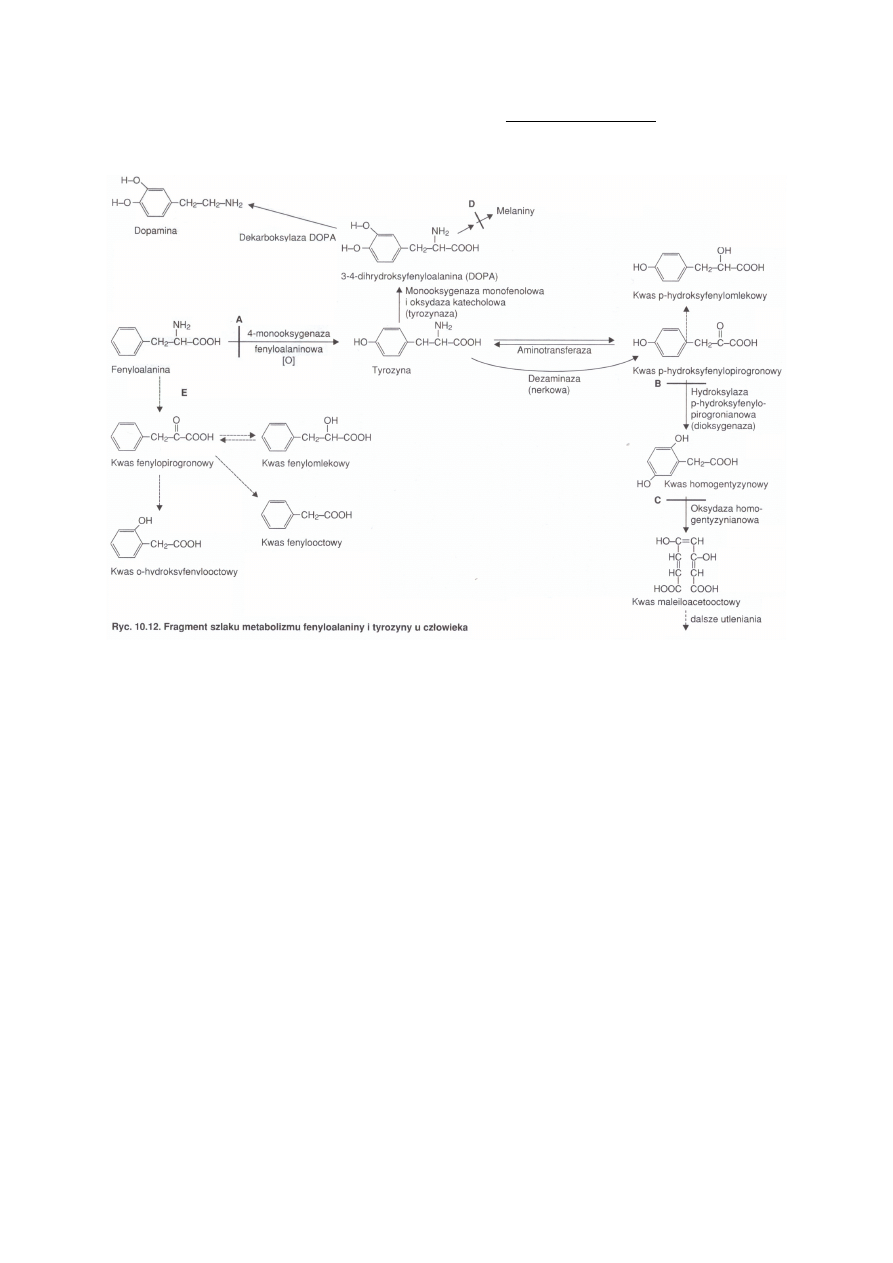
FENYLOKETONURIA 1:10000 urodzeń autosomalnie recesywnie
Loci: chromosom12q24.1 – częściowe delecje, mutacje punktowe, mutacje zmiany sensu
Objawy (zwiększonego poziomu fenyloalaniny):
•
Narastające upośledzenie umysłowe
•
Zahamowanie syntezy melanin – jasne włosy i jasna karnacja
•
Uporczywe wymioty
•
„mysi” zapach moczu i potu (obecność kwasu o-hydroksyfenylooctowego)
•
wzrost napięcia mięśniowego
•
niemożność chodzenia i mówienia
•
siedzenie na skrzyżowanych nogach
•
charakterystyczne kiwanie tułowia do przody i do tyłu
•
małogłowie (68%-94%)
Diagnostyka:
•
test przesiewowy u noworodków (Guthriego)
•
test z FeCl
3
•
oznaczanie fenyloalaniny we krwi i moczu
•
badanie prenatalne – analiza DNA
U noworodków poziom fenyloalaniny jest normalny
Leczenie - odpowiednia dieta (minimalna ilość fenyloalaniny w pożywieniu)
Organizm ‘przyzwyczaja’ się i po pewnym czasie można zmienić dietę. Kobieta w ciąży musi koniecznie do
niej wrócić.
