
luty 2000
POLSKI
KOMITET
NORMALIZACYJNY
POLSKA NORMA
PN-EN 1744-1
Badania chemicznych
w
łaściwości kruszyw
Analiza chemiczna
Zamiast:
patrz przedmowa krajowa
Grupa katalogowa
ICS 91.100.20
EN 1744-1:1998, IDT
This national document is identical with EN 1744-1:1998 and is published with
the permission of CEN; rue Stassart 36, B-1050 Bruxelles, Belgium
Niniejsza Polska Norma jest identyczna z EN 1744-1:1998 i jest publikowana
za zgod
ą CEN; rue Stassart 36, B-1050 Bruksela, Belgia
PRZEDMOWA KRAJOWA
Niniejsza norma jest oficjalnym t
łumaczeniem EN 1744-1:1998.
Niniejsza norma zast
ępuje niżej wymienione normy:
PN-78/B-06714/26
Kruszywa mineralne. Badania - Oznaczanie zawarto
ści zanieczyszczeń organicznych
PN-78/B-06714/28
Kruszywa mineralne. Badania - Oznaczanie zawarto
ści siarki metodą bromową
PN-90/B-06714/31
Kruszywa mineralne. Badania - Oznaczanie zawarto
ści związków rozpuszczalnych w wodzie
PN-78/B-06714/35
Kruszywa mineralne. Badania - Oznaczanie strat przy pra
żeniu
PN-78/B-06714/38
Kruszywa mineralne. Badania - Oznaczanie rozpadu wapniowego
PN-78/B-06714/39
Kruszywa mineralne. Badania - Oznaczanie rozpadu
żelazowego
PN-89/B-06714/49
Kruszywa mineralne. Badania - Oznaczanie chlorków
W normie stosowane s
ą odsyłacze krajowe oznaczone od
N1)
do
N10)
.
Norma zawiera za
łącznik krajowy NA (informacyjny), w którym podano wykaz aktualnych wyda ń norm powołanych w
normie europejskiej EN 1744-1:1998 i ich odpowiedników krajowych.
Powo
łane w niniejszej normie normy EN 196-2:1987 i EN 196-3:1987 zosta ły zastąpione, odpowiednio, EN 196-2:1994
i EN 196-3:1994.
Normy ISO 650:1977 i ISO 4788:1980 nie maj
ą odpowiedników krajowych. W przypadku wprowadzenia niniejszej
normy do obowi
ązkowego stosowania niezbędne jest uwzględnienie braku odpowiedników krajowych tych norm.
NORMA EUROPEJSKA
EUROPEAN STANDARD
NORME EUROPÉENNE
EUROPÄISCHE NORM
EN 1744-1
marzec 1998
ICS 71.040.40; 91.100.20
Deskryptory: kruszywa, w
łaściwości chemiczne, analiza chemiczna, oznaczanie, chlorki, siarczany, siarka, siarczyny,
sk
ładniki organiczne, wapno, rozpuszczalność, strata prażenia
Wersja polska
Badania chemicznych w
łaściwości kruszyw
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 1

Arkusz 1: Analiza chemiczna
Tests for chemical properties of
aggregates - Part 1: Chemical
analysis
Essais pour déterminer les propriétés
chimiques des granulats - Partie 1:
Analyse chimique
Prüfverfahren für chemische
Eigenschaften von
Gesteinskörnungen - Teil 1:
Chemische Analyse
Niniejsza norma jest polsk
ą wersją normy europejskiej EN 1744-1:1998. Zosta ła ona przetłumaczona przez Polski
Komitet Normalizacyjny i ma ten sam status co wersje oficjalne.
Norma europejska zosta
ła przyjęta przez CEN 15 lutego 1998 r.
Zgodnie z wewn
ętrznymi przepisami CEN/CENELEC, członkowie CEN są zobowiązani do nadania normie europejskiej
statusu normy krajowej bez wprowadzania jakichkolwiek zmian. Aktualne wykazy tych norm krajowych,
łącznie z ich
danymi bibliograficznymi, mo
żna otrzymać w Sekretariacie Centralnym CEN lub w krajowych jednostkach
normalizacyjnych b
ędących członkami CEN.
Norma europejska zosta
ła opracowana w trzech oficjalnych wersjach j ęzykowych (angielskiej, francuskiej, niemieckiej).
Wersja w ka
żdym innym języku, przetłumaczona na odpowiedzialność danego członka i zarejestrowana w Sekretariacie
Centralnym CEN, ma ten sam status co wersje oficjalne.
Cz
łonkami CEN są krajowe organizacje normalizacyjne nast ępujących państw: Austrii, Belgii, Czech, Danii, Finlandii,
Francji, Grecji, Hiszpanii, Holandii, Irlandii, Islandii, Luksemburga, Niemiec, Norwegii, Portugalii, Szwajcarii, Szwecji,
W
łoch i Zjednoczonego Królestwa.
CEN
Europejski Komitet Normalizacyjny
European Committee for Standardization
Comité Européen de Normalisation
Europäisches Komitee für Normung
Spis tre
ści
Przedmowa
1 Zakres normy
2 Normy powo
łane
3 Definicje
4 Odczynniki
5 Aparatura
6 Ogólne wymagania dotycz
ące badania
7 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą Volharda (metoda zalecana)
8 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą potencjometryczną (metoda alternatywna)
9 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą Mohra (metoda alternatywna)
10 Oznaczanie siarczanów rozpuszczalnych w wodzie
11 Oznaczanie zawarto
ści siarki całkowitej
12 Oznaczanie siarczanów rozpuszczalnych w kwasie
13 Oznaczanie siarczków rozpuszczalnych w kwasie
14 Oznaczanie sk
ładników wpływających na jakość powierzchni betonu
14.1 Sprawdzenie obecno
ści cząstek reaktywnego siarczku żelaza
14.2 Oznaczanie zanieczyszcze
ń lekkich
15 Oznaczanie sk
ładników organicznych wpływających na wiązanie i twardnienie cementu
15.1 Oznaczanie zawarto
ści humusu
15.2 Oznaczanie zawarto
ści kwasu fulvo
15.3 Oznaczanie zanieczyszcze
ń organicznych metodą zaprawy
16 Oznaczanie rozpuszczalno
ści w wodzie
17 Oznaczanie straty przy pra
żeniu
18 Oznaczanie wolnego wapna w
żużlu stalowniczym
18.1 Postanowienia ogólne
18.2 Oznaczanie wolnego wapna metod
ą kompleksometryczną (metoda zalecana)
18.3 Oznaczanie wolnego wapna metod
ą konduktometryczną (metoda alternatywna)
18.4 Oznaczanie wolnego wapna metod
ą acydymetryczną (metoda alternatywna)
19 Oznaczanie niesta
łości żużli wielkopiecowych i stalowniczych
19.1 Oznaczanie rozpadu krzemianu dwuwapniowego w
żużlu wielkopiecowym chłodzonym powietrzem
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 2

19.2 Oznaczanie rozpadu
żelaza w żużlu wielkopiecowym chłodzonym powietrzem
19.3 Oznaczanie p
ęcznienia żużla stalowniczego
Za
łącznik A (informacyjny) Dokładność
Za
łącznik B (informacyjny) Bibliografia
Przedmowa
Niniejsza norma europejska zosta
ła opracowana przez Komitet Techniczny CEN/TC 154 "Kruszywa"
N1)
, którego
sekretariat prowadzi BSI.
Niniejsza norma europejska jest jedn
ą z serii norm dotyczących badań chemicznych właściwości kruszyw. Metody
bada
ń innych właściwości kruszyw będą podane w następujących normach europejskich
N2)
:
EN 932 Tests for general properties of aggregates
EN 933 Tests for geometrical properties of aggregates
EN 1097 Tests for mechanical and physical properties of aggregates
EN 1367 Tests for thermal and weathering properties of aggregates
Innymi arkuszami EN 1744 b
ędą:
Arkusz 2 Determination of resistance to alkali reaction
Arkusz 3 Water leaching test
Arkusz 4 Determination of water susceptibility of fillers for bituminous mixtures
Niniejsza norma europejska powinna uzyska
ć status normy krajowej, przez opublikowanie identycznego tekstu lub
uznanie, najpó
źniej do września 1998 r., a normy krajowe sprzeczne z daną normą powinny być wycofane najpóźniej
do grudnia 1999 r.
Zgodnie z wewn
ętrznymi przepisami CEN/CENELEC do wprowadzenia niniejszej normy europejskiej s ą zobowiązane
nast
ępujące kraje członkowskie: Austria, Belgia, Czechy, Dania, Finlandia, Francja, Grecja, Hiszpania, Holandia,
Irlandia, Islandia, Luksemburg, Niemcy, Norwegia, Portugalia, Szwajcaria, Szwecja, W
łochy i Zjednoczone Królestwo.
1 Zakres normy
W niniejszej normie europejskiej podano metody chemicznych analiz kruszyw. Podano metody zalecane i w niektórych
przypadkach metod
ę alternatywną, którą można traktować jako metodę dającą równoważne wyniki.
Je
żeli zostały zastosowane inne metody, należy wówczas wykazać, że dają one wyniki równoważne z wynikami
otrzymywanymi metodami zalecanymi.
UWAGA: W przypadkach spornych, zaleca si
ę stosować tylko metody zalecane.
Je
żeli nie podano inaczej, metody bada ń wymienione w niniejszej normie europejskiej mog ą być użyte w fabrycznej
kontroli produkcji, badaniach auditowych lub badaniach typu.
2 Normy powo
łane
N3)
Do niniejszej normy europejskiej wprowadzono drog
ą datowanego lub niedatowanego powo łania się wymagania
zawarte w innych publikacjach. Powo
łania te znajdują się w odpowiednich miejscach w tek ście normy, a wykaz
publikacji podano poni
żej. W przypadku powołań datowanych późniejsze zmiany lub nowelizacje którejkolwiek z
wymienionych publikacji maj
ą zastosowanie do niniejszej normy europejskiej tylko wówczas, gdy zostan ą wprowadzone
do tej normy przez jej zmian
ę lub nowelizację. W przypadku powołań niedatowanych stosuje się ostatnie wydanie
powo
łanej publikacji.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 3

EN 196-1:1993
N4)
Methods of testing cement
Part 1: Determination of strength
EN 196-2:1987
N5)
Methods of testing cement
Part 2: Chemical analysis of cement
EN 196-3:1987
N6)
Methods of testing cement
Part 3: Determination of setting time and soundness
ENV 197-1:1992
Cement - composition, specifications and conformity criteria
Part 1: Common cements
EN 932-1:1996
Tests for general properties of aggregates
Part 1: Methods for sampling
prEN 932-2
Tests for general properties of aggregates
Part 2: Methods for reducing laboratory samples
prEN 932-5
Tests for general properties of aggregates
Part 5: Common equipment and calibration
EN 933-2:1995
Tests for geometrical properties of aggregates
Part 2: Determination of particle size distribution - Test sieves, nominal size of apertures
prEN 1015-4
Methods of test for mortar for masonry
Part 4: Determination of consistence of fresh mortar (by plunger penetration)
prEN 1015-9
Methods of test for mortar for masonry
Part 9: Determination of service life of fresh mortars
prEN 1015-11
Methods of test for mortar for masonry
Part 11: Determination of flexural and compressive strength of hardened mortar
prEN 1097-6
Tests for mechanical and physical properties of aggregates
Part 6: Determination of particle density and water absorption
ISO 384:1978
Laboratory glassware - Principles of design and construction of volumetric glassware
ISO 648:1977
Laboratory glassware - One-mark pipettes
ISO 650:1977
Relative density 60/60 degrees F hydrometers for general purposes
ISO 1042:1983
Laboratory glassware - One-mark volumetric flasks
ISO 4788:1980
Laboratory glassware - Graduated measuring cylinders
3 Definicje
W niniejszej normie zastosowano nast
ępujące definicje:
3.1 próbka analityczna: Próbka u
żyta w całości w pojedynczym badaniu.
3.2 próbka do badania: Je
żeli metoda badawcza wymaga więcej niż jednego oznaczania właściwości, próbkę do
badania stanowi próbka u
żyta w jednym oznaczaniu.
3.3 próbka laboratoryjna: Pomniejszona próbka pochodz
ąca z próbki ogólnej przeznaczona do badania
laboratoryjnego.
3.4 sta
ła masa: Masa próbki, która po kolejnych suszeniach co najmniej przez 1 h nie ró żni się więcej niż o 0,1 %.
UWAGA: W wielu przypadkach sta
łą masę można osiągnąć po wysuszeniu próbki analitycznej przez określony czas, w
okre
ślonej suszarce, w temperaturze (110 ± 5) °C. Na podstawie badania laboratoryjnego, w zale żności od wydajności
suszenia u
żytej suszarki można określić czas potrzebny do osiągnięcia stałej masy określonych typów i wielkości.
3.5 partia: Wyprodukowana ilo
ść, dostawa, część dostawy (wagon kolejowy, samochód ci ężarowy, barka) lub
sk
ładowisko kruszywa wyprodukowanego w danym czasie, w warunkach uznanych za jednolite.
UWAGA: W przypadku procesu ci
ągłego, ilość wyprodukowana w uzgodnionym czasie jest traktowana jako partia.
3.6 py
ły: Cząstki frakcji kruszywa, które przechodzą przez sito o wymiarze 0,063 mm.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 4

4 Odczynniki
Je
żeli nie podano inaczej, używać tylko odczynników analitycznej czystości i zdemineralizowanej wody lub wody
równowa
żnej czystości.
UWAGA 1: Je
żeli nie podano inaczej "%" znaczy "% masy".
UWAGA 2: Gdy nie podano
żadnych tolerancji objętości lub masy odczynników, podane wartości są przybliżone. W
tych przypadkach obj
ętości określone w cylindrach i masy oznaczane z użyciem zwykłych wag wymienionych w 5.2.4 i
5.2.5 s
ą wystarczająco dokładne na potrzeby niniejszej normy europejskiej.
UWAGA 3: Je
żeli nie podano inaczej roztwory odczynników mog ą być traktowane jako trwałe przez dłuższy czas.
UWAGA 4: Zaleca si
ę traktować wszystkie chemikalia jak potencjalne trucizny o toksycznych w łaściwościach i
przewidzie
ć odpowiednie środki ostrożności w trakcie ich używania. Zawsze przed rozpoczęciem każdej procedury
zaleca si
ę ocenić możliwe niebezpieczeństwa i zachować stałą uwagę przez wymagany czas.
4.1 Ogólne wymagania dotycz
ące gęstości
Wymienione w niniejszej normie st
ężone roztwory odczynników powinny charakteryzować się następującą gęstością
wyra
żoną w gramach na centymetr sześcienny w temperaturze 20 °C:
Kwas chlorowodorowy
Kwas azotowy
Kwas siarkowy
Wodorotlenek amonu
: od 1,18 do 1,19
: od 1,40 do 1,42
: 1,84
: od 0,88 do 0,91
Stopie
ń rozcieńczenia powinien być oznaczany jako suma objętości.
UWAGA 1: Na przyk
ład w 4.11.4 "kwas chlorowodorowy (1 + 1)" znaczy, że 1 objętość stężonego kwasu
chlorowodorowego zmiesza
ć z 1 objętością wody.
UWAGA 2: Roztwory gotowe dopuszcza si
ę jako alternatywne.
4.2 Odczynniki do oznaczania chlorków soli rozpuszczalnych w wodzie wed
ług Volharda (rozdział 7)
4.2.1 Roztwór 0,100 mol/l azotanu srebra (AgNO
3
), przygotowany z oko
ło 20 g azotanu srebra suszonego co najmniej
przez 1 h w temperaturze (110 ± 5) °C, a nast
ępnie ochłodzonego w eksykatorze. Odważyć (16,987 ± 0,001) g
wysuszonego azotanu srebra, rozpu
ścić go w wodzie i rozcieńczyć do 1 l w kolbie miarowej (5.3.6). Przechowywać
roztwór w szklanej brunatnej butelce do odczynników (5.2.14), chroni
ć przed długotrwałym przetrzymywaniem na
s
łońcu.
4.2.2 Roztwór oko
ło 0,1 mol/l rodanku (KSCN lub NH
4
SCN) przygotowany przez rozpuszczenie 9,7 g rodanku potasu
lub 7,6 rodanku amonu w wodzie i rozcie
ńczenie w kolbie miarowej o objętości 1 l.
Przenie
ść pipetą 25 ml roztworu azotanu srebra (4.2.1) do kolby miarowej (5.3.5) i dodać 5 ml kwasu azotowego (4.2.3)
i 1 ml roztworu wska
źnika siarczanu żelaza (III) amonu (4.2.5).
Dodawa
ć roztwór rodanku z biurety (5.2.13) aż wystąpi pierwsza trwała zmiana barwy z białej opalizującej na
jasnobrunatn
ą. Zapisać objętość dodanego roztworu rodanku.
Obliczy
ć stężenie roztworu rodanku c
T
(w molach na litr) z nast
ępującego wzoru:
c
T
= 2,5/V
1
w którym:
V
1
jest obj
ętością dodanego rodanku (w mililitrach)
Wzorcowa
ć roztwór co tydzień lub przed użyciem, jeżeli badania przeprowadza się rzadko.
4.2.3 Oko
ło 6 mol/l kwasu azotowego przygotowanego przez dodanie 100 ml kwasu azotowego (4.1) do 150 ml wody,
gotowanie pod wyci
ągiem (5.2.17) aż stanie się bezbarwny i pozostawienie do ostygnięcia do temperatury pokojowej.
4.2.4 Wolny od chlorków, techniczny 3,5,5,-trój-metylo-heksan-1-ol.
4.2.5 Roztwór wska
źnika siarczanu żelaza (III) amonu NH
4
Fe(SO
4
)
2
⋅
12H
2
O przygotowany przez dodanie 60 g wody do
50 g siarczanu
żelaza (III) amonu, podgrzanie do rozpuszczenia i dodanie 10 ml kwasu azotowego (4.2.3).
Ostudzi
ć roztwór do temperatury pokojowej i przechowywać w butelce szklanej (5.2.15).
4.3 Odczynniki do oznaczania chlorków soli rozpuszczalnych w wodzie metod
ą potencjo-metryczną (rozdział 8)
4.3.1 Roztwór 0,01 mol/l azotanu srebra (AgNO
3
) przygotowany z zastosowaniem procedury wymienionej w 4.2.1, lecz
rozcie
ńczony 1,699 g wysuszonego azotanu srebra w kolbie miarowej o obj ętości 1 l (5.3.6).
4.3.2 Roztwór 0,02 mol/l chlorku sodu (NaCl) przygotowany z oko
ło 2 g chlorku sodu wysuszonego w temperaturze
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 5

