminokwasy
te znajdują się w domenie zewnątrzkomórkowej, a wiąza-
nie pomiędzy nimi pełni funkcje regulatora struktury trze-
ciorzędowej cząsteczki wiążąc sąsiadujące ze sobą nici w β
kartce [10]. Okazało się, że aktywna prozakrzepowo forma
TF obecna w błonie komórkowej śródbłonka, wymaga utle-
nienia siarki i powstania mostku disiarczkowego pomię-
dzy resztami cystein [10,39]. Enzymem, który mógłby taką
reakcję katalizować jest izomeraza disulfidowa (PDI, ang.
Protein Disulfide Isomerase). Badania potwierdziły jej zdol-
ność do podwyższania aktywności prozakrzepowej TF [40],
ale sam mechanizm tej aktywacji wywołał wiele dyskusji,
głównie ze względu na umiejscowienie tego enzymu w ob-
rębie komórki [10,41]. PDI zawiera sekwencję sygnałową
KDEL (Lys-Asp-Glu-Leu), która powinna utrzymywać ją w
siateczce endoplazmatyczej. Wiadomo jednak, że znajduje
się ona w mikrodomenach błony komórek śródbłonka bo-
gatych w cholesterol [42]. Dotychczas nie udało się wyja-
śnić, w jaki sposób PDI przemieszcza się na powierzchnię
błony komórkowej.
Wyniki innych doświadczeń ujawniają obecność i rolę PDI
w miejscu powstawania skrzepu. Zahamowanie jej aktywno-
ści znacząco zmniejszyło polimeryzację fibryny [40]. Powstały
przypuszczenia, że PDI może być uwalniana do przestrzeni
zewnątrzkomórkowej i tam bezpośrednio oddziaływać z TF
na powierzchni śródbłonka i innych komórek. Wydają się one
zasadne choćby ze względu na fakt, że izomeraza disulfidowa
stanowi jedno z głównych białek występujących w błonie mi-
krocząstek [42]. Ich powstawanie jest zależne od tratw chole-
sterolowych [43], które są uwalniane z powierzchni komórek
Rycina 2. Struktura i ułożenie cząsteczki TF w błonie komórkowej. Na rycinie
oznaczono niektóre reszty aminokwasowe i ich modyfikacje występujące w czą-
steczce TF oraz mostki disiarczkowe pomiędzy resztami cystein.

278
www.postepybiochemii.pl
śródbłonka np. po stymulacji cytokinami [23]. Na podstawie
opisanych doniesień został zaproponowany model, w któ-
rym PDI zostaje uwolniona do krwi z zaktywowanych płytek
krwi i tam oddziałuje z TF znajdującym się zarówno na po-
wierzchni komórek śródbłonka, monocytów, jak i mikroczą-
stek [40,44]. Istnieją prace potwierdzające rolę i obecność PDI
w miejscu toczących się procesów prozakrzepowych, a więc
możliwe jest jej oddziaływanie z TF na komórkach śródbłonka.
Nie ustalono jednak dokładnego mechanizmu tego oddziały-
wania. Badania przeprowadzone na myszach wykazały, że
adhezja płytek krwi do ściany uszkodzonego naczynia nie jest
konieczna, aby PDI znalazła się w miejscu tworzenia skrzepu
[40]. Nie musi ona także wpływać na aktywność TF. Warto w
tym miejscu dodać, że naruszenie integralności naczynia po-
ciąga za sobą pojawienie się ujemnie naładowanych fosfolipi-
dów na powierzchni błony komórkowej, co aktywuje TF.
Wyniki badań mających na celu wyizolowanie TF pod-
dają jednak w wątpliwość istnienie w jego strukturze wol-
nych, poddających się utlenieniu przez PDI, grup tiolowych
[10]. Dokładniejsza analiza cząsteczki TF ujawniła jednak,
że reszta cysteiny 209 tego białka może być połączona z glu-
tationem (GSH) [40]. Powstanie tzw. mieszanych disiarcz-
ków (białko-S-S-glutation) ma na celu uchronienie grup -SH
przed nieodwracalnym utlenieniem. Przyłączenie glutatio-
nu może przebiegać według dwóch mechanizmów: utlenie-
nia grup sulfanilowych i ich reakcji z GSH lub reakcji utle-
nionego glutationu z grupami SH. Dla większości białek,
ich glutationylacja prowadzi do zahamowania aktywności
[45]. Wydaje się, że tak też się dzieje w przypadku TF [40].