1

ALBINIZM (11q) (bielactwo uogólnione) 1:30000 urodzeń autosomalnie recesywne
Chorobę powoduję brak tyrozynazy
Objawy (związane z zahamowaniem syntezy melanin w melanocytach oczu, włosów skórze):
•
skóra różowoczerwona
•
łatwo ulega oparzeniu po ekspozycji na promienie UV
•
Włosy białe, tęczówki niebieskie lub różowe
•
Ograniczona ostrość wzroku
•
Światłowstręt
Rozwój normalny.
GLAKTOZEMIA autosomalna recesywna 1:40000
Blok metaboliczny uniemożliwiający przekształcenie galaktozy w glukozę. Gromadzący się na skutek istnienia
bloku metabolicznego galaktozo-1-fosforan wywiera działanie toksyczne.
Galaktoza
>
aza
galaktokin
ATP
.
galaktozo-1-fosforan
>
−
−
fosforan
heksozo
sferaza
uidylotran
1
glukozo-1-fosforan glukoza
I blok typu GALK (17q23-25)
II blok typu GALT (9p13)
Objawy
•
Niechęć do jedzenia
•
Wymioty, wodne stolce
•
Spadek wagi
•
Żółtaczka, Powiększenie wątroby i śledziony
•
Skaza krwotoczna,
•
Uszkodzenia nerek
•
Znaczna podatność na zakażenia bakteryjne
•
Wzrost ciśnienia śródczaszkowego – wypukłe ciemiączko, zaćma
•
We krwi podwyższone stężenie galaktozy, obniżony poziom glukozy.
•
W moczu: galaktozuria, białkomocz, aminoaciduria
Nie leczona kończy się śmiercią w okresie niemowlęcym.
Leczenie: dieta bez galaktozy i laktozy; nie gwarantuje to w 100% prawidłowego rozwoju
ZESPÓŁ LESCHA-NYHAHA recesywny sprzężony z X puryny
Loci: chromosom X (Xq26-q27.2). gen HPRT1 kontrolujący syntezę enzymu fosfororybozylotransferazy
hipoksantynowej (HGPRT), biorącego udział w syntezie zasad purynowych.
Objawy:
•
znaczny wzrost stężenia kwasu moczowego w surowicy krwi
•
zaburzenia w rozwoju i funkcjonowaniu układu nerwowego
•
u dzieci niedorozwój umysłowy, przymusowe: ruchy rąk, gryzienie warg, śluzówek, jamy ustnej oraz
rąk – samookaleczanie
•
dziecko nie potrafi samo usiąść stać chodzić
Diagnostyka:
•
badanie stężenia HPRT w cebulkach włosowych
•
prenatalna
OSTRA PORFIRIA PRZERYWANA (AIP) hem i porfiryny
Loci: chromosom 11q23
AIP jest chorobą uwarunkowaną genetycznie dziedziczoną w sposób autosomalnie dominujący, wywołaną
częściowym niedoborem deaminazy porfobilinogenu (PBGD, E.C.4.3.1.8) , trzeciego enzymu na drodze
biosyntezy hemu. Enzym ten katalizuje kondensację czterech cząsteczek porfobilinogeny do
hydroxymetylbilanu.
