
PODSTAWOWE MECHANIZMY
N
ie jest ju˝ tajemnicà, jak po-
wstaje rak. W czasie ostatnich
dwóch dziesi´cioleci dokona-
no zdumiewajàcego post´pu w pozna-
niu najg∏´bszych podstaw procesu no-
wotworzenia, czyli identyfikacji zjawisk
zachodzàcych na poziomie molekular-
nym. Sà to odkrycia o niepodwa˝alnym
znaczeniu; sprostajà egzaminowi u
przysz∏ych pokoleƒ naukowców i stanà
si´ fundamentem nowych rewolucyj-
nych podejÊç leczniczych. Nie sposób
przewidzieç, kiedy terapie ukierunko-
wane na molekularne zmiany w komór-
kach rakowych znajdà powszechne za-
stosowanie, gdy˝ prze∏o˝enie nowej
wiedzy na praktyk´ klinicznà jest pro-
blemem z∏o˝onym, powolnym i kosz-
townym. Ale podj´to ju˝ wysi∏ki w tym
kierunku.
W istocie termin „rak”
1
odnosi si´ do
ponad 100 postaci choroby. Nieomal
ka˝da tkanka w organizmie mo˝e ulec
z∏oÊliwej transformacji (zez∏oÊliwieniu),
z niektórych rozwijajà si´ liczne typy
nowotworów. Co wi´cej, ka˝dy nowo-
twór z∏oÊliwy ma unikalne cechy. Jed-
nak podstawowe procesy, warunkujàce
powstawanie ró˝norodnych nowotwo-
rów, wydajà si´ ca∏kiem podobne. Z te-
go powodu w tym artykule b´d´ u˝y-
wa∏ terminu „rak” w sensie ogólnym,
przedstawiajàc wybrany jego rodzaj, aby
zilustrowaç ogólne zasady.
TrzydzieÊci bilionów komórek nor-
malnego, zdrowego organizmu ˝yje
w z∏o˝onej wspólnocie, wzajemnie re-
gulujàc swoje podzia∏y. Normalne ko-
mórki namna˝ajà si´ (proliferujà) tylko
wtedy, gdy dostanà sygna∏ od innych
komórek z sàsiedztwa. Dzi´ki takiemu
nieustannemu wspó∏dzia∏aniu ka˝da
tkanka utrzymuje w∏aÊciwe rozmiary
i odpowiednià budow´, stosownie do
potrzeb organizmu. Komórki rakowe
natomiast wy∏amujà si´ z tego sche-
matu; ignorujà systemy kontrolne pro-
liferacji i realizujà swój w∏asny program
reprodukcji. Majà nawet bardziej zdra-
dzieckà w∏aÊciwoÊç – sà zdolne do prze-
mieszczania si´ z miejsca, gdzie powsta-
∏y, czyli do inwazji na pobliskie tkanki,
i tworzenia guzów w odleg∏ych miej-
scach organizmu. Guzy z∏o˝one z takich
z∏oÊliwych komórek stajà si´ w miar´
up∏ywu czasu coraz agresywniejsze i
gdy niszczà tkanki i narzàdy niezb´dne
dla egzystencji organizmu jako ca∏oÊci,
prowadzà do jego Êmierci.
To nic nowego. Jednak w ciàgu ostat-
nich 20 lat naukowcy odkryli podstawo-
we prawa, które rzàdzà rozwojem raka.
Obecnie wiemy, ˝e komórki guza po-
chodzà od wspólnego komórkowego
przodka, który w pewnym momencie –
zwykle dziesiàtki lat przedtem, nim guz
sta∏ si´ wyczuwalny – zapoczàtkowa∏
program nieprawid∏owego namna˝ania.
Nast´pnie transformacja z∏oÊliwa takiej
komórki zachodzi poprzez akumulacj´
mutacji w specjalnych klasach jej genów.
Geny te sà kluczem do zrozumienia pro-
cesów powstawania ludzkiego raka.
NoÊnikiem genów sà czàsteczki chro-
mosomalnego DNA w jàdrze komórko-
wym. Gen wyznacza sekwencj´ amino-
kwasów, które muszà si´ po∏àczyç, by
powsta∏ okreÊlony rodzaj bia∏ka. A za-
tem bia∏ko wykonuje funkcj´ zapro-
gramowanà w genie. Gdy gen jest w∏à-
czony, komórka syntetyzuje kodowane
przez niego bia∏ko. Mutacje w genie mo-
gà zaburzyç biologi´ komórki przez
zmian´ iloÊci lub aktywnoÊci produktu
bia∏kowego.
Dwie klasy genów, które ∏àcznie sta-
nowià tylko ma∏à cz´Êç pe∏nego ich ze-
stawu, odgrywajà g∏ównà rol´ w zapo-
czàtkowaniu nowotworu. Normalnie
programujà one cykl ˝yciowy komórki –
z∏o˝onà sekwencj´ zdarzeƒ, dzi´ki któ-
Jak powstaje rak?
Naukowcy poznali ju˝ molekularne mechanizmy
powstawania nowotworów, co umo˝liwia
zastosowanie nowych metod leczniczych
Robert A. Weinberg
32 Â
WIAT
N
AUKI
Listopad 1996
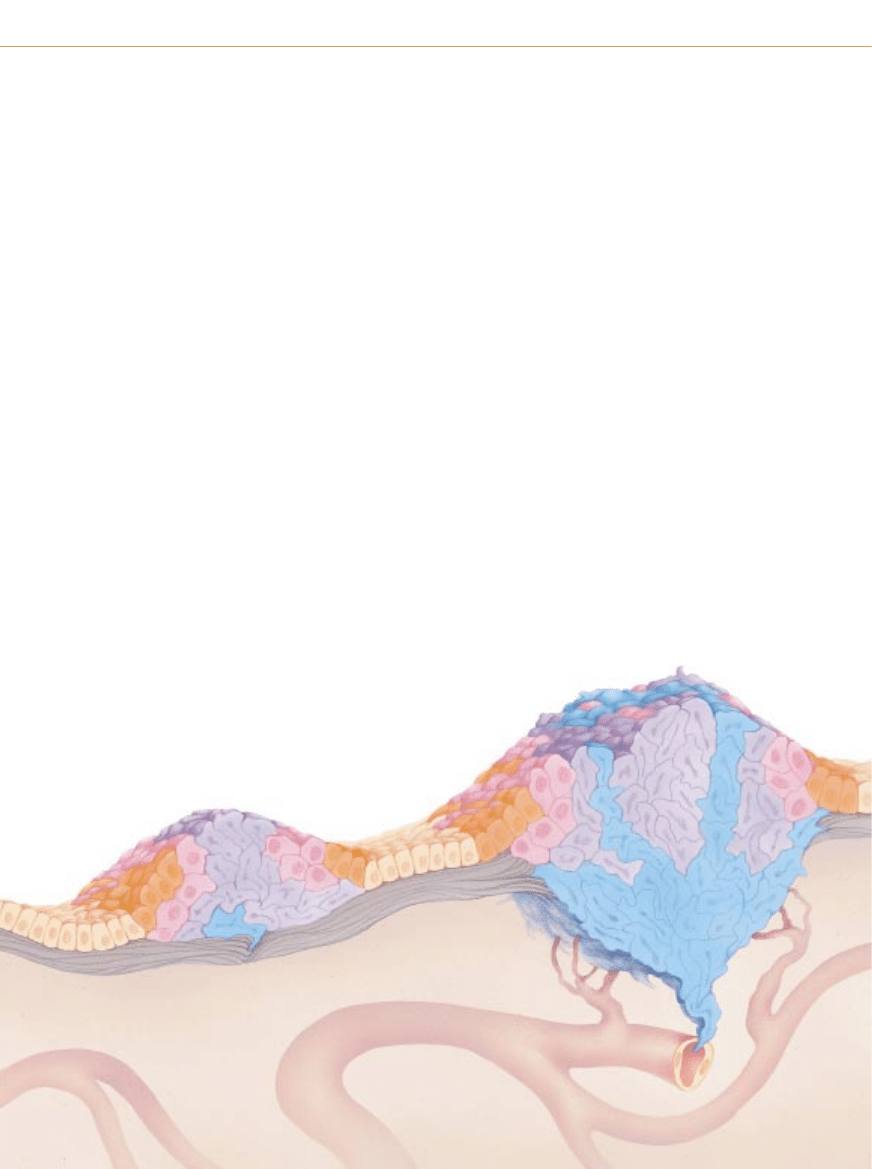
P
oni˝ej pokazano schematycznie rozwój nowotworu z∏oÊliwego w tkance na-
b∏onkowej. Nowotwory nab∏onkowe sà najcz´stszymi nowotworami z∏oÊliwymi
i nazywamy je nab∏oniakami (rakami, carcinoma). Masa komórek przedstawiona na
schemacie powstaje w wyniku mutacji w czterech genach, ale liczba genów zaan-
ga˝owanych w rozwój rzeczywistych nowotworów mo˝e byç zmienna.
