
Materiałoznastwo
metale i ich stopy
1. Wybór materiału inżynierskiego – istotne właściwości
Ekonomia
cena (im coś droższe tym bardziej „odpycha”)
dostępność (czy trzeba coś sprowadzać zza granicy, czy kupić „za rogiem”)
Mechanicznie
właściwości
objętościowe
gęstość
współczynnik sprężystości i tłumienia (by na drgania był odporny)
granica plastyczności, wytrzymałość na rozciąganie
odporność na pękanie
wytrzymałość zmęczeniowa
odporność na zmęczenie cieplne
odporność na pełzanie (żeby z czasem przy stałym obciążeniu nie nastąpiło odkształcenie
plastyczne)
Niemechaniczne
właściwości
objętościowe
właściwości
cieplne,
optyczne,
magnetyczne,
elektryczne
Właściwości
powierzchni
utlenianie i korozja
tarcie, ścieralność i zużycie
Właściwości
produkcyjne
łatwość wytwarzania, łączenia części, wykańczania
Właściwości
estetyczne
wygląd powierzchni, przyjemny dotyk
2. Metale i ich stopy – spośród wszystkich pierwiastków 80 to metale. Rzadko jednak stosuje się czyste metale, w
przeciwieństwie do stopów
stop – co najmniej dwuskładnikowe tworzywo metaliczne składające się z metalu (jako składnika podstawowego) i
innych pierwiastków
Najpopularniejsze stopy:
Fe: stale, żeliwa
Cu: brązy, mosiądze
Al.: durale, siluminy
Ni, Ti
Cechy metali i stopów:
sztywność
ciągliwość (zdolność do odkształceń trwałych)
odporność na pękanie
dobra przewodność elektryczna i cieplna
połysk metaliczny
czyste metale mają małą wytrzymałość mechaniczną, stopy dużą, dlatego stosowane są np. w budowie konstrukcji
przenoszących obciążenia
wadą metali i stopów jest mała odporność chemiczna i łatwość ulegania korozji
Materiały ceramiczne i szkła – to nic innego jak głównie tlenki lub związki chemiczne metali z takimi pierwiastkami jak C,
N, P i S. Podstawowymi składnikami materiałów ceramicznych są Al
2
O
3
, SiO
2
, MgO, SiC, Si
3
N
4
. Są naturalnie kruche,
posiadają ok. 15 razy mniejszą odporność na rozciąganie niż na ściskanie
Cechy materiałów ceramicznych:
mała przewodność elektryczna i cieplna
duża zdolność do przenoszenia obciążeń ściskających
bardzo mała ciągliwość i odporność na pękanie
odporne na korozję (nie mogą się utlenić, bo już są tlenkami ;])
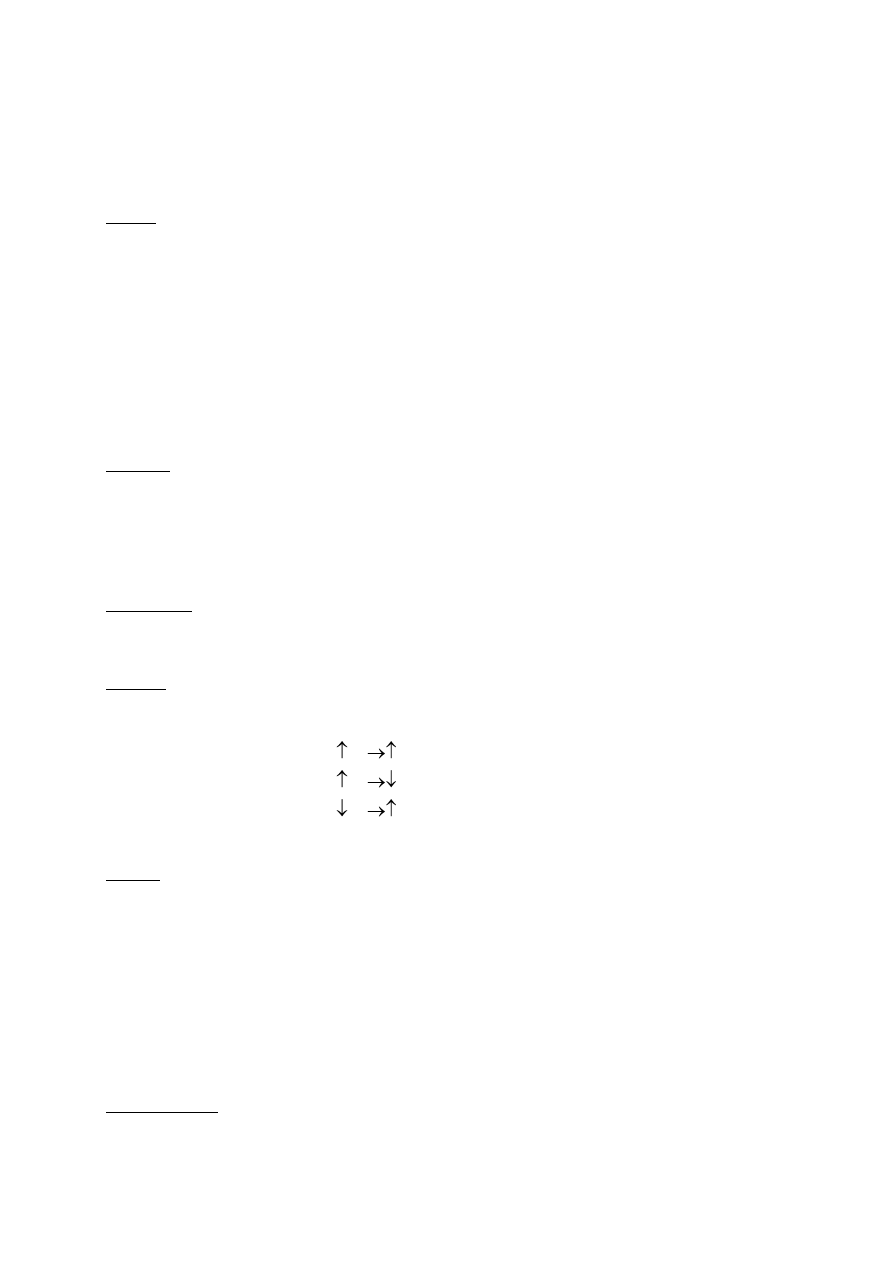
duża odporność na wysokie temperatury (wysokie temperatury topnienia np. Al
2
O
3
– 2020, gdy Al. - 660), przez co
bywają stosowane do budowy pieców przemysłowych
tak jak metale mają budowę krystaliczną (ułożenie atomów jest regularne i powtarzalne)
Cechy szkieł (materiałów amorficznych, bezpostaciowych):
posiadają strukturę niekrystaliczną
najczęściej stosowane są szkła krzemianowe, np. szyba okienna to 72% SiO
2
kruche
przezroczyste
odporne chemicznie
Polimery – zwane również tworzywami sztucznymi lub materiałami wielkocząsteczkowymi, to głównie związki chemiczne
węgla z wodorem
Cechy polimerów:
zbudowane z merów (z reguły ponad 500 w cząsteczce)
mała przewodność cieplna i energetyczna
odporność na korozję
estetyczny wygląd
mała gęstość
łatwość nadawania skomplikowanych kształtów
mała sztywność (wada!)
niska temperatura topnienia (wada!)
szybkie zmiany własności z temperaturą (do stosowania tylko w temperaturze nieodbiegającej od temperatury
naszego środowiska)
Kompozyty – kombinacje materiałów; składają się z przynajmniej dwóch materiałów należących do trzech uprzednio
wymienionych grup; przez odpowiedni dobór materiałów składowych można uzyskać kompozyt o żądanych właściwościach;
są one stosowane tam gdzie liczą się własności, a nie koszt, np. sprzęt sportowy oraz części samolotów.
Typowy podział kompozytów
wytwarzane przez człowieka – np. włókna szklane w osnowie polimeru
naturalne – np. drewno (włókna celulozowe, "sklejone" ligniną)
Półprzewodniki – wyróżniamy je jako osobny rodzaj ze względu na ich szczególne własności elektryczne; półprzewodniki
umożliwiły m.in. produkcję satelitów komunikacyjnych i nowoczesnych komputerów. (Opracował Dominik Kozłowski.)
3. Istniejące zależności między strukturą, procesem wytwarzania i własnościami pozwalają, przez właściwy dobór składu
chemicznego i sposób wytwarzania za uzyskanie struktury zapewniającej żądane własności użytkowe materiału.
Własności: np. moduły sprężystości, granica plastyczności, wytrzymałość na rozciąganie, odporność na pękanie, twardość i
ciągliwość, wytrzymałość na zmęczenie (pod wpływem obciążeń zmieniających się cyklicznie), wytrzymałość na pełzanie
(zachowanie się w wysokich temperaturach).
Zmiana temperatury ma istotny wpływ na zmianę właściwości materiału.
kruchosc
T
ściowe
wytrzymalo
wl
T
plastyczne
wl
T
.
.
Polimery b. szybko miękną ze wzrostem temperatury. Mogą być więc stosowane jedynie w niskich temperaturach, w
przeciwieństwie do np. ceramik lub kompozytu C-C.
Struktura: Należy ją rozpatrywać na kilku poziomach.
Najniższy stanowi struktura atomu. Liczba i rozmieszczenie elektronów wokół jądra atomu, a zwłaszcza liczba elektronów na
zewnętrznej powłoce elektronowej wpływa na rodzaj wiązania między atomami i w konsekwencji na ich własności
mechaniczne, elektryczne, cieplne, optyczne i magnetyczne.
Następny poziom dotyczy rozmieszczenia elektronów w przestrzeni. W metalach i wielu materiałach ceramicznych atomy
ułożone są w sposób krystaliczny (uporządkowany i powtarzalny). Niektóre materiały ceramiczne i większość polimerów nie
mają uporządkowanego ułożenia atomów, co wpływa na to, że wiele ich zachowań jest odmiennych od zachowań materiałów
krystalicznych.
W materiałach krystalicznych zawsze występują pewne zaburzenia (defekty struktury krystalicznej). Ich obecność wywiera
duży wpływ na własności (zwłaszcza mechaniczne). Materiały krystaliczne są zbudowane z małych kryształów nazywanych
ziarnami. Na powierzchni styku ziaren (granica ziaren) następuje zmiana ich orientacji krystalicznej, co wpływa na
własności. Na tym poziomie (nazywanym mikrostrukturą) istotne są: wielkość i kształt ziaren.
Większość materiałów składa się z więcej niż jednej fazy. Ponieważ fazy różnią się strukturą lub składem chemicznym to ich
własności są również różne.
Proces wytwarzania: W procesie wytwarzania z materiału o kształcie wstępnym uzyskuje się element o założonym kształcie i
własnościach. Rodzaj zastosowanego procesu wytwarzania zależy przynajmniej częściowo od własności przerabianego
materiału zależnych od jego struktury.
4. Atom składa się z dodatnio naładowanego jądra i zewnętrznej warstwy elektronowej. Jądro składa się natomiast z
protonów i elektronów. Protony są dodatnie, elektrony ujemne, a neutrony obojętne.

Protony, neutrony i mezony (w 1947 roku w promieniowaniu kosmicznym odkryto nowy rodzaj cząstek - tzw. mezony, które
są nośnikami oddziaływań jądrowych) zbudowane są z cząstek fundamentalnych, tzw. kwarków. Znamy dziś 6 różnych
kwarków:
· kwark u (up – górny)
· kwark d (down – dolny)
· kwark c (charm – powabny)
· kwark s (strange – dziwny)
· kwark b (bottom – denny)
· kwark t (top – szczytowy)
Kwarki posiadają ładunek elektryczny równy ułamkowi ładunku elementarnego.
Np. dodatni proton składa się z dwóch kwarków „u” i jednego kwarku „d”. Stąd właśnie wywodzi się jego ładunek:
1
3
1
3
2
3
2
Neutron zbudowany jest natomiast z dwóch kwarków „d” oraz jednego kwarku „u”. Wartość jego ładunku wynosi zatem:
0
3
2
3
1
3
1
Kwarki nie mogą nigdy występować pojedynczo, lecz zawsze w grupach po dwa lub trzy. Wiążą się w protony i neutrony
poprzez oddziaływanie silne przenoszone przez gluony
Istnieje także teoria superstrun. Materia składa się z atomów, które z kolei są zbudowane z kwarków i elektronów.