POSTACIE Wyróżnia się postać jawną i utajoną porfirii. Porfiria ujawnia się w postaci zaostrzeń lub ataków
pomiędzy którymi występują okresy remisji o zróżnicowanym okresie trwania (nawet kilka- kilkanaście lat). U
niektórych pacjentów zaostrzenia pojawiają się często – nawet kilka razy w roku (dotyczy to zwłaszcza kobiet w
wieku rozrodczym), u niektórych pojawiają się tylko jeden lub kilka razy w życiu. Chorzy z postacią utajoną AIP
2

nie mają żadnych dolegliwości klinicznych, stwierdza się jedynie obniżoną aktywność PBG-deaminazy oraz (u
części z nich- około 30%) podwyższone wydalanie porfiryn i ich prekursorów z moczem.
ROZPOZNANIE :w okresie zaostrzenia ze względu na bardzo różnorodne objawy kliniczne porfirię rozpoznaje
się na podstawie wyników badań biochemicznych. W czasie ataku pacjenci z AIP wydalają z moczem znaczne
ilości porfobilinogenu (15-100 razy więcej niż górna granica normy) i kwasu d-aminolewulinowego (5-20 razy
więcej niż górna granica normy). Wydalanie porfiryn jest również zwiększone ale w mniejszym stopniu. Mocz
ma zabarwienie brunatno-czerwone lub ciemnieje na świetle w ciągu 10-30 minut. W okresie zdrowienia
wydalanie porfiryn stopniowo zmniejsza się, ale w większości przypadków nie wraca do normy. Ilość porfiryn w
kale jest prawidłowa lub tylko nieznacznie zwiększona. W okresie ataku choroby aktywność PBGD wzrasta do
wartości prawidłowej i badanie nie ma wtedy wartości diagnostycznej.
Aktywność enzymu celem ostateczne potwierdzenia typu ostrej porfirii oznacza się kilka miesięcy po
przebytym ataku.
OBJAWY ZAOSTRZENIA / ATAKU PORFIRII
Ataki porfirii występują głównie w wieku 15-50 lat, zdecydowanie częściej u kobiet niż u mężczyzn. U
dzieci i osób starszych pojawiają się niezwykle rzadko, najczęściej są związane z dużym narażeniem na czynniki
porfirynogenne.
Do czynników indukujących atak porfirii należą :
•
leki
o
pochodnych kwasu barbituturowego (luminal, weronal, fenodorm)
o
wszystkie sulfonamidy
o
niektóre leki przeciwbólowe (piramidon, antypiryna, fenylobuzaton)
o
niektóre antybiotyki (chloramfenikol, gryzeofulwina)
o
chloropropamid
o
dihydroerogotamina (alkaloid sporyszu)
o
alkohol etylowy
o
halotan
o
doustne leki antykoncepcyjne
o
teofilina
•
środki chemiczne (zwłaszcza farby, lakiery, rozpuszczalniki organiczne, nawozy, środki ochrony roślin).
•
stres, skrajny wysiłek fizyczny, infekcje,
•
spożywanie alkoholu, niedobory kaloryczne (w tym nadmierne odchudzanie się).
PRZEBIEG Ostry atak porfirii zwykle rozpoczyna się silnymi, rozlanymi bólami brzucha z towarzyszącymi
nudnościami, wymiotami oraz tachykardią i wzrostem ciśnienia tętniczego. Zwykle występuje ciemne
zabarwienie moczu (lub jego ciemnienie pod wpływem światła). Często chorzy zauważają, że spożycie posiłku
(lub wypicie słodkiego płynu) przynosi złagodzenie objawów. Mimo nasilonych dolegliwości bólowych w
badaniu przedmiotowym nie stwierdza się (poza tachykardią i wahaniami RR) odchyleń od stanu prawidłowego.