Rozwój nowotworu
przechodzi ró˝ne stadia
KOMÓRKA ZMIENIONA GENETYCZNIE
HIPERPLAZJA
DYSPLAZJA
1
Rozwój nowotworu zaczyna si´ od genetycznej
mutacji niektórych komórek (pomaraƒczowy)
w obr´bie populacji normalnej (be˝owy);
mutacja zwi´ksza sk∏onnoÊç komórek
do namna˝ania si´, podczas gdy normalnie
pozosta∏yby one w stanie spoczynku.
DANA BURNS-PIZER
2
Zmieniona komórka i jej komórki potomne
nadal wyglàdajà prawid∏owo, ale dzielà si´
nadmiernie – wykazujà hiperplazj´ (nadmierny
rozrost). Po latach jedna na milion tych komórek
(ró˝owy) ulega innej mutacji, która rozluênia
kontrol´ wzrostu komórkowego.
3
Oprócz nadmiernej proliferacji potomstwo tej komórki odbiega
pod wzgl´dem kszta∏tu i orientacji od normalnych komórek,
czyli wykazuje dysplazj´. Po pewnym czasie zachodzi ponownie
rzadka mutacja, która zmienia zachowanie komórek (fioletowy).
rym komórka powi´ksza si´ i dzieli.
Protoonkogeny stymulujà taki wzrost,
podczas gdy geny supresorowe go ha-
mujà. ¸àcznie obie klasy genów odpo-
wiadajà za wi´kszoÊç zachodzàcych
w ludzkich nowotworach nie kontrolo-
wanych proliferacji komórek.
Gdy protoonkogeny ulegnà mutacji,
stajà si´ rakotwórczymi (kancerogenny-
mi) onkogenami, które nap´dzajà nad-
mierne namna˝anie si´ komórek. Mu-
tacje w protoonkogenie mogà powo-
dowaç, ˝e produkuje on zbyt du˝o ko-
dowanego przez siebie bia∏ka, które sty-
muluje wzrost, lub nadmiernie aktywne
jego odmiany. Natomiast geny supre-
sorowe (supresory nowotworów) przy-
czyniajà si´ do nowotworzenia, gdy
wskutek mutacji zostanà zinaktywo-
wane. Wynikajàca z tego utrata funk-
cjonalnych bia∏ek supresorowych po-
zbawia komórk´ g∏ównych hamul-
ców, które zapobiegajà niew∏aÊciwemu
wzrostowi.
Aby nastàpi∏ rozwój nowotworu, mu-
szà zajÊç mutacje w kilku genach kon-
trolujàcych wzrost komórek. W zapo-
czàtkowaniu procesu nowotworowego
mogà równie˝ braç udzia∏ zmienione
postacie jeszcze innych klas genów,
umo˝liwiajàc proliferujàcej komórce in-
wazyjnoÊç lub rozprzestrzenianie si´
(dawanie przerzutów) w organizmie.
Zak∏ócenie sygnalizacji
Podstawowy trop w wyjaÊnieniu,
w jaki sposób protoonkogeny i geny su-
presorowe biorà udzia∏ w kancerogene-
zie, nasun´∏y wyniki badaƒ nad rolà, ja-
kà w komórce odgrywajà normalne
odpowiedniki tych genów. Po prawie
dwóch dziesi´cioleciach wysi∏ków uczo-
nych funkcje genetyczne tych genów
znamy obecnie niezwykle szczegó∏owo.
Wiele protoonkogenów koduje bia∏-
ka szlaków molekularnych, na których
zewn´trzne sygna∏y stymulujàce wzrost
przekazywane sà do wn´trza komórki.
Wzrost komórki zostaje rozregulowa-
ny, gdy mutacja w jednym z jej proto-
onkogenów pobudza podstawowy szlak
stymulujàcy wzrost, utrzymujàc go
w ciàg∏ej aktywnoÊci, podczas gdy po-
winien byç on nieczynny.
Te szlaki wewnàtrz komórki otrzy-
mujà i przetwarzajà sygna∏y stymulu-
jàce wzrost wysy∏ane przez inne komór-
ki w tkance. Proces przekazywania syg-
na∏ów z komórki do komórki zaczyna
si´ zwykle wtedy, gdy jedna z nich
uwalnia czynniki wzrostowe. Bia∏ka te
przemieszczajà si´ nast´pnie w prze-
strzeniach mi´dzy komórkami i wià˝à
ze swoistymi receptorami (czàsteczkami
pe∏niàcymi funkcj´ anten) na powierzch-
ni innych pobliskich komórek. Recep-
tory w zewn´trznej b∏onie komórek do-
celowych tkwià jednym koƒcem w
przestrzeni pozakomórkowej, a drugim
– we wn´trzu komórki, w jej cytopla-
zmie. Gdy czynnik stymulujàcy wzrost
wià˝e si´ z receptorem, ten z kolei prze-
kazuje sygna∏ proliferacyjny do bia∏ek
w cytoplazmie. One wysy∏ajà nast´pnie
sygna∏y stymulujàce jeszcze innym bia∏-
kom szlaku sygna∏owego, który koƒczy
si´ w sercu komórki, tj. w jej jàdrze. W
jàdrze bia∏ka zwane czynnikami trans-
krypcyjnymi odpowiadajà na sygna∏ ak-
tywacjà zespo∏ów genów, które prowa-
dzà komórk´ przez jej cykl ˝yciowy.
Niektóre onkogeny zmuszajà komór-
ki do nadprodukcji czynników wzro-
stowych. Mi´saki i glejaki (nowotwory
z∏oÊliwe odpowiednio tkanki ∏àcznej
i komórek glejowych, czyli nienerwo-
wych podporowych komórek mózgu)
uwalniajà nadmierne iloÊci p∏ytkopo-
chodnego czynnika wzrostowego (pla-
telet-derived growth factor – PDGF).
Liczne inne rodzaje nowotworów wy-
dzielajà nadmiar transformujàcego
czynnika wzrostowego typu alfa (trans-
forming growth factor alfa – TGF
a).
Czynniki te dzia∏ajà zwykle na pobli-
skie komórki, ale co wa˝niejsze, mogà
one tak˝e zawracaç i trafiaç z powrotem
do tej samej komórki, która je produku-
je, i nap´dzaç jej proliferacj´.
Zidentyfikowano tak˝e onkogenne
wersje genów receptorów. Nieprawi-
d∏owe receptory kodowane przez takie
onkogeny wysy∏ajà ciàg sygna∏ów pro-
liferacyjnych do cytoplazmy komórki
nawet wówczas, gdy brak jest czynni-
ków wzrostowych naglàcych do replika-
cji. Na przyk∏ad komórki raka piersi cz´-
sto majà na swej powierzchni czàsteczki
receptora Erb-B2, które zachowujà si´
w ten w∏aÊnie sposób.