Powstała hipoteza, według której PDI mogłaby wpływać
na powstanie mostku disiarczkowego pomiędzy resztami
Cys186 i Cys 209 po uprzednim oderwaniu glutatnionu [40].
W oparciu o dostępne dane zaproponowane zostały dwa
mechanizmy oddziaływania TF z izomerazą disulfidową.
Pierwszy opiera się na stworzeniu niestabilnej formy po-
średniej po reakcji N-końcowej reszty cysteiny PDI z resztą
Cys 209 TF. Oddziaływanie to umożliwiłoby powstanie di-
sulfidu. Innym, możliwym mechanizmem jest reakcja izome-
ryzacji grup SH i S-S. Reakcja ta przebiegałaby bez zakłócenia
potencjału redoks PDI, co przyspieszyłoby pojawianie się ak-
tywnych form TF [40]. Jak jednak wskazuje analiza struktury
TF, utlenienie reszt tiolowych w momencie, gdy tworzy on
kompleks z czynnikiem VII, jest niemożliwe. Przyłączony
czynnik VII zasłania bowiem miejsce wiązania reszty cyste-
iny z PDI [46]. Wiadomo natomiast, że forma latentna TF
może tworzyć kompleks z VII czynnikiem krzepnięcia jesz-
cze przed wyrażeniem zdolności do proteolizy czynnika X. A
zatem zaproponowana hipoteza opierająca się na utlenianiu
reszt cystein 186 i 209 przez PDI wydaje się bezzasadna i nie
może wyjaśniać zjawiska uaktywniania TF.
W przedstawionych powyżej doniesieniach badacze sku-
piali się na mechanizmach regulujących przemianę form TF
z utajonej w uaktywnioną i odwrotnie. Istnieje jednak inna
hipoteza, według której w błonie komórkowej znajdują się
dwie, pełniące odmienne funkcje, frakcje TF. Cząsteczki biał-
ka zawierające disulfid miałyby być zaangażowane w procesy
krzepnięcia, zaś forma latentna brałaby udział w przekazywa-
niu sygnału szlakiem TF — aktywny czynnik VII — receptor
PAR2. Rozłączenie mostku disiarczkowego nie jest wymagane,
aby stworzyć kompleks TF-PAR2. Istnienie dwóch, różnych
funkcjonalnie form wyjaśniałoby, dlaczego TF może wyrażać
swoje właściwości prozakrzepowe nie inicjując jednocześnie
odpowiedzi wewnątrzkomórkowej i odwrotnie [39]. W opo-
zycji do powyższej hipotezy stoją dane, według których wią-
zanie pomiędzy resztami cystein w ogóle nie ma wpływu na
aktywację białka. Ma ono jednak znaczenie przy wytwarzaniu
i kotwiczeniu TF w błonie. W doświadczeniach polegających
na wprowadzeniu do komórek śródbłonka transkryptów TF,
w których reszty cysteiny 186 i 209 były zastąpione przez resz-
ty innych aminokwasów, na początku nie wykryto aktywności
protrombotycznych białka. Nabywały ich jednak w obecności
podwyższonego stężenia VII czynnika krzepnięcia. Ponad to
traktowanie tak zmodyfikowanych komórek jonoforem pro-
wadziło do pojawienia się aktywnej formy TF. Gdyby tworze-
nie wiązania disiarczkowego było kluczowym mechanizmem
prowadzącym do uaktywnienia TF opisane zjawiska by nie
wystąpiły [15]. Opisane badania sugerują, że na aktywację TF
w śródbłonku mają wpływ raczej ujemnie naładowane fosfoli-
pidy, a nie tworzące się wiązania disiarczkowe.