(110 ± 5) °C przez 1 h do 2 h i och
łodzonego, odważenie (1,169 ± 0,001) g chlorku sodu, rozpuszczenie w wodzie i
rozcie
ńczenie go w kolbie miarowej do obj ętości 1 l (5.3.6).
4.4 Wska
źnik do oznaczania chlorków soli rozpuszczalnych w wodzie metod ą Mohra (rozdział 9)
Roztwór chromianu potasu (K
2
CrO
4
) przygotowany przez rozpuszczenie 10 g chromianu potasu w 100 ml wody.
4.5 Odczynniki do oznaczania siarczanów rozpuszczalnych w wodzie (rozdzia
ł 10)
4.5.1 Roztwór kwasu chlorowodorowego (HCl) wykonany przez dodanie 200 ml st
ężonego kwasu chlorowodorowego (o
g
ęstości 1,18) do 800 ml wody.
4.5.2 Roztwór chlorku baru (BaCl
2
) wykonany przez rozpuszczenie 100 g chlorku baru (BaCl
2
⋅
2H
2
O) w 1 l wody, przed
u
życiem przefiltrowany przez średniej gęstości sączek.
4.6 Odczynniki do oznaczania zawarto
ści siarki całkowitej (rozdział 11)
4.6.1 Brom.
4.6.2 Wska
źnik czerwień metylowa (rozpuścić 20 mg proszku czerwieni metylowej w 50 ml etanolu i doda ć 50 ml wody).
4.7 Odczynniki do oznaczania zawarto
ści siarczków (rozdział 13)
4.7.1 Roztwór octanu o
łowiu (rozcieńczyć około 0,2 g octanu ołowiu {Pb(CH
3
COO)
2
⋅
3H
2
O} w wodzie i uzupe
łnić wodą
do 100 ml).
4.7.2 Amonowy roztwór siarczanu cynku (rozcie
ńczyć 50 g siarczanu cynku (ZnSO
4
⋅
7H
2
O) w 150 ml wody i doda
ć
350 ml st
ężonego wodorotlenku amonu (NH
4
OH)). Pozostawi
ć co najmniej przez 24 h i filtrować przez sączek średniej
twardo
ści.
4.7.3 Chlorek cyny (II) (SnCl
2
⋅
2H
2
O)
4.7.4 Metaliczny chrom Cr w proszku ( UWAGA: RAKOTWÓRCZY).
4.7.5 Roztwór wzorcowy jodanu potasu zawieraj
ący 0,0167 mol/l; sukcesywnie rozcieńczać w świeżo przegotowanej i
och
łodzonej wodzie w kolbie miarowej o obj ętości 1 l, (3,6 ± 0,1) g odważonego z dokładnością do 0,1 mg (6.3) jodanu
potasu KJO
3
wysuszonego w (110 ± 5) °C, dwie granulki (oko
ło 0,4 g) wodorotlenku sodu (NaOH) oraz 25 g jodku
potasu (KJ).
Uzupe
łnić do kreski świeżo przegotowaną i ostudzoną wodą.
UWAGA 1: Obecno
ść wodorotlenku sodu stabilizuje roztwór na d ługi czas; zaleca się nie stosować roztworu, gdy
zmieni kolor.
Wspó
łczynnik F tego roztworu oblicza się z następującego wzoru:
w którym:
m
1
masa próbki jodanu potasu
UWAGA 2: Je
żeli zawartość siarczku jest mała (mniej niż 0,1 %), zaleca się użyć roztworów dziesięć razy mniej
st
ężonych. Są one przygotowywane ze 100 ml roztworów (4.7.5 i 4.7.6) przeniesionych pipetą do kolb miarowych o
obj
ętości 1 l i uzupełnionych wodą do kreski.
4.7.6 Roztwór tiosiarczanu sodu o st
ężeniu około 0,1 mol/l.
Rozpu
ścić 24,82 g tiosiarczanu sodu (Na
2
S
2
O
3
⋅
5H
2
O) w wodzie i uzupe
łnić do 1 l. Przed każdą serią badań oznaczać
wspó
łczynnik f tego roztworu jak niżej.
Nastawi
ć miano roztworu tiosiarczanu jednym z następujących sposobów.
a) Nastawienie miana wobec wzorcowego roztworu jodanu potasu (4.7.5)
W tym przypadku przenie
ść pipetą 20 ml wzorcowego roztworu jodanu potasu do 500 ml kolby Erlenmeyera i uzupe łnić
wod
ą w ilości około 150 ml. Zakwasić 25 ml kwasu chlorowodorowego (1 + 1) i miareczkowa ć roztworem około 0,1 mol/l
tiosiarczanu sodu do osi
ągnięcia jasnożółtego koloru.
Nast
ępnie dodać 2 ml roztworu skrobi (4.7.7) i kontynuować miareczkowanie do czasu, aż kolor z niebieskiego zmieni
si
ę na bezbarwny.
Wspó
łczynnik f tego roztworu oblicza się z następującego wzoru:
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 6
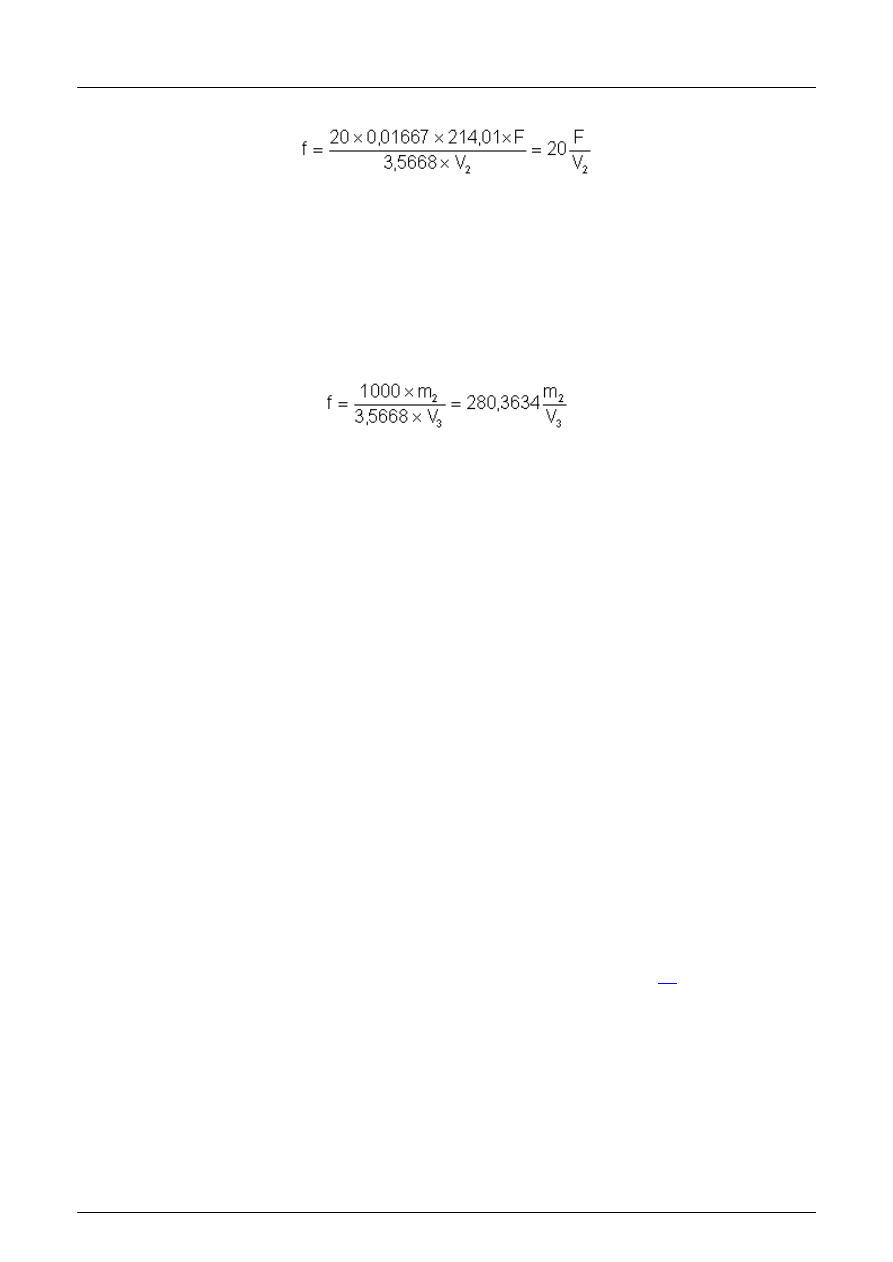
w którym:
F st
ężenie wzorcowego roztworu jodanu potasu (4.7.5), wyrażone w molach na litr;
V
2
obj
ętość około 0,1 mol/l roztworu tiosiarczanu sodu u żytego do miareczkowania;
3,5668 g/l jodanu potasu odpowiada roztworowi zawieraj
ącemu dokładnie 0,01667 mol/l jodanu potasu;
214,01 jest mas
ą cząsteczkową KJO
3
.
b) Nastawienie miana wobec znanej ilo
ści jodanu potasu.
W przypadku tego oznaczania umie
ścić w 500 ml kolbie Erlenmeyera (70 ± 5) mg jodanu potasu i rozcie ńczyć w około
150 ml wody.
Doda
ć około 1 g jodku potasu, zakwasić 25 ml kwasu chlorowodorowego (1 + 1) i miareczkowa ć roztworem
tiosiarczanu sodu oko
ło 0,1 mol/l do osiągnięcia jasnożółtego koloru. Następnie dodać 2 ml roztworu skrobi (4.7.7) i
miareczkowa
ć aż kolor z niebieskiego zmieni się na bezbarwny.
Wspó
łczynnik f tego roztworu oblicza się z następującego wzoru:
w którym:
m
2
masa jodanu potasu, w gramach;
V
3
obj
ętość około 0,1 mol/l roztworu tiosiarczanu sodu u żytego do miareczkowania;
3,5668 g/l jodanu potasu odpowiada roztworowi zawieraj
ącemu dokładnie 0,01667 mol/l jodanu potasu.
4.7.7 Roztwór skrobi (do 1 g skrobi (rozpuszczonej w wodzie) doda
ć 1 g jodku potasu KJ rozpuścić w wodzie i
uzupe
łnić wodą do 100 ml).
4.8 Odczynniki do oznaczania zanieczyszcze
ń lekkich (patrz 14.2)
4.8.1 Roztwór chlorku cynku otrzyma
ć rozpuszczając 7 kg ZnCl
2
w 3 l wody w celu uzyskania nasyconego roztworu o
g
ęstości (1,98 ± 0,02) g/cm
3
w (20 ± 3) °C. Wzgl
ędna gęstość roztworu po ochłodzeniu do temperatury pokojowej
powinna by
ć sprawdzona z użyciem odpowiedniego gęstościomierza (5.8.3)
UWAGA: Roztwór chlorku cynku umiarkowanie dra
żni skórę i błony śluzowe.
4.8.2 Roztwór poliwolframianu sodu (alternatywnie do 4.8.1), przygotowywany przez rozpuszczenie kryszta
łów
3Na
2
WO
4
⋅
9WO
3
⋅
H
2
O w wodzie, a
ż dobrze wymieszany roztwór nie będzie zawierać nierozpuszczonych kryształów tj.
do g
ęstości (1,98 ± 0,02) g/cm
3
w (20 ± 3) °C.
4.9 Odczynniki do oznaczania zawarto
ści humusu (15.1)
4.9.1 Roztwór wodorotlenku sodu, 3 % roztwór NaOH otrzymany przez rozpuszczenie 30 g pastylek wodorotlenku sodu
w wodzie, och
łodzenie do temperatury pokojowej, i rozcie ńczenie do 1 l w kolbie miarowej.
4.9.2 Roztwór o barwie wzorcowej przygotowuje si
ę przez rozpuszczenie 45,0 g FeCl
3
⋅
6H
2
O i 5,50 g CoCl
2
⋅
6H
2
O w
279,5 g wody i 1 ml st
ężonego HCl. Roztwór przechowuje się w szklanej butelce i jest on trwały co najmniej przez
2 tygodnie.
4.10 Odczynniki do oznaczania zawarto
ści kwasu fulvo (patrz 15.2)
4.10.1 Kwas chlorowodorowy (1 + 23).
4.10.2 Roztwór chlorku cyny (II). Rozpu
ścić 22,5 g SnCl
2
⋅
2H
2
O w 1 l kwasu chlorowodorowego (4.10.1). W
łaściwa
jako
ść tego roztworu utrzymuje się przez 2 tygodnie.
4.11 Odczynniki do oznaczania wolnego wapna metod
ą kompleksometryczną (patrz 18.1)
N7)
4.11.1 Etanodiol (glikol etylenowy),
świeży, bezwodny.
4.11.2 Propan-2-ol (Izopropanol), bezwodny.
4.11.3 Miazga z s
ączków w bezwodnym glikolu etylenowym.
4.11.4 Rozcie
ńczony kwas chlorowodorowy (1 + 1).
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 7
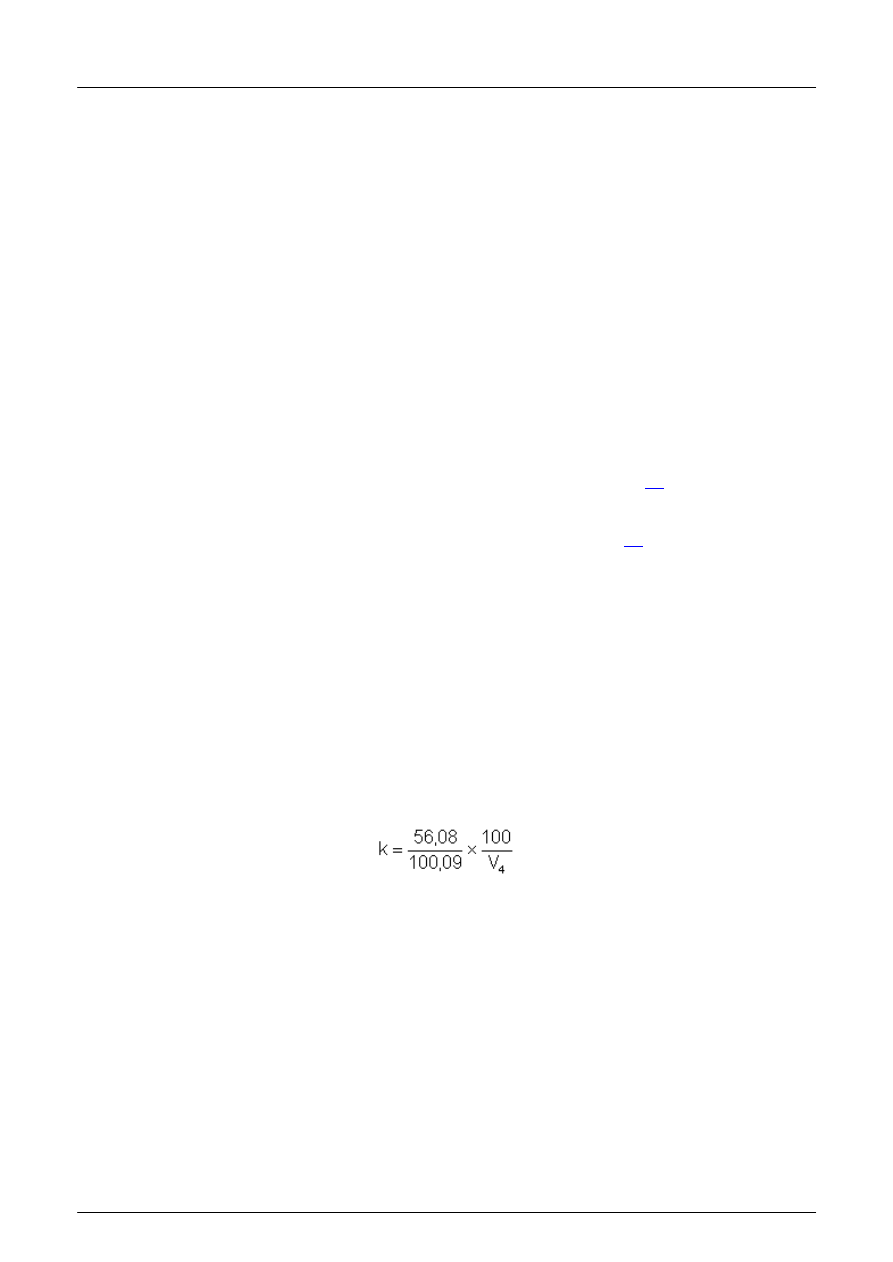
4.11.5 Trójetanoloamina.
4.11.6 m-Nitrofenol (0,1 g w 100 ml H
2
O).
4.11.7 Roztwór wodorotlenku sodu 2 mol/l, otrzymany w wyniku rozpuszczenia 80 g pastylek wodorotlenku sodu w
wodzie, ostudzony do temperatury pokojowej i rozcie
ńczony do 1 l w kolbie miarowej.
4.11.8 Wska
źnik, 1 g mureksydu (sól amonowa kwasu purpurowego) utrze ć w moździerzu z 100 g NaCl.
4.11.9 Roztwór EDTA 1/112 mol/l, (3,3 ± 0,1) g etylenodiaminy tetraoctowego kwasu soli disodowej wysuszonej do
sta
łej masy w 80 °C rozpuścić w wodzie i uzupełnić do 1 l. Należy oznaczać miano tego roztworu wobec roztworu o
znanej zawarto
ści wapnia (4.11.10).
4.11.10 Wzorcowy roztwór wapnia (1 ml = 1 mg tlenku wapnia). (1,785 ± 0,001) g czystego w
ęglanu wapnia (4.11.11)
wysuszonego w temperaturze (110 ± 5) °C rozpu
ścić w nieznacznym nadmiarze kwasu chlorowodorowego (1 + 4).
Gotowa
ć roztwór do usunięcia dwutlenku węgla, zakryć, ochłodzić do temperatury pokojowej i rozcieńczyć wodą do 1 l
w kolbie miarowej (5.3.6).
UWAGA: W handlu dost
ępne są wzorcowe roztwory, np. (1,000 ± 0,002) g CaO/l.
4.11.11 W
ęglan wapnia (CaCO
3
) str
ącony, wzorzec do oznaczania objętościowego.
4.11.12 Granulowane wapno sodowane
4.12 Odczynnik do oznaczania wolnego wapna metod
ą konduktometryczną (patrz 18.2)
N8)
4.12.1 Etanodiol (4.11.1).
4.13 Odczynniki do oznaczania wolnego wapna metod
ą acydymetryczną (patrz 18.3)
N9)
4.13.1 Acetylooctan etylu, bezwodny.
4.13.2 2-metylopropan-l-ol, (alkohol izobutylowy) bezwodny.
4.13.3 Wska
źnik błękit tymolowy (tymoloftaleina).
4.13.4 Kwas chlorowodorowy (4.1).
4.13.5 Roztwór rozpuszczalnika, 450 ml acetylooctanu etylu w 3 l alkoholu izobutylowego.
4.13.6 Wska
źnik (0,1 g proszku błękitu tymolowego rozpuszczonego w 100 ml alkoholu izobutylowego).
4.13.7 Roztwór kwasu chlorowodorowego w przybli
żeniu 0,2 mol/l.
Przygotowuj
ąc ten roztwór uzupełnić 17 ml kwasu chlorowodorowego (4.1) do 1 l alkoholu izobutylowego.
W celu oznaczania miana tego roztworu, odwa
żyć (100 ± 0,1) mg węglanu wapnia (4.11.11) i wyprażyć w tyglu (5.6.2)
przez 1 h w temperaturze 1000 °C. Ekstrahowa
ć wolne wapno i miareczkować zgodnie z 18.3.3.
Oznacza
ć współczynnik k z następującego wzoru:
w którym:
V
4
obj
ętość dodanego kwasu chlorowodorowego (w mililitrach);
k ilo
ść miligramów wolnego CaO w mililitrze mianowanego roztworu kwasu chlorowodorowego.
4.13.8 Wodorotlenek sodu na no
śniku, granulowany, o granulkach oko ło 0,8 mm do 1,6 mm do analizy elementarnej.
4.14 Odczynniki do oznaczania p
ęcznienia żużla stalowniczego (patrz 19.3)
4.14.1 Olej silikonowy.
4.14.2 Kwas chlorowodorowy, rozcie
ńczony (1 + 5).
5 Aparatura
5.1 Wymagania ogólne
Ca
ła aparatura powinna odpowiadać wymaganiom ogólnym prEN 932-5.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 8