Zgodnie z teorią strun wszystkie te cząstki mają w rzeczywistości postać maleńkich pętli drgających strun. Na dzień
dzisiejszy uważa się, że struny są absolutnie elementarnymi składnikami materii.
Różnice własności cząstek uważanych dotychczas za elementarne (masa, ładunek ) biorą się stąd, że ich struny drgają według
innych rezonansowych wzorów. (Opracowała Ewa Skiba).
5. WIĄZANIE CHEMICZNE - są to charakterystyczne oddziaływania występujące pomiędzy atomami, grupami
atomów, jonami lub cząsteczkami.
Wiązania chemiczne powstają w wyniku oddziaływania, przyjmowania lub uwspólniania elektronów walencyjnych
reagujących ze sobą atomów. Każdy atom pierwiastka składa się z jądra atomowego oraz elektronów znajdujących się na
tzw. powłokach elektronowych wokół jądra. Powłok tych jest kilka, a ich maksymalna ilość to siedem. W tworzeniu
wiązania bierze udział głównie ostatnia powłoka tzw. walencyjna.
Teorię powstawania wiązań wprowadzili Kossel i Lewis.
Wegług niej, pierwiastki dążą do osiągnięcia oktetu (lit i beryl - dubletu) elektronów na powłoce walencyjnej - dążą do
uzyskania struktury gazu szlachetnego.
Atomy gazów szlachetnych, różnią się od atomów pozostałych pierwiastków całkowicie zapełnionymi powłokami
elektronowymi. Taka konfiguracja powoduje, że helowce są najbardziej biernymi chemicznie pierwiastkami.
Atomy tworząc wiązanie chemiczne mogą uzyskać stabilną konfigurację elektronową - podobną do konfiguracji gazów
szlachetnych - (dubletową lub oktetową) na drodze:
przekazania elektronów jednego atomu drugiemu - powstaje wiązania heteropolarne - jonowe; uwspólnienia
(współużytkowania) elektronów walencyjnych - powstaje wiązanie kowalencyjne (atomowe) lub donorowo-akceptorowe
(koordynacyjne).
Rodzaj wiązania między atomami, zależy od właściwości pierwiastków tworzących związek chemiczny. Można je
scharakteryzować za pomocą elektroujemności, która jest umowną miarą "skłonności" atomu do przyciągania elektronów
podczas tworzenia wiązania. Pojęcie elektroujemności zostało wprowadzone przez L. Paulinga.
Elektroujemność to zdolność atomu do przyjmowania elektronów. Może być ona określana za pomocą liczb
bezwymiarowych - skala elektroujemności Paulinga.
Do pierwiastków elektroujemnych zalicza się te, których atomy wykazują wyższą tendencję do przyłączania elektronów
niż do jonizacji. Należą do nich niemetale (najbardziej elektroujemne są fluorowce).
Do pierwiastków elektrododatnich zalicza się te, których atomy wykazują wyższą tendencję do jonizacji (oddawania
elektronów) niż do przyłączania elektronów. Należą do nich metale.
Wg sklali Paulinga:
elektroujemność pierwiastków należących do tej samej grupy maleje nieznacznie, ze wzrostem liczby atomowej.
elektroujemność pierwiastków należących do tego samego okresu rośnie ze wzrostem liczby atomowej.
WIĄZANIE JONOWE - polega na przejściu jednego lub kilku elektronów walencyjnych z atomów
pierwiastka elektrododatniego do atomów pierwiastka elektroujemnego.
Atom pierwiastka oddający eletrony staje się kationem, a atom przyjmujący elektrony staje się anionem.
Powstałe różoimienne jony przyciągają się siłami elektrostatycznymi, tworząc wiązanie - sieć jonową.
Wiązania jonowe są wiązaniami mocnymi.
Powstają pomiędzy pierwiastkami, w których różnica elektroujemności jest większa od 1,7 (wg skali
Paulinga),
np.: chlorek sodu - Na Cl
Na - 1e --> Na
+
Cl + 1e --> Cl
-
Każdy jon sodu jest otoczony sześcioma jonami chlorkowymi, a każdy jon chlorkowy sześcioma jonami
sodowymi.
Nie można rozróżnić, który kation sodu do którego anionu chlorkowego należy, podobnie jak nie da się
określić, który anion chlorkowy należy do którego kationu sodu. Cały kryształ traktuje się więc jako jedną
makrocząsteczkę.

Związki
połączone
wiązaniem
jonowym
charakteryzuje:
krystaliczna
budowa
dobra
rozpuszczalność
w
wodzie
wysoka
temperatura
topnienia
przewodnictwo elektryczne - zarówno w roztworze jak i w stanie stopionym przewodzą prąd elektryczny.
Wiązanie jonowe tworzy się w litowcach (wykluczając wodór) oraz berylowcach w związkach z tlenowcami
i fluorowcami. WIĄZANIE KOWALENCYJNE (atomowe) - polega na utworzeniu wspólnej pary elektronów
(wiązanie pojedyncze), dwóch wspólnych par elektronów (wiązanie podwójne) lub trzech wspólnych par elektronów
(wiązanie potrójne), przez dwa atomy, z których każdy dostarcza do wytworzenia wspólnego dubletu (lub dubletów) taką
samą liczbę niesparowanych elektronów. Wiązania kowalencyjne występują pomiędzy pierwiastkami, w których różnica
elektroujemności jest równa 0. Przykładami takiego wiązania są cząsteczki dwuatomowe: H
2
, O
2
, N
2
, Cl
2
, Br
2
, I
2
. np.:
Warunkiem utworzenia wiązania kowalencyjnego przez dany atom, jest obecność przynajmniej jednego niesparowanego
elektronu. Uwspólnianie elektronów powoduje, że atomy uzyskują oktet elektronów, czyli konfigurację najbliższego
gazu
szlachetnego.
W rachunkach uwspólnione elektrony należy liczyć podwójnie: raz, że należą do jednego atomu, a drugi raz do drugiego
atomu.
Związki kowalencyjne (tworzące wiązania kowalencyjne), tworzą w stanie stałym sieć krystaliczną zbudowaną z
odrębnych
cząsteczek.
Mają niskie temperatury topnienia i wrzenia, rozpuszczają się w rozpuszczalnikach niepolarnych lub słabo polarnych.
Skroplone związki kowalencyjne i ich roztwory, nie przewodzą prądu elektrycznego, ponieważ nie ulegają dysocjacji.
Reakcje między związkami kowalencyjnymi polegają na rozerwaniu istniejących wiazań i tworzeniu nowych.
WIĄZANIA KOWALENCYJNE SPOLARYZOWANE - polega na uwspólnieniu pary elektronów, ale wspólna para
nie należy w jednakowym stopniu do obu atomów, lecz jest przesunięta w kierunku atomu bardziej elektroujemnego.
Zjawisko przesunięcia uwspólnionej pary elektronów w kierunku jednego z atomów, nosi nazwę polaryzacji wiązania.
Ma to miejsce w przypadku, kiedy atomy pierwiastków różnią się elektroujemnością, ale różnica nie przekracza 1,7 w
skali
Paulinga.
Polarność wiązania rośnie w miarę jak zwiększa się różnica miedzy elektroujemnościami pierwiastków. Jeśli przekroczy
wartość
1,
7
wiązanie
przyjmuje
charakter
jonowy.
Wiązanie spolaryzowane jest najbardziej pospolite dla związków nieorganicznych i organicznych w skład których
wchodzą
atomy
niemetali
różniących
się
dość
znacznie
wartością
elektroujemności.
Przykład
wiązania
kowalencyjnego
spolaryzowanego:
połączenie chloru i wodoru w cząsteczce chlorowodoru. WIĄZANIE KOORDYNACYJNE (semipolarne) - stanowi
szczególny przypadek wiązanie typu kowalencyjnego. Polega na utworzeniu wspólnej pary elektronowej z elektronów
dostarczonych przez jeden atom (tzw. donor). drugi atom (tzw. akceptor) uzupełnia własną powłokę walencyjną
elektronami
donora.
Warunkiem powstania wiazania koordynacyjnego jest zderzenie drobiny posiadającej wolną parę elektronową z drobiną
dysponującą
luką
elektronową
lub
wolnym
orbitalem
w
powłoce
walencyjnej.
Przykład: wiązania w tlenku siarki (IV):
WIĄZANIE METALICZNE - powstanie wiązania metalicznego polega na przekształceniu atomów tego samego
metalu lub atomów różnych metali w zbiór kationów i swobodnie poruszających się między nimi elektronów.
Wiązanie
metaliczne
może
istnieć
w
stanie
stałym
lub
ciekłym.
W stanie stałym węzły sieci krystalicznej metalu lub stopu są obsadzone przez kationy wykonujące wyłącznie ruchy
oscylacyjne wokół węzła, natomiast zdelokalizowane elektrony poruszają się swobodnie w obrębie całego kryształu,
podobnie jak drobiny substancji w stanie gazowym. Z tego względu mówi się o gazie elektronowym (chmurze
elektronowej)
wiązania
metalicznego.
Kationy stanowiące rdzenie atomowe utrzymują się w swoich położeniach dzięki przyciądaniu elektrostatycznemu
elektronów.
Właściwości
metali
wiążą
się
z
istniejącym
wiązaniem
metalicznym:
dobre przewodnictwo cieplne i elektryczne można uzasadnić ruchliwością elektronów należących do dazu
elektronowego;
połysk metaliczny wynika stąd, że pod wpływem światła widzialnego, elektrony znajdujące się na powierzchni kryształu
wykonują drgania o częstotliwości promieniowania padającego. Promienie odbite mają taką samą częstotliwość jak
promienie
padające,
co
postrzegamy
jako
charakterystyczny
połysk
metalu;
plastyczność - ciągliwość, kowalność metali, tłumaczy się brakiem w krysztale kierunków uprzywilejowanych, a więc
można przesuwać płaszczyzny sieciowe i powodować pęknięcia metali. Model budowy wewnętrznej metalu:
WIĄZANIE WODOROWE - jest to oddziaływanie (zwykle słabe) między kowalencyjnie związanym atomem wodoru
i należącym do innej cząsteczki atomem silnie elektroujemnym, dysponującym wolną parą elektronową.
Atom wodoru (proton) może byś związany równocześnie z dwoma atomami, jeśli mają one małe wymiary i dużą
elektroujemność. Wiązanie wodorowe występuje najczęściej w związkach wodoru z fluorem, chlorem, tlenem, azotem.
Wiązanie wodorowe występuje np.: między cząsteczkami wody. Substancje, w których występuje wiązanie wodorowe
charakteryzują
się
wysokimi
temperaturami
wrzenia
i topnienia. Energia wiązań Jonowe 600 - 1550 kJ/mol Kowalencyjne 500-1250 Metaliczne 100 -850 Van der Vaalsa
< 40 Komórka elementarna - w krystalografii - najmniejsza, powtarzalna część struktury kryształu, zawierająca
wszystkie rodzaje cząsteczek, jonów i atomów, które tworzą określoną sieć krystaliczną. Komórka elementarna powtarza
się we wszystkich trzech kierunkach, tworząc zamknięta sieć przestrzenną, której główną cechą jest symetria. Komórka
elementarna ma zawsze kształt równoległościanu.