WPŁYW UMIEJSCOWIENIA TF W MIKRODOMENACH
BŁONY KOMÓRKOWEJ NA JEGO UAKTYWNIENIE
Aktywacja latentnej formy TF jonoforami i silnymi utle-
niaczami, wywołuje napływ wapnia do komórki. Powoduje
to zmianę lokalizacji ujemnie naładowanych fosfolipidów (w
tym fosfatydyloseryny), które przechodzą z wewnętrznego
do zewnętrznego listka błony komórkowej. Naturalna asy-
metria w rozmieszczeniu lipidów w dwuwarstwie lipidowej
jest utrzymywana przez enzymy, które przenoszą fosfolipidy
z zewnętrznego listka do wewnętrznego (flipazy) i odwrotnie
(flopazy). Zwiększenie stężenia jonów wapnia w cytoplazmie
powoduje zahamowanie aktywności flipaz i uaktywnienie
dodatkowego enzymu transportującego, skramblazy. W wy-
niku tych procesów w listku zewnętrznym błony komórko-
wej zwiększa się frakcja ujemnie naładowanych fosfolipidów.
Przypuszcza się, że to m.in. ten mechanizm prowadzi do zmia-
ny fenotypu śródbłonka na prozakrzepowy. Dobrze znanym
przykładem obrazującym to zjawisko jest stymulacja komórek
cytokinami prozapalnymi. Wiadomo, że związki te zmieniają
właściwości błony komórkowej, powodując przeskok fosfoli-
pidów wewnętrznej warstwy błony, w tym fosfatydylosery-
ny, do listka zewnętrznego [4]. Niewiele jest prac badających
wpływ ujemnie naładowanych fosfolipidów na uaktywnienie
TF na powierzchni komórek śródbłonka. Wydaje się jednak, że
mechanizm ten powinien być uniwersalny. Warto zauważyć,
że obecność PS w listku zewnętrznym błony komórkowej od
dawna jest wykorzystywana do identyfikacji komórek apopto-
tycznych, w których, jak wiadomo, dochodzi do uaktywnienia
latentnej formy TF. Wiadomo również, że zablokowanie prze-
mieszczenia się cząsteczek PS, powoduje obniżenie poziomu
aktywnego TF na monocytach pomimo stymulacji jonoforem
wapniowym [3].
Nie jest jasne, w jaki sposób fosfolipidy miałyby regu-
lować aktywność TF. Być może ich obecność wpływa na
strukturę czwartorzędową lub na orientację białka w błonie
komórkowej poprzez bezpośrednie oddziaływania elektro-
statyczne. Doświadczenia polegające na zastąpieniu reszt
lizyny 165 i 166 resztą alaniny wpłynęły negatywnie na wła-
ściwości proteolityczne TF, pomimo obecności w środowi-

Postępy Biochemii 58 (3) 2012
279
sku PS. Modyfikacja ta nie zmieniła jednak charakterystycz-
nej dla latentnej formy TF zdolności do przyłączania swo-
jego liganda. Wymienione aminokwasy znajdują się blisko
zewnętrznej warstwy błony komórkowej, w pętli nie gra-
niczącej z miejscem wiązania czynnika VII. Umiejscowienie
takie wskazuje na możliwość oddziaływania między PS a
TF. Ujemnie naładowane fosfolipidy mogłyby usztywniać
powstały kompleks TF/czynnik VII, ułatwiając dostęp do
miejsc aktywnych czynnikowi X i zwiększając efektywność
zachodzących reakcji proteolitycznych [3].
Jedna z hipotez wyjaśniających zjawisko uśpienia TF opiera
się na zjawisku jego sekwestracji w mikrodomenach błono-
wych zawierających cholesterol [43] i w kaweolach [10,47].