Je
żeli nie podano inaczej, całe szkło laboratoryjne do oznaczań objętości powinno mieć klasę dokładności B, zgodnie z
ISO 384. Szk
ło laboratoryjne klasy A powinno być używane do badań auditowych i badań typu.
UWAGA: Je
żeli nie wymieniono tolerancji średnic, podane wartości są przybliżone.
5.2 Aparatura do ogólnych zastosowa
ń
5.2.1 Suszarka z dobr
ą wentylacją, wyposażona w urządzenie kontrolujące stałą temperaturę w przedziale od 40 °C do
150 °C z dok
ładnością ± 5 °C, wyposażona w tace z niekorodującego materiału.
5.2.2 Elektryczny piec muflowy wyposa
żony w urządzenie kontrolujące stałą temperaturę w przedziale od 800 °C do
1100 °C z dok
ładnością ± 25 °C.
5.2.3 Urz
ądzenie kruszące i rozdrabniające, aby pomniejszyć kruszywo do wymiarów przydatnych do poszczególnych
bada
ń, wytwarzające minimalną ilość pyłów.
5.2.4 Waga o zakresie wa
żenia do 10 kg, z dokładnością odczytu 1 g.
5.2.5 Waga o zakresie wa
żenia do 1 kg, z dokładnością odczytu 0,01 g.
5.2.6 Waga analityczna o zakresie wa
żenia do 100 g, z dokładnością odczytu 0,1 mg.
5.2.7 P
łyta grzejna z magnetycznym mieszadłem.
5.2.8 pH-metr, z dok
ładnością odczytu 0,1 pH.
5.2.9 Zlewki, sto
żkowe kolby, lejki sączki.
5.2.10 Pipety 25 ml, 50 ml i 100 ml, zgodne z wymaganiami ISO 648.
5.2.11 Cylindry pomiarowe z podzia
łką, pojemności 10 ml, 250 ml i 500 ml, zgodne z wymaganiami ISO 4788.
5.2.12 Tryskawki, zawieraj
ące zdemineralizowaną wodę.
5.2.13 Dwie biurety o pojemno
ści 50 ml z podziałką 0,1 ml.
5.2.14 Butelki na odczynniki z ciemnego szk
ła.
5.2.15 Butelki na odczynniki z bezbarwnego szk
ła.
5.2.16 Eksykatory.
5.2.17 Dygestoria.
5.3 Dodatkowa aparatura wymagana do oznaczania chlorków soli rozpuszczalnych w wodzie metod
ą Volharda
(rozdzia
ł 7)
5.3.1 Sito badawcze o otworach kwadratowych 16 mm z p
łyty perforowanej, zgodne z wymaganiami EN 933-2.
5.3.2 Dwie szklane plastikowe lub metalowe butelki, z szerokim wylotem i dobrze dopasowanymi korkami.
UWAGA: Zaleca si
ę aby butelka do badania kruszyw grubych lub lekkich mia ła objętość około 5 l oraz 2 l do badania
kruszyw drobnych.
5.3.3 Mechaniczna wstrz
ąsarka lub mieszalnik rolkowy, do butelek ekstrakcyjnych ( 5.3.2).
5.3.4 Dwa lejki filtracyjne o
średnicy około 100 mm z sączkami filtracyjnymi średniej i małej gęstości, o średnicy
odpowiedniej do wymiaru lejka.
5.3.5 Sto
żkowe kolby z korkami, o pojemności 100 ml i 250 ml.
5.3.6 Dwie kolby miarowe, o pojemno
ści 1 l, zgodne z wymaganiami ISO 1042.
5.4 Dodatkowa aparatura niezb
ędna do oznaczania chlorków soli rozpuszczalnych w wodzie metod ą
potencjometryczn
ą (patrz rozdział 8).
5.4.1 Urz
ądzenie do miareczkowania potencjometrycznego, odpowiednie do oznaczania st ężenia jonu chlorku, z
systemem elektrod sk
ładającym się z:
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 9

a) elektrody pomiarowej - albo srebrnej elektrody (najlepiej chlorowanej), albo jonoselektywnej.
b) elektrody odniesienia - albo siarczan rt
ęci, albo kombinowanej: srebro/chlorek srebra z bezchlorkowym elektrolitem w
zewn
ętrznej komorze.
5.5 Dodatkowa aparatura do fabrycznej kontroli produkcji niezb
ędna do oznaczania chlorków soli
rozpuszczalnych w wodzie metod
ą Mohra (patrz rozdział 9)
5.5.1 Dwie plastikowe butelki, o pojemno
ści 1 l, z szerokim wylewem i korkami.
5.6 Dodatkowa aparatura niezb
ędna do oznaczania siarczanów rozpuszczalnych w wodzie (patrz rozdzia ł 10)
5.6.1 Spiekane kwarcowe tygle filtruj
ące, o porowatości 4 stopnia, o średnicy około 35 mm i wysokości 40 mm.
5.6.2 Tygle do pra
żenia, jako alternatywa do 5.6.1, o średnicy około 35 mm i wysokości 40 mm, nie zmieniające masy w
czasie ogrzewania si
ę do temperatury 1100 °C.
UWAGA: Porcelana, kwarc lub platyna s
ą materiałami odpowiednimi na tygle do prażenia.
5.7 Dodatkowa aparatura wymagana do oznaczania zawarto
ści siarczków (patrz rozdział 13)
5.7.1 Typow
ą aparaturę do oznaczania zawartości siarczków przedstawiono na rysunku 1.
5.8 Dodatkowa aparatura wymagana do oznaczania zanieczyszcze
ń lekkich (patrz 14.2)
5.8.1 Sita 300
µ
m i 250
µ
m, zgodne z EN 933-2.
5.8.2 Parownice porcelanowe.
5.8.3 G
ęstościomierz do cieczy o zakresie 1,950 do 2,000, zgodny z wymaganiami ISO 650.
5.9 Dodatkowa aparatura wymagana do oznaczania zawarto
ści humusu (patrz 15.1)
5.9.1 Sito 4 mm, zgodne z EN 933-2.
5.9.2 Cylindryczna butelka z korkiem, ze szk
ła bezbarwnego. Pojemność butelki powinna wynosić około 450 ml, a
średnica zewnętrzna około 70 mm.
5.10 Dodatkowa aparatura wymagana do oznaczania zawarto
ści kwasu fulvo (patrz 15.2)
5.10.1 Szklany pr
ęt do mieszania.
5.10.2 Szklany lejek filtracyjny.
5.10.3 S
ączek filtracyjny średniej twardości o średnicy 180 mm.
5.10.4 P
łyta grzejna.
5.10.5 Normowe p
łyty kolorowe od (A do G)
1)
.
5.11 Dodatkowa aparatura wymagana do oznaczania zanieczyszcze
ń organicznych metodą zaprawy (patrz 15.3)
5.11.1 Stoper lub zegar, z dok
ładnością odczytu 1 s.
5.11.2 Porcelanowa lub kwarcowa parownica ogniotrwa
ła, o wymiarze odpowiednim do umieszczenia jej w piecu
muflowym.
5.11.3 Aparat do badania zanurzenia, zgodny z wymaganiami prEN 1015-4.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 10
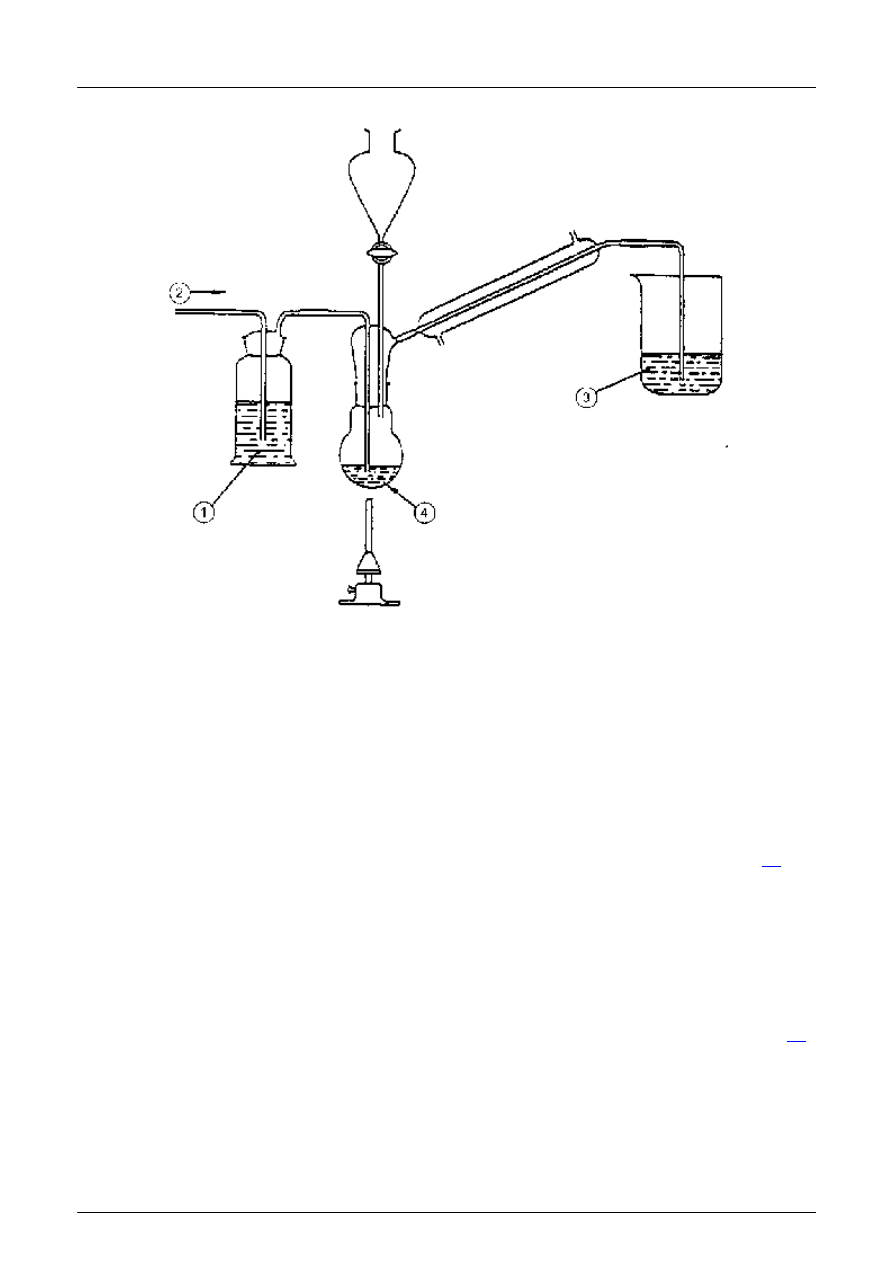
1 Roztwór octanu o
łowiu (4.7.1)
2 Azot lub argon
3 Amoniakalny roztwór siarczanu cynku ( 4.7.2)
4 Kolba reakcyjna
Rysunek 1: Przyk
ład aparatu do oznaczania siarczków
5.11.4 Mieszalnik, zgodny z wymaganiami EN 196-1.
5.11.5 Aparat do pomiaru twardnienia, zgodny z prEN 1015-9.
5.11.6 Aparat do oznaczania wytrzyma
łości na zginanie i ściskanie, zgodny z wymaganiami prEN 1015-11.
5.11.7 Elektryczny piec muflowy o wydajno
ści umożliwiającej prażenie 2 kg kruszywa, utrzymujący temperaturę
(480 ± 25) °C.
5.12 Dodatkowa aparatura wymagana do kompleksometrycznego oznaczania wolnego wapna (patrz 18.1)
N7)
5.12.1 Kolba sto
żkowa, o objętości 250 ml, ze szklanym korkiem.
5.12.2 Kolba miarowa, o obj
ętości 500 ml.
5.12.3 Mieszad
ło magnetyczne z kąpielą wodną, o regulowanej temperaturze.
5.12.4 Filtr ze szk
ła spiekanego, o średnicach porów 10 mm i 16 mm.
5.12.5 Urz
ądzenie do miareczkowania z galwanometrem do fotoelektrycznego oznaczania punktu ko ńcowego.
5.13 Dodatkowa aparatura wymagana do oznaczania wolnego wapna metod
ą konduktometryczną (patrz 18.2)
N8)
5.13.1 Naczynie pomiarowe (o obj
ętości około 160 ml) z termoplastyczną obudową i gwintowaną zakrętką oraz z
dwoma otworami NS 14 (patrz rysunek 2).
5.13.2 Elektroda konduktometryczna, o podstawie sto
żkowej NS 14.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 11
5.13.3 Termometr (50 °C do 100 °C): z dok
ładnością do 0,1 °C, o podstawie stożkowej NS 14.
5.13.4 Konduktometr
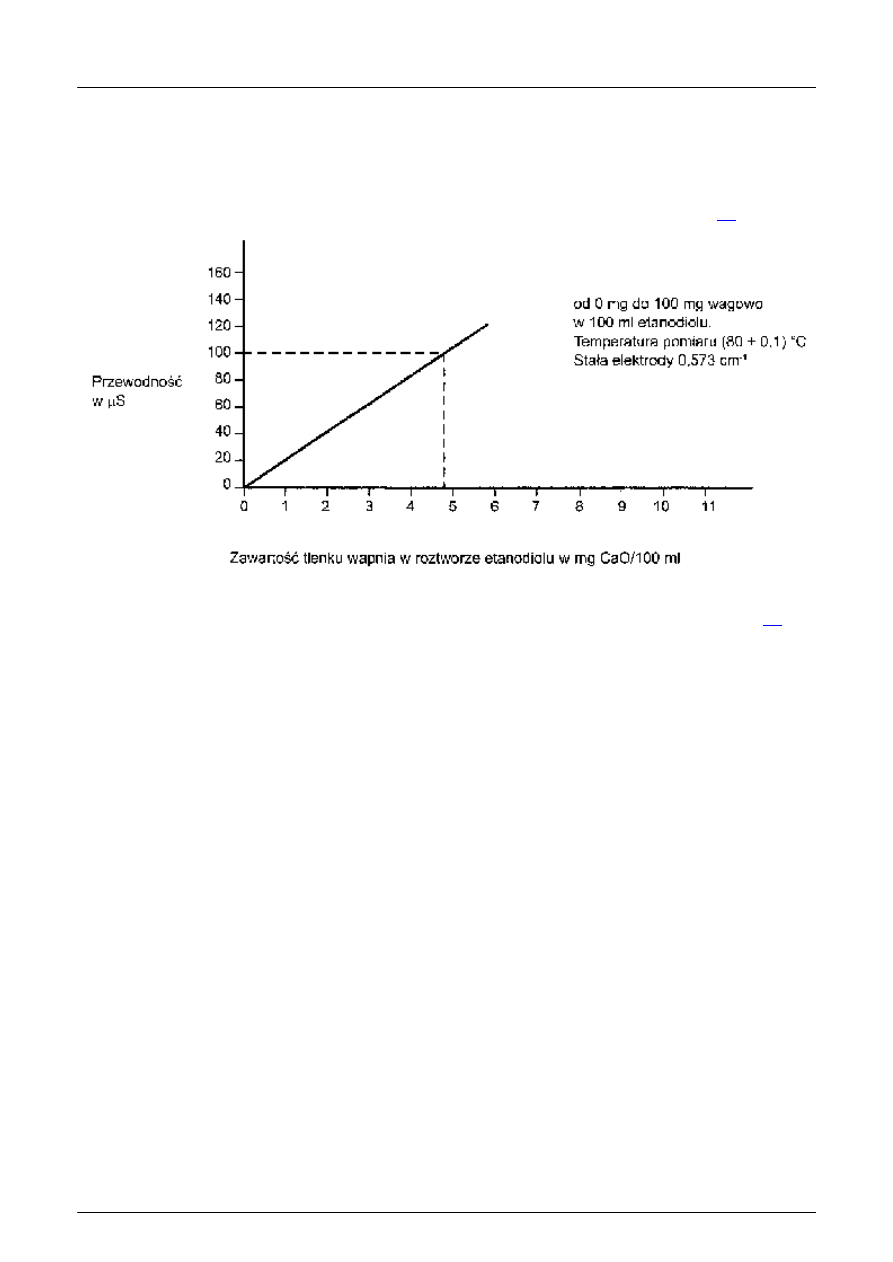
UWAGA 1: Zawarto
ść wolnego wapna jest określana na podstawie pomiaru przewodności z użyciem krzywej
wzorcowej. Jest ona sporz
ądzona przez rozpuszczanie znanych ilości wyprażonego CaO w etanodiolu i mierzenie
przewodno
ści tych roztworów. W tym celu zaleca się określić przewodność co najmniej pięciu różnych roztworów w
przedziale od 0 mg do 10 mg CaO/100 ml etanodiolu, w ka
żdym przypadku z trzech indywidualnych pomiarów.
UWAGA 2: U
żyte CaO otrzymuje się przy prażeniu CaCO
3
(4.11.11) w 1000 °C do osi
ągnięcia stałej masy, a następnie
och
łodzenie w eksykatorze, który zawiera materiał absorbujący wodę i dwutlenek węgla, np: wapno sodowane.
UWAGA 3: Zaleca si
ę, aby przewodność ślepego roztworu etanodiolu była każdorazowo ustalana i odejmowana od
roztworu badanego.
UWAGA 4: Na rysunku 3 przedstawiono wykres wzorcowania etanodiolu, zawieraj
ącego tlenek wapnia, w 80 °C, z
elektrod
ą o stałej 0,573 cm
-1
; w tym przypadku zmierzona przewodno
ść 100 mS odpowiada zawartości wolnego wapna
w ilo
ści 4,9 % masy.
5.13.5
Łaźnia wodna, z regulowaną temperaturą do (80 ± 0,1) °C.
1 termometr (5.13.3)
2 elektroda (5.13.2)
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 12
3 polipropylenowa pokrywa
4 naczynie wykonane ze szk
ła borokrzemianowego
5 oprawa plastikowa
6 dop
ływ wody
7 mieszad
ło magnetyczne (5.12.3)
Rysunek 2: Przekrój pionowy konduktometru do oznaczania wolnego wapna (5.13 i 18.2
N8)
)
Rysunek 3: Przyk
ład wykresu kalibracyjnego
5.14 Dodatkowa aparatura wymagana do oznaczania wolnego wapna metod
ą acydymetryczną (patrz 18.3)
N9)
5.14.1 Kolby Erlenmeyera, o obj
ętości 200 ml, 250 ml lub 300 ml, dopasowane z ch łodnicami chłodzonymi wodą za
pomoc
ą szlifów.
5.14.2 Rury absorpcyjne dopasowane do górnej cz
ęści chłodnic i zawierające wodorotlenek sodu (4.13.8) i sito
molekularne (5.14.3).
5.14.3 Sito molekularne 0,3 nm, o pere
łkach średnicy około 2 mm.
5.14.4 Filtry z mikrow
łókna szklanego z otworami 1,2 mm.
5.14.5 Urz
ądzenie do próżniowej filtracji.
5.15 Dodatkowa aparatura wymagana do oznaczania rozpadu ortokrzemianu dwuwapniowego w
żużlu
wielkopiecowym ch
łodzonym powietrzem (patrz 19.1)
5.15.1 O
świetlenie nadfioletowe, o długości fali od 300 nm do 400 nm, z maksymalnym nat ężeniem długości fali
366 nm.
5.16 Dodatkowa aparatura wymagana do oznaczania p
ęcznienia żużla stalowniczego (patrz 19.3)
5.16.1 Wytwornica pary z pomiarowym cylindrem i pomiarowym czujnikiem zegarowym lub przesuwnym miernikiem, o
zakresie pomiaru (10 ± 0,01) mm, jak przedstawiono na rysunkach 4 i 5.
UWAGA: Wytwornica pary sk
łada się z dwóch komór, w których woda jest podgrzewana do punktu wrzenia elementami
grzejnymi w czasie badania. Maksymalna moc elementów grzejnych jest 2 kW. Nad ogrzewan
ą komorą znajduje się
ściśnięta próbka żużla w cylindrze z perforowaną podstawą (średnica cylindra 210 mm, wysokość cylindra 100 mm).
Nagrzany strumie
ń pary unosi się i może równomiernie przenikać przez próbkę. W celu uniknięcia kondensacji
wewn
ątrz cylindra z powodu strat ciepła, cylinder jest ogrzewany do (120 ± 10) °C za pomoc ą obwodowego płaszcza
grzejnego przylegaj
ącego do zewnętrznej ściany (nominalna moc 250 W).
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 13
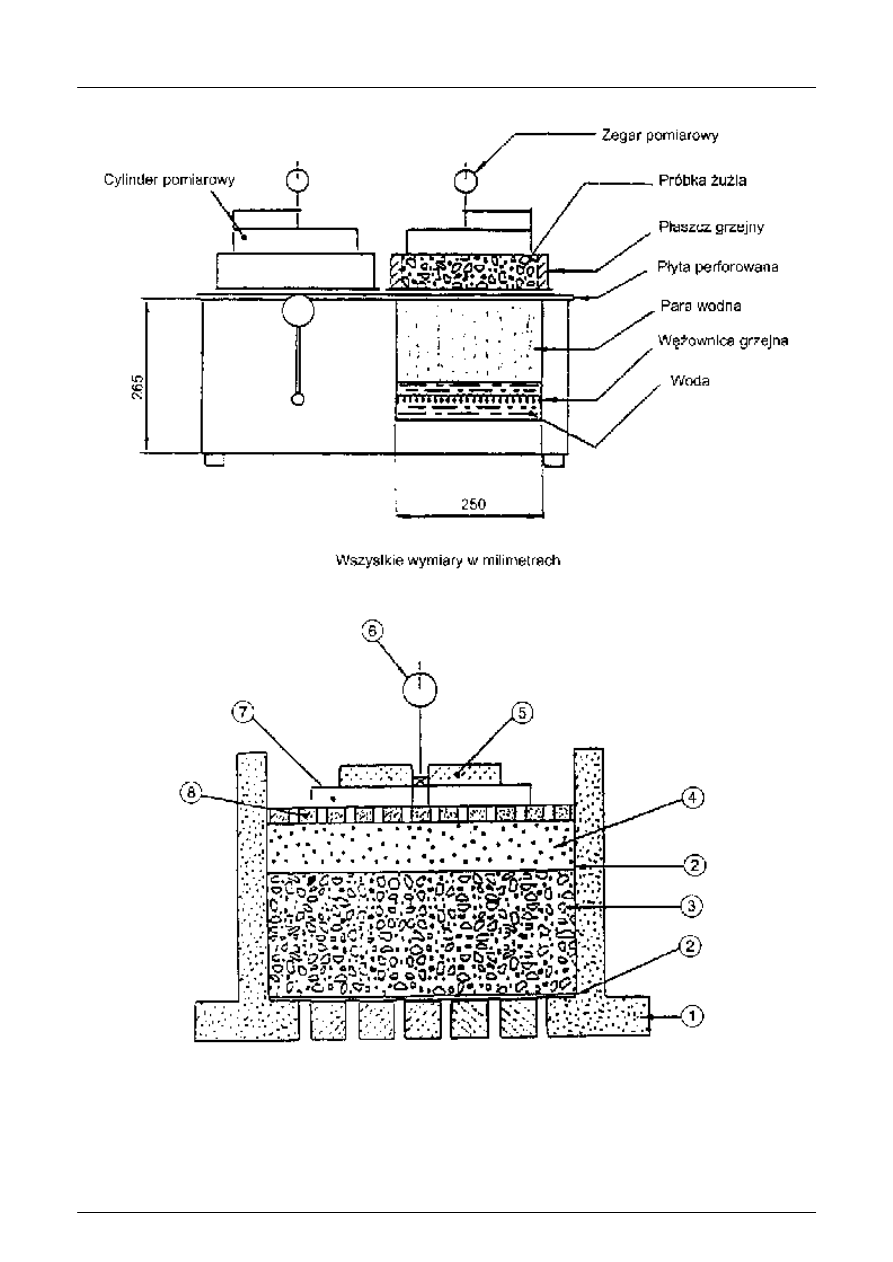
Rysunek 4: Przekrój pionowy typowego urz
ądzenia do wytwarzania pary
1 cylinder z perforowan
ą podstawą, 0,01 otworów na cm
2
,
np. 49 otworów o
średnicy 3 mm rozmieszczonych następująco:
na
środku 1 otwór
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 14

