Na ³amach poprzedniego numeru
„Problemów Kryminalistyki” [1] zosta-
³y opisane metody oznaczania pier-
wiastków technik¹ ICP-OES ze
szczególnym uwzglêdnieniem korek-
cji efektów matrycowych. We wcze-
œniejszych publikacjach omówiono
podstawy metody oraz badania
wstêpne [2, 3, 4]. W niniejszym
opracowaniu zaœ autorzy przedsta-
wi¹ zagadnienia zwi¹zane z wali-
dacj¹ prezentowanej wczeœniej
metody takie jak: dok³adnoœæ i pre-
cyzja metody, granica wykrywalno-
œci, granica oznaczalnoœci, zakres
prostoliniowy i roboczy krzywych
kalibracyjnych, selektywnoϾ me-
tody, szacowanie niepewnoœci me-
tody.
Dla przypomnienia nale¿y po-
daæ, ¿e w badaniach wykorzysty-
wany jest spektrometr ICP-OES
Optima 3100XL firmy Perkin El-
mer. Na podstawie przeprowadzo-
nych dotychczas badañ dalsze
prace prowadzone bêd¹ w warun-
kach odpornych (ang. robust) pra-
cy spektrometru, których parame-
try zamieszczono w tabeli 1.
Próbki ziela konopi przygotowy-
wane s¹ do badañ na drodze minera-
lizacji na mokro z wykorzystaniem
energii mikrofalowej w uk³adzie za-
mkniêtym za pomoc¹ systemu Multi-
wave firmy Anton Paar (Perkin El-
mer) z u¿yciem mieszaniny kwasu
azotowego i wody utlenionej.
Walidacja metody
Ka¿da nowo opracowana metoda
analityczna powinna byæ poddana
walidacji. Celem walidacji jest wyzna-
czenie i dokumentacja parametrów
charakteryzuj¹cych dan¹ metodê
analityczn¹. Nale¿¹ do nich: dok³ad-
noϾ, precyzja, selektywnoϾ, specy-
ficznoœæ, zakres prostoliniowoœci
krzywych kalibracji oraz zakres robo-
czy, granica wykrywalnoœci i ozna-
czalnoœci. Nale¿y podkreœliæ, ¿e nie
jest niezbêdne wyznaczanie wszyst-
kich wymienionych parametrów. Na
pocz¹tku konieczne jest zastanowie-
nie siê, które parametry najlepiej opi-
suj¹ metodê. Oczywiste jest, ¿e np.
przy walidacji metody oznaczania
wapnia w próbkach roœlinnych meto-
d¹ ICP-OES nie jest potrzebne wy-
znaczanie granicy
w y k r y w a l n o œ c i ,
gdy¿ zawartoœæ te-
go pierwiastka w ro-
œlinach wynosi kilka
procent w przelicze-
niu na masê próbki.
Wa¿ne bêdzie wy-
znaczenie zakresu
p r o s t o l i n i o w o œ c i
krzywych kalibracji.
Nale¿y zatem za-
planowaæ proces
walidacji tak, by jak
najmniejsza liczba
wyznaczonych pa-
rametrów ca³kowi-
cie charakteryzo-
wa³a stworzon¹
15
PROBLEMY KRYMINALISTYKI 253/06
Marzena Kuras
Marek Wachowicz
Profilowanie konopi
na podstawie sk³adu
pierwiastkowego
– cz. II (walidacja metody)
P
Paarraam
me
ettrr
W
Waarru
un
nk
kii
o
od
dp
po
orrn
ne
e
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
pllaazzm
mo
ow
we
eg
go
o [[ll//m
miin
n]]
15
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
po
om
mo
occn
niicczze
eg
go
o [[ll//m
miin
n]]
0,5
P
Prrzze
ep
p³³y
yw
w g
gaazzu
u p
prrzze
ezz rro
ozzp
py
yllaacczz [[ll//m
miin
n]]
0,5
M
Mo
occ p
pllaazzm
my
y [[W
W]]
1450
W
Wy
ysso
ok
ko
oœœææ o
ob
bsse
errw
waaccjjii p
pllaazzm
my
y [[m
mm
m]]
15
P
Prrzze
ep
p³³y
yw
w p
prró
ób
bk
kii [[m
mll//m
miin
n]]*
*
1,5
C
Czzaass o
op
pó
óŸŸn
niie
en
niiaa [[ss]]
60
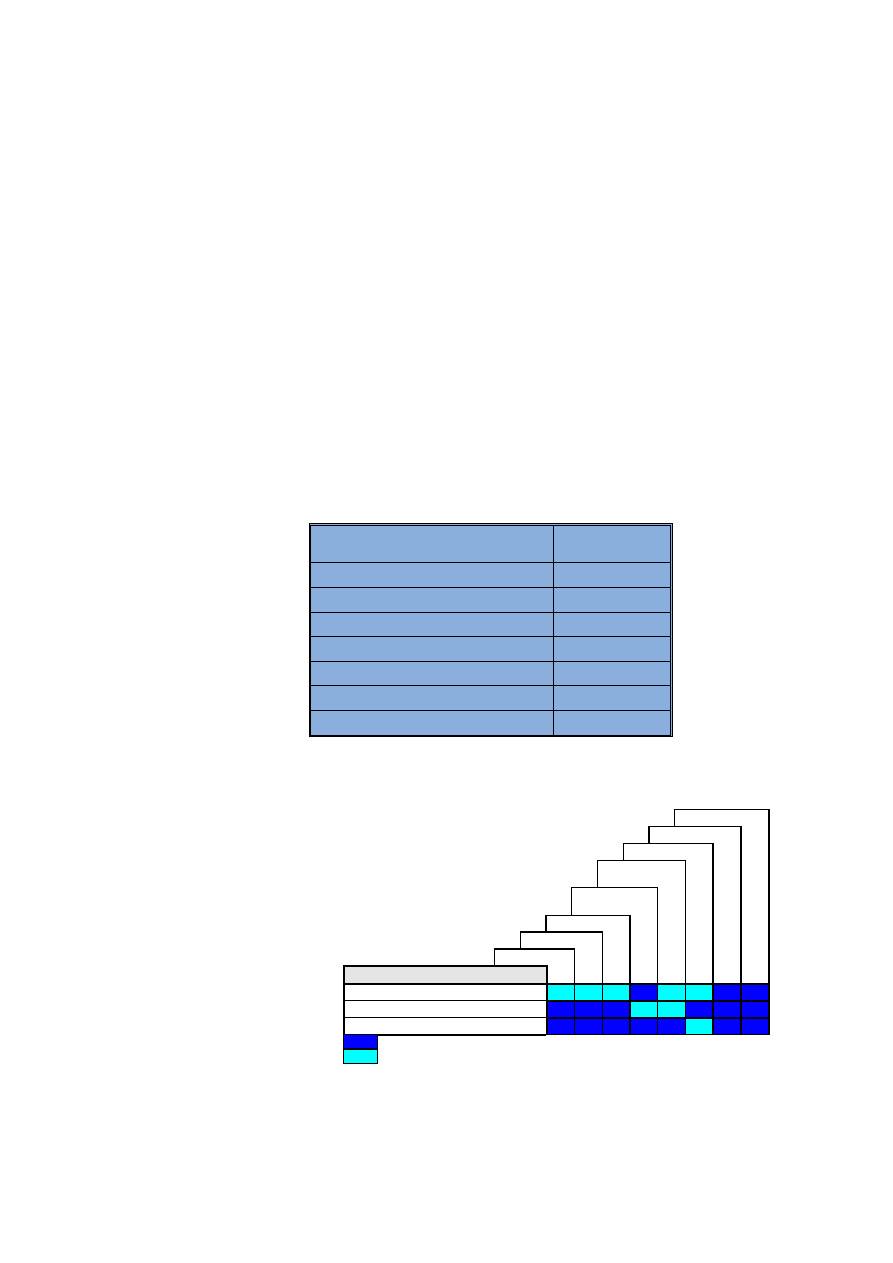
Tabela 1
Warunki operacyjne pracy spektrometru
Spectrometer set-up parameters
* W metodzie wzorca wewnêtrznego przep³yw próbki wynosi 0,65 ml/min,
a czas opóŸnienia 90 s
odporność
specyficzność
liniowość
granica
oznaczaln ości
granica
wykrywa lności
odtwarzalność
powtarzalność
dokładność
Metoda
Jakościowa (identyfikacja, potwie rdzenie)
Ilościowa - wysoki poziom stężeń
Ilościowa - niski poziom stężeń
- wymagane zawsze
- nie ma potrzeby wyznaczać
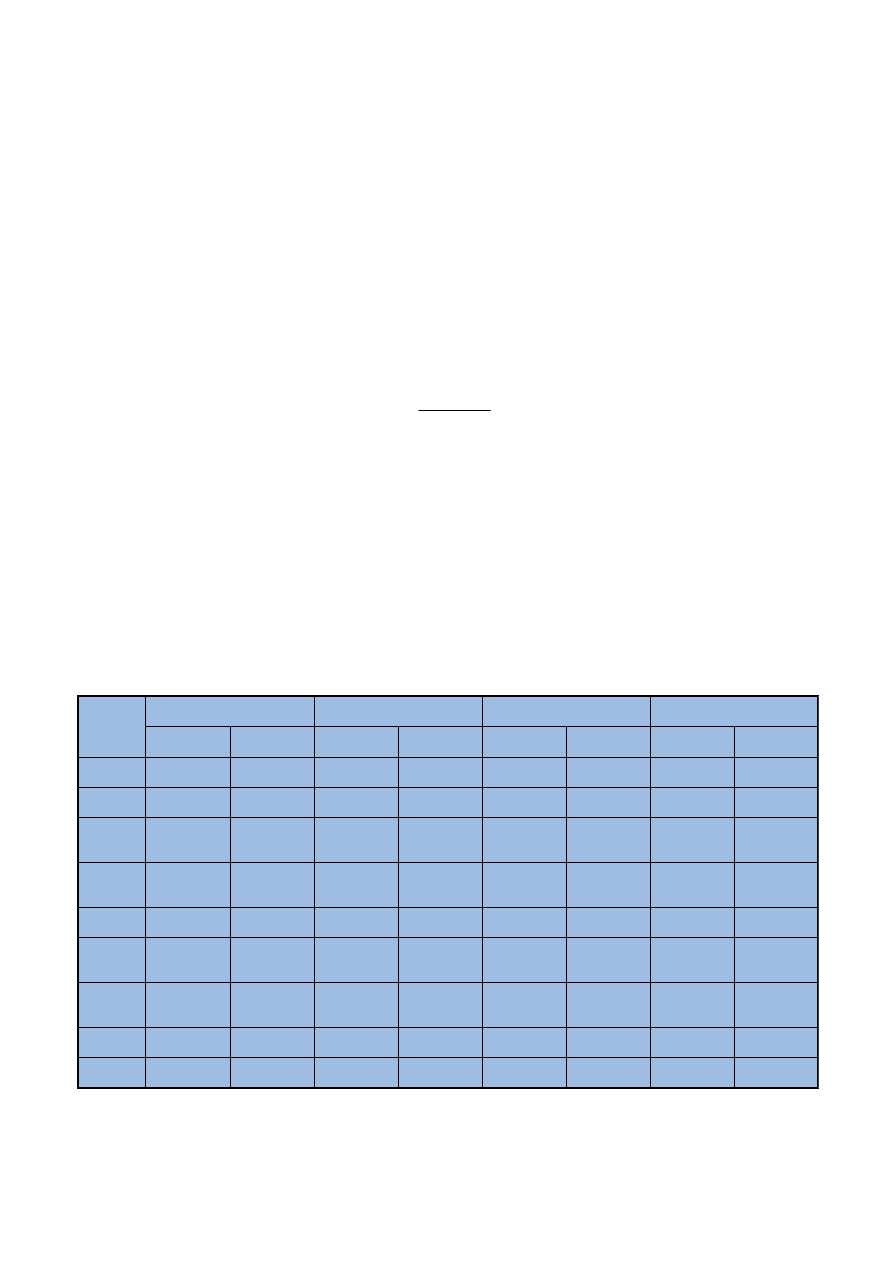
Ryc. 1. Elementy walidacji wymagane przy okreœlonych metodach analitycznych [5]
Fig. 1. Elements of validation required in certain analytical methods [5]
metodê. Parametry niezbêdne do wy-
znaczenia w procesie walidacji meto-
dy jakoœciowej i iloœciowej zamiesz-
czono na rycinie 1.
Istnieje wiele dokumentów, publi-
kacji i przewodników dotycz¹cych
walidacji i szacowania niepewnoœci
metod analitycznych, a najbardziej
znacz¹ce zosta³y opublikowane
przez AOAC International, Internatio-
nal Conference and Harmonization
(ICH) i Eurachem [6, 7, 8, 9, 10, 11].
Ze wzglêdu na znaczn¹ iloœæ publika-
cji zwi¹zanych z zagadnieniem wali-
dacji, jej parametry zostan¹ opisane
w skrócie.
Dok³adnoœæ
Celem pracy ka¿dego analityka
jest oznaczenie prawdziwego stê¿e-
nia substancji. W rzeczywistoœci jed-
nak mo¿liwe jest tylko pewne przybli-
¿enie tej wartoœci. Metodê analitycz-
n¹ uznaje siê za dok³adn¹, gdy war-
toœæ mierzona jest równa wartoœci
prawdziwej. Istnieje wiele sposobów
na sprawdzenie dok³adnoœci metody.