Cholesterol w błonie komórkowej pełni funkcje modulatora
aktywności wielu receptorów, m.in. EGFR (ang. Epidermal
Growth Factor Receptor), poprzez usztywnienie struktury biał-
ka i/lub bezpośrednie oddziaływanie [47]. Wykazano, że czą-
steczka TF zawiera sygnał dokowania w tratwach lipidowych,
jakim jest palmitylacja reszty cysteiny 245 znajdującej się w
domenie cytoplazmatycznej (Ryc. 2). Nie znany jest mecha-
nizm oddziaływania TF z cholesterolem, wiadomo natomiast,
że lokalizacja w mikrodomenach cholesterolowych sprzyja au-
toasocjacji podjednostek receptorów [3]. Według wspomnia-
nej hipotezy tratwy lipidowe miałyby stanowić rezerwuary
nieaktywnego, zdimeryzowanego TF [3,42,43]. Wyjaśniałoby
to, dlaczego krążące w krwi mikrocząstki z TF w mikrodo-
menach cholesterolowych, nie inicjują procesów krzepnięcia
[43]. Usunięcie cholesterolu z błony komórkowej wpływa na
zwiększenie właściwości prozakrzepowych TF [3,10]. Na pod-
stawie tych danych zasugerowano model, w którym forma
latentna TF występuje na terenie tratw lipidowych, natomiast
w odpowiedzi na specyficzny sygnał następuje jej uwolnienie,
oddziaływanie z fosfolipidami błony komórkowej i uaktyw-
nienie [3]. Badania przeprowadzone na fibroblastach nie po-
twierdzają jednak tego mechanizmu. Usunięcie cholesterolu z
powierzchni tych komórek zahamowało tworzenie komplek-
su TF/czynnik VII i w rezultacie nie doszło do aktywacji czyn-
nika X. Autorzy sugerują, że brak cholesterolu uniemożliwia
prawidłową asocjację i wzajemne oddziaływanie czynników
kaskady krzepnięcia [47]. Inne doniesienia wskazują z kolei,
że aktywująca TF fosfatydyloseryna jest obecna w tratwach
lipidowych [48,49]. Zmianę lokalizacji ujemnie naładowa-
nych fosfolipidów mogą kontrolować związane z tratwami
cholesterolowymi kinazy MAP. Umożliwiają one uwolnienie
jonów wapnia z siateczki endoplazmatycznej, w efekcie zaś
ekspozycję ujemnie naładowanych fosfolipidów. Hipotezę tą
potwierdzają badania przeprowadzone na komórkach ery-
troleukemicznych (HEL, ang. Human Erythroleukemia cell line).
Zostały one pozbawione cholesterolu z błony komórkowej, a
następnie były poddane działaniu jonoforu wapniowego. W
opisanych warunkach nie doszło do ujawnienia obecności PS
na powierzchni błony komórkowej i nie nastąpił napływ jo-
nów wapnia do cytoplazmy [3].
PODSUMOWANIE
TF jest białkiem, którego udział w procesach krzepnięcia
krwi znany jest od 1911 roku, czyli już od ponad stu lat. Jednak
badania przeprowadzone w ostatnim czasie doprowadziły do
obalenia szeregu utrwalonych dogmatów. Okazało się, że TF
może występować w formie nieaktywnej, a mechanizm jego
aktywacji nie został w pełni poznany. Odkryto także istnienie
dwóch izoform TF. Klasyczna, pełna forma kotwiczona jest
w błonie komórkowej, a alternatywna, krótsza postać, o nie
do końca poznanej funkcji, jest rozpuszczalna i wydzielana
przez komórki do krwi. Obie te formy są syntezowane przez
komórki śródbłonka człowieka. Najnowsze doniesienia suge-
rują, że rozpuszczalny TF bierze udział w procesach tworzenia
naczyń krwionośnych. Podwyższenie stężenia TF w osoczu
towarzyszy chorobom układu krążenia, posocznicy (sepsie),
zaburzeniom krzepnięcia krwi, jak również chorobom nowo-
tworowym. Ustalenie dokładnej roli obu izoform TF, a także
mechanizmów ich aktywacji i uwalniania przez komórki śród-
błonka jest niezwykle istotne klinicznie, m.in. ze względu na
nieustające poszukiwania markera rozwoju nowotworów.