Jeszcze inne onkogeny w nowotwo-
rach ludzkich zak∏ócajà te stopnie ka-
skady sygna∏owej, które znajdujà si´
Â
WIAT
N
AUKI
Listopad 1996 33
NACZYNIE W¸OSOWATE
RAK PRZEDINWAZYJNY
RAK INWAZYJNY
4
Komórki dotkni´te mutacjami sà coraz bardziej
nieprawid∏owe pod wzgl´dem wzrostu i wyglàdu.
JeÊli nowotwór nie naruszy∏ jeszcze granic
mi´dzy tkankami, mówimy, ˝e jest to rak in situ
(rak przedinwazyjny). Nowotwór ten mo˝e
pozostaç w takim stanie bardzo d∏ugo,
ale w niektórych komórkach czasami zachodzà
dodatkowe mutacje (niebieski).
5
Gdy zmiany genetyczne umo˝liwià nowotworowi
rozpocz´cie inwazji na tkank´ znajdujàcà si´
poni˝ej i rozsiewanie komórek do krwi lub limfy,
nowotwór jest w pe∏ni z∏oÊliwy. Zdradzieckie komórki
mogà doprowadzaç do powstawania w organizmie
nowych guzów (przerzutów); a przerzuty te
prowadziç do Êmierci wskutek zniszczenia
narzàdów wa˝nych dla ˝ycia.
w cytoplazmie. Najlepiej poznane on-
kogeny tego rodzaju nale˝à do rodziny
ras. Bia∏ka kodowane przez prawid∏o-
we geny ras przekazujà sygna∏y stymu-
lujàce od receptorów czynnika wzrosto-
wego do innych bia∏ek w dó∏ szlaku.
Jednak˝e bia∏ka kodowane przez zmu-
towane wersje tych genów stale na-
p´dzajà podzia∏y, nawet gdy nie sà
przynaglane przez receptory czynnika
wzrostowego. Nadmiernie aktywne
bia∏ka Ras znaleziono w oko∏o
1
/
4
no-
wotworów ludzkich z rakiem jelita gru-
bego, trzustki i p∏uca w∏àcznie. (Raki sà
najcz´stszà postacià nowotworów; wy-
wodzà si´ z komórek nab∏onkowych,
które wyÊcie∏ajà jamy cia∏a i tworzà ze-
wn´trznà warstw´ skóry.)
Poznano tak˝e takie onkogeny, na
przyk∏ad z rodziny myc, które zmienia-
jà aktywnoÊç czynników transkrypcyj-
nych w jàdrze. Normalnie komórki wy-
twarzajà czynniki transkrypcyjne Myc
wy∏àcznie po stymulacji czynnikami
wzrostowymi – stykajàcymi si´ z po-
wierzchnià komórki. Bia∏ka Myc akty-
wujà geny, które zmuszajà komórk´ do
wzrostu. Jednak˝e w wielu typach no-
wotworów, zw∏aszcza nowotworów
tkanki krwiotwórczej, iloÊç bia∏ka Myc
utrzymuje si´ stale na wysokim pozio-
mie, nawet wtedy gdy nie ma czynni-
ków wzrostowych.
Odkrycie mapy szlaków, które prze-
noszà sygna∏y proliferacji z powierzch-
ni komórki do jej jàdra, to nie tylko
satysfakcja intelektualna. Poniewa˝
szlaki te pobudzajà namna˝anie komó-
rek z∏oÊliwych, stanowià atrakcyjny
obiekt dla naukowców poszukujàcych
nowych typów leków przeciwno-
wotworowych. W wyniku tych ekscytu-
jàcych odkryç kilka firm farmaceutycz-
nych pracuje ju˝ nad lekami, majàcymi
wy∏àczaç receptory czynników wzro-
stowych stale nadajàce sygna∏y. Przy-
najmniej trzy inne firmy zmierzajà do
opracowania zwiàzków, które bloko-
wa∏yby syntez´ wadliwych bia∏ek Ras.
Obie grupy zwiàzków wstrzymujà nad-
mierne wysy∏anie sygna∏ów w komór-
kach nowotworowych w hodowli, ale
ich przydatnoÊci w zahamowaniu
wzrostu guzów u zwierzàt i ludzi jesz-
cze nie wykazano.
Hamowanie genów supresorowych
Nadmierna stymulacja maszynerii
odpowiedzialnej za wzrost nie wystar-
cza, aby komórki prawid∏owe sta∏y si´
z∏oÊliwe. Muszà one tak˝e znaleêç
sposób unikni´cia albo zignorowa-
nia sygna∏ów hamujàcych, wysy∏anych
przez normalne komórki z sàsiednich
tkanek. Informacje hamujàce odbierane
przez normalnà komórk´ docierajà do
jàdra (podobnie jak sygna∏y stymulujà-
ce) przez szlaki molekularne. W komór-
34 Â
WIAT
N
AUKI
Listopad 1996
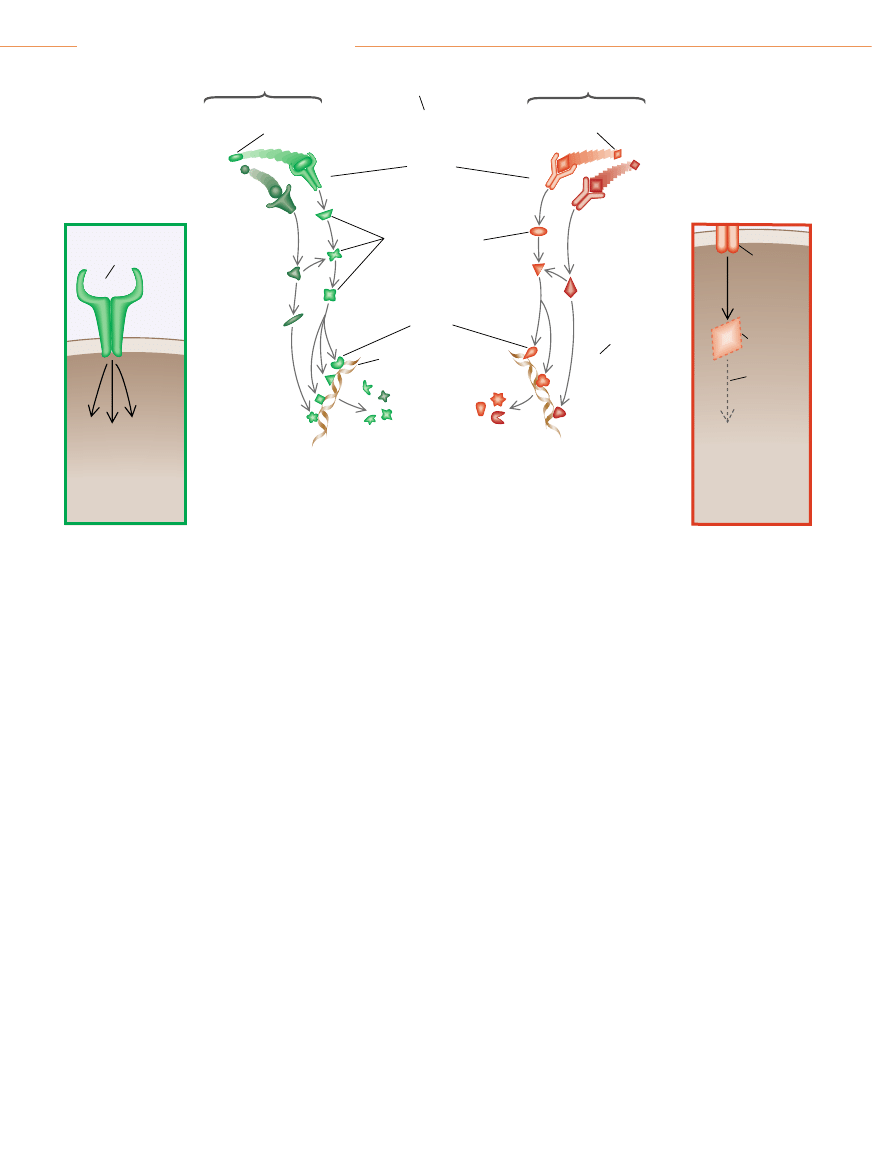
PODSTAWOWE MECHANIZMY
Zegar
cyklu komórkowego
decyduje, czy komórka
ma si´ podzieliç
Przekaênikowe bia∏ka
cytoplazmatyczne
Czynniki
transkrypcyjne
Bia∏ka
hamujàce
podzia∏
komórkowy
Jàdro
Bia∏ka
uruchamiajàce
podzia∏
komórkowy
DNA
Receptory
na powierzchni komórki
Czynnik wzrostowy
(sygna∏ „start")
Sàsiednie komórki