Poprzez translacje komórki elementarnej o wektory będące całkowitymi wielokrotnościami wektorów sieci krystalicznej
otrzymuje się całą sieć krystaliczną kryształu.Przykładowe komórki elementarne:
{kind=link}
komórka elementarna układu
regularnego
{kind=link}
komórka elementarna układu
tetragonalnego
komórka elementarna układu
heksagonalnego
komórka elementarna układu
trygonalnego
komórka elementarna układu
rombowego
{kind=link}
komórka elementarna układu
jednoskośnego
{kind=link}
komórka elementarna układu
trójskośnego
Stałe sieciowe(parametry sieciowe)
Długość krawędzi komórki( a,b,c)
Kąty między krawędziami( alfa, beta, gamma)
Siedem układów krystalograficznych:
Bravais'go sieci, czternaście rodzajów sieci krystalicznych wprowadzonych przez
, opisujących
rzeczywiste kryształy, sklasyfikowanych w 7 rodzajach układów. Są to układy (w nawiasach dane charakteryzujące
układ: proporcje boków a,b,c, kąty w narożniku α, β, γ i liczba sieci w danym układzie):
- trójskośny (a ≠ b ≠ c, α ≠ β ≠γ, 1),
- jednoskośny (a ≠ b ≠ c, α = β ≠ γ = 90°, 2),
- rombowy (a ≠ b ≠ c, α = β = γ = 90°, 4),
- tetragonalny (a = b ≠ c, α = β = γ = 90°, 2),
- regularny (a = b = c, α = β = γ = 90°, 3),
- romboedryczny (a = b = c, 90° ≠ α = β = γ < 120°, 1)
- heksagonalny (a = b ≠ c, α = β = 90°, γ = 120°, 1).
7.Większość materiałów ma jedną z trzech struktur krystalicznych:
- strukture regularną ściennie centrowaną (RSC, cF4, A1) taką strukturą krystaliczną mają miedzy innymi: Cu, Al, Ni, Fe-
(gama), Ag, Au i Pb
- strukturę regularną przestrzennie centrowaną (RPC, cI2, A2) taką strukturą krystaliczną mają miedzy innymi: Mo, W, Y,
Nb, Cr-(alfa)
-strukturę heksagonalną zwartą (HZ, hP2, A3 ) taką strukturą krystaliczną mają: Zn, Mg, Cd, Ti-(alfa) ,Zr-(alfa)
8.
Wsółczynnik Poissona jest to ujemny stosunek odkształcenia poprzecznego do odkształcenia liniowego w zakresie
odkształceń sprężystych. Jest to wielkość bezwymiarowa i nie określa sprężystości materiału, a jedynie sposób w jaki sie
odkształca. Definiujemy go wzorem:
ν= -ε
p
/ε
n
gdzie
ν - współczynnik Poissona
ε
p
- odkształcenie poprzeczne
ε
n
- odkształcenie liniowe
(Opracowała Ewelina Wróblewska).
9. Moduł Younga (tutaj M.Y.)
Moduły sprężystości zdefiniowane prawem Hooke’a:
-wartość odkształcenia jest wprost proporcjonalna do naprężenia rozciąg.
ζ=Eε, gdzie ζ-naprężenie rozciągające, E-moduł Younga, ε-odkształcenie liniowe.
Podobna zależność występuje przy ściskaniu.
Moduł Younga (E) inaczej moduł odkształcalności liniowej albo moduł sprężystości podłużnej (w układzie odniesienia SI).
Wielkość uzależniająca
ε materiału od
ζ jakie w nim występuje w zakresie odkształceń
sprężystych
Odkształcenie w zakresie liniowo sprężystym zachodzi dzięki zmianie odległości między atomami
Wartość M.Y,-miara oporu stawianego przez sąsiadujące atomy podczas zwiększenia odległości między nimi.
M.Y. jest proporcjonalny do pochylenia krzywej w punkcie odpowiadającym równowagowej odległości między atomami
(a
0
).
Duży moduł (E)- potrzeba dużej siły by nastąpiło odkształcenie sprężyste.
Mały moduł (?)-----------małej siły--------------------------------------------.
Moduł sprężystości (E)- określa sztywność materiału, tj. opór mat. przeciw wydłużaniu lub ściskaniu sprężystemu.
E- zależy od typu wiązań miedzy atomami, skł.chem., struktury kryst..
Obróbka cieplna i plastyczna mają mały wpływ na E, jeśli nie zmieniają parametrów wymienionych wyżej. Zależność nie jest
liniowa.(Opracował Paweł Bielecki).
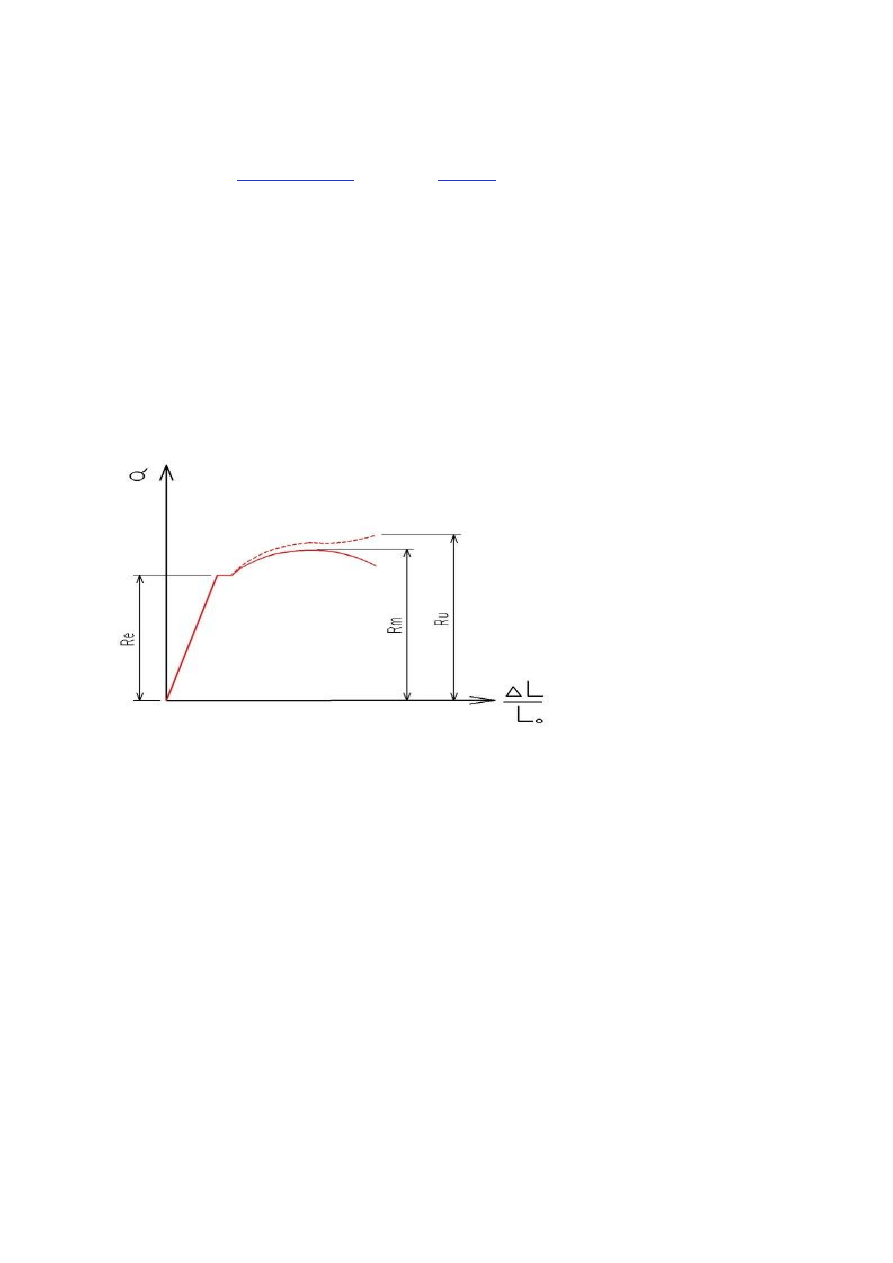
11. Statyczna próba rozciągania – podstawowa metoda badań wytrzymałościowych dla metalowych materiałów
konstrukcyjnych.
W statycznej próbie rozciągania rozciąga się odpowiednio wykonany pręt o przekroju okrągłym wykorzystując urządzenie
zwane zrywarką. W czasie próby rejestruje się zależność przyrostu długości próbki od wielkości siły rozciągającej oraz
rejestruje się granicę sprężystości, przewężenie próbki i siłę zrywającą próbkę.
Re - granica spręzystości
Rm - wytrzymałość na rozciąganie
Ru - naprężenie rozrywające
Typowy wykres naprężenie-odkształcenie pokazuje rysunek u góry. Początkowo wzrost naprężenia powoduje liniowy wzrost
odkształcenia. W zakresie tym obowiązuje prawo Hooke'a. Po osiągnięciu naprężenia Re materiał przechodzi w stan
plastyczności, a odkształcenie staje się nieodwracalne.
(Uwaga! Wykres przedstawia dwie linie. Przerywana pokazuje naprężenie rzeczywiste obliczane przy uwzględnieniu
przewężenia próbki. Linia ciągła pokazuje wykres naprężenia obliczanego przy uwzględnieniu pola wyjściowego próbki.
Czyni się tak, by zaobserwować wartość Rm, będącą lokalnym maksimum krzywej).
12. Granica sprężystości - to takie naprężenie, po przekroczeniu którego nie następuje powrót - po zdjęciu obciążenia - do
pierwotnej, nieodkształconej i wolnej od naprężeń postaci.
Prawo Hooke'a – prawo mechaniki określające zależność odkształcenia od naprężenia. Głosi ono, że odkształcenie ciała pod
wpływem działającej nań siły jest wprost proporcjonalne do tej siły.
Wytrzymałość na rozciąganie - jest to naprężenie odpowiadające największej sile niszczącej Fm uzyskanej w czasie
prowadzenia próby rozciągania, odniesionej do pierwotnego przekroju poprzecznego tej próbki.
Naprężenie rozrywające - jest to wartość siły powodującej zerwanie w odniesieniu do przekroju zerwanej próbki w jej
najwęższym miejscu.
Współczynnik Poissona - jest stosunkiem odkształcenia poprzecznego do odkształcenia podłużnego przy osiowym stanie
naprężenia. Współczynnik Poissona jest wielkością bezwymiarową i nie określa sprężystości materiału, a jedynie sposób w
jaki się on odkształca.(Opracował Rafał Kurowski).
13.Twardość- odporność na odkształcenie innymi materiałami.
Pomiar twardości odbywa się poprzez oddziaływanie twardszego ciała na bardziej miękkie.
Stosuje się trzy różne sposoby pomiaru twardości: Brinella (HB), Yickersa oraz Rockwella.
Stosuje się też inne metody gdy nie ma możliwości sprawdzenia twardości powyższymi sposobami.
Twardość podawana jest ze znakiem HB lub HV ale też MN/m
2
14.Udarność- odporność na działanie naprężeń dynamicznych. Miara odporności: energia konieczna do zniszczenia próbki
obciążonej udarowo.
-
energia potrzebna do złamania jest określona z różniczy położenia początkowego i końcowego wahadła.
-
Stopy o strukturze RSC-ciągliwy typ przełomu(dobra udarność) niezależnie od Temp.
-
O strukturze HZ-są zazwyczaj kruche

-
O strukturze RPC-sposób pękania zależy od T, w niskiej pękanie kruche,w wysokiej pękanie ciągliwe.
Temperatura przejścia- 100a + 100ºC -> przejście od przełomu kruchego do ciągliwego
a=TPSK (Opracowała Marzena Mikołajczyk).
15. Odporność na pękanie to właściwość mechaniczna materiału. Udarność - Jest to odporność materiału na pękanie przy
uderzeniu, miarą udarności jest energia wyrażona w [J] na złamanie znormalizowanej próbki.
Pękanie plastyczne
Występuje w materiałach plastycznych (metalach), przebiega przez uplastycznienie strefy wokół krawędzi istniejącego
pęknięcia. Przełom ma szorstką powierzchnię.
Kruche pękanie
Występuje w materiałach o wysokiej granicy plastyczności. Przełom ma gładką powierzchnię.(Opracowała Paulina Stojak).