Najpowszechniej stosowan¹ jest
analiza materia³ów referencyjnych.
S¹ to próbki o znanym stê¿eniu
sk³adników w niej zawartych. Ich
analiza pozwala na okreœlenie, jaki
jest rozrzut wyników miêdzy warto-
œciami certyfikowanymi a wyznaczo-
nymi doœwiadczalnie. Jedynym man-
kamentem jest fakt, ¿e dla wielu pró-
bek nie s¹ dostêpne materia³y refe-
rencyjne, np. próbki konopi z certyfi-
kowanymi zawartoœciami pierwiast-
ków. Dok³adnoœæ zwykle wyra¿a siê
jako procentowy odzysk (%R
c
):
(1),
gdzie:
Xexp – wartoœæ wyznaczona doœwiad-
czalnie,
Xcert РwartoϾ certyfikowana.
WartoϾ %R
c
równa 100% œwiad-
czy o idealnej zgodnoœci wyniku do-
œwiadczalnego z wartoœci¹ referen-
cyjn¹.
Analiza kilku materia³ów certyfiko-
wanych o ró¿nej zawartoœci sk³adni-
ka oznaczanego pozwala na oszaco-
wanie dok³adnoœci metody na ró¿-
nym poziomie stê¿eñ. Realizuje siê
to poprzez wykreœlenie zale¿noœci
wartoœci certyfikowanej od wyzna-
czonej doœwiadczalnie. Otrzymany
wykres powinien przedstawiaæ liniê
prost¹. Idealn¹ dok³adnoœæ metody
w ca³ym badanym zakresie stê¿eñ
uzyskuje siê przy wspó³czynniku ko-
relacji równym 1.
Innym sposobem wyznaczania do-
k³adnoœci jest porównanie wartoœci
wyznaczonych nowo opracowan¹ me-
tod¹ analityczn¹ i metod¹ sprawdzo-
n¹, która jest dok³adna, oraz przez po-
równanie miêdzylaboratoryjne.
Jak ju¿ zosta³o to podkreœlone
w niniejszym artykule, na rynku nie
s¹ dostêpne próbki konopi z certyfi-
kowanymi zawartoœciami pierwiast-
ków. Dlatego te¿ do okreœlenia do-
k³adnoœci metody wybrano cztery ro-
œlinne materia³y certyfikowane. Ze
PROBLEMY KRYMINALISTYKI 253/06
16
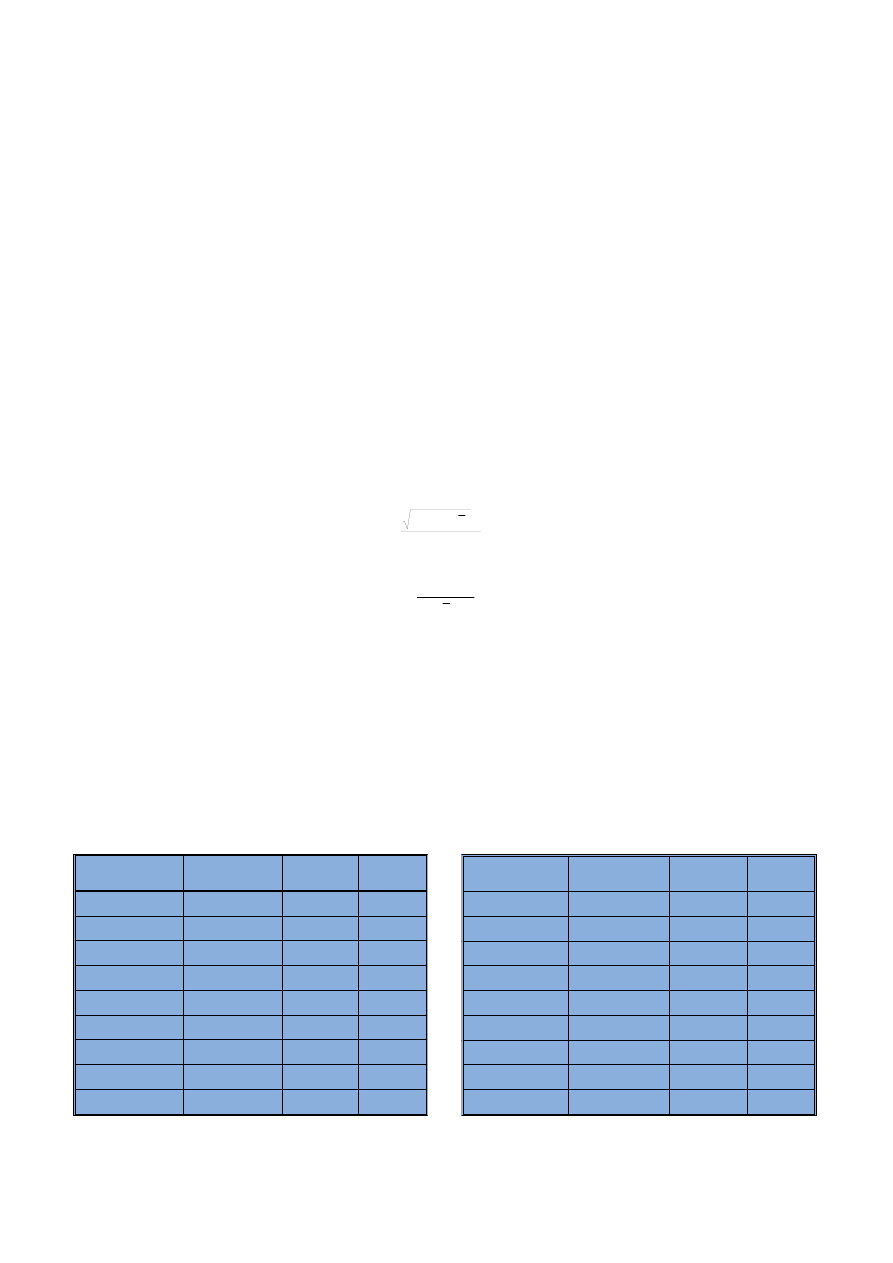
Tabela 2
Dok³adnoœæ metody przedstawiona na podstawie wyników analizy materia³ów certyfikowanych
Method accuracy presented on the basis of results of certified material analysis
IIN
NC
CT
T T
TL
L1
1
IIN
NC
CT
T M
MP
PH
H2
2
C
CT
TA
A V
VT
TL
L2
2
C
CT
TA
A O
OT
TL
L1
1
Z
Zaaw
waarrtto
oœœææ
p
piie
errw
wiiaass ttk
kaa
C
Ce
errtty
yffiik
kaatt
W
Waarrtto
oœœææ
o
ottrrzzy
ym
maan
naa
C
Ce
errtty
yffiik
kaatt
W
Waarrtto
oœœææ
o
ottrrzzy
ym
maan
naa
C
Ce
errtty
yffiik
kaatt
W
Waarrtto
oœœææ
o
ottrrzzy
ym
maan
naa
C
Ce
errtty
yffiik
kaatt
W
Waarrtto
oœœææ
o
ottrrzzy
ym
maan
naa
B
B [[
µµgg//gg]]
26*
29
B
Baa [[
µµgg//gg]] 43,2 ± 3,9
42,7 ± 1,5
32,5 ± 2,5
31,8 ± 0,8
42,7 ± 6,6
38,3 ± 1,6
84,2 ± 11,5
80,9 ± 0,8
C
Caa [[%
%]]
0,582 ±
0,052
0,578 ±
0,034
1,08 ± 0,07
1,08 ±
0,04
3,60 ± 0,15
3,58 ±
0,15
3,17 ± 0,12
3,25 ±
0,05
C
Cu
u [[
µµgg//gg]]
20,4 ± 1,5
21,1 ± 0,9
7,77 ± 0,53
7,70 ±
0,35
18,2 ± 0,9
17,2 ± 0,7
14,1 ± 0,5
13,8 ± 0,2
F
Fe
e [[
µµgg//gg]]
432*
448 ± 15
460*
459 ± 19
1083 ± 33
1075 ± 51
989*
1004 ± 24
M
Mg
g [[%
%]]
0,224 ±
0,017
0,220 ±
0,009
0,292 ±
0,018
0,296 ±
0,010
0,510 ±
0,023
0,519 ±
0,014
0,447 ±
0,021
0,439 ±
0,064
M
Mn
n [[
µµgg//gg]]
1570 ±
110
1559 ± 54
191 ± 12
195 ± 3
79,7 ± 2,6
78,1 ± 2,5
412 ± 14
398 ± 7
S
Srr [[
µµgg//gg]]
20,8 ± 1,7
22,0 ± 1,2
37,6 ± 1,1
38,2 ± 1,1
110 ± 12
119 ± 5
201 ± 20
184 ± 4
Z
Zn
n [[
µµgg//gg]] 34,7 ± 2,7
32,2 ± 1,4
33,5 ± 2,1
32,7 ± 1,4
43,3 ± 2,1
42,1 ± 1,9
49,9 ± 2,4
47,8 ± 0,8
* WartoϾ informacyjna
cert
c
X
X
R
100
*
%
exp
=
wzglêdu na podobn¹ matrycê próbek
za³o¿ono, ¿e próbki te bêd¹ dobrze
odzwierciedlaæ dok³adnoœæ opraco-
wywanej metody.
Dok³adnoœæ metody wyznaczono
na podstawie wyników analiz mate-
ria³ów certyfikowanych w warunkach
odtwarzalnych. Wyniki przedstawio-
ne w tabeli 2 to œrednie z oœmiu po-
miarów jednej próbki.
Metodê nale¿y uznaæ za dok³ad-
n¹, gdy¿ wyniki analiz czterech mate-
ria³ów certyfikowanych nie ró¿ni¹ siê
istotnie od wartoœci referencyjnych.
Nale¿y zwróciæ uwagê, ¿e zawartoœci
(stê¿enia) pierwiastków w poszcze-
gólnych materia³ach zmieniaj¹ siê
w szerokim zakresie, np. dla wapnia
jest to zakres od 0,582% do 3,60%,
a w przypadku strontu – od 20,8 mg/g
do 201 mg/g, zatem opracowana me-
toda jest dok³adna w szerokim zakre-
sie stê¿eñ dla ka¿dego oznaczanego
pierwiastka.
Precyzja
Na precyzjê metody sk³adaj¹ siê
dwa elementy: powtarzalnoϾ i od-
twarzalnoœæ wyników pomiarów.
PowtarzalnoϾ РS
R
– jest to pre-
cyzja metody wyznaczona w powta-
rzalnych warunkach. Obejmuje ona
tê sam¹ procedurê pomiaru, tego sa-
mego analityka, ten sam sprzêt po-
miarowy o takich samych warunkach
operacyjnych, to samo laboratorium
oraz powtarzanie badañ w krótkim
odstêpie czasu.
OdtwarzalnoϾ РS
r
– mo¿na
oszacowaæ w jednym laboratorium
lub poprzez badania miêdzylaborato-
ryjne. Jest to precyzja oznaczeñ da-
nego sk³adnika w zmiennych warun-
kach. Mo¿na zmieniaæ nastêpuj¹ce
warunki: metodê pomiarow¹, zasadê
pomiaru, analityka, aparat, odczynni-
ki, laboratorium i czas pomiaru.
Precyzjê metody zwykle wyra¿a
siê przez odchylenie standardowe
(SD) lub wzglêdne procentowe od-
chylenie standardowe (%RSD), na-
zywane coraz czêœciej wspó³czynni-
kiem zmiennoœci (ang. coefficient of
variation) o skrócie CV. Wzory po-
zwalaj¹ce wyznaczyæ SD i CV wygl¹-
daj¹ nastêpuj¹co:
(2)
(3),
gdzie:
xi – wynik analityczny,
x – œrednia z wyników analitycznych,
n – liczba wyników analitycznych
.
Na powtarzalnoœæ metody sk³ada
siê wiele elementów. Mo¿na wyzna-
czaæ np.:
РpowtarzalnoϾ aparatu Рodczy-
tu wyników podczas analizy po-
jedynczej próbki,
– powtarzalnoœæ obejmuj¹c¹ pre-
cyzjê mineralizacji próbki oraz
aparatu podczas jej analizy,
– powtarzalnoœæ ca³ej metody od
pobrania próbki poprzez homo-
genizacjê, mineralizacjê do ana-
lizy.
Precyzja analizy w nowoczesnych
aparatach analitycznych jest bardzo
du¿a i zwykle nie ma znacz¹cego
udzia³u w powtarzalnoœci ca³ej meto-
dy. Powtarzalnoœæ analizy metod¹
ICP-OES, któr¹ zamieszczono w ta-
beli 3, okreœlono na podstawie 8 po-
wtórzeñ analizy próbki CTA VTL2
(materia³ certyfikowany – liœcie tyto-
niu z Wirginii). PowtarzalnoϾ wyra-
¿ono za pomoc¹ wspó³czynnika
zmiennoœci (CV).
Powtarzalnoœæ analizy dla wiêk-
szoœci pierwiastków, wyra¿ona jako
CV, jest mniejsza od 1%. Tylko dla
manganu i strontu wartoœci CV s¹
wiêksze i wynosz¹ odpowiednio
1,1% i 3,8%. Nastêpnie wyznaczono
CV na podstawie wyników analizy
próbek materia³u certyfikowanego
CTA VTL2 przygotowanego w powta-
rzalnych warunkach. Wyniki przed-
stawiono w tabeli 4.