PIŚMIENNICTWO
1. Belting M, Ahamed J, Ruf W (2005) Signaling of the tissue factor co-
agulation pathway in angiogenesis and cancer. Arterioscler Thromb
Vasc Biol 25: 1545-1550
2. Bouchard BA, Mann KG, Butenas S (2010) No evidence for tissue fac-
tor on platelets. Blood 116: 854-855
3. Bach RR (2006) Tissue factor encryption. Arterioscler Thromb Vasc
Biol 26: 456-461
4. Szotowski B, Antoniak S, Rauch U (2006) Alternatively spliced tis-
sue factor: a previously unknown piece in the puzzle of hemostasis.
Trends Cardiovasc Med 16: 177-182
5. Rao LV, Pendurthi UR (2005) Tissue factor-factor VIIa signaling. Arte-
rioscler Thromb Vasc Biol 25: 47-56
6. Banfi C, Brioschi M, Barbieri SS, Eligini S, Barcella S, Tremoli E, Colli
S, Mussoni L (2009) Mitochondrial reactive oxygen species: a common
pathway for PAR1- and PAR2-mediated tissue factor induction in hu-
man endothelial cells. J Thromb Haemost 7: 206-216
7. Banfi C, Brioschi M, Barcella S, Pignieri A, Parolari A, Biglioli P, Tre-
moli E, Mussoni L (2007) Tissue factor induction by protease-activated
receptor 1 requires intact caveolin-enriched membrane microdomains
in human endothelial cells. J Thromb Haemost 5: 2437-2444
8. Chu AJ (2005) Tissue factor mediates inflammation. Arch Biochem
Biophys 440: 123-132
9. Kasthuri RS, Taubman MB, Mackman N (2009) Role of tissue factor in
cancer. J Clin Oncol 27: 4834-4838
10. Pendurthi UR, Rao LV (2008) Role of tissue factor disulfides and lipid
rafts in signaling. Thromb Res 122 Suppl 1: S14-18
11. Owens AP, 3rd, Mackman N (2010) Tissue factor and thrombosis: The
clot starts here. Thromb Haemost 104: 432-439
12. Key NS (2008) Platelet tissue factor: how did it get there and is it im-
portant? Semin Hematol 45: S16-20
13. Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C
(2010) Microvesicles: mediators of extracellular communication du-
ring cancer progression. J Cell Sci 123: 1603-1611
14. Mezzano D, Matus V, Saez CG, Pereira J, Panes O (2008) Tissue factor
storage, synthesis and function in normal and activated human plate-
lets. Thromb Res 122 Suppl 1: S31-36
15. Kothari H, Nayak RC, Rao LV, Pendurthi UR (2010) Cystine 186-cysti-
ne 209 disulfide bond is not essential for the procoagulant activity of
tissue factor or for its de-encryption. Blood 115: 4273-4283
16. Henriksson CE, Klingenberg O, Hellum M, Landsverk KS, Joo GB,
Westvik AB, Kierulf P (2007) Calcium ionophore-induced de-encryp-
tion of tissue factor in monocytes is associated with extensive cell de-
ath. Thromb Res 119: 621-630
17. Basi DL, Ross KF, Hodges JS, Herzberg MC (2003) The modulation of
tissue factor by endothelial cells during heat shock. J Biol Chem 278:
11065-11071
18. Parry GC, Mackman N (1995) Transcriptional regulation of tissue fac-
tor expression in human endothelial cells. Arterioscler Thromb Vasc
Biol 15: 612-621

280
www.postepybiochemii.pl
Tissue factor in endothelial cells — its structure and
function according to the current literature
Joanna Drozdowska
*
Medical University of Warsaw, Department of General and Nutritional Biochemistry, 61 Żwirki i Wigury St., Warsaw, Poland
*
e-mail: jdrozdowska@wum.edu.pl
Key words: tissue factor, TF, asTF, encrypted TF, endothelium, HUVEC
ABSTRACT
Tissue factor (TF) is a glycosylated transmembrane protein, expressed by endothelial and other cells. Its best known function is the initiation
of thrombus formation, but TF also participates in inflammation processes. This article presents the established knowledge of TF expression
in endothelial cells and current opinions on encrypted TF form and mechanisms of its activation.