16. Zmęczenie materiału- to jego pękanie pod wpływem cyklicznie zmieniających się naprężeń. Podczas eksploatacji
ruchomych części maszyn 90% stanowią zniszczenia zmęczeniowe. Wynikiem pojedyńczego badania zmęczeniowego
materiału jest liczba cykli obciążeń aż do wystąpienia zniszczenia (N) przy zadanej amplitudzie napreżeń (ζ). W przypadku
stali, poniżej pewnej amplitudy naprężeń, próbka nie ulega zniszczeniu nawet przy bardzo dużej liczbie cykli.Graniczną
amplitudę naprężeń nazywamy wytrzymałością zmęczeniową. W przyapdku innych stopów amplituda naprezeń maleje ze
wzrostem liczby cykli powodujących zniszczenie. W praktyce wytrzymałość zmęczeniową stopów niezależnych definiuje się
jako największą wartość amplitudy naprężeń nie powodującej zniszczenia materiału podczas abitralnie dużej liczby
cykli(zwykle N=10
8
). Zwiększenie wytrzymałości zmęczeniowej materiału jest możliwe przez eliminację lub ograniczenie
miejsc zarodkowania pęknięć zmęczeniowych. Innym sposobem poprawy wytrzymałości zmęczeniowej jest wprowadzenie
naprężeń ściskających w warstwę powierzchniową materiału lub jej umocnienie. Wykonuje się to zwykle przez śrutowanie,
nawęglanie, azotowanie lub hartowanie. Powierzchnia przełomu zmeczeniowego składa się z dwóch części: jednej gładkiej z
koncentrycznymi liniami względem miejsca od którego rozwijało się pęknięcie, drugiej szorstkiej i ziarnistej. Część pierwsza
powstaje powstaje powoli, a linie koncentryczne wskazują na zmianę jej wielkości z upływem czasu eksploatacji elementu.
Druga część odpowiada gwałtownemu zniszczeniu elementu.
19. Defektami punktowymi nazywa się zakłócenia budowy krystalicznej umiejscowione
wokół punktu. Najprostszym defektem tego typu jest brak atomu w węźle sieci przestrzennej, zwany wakansem albo luką.
Wakanse powstają przede wszystkim wskutek drgań cieplnych sieci, które są tym większe,
im wyższa jest temperatura. Przy określonej amplitudzie drgań atom może wypaść ze swego
średniego położenia w węźle sieci i zająć pozycję międzywęzłową. Powstaną wówczas
jednocześnie dwa defekty punktowe: wakans i atom wtrącony między węzłowo. Oba
wywołują lokalne zakłócenie budowy sieciowej, gdyż obecność wakansu powoduje większe
od normalnego zbliżenie sąsiednich atomów (rys. 2.15b)*, natomiast atom wtrącony powoduje rozsunięcie sąsiednich
atomów na odległość większą od normalnej. Opisany defekt nosi nazwę defektu Frenkla i może powstawać tylko w
strukturach metali alkalicznych, w których odległości między atomami są wystarczająco duże, by atom mógł zająć pozycję
międzywęzłową (rys. 2.15b)*. Natomiast w zwarcie wypełnionych sieciach krystalicznych
tworzą się, defekty punktowe, polegające na powstawaniu wakansu i wywędrowaniu atomu,
który ten wakans utworzył, na powierzchnię kryształu. Ten typ defektu nazywa się defektem
Schottky'ego i jest powszechny w kryształach metali – rys. 2.15a*. Wakanse powstające w
sieci mogą wędrować wewnątrz kryształu przez zamianę miejsc z węzłami obsadzonymi
atomami. Mogą wywędrować na powierzchni kryształu, co prowadzi do zmniejszenia się
ogólnej liczby wakansów. Mogą wreszcie się łączyć, tworząc tzw. zgrupowania wakansów.
Rys.2.15. Punktowe defekty sieci krystalicznej wywołane drganiami cieplnymi: a) defekt
Schottky’ego, b) defekt Frenkla
Punktowe defekty sieci tworzą również znajdujące się w niej obce atomy. Możliwe są tu
następujące przypadki. Jeśli obcy atom ma średnicę atomową dużo mniejszą od średnicy
atomowej atomów metalu, to zajmuje on położenie między węzłowe, wywołując lokalne
rozsunięcie sąsiednich atomów i powiększenie parametrów sieci (rys.2.16b)*. W typowych
sieciach krystalicznych metali przestrzenie międzywęzłowe są niewielkie, toteż położenie
międzywęzłowe mogą zajmować w nich tylko atomy azotu, wodoru, węgla i boru, mające
najmniejsze średnice atomowe. Wtrącone atomy innych pierwiastków mogą zajmować
wyłącznie pozycje węzłowe zastępując atomy metalu podstawowego. W tym przypadku
rodzaj zniekształcenia sieci krystalicznej zależy od tego czy obcy atom ma mniejszą, czy
większą średnicę od atomu metalu podstawowego (rys. 2.16b i c)*. Jeśli większą — występuje lokalne rozsunięcie sąsiednich
atomów (powiększenie parametrów sieci), jeśli mniejszą — lokalne zbliżenie atomów (zmniejszenie parametrów sieci)
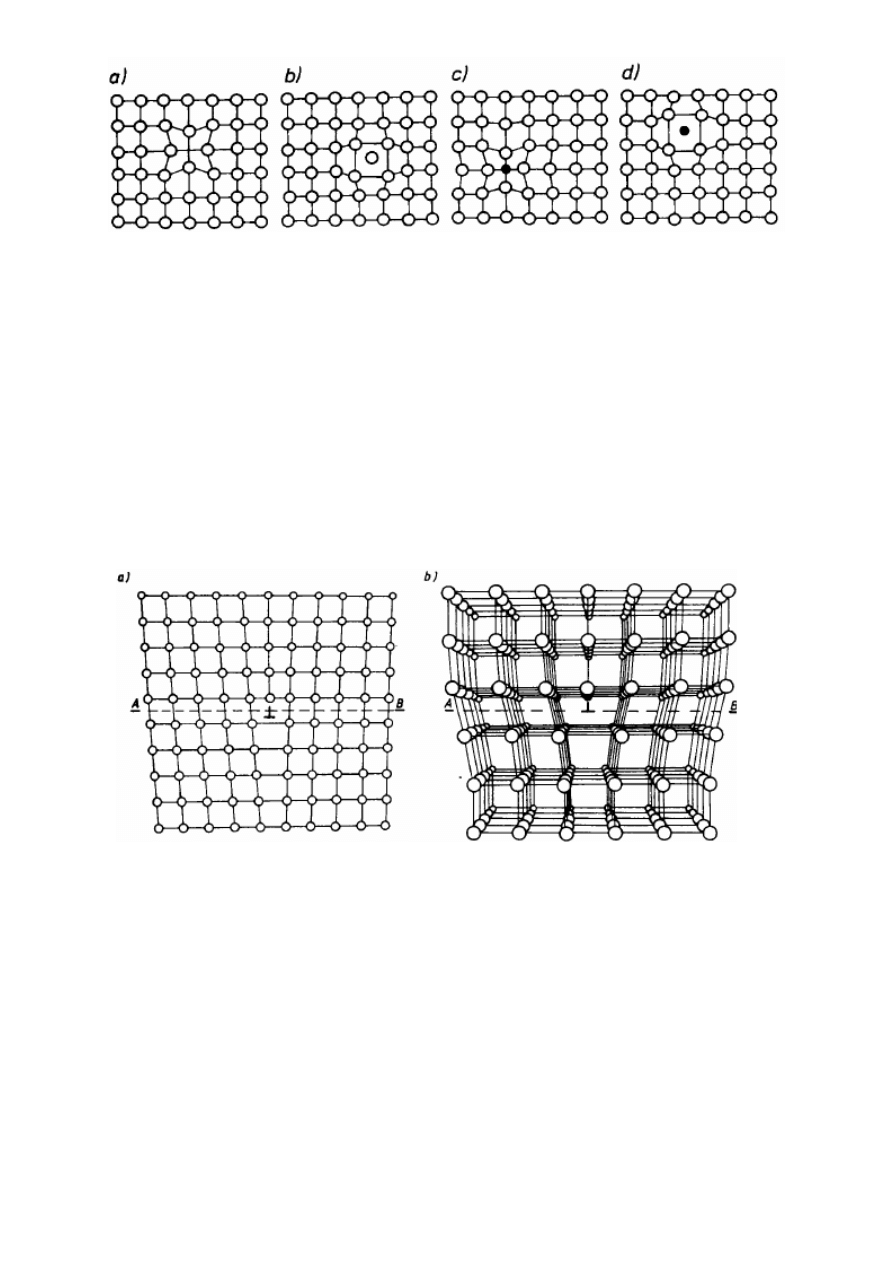
Rys.2.16. Defekty punktowe: a) wakans, b) atom międzywęzłowy, c) atom obcy
węzłowy, d) atom obcy międzywęzłowy
Odkształcenie sieci wywołane wakansem polega na kontrakcji, a atomem
międzywęzłowym — na ekspansji. Atom obcy węzłowy powoduje kontrakcję, jeżeli jego
promień jest mniejszy, albo ekspansję, jeżeli jego promień jest większy od promienia atomu
bazowego, natomiast atom obcy międzywęzłowy zawsze powoduje ekspansję sieci.
Wzajemne oddziaływanie defektów punktowych, przy większym ich stężeniu
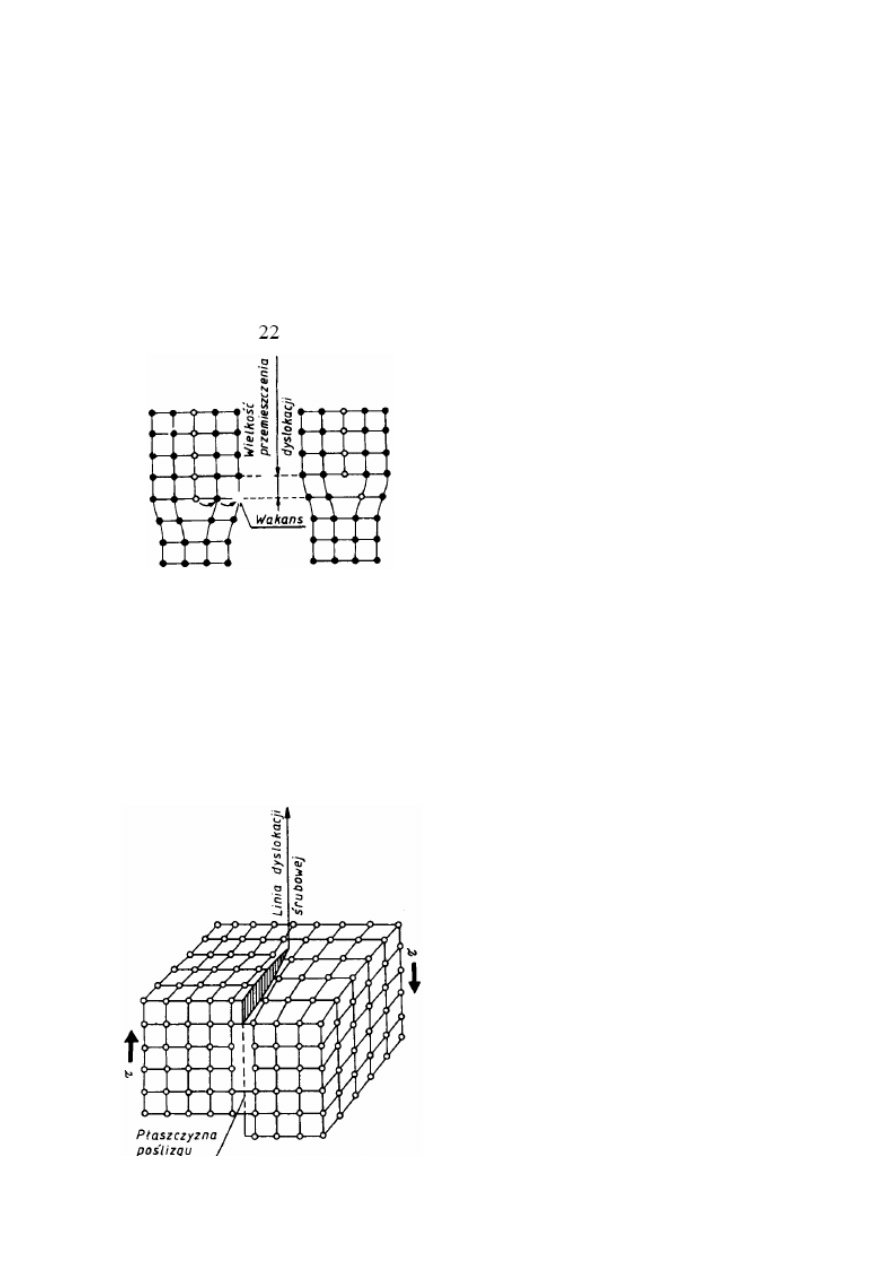
20. Defektami liniowymi nazywa się zakłócenia budowy krystalicznej, które w jednym
kierunku mają wymiar kilku odległości atomowych, a w drugim — całego ziarna lub znacznej
jego części. Rozróżnia się dwa zasadnicze rodzaje defektów liniowych: dyslokację
krawędziową i dyslokację śrubową
Dyslokację krawędziową wywołuje obecność w przestrzennej sieci krystaliczne
dodatkowej półpłaszczyzny obsadzonej atomami (zw. ekstrapłaszczyzną), które krawędź
stanowi dowolna linia brzegowa, nazywana linią dyslokacji. W zależność od usytuowania
dodatkowej półpłaszczyzny rozróżnia się dyslokację dodatnią, oznaczoną symbolem - ⊥ i
ujemną, oznaczoną symbolem T (pionowa kreska w symbolu dyslokacji oznacza dodatkową
półpłaszczyznę, pozioma — płaszczyznę poślizgu). Na rysunku 2.17* pokazano dyslokację
krawędziową dodatnią
Rys. 2.17. Schemat dyslokacji krawędziowej w krysztale o sieci regularnej: a) przekrój
poprzeczny kryształu zawierającego dyslokację dodatnią, b) perspektywiczny obraz
rozmieszczenia atomów wokół dyslokacji dodatniej; AB — płaszczyzna poślizgu
Wokół linii dyslokacji istnieje pole naprężeń sprężystych; ściskających w części kryształu zawierającej dodatkową
półpłaszczyznę (odległości między sąsiednimi atomami są mniejsze od stałych sieciowych), rozciągających — w pozostałej
części kryształu (odległości między sąsiednimi atomami są większe od stałych sieciowych). Wynika z tego, że wokół
dyslokacji krawędziowej występuje jednocześnie postaciowe i objętościowe odkształcenie kryształu. Dyslokacje
krawędziowe charakteryzują się określonymi własnościami dynamicznymi, m.in. mają możliwość poruszania się w
płaszczyźnie poślizgu pod wpływem naprężeń
wewnętrznych lub zewnętrznych, w wyniku czego następuje poślizg części kryształu wzdłuż
określonej płaszczyzny sieciowej. Obliczono, że naprężenie potrzebne do wywołania
przesuwania się dyslokacji jest bardzo małe, rzędu l MPa pod warunkiem, że siły wiązań w
krysztale nie zależą od kierunków.