W przypadku niepodjęcia właściwego leczenia lub podawania leków szkodliwych w porfirii, dochodzi do
dalszego postępu choroby – pojawiają się wzdęcia, zaparcia (porażenna niedrożność jelit), bóle i drętwienia
kończyn dolnych, następnie symetryczne niedowłady kończy dolnych i górnych, trudności w połykaniu.
Ostatecznie dochodzi do porażenia mięśni oddechowych co stanowi realne zagrożenie dla życia pacjenta.
OKRES REMISJI
Po napadzie porfirii następuje kilkumiesięczny okres nadmiernej wrażliwości na czynniki
porfirynogenne. W tym czasie wydalanie w moczu porfiryn i ich prekursorów jest podwyższone. Konieczne jest
unikanie kontaktu z czynnikami indukującymi atak. W tym okresie mogą niekiedy pojawiać niezbyt nasilone
dolegliwości pod postacią bólów brzucha, nudności, drętwienia kończyn dolnych, ciemnego moczu. Z wiekiem
wrażliwość na czynniki indukujące maleje (zwłaszcza po okresie pokwitania). Pacjent w okresie remisji nie
wymaga specjalnego leczenia, zwykle jest zdolny do powrotu do pracy zawodowej (o ile nie wiąże się to z
narażeniem na czynniki porfirynogenne). Jedynie u kobiet z nawracającymi zaostrzeniami przedmiesiączkowymi
podejmuje się próby leczenia analogami gonadoliberyn (leczenie takie należy prowadzić pod kontrolą wydalania
porfiryn i ich prekurów z moczem).
PROFILAKTYKA
Zapobieganie atakom porfirii polega na unikaniu kontaktu z czynnikami porfirynogennymi.
BADANIA RODZINNE
3

Ważną rolę w profilaktyce porfirii odgrywają badania rodzinne. Wykrycie wady metabolicznej pozwala
na wdrożenie postępowania zapobiegającego wystąpieniu objawów klinicznych u kolejnych osób obciążonych
chorobą, a w przypadku zaostrzenia umożliwia szybkie rozpoczęcie właściwego leczenia.
Choroba WILSONA (zwyrodnienie wątrobowo-soczewkowe) - Cu
Obniżenie aktywności enzymatycznej ceruloplazminy (białko zawierające miedź) – loci 3. Enzym
przyłączający do ceruloplazminy miedź ma charakter ATP-azy.
Jest zapisany loci 13q14.
Autosomalna recesywna.
Choroba genetyczna charakteryzująca się nadmiernym gromadzeniem miedzi w różnych tkankach organizmu,
głównie w wątrobie, ośrodkowym układzie nerwowym, rogówce oka. Akumulacja ta spowodowana jest
defektem wydalania miedzi z żółcią. Choroba zazwyczaj ujawnia się w 11- 25 rż.
Objawy kliniczne :
•
Objawy wątrobowe obejmujące zapalenie wątroby, marskość, lub ostrą niewydolność wątroby
•
Objawy neurologiczne obejmujące zaburzenia mowy, drżenia, brak koordynacji ruchowej, trudności w
połykaniu; porażenie górnych kończyn (trzepotanie), sztywność mięśni, opad żuchwy.
•
Objawy psychiczne obejmujące chwiejność emocjonalną (nagłe wybuchy złości, skłonność do płaczu,
depresja). Wyjątkowo występuje paranoja lub halucynacje.
Wiele z tych objawów występuje także w innych jednostkach chorobowych, co powoduje, że pacjenci mogą być
błędnie zdiagnozowani. Choroba Wilsona powinna być podejrzewana, jeśli u pacjenta w wieku 40 lat lub
młodszego, występują jakiekolwiek zaburzenia funkcji wątroby lub neurologiczne.
1-3 lat po wystąpieniu objawów śmierć z powodu niewydolności wątroby.