wydzielajà czynniki
stymulujàce wzrost
Sàsiednie komórki
wydzielajà czynniki
hamujàce wzrost
SZLAK
STYMULUJÑCY
SZLAK
HAMUJÑCY
Prawid∏owa komórka
PRZYK¸AD NIEPRAWID¸OWOÂCI
OBSERWOWANEJ W NOWOTWORACH
NA SZLAKU STYMULUJÑCYM
Receptor
mimo to
si´ aktywuje
Komórka si´ dzieli,
mimo ˝e nie jest
stymulowana
czynnikami
wzrostowymi
z zewnàtrz
Czynnik wzrostowy
nie wià˝e si´
z receptorem
Czynnik hamujàcy
(sygna∏ „stop” )
PRZYK¸AD
NIEPRAWID¸OWOÂCI
NA SZLAKU HAMUJÑCYM
Receptor
Brak
przekaênikowej
czàsteczki
w cytoplazmie
Przerwanie
sygnalizacji
Komórka si´ dzieli,
poniewa˝ sygna∏y
hamujàce nie mogà
dotrzeç do jàdra
SZLAKI SYGNALIZACYJNE W PRAWID¸OWEJ KOMÓRCE przenoszà informacje kontrolujàce wzrost z zewn´trznej powierzchni
komórki do wn´trza jàdra. MieÊci si´ tam aparat molekularny zwany zegarem cyklu komórkowego, który zbiera informacje i decyduje,
czy komórka powinna si´ podzieliç. Komórki rakowe cz´sto namna˝ajà si´ nadmiernie, poniewa˝ z powodu mutacji genetycznych szla-
ki stymulujàce
(zielony) wysy∏ajà zbyt du˝o sygna∏ów „start”, albo szlaki hamujàce (czerwony) nie mogà przekazywaç sygna∏ów „stop”.
Szlak stymulujàcy staje si´ hiperaktywny, gdy wskutek mutacji jakiegokolwiek jego komponentu, na przyk∏ad receptora czynnika wzro-
stowego
(ramka z lewej), autonomicznie wysy∏a informacje stymulujàce, nie czekajàc na komend´ „start” od swoich poprzedników na
szlaku. Odwrotnie, szlaki hamujàce zostajà zamkni´te, gdy jakiÊ komponent, na przyk∏ad przekaênik cytoplazmatyczny, wypada
(ramka z prawej), przez co ∏aƒcuch sygnalizacyjny p´ka.
DIMITRY SCHIDLOVSKY

kach nowotworowych szlaki te mogà
byç zniszczone, umo˝liwiajàc komórce
zignorowanie silnych zazwyczaj sy-
gna∏ów hamujàcych docierajàcych do
powierzchni komórki. W wielu rodza-
jach komórek rakowych najwa˝niejsze
komponenty tych szlaków (czyli bia∏ka
– przyp. t∏um.) kodowane przez geny
supresory sà nieobecne albo nieaktywne.
Wzrost ró˝nych rodzajów komórek
prawid∏owych zatrzymuje czynnik se-
krecyjny, zwany transformujàcym czyn-
nikiem wzrostowym typu beta (trans-
forming growth factor beta – TGF-
b).
Niektóre komórki raka jelita grubego
stajà si´ niewra˝liwe na TGF-
b wskutek
inaktywacji genu, który koduje receptor
powierzchniowy dla tego czynnika.
W niektórych rakach trzustki inakty-
wacji ulega gen DPC4, którego produkt
bia∏kowy dzia∏a na szlaku poni˝ej recep-
tora czynnika wzrostowego. Liczne in-
ne nowotwory sà pozbawione genu p15
kodujàcego bia∏ko, które w odpowiedzi
na sygna∏ TGF-
b wy∏àcza mechanizm
prowadzàcy komórk´ poprzez cykl
˝yciowy.
Bia∏ka supresorowe mogà ograniczaç
proliferacj´ komórki tak˝e w inny spo-
sób. Niektóre blokujà na przyk∏ad prze-
p∏yw sygna∏ów przez szlaki stymulujà-
ce wzrost. Jedno z takich bia∏ek jest
produktem genu NF-1. Ta czàsteczka,
znajdujàca si´ w cytoplazmie, przechwy-
tuje bia∏ko Ras, zanim wyda ono instruk-
cje decydujàce o wzroÊcie komórki. A
zatem komórki, którym brak genu NF-1,
sà pozbawione istotnej przeciwwagi dla
bia∏ka Ras i nara˝one na nie kontrolo-
wanà proliferacj´.
Ró˝ne badania wykaza∏y, ˝e wprowa-
dzenie genu supresorowego do komó-
rek nowotworowych (które go nie ma-
jà) mo˝e im przywracaç w pewnym
stopniu „normalnoÊç”. Tego typu odpo-
wiedê nasuwa pomys∏ zwalczania raka
przez wyposa˝enie komórek nowotwo-
rowych w nietkni´te wersje genów su-
presorowych, które komórki te utraci∏y
podczas rozwoju nowotworu. Na dro-
dze stojà jednak przeszkody techniczne
wcià˝ utrudniajàce terapi´ genowà wie-
lu chorób. Obecne metody nie pozwala-
jà na dostarczenie genów do du˝ej cz´-
Êci komórek guza. Dopóki nie pokona
si´ tych problemów, zastosowanie tera-
pii genowej do leczenia raka pozostanie
bardzo pociàgajàcà, ale nierealnà ideà.