w okr
ęgu o średnicy 65 mm: 8 otworów
w okr
ęgu o średnicy 125 mm: 16 otworów
w okr
ęgu o średnicy 185 mm: 24 otwory
2 mata tkaninowa
3 zag
ęszczona próbka żużla do badania
4 szklane pere
łki, o średnicy 5 mm
5 obci
ążnik wagowy
6 czujnik pomiarowy
7 obci
ążnik krzyżakowy
8 perforowana p
łyta, 0,3 otworów na cm
2
,
np. otwory o
średnicy 3 mm koncentrycznie rozmieszczone na okr ęgach co 6 mm, przy odległości między okręgami od
6,5 mm do 7 mm
Rysunek 5: Schematyczna ilustracja urz
ądzenia do oznaczania pęcznienia
5.16.2 Sita, zgodne z EN 933-2, z otworami 0,5 mm, 2,0 mm, 5,6 mm, 8,0 mm, 11,2 mm, 16,0 mm i 22,4 mm.
5.16.3 Pere
łki szklane, o średnicy 5 mm.
5.16.4 S
ączki filtracyjne średniej twardości, o średnicy 240 mm.
5.16.5 Stó
ł wibracyjny z przybliżoną częstotliwością (48 ± 3) Hz i amplitudą ± 1,5 mm lub ręczny zagęszczacz, np.
m
łotek Proktora lub ręczny młotek, który zapewnia końcowe zagęszczenie próbki analitycznej do zawartości wolnych
przestrzeni pomi
ędzy 20 % i 25 % objętości (patrz EN 196-1).
5.16.6 G
łębokościomierz prętowy z podziałką milimetrową o minimalnej całkowitej skali 200 mm.
5.16.7 Obci
ążnik wagowy o średnicy zewnętrznej mniejszej niż 210 mm (np. 180 mm) mający centralny otwór (np. o
średnicy 15 mm), stanowiący trzon dla czujnika pomiarowego i drog ę dla strumienia pary; całkowita masa "obciążnika
krzy
żakowego plus szklane perełki" powinna wynosić 6 kg.
6 Ogólne wymagania dotycz
ące badania
6.1 Liczba bada
ń
Je
żeli nie podano inaczej, przyjęto podwójną liczbę pojedynczych oznaczań dla poszczególnych metod badań (patrz
rozdzia
ły 7 do 19.3) (patrz również 6.3).
6.2 Powtarzalno
ść i odtwarzalność
Odchylenie standardowe powtarzalno
ści podaje zakres zgodności pomiędzy kolejnymi wynikami otrzymywanymi tą
sam
ą metodą na identycznym badanym materiale, w tych samych warunkach (ten sam operator, ten sam aparat, to
samo laboratorium i krótki przedzia
ł czasu).
Odchylenie standardowe odtwarzalno
ści podaje zakres zgodności pomiędzy indywidualnymi wynikami otrzymywanymi
t
ą samą metodą na identycznym materiale lecz badanym w różnych warunkach (różni wykonawcy, inna aparatura, inne
laboratorium i/lub ró
żne czasy) (patrz prEN 932-6).
Odchylenie standardowe powtarzalno
ści i odtwarzalności podaje się w procentach bezwzględnych.
6.3 Wyra
żanie masy, objętości, współczynników i wyników
Mas
ę ze wskazań wagi analitycznej zapisać (5.2.6) w gramach, z dokładnością do 0,1 mg a objętość ze wskazań biuret
(5.2.13) w mililitrach, z dok
ładnością do 0,05 ml.
Mas
ę ze wskazań wagi zwykłej podanej w 5.2.4 zapisać w gramach, z dokładnością do 1 g lub z wagi podanej w 5.2.5 z
dok
ładnością do 0,01 g.
Miana roztworów (4.7.5, 4.7.6 i 4.13.7), wyra
żonych jako średnie z trzech oznaczań, podawać do trzeciego miejsca po
przecinku.
Wyniki bada
ń, wyrażone jako średnia z dwóch oznaczań, podawać w procentach z dokładnością do 0,01 %, jeżeli nie
podano inaczej.
Je
żeli różnica pomiędzy dwoma oznaczaniami jest większa niż podwójne odchylenie standardowe powtarzalno ści,
badanie powtórzy
ć i przyjąć średnią z dwóch najbliższych wartości.
6.4 Suszenie materia
łów
Suszenie nale
ży przeprowadzić w suszarce z dobrą wentylacją (5.2.1), w temperaturze (110 ± 5) °C.
6.5 Oznaczanie sta
łej masy po suszeniu
Sta
łą masę oznacza się po suszeniu próbki analitycznej przez 24 h.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 15

6.6 Spalanie osadów
Spalanie osadów nale
ży przeprowadzić następująco.
Umie
ścić bibułę filtracyjną z zawartością w tyglu, który został uprzednio wyprażony i wytarowany. Wysuszyć i spalić na
popió
ł powoli w atmosferze utleniającej, zapewniając całkowite spalanie bez płomienia.
Pra
żyć co najmniej przez 1 h w stałej temperaturze. Ostudzić tygiel z jego zawartością do temperatury pokojowej w
eksykatorze. Zwa
żyć tygiel z jego zawartością.
6.7 Sprawdzenie braku jonów chlorkowych (badanie azotanem srebra)
Zwykle po pi
ęciu do sześciu przemyciach osadu, przepłukać podstawę sączka kilkoma kroplami wody.
Przemy
ć sączek i jego zawartość kilkoma mililitrami wody i zachowa ć przesącz w probówce. Dodać kilka kropel
st
ężonego kwasu azotowego (4.1) i roztworu azotanu srebra (4.2.1). Sprawdzić, czy brak zmętnienia lub osadu w
roztworze. Je
żeli występuje, kontynuować przemywanie, okresowo sprawdzając. Brak zmętnienia w badanym azotanie
srebra wskazuje,
że przemyta pozostałość nie zawiera jonów chlorkowych.
7 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą Volharda (metoda zalecana)
7.1 Zasada metody
Niniejsze badanie jest przydatne dla kruszyw, w których chlorki powsta
ły w wyniku bezpośredniego kontaktu z zasoloną
wod
ą lub zanurzenia w niej, np. dotyczy to kruszyw wydobywanych z morza. Niektóre kruszywa, np. wydobywane z
niektórych obszarów pustynnych, przy badaniu ekstraktów z kwasem azotowym mog
ą wykazywać znacznie wyższe
poziomy chlorków ni
ż przy badaniu metodą ekstraktów wodnych.
Próbka analityczna kruszywa jest ekstrahowana wod
ą do usunięcia jonów chlorkowych. Metoda analizy ekstraktu jest
oparta na miareczkowaniu Volharda, gdzie dodaje si
ę nadmiar roztworu azotanu srebra do roztworu chlorku, a
nieprzereagowana cz
ęść jest miareczkowana wzorcowym roztworem rodanku potasu wobec wska źnika siarczanu
żelaza (III) amonu.
Chlorki s
ą wyrażane jako zawartość jonu chlorku i podawane w procentach masy kruszywa.
7.2 Pobieranie próbek
Próbki laboratoryjne powinny by
ć pobrane zgodnie z procedurami wymienionymi w EN 932-1.
Upewni
ć się, czy próbka laboratoryjna zawiera taką samą wilgoć jak cała masa kruszywa.
7.3 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną według procedur wymienionych w prEN 932-2 do wielko ści nie mniejszej niż masa
podana w tablicy 1, w
łaściwa dla nominalnej wielkości kruszywa.
Wysuszy
ć podpróbkę w temperaturze (110 ± 5) °C do stałej masy (6.5).
Przesia
ć podpróbkę przez sito 16 mm (patrz 5.3.1) i przekruszyć całe nadziarno tak, aby przeszło przez sito, unikając
nadmiernego rozdrobnienia. Po
łączyć i wymieszać oraz stosując procedury wymienione w prEN 932-2, przygotować
dwie próbki analityczne ka
żda o masie około (2 ± 0,3) kg dla kruszyw grubych lub dwie próbki analityczne o masie
oko
ło (500 ± 75) g dla kruszyw drobnych.
W przypadku kruszyw lekkich, dwie próbki analityczne b
ędą stanowić objętość około 1 l.
Tablica 1: Minimalna pocz
ątkowa masa podpróbki
Nominalny maksymalny wymiar ziarna kruszywa
mm
Minimalna masa podpróbki
kg
63
45
22,4 lub mniej
50
35
15
5
7.4 Przygotowanie ekstraktów
W przypadku kruszyw grubych i kruszyw lekkich u
żyć dwóch szklanych, plastikowych lub metalowych butli o obj ętości
5 l, z szerok
ą szyjką, a dla kruszyw drobnych użyć butli o objętości 2 l (5.3.2). Zważyć każdą butlę i zapisać ich masy z
dok
ładnością do 1 g.
Przenie
ść próbki analityczne, otrzymane wg 7.3, do butli, zważyć butle z zawartościami i zapisać ich masy z
dok
ładnością do 1 g. Z różnicy obliczyć masę kruszywa w każdej butli.
Doda
ć do każdej butli wodę w ilości równej masie próbki analitycznej. W przypadku kruszyw lekkich doda ć 1 l wody.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 16

Miesza
ć zawartość butli w sposób ciągły przez 60 min z użyciem wstrząsarki lub mieszalnika rolkowego ( 5.3.3).
Nast
ępnie filtrować ekstrakty przez suche, średniej twardości, sączki filtracyjne (5.3.4) aż do uzyskania co najmniej
100 ml klarownego lub jasnoopalizuj
ącego przesączu w suchych czystych zlewkach. (5.2.9).
7.5 Procedura oznaczania zawarto
ści chlorków w ekstraktach
Pobra
ć 100 ml przefiltrowanego ekstraktu (7.4) z użyciem pipety o objętości 100 ml (5.2.10) i przenieść do kolby o
pojemno
ści 250 ml (5.3.5). Dodać do kolby 5 ml kwasu azotowego (4.2.3), następnie dodać z biurety roztworu azotanu
srebra (4.2.1) a
ż całość jonów chlorkowych zostanie strącona, a następnie dodać nadmiar.
UWAGA: Je
żeli analizowane są kruszywa zawierające siarczki (np. żużle), roztwór przenieść pod dygestorium na 3 min
lub 5 min do temperatury poni
żej wrzenia. Może wystąpić biały osad siarki, którego nie trzeba koniecznie ods ączyć.
Och
łodzić i dodać roztwór azotanu srebra.
Wymagana jest ilo
ść azotanu srebra wystarczająca dla zapewnienia miareczkowania co najmniej 3 ml roztworu
rodanku.
Zanotowa
ć całkowitą objętość V
5
dodanego roztworu azotanu srebra.
Doda
ć 2 ml 3,5,5 trój-metylo-heksan-1-ol ( 4.2.4), zakorkować i wstrząsnąć energicznie kolbą w celu koagulacji osadu.
Ostro
żnie odkorkować, unikając strat roztworu, przemyć korek wodą, a wodę z przemycia dodać do roztworu.
Doda
ć jako wskaźnika 5 ml roztworu siarczanu żelaza (III) amonu (4.2.5), a następnie z biurety mianowanego roztworu
rodanku (4.2.2), a
ż wystąpią pierwsze trwałe zmiany koloru, z białego opalizującego na jasnobrązowy, a roztwór
osi
ągnie taki sam odcień jaki był przy wzorcowaniu 4.2.4.
Zanotowa
ć objętość V
6
dodanego roztworu rodanku.
Powtórzy
ć procedurę z ekstraktem sporządzonym z drugiej próbki badanej.
Oznaczania na ka
żdym ekstrakcie zapisuje się jako jedno. Wynik badania otrzymuje si ę jako średnia z oznaczań na
dwóch ekstraktach.
7.6 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość chlorku C w kruszywie z następującego wzoru:
C = 0,003546
.
W
.
{V
5
- (10 × c
T
× V
6
)} (w %)
w którym:
V
5
obj
ętość roztworu azotanu srebra (w mililitrach);
V
6
obj
ętość dodanego roztworu wzorcowego rodanku (w mililitrach);
c
T
st
ężenie rodanku w roztworze wzorcowym (w molach na litr);
W jest stosunkiem wody do kruszywa (w gramach na gram), dla kruszyw lekkich W jest to 1000 g/masa kruszywa w
gramach.
UWAGA: Ustalenie dotycz
ące dokładności oznaczania chlorków rozpuszczalnych w wodzie podano w za łączniku A.
8 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą potencjometryczną (metoda alternatywna)
8.1 Zasada metody
Próbki analityczne kruszywa s
ą wydzielane tak samo jak w 7.4. Jony chlorkowe są strącane z ekstraktów z użyciem
mianowanego roztworu azotanu srebra.
Miareczkowanie przeprowadza si
ę za pomocą potencjometru, używając odpowiedniej elektrody jako wska źnika.
UWAGA: Chlorkowe elektrody jonoselektywne oraz u
życie wykresu Grana jest także dopuszczalne (patrz załącznik B).
8.2 Pobieranie próbek, przygotowanie próbek analitycznych i ekstraktów
Zastosowa
ć procedury wymienione w 7.2, 7.3 i 7.4.
8.3 Procedura oznaczania zawarto
ści chlorku w ekstraktach
Pobra
ć 50 ml przefiltrowanego ekstraktu ( 7.4) z użyciem pipety o objętości 50 ml (5.2.10) i przenieść do 250 ml zlewki.
Zakwasi
ć kwasem azotowym (HNO
3
) (4.2.3) do warto
ści pH od 2 do 3. Dodać pipetą 5 ml roztworu chlorku sodu ( 4.3.2).
UWAGA: Je
śli badane są kruszywa zawierające siarczki (np. żużle), ekstrahować roztwór przez 3 min do 5 min w
temperaturze tu
ż poniżej wrzenia. Może utworzyć się biały osad siarki, lecz nie trzeba koniecznie go ods ączać.
Ostudzi
ć i kontynuować miareczkowanie.
U
żywając potencjometru (5.4.1) miareczkować roztworem azotanu srebra (4.3.1). Zawartość chlorku w roztworze
oznacza si
ę przez zużycie roztworu azotanu srebra odpowiadające punktowi przegięcia krzywej. Ilość chlorku sodu
(4.3.2), dodana w celu
łatwiejszego rozpoznania punktu końcowego, powinna być odliczona.
Powtórzy
ć procedurę z ekstraktem sporządzonym z drugiej próbki analitycznej.
Nale
ży wykonać ślepą próbę dla zweryfikowania dodanego chlorku sodu.
8.4 Obliczanie i wyra
żenie wyników
Obliczy
ć zawartość chlorku C w kruszywie z następującego wzoru:
C = 0,000709V
7
× W (w %)
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 17

w którym:
V
7
zu
życie roztworu azotanu srebra, w mililitrach, po odj ęciu 10 ml na dodany roztwór chlorku;
W jest stosunkiem wody do kruszywa (w gramach/gram) - dla kruszyw lekkich W jest to 1000 g/masa kruszywa, w
gramach.
UWAGA: Ustalenia dotycz
ące dokładności oznaczania chlorków rozpuszczalnych w wodzie metod ą potencjometryczną
podano w za
łączniku A.
9 Oznaczanie chlorów soli rozpuszczalnych w wodzie metod
ą Mohra (metoda alternatywna)
9.1 Postanowienia ogólne
Niniejsza metoda podaje szybsz
ą metodę ekstrakcji niż metoda wymieniona w rozdziale 7. Jest ona bardziej zalecana
jako wst
ępna ocena przed przystąpieniem do badania wymienionego w punkcie 7, które mo że być potrzebne dla
sprawdzenia zgodno
ści z normą. Procedura ta powinna być użyta wyłącznie do fabrycznej kontroli produkcji.
St
ężenie jonu chlorkowego w wodnym ekstrakcie kruszywa naturalnego mo że być oznaczone z użyciem
instrumentalnych technik opartych na pomiarach przewodnictwa.
9.2 Zasada metody
Próbka analityczna kruszywa jest szybko ekstrahowana wod
ą w temperaturze pokojowej w celu usuni ęcia jonów
chlorkowych. Analiz
ę ekstraktu przeprowadza się metodą Mohra, w której chlorek jest miareczkowany azotanem srebra,
a jako wska
źnik użyty jest chromian potasu. Stężenie jonów chlorkowych może być także oznaczane z użyciem metod
instrumentalnych opartych na przewodno
ści.
9.3 Pobieranie próbek
Próbki laboratoryjne powinny by
ć pobrane zgodnie z procedurami wymienionymi w EN 932-1.
9.4 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną do próbki analitycznej 250 g kruszywa (1 l w przypadku kruszywa lekkiego) wed ług
procedury wymienionej w prEN 932-2.
9.5 Przygotowanie ekstraktów
Zarówno dla kruszyw grubych, jak i drobnych nale
ży zastosować plastikowe butelki o pojemno ści 1 l, z szeroką szyjką
(5.5.1). Dla kruszyw lekkich nale
ży użyć butli o pojemności 5 l (5.3.2). Zważyć każdą butlę i zapisać jej masę z
dok
ładnością do 1 g.
Przenie
ść próbkę analityczną do butli, zważyć butle z zawartością i zapisać ich masy z dokładnością do 1 g.
Z ró
żnicy obliczyć masę kruszywa w każdej butli.
Doda
ć do każdej butli wody w ilości równej masie kruszywa. W przypadku kruszywa lekkiego doda ć 1 l wody.
Zamkn
ąć butle i wymieszać zawartość, potrząsając co najmniej 20 razy. Pozwolić odstać się roztworowi, aby woda na
powierzchni by
ła w miarę klarowna.
9.6 Procedura oznaczania zawarto
ści chlorku w ekstraktach
UWAGA: Je
żeli będzie zastosowany pomiar przewodności, zlać około 100 ml do 250 ml zlewki i zmierzyć stężenie.
Pobra
ć 25 ml sklarowanej wody (9.5) za pomocą 25 ml pipety (5.2.10) i przenieść do kolby o pojemności 100 ml (5.3.5).
Doda
ć od 4 ml do 6 ml roztworu chromianu potasu ( 4.4.1) i wymieszać. Miareczkować roztworem azotanu srebra o
st
ężeniu 0,01 mol/l (4.3.1) do osiągnięcia koloru jasnoczerwonego. Zanotowa ć objętość V
8
u
żytego roztworu azotanu
srebra.
9.7 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość chlorków C w kruszywie z następującego wzoru:
C = 0,01 × 0,03545 × V
8
× W × 4 (w %)
w którym:
V
8
u
żyta objętość roztworu 0,01 mol/l azotanu srebra;
W stosunk wody do kruszywa (w gramach/gram) - dla kruszyw lekkich W jest to 1000 g/masy kruszywa, w gramach.
10 Oznaczanie siarczanów rozpuszczalnych w wodzie
10.1 Zasada metody
Próbk
ę analityczną kruszywa ekstrahuje się wodą celem uwolnienia jonów siarczanowych rozpuszczalnych w wodzie.
Zawarto
ść siarczanów rozpuszczalnych w wodzie jest oznaczana poprzez wytrącenie roztworem chlorku baru przy pH
pomi
ędzy 1 i 1,5 w temperaturze wrzenia.
Oznaczanie jest zako
ńczone podaniem wagowej zawartości jonu siarczanu wyrażonej jako procent masy kruszywa.
Metoda ta jest przydatna, gdy badane kruszywa zawieraj
ą siarczki, np. żużle.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 18