Dyslokacje krawędziowe mogą się przemieszczać w krysztale również przez wspinanie,
polegające na odłączeniu się atomów od krawędzi dodatkowej półpłaszczyzny i ich migracji
do wakansów (rys. 2.18)*. Oczywiście możliwe jest także zjawisko odwrotne, polegające na
opuszczaniu pozycji węzłowych przez atomy i ich dołączaniu do krawędzi półpłaszczyzny.
Przemieszczanie się dyslokacji krawędziowych przez wspinanie zależy od ilości wakansów w
krysztale i zachodzi bardziej intensywnie w temperaturach podwyższonych, np. podczas
pełzania metali. Innym przejawem własności dynamicznych jest przyciąganie się dyslokacji
różnoimiennych i odpychanie się dyslokacji jednoimiennych. W pierwszym przypadku
możliwa jest anihilacja (zanik) dyslokacji, jeśli leżą one w tej samej płaszczyźnie poślizgu lub
w płaszczyznach równoległych.
Określone oddziaływanie występuje także między dyslokacjami krawędziowymi atomami
obcych pierwiastków znajdujących się w metalu. Atomy o większych średnicach zajmujące
położenia węzłowe oraz atomy o małych średnicach zajmujące położenia międzywęzłowe
(węgiel, azot, wodór) migrują do rozciągniętej strefy kryształu, leżącej bezpośrednio pod
krawędzią dodatkowej półpłaszczyzny. Natomiast atomy o małych średnicach, zajmujące
położenia węzłowe migrują do ściskanej części kryształu, gdzie zastępując większe atomy
metalu osnowy, obniżają energię odkształcenia kryształu
Drugim prostym rodzajem dyslokacji jest dyslokacja śrubowa, wyznaczająca granicę
między przesuniętą i nieprzesuniętą częścią kryształu. Granica ta przebiega równolegle do
kierunku poślizgu a nie prostopadle, jak to ma miejsce w przypadku dyslokacji krawędziowej.
Dyslokację śrubową najlepiej wyjaśnić na perspektywicznym modelu fragmentu kryształu,
którego jedna część jest przesunięta względem drugiej o jedną odległość atomową (rys. 2.19).
Rys. 2.18. Schemat przemieszczania się dyslokacji krawędziowej przez wspinanie
W wyniku tego przesunięcia poszczególne płaszczyzny atomowe przekształcają się w
powierzchnie śrubowe. Rozróżnia się dyslokacje prawo-skrętne wywołujące poślizg w
kierunku pokazanym na rys. 2.19, i dyslokacje lewo-skrętne wywołujące poślizg w kierunku
przeciwnym.
Podobnie jak dyslokacje krawędziowe, dyslokacje śrubowe mogą przemieszczać się przy
małych naprężeniach stycznych, jeśli w płaszczyźnie poślizgu nie ma przeszkód hamujących
ich ruch. W przypadku obecności takich przeszkód (np. obcych atomów), naprężenie
potrzebne do uruchomienia dyslokacji jest tym większe, im mniejsza jest odległość między
sąsiednimi przeszkodami. Zjawisko to ma oczywisty wpływ na własności wytrzymałościowe
stopów. Równoległe dyslokacje śrubowe jednoimienne odpychają się, różnoimienne-
przyciągają. Te ostatnie mogą się także w określonych przypadkach anihilować.
Rys. 2.19. Schemat dyslokacji śrubowej
Proste typy dyslokacji występują w sieci krystalicznej rzadko. Większość dyslokacji

stanowi kombinację dyslokacji krawędziowych i śrubowych.
Omówione defekty dotyczyły zakłóceń budowy sieci krystalicznej występujących w
pojedynczym krysztale. Metale i stopy techniczne, jak już wiadomo, są jednak materiałami
wielokrystalicznymi, złożonymi z wielkiej liczby ziarn. Orientacja krystalograficzna tych
ziarn jest w zasadzie chaotyczna, toteż na granicy ziarn spotykają się różnie
zorientowane sieci przestrzenne, ukierunkowane względem siebie pod dużymi kątami,
wynoszącymi najczęściej kilkanaście do kilkudziesięciu stopni (dlatego granice ziarn nazywa
się także granicami dużego kąta). Jest rzeczą oczywistą, że ułożenie atomów na granicy ziarn
jest uzależnione od działania obu stykających się sieci krystalograficznych, w wyniku czego
stanowi pewną mikrostrukturę przejściową, nie odpowiadającą orientacji ani jednego, ani
drugiego ziarna.
Odkrycie dyslokacji umożliwiło wyjaśnienie dwojakiego wpływu defektów sieci
krystalicznej na wytrzymałość kryształu .
Z jednej strony defekty sieci krystalicznej osłabiają kryształ, a odkształcenie plastyczne
jest wynikiem przemieszczania się w nim dyslokacji bądź już istniejących, bądź powstających
podczas odkształcania (czemu sprzyjają niektóre inne defekty sieciowe).
Z drugiej jednak strony wiadomo, że wytrzymałość pojedynczych kryształów jest
mniejsza niż materiałów polikrystalicznych, ponieważ zaburzenia budowy sieciowej na
granicach ziarn umacniają metal. Wiadomo też, że kryształy zawierające dużą liczbę
defektów są bardziej wytrzymałe od kryształów z małą liczbą defektów. Dzieje się tak
dlatego, że w przypadku dużej liczby defektów sieciowych ruch dyslokacji jest hamowany na
skutek wzajemnego przecinania się dyslokacji (powstają dyslokacje nie tylko równolegle do
siebie, ale również umiejscowione w różnych płaszczyznach i o różnych kierunkach), ich
grupowania się, a także obecności przeszkód w postaci innych defektów sieciowych, np. obcych atomów.
Wynika z tego, że wytrzymałość rzeczywista metali zmniejsza się wraz ze zwiększaniem liczby (gęstości) dyslokacji i innych
defektów sieciowych, tylko do pewnej granicy i po
osiągnięciu minimalnej wartości, przy tzw. krytycznej gęstości dyslokacji zaczyna ponownie wzrastać
Wynika z tego również, że warunkiem podwyższenia wytrzymałości metalu jest albo
całkowite usunięcie z niego wszelkich nieprawidłowości budowy krystalicznej, albo
zwiększenie oporu ruchu dyslokacji poprzez wytworzenie w nim odpowiedniej liczby
dyslokacji i innych defektów.(Opracowała Paulina Kizińska).
21. Odmiany alotropowe żelaza
Żelazo wykazuje dwie odmiany alotropowe:
- w temperaturze niższej od 912°C oraz w zakresie temperatury od 1394 do 1538°C występuje odmiana alotropowa
oznaczana α, a w zakresie wysokotemperaturowym oznaczana niekiedy również α (δ) lub δ.
Roztwory stałe w żelazie α są nazywane ferrytem. W temperaturze niższej od temperatury 770°C, zwanej temperaturą Curie,
żelazo α jest ferromagnetyczne, a w temperaturze wyższej paramagnetyczne.
- w zakresie temperatury od 912 - 1394°C stabilna jest odmiana żelaza γ o sieci ściennie centrowanej. Roztwory stałe w
żelazie γ są nazywane austenitem. Parametr sieci a każdej odmiany alotropowej żelaza zwiększa się wraz z podwyższeniem
temperatury. Przemianie żelaza α w żelazu γ towarzyszy zmniejszenie objętości właściwej, a przemianom odwrotnym -
wzrost objętości.
Ferryt jest roztworem stałym, międzywęzłowym węgla w żelazie c wstaje przez wchodzenie atomów węgla do luk
oktaedrycznych, które są spłaszczone, i tetraedrycznych.. Ze względu na małą zawartość węgla własności ferrytu niewiele
różnią się od własności czystego żelaza i tak Rm = ok. 300 MPa, 80 HB, A
10
= 40 , KC = ok. 180 J/cm
2
. Na zgładach
metalograficznych jest widoczny jako jasny składnik. Ferryt przedeutektoidalny występuje w postaci oddzielnych ziarenek
na przemian z ziarnami perlitu (struktura komórkowa) lub na granicach ziaren perlitu.
Ferryt jest roztworem stałym węgla w wysokotemperaturowej odmianie żelaza . Wykazuje on większą
rozpuszczalność węgla niż ferryt (do 0,09 ), ma również większy parametr sieci niż ferryt .
Austenit jest roztworem stałym, międzywęzłowym węgla w Fe- o maksymalnej rozpuszczalności węgla 2,11 . Większa
rozpuszczalność węgla wiąże się z kulistym kształtem luk oktaedrycznych. Ze względu na typ sieci A1 ma największą
gęstość spośród wszystkich faz układu. W warunkach równowagi nie może istnieć poniżej temperatury A
1
(727°C). Wpro-
wadzenie pierwiastków austenitotwórczych (np. Mn, Ni) obniża zakres istnienia austenitu do temperatury pokojowej.
Własności mechaniczne austenitu w temperaturze pokojowej są następujące: Rm = ok. 700-800 MPa, Re = ok. 250 MPa, 200
HB, A
10
= 40=60%, KC = ok. 200=300 J/cm
2
. W próbie rozciągania odkształca się równomiernie (nie tworzy się szyjka). Na
zgładach metalograficznych występuje jako składnik z charakterystycznymi, prostoliniowymi granicami bliźniaczymi.