Pora˝ony zegar
W ciàgu ostatnich 5 lat dzi´ki przeko-
nujàcym dowodom ustalono miejsca
przeznaczenia wszystkich stymulujà-
Â
WIAT
N
AUKI
Listopad 1996 35
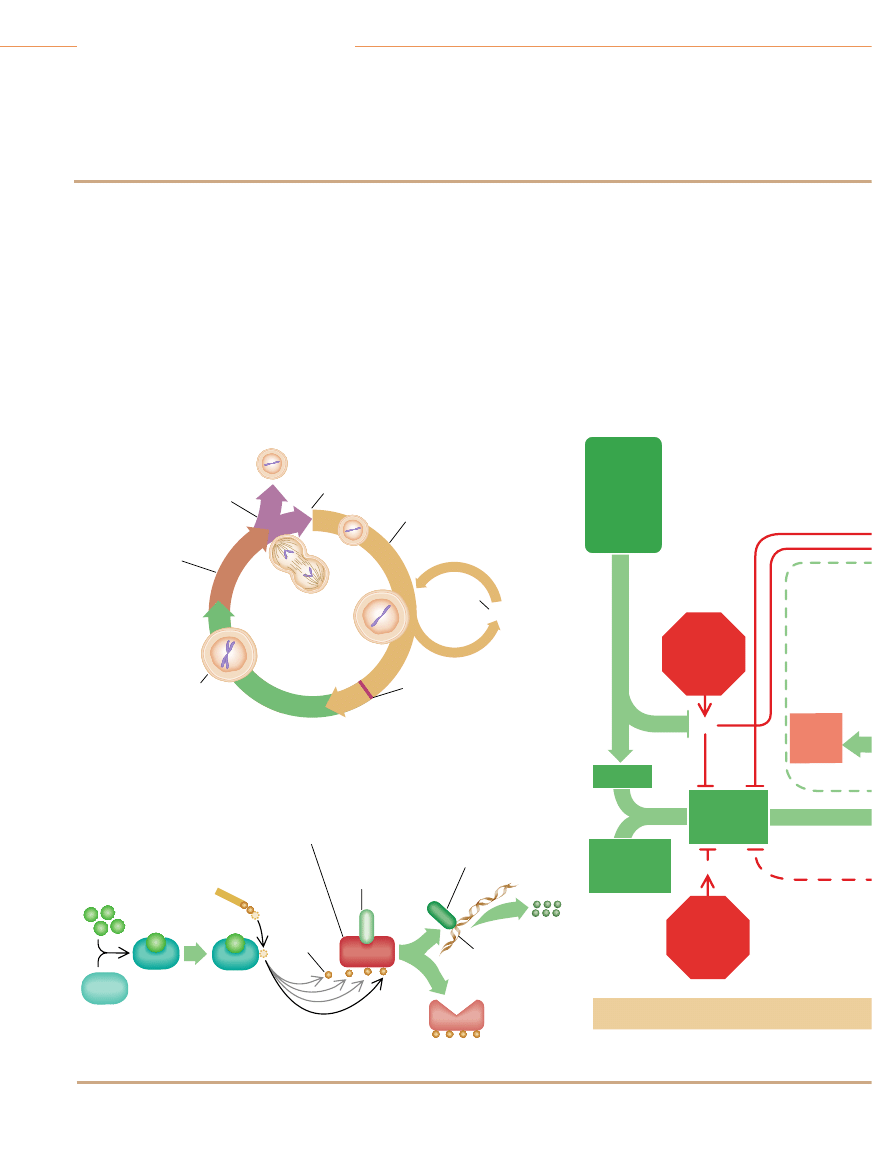
Niektóre geny warunkujàce podatnoÊç na raka
G
eny zwane protoonkogenami kodujà bia∏ka, które stymulujà podzia∏ komórek: zmu-
towane postacie tych genów (zwanych wówczas onkogenami) dajà bia∏ka o
zwi´kszonej aktywnoÊci, w wyniku czego komórki nadmiernie proliferujà. Geny supreso-
rowe nowotworów kodujà bia∏ka, które hamujà podzia∏ komórek. Mutacje genów supre-
sorowych powodujà, ˝e ich bia∏ka sà nieaktywne, co pozbawia komórki niezb´dnych
ograniczeƒ w proliferacji. Badacze nadal usi∏ujà wyjaÊniç swoiste funkcje wielu genów su-
presorowych nowotworów.
ONKOGENY
Geny czynników wzrostowych i ich receptorów
PDGF
Koduje czynnik wzrostowy p∏ytek krwi. Bierze udzia∏ w powstawaniu gleja-
ków (rodzaj nowotworów mózgu)
erb-B
Koduje receptor naskórkowego czynnika wzrostowego. Bierze udzia∏ w po-
wstawaniu glejaków zarodziowych (nowotwór mózgu) i raka piersi
erb-B2
Zwany tak˝e HER-2 lub neu. Koduje receptor czynnika wzrostowego. Bie-
rze udzia∏ w powstawaniu raków piersi, Êlinianek, jajnika
RET
Koduje receptor czynnika wzrostowego. Bierze udzia∏ w powstawaniu raka
tarczycy
Geny przekaêników cytoplazmatycznych na stymulujàcych szlakach sygnalizacyjnych
Ki-ras
Bierze udzia∏ w powstawaniu raków p∏uca, jajnika, jelita grubego, trzustki
N-ras
Bierze udzia∏ w powstawaniu bia∏aczek
Geny czynników transkrypcyjnych aktywujàcych geny pobudzajàce wzrost
c-myc
Bierze udzia∏ w powstawaniu bia∏aczek i raków piersi, ˝o∏àdka, p∏uca
N-myc
Bierze udzia∏ w powstawaniu nerwiaków zarodziowych (nowotwór komórek
nerwowych) i glejaków zarodziowych
L-myc
Bierze udzia∏ w powstawaniu raka p∏uca
Geny kodujàce inne rodzaje czàsteczek
Bcl-2
Koduje bia∏ko, które w warunkach prawid∏owych zapobiega apoptozie ko-
mórek. Bierze udzia∏ w powstawaniu folikularnej postaci ch∏oniaka typu B
Bcl-1
Zwany tak˝e PRAD1. Koduje cyklin´ D1, stymulujàcy komponent zegara
cyklu komórkowego. Bierze udzia∏ w powstawaniu raków piersi oraz g∏owy
i szyi
MDM2
Koduje bia∏ko antagonist´ bia∏ka supresorowego p53. Bierze udzia∏ w po-
wstawaniu mi´saków (nowotworów tkanki ∏àcznej) i innych nowotworów
GENY SUPRESOROWE NOWOTWORÓW
Geny bia∏ek cytoplazmatycznych
APC
Bierze udzia∏ w powstawaniu raków jelita grubego i ˝o∏àdka
DPC4
Koduje czàsteczk´ przekaênika na szlaku sygnalizacyjnym hamujàcym
podzia∏ komórkowy. Bierze udzia∏ w powstawaniu raka trzustki
NF-1
Koduje bia∏ko, które hamuje aktywnoÊç bia∏ka stymulujàcego Ras. Bierze
udzia∏ w powstawaniu nerwiako-w∏ókniaków i feochromocytoma (nowotwory
obwodowego systemu nerwowego) oraz bia∏aczki szpikowej
NF-2
Bierze udzia∏ w powstawaniu oponiaków i ependymoma (nowotwory mó-
zgu) oraz schwannoma (nowotwory wywodzàce si´ z komórek os∏aniajà-
cych nerwy obwodowe)
Geny bia∏ek jàdrowych
MTS1
Koduje bia∏ko p16, komponent hamujàcy zegar cyklu komórkowego. Bierze
udzia∏ w powstawaniu wielu ró˝nych rodzajów nowotworów
RB
Koduje bia∏ko pRB, g∏ówny hamulec cyklu komórkowego. Bierze udzia∏
w powstawaniu siatkówczaków oraz nowotworów koÊci, raków p´cherza
moczowego, piersi i drobnokomórkowego raka p∏uca
p53
Koduje bia∏ko p53, które mo˝e zatrzymaç podzia∏ komórkowy i indukowaç
samobójczà Êmierç komórek nieprawid∏owych. Bierze udzia∏ w powstawaniu
wielu ró˝nych rodzajów nowotworów
WT1
Bierze udzia∏ w powstawaniu guza Wilmsa w nerkach
Geny bia∏ek o nie znanej jeszcze lokalizacji komórkowej
BRCA1
Bierze udzia∏ w powstawaniu raków piersi i jajnika
BRCA2
Bierze udzia∏ w powstawaniu raków piersi
VHL
Bierze udzia∏ w powstawaniu raka nerki
cych i hamujàcych szlaków w komórce.
Zbiegajà si´ one w molekularnym apa-
racie jàdra komórkowego, który cz´sto
jest okreÊlany jako zegar cyklu komór-
kowego. Regulacyjny zegar podejmuje
decyzje oraz nadzoruje ich wykonanie
i najwyraêniej dostaje ob∏´du we
wszystkich rodzajach ludzkich nowo-
tworów z∏oÊliwych. W komórce prawi-
d∏owej integruje on otrzymywane przez
nià sygna∏y regulujàce wzrost, a nast´p-
nie decyduje, czy powinna ona przejÊç
przez swój cykl ˝yciowy. JeÊli odpo-
wiedê jest pozytywna, zegar kieruje tym
procesem.