10.2 Pobieranie próbek
Próbki laboratoryjne powinny by
ć pobrane zgodnie z procedurami wymienionymi w EN 932-1.
10.3 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną według procedur wymienionych w prEN 932-2 celem otrzymania mas nie mniejszych
ni
ż podano w tablicy 1 (7.3), odpowiednio do nominalnych wymiarów kruszywa.
Wysuszy
ć podpróbkę w temperaturze (110 ± 5) °C do stałej masy (6.5).
Przesia
ć podpróbkę przez sito 16 mm (patrz 5.3.1) i przekruszyć nadziarno, tak aby przeszło przez sito bez
nadmiernego rozdrobnienia. Po
łączyć i wymieszać według procedur wymienionych w prEN 932-2. Pobrać dwie próbki
do badania ka
żda (2 ± 0,3) kg masy, w przypadku kruszyw grubych lub dwie próbki do badania, ka żda około
(500 ± 75) g, w przypadku kruszyw drobnych.
W przypadku kruszyw lekkich obie próbki badane b
ędą stanowiły około 1 l.
10.4 Przygotowanie ekstraktów
W przypadku kruszyw grubych lub lekkich zastosowa
ć dwie butle plastikowe lub metalowe o pojemno ści 5 l, a dla
kruszywa drobnego zastosowa
ć dwie butle o pojemności 2 l (5.3.2). Zważyć każdą butlę i zapisać ich masę z
dok
ładnością do 1 g.
Próbki badane, otrzymane wg 10.3, przenie
ść do butli. Zważyć butle z zawartościami i zapisać ich masy z dokładnością
do 1 g. Obliczy
ć masę kruszywa w każdej butli.
Doda
ć do każdej butli dwukrotną masę wody w stosunku do masy kruszywa. W przypadku kruszywa lekkiego doda ć 1 l
wody. Zamkn
ąć butle i wymieszać zawartość, stale potrząsając lub stosując mieszalnik rolkowy (5.3.3) przez minimum
24 h.
UWAGA: Je
żeli nie użyto mechanicznego wstrząsania i kruszywo jest tylko w kontakcie z wod ą i jest rzadko
wstrz
ąsane, występuje możliwość (szczególnie gdy złoże jest siarczanowe, na przykład duże kryształy gipsu), że sól
siarczanowa, która teoretycznie rozpu
ściła się, nie ulegnie całkowitej ekstrakcji w ciągu 24 h.
Filtrowa
ć ekstrakty przez suche, średniej twardości sączki filtracyjne (5.3.4) aż co najmniej 100 ml czystego filtratu
zgromadzi si
ę w czystych suchych zlewkach (5.2.9).
10.5 Procedura oznaczania zawarto
ści siarczanu w ekstraktach
Za pomoc
ą pipety (5.2.10) przenieść 50 ml przefiltrowanego ekstraktu do 500 ml kolby, rozcie ńczyć wodą do 300 ml,
doda
ć 10 ml roztworu kwasu chlorowodorowego (4.5.1).
Doprowadzi
ć do wrzenia i gotować przez 5 min.
UWAGA: Je
żeli kruszywo zawiera siarczki, np. żużle, po gotowaniu przez 5 min, pozostawić roztwór w ciepłym miejscu
przez 30 min. Je
żeli powstał biały osad, odsączyć go przez sączek filtracyjny średniej twardości i przemyć dokładnie
ciep
łą destylowaną wodą, odrzucając pozostałość.
Utrzymywa
ć roztwór w punkcie wrzenia energicznie mieszaj ąc, dodać kroplę po kropli 5 ml roztworu chlorku baru (4.5.2)
ogrzewanego do temperatury tu
ż poniżej wrzenia. Kontynuować gotowanie przez 15 min, tak aby nastąpiło całkowite
str
ącenie.
Pozostawi
ć tuż poniżej wrzenia przez 30 min, a następnie pozostawić na noc w ciepłym miejscu.
Wytr
ącony siarczan baru przenieść ze szczególną ostrożnością, za pomocą zasysania, do uprzednio wyprażonego i
zwa
żonego tygla ze spiekanym filtrem krzemianowym ( 5.6.1). Zastępczo można przenieść osad, ze szczególną
ostro
żnością, na miękki sączek filtracyjny w szklanym lejku i filtrowa ć. W każdym przypadku przemywać osad kilka razy
gor
ącą wodą, aż woda z płukania nie będzie zawierała chlorków (6.7).
Je
żeli użyto tygla ze spiekanym filtrem krzemianowym, zdjąć go z kolby filtracyjnej i wysuszyć w (110 ± 5) °C przez
oko
ło 30 min, stopniowo podnosząc temperaturę do (925 ± 25) °C, w elektrycznym piecu muflowym ( 5.2.2), do
osi
ągnięcia stałej masy; 15 min w tej temperaturze wystarczy.
Och
łodzić tygiel w eksykatorze (5.2.16) i zważyć z dokładnością do 0,1 mg, obliczyć masę osadu m
3
ze wzrostu masy
tygla.
Je
żeli osad jest filtrowany przez sączek filtracyjny, przenieść sączek i osad do uprzednio wyprażonego i zważonego
tygla (5.6.2). Umie
ścić tygiel z zawartością w elektrycznym piecu muflowym (5.2.2) i postępować zgodnie z procedurą
wymienion
ą w 6.6.
Obliczy
ć masę osadu m
3
ze wzrostu masy tygla (z dok
ładnością do 0,1 mg).
10.6 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość rozpuszczalnych siarczanów w kruszywie, wyrażoną jako SO
3
z nast
ępującego wzoru:
Rozpuszczalne SO
3
= 2 × W × 0,343 × m
3
(w %)
w którym:
m
3
masa osadu siarczanu baru, w gramach;
W stosunek wody do kruszywa (w gramach na gram) - dla kruszyw lekkich;
W wynosi 1000 g masy kruszywa, w gramach.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 19

11 Oznaczanie zawarto
ści siarki całkowitej
11.1 Zasada metody
Próbka analityczna kruszywa jest traktowana bromem i kwasem azotowym, aby wyst
ępujące składniki siarki związać w
siarczany. Siarczany s
ą strącane w postaci BaSO
4
i wa
żone. Zawartość siarki jest wyrażana jako procent masy
kruszywa.
11.2 Pobieranie próbek
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, by próbka laboratoryjna miała zawartość wilgotności taką samą jak cała partia kruszywa.
11.3 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną, według procedur wymienionych w prEN 932-2, do wielko ści nie mniejszych niż masa
wymieniona w tablicy 1, przewidziana dla nominalnej wielko
ści kruszywa.
Je
żeli potrzeba, suszyć próbkę w temperaturze nie przekraczającej (110 ± 5) °C, aby uniknąć utlenienia siarczków.
Rozdrobni
ć i zmniejszyć podpróbkę do masy około 20 g, rozdrobnić tę próbkę tak, aby przeszła przez sito 125
µ
m.
Pobra
ć około 1 g tego materiału jako próbkę analityczną.
11.4 Procedura oznaczania
Zwa
żyć próbkę analityczną z dokładnością do 0,1 mg (m
4
) i przenie
ść ją do kolby Erlenmeyera z (5.12.1) szeroką
szyjk
ą, o którą opiera się lejek z krótkim trzonem. Dodać do kolby (5.2.17) 3 ml wody i 1 ml bromu (4.6.1) pod
dygestorium i ostro
żnie mieszać mieszaninę przez 1 min, aby zapobiec nadmiernemu tworzeniu si ę grudek. Następnie
powoli doda
ć, przez lejek 15 ml, stężonego kwasu azotowego nie zawierającego jonów siarczanowych (4.1). Umieścić
mieszanin
ę na łaźni parowej na 1 h i rozbijać żel co pewien czas za pomocą płaskiego pręta szklanego (pozostawiając
na sta
łe pręt w kolbie). Dodać 30 ml wody i ostrożnie gotować mieszaninę na gorącej płycie (5.2.7) do czasu aż
przestan
ą wydzielać się gęste brunatne opary. Dodać 5 ml stężonego kwasu chlorowodorowego ( 4.1) i 10 ml wody i
odparowa
ć mieszaninę do małej objętości. Powtórzyć dodawanie i odparować do małej objętości. Przenieść zawartość
kolby do 250 ml zlewki (5.2.9) i przemy
ć kolbę, aż całkowita objętość w zlewce wyniesie około 100 ml.
Doda
ć trochę miazgi sączkowej, doprowadzić zawartość zlewki prawie do wrzenia, zwiększyć alkaliczność dodając
amoniak, sprawdzi
ć alkaliczność używając jako wskaźnika czerwieni metylowej (4.6.2) lub używając pH-metru (5.2.8).
Gotowa
ć na wolnym ogniu przez 30 s, filtrować pod łagodną próżnią (używając sączka filtracyjnego średniej
porowato
ści), jednokrotnie przemyć niewielką ilością ciepłej destylowanej wody, zachować przesącz. Przenieść bibułę
filtracyjn
ą do zlewki, ponownie rozpuścić ją w 5 ml stężonym kwasie chlorowodorowym, do którego dodano 70 ml
gor
ącej wody.
Post
ępując jak wyżej, gotować, wytrącać osad, filtrować i przepłukiwać wodą, odrzucając osad. Zakwasić całość
przes
ączu i wody z płukania (których powinno być ogółem 220 ml) 1 ml stężonego kwasu chlorowodorowego,
doprowadzi
ć do wrzenia i gotować przez 5 min. Gdy roztwór osiągnie punkt wrzenia, mieszając energicznie dodać
kroplami 10 ml roztworu chlorku baru ( 4.5.2) ogrzanego do temperatury poni
żej wrzenia.
Dojrzewanie, filtrowanie i spalanie siarczanu baru jak podano w 10.5.
Zwa
żyć z dokładnością do 0,1 mg i obliczyć masę osadu m
5
.
11.5 Obliczanie i przedstawienie wyników
Obliczy
ć całkowitą zawartość siarki w kruszywie, wyrażoną jako S z następującego wzoru:
S = m
5
/m
4
× 13,74 (w %)
w którym:
m
5
masa osadu, w gramach;
m
4
masa próbki analitycznej, w gramach.
UWAGA: Ustalenia dotycz
ące dokładności oznaczania zawartości siarki całkowitej podano w załączniku A.
12 Oznaczanie siarczanów rozpuszczalnych w kwasie
12.1 Zasada metody
Siarczany ekstrahowane z próbki analitycznej kruszywa za pomoc
ą rozcieńczonego kwasu chlorowodorowego są
oznaczane wagowo. Jon siarczanowy jest wyra
żony jako procent masy kruszywa.
12.2 Pobieranie próbek
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, aby próbka laboratoryjna miała taką samą wilgotność jak cała masa kruszywa.
12.3 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną, według procedur wymienionych w prEN 932-2, do wielko ści nie mniejszej niż masa
wymieniona w tablicy 1 przewidziana dla nominalnej wielko
ści kruszywa. Rozkruszyć i stopniowo pomniejszyć
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 20

podpróbk
ę. Następnie rozetrzeć i dalej zmniejszać aż masa około 20 g przejdzie przez sito badawcze 0,125 mm.
Pobra
ć około 2 g materiału jako próbkę analityczną.
Je
żeli konieczne jest suszenie, temperatura nie powinna przekracza ć (110 ± 5) °C, aby nie dopuścić do utlenienia
siarczków.
12.4 Metoda badania
Zwa
żyć próbkę analityczną z dokładnością do 0,1 mg (m
6
), umie
ścić w 250 ml zlewce i dodać 90 ml zimnej wody.
Mieszaj
ąc energicznie dodać 10 ml stężonego kwasu chlorowodorowego. Delikatnie podgrza ć roztwór i rozbić grudki za
pomoc
ą płaskiego pręta szklanego. Pozostawić roztwór na 15 min w temperaturze tuż poniżej wrzenia.
UWAGA: Kruszywa zawieraj
ące znaczne ilości węglanów będą burzyć się po dodaniu kwasu. W tych przypadkach
dodawa
ć kwas powoli, ciągle mieszając. Kruszywa zawierające siarczki będą uwalniać H
2
S przy zakwaszeniu, co
b
ędzie zauważalne po zapachu. W tych przypadkach występuje niebezpieczeństwo, że metoda doprowadzi do
przeszacowania zawarto
ści siarczanów z powodu utleniania siarczków. Aby zapobiec utlenianiu, umie ścić w 250 ml
zlewce 90 ml wody i 10 ml st
ężonego kwasu chlorowodorowego i ogrzać do punktu wrzenia. Zdjąć ze źródła ciepła i
mieszaj
ąc rozprowadzić próbkę analityczną w roztworze kwasu.
Przefiltrowa
ć pozostałość przez sączek filtracyjny średniej twardości do 400 ml zlewki. Przemyć gorącą wodą.
Sprawdzi
ć przesącz po płukaniu na obecność jonów chlorkowych badając azotanem srebra (6.7).
Uzupe
łnić objętość do około 250 ml i, jeżeli to konieczne zakwasić kwasem chlorowodorowym (1 + 11) do czerwonego
koloru wska
źnika czerwieni metylowej (4.6.2).
Doprowadzi
ć do wrzenia i gotować przez 5 min. Sprawdzić, czy roztwór jest klarowny, jeżeli nie, rozpocząć badanie od
nowa z now
ą próbką analityczną. Intensywnie mieszając, doprowadzić roztwór do punktu wrzenia, dodać kroplami
10 ml roztworu chlorku baru (4.5.2) ogrzanego do temperatury tu
ż poniżej wrzenia.
Dojrzewanie, filtrowanie i spalanie siarczanu baru jak podano w 10.5.
Zwa
żyć z dokładnością do 0,1 mg i obliczyć masę osadu (m
7
).
12.5 Obliczanie i przedstawienie wyników
Obliczy
ć w kruszywie zawartość siarczanów rozpuszczalnych w kwasie, wyrażoną jako SO
3
z nast
ępującego wzoru:
Zawarto
ść siarczanu = m
7
/m
6
× 34,30 (w %)
w którym:
m
7
masa osadu, w gramach;
m
6
masa próbki analitycznej, w gramach.
UWAGA: Ustalenia dotycz
ące dokładności oznaczania zawartości siarczanów rozpuszczalnych w kwasie podano w
za
łączniku A.
13 Oznaczanie siarczków rozpuszczalnych w kwasie
13.1 Zasada metody
Próbka analityczna jest rozk
ładana pod wpływem kwasu chlorowodorowego w warunkach redukcyjnych. Siarczki s ą
zamieniane w siarkowodór, który jest przenoszony przez strumie
ń gazów do amoniakalnego roztworu siarczanu cynku.
Str
ącony siarczek cynku oznacza się metodą jodometrii.
13.2 Pobieranie próbki
Próbk
ę laboratoryjną należy pobrać zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, aby próbka laboratoryjna miała taką samą wilgotność, jak cała masa kruszywa.
13.3 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną według procedur wymienionych w prEN 932-2 do wielko ści nie mniejszej niż masa
wymieniona w tablicy 1, przewidziana do nominalnej wielko
ści kruszywa. Rozkruszyć i stopniowo pomniejszyć
podpróbk
ę. Kontynuować rozdrabnianie i pomniejszanie, aż masa około 20 g przejdzie przez sito badawcze 0,125 mm.
Pobra
ć około 1 g tego materiału jako próbkę analityczną.
Je
żeli potrzebne jest suszenie, temperatura nie powinna przekracza ć (110 ± 5) °C, aby uniknąć utleniania siarczków.
13.4 Metoda badania
U
żyć aparatury opisanej w 5.7.1 i pokazanej na rysunku 1. Zważyć próbkę analityczną z dokładnością do 0,1 mg (m
8
) i
przenie
ść do 250 ml kolby okrągłodennej, zamykanej korkiem ze szlifem.
UWAGA 1: Je
żeli zawartość siarczków jest mała (< 0,1 %), zaleca się użycie roztworów odczynników o
dziesi
ęciokrotnym rozcieńczeniu (4.7.5 i 4.7.6).
Doda
ć około 2,5 g chlorku cyny (II) (4.7.3) i 0,1 g chromu (4.7.4).
UWAGA 2: Chrom bierze udzia
ł w rozkładzie pirytu (FeS
2
), który mo
że być w kruszywie.
Rozprowadzi
ć mieszaninę w 50 ml wody. Zamocować do szyjki kolby uchwyt podtrzymujący lejek i połączyć szyjkę do
wlotu ch
łodnicy, połączyć wylot chłodnicy do szklanej rurki, która zanurza si ę w kolbie zawierającej 15 ml
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 21
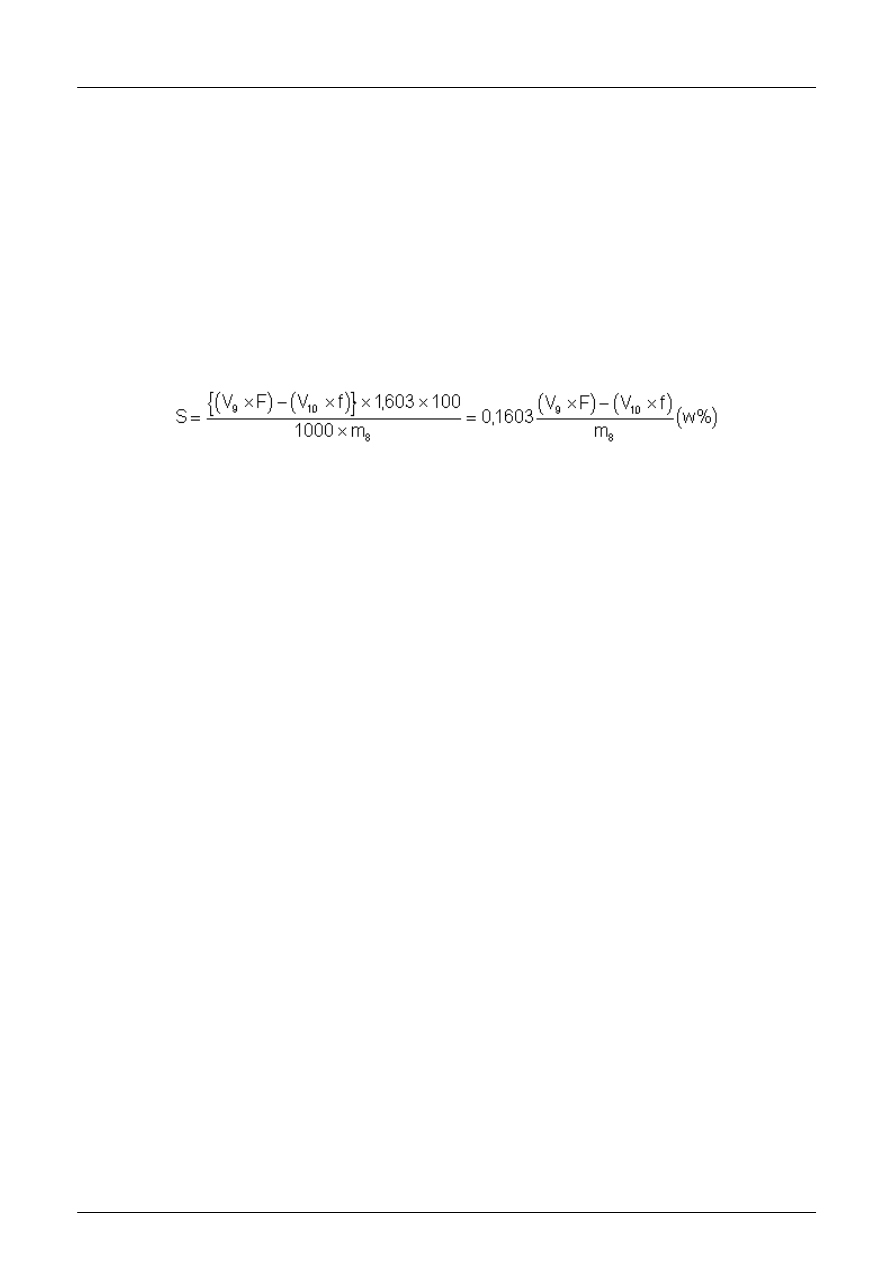
amoniakalnego roztworu siarczanu cynku ( 4.7.2) i 285 ml wody. Pod
łączyć gaz (azot lub argon) i uregulować przepływ
do oko
ło 10 ml na minutę. Zatrzymać przepływ gazu. Spuścić z oddzielnego lejka 50 ml kwasu chlorowodorowego
(1 + 1), uwa
żając aby mała ilość kwasu pozostała w oddzielnym lejku, zapobiegając przeciekaniu. Ponownie włączyć
urz
ądzenie gazowe, ogrzewać zawartość kolby do zagotowania i gotować przez 10 min. Odłączyć wylot, który będzie
s
łużyć jako mieszalnik podczas miareczkowania.
UWAGA 3: Niektóre kruszywa o podwy
ższonej zawartości siarczków mogą potrzebować więcej czasu niż 10 min dla
ca
łkowitego przejścia wszystkich siarczków w osad siarczku cynku. Sprawdzi ć, czy ekstrakcja jest zakończona przez
zanurzenie wylotu w
świeżym roztworze amoniakalnego siarczanu cynku. Je żeli nie wystąpi wytrącanie, ekstrakcja jest
zako
ńczona.
Och
łodzić zawartość do 20 °C i dodać pipetą 0,0166 mol/l roztworu jodanu potasu ( 4.7.5) i 25 ml stężonego kwasu
chlorowodorowego. Miareczkowa
ć roztworem tiosiarczanu sodu (4.7.6) do koloru jasnożółtego. Następnie dodać 2 ml
roztworu skrobi (4.7.7) i miareczkowa
ć do zmiany koloru z niebieskiego na bezbarwny.
13.5 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość siarczku w kruszywie wyrażonego jako S, z następującego wzoru:
w którym:
V
9
obj
ętość roztworu jodanu potasu w mililitrach;
F miano roztworu jodanu potasu wymienionego w 4.7.5;
V
10
obj
ętość roztworu tiosiarczanu sodu użytego do miareczkowania, w mililitrach;
f st
ężenie roztworu tiosiarczanu sodu wymienionego w 4.7.6;
m
8
masa próbki analitycznej, w gramach.
14 Oznaczanie sk
ładników wpływających na jakość powierzchni betonu
14.1 Sprawdzenie obecno
ści cząstek reaktywnego siarczku żelaza
14.1.1 Postanowienia ogólne
W niniejszym rozdziale wyszczególnia si
ę metody wykrywania cząstek siarczku żelaza, które występując na, lub blisko,
powierzchni betonu, mog
ą być przyczyną brunatnych plam. Plamy te mogą być niemożliwe do usunięcia inaczej jak
przez wyci
ęcie.
14.1.2 Pobieranie próbek
Pryzma powinna by
ć sprawdzona i około 50 ziarn powinno być pobrane do badania, w którym spodziewana jest
obecno
ść siarczku żelaza.
14.1.3 Metoda badania
Oznacza
ć reaktywność ziarn, umieszczając je w nasyconym roztworze wodorotlenku wapnia.
UWAGA: Jasnozielony galaretowaty osad siarczanu
żelaza (II) ukształtuje się w czasie 5 min. Osad po wystawieniu na
powietrze i
światło zmienia się szybko w brązowy wodorotlenek żelaza (II). Reakcja ta zakończy się w ciągu 30 min i
wskazuje na obecno
ść szybko reagującego siarczku żelaza.
Je
żeli nie powstał brązowy galaretowaty osad, gdy ziarna zostały umieszczone w nasyconej wodzie wapiennej, mog ą
by
ć one ziarnami wolno reagującymi. Jeżeli spodziewana jest obecność wolno reagujących ziarn należy przeprowadzić
nast
ępującą procedurę.
Sprawdzi
ć ziarna wizualnie i oszacować, na ile to jest możliwe, ich skłonność do barwienia w zaprawie lub betonie.
UWAGA: Je
śli dany zakład w przeszłości sporadycznie produkował kruszywo zawierające siarczek żelaza, sprawdzenie
przez technologa z do
świadczeniem, z użyciem mikroskopu o małym powiększeniu może wystarczyć do określenia, czy
materia
ł jest lub nie jest szkodliwy.
W innych przypadkach zmiesza
ć wybrane ziarna w zaczynie z cementu portlandzkiego, przechowywa ć przez 28 dni w
wilgotnych warunkach, a nast
ępnie sprawdzić powierzchnię cementu pod kątem barwienia.
14.2 Oznaczanie zanieczyszcze
ń lekkich
14.2.1 Postanowienia ogólne
Niniejsze badanie jest metod
ą oszacowania masy ziaren lekkich w kruszywach drobnych, w procentach. Metoda
okre
śla zawartość substancji takich jak lignit i w ęgiel, które mogą być przyczyną barwienia lub odprysków na
powierzchni betonu lub zaprawy. Je
żeli zachodzi potrzeba, metoda może być zaadaptowana do zastosowania do
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 22