Postępowanie
Leczenie preparatami wiążącymi miedź. Może to być realizowane za pomocą substancji, które wiążą miedź i
powodują jej wydalanie z moczem lub substancji, które ułatwiają jej wydalanie z kałem.
HOMOCYSTYNURIA autosomalny recesywny aminokwasy
Loci: 21q22.3
Defekt związany z niedoborem
β
syntetazy cystioniny (typ I); lub zaburzenia syntezy metylokobalaniny (typ II).
Zaburzenia dotyczą metabolizmu metioniny. Defekt genetyczny związany z niedoborem w/w enzymów jest
bardzo heterogenny, wśród rodzeństwa efekt jest zawsze jednakowy.
Objawy:
•
Niskie IQ
•
Zaburzenia psychiczne: stany depresyjne, zaburzenia zachowanie obsesje
•
U niektórych pacjentów udar mózgu, zawał serca, zatorowość tętnic, żył obwodowych, zatorowość płuc
•
Zaburzenia kostne, osteoporoza
Homocystynuria jest największą przyczyną udaru mózgu u dzieci. Homocystynuria typu II może być wywołana
przez różne bloki metaboliczne powodując powstawanie dużych erytrocytów.
4

CHOROBY MONOGENOWE
Dziedziczenie autosomalne dominujące
CHOROBA HUNGTINKTONA
Obecność niestabilnej liczby sekwencji CAG na końcu 5`kodujące białko huntingina (loci 4p16.3). Osoby
zdrowe 13-29, chore 36-120 powtórzeń. Im większa liczba powtórzeń CAG tym wcześniejsze objawy choroby.
Mutacja przekazana przez matkę – liczba powtórzeń się nie zwiększa
Mutacja przekazana przez ojca – liczba powtórzeń się zwiększa!!!
Objawy:
•
Zanik komórek nerwowych jądra ogoniastego i skorupy, oraz komórek nerwowych w gałce bladej jądra
soczewkowatego
•
Początek 30-45rż, 6% przed 15rż – zachorowanie przed 20 rż związane jest z ciężkim przebiegiem
choroby
•
Zaburzenia psychiczne
•
Pląsawiczne ruchy mimowolne (szyi, kk. górnych, tułowia)
•
Postępujące otępienie
•
Nieznanie przyczyny upadków, lekka niestabilność
Żyje się ok. 15-20lat
DYSTROFIA MIOTONICZNA 1:7500
Loci: 19q13.3
Przyczyną są mutacje dynamiczne – niestabilna długość sekwencji 3 nukleotydów CTG w końcu 3` genu DM.
Osoby zdrowe 5-37 powtórzeń, chore 50-2000.
Postać lekka 50-99 ; klasyczna 100-1000; ciężka 1000-2000.
Długość powtórzeń zwiększa się w obrębie kolejnych pokoleń, ale może się również skrócić.
Jeżeli choroba jest przekazywana przez ojca to objawy występują później.
Objawy:
•
Postępujące osłabienie mięśnie
•
Trudności w rozluźnieniu zaciśniętej pięści
•
Zaćma 85% przypadków
•
Zanik jąder i jajników
•
Skrytość i podejrzliwość IQ w normie
•
Uszkodzenia m. sercowego
•
U noworodków wiotkość, zaburzenia oddychania
Diagnostyka:
•
Objawy kliniczne
•
Elektromiografia
•
Badanie molekularne
•
Diagnostyka prenatalna
ZESPÓŁ MARFANA 1:10000
Loci 15q21.1 – gen dla fibryliny 1 (FBN1). 25% mutacji tego genu to mutacje de novo
Objawy kliniczne:
•
Długie i cienkie kończyny, nieprawidłowe proporcje ciała
•
Krótki korpus
•
Tętniak aorty
•
Podwichnięcie soczewki
HIPERCHOLESTEROLEMIA RODZINNA
Loci: 19q13.1
Chorobę wywołuje defekt receptora LDL, będącego glikoproteidem. Cały gen ma 45 k nukleotydów i jest
skupione w 18 eksonach, podzielonych intronami.