36 Â
WIAT
N
AUKI
Listopad 1996
PODSTAWOWE MECHANIZMY
W
i´kszoÊç, a byç mo˝e wszystkie ludzkie nowotwory rosnà
nie tylko z powodu zaburzeƒ na szlakach sygnalizacyjnych
w komórkach, lecz tak˝e wskutek rozregulowania tzw. zegara cy-
klu komórkowego. Zegar – na który sk∏adajà si´ wspó∏dzia∏ajàce
ze sobà bia∏ka obecne w jàdrze komórkowym – normalnie inte-
gruje informacje docierajàce ze szlaku stymulujàcego i hamujàce-
go. JeÊli przewa˝ajà pierwsze z nich, zegar programuje przejÊcie
komórki przez cykl wzrostu i podzia∏u. PrzejÊcie przez cztery fa-
zy cyklu komórkowego (a) jest nap´dzane w du˝ej mierze podnie-
sieniem poziomu bia∏ek zwanych cyklinami: najpierw cykliny typu
D, nast´pnie E, A i w koƒcu B.
Kluczowym momentem cyklu jest tzw. punkt restrykcyjny (R)
póênej fazy G
1
: wówczas zapada decyzja, czy komórka ukierunku-
je si´ na ukoƒczenie cyklu. Aby komórka mog∏a przejÊç przez
punkt R i wejÊç w faz´ S, molekularny prze∏àcznik musi byç prze-
stawiony z pozycji „wy∏àczony” na pozycj´ „w∏àczony”. Prze∏àcznik
pracuje w nast´pujàcy sposób (b): Gdy wzrasta poziom cykliny
D i póêniej cykliny E, bia∏ka te ∏àczà si´ z cyklinozale˝nymi kina-
G
2
M
Poczàtek
cyklu
Komórka
dzieli si´
(mitoza)
Faza
spoczynkowa
Komórka
replikuje
swój DNA
Komórka powi´ksza si´
i produkuje nowe bia∏ka
Komórka
przygotowuje si´
do mitozy
Punkt restrykcyjny:
komórka decyduje,
czy nadal odpoczywaç,
czy wejÊç w cykl
S
R
G
1
G
0
1
2
3
Reszta
fosforanowa
Cyklinozale˝na
kinaza
Kompleks
aktywny
Cyklina D lub E
ATP
Nieaktywne pRB
Aktywne pRB
(hamulec
g∏ówny)
Nieaktywny
czynnik
transkrypcyjny
Aktywny
czynnik
transkrypcyjny
Gen
Bia∏ka
potrzebne
do przejÊcia
przez cykl
komórkowy
Sygna∏y
z sàsiednich
komórek
hamujàce
wzrost
Transformujàcy
czynnik
wzrostowy beta
(inhibitor)
p27
p15*
Kompleks
cykliny D
z CDK4/6
Sygna∏y
z sàsiednich
komórek
stymulujàce
wzrost
Cyklina D*
Cyklinozale˝ne
kinazy 4* lub 6
(CDK4/6)
Wczesna G
1
FAZY CYKLU KOMÓRKOWEGO
Nieaktywne
bia∏ko
pRB
Zegar cyklu komórkowego i rak
a
FAZY CYKLU KOMÓRKOWEGO
b
MOLEKULARNY PRZE¸ÑCZNIK
c
ZEGAR CYKLU KOMÓRKOWEGO W AKCJI
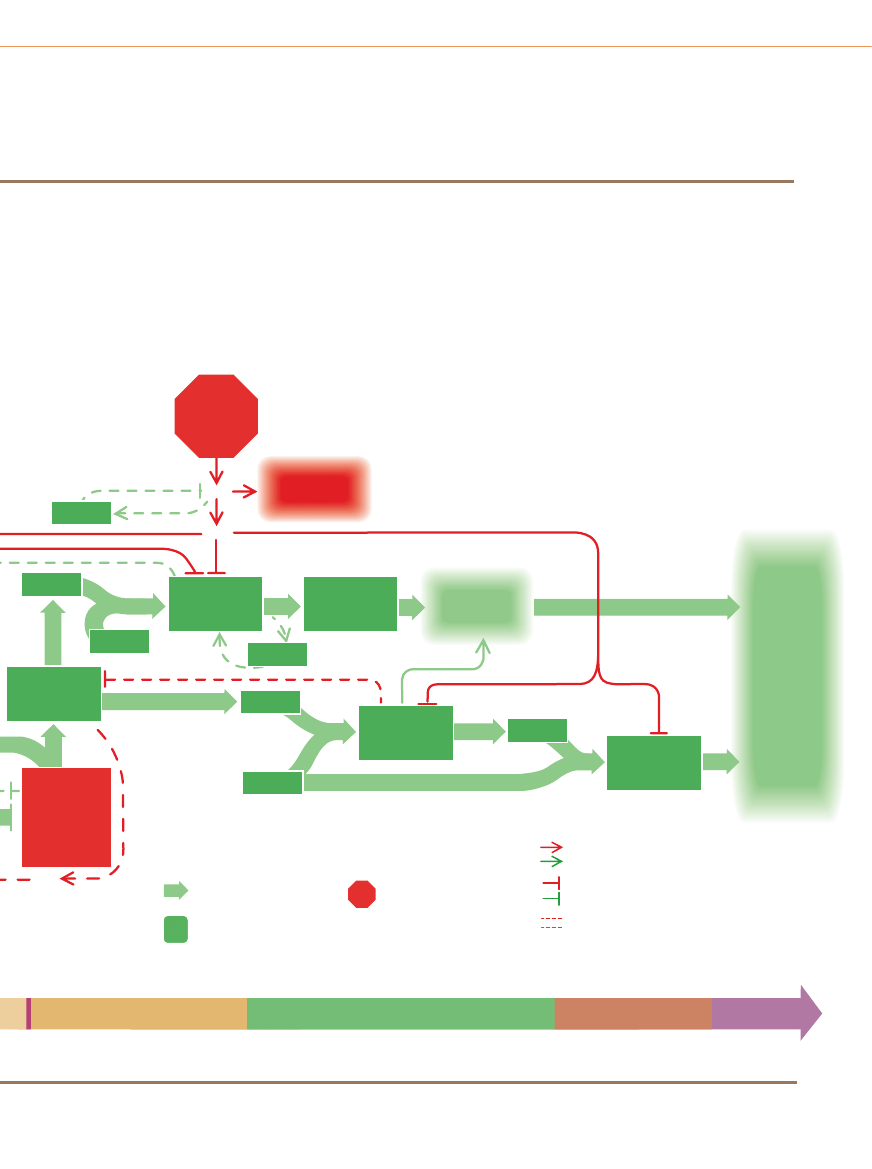
Cykl komórkowy sk∏ada si´ z czte-
rech faz. W fazie G
1
(gap 1 – przerwa 1)
komórka zwi´ksza swojà mas´ i przy-
gotowuje si´ do kopiowania (replikacji)
DNA. Replikacja zachodzi w fazie na-
st´pnej zwanej S (synthesis – synteza)
i umo˝liwia komórce precyzyjne po-
dwojenie jej zestawu chromosomów.
Gdy chromosomy sà ju˝ zduplikowa-
ne, nast´puje druga przerwa zwana G
2
,
w czasie której komórka przygotowuje
si´ do fazy M (mitosis – mitoza). Pod-
czas mitozy powi´kszona komórka ma-
cierzysta ostatecznie dzieli si´ na pó∏,
na dwie komórki potomne, z których
Â
WIAT
N
AUKI
Listopad 1996 37
zami – CDK (1). Zaktywowane w ten sposób kinazy (dzia∏ajàce
jako cz´Êç kompleksu cyklina–kinaza) odbierajà reszty fosforano-
we (2) z czàsteczek ATP (adenozynotrifosforanu) i przenoszà je na
bia∏ko zwane pRB – g∏ówny hamulec zegara cyklu komórkowego.