kruszyw grubych poprzez badanie wi
ększych próbek analitycznych (patrz tablica 1).
14.2.2 Zasada metody
Nieorganiczne minera
ły, z których składają się kruszywa drobne dostępne w handlu, do betonu i zaprawy, na ogół mają
g
ęstość objętościową powyżej 2,0. Przy zanurzeniu próbki analitycznej kruszyw drobnych w cieczy g ęstości tuż poniżej
2,0, ziarna o ni
ższej gęstości wypłyną na powierzchnię cieczy, ułatwiając ich usunięcie w celu sprawdzenia ilości i
kwalifikacj
ę.
UWAGA: Ten proces flotacyjny jest nieprzydatny dla kruszyw lekkich, oznaczanie zanieczyszcze
ń lekkich w
kruszywach lekkich zaleca si
ę wykonywać ręcznie.
14.2.3 Metoda badania
Minimalna wielko
ść próbki laboratoryjnej kruszywa drobnego powinna wynosi ć 5 kg, którą poprzez kwartowanie należy
zmniejszy
ć do próbki analitycznej o masie (350 ± 50) g.
Rozsypa
ć próbkę analityczną na tacę (5.2.1) i przenieść do suszarki o temperaturze (110 ± 5) °C (6.4 i 6.5). Zapisać
mas
ę wysuszonego piasku m
9
z dok
ładnością do 0,1 g. Rozdzielić kruszywo na sicie 300
µ
m (5.8.1) i odrzuci
ć frakcję
drobniejsz
ą.
Wla
ć około 1 l roztworu chlorku cynku (4.8.1) lub poliwolframianu sodu ( 4.8.2) do 2 l zlewki, a następnie wsypać
kruszywo do roztworu.
Łagodnie mieszać szklanym prętem po dnie kruszywa, aby ziarna lekkie mog ły wypłynąć na
powierzchni
ę roztworu. Łagodnie mieszać pływające ziarna za pomocą szklanego pręta celem usunięcia pęcherzyków
powietrza z p
ływających ziarn kruszywa i pozwolić tym ziarnom opaść na dno.
Dekantowa
ć roztwór znad powierzchni do drugiej 2 l kolby, przepuszczaj ąc ciecz przez sito 250
µ
m (5.8.1), pozwalaj
ąc
p
ływającym ziarnom spłynąć, aby zebrać je z sita. Upewnić się, że żadne ziarno nie przeszło na sito. Przelać roztwór do
pierwszej zlewki i ponownie wymiesza
ć kruszywo. Jeżeli następne ziarna wypłyną na wierzch, ponownie dekantować
przez sito i powtarza
ć proces do czasu aż wszystkie pływające ziarna zostaną zebrane na sicie.
Przemy
ć wodą sito i ziarna na nim do całkowitego usunięcia chlorku cynku lub poliwolframianu sodu. Suszy ć sito i
zawarto
ść przez (20 ± 4) h w (40 ± 5) °C a następnie zawartość przesypać do parownicy (5.8.2) i dosuszyć w
(110 ± 5) °C przez (4 ± 0,25) h.
Ostudzi
ć parownicę i zważyć ziarna lekkie (m
10
) z dok
ładnością do 0,1 g.
14.2.4 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość ziarn lekkich w kruszywie, w procentach, wyra żoną jako m
LPC
, z nast
ępującego wzoru:
m
LPC
= m
10
/m
9
× 100 (w %)
w którym:
m
9
masa wysuszonej w suszarce próbki analitycznej, w gramach;
m
10
masa wysuszonych w suszarce ziarn lekkich, wydzielonych z próbki analitycznej, w gramach.
Wynik poda
ć z dokładnością do 0,1 %.
15 Oznaczanie sk
ładników organicznych wpływających na wiązanie i twardnienie cementu
15.1 Oznaczanie zawarto
ści humusu
15.1.1 Zasada metody
Humus jest substancj
ą organiczną, która tworzy się w gruncie w wyniku rozkładu pozostałości zwierząt i roślin.
Zawarto
ść humusu jest oceniana na podstawie koloru roztworu wodorotlenku sodu, w którym próbka analityczna jest
wstrz
ąsana.
UWAGA: Metoda jest oparta na tym,
że humus reagując z NaOH wytwarza ciemny kolor. Intensywność koloru zależy
od zawarto
ści humusu. Jeżeli roztwór nie jest zabarwiony albo jest s łabo zabarwiony, kruszywo nie zawiera znacznej
ilo
ści humusu. Intensywny kolor roztworu po reakcji przewa żnie wynika z dużej zawartości humusu, ale może także być
spowodowany innymi przyczynami. Dlatego w tym przypadku metoda ta nie daje definitywnej odpowiedzi.
15.1.2 Pobieranie próbki
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, aby próbka laboratoryjna miała taką samą wilgotność jak cała masa kruszywa.
15.1.3 Przygotowanie próbki analitycznej
Wysuszy
ć podpróbkę (7.3) rozsypaną na tacy w suszarce (5.2.1) w (55 ± 5) °C a nie w (110 ± 5) °C. Przesiać próbkę
na sicie 4 mm (5.9.1) i zatrzyma
ć frakcję pozostającą na sicie. Pokruszyć frakcję pozostającą na sicie do mniejszej niż
4 mm i po
łączyć z materiałem przechodzącym przez 4 mm.
15.1.4 Metoda badania
Wla
ć 3 % roztwór NaOH (4.9.1) do szklanej butelki (5.9.2) do wysokości 80 mm. Następnie wsypać próbkę analityczną
tak, aby wysoko
ść kruszywa i roztworu wynosiła 120 mm. Wstrząsnąć butlą, aby uwolnić pęcherzyki powietrza.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 23

Zamkn
ąć butlę i energicznie wstrząsać przez 1 min a następnie odstawić. Po 24 h porównać kolor roztworu z kolorem
roztworu wzorcowego (4.9.2) umieszczonego w podobnej butli.
15.1.5 Wyra
żenie wyników
Wynik badania powinien okre
ślić, czy kolor roztworu jest jaśniejszy, czy ciemniejszy niż kolor roztworu wzorcowego
(4.9.2).
15.2 Oznaczanie zawarto
ści kwasu fulvo
15.2.1 Postanowienia ogólne
W tym rozdziale podano metod
ę oznaczania zawartości kwasu fulvo, który może występować w kruszywach drobnych,
szczególnie w glebach piaszczystych stabilizowanych cementem.
15.2.2 Zasada metody
Kwasy fulvo s
ą składnikami kwasów humusowych, które maj ą wpływ na opóźnienie hydratacji cementów. Kwasy fulvo
rozpuszczone w kwasie chlorowodorowym wywo
łują żółte zabarwienie. Wzrost stężenia kwasu fulvo wywołuje wzrost
intensywno
ści koloru. Składniki zawierające Fe(III) wywołują brązowy kolor kwasu chlorowodorowego. Ten kolor jest
eliminowany przez przemian
ę składników Fe(III) w bezbarwne składniki Fe(II) po zastosowaniu roztworu chlorku cyny(II).
15.2.3 Pobieranie próbek
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, aby próbka laboratoryjna reprezentowała zawartość wilgoci w całej masie kruszywa.
15.2.4 Przygotowanie próbki analitycznej
Pomniejszy
ć próbkę laboratoryjną do próbki analitycznej (100 ± 0,5) g według procedur wymienionych w prEN 932-2.
Zawarto
ść wilgoci w próbce analitycznej nie powinna przekracza ć 10 % masy próbki. Próbki analityczne o wilgotno ści
wi
ększej niż 10 % powinny być wysuszone w (40 ± 5) °C do wilgotności 10 % lub mniejszej.
15.2.5 Metoda badania
Badanie przeprowadza si
ę w temperaturze (20 ± 2) °C. Próbkę analityczną umieścić w 250 ml lub 300 ml kolbie
Erlenmeyera (5.2.9). Doda
ć 100 ml kwasu chlorowodorowego (4.10.1). Odstawić kolbę z zawartością na 4 h,
potrz
ąsając od czasu do czasu. Z kolby przenieść 75 ml roztworu filtratu do 250 ml cylindra miarowego (5.2.11).
U
żywając 10 ml cylindra miarowego ( 5.2.11) dodać 10 ml sklarowanego roztworu chlorku cyny(II) ( 4.10.2). Odstawić
250 ml cylinder miarowy z zawarto
ścią na kolejną 1 h, a następnie po tym czasie uzupełnić do 100 ml kwasem
chlorowodorowym (4.10.1). Wymiesza
ć zawartość w cylindrze z użyciem pręta (5.10.1).
Je
żeli po dodaniu chlorku cyny(II) roztwór zmętnieje, siarczki są obecne, powinno się badanie powtórzyć gotując przez
5 min na gor
ącej płycie (5.10.4) przed dodaniem chlorku cyny(II).
15.2.6 Wyra
żenie wyników
Okre
ślić zawartość kwasu fulvo przez wybranie płytki o wzorcowym kolorze takim jak kolor roztworu ( 5.10.5). W
tablicy 2 podano poziomy dopuszczalne zawarto
ści kwasu fulvo przez porównanie z płytką we wzorcowym kolorze.
Tablica 2: Dopuszczalne zawarto
ści kwasu fulvo
Kolor p
łytki
(5.10.5)
Przydatno
ść piasku do betonu
Przydatno
ść do
stabilizacji gruntu
Wytrzyma
łość po 3 dniach
Wytrzyma
łość po 28 dniach
A
B
C
D
E
F
G
nie wp
ływa
nie wp
ływa
umiarkowane obni
żenie
umiarkowane obni
żenie
silny spadek
silny spadek
silny spadek
nie wp
ływa
nie wp
ływa
nie wp
ływa
nie wp
ływa
nie wp
ływa
umiarkowane obni
żenie
silny spadek
dobra
dobra
umiarkowanie dobra
umiarkowana
umiarkowanie z
ła
z
ła
z
ła
15.3 Oznaczanie zanieczyszcze
ń organicznych metodą zaprawy
15.3.1 Zasada metody
Metoda zaprawy jest badaniem przeznaczonym do przedstawienia i okre
ślenia wpływu na wiązanie i twardnienie
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 24

betonu, jaki mog
ą wywołać zanieczyszczenia organiczne w kruszywie. Metoda polega na wykonaniu dwóch nominalnie
identycznych zapraw i badaniu ich w
łasności wiążących i wytrzymałości na ściskanie. Jedna zaprawa zawiera kruszywo
w stanie naturalnym, podczas gdy druga mieszanka jest wykonana z drugiej próbki analitycznej, która by
ła prażona
celem zniszczenia cz
ęści organicznych. Prażone kruszywo traktuje się jako porównawcze w stosunku do kruszywa
oryginalnego. Badanie okre
śla przyspieszenie lub opóźnienie czasu wiązania zaprawy, podczas gdy 28-dniowa
wytrzyma
łość wskazuje na efekt późniejszy.
15.3.2 Pobieranie próbek
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1. Minimalna ilo ść
kruszywa w próbce laboratoryjnej powinna wynosi
ć 15 kg, to znaczy więcej niż podano w tablicy 1.
15.3.3 Przygotowanie próbki analitycznej
Wysuszy
ć próbkę laboratoryjną, rozkładając ją na tacach i pozostawić do wyschnięcia w temperaturze laboratoryjnej.
Nast
ępnie według procedur wymienionych w prEN 932-2 pomniejszy ć wysuszoną próbkę laboratoryjną i przygotować
próbki analityczne, ka
żdą po (1900 ± 100) g.
W czasie pomniejszania wysuszonych próbek laboratoryjnych w celu otrzymania próbek analitycznych do metody
zaprawy, zaleca si
ę utworzyć dwie próbki analityczne, każda o masie mniejszej niż 1,8 kg, następnie jedna z próbek
analitycznych powinna by
ć podzielona jedno-, dwu- albo, jeśli potrzeba, trzykrotnie dla uzyskania podpróbki, która po
dodaniu do pierwszej próbki analitycznej utworzy
łączną masę nie większą niż 2 kg.
UWAGA: W przypadku kruszyw lekkich ka
żda próbka analityczna zawiera 1 l kruszywa.
15.3.4 Post
ępowanie z kruszywem
Pozostawi
ć dwie z czterech próbek analitycznych otrzymanych zgodnie z 15.3.3 w ich stanie naturalnym, a dwie
pozosta
łe poddać następującej procedurze:
(a) Umie
ścić próbkę analityczną w zważonej porcelanowej lub kwarcowej parownicy (5.11.2), zważyć i umieścić w piecu
muflowym (5.1.7) o temperaturze otoczenia.
UWAGA: Je
śli tylko mały piec jest dostępny, każda próbka analityczna może być podzielona na dwie lub więcej części,
które s
ą zważone, wyprażone, ponownie oddzielnie zważone, a następnie połączone, gdy ostygną.
(b) Zwi
ększyć temperaturę pieca do (480 ± 25) °C w ciągu (4 ± 0,25) h.
Utrzymywa
ć temperaturę (480 ± 25) °C przez (4 ± 0,25) h i pozwolić ostygnąć piecowi przez noc. Zważyć parownicę i
kruszywo w temperaturze otoczenia i zapisa
ć ubytek masy.
Post
ąpić z drugą próbką analityczną w ten sam sposób.
15.3.5 Sk
ładniki
Cement powinien by
ć CEM I, zgodnie z wymaganiami EN 197-1. Powinien być przechowywany w hermetycznym
pojemniku.
15.3.6 Liczba mieszanek
15.3.6.1 Ogólne wymagania i próbne mieszanki
Ka
żda mieszanka zaprawy powinna zawierać albo próbkę analityczną kruszywa uprzednio nie wyprażonego, albo
próbk
ę analityczną kruszywa wyprażonego wg 15.3.4. Każda mieszanka zaprawy powinna także zawierać cement
CEM I w ilo
ści czwartej części masy kruszywa zawartego w mieszance. Cement nale ży zważyć z dokładnością do ± 1 g.
Upewni
ć się, że woda zawarta w zaprawach zawierających kruszywo nie wyprażone zapewnia normową konsystencję,
okre
śloną średnim zagłębieniem stożka pomiarowego (5.11.3) (23 ± 0,5) mm według metody podanej w EN 1015-4.
W przypadku kruszyw lekkich u
żyć 300 g cementu do każdej mieszanki zaprawy i 30 g do każdego badania wstępnego.
Dla ustalenia niezb
ędnej zawartości wody przygotować serię próbnych mieszanek, używając kruszyw nie wyprażonych,
zmieniaj
ąc sukcesywnie zawartość wody i mierząc konsystencję każdej, aż zostanie osiągnięta prawidłowa wartość
konsystencji. Zanotowa
ć masę wody zawartej w ostatniej mieszance i obliczy ć stosunek wodno-cementowy mieszanki.
Odrzuci
ć próbne mieszanki.
Podczas wa
żenia przed prażeniem kruszywo nie wyprażone ma takie same warunki wilgotno ściowe jak kruszywo
kontrolne. Dlatego nale
ży wykonać próbne mieszanki i przygotować próbki do badania zaprawy z badanego kruszywa w
tym samym dniu, gdy rozpoczyna si
ę wyprażanie kontrolnego kruszywa.
UWAGA: Kontrolne mieszanki b
ędą tym sposobem przygotowane jeden dzień po mieszankach badanych, zaleca si ę
aby warunki laboratoryjne by
ły podobne, na ile to jest możliwe, podczas obu dni przygotowywania mieszanek.
15.3.6.2 Mieszanki badane
Maj
ąc obliczoną masę cementu właściwą dla każdej próbki analitycznej kruszywa nie wyprażonego, ze stosunku
wodno-cementowego uzyskanego w 15.3.6.1 obliczy
ć ilość wody niezbędnej dla każdej mieszanki i odważyć ją z
dok
ładnością do ± 0,5 g.
15.3.6.3 Mieszanki kontrolne
Upewni
ć się, że stosunek wodno-cementowy zapraw kontrolnych z wypra żonego kruszywa jest taki sam, jak ten dla
zapraw z kruszywem nie wypra
żonym, patrz wstępne obliczenia wymaganej masy wody dla ka żdej mieszanki wg
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 25