22. Reguła faz Gibbsa
Reguła faz lub reguła faz Gibbsa to zależność obowiązująca dla każdego układu będącego w równowadze
termodynamicznej, łącząca liczbę faz w układzie, liczbę składników niezależnych oraz liczbę stopni swobody.
s = α − β + 2
gdzie:
s - liczba stopni swobody, czyli liczba zmiennych intensywnych, które można zmieniać bez jakościowej zmiany układu (bez
zmiany liczby faz w równowadze)
α - liczba niezależnych składników, a więc takich, które nie dają się określić za pomocą zależności chemicznych poprzez
stężenia innych składników (niezależnych).
β - liczba faz, a więc postaci materii jednorodnej chemicznie i fizycznie (np. roztwór, faza gazowa, kryształy o określonym
składzie)
Dla układów w których zachodzą reakcje chemiczne często podaje się również inną (w pewnym sensie uproszczoną) wersję
reguły faz:
s = α − β + 2 + r
gdzie:
r - ilość reakcji chemicznych zachodzących w układzie
Ważnym pojęciem jest tzw. punkt niezmienniczy albo inwariantny dla którego:

s = 0
W takim punkcie nie można zmienić żadnej zmiennej intensywnej bez zmiany ilości faz w układzie - przykładem takiego
punktu jest
dla układu jednoskładnikowego lub punkt poczwórny dla układu dwuskładnikowego.
Punkt potrójny - to stan w jakim dana
może istnieć w trzech stanach skupienia równocześnie w równowadze
termodynamicznej. Na wykresie stanów równowagi jest to punkt przecięcia krzywych równowagi fazowej
odpowiadający stanowi równowagi trwałej trzech stanów skupienia (
Punkt potrójny jest wielkością charakterystyczną dla danej substancji, podawany jest w opisach substancji. Punkty potrójne
niektórych substancji są używane jako wzorce
temperatur.Układ jednoskładnikowy- liczba składników dla takiego
układ, α = 1, stąd liczba stopni swobody s = 3 - β gdzie β to liczba faz w stanie równowagi. Z reguły faz wynika, że w
układzie jednoskładnikowym mogą występować maksymalnie trzy fazy i to tylko w jednych, ściśle określonych warunkach
temperatury i ciśnienia. W punkcie potrójnym liczba faz w stanie równowagi β = 3, stąd liczba stopni swobody s = 1 - 3 + 2 =
0. Z reguły tej wynika też, że dla czystej chemicznie substancji nie może istnieć punkt poczwórny, czyli punkt gdzie w
Wykresy fazowe dla układów jednoskładnikowych Krzywe równowagi, linie równowagi albo krzywe współistnienia
określają współrzędne ciśnienia i temperatury (p,T) punktów na wykresie fazowym oznaczających β = 2 fazy w równowadze
termodynamicznej. Dla krzywych równowagi w układzie jednoskładnikowym otrzymujemy s = 1 - 2 + 2 = 1 stopień
swobody, a więc istnieje możliwość zmiany ciśnienia albo temperatury, ale nie obu naraz. Jednoczesna zmiana ciśnienia i
temperatury musi prowadzić zmiany liczby faz w układzie jednoskładnikowym.
W układzie ciecz-para w równowadze są 2 fazy (β = 2), stąd liczba stopni swobody s = 1 - 2 + 2 = 1. Można wówczas
zmienić temperaturę albo ciśnienie - jeżeli zmienimy temperaturę, ciśnienie musi zmienić się samo, jeżeli zmienimy
ciśnienie, wówczas temperatura układu musi się odpowiednio dostosować - nie można zmienić dowolnie (nawet o niewielkie
wartości) naraz obu parametrów intensywnych bez opuszczenia krzywej równowagi ciecz-para.
Po przekroczeniu punktu krytycznego (końcowy punkt krzywej ciecz-para od strony wysokich ciśnień i temperatur), mamy
do czynienia z 1 fazą (gazową - nie mogą istnieć ani ciecz ani ciało stałe), a więc liczba stopni swobody s = 1 - 1 + 2 = 2,
czyli można zmieniać równocześnie 2 zmienne intensywne: ciśnienie i temperaturę.
Dokładnie tak samo jest w układzie para - ciało stałe w równowadze są również 2 fazy a zatem liczba stopni swobody = 1.
Oznacza to, że można zmieniać temperaturę albo ciśnienie, ale nie obie zmienne równocześnie.
Na wykresie równowagi fazowej dla układu jednoskładnikowego obszar poza krzywymi współistnienia i punktem potrójnym
oznacza zawsze czystą pojedynczą fazę:
powierzchnia ograniczona krzywą ciało stałe-gaz i krzywą ciecz-gaz (niskie ciśnienia i wysokie temperatury) określa obszar
występowania pary (a powyżej punktu krytycznego - gazu)
powierzchnia ograniczona krzywą ciało stałe-ciecz i krzywą ciecz-gaz (wysokie ciśnienia i wysokie temperatury) określa
obszar występowania cieczy (tylko poniżej punktu krytycznego - powyżej jest tylko 1 faza gazowa)
powierzchnia ograniczona krzywą ciało stałe-gaz i krzywą ciało stałe-ciecz (wysokie ciśnienia i niskie temperatury) określa
obszar występowania ciała stałego.
23. WYKRESY FAZOWE
Analizą
termiczną nazywa się badania polegające na określeniu temperatury początku i końca krzepnięcia (lub topnienia) oraz
temperatury przemian zachodzących w stanie stałym podczas ochładzania (lub ogrzewania) metali i ich stopów. Badania te
mają na celu wykreślenie
krzywych przebiegu ogrzewania lub chłodzenia, na których wszelkie efekty cieplne występujące podczas zmian fazowych
uwidaczniają się w postaci przystanków temperaturowych i załamań. Te z kolei służą do zbudowania tzw. wykresów
równowagi układów pierwiastków przedstawiających położenie granic faz w funkcji temperatury i składu chemicznego.
Ogólnie wykresy równowagi obrazują:
a) przemiany w stanie ciekłym (zmiany rozpuszczalności),
b) zmiany stanu skupienia (krzepnięcie, topnienie),
c) przemiany w stanie stałym (zmiany rozpuszczalności, przemiany alotropowe,
eutektoidalne itp.),
d) tworzenie się lub rozpad faz międzymetalicznych w stanie ciekłym, w czasie zmiany stanu skupienia lub w stanie
stałym.
Wykresy równowagi podają więc w zasadzie tylko budowę fazową stopów, tzn. można z nich wnioskować, z jakich faz
stop jest zbudowany, w jakim stosunku ilościowym te fazy pozostają i jaki jest ich skład chemiczny. Inaczej mówiąc,
wykresy równowagi przedstawiają graficznie stan stopu. Jeśli zachodzi zmiana jego składu chemicznego, temperatury lub
ciśnienia, zmienia się również stan stopu, co znajduje odwzorowanie graficzne na wykresie równowagi.
Wykres równowagi obrazuje stany trwałe stopu, tj. stany, którym w danych warunkach
odpowiada najmniejszy zasób energii swobodnej układu. Również zmiany stanu stopu
odwzorowane na wykresie odpowiadają warunkom równowagi, tzn. nie uwzględniają zjawisk
przegrzania lub przechłodzenia, które w rzeczywistości mają zawsze miejsce (oczywiście w
różnym stopniu). Dlatego wykres równowagi stopu jest wykresem teoretycznym, a posługiwanie
się nim jest uwarunkowane rozpatrywaniem przemian przy małych szybkościach nagrzewania
lub chłodzenia, gdy zjawiska przegrzania lub przechłodzenia występują w minimalnym stopniu i praktycznie można je
pominąć.
Ogólne zasady określające współistnienie trwałych faz odpowiadających teoretycznym
warunkom równowagi wyraża matematycznie tzw. reguła faz, zwana także regułą Gibbsa.
Podaje ona ilościową zależność między liczbą stopni swobody danego stopu czy metalu, a liczbą jego faz i składników.
Posługując się regułą faz można przewidzieć, w jakich warunkach temperaturowych
przebiegają przemiany fazowe (w stałej temperaturze, czy w zakresie temperatur) i jakie
czynniki mogą ulegać pewnym zmianom bez naruszenia stanu równowagi.
Przy omawianiu reguły faz konieczne jest dokładne zdefiniowanie pojęć: fazy, składnika oraz liczby stopni swobody.
Faza jest to jednorodna część układu oddzielona od innych jego części (faz) powierzchnią
rozdziału, czyli granicą fazy, po przekroczeniu której własności fizyczne czy też struktura
zmieniają się w sposób nieciągły.
Składnikami układu nazywa się substancje tworzące dany układ. Dla przykładu czysty meta
tworzy układ jednoskładnikowy, stop dwóch metali — układ dwuskładnikowy itd. Fazy
międzymetaliczne uważa się także za składniki, jeśli w rozpatrywanym zakresie temperatury ni
rozkładają się na pierwiastki składowe.
Liczba stopni swobody układu jest to liczba zewnętrznych i wewnętrznych czynników
(temperatura, ciśnienie i skład chemiczny), które można zmieniać bez spowodowania zmiany
liczby faz w danym układzie.
Przy założeniu, że w rozpatrywanym układzie wszystkie przemiany zachodzą przy stałym i
niezmiennym ciśnieniu, reguła faz wyraża się wzorem:
s=m–f+l
gdzie: s - liczba stopni swobody,
m - liczba składników,
f - liczba faz.
Dla rzeczywistych układów wielofazowych liczba stopni swobody wynosi zwykle 0, l lub 2.
Gdy S = 0, układ jest niezmienny, czyli nie można zmieniać ani temperatury, ani składu
chemicznego bez spowodowania zmiany liczby faz w układzie.
Gdy S = l, układ jest jednozmienny. Oznacza to, że nie zmieniając liczby faz w układzie
można zmienić (w pewnych granicach) temperaturę bądź skład chemiczny.
Gdy S = 2, układ jest dwuzmienny. Oznacza to, że nie zmieniając liczby faz w układzie
można zmienić (w pewnych granicach) temperaturę i skład chemiczny.
Budowa stopów podwójnych
Jak już wspomniano, przy badaniu struktury stopów bardzo ważne znaczenie ma znajomość
ich wykresów równowagi fazowej. Jeżeli dwa składniki stopu nie oddziałują na siebie
chemicznie i nie podlegają żadnym przemianom w stanie stałym, to ich wykresy równowagi
podają dla dowolnego składu chemicznego stopu temperaturę początku krzepnięcia (linia
likwidusu) i jego końca (linia solidusu).