PROBLEMY KRYMINALISTYKI 253/06
17
(
)
1
2
−
−
=
∑
n
x
x
SD
i
x
*
SD
CV
100
=
Pierwiastek
Średnia
zawartość
SD
CV
B [µg/g]
22
0,2
0,7
Ba [µg/g]
38,3
0,1
0,2
Ca [%]
3,58
0,03
0,9
Cu [µg/g]
17,2
0,1
0,5
Fe [µg/g]
1075
3
0,3
Mg [%]
0,517
0,01
1,1
Mn [µg/g]
78,1
0,2
0,3
Sr [µg/g]
119
5
3,8
Zn [µg/g]
42,1
0,1
0,3
Pierwiastek
Średnia
zawartość
SD
CV
B [µg/g]
23
1
4,3
Ba [µg/g]
38,8
1,6
4,2
Ca [%]
3,60
0,15
4,3
Cu [µg/g]
17,3
0,7
3,8
Fe [µg/g]
1100
51
4,6
Mg [%]
0,512
0,014
2,6
Mn [µg/g]
78,6
2,5
3,1
Sr [µg/g]
112
5
4,3
Zn [µg/g]
42,5
1,9
4,5
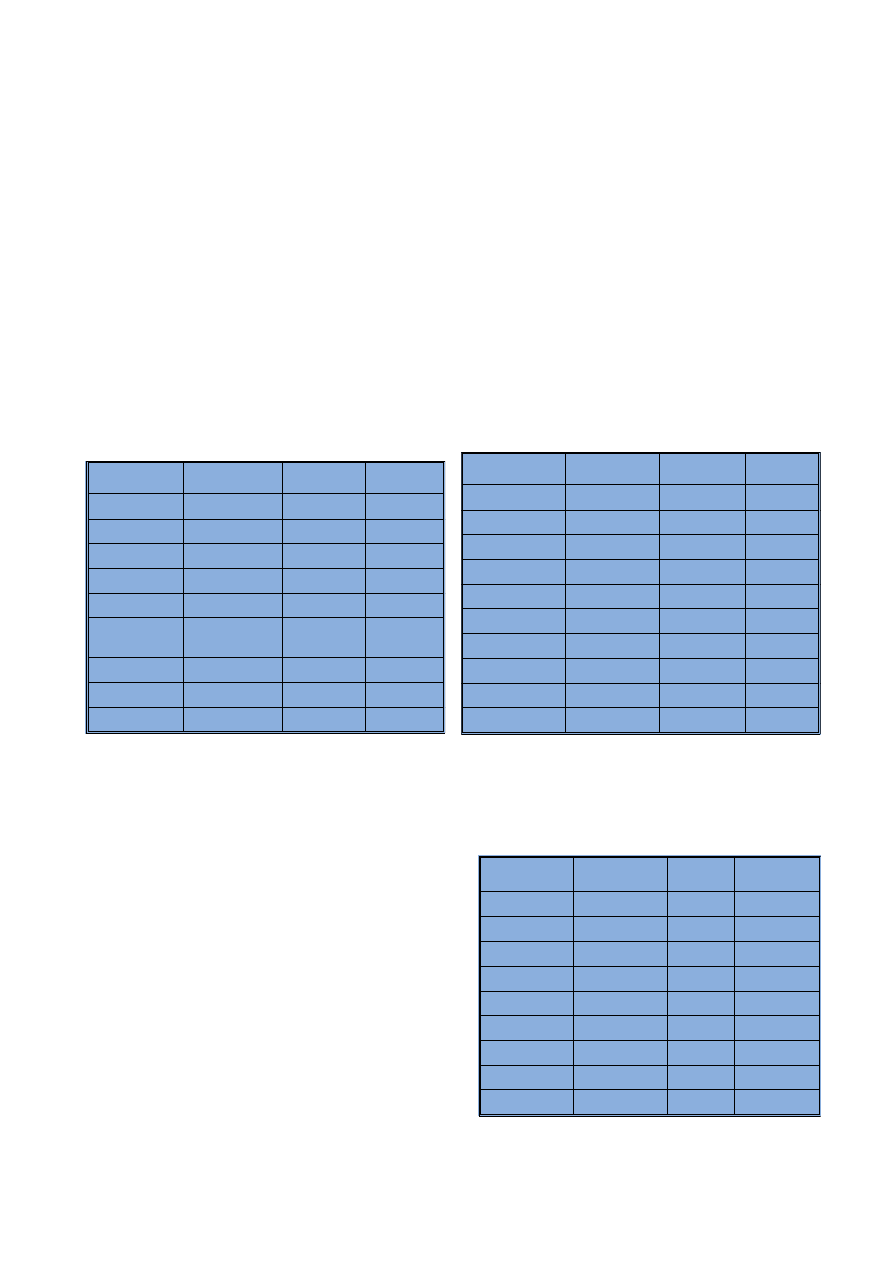
Tabela 3
Powtarzalnoœæ analizy wykonywanej metod¹ ICP-OES
Repeatability of analysis performed by ICP-OES
Tabela 4
Powtarzalnoœæ wyra¿ona jako CV wyznaczona na podsta-
wie analizy materia³u certyfikowanego CTA VTL2
Repeatability expressed as CV determined as the result
of analysis of CTA VTL2 certified material
Wszystkie wartoœci CV mieszcz¹
siê w granicach 2,6
÷4,6%.
Nale¿y zauwa¿yæ, ¿e materia³ cer-
tyfikowany to próbka idealnie jedno-
rodna. Z tego wzglêdu przy wyzna-
czaniu powtarzalnoœci metody na
podstawie jego analizy pomijane s¹
takie etapy jak pobranie próbki i jej
homogenizacja, które, jak mo¿na
przypuszczaæ, maj¹ istotny wk³ad
w powtarzalnoœæ ca³ej metody. Po-
branie próbki reprezentatywnej roœli-
ny konopi jest niezwykle trudnym eta-
pem. Otó¿ do analizy trafiaj¹ zwykle
b¹dŸ ca³e roœliny konopi, b¹dŸ próbki
konopi wstêpnie rozdrobnione. W ce-
lu wykonania analiz porównawczych
niezbêdne jest opracowanie proce-
dury poboru próbki, która umo¿liwia-
³aby wiarygodne porównywanie wyni-
ków uzyskanych dla ró¿nych próbek.
Zbadanie zawartoœci pierwiastków
w poszczególnych czêœciach roœliny
u³atwi³o opracowanie procedury po-
boru próbek konopi.
W tym celu z próbki konopi pobra-
no 8 próbek analitycznych. Poddano
je ujednorodnieniu w oddzielnych po-
jemnikach do homogenizacji m³ynka
planetarnego. Nastêpnie wykonano
mineralizacjê i analizowano. Taki pro-
ces wyznaczania precyzji w warun-
kach powtarzalnych obejmuje
wszystkie etapy postêpowania anali-
tycznego od pobrania próbki, po-
przez jej ujednorodnienie i minerali-
zacjê a¿ do analizy. Wyznaczone
wartoœci CV na podstawie wyników
analizy próbki konopi zamieszczono
w tabeli 5.
Jak wynika z danych zamieszczo-
nych w tabeli 3, dla baru i manganu
wspó³czynniki zmiennoœci wynios³y
odpowiednio 6,2 i 5,9%. Dla pozosta-
³ych pierwiastków nie przekroczy³ on
5%.
Bardzo istotn¹ sk³adow¹ powta-
rzalnoœci metody jest powtarzalnoœæ
procesu kalibracji. By j¹ wyznaczyæ,
przygotowano trzy niezale¿ne krzywe
kalibracyjne. Jedn¹
z wielkoœci charakte-
ryzuj¹cych krzyw¹
kalibracji jest jej na-
chylenie. Porównano
nachylenia trzech
wyznaczonych krzy-
wych. Uzyskane wy-
niki zamieszczono
w tabeli 6.
Wyniki zamiesz-
czone w tab. 4 wska-
zuj¹, ¿e kalibracja to
proces charakteryzu-
j¹cy siê du¿¹ powta-
rzalnoœci¹. Dla wiêk-
szoœci pierwiastków
wartoœci wspó³czyn-
nika zmiennoœci nie
przekracza³y 2%.
W celu wyznaczenia odtwarzalno-
œci wyra¿onej za pomoc¹ CV anali-
zie poddano równie¿ 8 próbek mate-
ria³u certyfikowanego CTA VTL2.
Próbki by³y pobierane, mineralizowa-
ne i analizowane w ró¿nych dniach
analitycznych, w tych dniach przygo-
towywano równie¿ wzorce i wykony-
wano kalibracjê. Nastêpnie spraw-
dzano kalibracjê za pomoc¹ próbki
kontrolnej o certyfikowanych zawar-
toœciach oznaczanych pierwiastków.
Kalibracjê akceptowano, gdy odchy-
lenie od stê¿enia certyfikowanego
nie przekracza³o 5%. W przeciwnym
PROBLEMY KRYMINALISTYKI 253/06
18
Pierwiastek
Średnia
zawartość
SD
CV
B [µg/g]
77
3
3,4
Ba [µg/g]
27,3
1,7
6,2
Ca [%]
3,94
0,14
3,5
Cu [µg/g]
21,4
0,2
1,0
Fe [µg/g]
900
43
4,8
Mg [%]
0,584
0,01
2
2,1
Mn [µg/g]
128
8
5,9
Sr [µg/g]
87
4
4,3
Zn [µg/g]
70,8
2,2
3,1
Pierwiastek
Średnie
nachylenie
SD
CV
B
77000
2000
2,6
Ba
152000
1000
0,7
Ca
95000
2000
2,1
Cu
153000
2000
1,3
Ca
97800
900
0,9
Fe
181000
6000
3,3
Mg
382000
5000
1,3
Mn
599000
1000
0,2
Sr
4870
10
0,2
Zn
131000
4000
3,1
Tabela 5
CV wyznaczone na podstawie wyników analizy próbki konopi
CV determined basing on results of hemp sample analysis
Tabela 6
Powtarzalnoœæ procesu kalibracji metody wyra¿ona jako CV
Repeatability of process of method calibration expressed as CV
Pierwiastek
Średnia
zawartość
SD
CV
B [µg/g]
20,5
1,2
5,6
Ba [µg/g]
31,8
0,8
2,5
Ca [%]
1,08
0,04
3,7
Cu [µg/g]
7,70
0,35
4,5
Fe [µg/g]
459
19
4,2
Mg [%]
0,296
0,010
3,3
Mn [µg/g]
195
3
1,3
Sr [µg/g]
38,2
1,1
2,8
Zn [µg/g]
32,7
1,4
4,2
Tabela 7
Odtwarzalnoœæ wyra¿ona jako CV wyznaczona
na podstawie analizy materia³u certyfikowanego
Reproducibility expressed as CV determined as the result
of analysis of certified material
razie przygotowywano now¹ krzyw¹
kalibracji. Wyniki przedstawiono
w tabeli 7.
Podobnie jak w przypadku powta-
rzalnoœci, wyznaczono nastêpnie od-
twarzalnoœæ metody, poddaj¹c anali-
zie próbkê konopi pobran¹, homoge-
nizowan¹, mineralizowan¹ i analizo-
wan¹ w ró¿nych dniach analitycz-
nych. Uzyskane wyniki zamieszczo-
no w tabeli 8.
Granica wykrywalnoœci
i oznaczalnoœci
Granicê wykrywalnoœci, zgodnie
z zaleceniem organizacji IUPAC, de-
finiuje siê jako najmniejsze stê¿enie,
któremu odpowiada sygna³ ró¿ni¹cy
siê statystycznie (istotnie) od sygna³u
œlepej próby. Najczêœciej stosowane
s¹ podejœcia oparte albo na stosunku
sygna³u do szumu, albo na stosunku
sygna³u do t³a i wzglêdnym odchyle-
niu standardowym t³a. W pierwszym
przypadku wyra¿enie na granicê wy-
krywalnoœci GW mo¿na przedstawiæ
jako [12]:
(4),
gdzie:
k – sta³a,
c0 – stê¿enie odpowiadaj¹ce sygna³o-
wi analitu ya,
SDb – odchylenie standardowe œlepej
próby.
W drugim przypadku wyra¿enie to
przyjmuje postaæ:
(5),
gdzie:
k – sta³a,
c0 – stê¿enie odpowiadaj¹ce sygna³o-
wi analitu ya,
RSDb – wzglêdne odchylenie standar-
dowe œlepej próby,
Ra-b – stosunek sygna³ów t³a i analitu
o stê¿eniu c0.
W kolejnej metodzie do wyznacza-
nia granicy wykrywalnoœci stosowany
jest nastêpuj¹cy wzór:
(6),
gdzie:
k – sta³a,
Xblank – wartoœæ œlepej próby,
SDb – odchylenie standardowe œlepej
próby.
Nale¿y zaznaczyæ, ¿e dla granicy
wykrywalnoœci wartoœæ sta³ej k wyno-
si 3.
Kolejnym sposobem wyznaczania
granicy wykrywalnoœci jest metoda
wizualna. Polega ona na ocenie wi-
zualnej sygna³ów uzyskanych dla
ró¿nych stê¿eñ oznaczanej substan-
cji. Stê¿enia te powinny byæ tak do-
brane, by obejmowa³y zakres niewy-
krywany i wykrywany przez aparat.