15.3.6.2. Nast
ępnie dodać do każdej obliczonej masy ubytek masy przez analogiczn ą porcję kruszywa podczas
wypra
żania podanego w 15.3.4 (c).
Zwa
żyć całkowitą ilość wody z dokładnością do ± 0,5 g dla każdej mieszanki.
15.3.7 Mieszanie
Potrzebne s
ą cztery mieszanki; dwie wykonane z otrzymanego kruszywa wysuszonego na powietrzu i dwie z kruszywa
wypra
żonego. Doprowadzić temperaturę wszystkich materiałów do (20 ± 2) °C przed rozpoczęciem mieszania zaprawy.
Miesza
ć w pomieszczeniu lub innym kontrolowanym środowisku mającym temperaturę (20 ± 2) °C i wilgotność
wzgl
ędną nie mniejszą niż 50 %.
Umie
ścić całe kruszywo, a następnie cement, w suchej mieszarce ( 5.11.4) i mieszać przez 30 s. Kontynuować
mieszanie i dodawa
ć wody przez następne 30 s. Kontynuować mieszanie przez 60 s po dodaniu całkowitej ilości wody.
Zatrzyma
ć mieszarkę i oczyścić z materiału przyklejonego do mieszadła i boków mieszarki za pomocą skrobaka do
misy, zwróci
ć szczególną uwagę na to, czy na dnie misy nie ma nie wymieszanych materia łów. Czynności te zakończyć
w ci
ągu 60 s. Nakryć misę wilgotną ścierką i odczekać 5 min.
Umie
ścić misę w mieszarce i mieszać zaprawę przez dalsze 60 s.
15.3.8 Pomiar czasu wi
ązania
Niezw
łocznie po ukończeniu mieszania każdej zaprawy oznaczać czas wiązania zgodnie z EN 1015-9. Zapisać czasy
wi
ązania podwójnych próbek analitycznych wyprażonych i nie wyprażonych kruszyw.
15.3.9 Wytrzyma
łość na ściskanie stwardniałej zaprawy
Przygotowa
ć z każdej zaprawy trzy beleczki o wymiarach 160 mm × 40 mm × 40 mm wed ług procedury podanej w
EN 1015-11. Zbada
ć wytrzymałość beleczek na ściskanie po 28 dniach. Zapisa ć wszystkie 12 wytrzymałości na
ściskanie podwójnych próbek analitycznych wypra żonych i nie wyprażonych kruszyw. Oznaczać gęstość każdej
beleczki po rozformowaniu.
15.3.10 Obliczanie i przedstawienie wyników
15.3.10.1 Czas wi
ązania
Obliczy
ć, z dokładnością do 15 min, zmianę w czasie wiązania przez odjęcie średniego czasu wiązania zaprawy z
kruszywa wypra
żonego od średniego czasu wiązania zaprawy z kruszywa nie wyprażonego.
UWAGA: Wyniki negatywne wskazuj
ą, że zanieczyszczenia przyspieszają wiązanie zaprawy
15.3.10.2 Wytrzyma
łość na ściskanie
Obliczy
ć, z dokładnością do 1 %, względną wytrzymałość na ściskanie S % zaprawy z nie wyprażonego kruszywa z
nast
ępującego równania:
S = A/B × 100 %
w którym:
A
średnia wytrzymałość na ściskanie 6 beleczek z kruszywa nie wyprażonego, w niutonach na milimetr kwadratowy;
B
średnia wytrzymałość na ściskanie 6 beleczek z kruszywa wyprażonego, w niutonach na milimetr kwadratowy.
UWAGA: Zanieczyszczenia organiczne mog
ą w mieszankach zaprawy powodować napowietrzanie lub odpowietrzanie.
Napowietrzanie wp
ływa na wytrzymałość na ściskanie, niezależnie od efektu chemicznego, którego zanieczyszczenia
organiczne mog
ą mieć wpływ na przebieg hydratacji cementu. Obecność wprowadzonego powietrza może być
wyznaczona przez zwa
żenie beleczek zaprawy z kruszywa nie wyprażonego, które powinny być lżejsze od beleczek z
zaprawy z kruszywa wypra
żonego.
15.3.11 Sprawozdanie z badania
Sprawozdanie z badania powinno zapewni
ć, że badania zanieczyszczeń wpływających na wiązanie i twardnienie
zapraw z cementu CEM I by
ły przeprowadzone zgodnie z wymaganiami EN 196-3 i poda ć, jeśli jest to możliwe, protokół
z pobrania próbki. Je
żeli jest osiągalny, kopię certyfikatu z pobrania próbki należy załączyć. Sprawozdanie z badania
powinno zawiera
ć, tam gdzie to możliwe, następujące dodatkowe informacje:
a) dane identyfikuj
ące próbkę;
b) zmian
ę czasu wiązania i względną wytrzymałość zaprawy z kruszywa nie wyprażonego.
16 Oznaczanie rozpuszczalno
ści w wodzie
16.1 Zasada metody
Próbk
ę analityczną kruszywa ekstrahuje się z dwukrotnie większą ilością wody w stosunku do masy próbki analitycznej,
zgodnie z procedur
ą wymienioną w 10.4. Po ekstrakcji kruszywo jest suszone i wa żone.
16.2 Pobieranie próbek
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
Zapewni
ć, aby próbka laboratoryjna miała taką wilgotność jak cała masa kruszywa.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 26

16.3 Przygotowanie próbki analitycznej
Post
ępować jak podano w 7.3. Zważyć wysuszoną próbkę analityczną z dokładnością do 0,1 g (m
11
).
W przypadku wype
łniaczy, zmniejszyć próbkę analityczną do (10 ± 0,2) g i zważyć z dokładnością do 0,1 g.
16.4 Ekstrahowanie sk
ładników rozpuszczalnych
Po 24 h ekstrahowania, jak podano w 10.4, pozostawi
ć do opadnięcia głównej części ciał stałych. Odfiltrować
maksymaln
ą ilość sklarowanej cieczy przez wstępnie zważony sączek filtracyjny średniej twardości (5.3.4). Przenieść
ilo
ściowo kruszywo za pomocą małej ilości wody do wstępnie zważonej porcelanowej parownicy ( 5.8.2). Dodać sączek
filtracyjny z zatrzymanymi ziarnami do zawarto
ści parownicy. Wysuszyć i ostudzić według procedur wymienionych w 6.4
i 6.5. Zwa
żyć z dokładnością do 0,1 g i obliczyć masę kruszywa przez odjęcie mas parownicy i sączka filtracyjnego
(m
12
).
UWAGA: W przypadku wype
łniaczy zaleca się użyć szklanych butli, umożliwiających dostateczne mieszanie,
zapobiegaj
ące sedymentacji.
16.5 Obliczanie i przedstawienie wyników
Obliczy
ć rozpuszczalność kruszywa w wodzie z następującego wzoru:
w którym:
m
11
masa kruszywa przed ekstrakcj
ą, w gramach;
m
12
masa kruszywa po ekstrakcji, w gramach.
Zapisa
ć wynik z dokładnością do 0,1 %.
17 Oznaczanie straty przy pra
żeniu
17.1 Zasada metody
Strata przy pra
żeniu jest oznaczana w atmosferze utleniaj ącej (powietrze). Przy spalaniu w powietrzu w temperaturze
(975 ± 25) °C dwutlenek w
ęgla i woda, które nie wyparowały w czasie suszenia, są usuwane podobnie jak wszystkie
utleniaj
ące się lotne składniki.
UWAGA: Je
żeli kruszywa zawierają utleniające się nielotne składniki, jak w przypadku żużli wielkopiecowych, zaleca
si
ę stratę przy prażeniu skorygować zgodnie z EN 196-2:1987 p. 7.4.
17.2 Pobranie próbki i przygotowanie próbki analitycznej
Post
ępując jak podano w 11.2 i 11.3, przygotować próbkę analityczną o masie (1 ± 0,05) g.
17.3 Procedura oznaczania straty przy pra
żeniu
Zwa
żyć próbkę analityczną z dokładnością do 0,1 mg (m
13
) i umie
ścić w tyglu (5.6.2) uprzednio wyprażonym i
wytarowanym. Umie
ścić tygiel w piecu elektrycznym (5.2.2) z regulowaną temperaturą (975 ± 25) °C. Pozostawić tygiel
w piecu co najmniej na 50 min, a nast
ępnie tygiel schłodzić do temperatury pokojowej w eksykatorze (5.2.16) i
ponownie zwa
żyć (m
14
).
UWAGA: W przypadku kruszyw wapiennych ogrzewanie do 975 °C przeprowadza
ć powoli, w celu zminimalizowania
ryzyka gwa
łtownego rozpadu.
17.4 Obliczanie i wyra
żenie wyników
Obliczy
ć stratę przy prażeniu kruszywa z następującego wzoru:
w którym:
m
13
masa próbki analitycznej, w gramach;
m
14
masa wypra
żonej próbki analitycznej, w gramach.
18 Oznaczanie wolnego wapna w
żużlu stalowniczym
18.1 Postanowienia ogólne
Ka
żda z metod wymienionych w niniejszym rozdziale służy do oznaczania obecności wolnego wapna (CaO), które jest
potencjalnie ekspansywne i uwodnionego wapna (Ca(OH)
2
), które nie jest ekspansywne. Do rozró
żnienia tych dwóch
form wapna potrzebne s
ą dodatkowe badania, takie jak analizy: termograwimetryczna lub rentgenowska.
18.2 Oznaczanie wolnego wapna metod
ą kompleksometryczną (metoda zalecana)
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 27
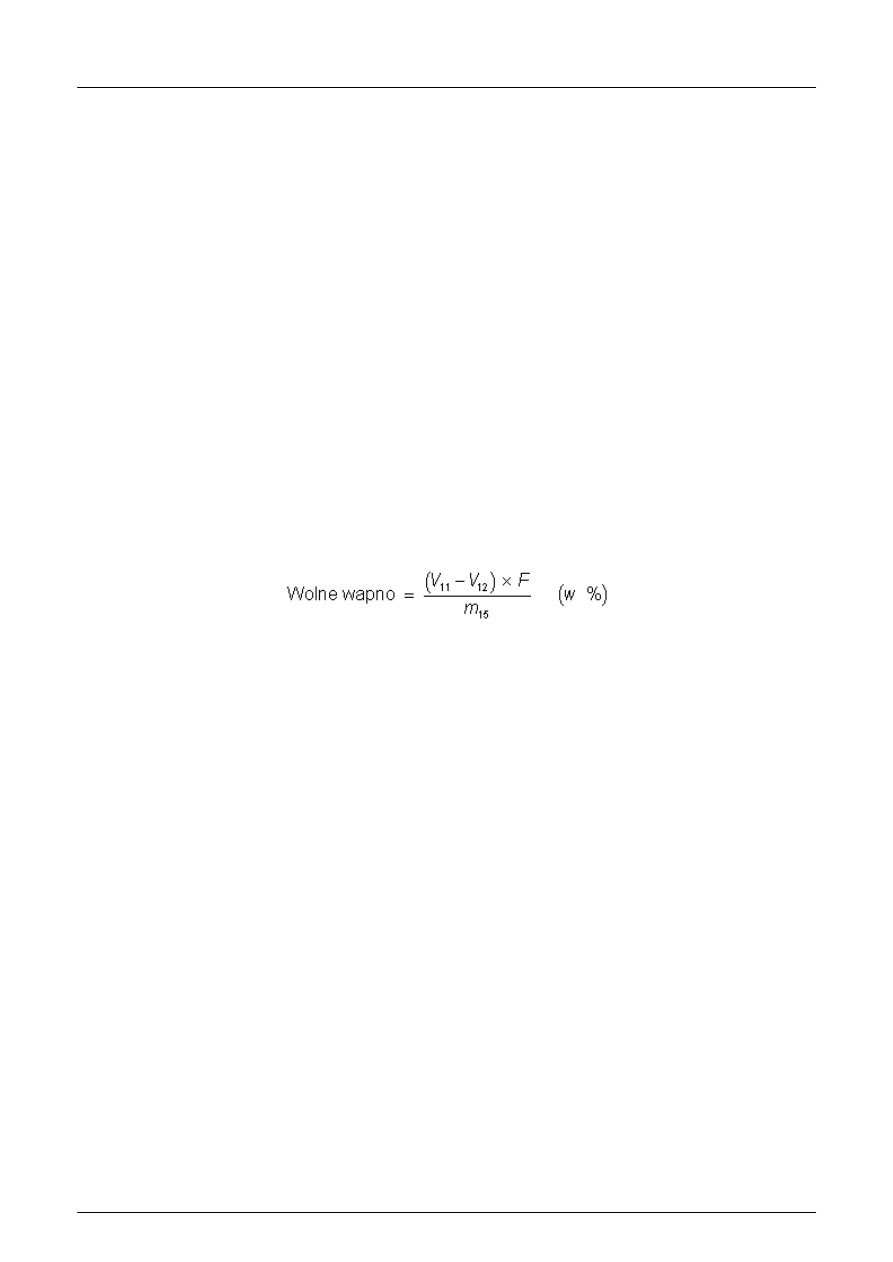
18.2.1 Zasada metody
Wolne wapno jest ekstrahowane z próbki kruszywa za pomoc
ą gorącego etanodiolu. Zawartość jonów wapna w
ekstrakcie jest nast
ępnie oznaczana miareczkowaniem kompleksometrycznym.
18.2.2 Pobieranie i przygotowanie próbki analitycznej
Post
ępować jak podano w 11.2 i 11.3 lecz rozdrobnić co najmniej 20 g, tak aby całość przeszła przez sito 63
µ
m i
pobra
ć około 0,5 tego materiału jako próbkę analityczną. Duże fragmenty żelaza pozostające na sicie powinny być
usuni
ęte.
18.2.3 Wykonanie badania
Zwa
żyć próbkę analityczną z dokładnością do 0,1 mg (m
15
) i przenie
ść do stożkowej kolby (5.12.1) zawierającej
mieszad
ło PTFE. Odmierzyć 50 ml bezwodnego etanodiolu ( 4.11.1) i przenieść do kolby. Zamknąć kolbę szklanym
korkiem i miesza
ć na łaźni wodnej o temperaturze 70 °C przez 30 min po osi ągnięciu temperatury, używając
magnetycznego mieszad
ła od 300 obr/min do 400 obr/min. Nast ępnie niezwłocznie filtrować przez spiekany szklany
s
ączek (5.12.4), który ma warstwę (około 4 mm do 5 mm) z mocno ubitej papki z bibu ły filtracyjnej w etanodiolu. Obmyć
kolb
ę trzykrotnie, łącznie 50 ml propanem-2-ol (4.11.2).
Zakwasi
ć czysty przesącz, zawierający rozpuszczone wolne wapno, 10 ml kwasu chlorowodorowego (1 + 1) ( 4.11.4) i
przemy
ć wodą do zlewki pomiarowej (5.12.2). Dopełnić do kreski i homogenizować, potrząsając. Zgodnie z
przewidywan
ą ilością przenieść za pomocą pipety 50 ml lub 100 ml do szklanej kolby. Doda ć 10 kropli roztworu
m-nitrofenolu (4.11.6) i 10 kropli trójetanoloaminy ( 4.11.5) (do oddzielenia jonów Mn i Fe), a nast
ępnie zneutralizować
roztworem 2 mol/l NaOH (4.11.7); rozcie
ńczyć wodą do około 500 ml i doprowadzić pH do wartości powyżej 13, dodając
oko
ło 10 ml roztworu 2 mol/l NaOH. Dodać wskaźnik mureksydu (4.11.8) i miareczkować roztworem EDTA (4.11.9) aż
kolor ró
żowofioletowy zamieni się na niebieskawy. Oznaczać koniec miareczkowania za pomocą urządzenia do
fotoelektrycznego miareczkowania ( 5.12.5). Powinna by
ć zawsze wykonana ślepa próba dla etanodiolu i odczynników.
18.2.4 Obliczanie i wyra
żenie wyników
Obliczy
ć wolne wapno zawarte w kruszywie z następującego wzoru:
w którym:
V
11
obj
ętość dodanego roztworu EDTA (w mililitrach)
V
12
obj
ętość dodanego roztworu EDTA do ślepej próby (w mililitrach)
F st
ężenie roztworu EDTA, w miligramach CaO na mililitr; w przypadku pobrania pipet ą 100 ml z kolby (5.12.2),
pomno
żyć przez 0,5
m
15
masa próbki analitycznej (w gramach).
Zawarto
ść wolnego wapna podać z dokładnością do 0,1 %.
18.3 Oznaczanie wolnego wapna metod
ą konduktometryczną (metoda alternatywna)
18.3.1 Zasada metody
Wolne wapno jest ekstrahowane z próbki podstawowej kruszywa za pomoc
ą gorącego etanodiolu. Zawartość jonów
wapna w ekstrakcie jest nast
ępnie oznaczana za pomocą pomiarów przewodności.
18.3.2 Pobieranie i przygotowanie próbki analitycznej
Post
ępując jak podano w 18.2.2 pobrać (100 ± 0,1) mg materiału przechodzącego przez 63 mm jako próbkę
analityczn
ą.
18.3.3 Wykonanie badania
Ogrza
ć w naczyniu pomiarowym (patrz rysunek 2) z termostatem 100 ml etanodiolu ( 4.11.1) do temperatury
(80 ± 0,1) °C, mieszaj
ąc za pomocą magnetycznego mieszadła. Dodać do tego rozpuszczalnika próbkę analityczną i
wprowadzi
ć elektrodę pomiarową.
UWAGA: Przez pomiar przewodno
ści podczas ekstrakcji, rozpuszczanie wolnego wapna mo że być monitowane
bezpo
średnio.
Proces ekstrakcji jest zako
ńczony co najmniej po 10 min ekstrahowania i gdy dalsza zmiana przewodno ści nie
zachodzi. Wówczas odczyta
ć końcową wartość przewodności.
18.3.4 Ocena i przedstawienie wyników
Wyznaczy
ć zawartość wolnego wapna ze zmierzonej zmiany przewodności za pomocą krzywej wzorcowej (patrz
rysunek 3). Wyrazi
ć zawartość wolnego wapna, w procentach, z dokładnością do 0,1 %.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 28

18.4 Oznaczanie wolnego wapna metod
ą acydymetryczną (metoda alternatywna)
18.4.1 Zasada metody
Wolne wapno jest ekstrahowane z próbki podstawowej kruszywa przez gotowanie w acetylooctanie etylu (metoda
Franka); ekstrakt jest miareczkowany wzorcowym 0,2 mol/l roztworem kwasu chlorowodorowego.
18.4.2 Pobranie i przygotowanie próbki analitycznej
Post
ępując jak podano w 18.2.2 pobrać około 1 g materiału przechodzącego przez 63 mm jako próbkę analityczną.
18.4.3 Wykonanie badania
Zmierzy
ć 70 ml przygotowanego roztworu rozpuszczalnika (4.13.5) zawierającego acetylooctan etylu i
2-metylo-propan-1-ol w proporcji 3 do 20 i przenie
ść do kolby Erlenmeyera (5.14.1). Zważyć próbkę analityczną z
dok
ładnością do 0,1 mg (m
16
) i przenie
ść do kolby.
Ustawi
ć kolbę w odpowiedniej pozycji do chłodnicy dopasowanej w górnej części, łącznikiem do rury absorpcyjnej
zawieraj
ącej wodorotlenek sodu (4.13.8) i sito molekularne (5.14.3), gotować w temperaturze wrzenia pod chłodnicą
zwrotn
ą, mieszając przez 3 h, na gorącej płycie (5.2.7). Wyłączyć gorącą płytę, ostudzić, a następnie filtrować pod
pró
żnią przez szklany mikrowłóknisty filtr (5.14.4), zatrzymując przesącz w drugiej kolbie. Wymyć pierwszą kolbę wraz z
pozosta
łością 50 ml 2-metylopropanu-1-ol (4.13.2), wykorzystując pręt do mieszania wyposażony w gumkę do
kierowania przep
ływem.
Doda
ć do przesączu od 10 do 12 kropel roztworu wskaźnika (4.13.6) i miareczkować wzorcowym 0,2 mol/l roztworem
kwasu chlorowodorowego ( 4.13.7) do uzyskania wyra
źnego odcienia czerwonego.
UWAGA: Je
żeli miareczkowanie jest prowadzone z użyciem pH-metru z rejestracją, filtracja ekstraktu jest zbędna.
18.4.4 Obliczanie i przedstawienie wyników
Obliczy
ć zawartość wolnego wapna w kruszywie z następującego wzoru:
Wolne wapno = k/1000 × V
12
/m
16
× 100 (w %)
w którym:
V
12
obj
ętość dodanego kwasu chlorowodorowego (w mililitrach);
m
16
masa próbki analitycznej (w gramach);
k wspó
łczynnik podany w 4.13.7 przedstawiający liczbę miligramów wolnego CaO w mililitrze wzorcowego roztworu
kwasu chlorowodorowego.
19 Oznaczanie niesta
łości żużli wielkopiecowych i stalowniczych
19.1 Oznaczanie rozpadu krzemianu dwuwapniowego w
żużlu wielkopiecowym chłodzonym powietrzem
19.1.1 Postanowienia ogólne
W niniejszym rozdziale podano metod
ę oznaczania podatności na rozpad kruszonych kawałków żużla
wielkopiecowego, spowodowanego inwersj
ą niestabilnej formy
β
krzemianu dwuwapniowego w form
ę
γ
. Zjawisko to jest
cz
ęsto nazywane niewłaściwie "rozpadem wapniowym".
19.1.2 Zasada metody
Podda
ć łamane powierzchnie żużla fluoryzacji w ultrafioletowym świetle, w zakresie światła widzialnego. Postać i kolor
fluorescencji umo
żliwia wykrycie podatności żużla na rozpad krzemianowy.
19.1.3 Pobieranie próbki
Post
ępować jak podano w 11.2.
19.1.4 Przygotowanie próbki analitycznej
Zmniejszy
ć próbkę laboratoryjną do próbki analitycznej składającej się co najmniej z 30 kawałków, wymyć i wysuszyć
próbk
ę, a następnie rozłupać każdy kawałek, aby uzyskać świeże powierzchnie łamane.
19.1.5 Metoda badania
Oznacza
ć rozpad krzemianu dwuwapniowego w świetle ultrafioletowym (5.15.1).
19.1.6 Przedstawianie wyników
Zapisa
ć obserwacje wykonane na świeżo łamanych powierzchniach. Żużle z licznymi widocznymi lub zgromadzonymi
du
żymi i małymi plamami żółtymi, brązowymi lub koloru cynamonowego na tle fioletu powinny by ć uznane za mające
zwi
ązek z rozpadem.
Żużle o jednolitym połysku, w różnorodnych odcieniach fioletu i żużle z jasnymi plamami występującymi tylko w
ograniczonej ilo
ści i nieregularnie, mogą być uznane za pewne.
19.2 Oznaczanie rozpadu
żelaza w żużlu wielkopiecowym chłodzonym powietrzem
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 29