I klasa – delecje w obrębie genu
II klasa – wadliwy transport receptora z reticulum endoplazmatycznego do aparatu Golgiego. Mechanizm nie
znany.
III klasa – zaburzenie wiązaniu LDL przez receptor
IV klasa – zaburzenie w transporcie do wnętrza komórki
5

ACHONDROPLAZJA 1:15000-1:77000
Loci:4p16.3 Jest to gen receptora wzrostu fibroblastów (FGFR3). Mutacja punktowa (G380R)
Objawy:
•
Skrócenie kończyn szczególnie w odcinku proksymalnym
•
Długość tułowia prawidłowa
•
Lordoza lędźwiowa
•
Dłonie kształtu trójdzielnego (widać na RTG)
•
Wąski kanał kręgowy
•
Rozwój umysłowy prawidłowy
•
Wzrost 132 M 123K
NERWIAKOWŁÓKNIAKOWATOŚĆ 1:3500
Loci 17q11.2.7 (NF1) – białko neurofibronina
Gen warunkujący przejawia pełną penetrację, ale zmienną ekspresję. Mutacje de novo pochodzenia ojcowskiego
50% nowych przypadków
Objawy:
•
We wczesnym dzieciństwie plamy na skórze ‘cafe au lait’ u99% chorych pręgi 75%
•
W okresie dojrzewania rozwijają się liczne guzki wywodzące się z nerwów
•
Niedorozwój umysłowy
•
Skrzywienie kręgosłupa
Diagnostyka:
•
Więcej niż 6 plam większych niż 5mm po urodzeniu
•
Więcej niż 6 plam większych niż 15mm u dorosłych
•
Analiza DNA
•
Wykrywanie wadliwego białka
•
Możliwość diagnostyki prenatalnej
Loci 22q12.2 – białko merlina (składnik cytoszkieletu)
Objawy:
•
Nerwiaki osłonowe nerwu słuchowego
•
Zmętnienie soczewek
•
Objawy w wieku dojrzewania
Dziedziczenie dominujące sprzężone z chromosomem X
ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X 1:1250M 1:2000K
Loci Xq27.3 gen obejmuje 38kb i ma 17eksonów. W eksonie 1 znajduje się sekwencja polimorficzna złożona z
powtórzeń CGG. Osoby zdrowe 6-60 powt.; nosiciele 60-200 powt, chorzy 250-1500 powt.
Objawy:
•
odchylenia od normalnych zachowań (autyzm, obgryzanie paznokci, niedorozwój)
•
mały obwód głowy, zwiększona objętość jąder
•
u dorosłych: wydatne guzy czołowe, duże i odstające uszy
•
bladoniebieskie tęczówki
•
zniekształcenie kręgosłupa, szerokie, krótkie palce
•
encefalopatia
•
napady padaczki
Liczba powt CGG nie zmienia się gdy przekazuje ojciec
6

Dziedziczenie autosomalne recesywne
MUKOWISCYDOZA
Biali 1:2500; czarni 1:17000 żółci 1:90000
Loci 7q31-32 wielkość ok. 250kb
Białko kodowanie przez ten gen ma wielkość 170kD. Pełni rolę w regulacji kanału chlorkowego w komórkach
nabłonka dróg oddechowych i gruczołów wydzielania wewnętrznego.