Na podstawie oceny wizualnej wybie-
ramy sygna³, który jest wyraŸnie wy-
odrêbniony z t³a, a stê¿enie mu odpo-
wiadaj¹ce to granica wykrywalnoœci.
Granica oznaczalnoœci to naj-
mniejsze stê¿enie, jakie mo¿na wy-
kryæ dan¹ metod¹ z odpowiedni¹ pre-
cyzj¹. By wyznaczyæ granicê ozna-
czalnoœci (GO), nale¿y we wzorach
4
÷6 przyj¹æ wartoœæ sta³ej k=6.
Istnieje wiele metod wyznaczania
granicy wykrywalnoœci i oznaczalno-
œci. W realizowanych badaniach po-
stanowiono przedstawiæ wartoœci
granicy wykrywalnoœci i oznaczalno-
œci uzyskane ró¿nymi metodami. Ja-
ko pierwsz¹ wybrano wyznaczanie
tych parametrów na podstawie analiz
œlepej próby. Wyniki dla 10 œlepych
prób uzyskano w warunkach odtwa-
rzalnych. Na ich podstawie wyzna-
czono wartoœæ granicy wykrywalnoœci
(GW) i oznaczalnoœci (GO) wed³ug
wzorów 4
÷6. Uzyskane wyniki przed-
stawiono w tabeli 9.
Kolejn¹ metod¹ wyznaczania GW
i GO jest metoda wizualna. Jak
PROBLEMY KRYMINALISTYKI 253/06
19
Pierwiastek
Średnia
zawartość
SD
CV
B [µg/g]
26
1
5,7
Ba [µg/g]
43,1
1,3
3,0
Ca [%]
1,25
0,05
3,9
Cu [µg/g]
10,1
0,4
4,3
Fe [µg/g]
512
23
4,5
Mg [%]
0,273
0,011
4,0
Mn [µg/g]
205
4
2,0
Sr [µg/g]
58,1
2,0
3,5
Zn [µg/g]
50,8
2,4
4,8
Tabela 8
OdtwarzalnoϾ wyznaczona na podstawie analizy
próbki konopi
Reproducibility determined basing on analysis
of hemp sample
a
b
y
SD
kc
GW
0
=
b
a
b
R
RSD
kc
GW
−
=
0
b
blank
kSD
X
GW
+
=
Pierwiastek
Średnie stężenie
[mg/l]
SD
[mg/l]
GW
[mg/l]
GO
[mg/l]
B
0,05
0,02
0,10
0,21
Ba
0,01
0,00
0,02
0,03
Ca
0,82
0,22
1,47
2,99
Cu
0,01
0,00
0,03
0,06
Fe
0,06
0,04
0,17
0,44
Mg
0,02
0,01
0,04
0,07
Mn
0,002
0,001
0,006
0,015
Sr
0,37
0,04
0,49
0,76
Zn
0,02
0,01
0,03
0,07
Tabela 9
Wartoœci granicy wykrywalnoœci i oznaczalnoœci metody
wyznaczone na podstawie wyników analizy œlepych prób
Values of detection and determination limits obtained basing
on results of analysis of blind samples
wspomniano, polega ona na zareje-
strowaniu i analizie widm pierwiast-
ków w próbkach wzorców o ró¿nym
stê¿eniu. Przygotowywana jest seria
roztworów o wzrastaj¹cym stê¿eniu
oznaczanego pierwiastka. Nale¿y za-
znaczyæ, ¿e stê¿enia wzorców powin-
ny byæ dobrane tak, by obejmowa³y
poziom, którego aparat nie jest w sta-
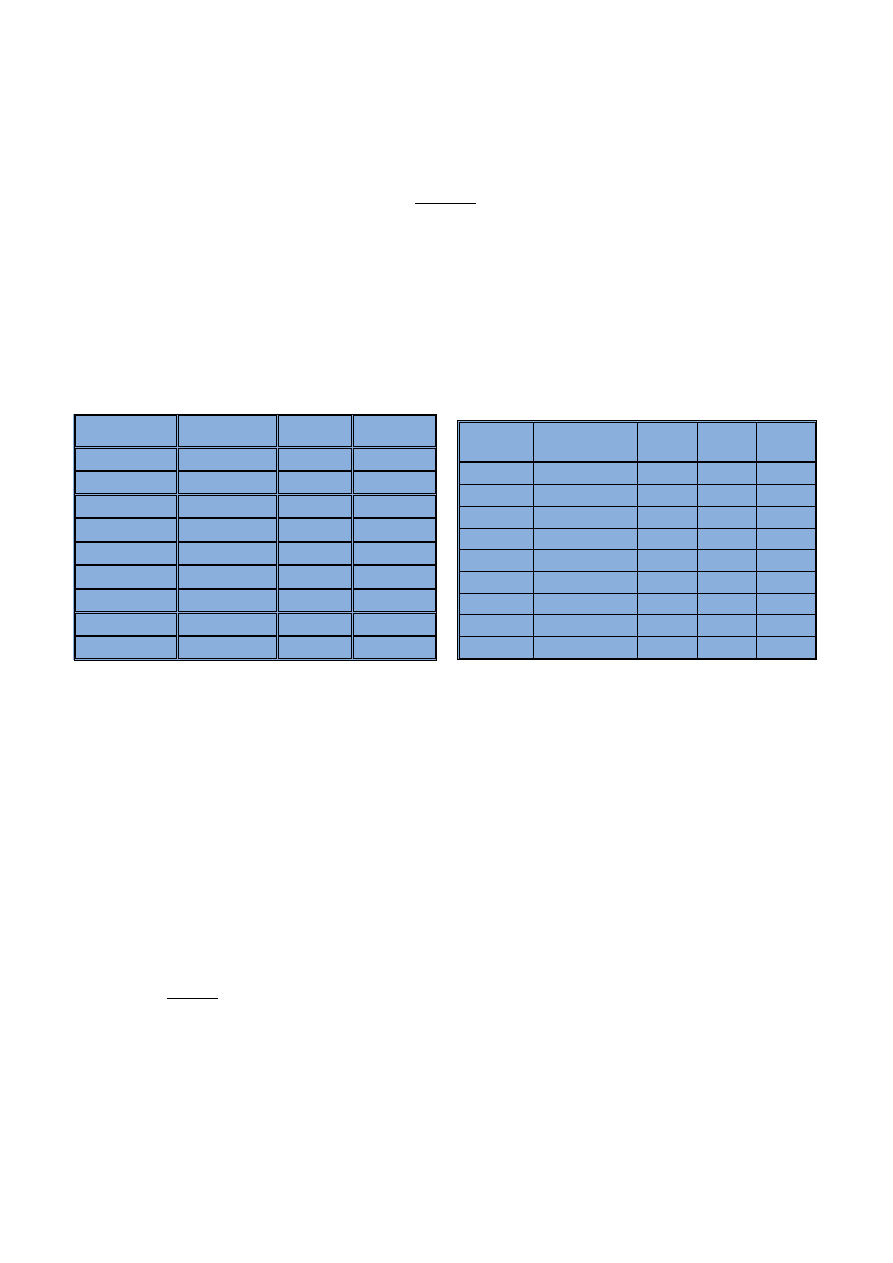
nie wykryæ. W badaniach przygotowa-
no serie wzorców pierwiastków o stê-
¿eniach: 0; 0,01; 0,02; 0,05; 0,1; 0,2;
0,5; 1 i 2 mg/l. Matryc¹ wszystkich
wzorców jest 20-procentowy kwas
azotowy. Widma pierwiastków o wzra-
staj¹cym stê¿eniu zamieszczono na
rycinach 2
÷10. Nale¿y zaznaczyæ, ¿e
zamieszczono widma tylko dla tych
stê¿eñ pierwiastków, które s¹ nie-
zbêdne do wyodrêbnienia granicy wy-
krywalnoœci i oznaczalnoœci.
PROBLEMY KRYMINALISTYKI 253/06
20
B
26000
29000
32000
35000
38000
41000
44000
47000
249.65
249.66
249.67
249.68
249.69
249.7
249.71
D łu go ść fal i [ n m ]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
0,2 mg/l
0,5 mg/l
Ca
49000
51000
53000
55000
57000
59000
61000
63000
315.86
315.87
315.88
315.89
315.9
315.91
315.92
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
0,2 mg/l
Fe
18000
20000
22000
24000
26000
28000
30000
238.18
238.19
238.2
238.21
238.22
238.23
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
Mg
46000
51000
56000
61000
66000
71000
76000
285.17
285.19
285.21
285.23
285.25
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
Cu
100000
105000
110000
115000
120000
125000
130000
135000
327.36
327.38
327.4
327.42
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
0,2 mg/l
0,5 mg/l
Ba
10000
11000
12000
13000
14000
15000
16000
17000
18000
233.505
233.515
233.525
233.535
233.545
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
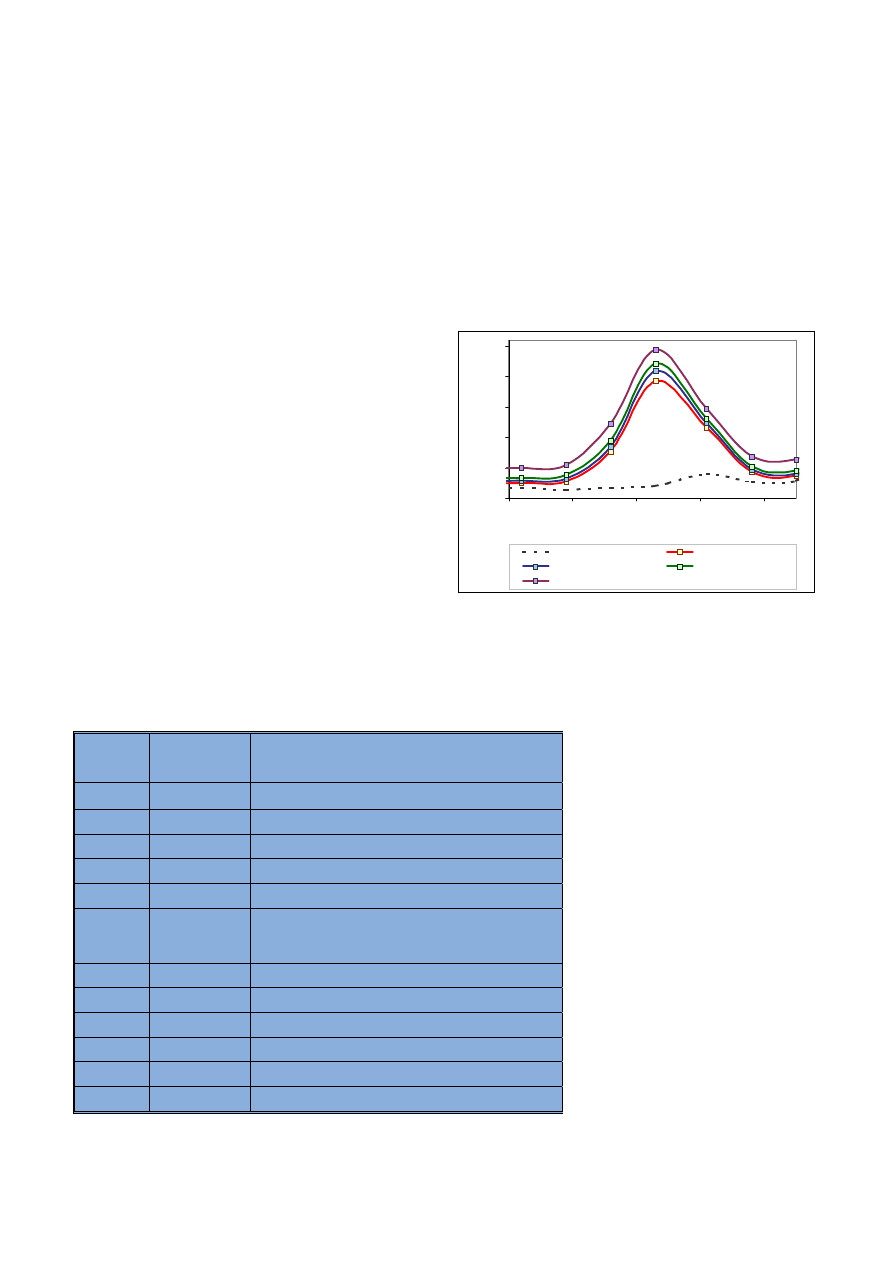
Ryc. 2. Widma uzyskane dla boru o ró¿nych stê¿eniach
Fig. 2. Spectra of boron in various concentrations
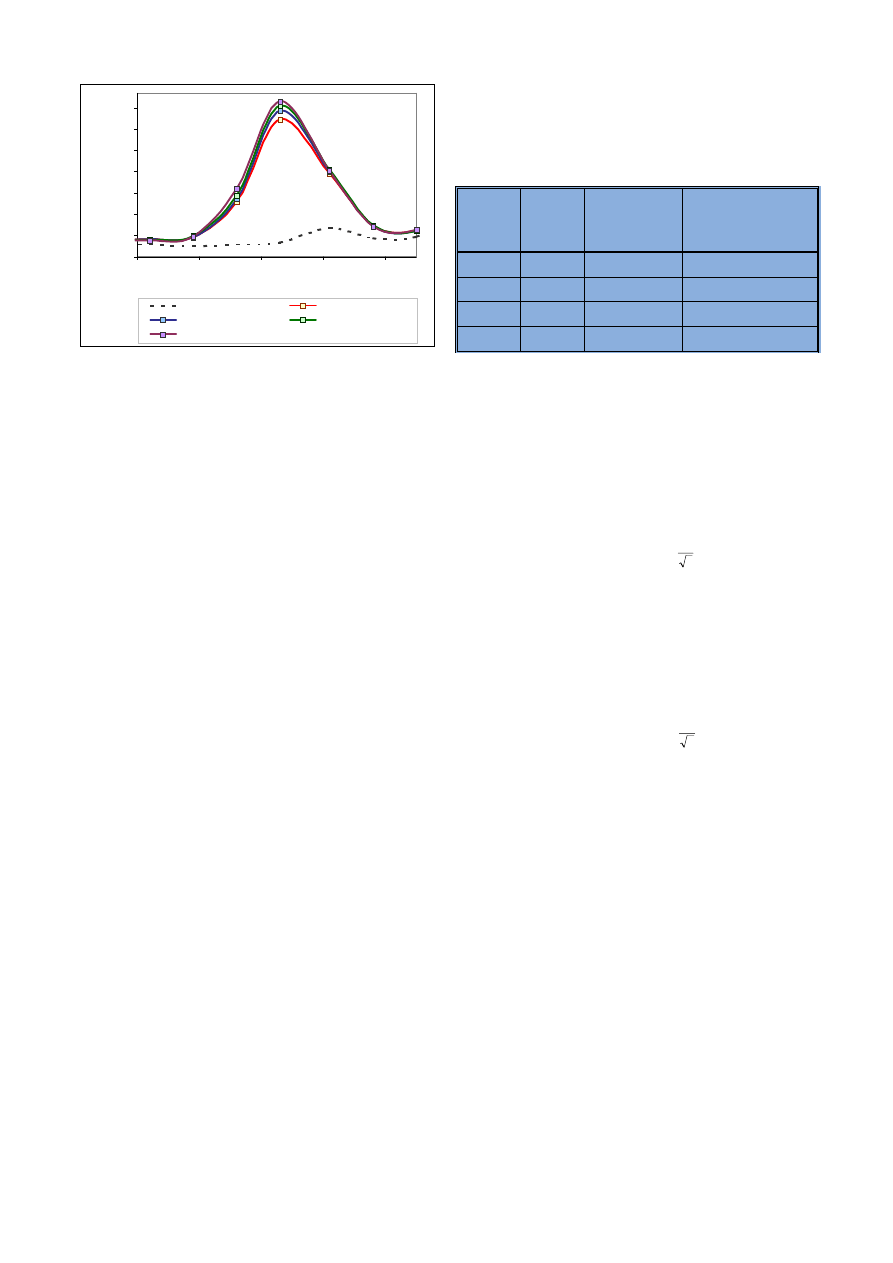
Ryc. 4. Widma uzyskane dla wapnia o ró¿nych stê¿eniach
Fig. 4. Spectra of calcium in various concentrations
Ryc. 5. Widma uzyskane dla miedzi o ró¿nych stê¿eniach
Fig. 5. Spectra of copper in various concentrations
Ryc. 3. Widma uzyskane dla baru o ró¿nych stê¿eniach
Fig. 3. Spectra of barium in various concentrations
Ryc. 6. Widma uzyskane dla ¿elaza o ró¿nych stê¿eniach
Fig. 6. Spectra of iron in various concentrations
Ryc. 7. Widma uzyskane dla magnezu o ró¿nych stê¿eniach
Fig. 7. Spectra of magnesium in various concentrations
W tabeli 10 podano wartoœci grani-
cy wykrywalnoœci i oznaczalnoœci wy-
znaczone na podstawie oceny wizu-
alnej widm.