Nieufosforylowane bia∏ko pRB aktywnie blokuje cykl (trzymajàc
prze∏àcznik w pozycji „wy∏àczony”) dzi´ki wychwytywaniu innych
bia∏ek zwanych czynnikami transkrypcyjnymi. Jednak gdy kom-
pleksy cyklinokinazowe dodadzà do bia∏ka pRB odpowiednià iloÊç
reszt fosforanowych, hamulec puszcza (3; na dole); uwalnia czyn-
niki transkrypcyjne zdolne teraz oddzia∏ywaç z genami (3; u gó-
ry)
. Uwolnione czynniki wzmagajà produkcj´ ró˝nych bia∏ek po-
trzebnych do kontynuowania cyklu komórkowego.
Na rycinie (c) prze∏àcznik ten pokazano w Êrodowisku oddzia-
∏ywaƒ czàsteczkowych, które regulujà cykl komórkowy. Prze∏àcz-
nik w pozycji „w∏àczony” (a wi´c takiej, w której hamulec podzia∏ów
jest zwolniony) widoczny jest na schemacie ponad punktem R.
W niektórych ludzkich nowotworach wyst´puje nadmierna aktyw-
noÊç bia∏ek stymulujàcych, takich jak cyklina D, cyklina E i CDK4.
Udokumentowano tak˝e inaktywacj´ ró˝nych bia∏ek inhibitoro-
wych, na przyk∏ad p53 (nieobecnego albo nieaktywnego w po-
nad po∏owie wszystkich rodzajów nowotworów), pRB, p16 i p15.
Cz´stym wynikiem jakiejkolwiek z tych zmian jest rozregulowa-
nie zegara cyklu komórkowego i w nast´pstwie nadmierna proli-
feracja komórek.
Uszkodzenie
DNA lub
brak tlenu
Podzia∏
komórki
p53*
MDM2
p21
p16*
AktywnoÊç, która stymuluje
podzia∏ komórkowy
G∏ówny szlak prowadzàcy
do podzia∏u komórkowego
Zewn´trzny sygna∏,
który stymuluje
podzia∏ komórkowy
AktywnoÊç, która powstrzymuje
podzia∏ komórkowy
Zewn´trzny sygna∏,
który powstrzymuje
podzia∏ komórkowy
Cyklina B
CDC25A
Synteza
DNA
Kompleks cykliny A
z CDK1
Kompleks cykliny B
z CDK1
Bia∏ka
zaanga˝owane
w syntez´ DNA
Kompleks
cykliny E
z CDK2
Uwolnione
czynniki
transkrypcyjne
Samobójstwo
komórki
(apoptoza)
Cyklina E*
CDK2
Cyklina A
CDK1
G
2
Póêna G
1
R
S
M
zielony
*
czerwony
Sygna∏ pozytywny (zwi´kszajàcy iloÊç
lub aktywnoÊç okreÊlonych czàsteczek),
Sygna∏ negatywny (zmniejszajàcy iloÊç
lub aktywnoÊç okreÊlonych czàsteczek)
P´tla zwrotna
Mutacje lub rozregulowanie dzia∏ania
genów tych bia∏ek znaleziono
w ludzkich komórkach nowotworowych
Aktywne
bia∏ko pRB*
w kompleksie
z nieaktywnymi
czynnikami
transkrypcji
(g∏ówny hamulec)

ka˝da jest wyposa˝ona w kompletny
zestaw chromosomów. Komórki po-
tomne natychmiast wchodzà w faz´ G
1
i mogà ponownie przejÊç ca∏y cykl albo
te˝ zatrzymaç go na jakiÊ czas lub na
sta∏e.
Zegar cyklu komórkowego progra-
muje ten szczegó∏owo opracowany ciàg
zdarzeƒ za pomocà rozmaitych czàste-
czek. Dwa podstawowe komponenty
zegara, cykliny i cyklinozale˝ne kinazy
(CDK – cyclin-dependent kinases), aso-
cjujà ze sobà i inicjujà wejÊcie w poszcze-
gólne fazy cyklu komórkowego. Na
przyk∏ad podczas fazy G
1
cykliny ty-
pu D wià˝à si´ z CDK4 lub CDK6, a po-
wsta∏e kompleksy czàsteczkowe dzia-
∏ajà na czàsteczk´ silnego inhibitora
wzrostu – bia∏ka zwanego pRB. Dzia∏a-
nie to znosi hamujàcy efekt pRB i umo˝-
liwia komórce wejÊcie w póênà faz´ G
1
,
a nast´pnie w faz´ S [b na poprzedniej
stronie].
ZdolnoÊç komórki do przejÊcia ca∏ego
cyklu mogà ograniczaç ró˝ne bia∏ka ha-
mujàce, wÊród nich p15 (wspomniane
wczeÊniej) i p16. Blokujà one aktywnoÊç
kinaz zale˝nych od cykliny D i w ten
sposób uniemo˝liwiajà przejÊcie komór-
ki z fazy G
1
do fazy S. Inny inhibitor cy-
klinozale˝nych kinaz, bia∏ko p21, mo˝e
dzia∏aç w ciàgu ca∏ego cyklu komórko-
wego. Znajduje si´ ono pod kontrolà
bia∏ka supresorowego p53, które moni-
toruje „stan zdrowia” komórki, integral-
noÊç jej chromosomalnego DNA i po-
myÊlne zakoƒczenie ró˝nych faz cyklu.
Komórki raka piersi cz´sto produku-
jà nadmiar cykliny D i cykliny E. W wie-
lu przypadkach czerniaka komórki skó-
ry tracà gen kodujàcy inhibitorowe bia∏-
ko p16. Po∏owa wszystkich typów ludz-
kich nowotworów traci funkcjonalne
bia∏ko p53. W raku szyjki macicy – indu-
kowanym przez infekcj´ komórek wi-
rusem brodawczaka ludzkiego – za-
równo bia∏ko pRB, jak i bia∏ko p53 sà
cz´sto unieczynnione, co eliminuje dwa
najbardziej podstawowe ograniczniki
zegara cyklu komórkowego. W rezul-
tacie we wszystkich tych przypadkach
zegar si´ rozregulowuje i ignoruje
wszelkie zewn´trzne sygna∏y nakazu-
jàce mu zatrzymanie. JeÊli naukowcy
zdo∏ajà opracowaç sposób zabloko-
wania cyklin i cyklinozale˝nych kinaz
uczestniczàcych w cyklu komórkowym,
b´dzie mo˝na powstrzymaç rakowa-
ciejàce komórki przed podzia∏em.
Dotychczas opisa∏em dwa sposoby,
które pozwalajà naszym prawid∏owym
tkankom hamowaç proliferacj´ komórek
i unikaç nowotworzenia. Zapobiegajà
one nadmiernemu namna˝aniu komó-
rek albo pozbawiajàc komórk´ czynni-
ków stymulujàcych wzrost, albo zalewa-
jàc jà czynnikami, które ten wzrost
hamujà. Jednak˝e jak widzimy, komórki
na drodze do rakowacenia cz´sto wy-
mykajà si´ spod kontroli, same si´ sty-
mulujà i g∏uchnà na sygna∏y hamujàce.
Organizm cz∏owieka przygotowany na
takà ewentualnoÊç wyposa˝y∏ komórki
w pewne systemy zabezpieczajàce przed
nie kontrolowanymi podzia∏ami. Dodat-
kowe mutacje w genetycznym wyposa-
˝eniu komórki mogà jednak pokonywaç
nawet te mechanizmy, przyczyniajàc si´
do nowotworzenia.