Objawy:
•
Uszkodzenie wątroby, trzustki
•
Nawracające infekcje górnych dróg oddechowych
•
Nieprawidłowa sekrecja seromukoidu
•
U mężczyzn wrodzony brak nasieniowodów
•
U noworodków niedrożność smółkowa, słony pot
Diagnostyka:
•
Badanie DNA
•
Diagnostyka prenatalna
•
Stężenie w pocie :Na
+
>60mmol/l (n,20-25mmol/l), Cl
-
>70mmol/l (n16-18mmol/l)
WRODZONY PRZEROST NADNERCZY (NIEDOBÓR 21-HYDROKSYLAZY)
MUKOPOLISACHARYDOZY
Typ I – zespół Hurler
Typ II – zespół Huntera
Typ III – zespół Sanfilippo
Typ IV – choroba Morquio
Typ VI - chorobę Maroteaux – Lamy
CHOROBA TAYA-SACHSA
Loci 15chromosom
Objawy:
•
Postępujące nieprawidłowości neurologiczne od późnego niemowlęctwa
•
Malinka w okolicy plamki żółtej
•
Zmniejszenie stężenia heksaminidazy A w surowicy (można wykryć nosicielstwo)
•
Zgon w wieku 3-4lat
Diagnostyka badanie surowicy (nosicielstwo), diagnostyka prenatalna - stężenie heksaminidazy A
Dziedziczenie recesywne sprzężone z chromosomem X
DYSTROFIA MIĘSNIOWA DUCHENNE`A 1:3500
Loci Xp21.3 – koduje dystrofinę (białko strukt. Membrany włókien mięśniowych)- największy poznany gen
Objawy:
•
Kardiomoipatia rozszerzeniowa
•
U dzieci ‘kaczkowaty chód’
•
Przerost łydek
•
Wysoki poziom fosforokinazy kreatynowej w surowicy
•
Symetryczny zanik mięśni obręczy miednicy a później barków
DMD letalna po 10 latach, BMD objawy później i nie jest letalna
ZESPÓŁ NIEWRAŻLIWOŚCI NA ANDROGENY
Loci Xq11 – mutacja genu receptora androgenowego.
Objawy:
•
Kariotyp tylko 46,XY
•
Zaburzenia w produkcji gametotropin
•
Wzrost ok. 170cm
•
Atrakcyjne kobiety, dobrze wykształcone piersi, brak lub skąpe owłosienie łonowe
•
Krótka i ślepo zakończona pochwa
•
Jądra w wargach sromowych
•
Absolutna bezpłodność
7
Document Outline
- GLAKTOZEMIA autosomalna recesywna 1:40000
- ZESPÓŁ LESCHA-NYHAHA recesywny sprzężony z X puryny
- OSTRA PORFIRIA PRZERYWANA (AIP) hem i porfiryny
- Choroba WILSONA (zwyrodnienie wątrobowo-soczewkowe) - Cu
- CHOROBA HUNGTINKTONA
- DYSTROFIA MIOTONICZNA 1:7500
- ZESPÓŁ MARFANA 1:10000
- HIPERCHOLESTEROLEMIA RODZINNA
- ACHONDROPLAZJA 1:15000-1:77000
- NERWIAKOWŁÓKNIAKOWATOŚĆ 1:3500
- ZESPÓŁ ŁAMLIWEGO CHROMOSOMU X 1:1250M 1:2000K
- MUKOWISCYDOZA
- WRODZONY PRZEROST NADNERCZY (NIEDOBÓR 21-HYDROKSYLAZY)
- MUKOPOLISACHARYDOZY
- CHOROBA TAYA-SACHSA
- DYSTROFIA MIĘSNIOWA DUCHENNE`A 1:3500
- ZESPÓŁ NIEWRAŻLIWOŚCI NA ANDROGENY
Wyszukiwarka
Podobne podstrony:
CHOROBY GENETYCZNEp id 115004 Nieznany
choroby genetyczne id 114957 Nieznany
Biologia część V, Zespół Turnera choroba genetyczna
318 choroby genetyczne czlowiek Nieznany
Biologia część V Zespół Downa-choroba genetyczna
DO TEL! 5= Genetyka nadci nieni Nieznany
Choroba Aujeszkyego id 114534 Nieznany
Genetyczny id 187377 Nieznany
Genetyczna regulacja embriogene Nieznany
pbfd choroba nie tylko duzych p Nieznany
diagnostyka chorob nerek id 134 Nieznany
więcej podobnych podstron