19.2.1 Postanowienia ogólne
W niniejszym rozdziale podano metod
ę oznaczania podatności na rozpad kruszonego żużla wielkopiecowego
wynikaj
ącego z hydrolizy siarczków żelaza i manganu.
19.2.2 Zasada metody
Rozpad
żelaza występuje pod wpływem działania atmosfery wilgotnej lub deszczu, lecz bardziej intensywnie pod
wp
ływem wody. Dlatego jest obserwowany przy badaniu zachowania kawa łków żużla, zanurzonych w wodzie.
19.2.3 Pobieranie próbki
Post
ępować jak podano w 11.2.
19.2.4 Metoda badania
Umie
ścić 30 kawałków żużla o nominalnych rozmiarach pomiędzy 40 mm i 150 mm w wodzie o temperaturze
(20 ± 2) °C na dwa dni.
19.2.5 Przedstawianie wyników
Zapisa
ć wszystkie pęknięcia lub rozpad. Jeżeli żaden kawałek nie rozpadł się lub nie popękał, próbkę należy uznać za
spe
łniającą warunki badania. Jeżeli jeden lub dwa kawałki ulegną rozpadowi lub pęknięciom, badanie należy powtórzyć
z nast
ępnymi 30 kawałkami. Jeżeli jakieś kawałki rozpadną się lub pękną w drugim badaniu, próbkę należy traktować
jako nie spe
łniającą warunków badania.
19.3 Oznaczanie p
ęcznienia żużla stalowniczego
19.3.1 Postanowienia ogólne
W niniejszym rozdziale podano metod
ę oznaczania podatności na pęcznienie kruszonego żużla stalowniczego,
wynikaj
ące z opóźnionej hydratacji przepalonego wolnego wapna i/lub wolnego tlenku magnezu.
19.3.2 Zasada metody
Zag
ęszczona próbka żużla składająca się z ziaren o znanej wielkości, jest poddana pod ciśnieniem otoczenia
przep
ływowi pary o temperaturze 100 °C w urządzeniu do wytwarzania pary. W tych warunkach wilgoć potrzebna do
reakcji wolnego wapna lub wolnego tlenku magnezu jest w sposób ci
ągły doprowadzana do próbki badanej. Każda
zmiana obj
ętości spowodowana tą reakcją jest odczytywana bezpośrednio z zegarowego wskaźnika umieszczonego na
wierzchu próbki. Za wynik przyjmuje si
ę wzrost objętości, obliczony w % objętości w stosunku do objętości początkowej
zag
ęszczonej próbki żużla.
19.3.3 Pobieranie próbki
Próbka laboratoryjna powinna by
ć pobrana zgodnie z procedurami wymienionymi w EN 932-1.
19.3.4 Przygotowanie i zag
ęszczanie próbek
Pobrana próbka laboratoryjna powinna by
ć wysuszona niezwłocznie w laboratorium w (temperaturze 110 ± 5) °C do
sta
łej masy (6.5).
Do badania par
ą użyć próbki analityczne o uziarnieniu od 0 mm do 22 mm, wysuszonych mieszanek żużla, które były
po
łączone zgodnie z parabolą Fullera. Proporcje mas poszczególnych wymiarów klas ziarnowych podano w tablicy 3:
Tablica 3: Proporcje mas poszczególnych klas ziarnowych
Wymiary
mm
Procent
w masie
od 0 do 0,5
od 0,5 do 2
od 2 do 5,6
od 5,6 do 8
od 8 do 11,2
od 11,2 do 16
od 16 do 22
15
15
19
10
11
15
15
Ca
łość
100
Poszczególne klasy ziarnowe powinny by
ć pobrane z kruszyw przekruszonych.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 30

Zmniejszanie próbki powinno by
ć przeprowadzone zgodnie z procedurami wymienionymi w prEN 932-2.
P
ęcznienie powinno być oznaczane co najmniej na dwóch pojedynczych próbkach do badania. Ka żda indywidualna
próbka do badania powinna by
ć oddzielnie złożona z wyżej wymienionego składu ziarnowego. Ilość materiału
potrzebnego do ka
żdej indywidualnej próbki wynosi 4,5 kg. Dodatkowo próbki powinny by ć przygotowane do
oznaczania g
ęstości objętościowej, zgodnie z prEN 1097-6.
Nakry
ć perforowaną podstawę cylindra badawczego okrągłym filtrem (tkaninowa mata) i za pomoc ą laboratoryjnego
czerpaka przenie
ść przygotowaną próbkę analityczną do cylindra (5.16.1) przedstawionego na rysunku 5. Następnie
dynamicznie zag
ęścić próbkę na stole wibracyjnym z częstotliwością (48 ± 3) Hz (amplituda ±1,5 mm) (5.16.5) przez
6 min pod statycznym obci
ążeniem 0,035 N/mm
2
N10)
, wytworzonym na przyk
ład przez prasę hydrauliczną. W tych
warunkach praktycznie zawarto
ść wolnych przestrzeni w żużlowej próbce wynosi pomiędzy 20 % objętości i 25 %
obj
ętości, wartości zgodne z praktyką. Alternatywnie, próbka może być zagęszczona inną metodą przydatną do
uzyskania takiego samego stopnia zag
ęszczenia, np. młotkiem Proctora lub ręcznym młotkiem. Jeżeli dodano wodę do
próbki dla u
łatwienia zagęszczenia, badanie powinno rozpocząć się w ciągu 24 h po ukończeniu procesu zagęszczania.
Po zag
ęszczaniu oznaczać objętość próbki żużla V
S
, która jest ró
żnicą pomiędzy objętością cylindra V
C
a obj
ętością
powietrza V
A
pomi
ędzy próbką żużla a górną krawędzią cylindra. Obliczyć V
C
i V
A
z pomiarów wysoko
ści wykonanych
za pomoc
ą pręta z podziałką (5.16.6) i przyjęciu średniej z czterech odczytów na skrajnych dwóch średnicach cylindra
przecinaj
ących się pod kątem prostym.
Nast
ępnie nakryć powierzchnię próbki żużla okrągłym filtrem (mata tkaninowa) i położyć warstwę szklanych kulek
(5.16.3) naoliwionych przed badaniem olejem silikonowym w celu zmniejszenia tarcia pomi
ędzy poszczególnymi
kulkami. Ca
łkowita masa położonych kulek wynosi (1,5 ± 0,01) kg. Rozłożyć szklane kulki w cylindrze badawczym
równomiernie na powierzchni.
UWAGA: 1,5 g oleju silikonowego wystarcza do partii 1,5 kg kulek.
Powtarza
ć oliwienie szklanych kulek po ka żdym badaniu parą. W przypadku osadzania się wapna na szklanych
kulkach podczas badania par
ą, niezbędne jest czyszczenie ich z wapna za pomocą rozcieńczonego kwasu
chlorowodorowego po ka
żdym czterokrotnym użyciu.
19.3.5 Metoda badania par
ą
Po przykryciu badanej próbki
żużla warstwą szklanych kulek wstawić cylinder badawczy do urządzenia wytwarzającego
par
ą i włączyć płaszcz grzejny wokół ścianek cylindra. Następnie nakryć szklane kulki perforowaną płytą, obciążnikiem
krzy
żakowym i obciążnikiem. Zamocować zegarowy czujnik lub miernik, który rejestruje ruchy podnoszenia si ę
powierzchni próbki pod wp
ływem pary. Po włączeniu płaszcza grzejnego i wytwornicy pary następuje nagrzewanie
badanej próbki
żużla i ekspansja jej objętości. W celu wyeliminowania zapisu ruchów unoszenia zwi ązanych z
w
łączeniem nagrzewania, nie rozpoczynać zapisywania zmian położenia do momentu, gdy para zacznie swobodnie
przechodzi
ć przez próbkę.
Badanie w parze trwa
łącznie 24 h w przypadku żużli LD (wielkopiecowych) i 168 h (7 dni) w przypadku żużli z pieców
elektrycznych i
żużli z pieców stalowniczych (żużle stalownicze). Na koniec tego okresu nale ży odczytać podniesienie
si
ę powierzchni próbki i obliczyć w % objętości w stosunku do objętości początkowej (patrz 19.3.6).
Zbiornik wody w wytwornicy pary ma tak
ą wielkość, żeby para mogła być wytwarzana przez 24 h. Gdy zachodzi
potrzeba uzupe
łniania wody, należy uważać, aby nie nastąpił spadek temperatury.
UWAGA: W wielu przypadkach celowe jest zapisanie post
ępującego wzrostu objętości w zależności od czasu.
Poniewa
ż zmiany w początkowym okresie badania następują gwałtownie, zaleca się wykonywanie odczytu co 15 min.
Po 4 h odst
ępy między odczytami mogą być zwiększone do 60 min. Jeżeli wzrost objętości jest funkcją czasu,
szczegó
łowa interpretacja wyników badania mo że być przedstawiona graficznie (początek wzrostu, okres
asymptotyczny).
19.3.6 Obliczanie i przedstawienie wyników
Obliczy
ć objętość próbki analitycznej żużla V
S
, przed badaniem par
ą, z następującego wzoru:
V
S
= V
C
- V
A
w którym:
V
S
obj
ętość próbki badanego żużla po zagęszczeniu w cylindrze badawczym (w centymetrach sześciennych);
V
C
obj
ętość cylindra (w centymetrach sześciennych);
V
A
obj
ętość powietrza między próbką żużla i górną krawędzią cylindra (w centymetrach sześciennych).
V
C
i V
A
s
ą obliczane z pomiarów wysokości za pomocą pręta pomiarowego i średnicy cylindra (210 mm).
Po zag
ęszczeniu oznaczać gęstość nasypową na sucho i zawartość wolnych przestrzeni zagęszczonej mieszanki
nast
ępująco:
V
M
= (1 - r
M
/ r) × 100 (w % obj
ętości)
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 31

gdzie:
r
M
g
ęstość nasypowa zagęszczonej mieszanki (w megagramach na centymetr sze ścienny);
W masa zag
ęszczonej mieszanki (w gramach);
V
M
zawarto
ść wolnych przestrzeni w zagęszczonej mieszance (w % objętości);
V
S
obj
ętość badanej próbki żużla po zagęszczeniu w cylindrze (w centymetrach sześciennych);
r g
ęstość cząstek żużla, oznaczanych wg prEN 1097-6 (w megagramach na metr sze ścienny);
w woda zawarta w próbce (w procentach masy).
Po przeprowadzeniu badania obliczy
ć pęcznienie w % objętości ze wzrostu próbki odczytanego ze wskaźnika
zegarowego lub miarki i z wewn
ętrznej średnicy cylindra badawczego (210 mm) z nast ępującego wzoru:
w którym:
h przyrost próbki po badaniu par
ą (w milimetrach);
d
średnica wewnętrzna cylindra badawczego (210 mm).
Zapisa
ć wynik jako średnia arytmetyczna pęcznienia 2 próbek do badania, w zaokrągleniu do 0,1 % objętości.
Za
łącznik A (informacyjny)
Dok
ładność
A.1 Symbole
r
i
granica powtarzalno
ści, jak podano w prEN 932-6.
R
i
granica odtwarzalno
ści, jak podano w prEN 932-6.
X
średnia z wyników badania.
A.2 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą Volharda (metoda zalecana) (patrz rozdział 7)
Dok
ładność oznaczania chlorków rozpuszczalnych w wodzie jest wyznaczana (jako zawarto ść jonu chlorku w
procentach masy kruszywa) jako:
r
1
= 0,0004 + 0,029 X i R
1
= 0,0006 + 0,124 X
A.3 Oznaczanie chlorków soli rozpuszczalnych w wodzie metod
ą potencjometryczną (metoda alternatywna)
(patrz rozdzia
ł 8)
Odchylenie standardowe powtarzalno
ści r wynosi 0,001 %.
Odchylenie standardowe odtwarzalno
ści R wynosi 0,003 %.
A.4 Oznaczanie zawarto
ści siarki całkowitej (metoda zalecana) (patrz rozdział 11)
Dok
ładność oznaczania zawartości siarki całkowitej jest wyznaczana (jako zawartość siarki w procentach masy
kruszywa) jako:
r
1
= 0,017 + 0,081 X i R
1
= 0,062 + 0,204 X
A.5 Oznaczanie siarczanów rozpuszczalnych w kwasie (metoda zalecana) (patrz rozdzia
ł 12)
Dok
ładność oznaczania zawartości siarczanów rozpuszczalnych w kwasie jest wyznaczana (jako procent SO
3
w masie
kruszywa) jako:
r
1
= 0,021 + 0,200 X i R
1
= 0,000 + 0,812 X
Za
łącznik B (informacyjny)
Bibliografia
Wykaz podstawowych dokumentów, które by
ły podstawą do opracowania normy.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 32

prEN 932-6
Tests for general properties of aggregates
Part 6: Definitions of repeatability and reproducibility
BS 812
Testing aggregates:
Part 117:1988 Method for determination of water-soluble chloride salts
Part 118:1988 Method for determination of sulfate content
Section 122.1 Organic contaminators which influence the setting and hardening of
Portland cement mortars (draft)
Section 122.2 Lightweight contaminators which may disfigure concrete and mortar
(draft)
BS 1047:1983
Air-cooled blast-furnace slag aggregate for use in construction - Appendix B.2.
Method for determination of total sulfur
DIN 4226:Teil 3:April 1983
Zuschlag für Beton - Prüfung von Zuschlag mit dichtem oder porigem Gefüge
DS 405.3 (1978)
Prfvningsmetoder for Sand-, grus- og stenmaterialer- Humusindhold
NEN 5920:November 1988
Toeslagmaterialen voor beton. Bepaling van het gehalte aan fulvozuur
Clemena G.G. et al
Transportation Research Record nr 651 (1977), pp1 - 6, publ. by National
Academy of Sciences USA. Gran method of endpoint determination in chloride
analysis by potentiometric titration
Keil, F.
Hochofenschlacke, Verlag Stahleisen M.B.H./Düsseldorf, pp 156, 163 (1949)
Lees. T.P.
C and CA Guide: Impurities in cocreting aggregates, Cement and Concrete
Association Publication 45.016
Midgley, H.G.
Magazine of Concrete Research: August 1958, pp, 75-78 The Staining of
concrete by pyrite
Pressle, E.E. et al
Analytical Chemistry: (1956) 28, p. 896. Investigation of the FRANKE method of
determining free calcium hydroxide and free calcium oxide.
Forschungsgemeinschaft Eisenhüttenschlacken. Vorläufige technische Lieferbedingungen für LD-Schlacke in
Tragschichten ohne Bindemittel (Ausgabe 1988).
Za
łącznik krajowy NA
(informacyjny)
NORMY POWO
ŁANE W TREŚCI NORMY EUROPEJSKIEJ I ICH ODPOWIEDNIKI KRAJOWE
UWAGA - Zaleca si
ę sprawdzić, czy podane w wykazie normy i ich odpowiedniki krajowe nie zosta ły zaktualizowane.
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 33

Normy powo
łane w EN
Odpowiedniki krajowe
EN 196-1:1993
zast
ąpiona
EN 196-1:1994
- Brak odpowiednika
PN-EN 196-1:1996
Metody badania cementu - Oznaczanie wytrzyma
łości
EN 196-2:1987
zast
ąpiona
EN 196-2:1994
- Brak odpowiednika
PN-EN 196-2:1996
Metody badania cementu - Analiza chemiczna cementu
EN 196-3:1987
zast
ąpiona
EN 196-3:1994
- Brak odpowiednika
PN-EN 196-1:1996
Metody badania cementu - Oznaczanie czasów wi
ązania i
sta
łości objętości
ENV 197-1:1992
- Oficjalne t
łumaczenie ENV dostępne w Ośrodku Informacji i Dokumentacji Biura
PKN jako publikacja. Wersja polska ENV 197-1:1992 Cement - Sk
ład wymagania
i kryteria zgodno
ści - Arkusz 1: Cement powszechnego użytku ENV 197-1:1992
zosta
ła wykorzystana do opracowania normy
PN-B-19701:1997
Cement - Cement
powszechnego u
żytku - Skład, wymagania i ocena zgodności
EN 932-1:1996
- PN-EN 932-1:1999 Badania podstawowych w
łaściwości kruszyw - Metody
pobierania próbek
prEN 932-2
- (Norma opracowywana w CEN/TC 154; PN w przygotowaniu w NKP nr 108)
prEN 932-5
- (Norma opracowywana w CEN/TC 154; PN w przygotowaniu w NKP nr 108)
EN 933-2:1995
- PN-EN 933-2:1999 Badania geometrycznych w
łaściwości kruszyw - Oznaczanie
sk
ładu ziarnowego. Normalne wymiary otworów sit badawczych
prEN 1015-4
- (Norma opracowywana w CEN/TC 125; PN w przygotowaniu w NKP nr 233)
prEN 1015-9
- (Norma opracowywana w CEN/TC 125; PN w przygotowaniu w NKP nr 233)
prEN 1015-11
- (Norma opracowywana w CEN/TC 125; PN w przygotowaniu w NKP nr 233)
prEN 1097-6
- (Norma opracowywana w CEN/TC 154; PN w przygotowaniu w NKP nr 108)
ISO 384:1978
- PN-B-13014:1992 Szklany sprz
ęt laboratoryjny - Zasady projektowania i
konstruowania naczy
ń do pomiaru objętość
ISO 648:1997
- PN-B-13023:1990 Szklany sprz
ęt laboratoryjny - Naczynia pomiarowe. Pipety
jednomiarowe
ISO 650:1977
- Brak odpowiednika krajowego. Orygina
ł normy jest dostępny w Ośrodku
Informacji i Dokumentacji Biura PKN, w przypadku wprowadzenia niniejszej normy
do obowi
ązkowego stosowania niezbędne jest uwzględnienie braku odpowiednika
krajowego tej normy
ISO 1042-1:1983
- PN-74/B-13017 Szklany sprz
ęt laboratoryjny - Naczynia pomiarowe - Kolby
pomiarowe z jedn
ą kreską
ISO 4788:1980
- Brak odpowiednika krajowego. Orygina
ł normy jest dostępny w Ośrodku
Informacji i Dokumentacji Biura PKN, w przypadku wprowadzenia niniejszej normy
do obowi
ązkowego stosowania niezbędne jest uwzględnienie braku odpowiednika
krajowego tej normy
PN-EN 1744-1:2000 Badania chemicznych w
łaściwości kruszyw Analiza chemiczna
Powielanie dokumentu zabronione. Wszelkie prawa zastrze
żone.
INTEGRAM BUDOWNICTWO
Strona 34
Wyszukiwarka
Podobne podstrony:
PN EN 933 1 2000 Badania geometrycznych wl kruszyw Oznaczanie skladu ziarnowego Metoda przesiewan
PN EN 933 1 2000 Badania geometrycznych wl kruszyw Oznaczanie skladu ziarnowego Metoda przesiewan
1 5 PN EN 1097 2 2000 Badania mechanicznych i fizycznych wl kruszyw Metoda oznaczania odpornosci na
PN EN 933 4 2001 Badania geometrycznych wl kruszyw Oznaczanie ksztaltu ziarn Wskaznik ksztaltu
PN EN 933 8 2001 Badania geometrycznych wl kruszyw Ocena zawartosci drobnych czastek Badanie wska
PN EN 933 4 2001 Badania geometrycznych wl kruszyw Oznaczanie ksztaltu ziarn Wskaznik ksztaltu
PN EN 206 1 2000 Beton Wymagania wlasciwosci produkcja i zgodnosc
PN EN 933 8 2001 Badania geometrycznych wl kruszyw Ocena zawartosci drobnych czastek Badanie wska
1 7 PN EN 1097 8 2002 Badanie mechanicznych i fizycznych wl kruszyw Oznaczanie polerowalnosci kamien
1 9 PN EN 1367 3 2002 Badania wl cieplnych i odpornosci kruszyw na dzialanie czynnikow atm Badanie b
PN EN 1097 6 2002 Badania mechanicznych i fizycznych wl kruszyw Oznaczanie gestosci ziarn i nasia
PN EN 1097 6 2002 Badania mechanicznych i fizycznych wl kruszyw Oznaczanie gestosci ziarn i nasia
PN EN 1097 6 2002 Badania mechanicznych i fizycznych wl kruszyw Oznaczanie gestosci ziarn i nasia
PN EN 1097 6 2002 Badania mechanicznych i fizycznych wl kruszyw Oznaczanie gestosci ziarn i nasia
1 9 PN EN 1367 3 2002 Badania wl cieplnych i odpornosci kruszyw na dzialanie czynnikow atm Badanie b
1 7 PN EN 1097 8 2002 Badanie mechanicznych i fizycznych wl kruszyw Oznaczanie polerowalnosci kamien
PN EN 206 1 Beton Część 1 Wymagania, właściwości, produkcja i zgodność
więcej podobnych podstron