19. Moll T, Czyz M, Holzmuller H, Hofer-Warbinek R, Wagner E, Winkler
H, Bach FH, Hofer E (1995) Regulation of the tissue factor promoter in
endothelial cells. Binding of NF kappa B-, AP-1-, and Sp1-like trans-
cription factors. J Biol Chem 270: 3849-3857
20. Bierhaus A, Zhang Y, Deng Y, Mackman N, Quehenberger P, Haase
M, Luther T, Muller M, Bohrer H, Greten J, et al. (1995) Mechanism
of the tumor necrosis factor alpha-mediated induction of endothelial
tissue factor. J Biol Chem 270: 26419-26432
21. Rutkowski R, Pancewicz SA, Skrzydlewska E, Hermanowska-Szpako-
wicz T (2005) Właściwości biologiczne czynnika transkrypcji jądrowej
NF-κB. Alergia Astma Immunologia 10: 125-131
22. Mackman N (1995) Regulation of the tissue factor gene. FASEB J 9:
883-889
23. Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U (2005)
Procoagulant soluble tissue factor is released from endothelial cells in
response to inflammatory cytokines. Circ Res 96: 1233-1239
24. Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Ne-
merson Y (2003) Alternatively spliced human tissue factor: a circula-
ting, soluble, thrombogenic protein. Nat Med 9: 458-462
25. Butenas S, Bouchard BA, Brummel-Ziedins KE, Parhami-Seren B,
Mann KG (2005) Tissue factor activity in whole blood. Blood 105: 2764-
70
26. Butenas S, Orfeo T, Mann KG (2008) Tissue factor activity and function
in blood coagulation. Thromb Res 122 Suppl 1: S42-6
27. van den Berg YW, Versteeg HH (2010) Alternatively spliced tissue fac-
tor. A crippled protein in coagulation or a key player in non-haemosta-
tic processes? Hamostaseologie 30: 144-149
28. van den Berg YW, van den Hengel LG, Myers HR, Ayachi O, Jorda-
nova E, Ruf W, Spek CA, Reitsma PH, Bogdanov VY, Versteeg HH
(2009) Alternatively spliced tissue factor induces angiogenesis through
integrin ligation. Proc Natl Acad Sci USA 106: 19497-19502
29. Hobbs JE, Zakarija A, Cundiff DL, Doll JA, Hymen E, Cornwell M,
Crawford SE, Liu N, Signaevsky M, Soff GA (2007) Alternatively spli-
ced human tissue factor promotes tumor growth and angiogenesis in a
pancreatic cancer tumor model. Thromb Res 120 Suppl 2: S13-21
30. Eisenreich A, Boltzen U, Malz R, Schultheiss HP, Rauch U (2011)
Overexpression of alternatively spliced tissue factor induces the pro-
-angiogenic properties of murine cardiomyocytic HL-1 cells. Circ J 75:
1235-1242
31. Szcześniak M, Szweykowska-Kulińska Z (2009) Regulacja alternatyw-
nego splicingu. Post Biol Kom 36: 23-35
32. Eisenreich A, Bogdanov VY, Zakrzewicz A, Pries A, Antoniak S, Poller
W, Schultheiss HP, Rauch U (2009) Cdc2-like kinases and DNA to-
poisomerase I regulate alternative splicing of tissue factor in human
endothelial cells. Circ Res 104: 589-599
33. Eisenreich A, Malz R, Pepke W, Ayral Y, Poller W, Schultheiss HP,
Rauch U (2009) Role of the phosphatidylinositol 3-kinase/protein
kinase B pathway in regulating alternative splicing of tissue factor
mRNA in human endothelial cells. Circ J 73: 1746-1752
34. Chandradas S, Deikus G, Tardos JG, Bogdanov VY (2010) Antagonistic
roles of four SR proteins in the biosynthesis of alternatively spliced
tissue factor transcripts in monocytic cells. J Leukoc Biol 87: 147-152
35. Jiang K, Patel NA, Watson JE, Apostolatos H, Kleiman E, Hanson O,
Hagiwara M, Cooper DR (2009) Akt2 regulation of Cdc2-like kinases