Zakres prostoliniowy i roboczy
krzywych kalibracyjnych
LiniowoϾ to niezwykle istotny pa-
rametr metody analitycznej. Okreœla
ona zakres prostoliniowej zale¿noœci
pomiêdzy stê¿eniem oznaczanego
sk³adnika i uzyskanym sygna³em
analitycznym. Ze wzglêdów praktycz-
nych po¿¹dane jest, by zakres pro-
stoliniowoœci by³ jak najwiêkszy.
Umo¿liwia to oznaczanie ma³ych
i du¿ych stê¿eñ sk³adnika bez ko-
niecznoœci rozcieñczania próbki. Ko-
relacjê pomiêdzy stê¿eniem a sygna-
³em mo¿na opisaæ za pomoc¹ wspó³-
czynnika korelacji – r.
(7),
gdzie:
xi, yi – wspó³rzêdne kolejnych punk-
tów w analizowanym zbiorze,
n – liczba punktów,
x, y – odpowiednio wartoœci œrednie
wspó³rzêdnych.
Zale¿noœæ miêdzy y (zmienna za-
le¿na) a x (zmienna niezale¿na) bê-
dzie idealnie liniowa, gdy wspó³czyn-
nik korelacji wyniesie 1 albo –1.
W analizie chemicznej oczekiwane
s¹ korelacje lepsze od 0,99.
By wyznaczyæ zakres prostolinio-
wy metody, nale¿y poddaæ analizie
seriê co najmniej piêciu roztworów
analitu o ró¿nym stê¿eniu, a nastêp-
nie obliczyæ wspó³czynnik korelacji.
Zakres roboczy krzywej kalibracji
to zakres stê¿eñ, z jakiego korzysta
siê w danej metodzie analitycznej.
Musi charakteryzowaæ siê dobrym
wspó³czynnikiem korelacji.
Ró¿nicê miêdzy zakresem prosto-
liniowym a roboczym metody przed-
stawiono na rycinie 11.
Metoda ICP-OES ma du¿y zakres
prostoliniowoœci krzywych kalibracyj-
nych, zwykle siêgaj¹cy kilku rzêdów
PROBLEMY KRYMINALISTYKI 253/06
21
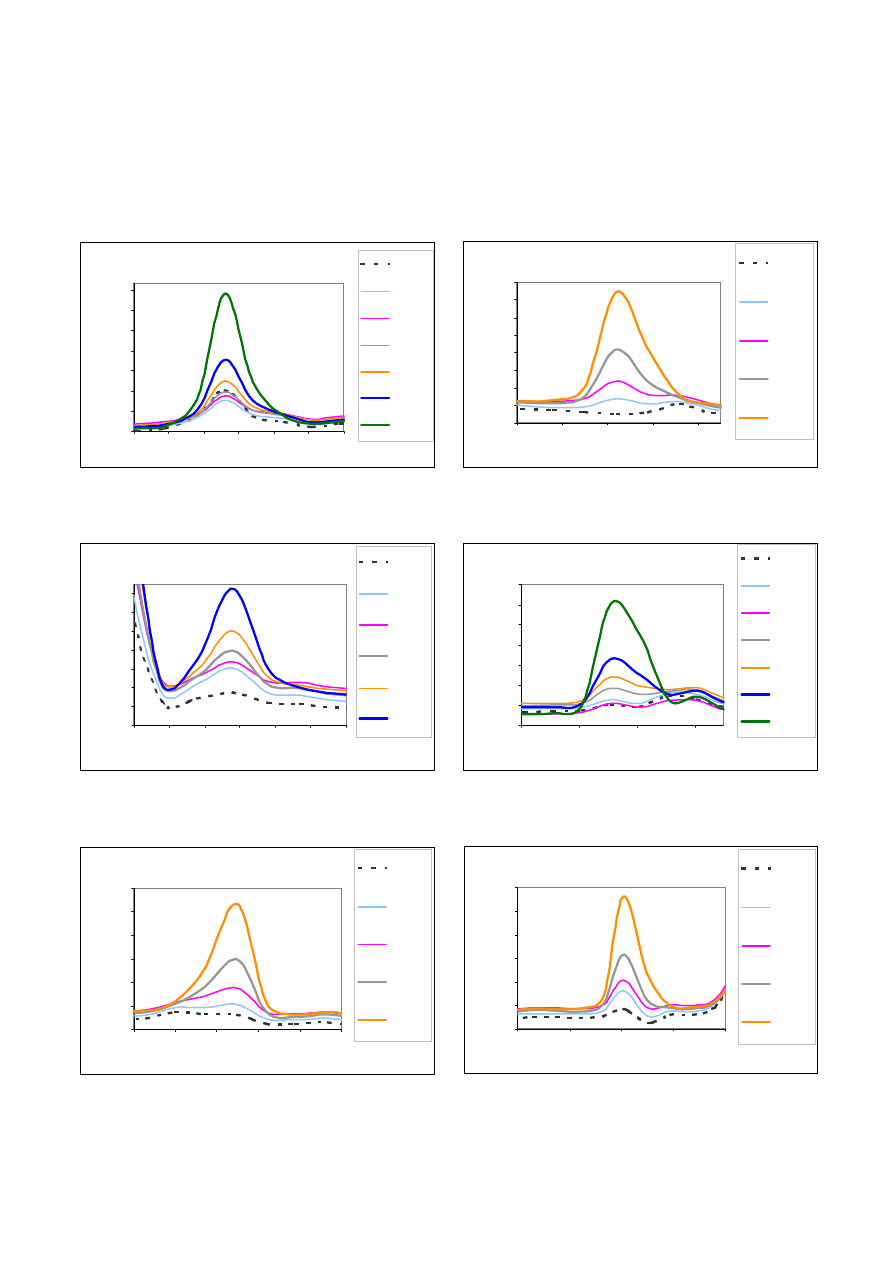
Mn
16500
17500
18500
19500
20500
21500
22500
23500
24500
257.57
257.59
257.61
257.63
257.65
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
Zn
6000
6500
7000
7500
8000
8500
9000
9500
10000
10500
206.176
206.186
206.196
206.206
206.216
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
Sr
16500
17500
18500
19500
20500
21500
232.21
232.22
232.23
232.24
232.25
232.26
D ługość fal i [ nm]
Intensywność [cps]
0 mg/l
0,01 mg/l
0,02 mg/l
0,05 mg/l
0,1 mg/l
0,2 mg/l
0,5 mg/l
1 mg/l
2 mg/l
Ryc. 8. Widma uzyskane dla manganu o ró¿nych stê¿eniach
Fig. 8. Spectra of manganese in various concentrations
Ryc. 9. Widma uzyskane dla strontu o ró¿nych stê¿eniach
Fig. 9. Spectra of strontium in various concentrations
Ryc. 10. Widma uzyskane dla cynku o ró¿nych stê¿eniach
Fig. 10. Spectra of zinc in various concentrations
Tabela 10
Wartoœci granicy wykrywalnoœci i oznaczalnoœci metody
wyznaczone na podstawie wizualnej oceny widm
Values of detection and determination limits obtained basing
on visual assessment of spectra
P
Piie
errw
wiiaasstte
ek
k
G
GW
W
[[m
mg
g//ll]]
G
GO
O
[[m
mg
g//ll]]
B
B
0,2
0,5
B
Baa
0,05
0,1
C
Caa
0,05
0,2
C
Cu
u
0,2
0,5
F
Fe
e
0,05
0,1
M
Mg
g
0,05
0,1
M
Mn
n
0,01
0,02
S
Srr
1
2
Z
Zn
n
0,02
0,05
(
)(
)
(
) (
)
∑
∑
∑
=
=
=
−
−
−
−
=
n
i
n
i
i
i
n
i
i
i
y
y
x
x
y
y
x
x
r
1
1
2
2
1
wielkoœci. Jednak ka¿da krzywa kali-
bracji odznacza siê ograniczonym
zakresem.
W przypadku analizy próbek kono-
pi najwiêksze stê¿enia oznaczane s¹
dla wapnia i magnezu – do odpo-
wiednio 2100 mg/l i 1270 mg/l. Jedy-
nie w przypadku tych dwóch pier-
wiastków jest uzasadnione wyzna-
czanie prostoliniowego zakresu krzy-
wych kalibracji. W tym celu przygoto-
wano i analizowano serie roztworów
kalibracyjnych zawieraj¹cych:
– dla wapnia 0, 100, 200, 400,
600, 700, 800, 900, 1000 mg/l,
– dla magnezu 0, 50, 100, 150,
175 i 200 mg/l.
Uzyskane wyniki zamieszczono
na rycinach 12 i 13.