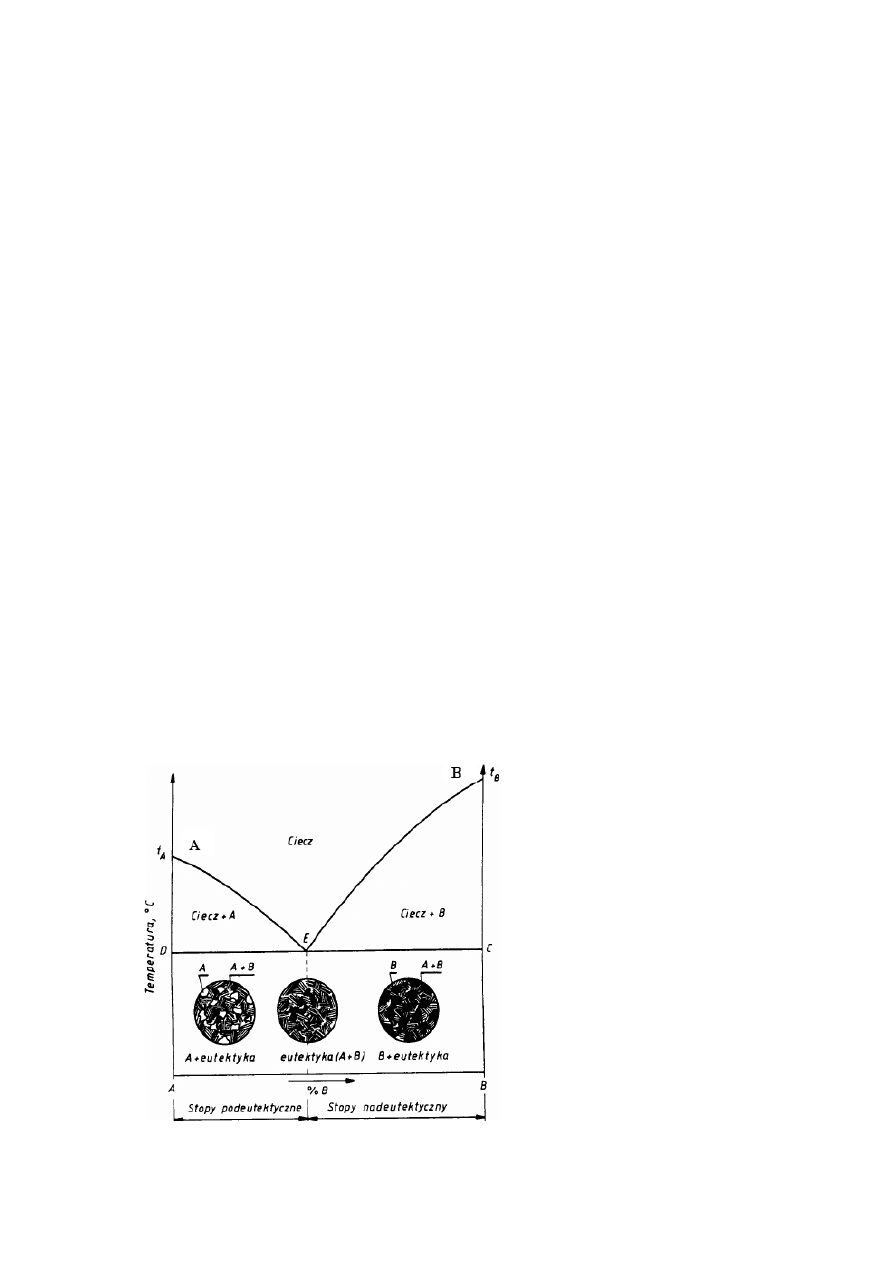
Na rysunku 3.5 przedstawiono wykres równowagi typu I, odpowiadający całkowitej
wzajemnej rozpuszczalności składników w stanie ciekłym i jej brakowi w stanie stałym.
Składniki nie tworzą również związków chemicznych. Linia AEB jest linią likwidusu, a linia
DEC linią solidusu. Na linii AE przy chłodzeniu zaczynają się wydzielać z cieczy kryształy
metalu A, na linii EB — kryształy metalu B. Na linii DEC z cieczy o składzie E wydzielają się jednocześnie bardzo drobne
krysztale A i B tworzące mieszaninę, która nazywa się eutektyką (po grecku łatwo topliwa) a stop o tym składzie - stopem
eutektycznym (stopy o składzie odpowiadającym zakresowi DE są stopami podeutektycznymi, pozostałe nadeutektycznymi).
Na rysunku podano również schematy struktur stopów podeutektycznych, eutektycznego i
nadeutektycznych. Jak widać, w pierwszym przypadku na tle eutektyki występują kryształy
metalu A, w drugim - sama eutektyką, w trzecim - na tle eutektyki kryształy metalu B.
Oczywiście skrajne odcięte wykresu odpowiadają czystym składnikom A i B. Taki wykres
równowagi tworzą m.in. stopy bizmut-kadm.
Trzeba podkreślić, że zawartość eutektyki w stopie zmienia się liniowo, od 0 dla czystych
składników A i B do 100% dla stopu eutektycznego.
Wykres równowagi typu II, odpowiadający całkowitej rozpuszczalności wzajemnej
składników zarówno w stanie ciekłym, jak i stałym (przy braku faz międzymetalicznych),
podano na rys. 3.6.
Rys. 3.5. Wykres równowagi odpowiadający
brakowi rozpuszczalności w stanie stałym oraz
schematy struktur poszczególnych grup stopów.
W tym przypadku eutektyka nie występuje, a
wszystkie stopy w stanie stałym: mają jednakową
strukturę złożoną z kryształów roztworu stałego α
o składzie odpowiadającym składowi stopu.
Rys. 3.6. Wykres równowagi odpowiadający całkowitej
rozpuszczalności wzajemnej składników zarówno w stanie ciekłym,
jak i w stanie stałym oraz schemat struktury stopów.
24. WYKRESY FAZOWE (WERSJA II)
Dwuskładnikowe układy równowagi fazowej
DWUSKŁADNIKOWY UKŁAD RÓWNOWAGI FAZOWEJ
O ZUPEŁNEJ ROZPUSZCZALNOŚCI SKŁADNIKÓW W STANIE STAŁYM
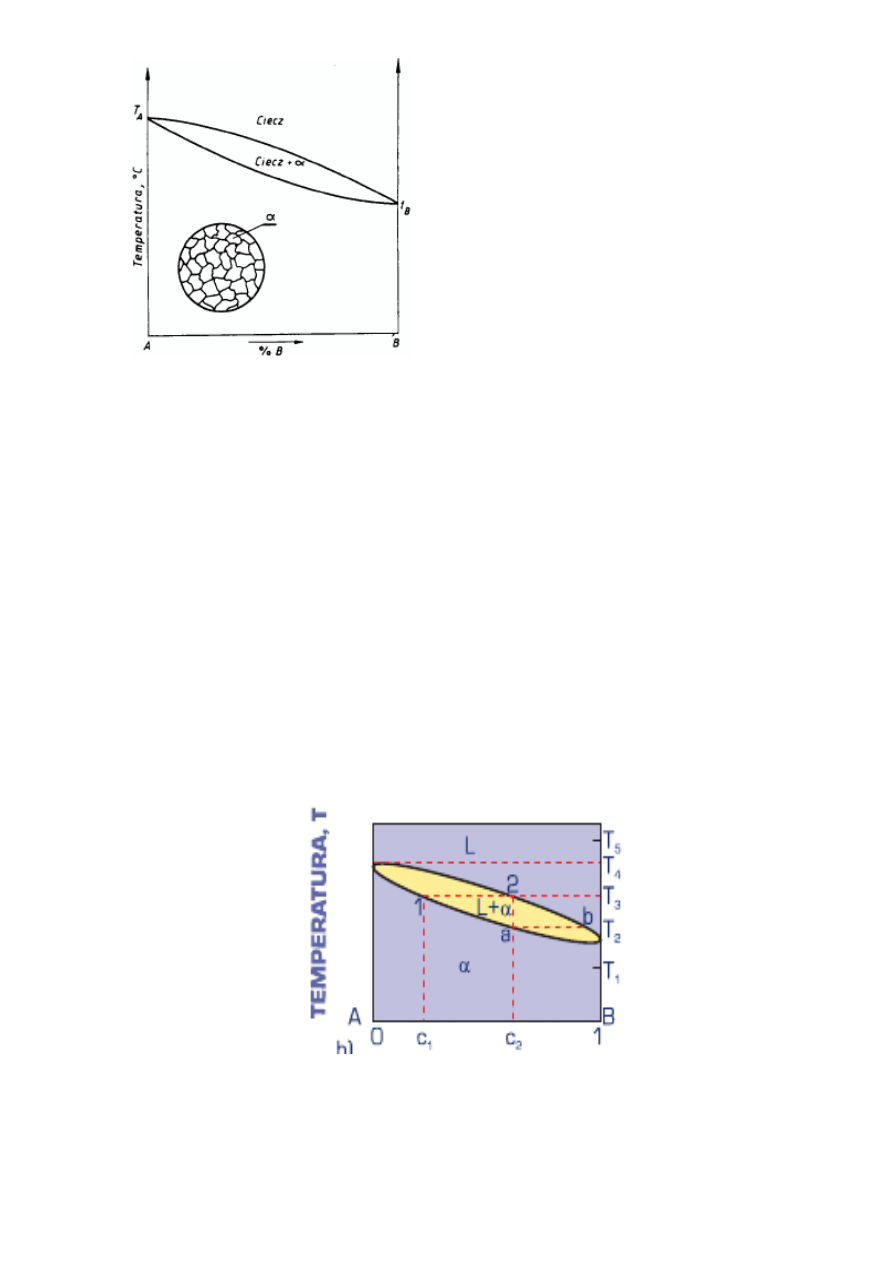
Układy dwuskładnikowe o zupełnej rozpuszczalności składników w stanie
stałym (rys. 3.65) są tworzone przez izomorficzne składniki A i B rozpuszczające
się wzajemnie przy dowolnych stosunkach ilościowych – zarówno w stanie ciekłym, jak i stałym. Proces krzepnięcia
roztworu stałego przebiega podobnie dla wszystkich stopów układu. Krzepnięcie stopu o stężeniu c2 rozpoczyna się w
temperaturze T3 przecięcia się linii składu stopu c2 z linią likwidusu (rys.3.65). Likwidusem jest nazywana linia
przedstawiająca na wykresie równowagi wartości temperatury, powyżej których stopy w całym zakresie stężeń są ciekłe. W
temperaturze T3 z cieczy wydzielają się pierwsze zarodki roztworu stałego α o stężeniu c1 odpowiadającym punktowi 1. W
miarę dalszego obniżania temperatury skład krzepnącego stopu zmienia się wzdłuż solidusu od punktu 1 do α, cieczy zaś
wzdłuż likwidusu od 2 do b. Solidusem jest nazywana linia na wykresie równowagi oznaczająca wartości
temperatury, poniżej których stopy w całym zakresie stężeń występują w stanie stałym.
Ilościowe udziały faz stałej i ciekłej w przedziale krzepnięcia między likwidusem a solidusem określa prawo dźwigni.
Po zakończeniu krzepnięcia (punkt a) wszystkie ziarna roztworu stałego powinny osiągnąć skład c2, co następuje w
wyniku dyfuzji między fazą stałą i ciekłą, zachodzącej podczas krzepnięcia. W rzeczywistości wyrównanie składu
chemicznego wszystkich ziarn w wyniku dyfuzji przebiega znacznie wolniej niż krzepnięcie. Temperatura zakończenia
krzepnięcia jest również niższa od
odpowiadającej punktowi a, a skład ostatnich krzepnących kropel cieczy – bogatszy w składnik B bardziej, niż wynika to z
położenia punktu b w stanie równowagi. Stopy krzepnące w warunkach technicznych charakteryzują się więc znaczną
segregacją (niejednorodnością) składu chemicznego
w obrębie ziarn.
Rysunek 3.65
Wykres równowagi faz
podwójnego układu z
nieograniczoną
rozpuszczalnością składników w stanie stałym.