(Clk/Sty), serine/arginine-rich (SR) protein phosphorylation, and in-
sulin-induced alternative splicing of PKCbetaII messenger ribonucleic
acid. Endocrinology 150: 2087-2097
36. Caldwell JA, Dickhout JG, Al-Hashimi AA, Austin RC (2010) Deve-
lopment of a continuous assay for the measurement of tissue factor
procoagulant activity on intact cells. Lab Invest 90: 953-962
37. Wolberg AS, Kon RH, Monroe DM, Ezban M, Roberts HR, Hoffman
M (2000) Deencryption of cellular tissue factor is independent of its
cytoplasmic domain. Biochem Biophys Res Commun 272: 332-6
38. Wolberg AS, Monroe DM, Roberts HR, Hoffman MR (1999) Tissue
factor de-encryption: ionophore treatment induces changes in tissue
factor activity by phosphatidylserine-dependent and -independent
mechanisms. Blood Coagul Fibrinolysis 10: 201-210
39. Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ,
Ruf W (2006) Disulfide isomerization switches tissue factor from co-
agulation to cell signaling. Proc Natl Acad Sci USA 103: 13932-13937
40. Manukyan D, von Bruehl ML, Massberg S, Engelmann B (2008) Pro-
tein disulfide isomerase as a trigger for tissue factor-dependent fibrin
generation. Thromb Res 122 Suppl 1: S19-22
41. Pendurthi UR, Ghosh S, Mandal SK, Rao LV (2007) Tissue factor ac-
tivation: is disulfide bond switching a regulatory mechanism? Blood
110: 3900-3908
42. Popescu NI, Lupu C, Lupu F (2010) Extracellular protein disulfide iso-
merase regulates coagulation on endothelial cells through modulation
of phosphatidylserine exposure. Blood 116: 993-1001
43. Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA (2005) Tissue-
-factor-bearing microvesicles arise from lipid rafts and fuse with acti-
vated platelets to initiate coagulation. Blood 106: 1604-1611
44. Versteeg HH, Ruf W (2007) Tissue factor coagulant function is enhan-
ced by protein-disulfide isomerase independent of oxidoreductase ac-
tivity. J Biol Chem 282: 25416-25424
45. Bilska A, Kryczyk A, Włodek L (2007) Różne oblicza biologicznej roli
glutationu. Postepy Hig Med Dośw 61: 438-453
46. Bach RR, Monroe D (2009) What is wrong with the allosteric disulfide
bond hypothesis? Arterioscler Thromb Vasc Biol 29: 1997-1998
47. Mandal SK, Iakhiaev A, Pendurthi UR, Rao LV (2005) Acute chole-
sterol depletion impairs functional expression of tissue factor in fibro-
blasts: modulation of tissue factor activity by membrane cholesterol.
Blood 105: 153-160
48. Clark MR (2011) Flippin’ lipids. Nat Immunol 12: 373-375
49. Ishii H, Mori T, Shiratsuchi A, Nakai Y, Shimada Y, Ohno-Iwashita Y,
Nakanishi Y (2005) Distinct localization of lipid rafts and externalized
phosphatidylserine at the surface of apoptotic cells. Biochem Biophys
Res Commun 327: 94-99
Wyszukiwarka
Podobne podstrony:
II CKN 273 97 id 209806 Nieznany
280 284 id 32002 Nieznany
Abolicja podatkowa id 50334 Nieznany (2)
4 LIDER MENEDZER id 37733 Nieznany (2)
katechezy MB id 233498 Nieznany
metro sciaga id 296943 Nieznany
perf id 354744 Nieznany
interbase id 92028 Nieznany
Mbaku id 289860 Nieznany
Probiotyki antybiotyki id 66316 Nieznany
miedziowanie cz 2 id 113259 Nieznany
LTC1729 id 273494 Nieznany
D11B7AOver0400 id 130434 Nieznany
analiza ryzyka bio id 61320 Nieznany
pedagogika ogolna id 353595 Nieznany
więcej podobnych podstron