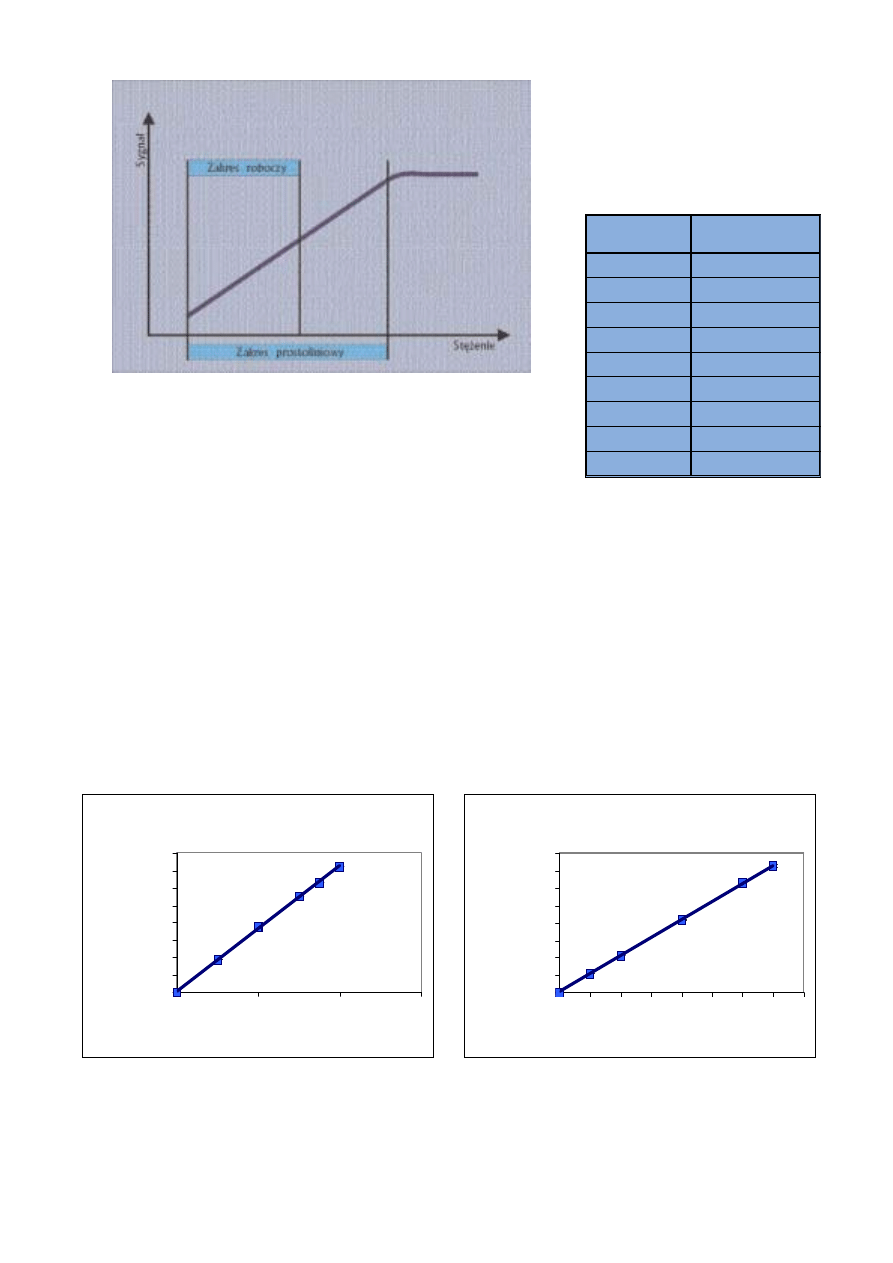
Powy¿ej stê¿enia 200 mg/l nastê-
puje prze³adowanie linii Mg 285,213.
Zatem oznaczanie wy¿szych stê¿eñ
za pomoc¹ tej linii nie jest mo¿liwe.
Wartoœæ wspó³czynnika (0,9995)
wskazuje na du¿¹ prostoliniowoœæ
krzywej kalibracji.
W przypadku wapnia krzywa jest
prostoliniowa w zakresie 0
÷700 mg/l.
Wspó³czynnik korelacji wynosi
0,9998. Powy¿ej 700 mg/l nastêpuje
prze³adowanie linii wapnia, co unie-
mo¿liwia oznaczanie wy¿szych stê-
¿eñ.
W tabeli 11 zamieszczono zakresy
robocze krzywych kalibracji dla
wszystkich oznaczanych pierwiast-
ków. Czu³oœæ metody wyra¿ona jako
nachylenie krzywej kalibracji za-
mieszczono wczeœniej w tabeli 6.
SelektywnoϾ
SelektywnoϾ to zdolnoϾ metody
do odró¿niania oznaczanego analitu
od innych substancji. Badanie selek-
tywnoœci zwykle przeprowadza siê
poprzez analizê próbek analitów, do
których dodano potencjalne interfe-
renty, i obserwacjê uzyskiwanych sy-
gna³ów.
Metoda ICP-OES nie odznacza
siê niestety dobr¹ selektywnoœci¹ ani
specyficznoœci¹. Mo¿liwoœæ jedno-
czesnego oznaczania zdecydowanej
wiêkszoœci pierwiastków uk³adu
okresowego jest du¿¹ zalet¹, lecz
PROBLEMY KRYMINALISTYKI 253/06
22
Ryc. 11. Zakres prostoliniowy i roboczy metody analitycznej
Fig. 11. Linear and working range of analytical method
Mg 285.213
R
2
= 0.9995
0
10000000
20000000
30000000
40000000
50000000
60000000
70000000
80000000
0
100
200
300
st ężeni e Mg [ mg/l ]
intensywność [cps]
Ca 315.877
R
2
= 0.9998
0
10000000
20000000
30000000
40000000
50000000
60000000
70000000
80000000
0
100
200
300
400 500
600
700
800
st ężeni e C a [ mg/l ]
intensywność [cps]
Ryc. 12. Zakres prostoliniowy krzywej kalibracji wyznaczonej dla magnezu
Fig. 12. Rectilinear range of calibration curve for magnesium
Ryc. 13. Zakres prostoliniowy krzywej kalibracji wyznaczonej dla wapnia
Fig. 13. Rectilinear range of calibration curve for calcium
Pierwiastek
Zakres roboczy
[mg/l]
B
0÷10
Ba
0÷10
Ca
0÷500
Cu
0÷10
Fe
0÷100
Mg
0÷200
Mn
0÷50
Sr
0÷10
Zn
0÷10
Tabela 11
Zakres roboczy krzywych kalibracji
Working range of calibration curves
mo¿e równie¿ byæ wad¹. Wystêpo-
wanie sygna³ów pierwiastka przy
wielu ró¿nych d³ugoœciach fali mo¿e
prowadziæ do ich nak³adania siê z sy-
gna³ami innych pierwiastków. Naj-
wiêksze b³êdy zwi¹zane z nak³ada-
niem siê sygna³ów obserwowane s¹,
gdy obok siebie wystêpuj¹ silne linie
pierwiastka oznaczanego i interferen-
ta. Emitowanie wielu fal daje mo¿li-
woϾ prostej eliminacji interferencji
spektralnych poprzez wybranie innej
– nieobarczonej interferencjami linii
analitycznej. Jednak czêsto takie linie
s¹ mniej czu³e i wykrycie stê¿eñ œla-
dowych za ich pomoc¹ jest utrudnio-
ne.
W trakcie walidacji metody ozna-
czania pierwiastków w próbkach ko-
nopi sprawdzono, jakie potencjalne
interferenty emituj¹ sygna³y w pobli-
¿u linii analitycznych oznaczanych
pierwiastków. Zestawienie tych infor-
macji zamieszczono w tabeli 12. Bra-
no pod uwagê linie interferentów
znajduj¹ce siê w odleg³oœci mniejszej
ni¿ 0,005 nm od linii oznaczanego
pierwiastka.
W wyznaczaniu interferencji spek-
tralnych najwiêksz¹ uwagê zwrócono
na liniê strontu. Spektrometr, za po-
moc¹ którego wykonywano analizy,
wyposa¿ony jest w detektor w zakre-
sie UV. W tym zakresie stront emituje
tylko jedn¹ falê o stosunkowo ma³ej
czu³oœci. Interferencja spektralna
mog³aby uniemo¿liwiæ dok³adne
oznaczanie tego pierwiastka w prób-
kach konopi. Jedynym pierwiastkiem,
który móg³by zak³ócaæ sygna³ Sr, jest
¿elazo. Linia Fe
232,233
jest bardzo
s³aba, lecz ze wzglêdu na idealne na-
³o¿enie z lini¹ Sr postanowiono
sprawdziæ jej
wp³yw. Dodatko-
wym czynnikiem
przemawiaj¹cym
za sprawdzeniem
wystêpowania tej
interferencji jest
fakt, ¿e stront
w próbkach kono-
pi wystêpuje na
poziomie stê¿eñ
kilkakrotnie ni¿-
szym od ¿elaza.
Zatem ¿elazo mo-
¿e mieæ faktyczny
wp³yw na wyniki
oznaczeñ. W celu
sprawdzenia, czy
tak jest w istocie,
przygotowano seriê czterech roztwo-
rów wzorcowych oraz œlep¹ próbê.
W ka¿dym roztworze stê¿enie strontu
by³o sta³e i wynosi³o 2,5 mg/l. Stê¿e-
nie ¿elaza wynosi³o odpowiednio 0,
10, 20 i 50 mg/l. Nale¿y zaznaczyæ,
¿e stê¿enia strontu i ¿elaza dobrano
tak, by odzwierciedla³y poziom wy-
stêpuj¹cy w próbkach konopi. Wyniki
analizy przedstawiono na rycinach
14, 15 oraz w tabeli 13.
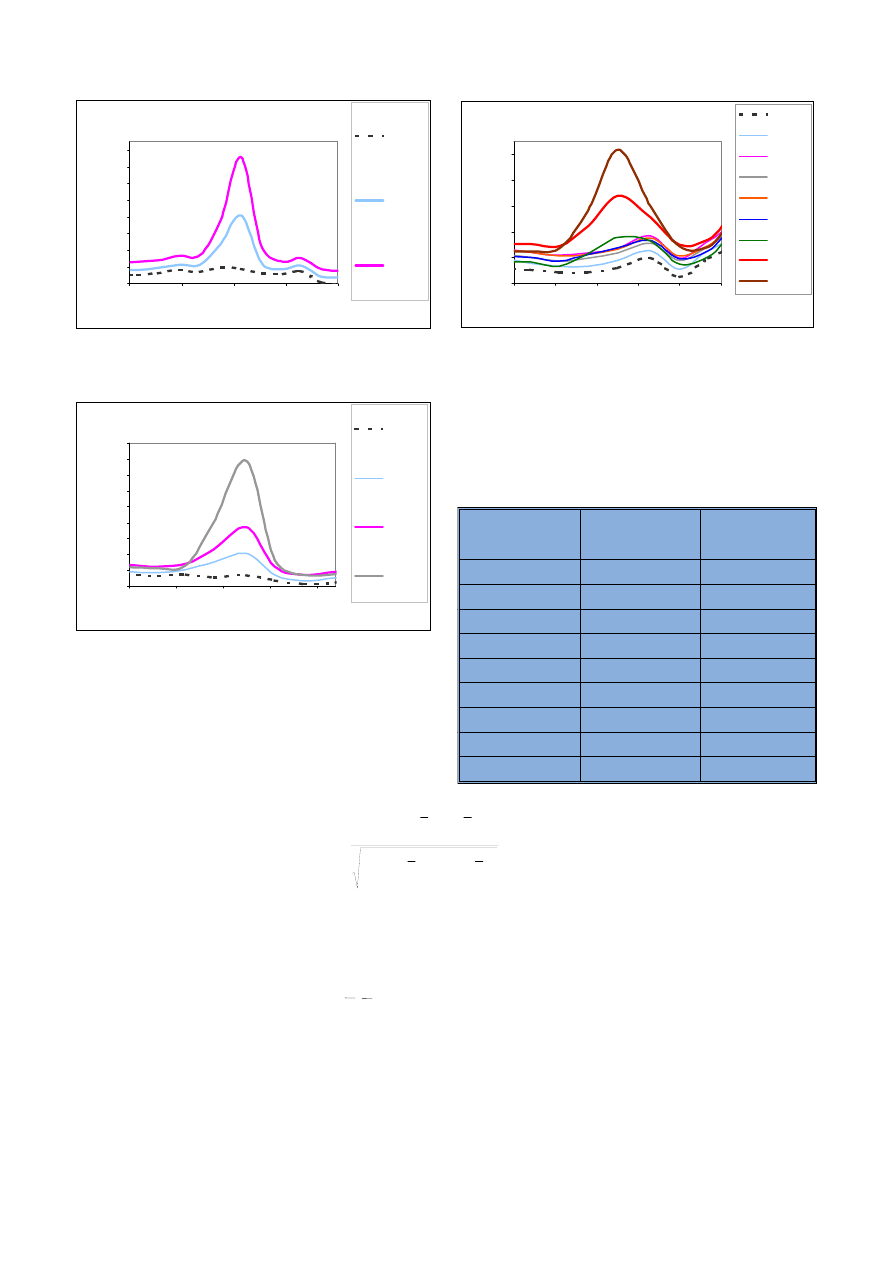
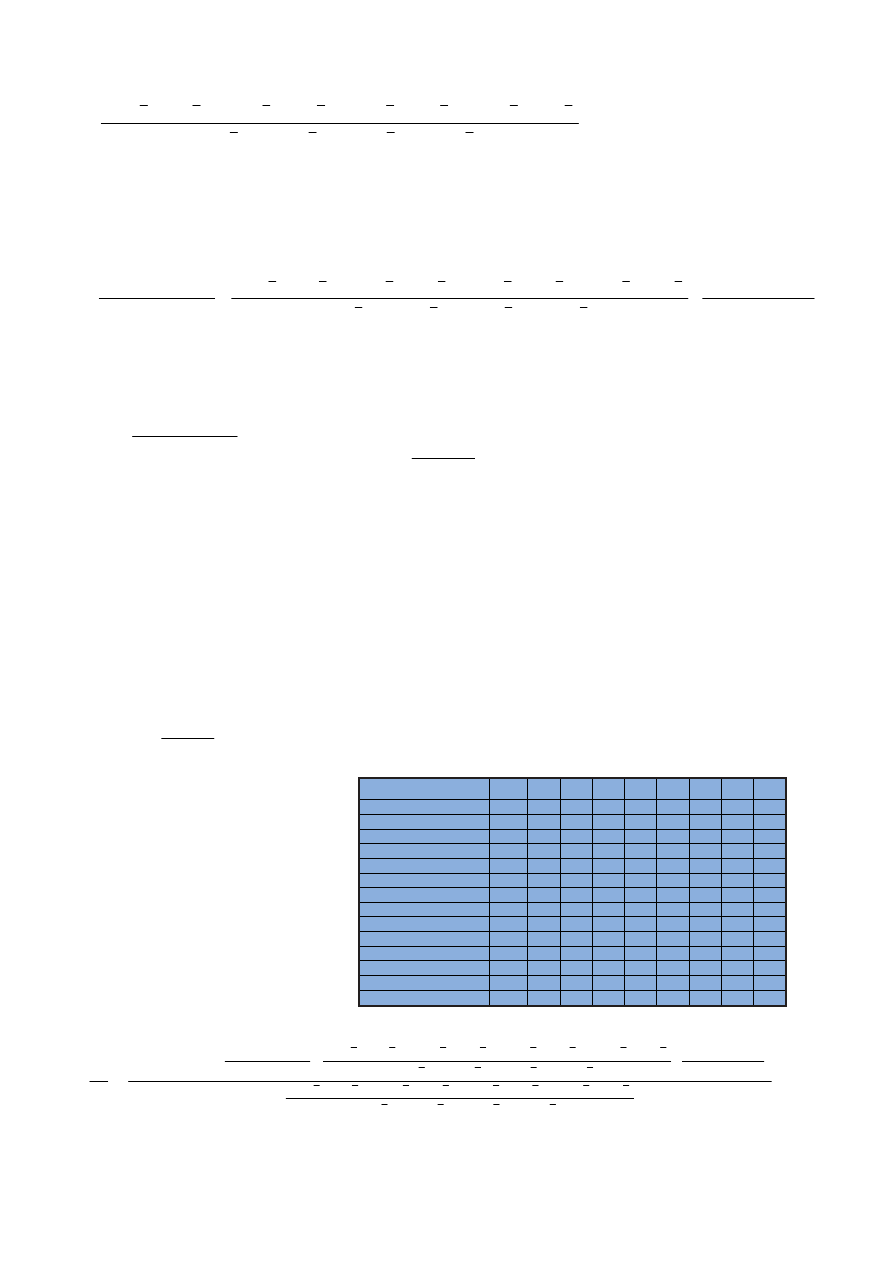
Analiza widm zamieszczonych na
rycinie 14 wskazuje, ¿e obecnoœæ ¿e-
laza powoduje przede wszystkim
wzrost poziomu t³a, który jest propor-
cjonalny do stê¿enia ¿elaza w prób-
ce. Na podstawie tych widm trudno
okreœliæ, czy wystêpuj¹ interferencje
spektralne, czyli czy nastêpuje
wzrost intensywnoœci sygna³u strontu
w obecnoœci coraz wiêkszych stê¿eñ
¿elaza.