DWUSKŁADNIKOWY UKŁAD RÓWNOWAGI FAZOWEJ O CAŁKOWITYM BRAKU
ROZPUSZCZALNOŚCI SKŁADNIKÓW W STANIE STAŁYM Z EUTEKTYKĄ
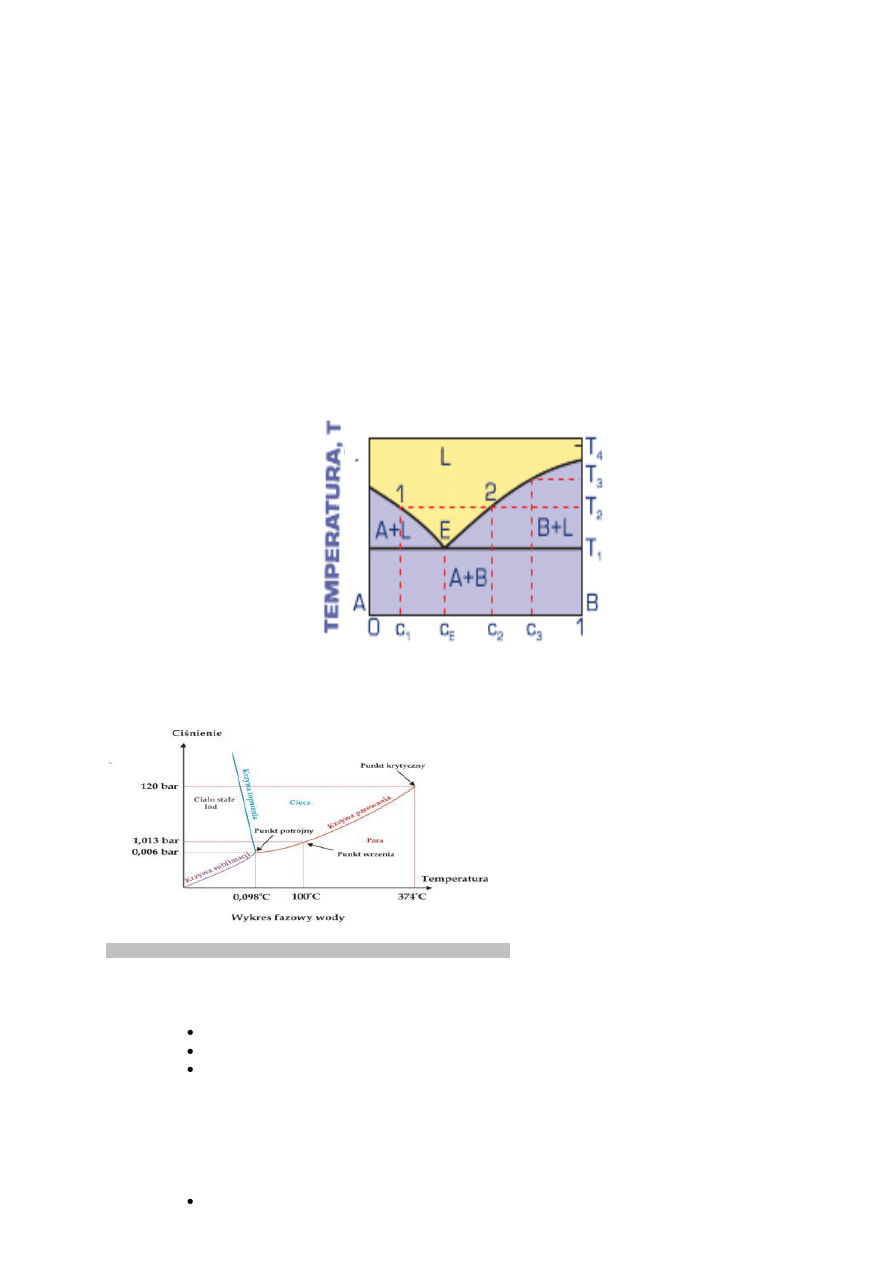
Cechą charakterystyczną dwuskładnikowego układu równowagi o całkowitym braku rozpuszczalności składników w stanie
stałym z eutektyką jest występowanie mieszaniny
czystych składników A + B o energii swobodnej, której obrazem jest prosta łącząca energie swobodne czystych składników
(rys.3.66). W temperaturze T1, zwanej temperaturą eutektyczną
(i oznaczanej TE), najmniejszą energią swobodną w całym zakresie stężeń charakteryzuje się mieszanina fazy ciekłej L i
składników A + B. W temperaturze niższej od T1 najmniejsza jest energia swobodna mieszaniny eutektycznej A + B.
Proces krzepnięcia roztworu ciekłego o stężeniu cE, zwanym składem eutektycznym, następuje przez wydzielanie
kryształów obu składników A i B z jednorodnej cieczy LE (rys. 3.66). Proces ten przebiega w stałej temperaturze
eutektycznej TE aż do chwili, gdy cała ciecz ulegnie zakrzepnięciu.
Przemianę eutektyczną można zapisaćw następujący sposób:
LE → A + B
Podczas krzepnięcia stopów podeutektycznych – o stężeniu c < cE – z jednorodnej cieczy wydzielają się początkowo
kryształy metalu A, a skład cieczy zmienia się wzdłuż linii likwidusu od punktu 1 do E (rys. 3.66). Dopiero wówczas reszta
cieczy krzepnie w stałej temperaturze eutektycznej TE, jako mieszanina kryształów metali A i B. Strukturę takiego stopu
stanowi mieszanina pierwotnie wydzielonych dużych kryształów metalu A i drobnoziarnista mieszanina
eutektyczna metali A i B.
Stopy nadeutektyczne – o stężeniu c > cE – krzepną podobnie jak stopy podeutektyczne, z tym że początkowo wydzielają
się kryształy metalu B.
Układy równowagi metali o całkowitym braku rozpuszczalności składników praktycznie nie występują, jednak
wykorzystuje się je do analizy stopów metali o bardzo małej wzajemnej rozpuszczalności składników w stanie stałym.
Rysunek 3.66
Wykres równowagi faz
podwójnego układu o zupełnym
braku rozpuszczalności składników w stanie stałym z eutektyką.
31.Składniki zwykłe zanieczyszczenia i pierwiastki stopowe w stalach.
Stal jest przerobionym technicznie plastycznym stopem żelaza z węglem zawierającym do 2,11% węgla oraz inne pierwiastki
pochodzące z surowców i paliw stosowanych podczas otrzymywania stali lub dodawane celowo .Pierwiastki występujące w
stalach można podzielić na trzy grupy
1.
Składniki zwykłe-pierwiastki konieczne ze względów metalurgicznych. Należą do nich:
Mangan (do 1,65%)
Krzem (do 0,5%)
Aluminium (nie konieczny)
Pierwiastki te są dodawane w celu odtlenienia stali ponieważ podczas wytwarzania stali w procesie stalowniczym jest
konieczny pewien nadmiar tlenu. Są one dodawane pod koniec procesu stalowniczego do ciekłej stali w celu związania tlenu
w tlenki. Najsilniejszym utleniaczem jest Al lecz jego nadmiar łączy się z występującym w stali azotem hamując rozrost
ziarna austenitu z tego względu stale odtleniane Al nazywa się stalami drobnoziarnistymi.
2.
Zanieczyszczenia – pierwiastki, których usuwanie poniżej pewnych granic jest nieopłacalne ekonomicznie lub
Worgule nie możliwe. Są to:
Siarka –Dostaje się do stali z koksu i rudy. Jest zwykle niepożądana w stali ze względu na tworzenie
wtrąceń MnS działających jako miejsca zarodkowania pęknięć.
Fosfor- Dostaje się do stali z rudy. Powoduje kruche pękanie wzdłuż granic ziarn.
Tlen- Występuje w stali w postaci wtrąceń tlenkowych gdyż jego rozpuszczalność w stali w stanie stałym
jest bardzo mała. Zmniejsza ciągliwość i udarność stali.
Azot – Dostaje się do stali w procesie wytapiania z powietrza. Powoduje wzrost wytrzymałości i
zmniejszenie plastyczności stali odkształconej w miarę upływu czasu, występowanie wyraźnej granicy
plastyczności oraz zmniejszenie ciągliwości i udarności.
Wodór- Dostaje się z pary wodnej w piecu stalowniczym. Powoduje powstanie mikropęknięć i w
rezultacie pękanie stali.
3.
Pierwiastki stopowe – wprowadzane są do stali celowo, dla nadania jej własności związanych z zawartością danego
pierwiastka. Najczęściej stosowane w tym celu są:
Mangan –W ilościach 1-1.5% mangan jest dodawany w celu umocnienia roztworowego stali
zmniejszenia wielkości ziarna ferrytu w blachach walcowanych na gorąco oraz zwiększenia hartowności.
Krzem-W małych ilościach stosowany jako odtleniacz. W większych powoduje zwiększenie oporu
elektrycznego oraz zmniejszenie stratności stali magnetycznie miękkich. Zwiększa żaroodporność stali
Nikiel- w małych ilościach ma na celu zwiększenie hartowności. Większe ilości>8% są dodawane w celu
stabilizacji austenitu w stalach odpornych na korozję i żaroodpornych.
Chrom-duże ilości chromu zapewniają stali odporność na korozję oraz utlenianie. Chrom jest więc
podstawowym pierwiastkiem stali odpornych na korozję, żaroodpornych i żarowytrzymałych.
Molibden-Powoduje zwiększenie odporności na korozję stali nierdzewnych, szczególnie w obecności
jonów Cl
-
, oraz opóźnienie mięknięcia stali przy wzroście temperatury dzięki tworzeniu się węglika
Mo
2
C.
Wolfram-Tworząc węglik W
2
C zapewnia dużą odporność na ścieranie oraz twardość wtórną.
Wanad –zwiększenie odporności na ścieranie
Miedź- Zwiększa odporność na korozję atmosferyczną
Bor-jest dodawany w niewielkich ilościach(od 0,0003 do 0,003%) w celu poprawy hartowności.
Węgiel nie jest pierwiastkiem stopowym jest natomiast z definicji pierwiastkiem obecnym w stalach, ze wzrostem zawartości
węgla wzrasta wytrzymałość i twardość natomiast zmniejsza się ciągliwość i spawalność stali .
32. Stal uspokojona i nieuspokojona
Stal nieuspokojona- to stal nie odtleniona w wystarczającym stopniu. Przy obniżaniu temperatury , na skutek wzrostu
aktywności tlenu i węgla, następuje wydzielanie się CO. Reakcja wydzielania się CO rozpoczyna się z chwila wlania stali do
wlewnicy. Krzepnięciu wlewka z takiej stali towarzyszy intensywne wydzielanie się gazów (tzw. Gotowanie się stali). W
zakrzepłym wlewku występują liczne pęcherze. Ze względu na turbulencje w ciekłej stali wywołane intensywnym
wydzielaniem się gazów , występuje silna segregacja siarki, fosforu i węgla, a w przypadku stali stopowych- również
dodanych pierwiastków. Z tej przyczyny można produkować jako nieuspokojone jedynie stale niestopowe do zawartości
0.25%C. W górnej części wlewka stali nieuspokojonej nie tworzy się zagłębienie nazywane jamą skurczową gdyż związany z
krystalizacją skurcz jest kompensowany przez tworzące się pęcherze. W ostatnich czasach wzrasta dominacja odlewania
ciągłego w procesie wytwarzania stali. W odlewaniu ciągłym wydzielanie gazu jest niedopuszczalne podczas krzepnięcia
stali. Więc tale nieuspokojone mogą mieć wkrótce znaczenie tylko historyczne.
Stal uspokojona- jest odtleniona w takim stopniu aby podczas krzepnięcia nie następowało wydzielanie gazów. Odtlenianie
prowadzi się Mn Si a niekiedy również Al. Zawartość krzemu w tych stalach jest większa niż 0.17%. Górna część wlewka
stali uspokojonej, w której znajduje się jama skurczowa, spowodowana skurczem stali podczas krystalizacji, stanowi odpad
wynoszący ok. 15% do 18% wlewka. Jest to podstawową wadą odlewania stali uspokojonej do wlewnic.
33. stale konstrukcyjne – zawartość pierwiastków stopowych nie przekracza kilku procent; zawartość C < 0,7% ( a zwykle <
0,5%)
niestopowe (węglowe)
niskostopowe o podwyższonej wytrzymałości
do nawęglania lub azotowania
do ulepszania cieplnego
sprężynowe i łożyskowe (zawartość C ok. 1%)
1.
stale narzędziowe ( stale wyżej węglone)
niestopowe (węglowe)
do pracy na zimno
do pracy na gorąco
szybkotnące (na narzędzia skrawające z dużymi szybkościami; temp. pracy ok. 650°C
2.
stale o szczególnych właściwościach
odporne na korozję i utlenianie
o szczególnych właściwościach magnetycznych
Oznaczenie stali wg zastosowania i właściwości:
P- stale pracujące pod ciśnieniem
L- stale na rury przewodowe
B- stale do zbrojenia betonu
R- stale na szyny

Wyszukiwarka
Podobne podstrony:
edema biotech materialy id 1501 Nieznany
Budowa materii id 94290 Nieznany (2)
opacow materialy id 335809 Nieznany
materialy 2 id 284532 Nieznany
Egzamin materialy id 153600 Nieznany
nauka o materialach 1 id 315348 Nieznany
inz materialowa id 212380 Nieznany
Cechy fizyczne materialow id 10 Nieznany
biofizyka materialy id 87015 Nieznany
materials4 id 285958 Nieznany
material 2 id 284353 Nieznany
materialy 9 2 id 284622 Nieznany
klasyfikacja materialow id 2359 Nieznany
Egzamin material id 152443 Nieznany
materialy 2 id 284478 Nieznany
3 Klasyfikacja materialow id 33 Nieznany (2)
Powtorzenie materialu id 379879 Nieznany
materialoznastwo id 285886 Nieznany
BHP pytania materialy id 6360 Nieznany (2)
więcej podobnych podstron