Na rycinie 15 zamieszczono wid-
ma takie same jak na rycinie 14, lecz
z przesuniêtym poziomem t³a. Wy-
stêpowanie t³a na podobnym pozio-
mie u³atwia interpretacjê wizualn¹
widm. Okazuje siê, ¿e wysokoœæ sy-
gna³u Sr roœnie wraz ze wzrostem za-
wartoœci ¿elaza w próbce. Œwiadczy
to o wystêpowaniu interferencji spek-
tralnych.
Dane zestawione w tabeli 13 po-
zwalaj¹ na oszacowanie wielkoœci tej
interferencji. Okazuje siê, ¿e stê¿enie
¿elaza w wysokoœci 10 mg/l powodu-
PROBLEMY KRYMINALISTYKI 253/06
23
Pierwiastek
Długość fali
[nm]
Potencjalny interferent/długość fali [nm]
B
249,677
Tc 249,677; Sn 249,677; Hg 249,678; Cr 249,681
Ba
233,527
Nb 233,531; V 233,533
Be
234,861
Mo 234,858; Ta 234,859; Zr 234,859
Be
313,107
Th 313,107; Zr 313,111
Ca
315,887
Ce 315,888
Cu
327,393
Nb 327,389; U 327,390; Th 327,392; Ce 327,393;
Co 327,393; Ta 327,396; Mo 327,396; Ce 327,396;
Sb 327,397
Fe
238,204
V 238,203; Hg 238,206
Mg
285,213
U 285,209; W 285,210; Ce 285,212; Mo 285,213;
Mn
257,610
Ru 257,609; Co 257,610; Zr 257,610
Sr
232,235
Fe 232,233
Zn
206,200
Nb 206,297; In 206,200; V 206,200
Y
371,029
Ce 371,025; W 371,029; U 371,031
Tabela 12
Potencjalne interferenty [13]
Possible interferents
Sr
12000
14500
17000
19500
22000
24500
232.22
232.23
232.24
232.25
232.26
długość fal i [ nm]
intensywność [cps]
Sr 0 mg/l + Fe 0 mg/l
Sr 2.5 mg/l
Sr 2.5 mg/l + Fe 10 mg/l
Sr 2.5 mg/l + Fe 20 mg/l
Sr 2.5 mg/l + Fe 50 mg/l
Ryc. 14. Widma uzyskane dla strontu w matrycy ¿elaza o ró¿nym stê¿eniu
Fig. 14. Spectra of strontium in ferrous matrix in various concentrations
je wzrost intensywnoœci strontu o 8%,
a w wysokoœci 50 mg/l a¿ o 22%
w stosunku do sygna³u uzyskanego
bez dodatku interferenta.
Wyniki uzyskane dla 105 próbek
konopi wskazuj¹, ¿e oznaczane stê-
¿enie ¿elaza nie przekracza 40 mg/l.
Nale¿y zaznaczyæ, ¿e dla 72% pró-
bek oznaczone stê¿enie tego pier-
wiastka nie przekracza³o 10 mg/l,
a tylko w 5% próbek wynios³o wiêcej
ni¿ 20 mg/l. Te informacje wskazuj¹,
¿e efekt interferencyjny dla strontu
jest istotny dla 28% badanych pró-
bek.
Szacowanie niepewnoœci metody
Na niepewnoϾ metody oznacza-
nia pierwiastków w próbce konopi
technik¹ ICP-OES wp³ywa wiele
czynników.
Niepewnoœæ wynikaj¹ca z procesu
przygotowania próbki obejmuje na-
stêpuj¹ce sk³adniki:
РniepewnoϾ wyznaczania masy
próbki (niepewnoœæ wagi analitycz-
nej),
– niepewnoœæ kolby, w której przy-
gotowywana jest próbka.
NiepewnoϾ wyznaczania kalibra-
cji i oznaczania pierwiastka w próbce
konopi obejmuje cztery rodzaje
sk³adników:
– sk³adniki niepewnoœci zwi¹zane
z
przygotowywaniem roztworów
wzorcowych,
– niepewnoœæ wzorca wyjœciowe-
go, z którego w wyniku rozcieñczenia
przygotowywane s¹ wzorce robocze,
– niepewnoœæ objêtoœci pipetowa-
nego wzorca (niepewnoϾ pipety),
– niepewnoœæ kolb, w których
przygotowywane s¹ wzorce robocze.
Sk³adniki niepewnoœci zwi¹zane
z analiz¹ wzorców roboczych metod¹
ICP-OES:
РniepewnoϾ wyznaczenia inten-
sywnoœci sygna³u dla pierwiastków
we wzorcach roboczych,
– sk³adniki niepewnoœci zwi¹zane
z analiz¹ próbki konopi,
РniepewnoϾ wyznaczenia inten-
sywnoœci sygna³u dla pierwiastków
w próbce konopi,
РniepewnoϾ wyznaczenia inten-
sywnoœci sygna³u dla pierwiastków
w œlepej próbie.
Sk³adniki niepewnoœci nale¿y po-
grupowaæ w zale¿noœci od tego, ja-
kim typom szacowania niepewnoœci
podlegaj¹ (A czy B).
W tym przypadku typ A szacowa-
nia niepewnoœci obejmuje sk³adniki,
które zosta³y wyznaczone doœwiad-
czalnie, tj. intensywnoœci pierwiast-
ków dla próbki, intensywnoœci pier-
wiastków dla œlepej próby i intensyw-
noœci pierwiastków dla wzorców. Dla
tych sk³adników niepewnoœæ standar-
dowa (U) jest równa odchyleniu stan-
dardowemu.
Sk³adniki podlegaj¹ce typowi B
szacowania niepewnoœci to: objêtoœæ
kolb, w których przygotowywano prób-
kê i wzorce robocze, objêtoœæ pipeto-
wanego wzorca oraz masa próbki.
Prostok¹tnemu rozk³adowi praw-
dopodobieñstwa, dla którego niepew-
noœæ standardowa jest równa
U=
podlegaj¹ objêtoœci kolb, w któ-
rych przygotowywano próbkê i wzor-
ce robocze, a tak¿e objêtoœæ pipeto-
wanego wzorca. Natomiast masa
próbki, czyli niepewnoœæ wagi, bêdzie
podlegaæ trójk¹tnemu rozk³adowi
prawdopodobieñstwa, dla którego
U =
Aby opracowaæ model szacowa-
nia niepewnoœci, nale¿y równie¿
znaæ model matematyczny, wed³ug
którego spektrometr oblicza stê¿enie
analitu na podstawie zmierzonej in-
tensywnoœci sygna³u.
Równanie krzywej wzorcowej
mo¿na przedstawiæ nastêpuj¹co:
y = ax + b
(8),
gdzie:
y – intensywnoœæ sygna³u analityczne-
go,
x – stê¿enie,
a – nachylenie krzywej,
b – punkt przeciêcia krzywej z osi¹ y.
WartoϾ a obliczana jest ze wzoru 9.
WartoϾ b obliczana jest ze wzoru 10:
PROBLEMY KRYMINALISTYKI 253/06
24
Ryc. 15. Wystandaryzowane widma uzyskane dla strontu w matrycy ¿elaza
o ró¿nym stê¿eniu
Fig. 15. Standardised spectra of strontium in ferrous matrix in various
concentrations
Tabela 13
Intensywnoœci emisji uzyskane dla strontu w obecnoœci
matrycy ¿elaza o ró¿nym stê¿eniu
Emission intensities obtained for strontium in presence
of ferrous matrix
Sr
12000
13500
15000
16500
18000
19500
21000
22500
232.22
232.23
232.24
232.25
232.26
długość fal i [ nm]
intensywność [cps]
Sr 0 mg/l + Fe 0 mg/l
Sr 2.5 mg/l
Sr 2.5 mg/l + Fe 10 mg/l
Sr 2.5 mg/l + Fe 20 mg/l
Sr 2.5 mg/l + Fe 50 mg/l
Stężenie
Sr
[mg/l]
Stężenie
Fe
[mg/l]
Intensywność
dla Sr
[cps]
Intensywność względna
dla Sr
[%]
2,5
0
10537
100
2,5
10
11330
108
2,5
20
12037
114
2,5
50
12805
122
3
Sd
6
Sd
gdzie:
xi – wartoœæ stê¿enia wzorca,
yi – wartoœæ sygna³u analitycznego.
Stê¿enie nieznanej próbki w mg/l
obliczane jest z zale¿noœci 11.
(11),
gdzie:
x0 – wartoœæ stê¿enia [mg/l],
y0x – wartoœæ œrednia sygna³u anali-
tycznego dla nieznanej próbki [mg/l],
y0sp – wartoœæ œrednia sygna³u anali-
tycznego dla œlepej próby [mg/l].
Stê¿enie analitu wyra¿one w mg/l
przekszta³cane jest na mg/kg wed³ug
wzoru (12) z uwzglêdnieniem masy
próbki oraz objêtoœci kolby, w której
j¹ przygotowano:
(12),
gdzie:
C – wartoœæ stê¿enia [mg/kg],
x0 – wartoœæ stê¿enia [mg/l],
Vp – objêtoœæ kolby, w której przygoto-
wano próbkê [ml],
m – masa próbki [g].
Po po³¹czeniu tych wzorów 8–12
otrzymujemy model matematyczny
opisany wzorem 13.
Jednak nale¿y dodatkowo
uwzglêdniæ fakt, ¿e wzorce kalibra-
cyjne zosta³y przygotowane przez
rozcieñczanie z
odpowiedniego
wzorca wyjœciowego. Stê¿enie wzor-
ca do kalibracji mo¿na wyraziæ wzo-
rem 14.
(14),
gdzie:
Cwz – stê¿enie wzorca wyjœciowego
[mg/l],
Vi – objêtoœæ odpipetowanego wzorca
[ml],
Vk – objêtoœæ kolby, w której przygoto-
wywano wzorzec [ml].
T¹ zale¿noœæ nale¿y wstawiæ
w miejsce x
1
, x
2
, x
3
do modelu mate-
matycznego (wzór 13). Poniewa¿ to
jeszcze bardzie komplikuje model ma-
tematyczny, zrezygnowano z wsta-
wiania tego wzoru w tym miejscu pra-
cy. Pe³ny model matematyczny jest
uwzglêdniony w skoroszycie Microsoft
Excel, który by³ wykorzystywany w ob-
liczeniach.
W tabeli 14 podano wzglêdny
udzia³ poszczególnych elementów
w niepewnoœci z³o¿onej.
Dla pierwiastków B, Ba, Cu, Sr i Zn
najwiêkszy udzia³ w niepewnoœci me-
tody ma stê¿enie wzorca g³ównego.
NiepewnoϾ tego wzorca stanowi od
39,9% ca³kowitej niepewnoœci dla
miedzi do 61,0% dla cynku. Otó¿ jest
to zwi¹zane z faktem, ¿e roztwór tego
wzorca przygotowano poprzez roz-
cieñczenie wzorca wyjœciowego
o stê¿eniu ka¿dego z pierwiastków
1000 mg/l. Oczywiste jest, ¿e wraz
z rozcieñczaniem próbki (wzorca) ro-
œnie niepewnoœæ metody. By zmniej-
szyæ zatem niepewnoœæ oznaczania
tych pierwiastków, nale¿y zastosowaæ
roztwór wzorca o stê¿eniu 100 mg/l.
PROBLEMY KRYMINALISTYKI 253/06
25
(
)(
) (
)(
) (
)(
) (
)(
)
(
) (
) (
) (
)
2
4
2
3
2
2
2
1
4
4
3
3
2
2
1
1
x
x
x
x
x
x
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
x
x
a
−
+
−
+
−
+
−
−
−
+
−
−
+
−
−
+
−
−
=
(
)(
) (
)(
) (
)(
) (
)(
)
(
) (
) (
) (
)
4
4
4
3
2
1
2
4
2
3
2
2
2
1
4
4
3
3
2
2
1
1
4
3
2
1
x
x
x
x
x
x
x
x
x
x
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
y
y
b
+
+
+
∗
−
+
−
+
−
+
−
−
−
+
−
−
+
−
−
+
−
−
−
+
+
+
=
(9)
(
)
a
b
y
y
x
osp
ox
o
−
−
=
m
V
x
C
p
∗
=
0
Vk
V
Cwz
x
i
i
∗
=
(
)
(
)(
) (
)(
) (
)(
) (
)(
)
(
) (
) (
) (
)
(
)(
) (
)(
) (
)(
) (
)(
)
(
) (
) (
) (
)
2
4
2
3
2
2
2
1
4
4
3
3
2
2
1
1
4
3
2
1
2
4
2
3
2
2
2
1
4
4
3
3
2
2
1
1
4
3
2
1
0
4
4
x
x
x
x
x
x
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
x
x
m
x
x
x
x
x
x
x
x
x
x
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
x
x
y
y
y
y
y
y
V
kg
mg
C
sp
ox
p
−
+
−
+
−
+
−
−
−
+
−
−
+
−
−
+
−
−
∗
+
+
+
∗
−
+
−
+
−
+
−
−
−
+
−
−
+
−
−
+
−
−
−
+
+
+
−
−
∗
=
(13)
Element metody
B
Ba
Ca
Cu
Fe
Mg
Mn
Sr
Zn
Intensywność próbki [cps]
26,2
23,7
16,3
14,57
0,2
18,8
1,8
30,6
17,8
Intensywność ślepej próbki [cps]
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
Intensywność wzorca 1 [cps]
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
3,2
<0,1
Intensywność wzorca 2 [cps]
0,9
0,4
<0,1
2,42
<0,1
0,5
0,11
<0,1
0,1
Intensywność wzorca 3 [cps]
5,6
4,5
0,7
10,4
3,6
1,6
2,13
6,9
7,1
Intensywność wzorca 4 [cps]
0,03
0,5
7,2
2,3
4,0
6,2
5,3
1,2
0,7
Objętość próbki [ml]
0,3
0,3
1,3
0,20
2,6
1,0
1,50
0,2
0,3
Masa próbki [g]
<0,1
<0,1
0,2
<0,1
0,2
0,1
0,1
<0,1
<0,1
Stężenie wzorca głównego [mg/l]
55,0
58,3
0,2
39,9
0,4
0,4
0,2
49,1
61,0
Objętość wzorca 1 [ml]
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
<0,1
Objętość wzorca 2 [ml]
3,6
3,5
<0,1
9,7
15,2
<0,1
0,1
1,7
3,7
Objętość wzorca 3 [ml]
7,3
7,5
2,3
16,6
37,8
11,0
28,1
4,1
8,0
Objętość wzorca 4 [ml]
0,7
1,0
70,4
3,6
33,4
59,5
59,1
2,7
1,0
Objętość roztworu wzorca [ml]
0,3
0,3
1,3
0,2
2,6
1,0
1,5
0,2
0,3
Tabela 14
Wzglêdny udzia³ poszczególnych elementów metody w niepewnoœci z³o¿onej
Relative contribution of individual method components in complex uncertainty
(10)
Dla pozosta³ych pierwiastków
g³ównym elementem determinuj¹cym
niepewnoϾ metody jest dozowanie
objêtoœci roztworu wzorca g³ównego,
czyli by zmniejszyæ niepewnoœæ me-
tody, nale¿y kupiæ pipety o wiêkszej
precyzji dozowania objêtoœci.
W tabeli 15 podano wzglêdne pro-
centowe wartoœci niepewnoœci z³o¿o-
nej (U) metody oznaczania ka¿dego
pierwiastka. Zamieszczono w niej
równie¿ wartoœæ stê¿enia, gdy¿ wa¿-
ny jest poziom stê¿eñ, dla którego
wyznaczono niepewnoϾ. Podana
wartoœæ Ur to wartoœæ wzglêdna nie-
pewnoœci rozszerzonej. Obliczono j¹,
mno¿¹c wartoœæ U przez 2. Niepew-
noœæ rozszerzona oddaje pe³niej war-
toœæ niepewnoœci metody. Jest to
spowodowane tym, ¿e przy szacowa-
niu niepewnoœci czêsto nie mo¿na
przewidzieæ i okreœliæ wszystkich
czynników, które na ni¹ wp³ywaj¹.
Jak mo¿na siê by³o spodziewaæ,
najwiêksz¹ wzglêdn¹ niepewnoœæ
metody uzyskano dla pierwiastków,
które oznaczano na poziomie œlado-
wym, tj. B, Ba, Cu, Sr i Zn, i mieœci³a
siê ona w zakresie 10,4
÷12,9%. Dla
pierwiastków, których stê¿enia pier-
wiastków w próbkach konopi s¹
znacznie wy¿sze, niepewnoœæ roz-
szerzona wynosi od 5,1% dla wapnia
do 5,8% dla manganu.
Podsumowanie
W opublikowanych ju¿ artyku³ach
zwi¹zanych z opracowywaniem me-
tody profilowania konopi na podsta-
wie sk³adu pierwiastkowego przed-
stawiono: badania wstêpne zwi¹zane
z analiz¹ materia³u roœlinnego, stabil-
noœci¹ stosowanych roztworów, efek-
tami matrycowymi i walidacj¹ meto-
dy. Doprowadzi³y one do opracowa-
nia wiarygodnej i pewnej metody wy-
znaczania sk³adu pierwiastkowego
konopi. Na podstawie wykonanej pra-
cy okreœlone zosta³y zasady i para-
metry mineralizacji konopi oraz wa-
runki analizy technik¹ ICP-OES.
Wyniki opisanych etapów pracy
autorzy wykorzystali w nastêpnych
badaniach zwi¹zanych ju¿ z w³aœci-
wym, bezpoœrednim badaniem ziela
konopi pod k¹tem rozk³adu pierwiast-
ków w roœlinie (korzeniach, ³odygach,
liœciach, kwiatostanach oraz nasio-
nach). Ponadto przeprowadzono
analizy próbek kono-
pi w³óknistych pobra-
nych z plantacji usy-
tuowanych w
ró¿-
nych rejonach Polski,
a tak¿e próbek kono-
pi „narkotycznych”,
które by³y jednocze-
œnie przedmiotem
badañ w eksperty-
zach opracowywa-
nych w CLK KGP.
Otrzymane wyniki
zosta³y poddane za-
awansowanej anali-
zie statystycznej
w celu oceny mo¿li-
woœci grupowania
próbek konopi.
Opracowana me-
toda jest równie¿
w y k o r z y s t y w a n a
w
bie¿¹cej pracy
Wydzia³u Chemii CLK KGP przy bez-
poœrednim porównywaniu konopi do-
wodowych (u¿ytkownik) i porównaw-
czych (hurtownik) pod k¹tem ich
wspólnego pochodzenia. Wyniki
z tych etapów pracy autorzy przed-
stawi¹ w nastêpnym numerze „Pro-
blemów Kryminalistyki”.
BIBLIOGRAFIA
1. M. Kuras, M. Wachowicz: Profilo-
wanie konopi na podstawie sk³adu pier-
wiastkowego – cz. I (efekty matrycowe),
„Problemy Kryminalistyki” 2006, nr 252,
s. 21–30.
2. M. Kuras, praca magisterska „Ana-
liza elementarna wybranych narkotyków
oraz pó³produktu i produktu syntezy siar-
czanu 4-etoksyamfetaminy”, Wydzia³
Chemii UW, Warszawa 2002.
3. M. Wachowicz, M. Kuras: Wstêp
do profilowania konopi na podstawie sk³a-
du pierwiastkowego, „Problemy Krymina-
listyki” 2003, nr 240, s. 10–19.
4. M. Wachowicz, M. Kuras: Minerali-
zacja mikrofalowa jako jedna z technik
przygotowania próbek do badañ porów-
nawczych, „Problemy Kryminalistyki”
2002, nr 238, s. 8–22.
5. P. van Zoonen, R. Hoogerbrugge,
S.M. Gort, H.J. van de Wiel, H.A. van`t
Klooster: Some practical examples of
method validation in the analytical labora-
tory, „Trends in Analytical Chemistry”, 18
(1999), s. 584–593.
6. Eurachem/CITAC Guide: Quantify-
ing uncertainty in analytical measure-
ment, Second edition, 2000.
7. B.N. Taylor, C.E. Kuyatt: Guideli-
nes for evaluating and expressing the un-
certainty of NIST measurement results,
„NIST Technical Note” 1297, 1994 Edition.
8. M. Thompson, S.L.R. Ellison,
R. Wood: Harmonized guidelines for sin-
gle – laboratory validation of methods of
analysis (IUPAC Technical Report), „Pure
Apel. Chem.”, vol. 74, nr 5 (2002), s. 835–855.
9. Eurachem/CITAC Guide: Traceabili-
ty in chemical measurement, a guide to
achieving comparable results in chemical
measurement, 2003.
10. Analytical Methods Committee:
Uncertainty of measurement: implications
of its use in analytical science, „Analyst”
1995, vol. 120, s. 2303–2308.
11. R.J.N. Bettencourt da Silva, M. Fi-
lomena, G.F.C. Camões, J. Seabra e
Barros: Validation and quality control
schemies based on the expression of re-
sults with uncertainty, „Analytica Chimica
Acta” 1999, 393, s. 167–175.
12. W. Hyk, Z. Stojek: Analiza statystycz-
na w laboratorium analitycznym, Komitet
Chemii Analitycznej PAN, Warszawa 2000.
13. ICP WinLab Bonus Pack, Perkin Elmer.
PROBLEMY KRYMINALISTYKI 253/06
26
Tabela 15
Wartoœci niepewnoœci standardowej (U) i rozszerzonej
(Ur) metody oznaczania pierwiastków technik¹ ICP OES
Values of standard (U) and extended (Ur) uncertainty
for element determination method by ICP OES
Pierwiastek
Stężenie
[mg/kg]
U
[%]
U
r
[%]
B
29,6
5,5
11,0
Ba
31,4
5,3
10,6
Ca
11428
2,5
5,1
Cu
7,27
6,4
12,9
Fe
423
2,8
5,6
Mg
2952
2,8
5,6
Mn
184
2,9
5,8
Sr
39,1
5,8
11,6
Zn
31,4
5,2
10,4
Wyszukiwarka
Podobne podstrony:
Profilowanie konopii na podstawie składu pierwiastkowego Część I efekty matrycowe
Profilowanie konopii na podstawie składu pierwiastkowego Część I efekty matrycowe
Ocena stanu srodowiska na podstawie szaty roslinnej wyklad II
Pytania na Fizyke, Pytania FIZYKA2, Część II
0krślanie struktury stali specjalnych na podstawie składu chemicznego
ADORACJA na podstawie tekstów Jana Pawła II o EUCHARYSTII 2
Bawimy się w lustra – na podstawie „Lustereczka” D Gellner (konspekt zajęć dramowych dla klasy II)
Mikrobiologia opracowanie na podstawie części II Skryptu WAM wersja ostateczna wreszcie kurna!!! , Z
Bankowość II, Charakterystyka banku na podstawie bilansu
Nie - Boska komedia, W14, Na podstawie części I i II dramatu zreferuj zasadnicze myśli o poezji i po
Podstawowe zasady tworzenia projektu dla STM32F4 w środowisku uVision 4 czesc II
Pytania egzaminacyjne Biofizjologiczne podstawy zachowania Część II, Psychologia, psychologia stosow
Wykład 9 2 Podstawy biologicznego oczyszczania ścieków część II
I . ANALIZA POJĘĆ NA PODSTAWIE LITERATURY, szkoła, Rady Pedagogiczne, wychowanie, profilaktyka
9.Rysunki złożeniowe, Rysunek zlozeniowy, Rysunek wykonawczy-rysunek na podstawie ktorego wykonana b
Część II, Szkoła, Semestr 2, Podstawy Budowy Maszyn I, Spawanko, Spawanie, Sprawko Spawanie, Sprawko
więcej podobnych podstron