Uszkodzenie zabezpieczeƒ
Jeden z takich systemów obecny w
ka˝dej ludzkiej komórce sk∏ania jà do
pope∏nienia samobójstwa (wejÊcia w
apoptoz´) w przypadku uszkodzenia
podstawowych komponentów zegara
lub rozregulowania systemów kontrol-
nych. Apoptoz´ mo˝e na przyk∏ad in-
dukowaç uszkodzenie chromosomal-
nego DNA.
Co wi´cej, ostatnie wyniki wielu prac
z ró˝nych laboratoriów wskazujà, ˝e ta-
kà odpowiedê mo˝e tak˝e wywo∏ywaç
uaktywnienie onkogenu lub unieczyn-
nienie genu supresorowego. Rozpad
uszkodzonej komórki jest dla niej samej
niekorzystny, ale ma znaczenie dla orga-
nizmu jako ca∏oÊci: potencjalne niebez-
pieczeƒstwo ze strony kancerogennych
mutacji jest daleko wi´ksze ni˝ niewiel-
ka cena p∏acona za utrat´ jednej komór-
ki. Pojawiajàce si´ w naszych tkankach
guzy nowotworowe wydajà si´ zatem
wywodziç z rzadko wyst´pujàcej, ge-
netycznie uszkodzonej komórki, której
w jakiÊ sposób uda∏o si´ uniknàç reali-
zacji programu apoptozy wbudowane-
go w jej obwód kontrolny.
Rozwijajàce si´ komórki rakowe wy-
nalaz∏y kilka sposobów unikni´cia sa-
mobójczej Êmierci. W ró˝nych rodzajach
komórek nowotworowych inaktywacja
bia∏ka p53 (które pe∏niàc wiele funkcji,
pomaga m.in. w indukcji apoptozy)
zmniejsza szanse, ˝e genetycznie uszko-
dzone komórki zostanà wyeliminowa-
ne. Komórki rakowe mogà tak˝e produ-
38 Â
WIAT
N
AUKI
Listopad 1996
PODSTAWOWE MECHANIZMY
CHROMOSOMY LUDZKIE w normalnej dzielàcej si´ komórce
(u góry) wyst´pujà w po-
staci identycznych par; na ilustracji widoczne sà chromosomy od 8 do 18. Natomiast chro-
mosomy z komórki raka szyjki macicy wykazujà liczne nieprawid∏owoÊci
(na dole). Na
przyk∏ad w chromosomie 8 widaç trzy zaburzenia – zwi´kszenie liczby kopii, utrat´ ma-
teria∏u genetycznego przez niektóre kopie i po∏àczenie nie nale˝àcych do siebie segmen-
tów powsta∏ych na skutek uprzedniego p´kni´cia
(chromosom 8 najbardziej na prawo).
Utrata kopii, tak jak w chromosomie 13, jest tak˝e powszechna. Obrazy chromosomów
otrzymano dzi´ki kariotypowaniu widmowemu – nowej metodzie analizy chromosomów
(komputerowa analiza widma Êwiat∏a emitowanego przez sondy fluorescencyjne rozpozna-
jàce swoiste regiony chromosomów – przyp. t∏um.).
KOMÓRKA PRAWID¸OWA
KOMÓRKA RAKOWA
8q
8q
8
8
Kopie skrócone
9
9
10
11
12
13
14
15
15
14
13
12
11
10
MERRYN MACVILLE i THOMAS REID
National Center for Human Genome Research, NIH

kowaç nadmierne iloÊci bia∏ka Bcl-2, któ-
re skutecznie chroni przed apoptozà.
Obecnie naukowcy uÊwiadomili so-
bie, ˝e ta zdolnoÊç komórek do unikni´-
cia apoptozy nara˝a pacjentów nie tyl-
ko na niebezpieczeƒstwo rozrostu guza,
ale i na to, ˝e stanie si´ on oporny na le-
czenie. Przez wiele lat uwa˝ano, ˝e tera-
pia radiacyjna i liczne leki zabijajà z∏o-
Êliwe komórki bezpoÊrednio, wywo∏ujàc
rozleg∏e uszkodzenia w ich DNA. Obec-
nie wiemy, ˝e takie post´powanie cz´sto
tylko nieznacznie uszkadza DNA. Nie-
mniej tak dotkni´te komórki „stwier-
dzajà”, ˝e uszkodzenie nie mo˝e byç ∏a-
two naprawione i pope∏niajà samo-
bójstwo. Oznacza to, ˝e komórki rakowe
zdolne uniknàç apoptozy b´dà znacz-
nie mniej podatne na leczenie oraz ˝e
terapia pozwalajàca przywróciç komór-
kom zdolnoÊç do samobójstwa mog∏aby
niszczyç nowotwór, podnoszàc efek-
tywnoÊç dotychczasowych strategii
walki z rakiem poprzez radio- i
chemioterapi´.
NieÊmiertelnoÊç komórek
W nasze komórki wbudowany jest
tak˝e drugi mechanizm zabezpieczajà-
cy je przed nie kontrolowanymi po-
dzia∏ami, ca∏kiem odmienny od progra-
mu apoptozy. Liczy on i ogranicza
liczb´ wszystkich podzia∏ów, jakie ko-
mórka przechodzi w ciàgu ca∏ego swo-
jego ˝ycia. Znaczna cz´Êç wiedzy o tym
mechanizmie pochodzi z badaƒ nad ko-
mórkami hodowanymi na p∏ytkach Pe-
triego. Komórki pobrane z zarodka my-
siego lub ludzkiego, hodowane in vitro,
mniej wi´cej podwajajà swà populacj´
ka˝dego dnia. Ale po pewnej przewi-
dywalnej liczbie podzia∏ów – 50 do 60
w przypadku komórek ludzkich – za-
trzymujà si´; mówimy wtedy, ˝e komór-
ki zaczynajà si´ starzeç. Tak w∏aÊnie si´
dzieje, jeÊli tylko majà one nienaruszo-
ne geny RB i p53. Natomiast komórki,
które prze˝y∏y inaktywujàce mutacje
w jednym z tych genów, kontynuujà po-
dzia∏y. Takie prze˝ywajàce, ciàgle dzie-
làce si´ komórki osiàgajà w koƒcu dru-
gà faz´ zwanà kryzysem, w której
przewa˝ajàca ich liczba ginie. Zdarza
si´ jednak, ˝e jakaÊ przypadkowa ko-
mórka z tej wymierajàcej populacji
przetrwa kryzys i zyska zdolnoÊç do na-
mna˝ania si´ w nieskoƒczonoÊç. Ko-
mórki takie i ich potomstwo stajà si´
nieÊmiertelne.
Wynika z tego, ˝e istnieje mechanizm
zliczajàcy liczb´ pokoleƒ, przez które
populacja komórek ju˝ przesz∏a. W cià-
gu ostatnich kilku lat naukowcy wy-
kryli jego molekularne elementy. To
segmenty DNA na koƒcu chromoso-
mów, zwane telomerami, decydujà o
liczbie podzia∏ów komórkowych i w
odpowiednim czasie inicjujà proces sta-
rzenia i kryzysu. W ten sposób ograni-
czajà one zdolnoÊç populacji komórko-
wych do nie kontrolowanej ekspansji
[patrz: Carol W. Greider i Elizabeth H.
Blackburn, „Telomery, telomeraza
i rak”; Âwiat Nauki, kwiecieƒ 1996].
Kapturki telomerowe niczym plasti-
kowe koƒcówki sznurowade∏ chronià
koƒce chromosomów przed uszkodze-
niem. W wi´kszoÊci komórek cz∏owieka
telomery nieznacznie si´ skracajà pod-
czas ka˝dego podwajania si´ chromo-
somów zachodzàcego w fazie S cyklu
komórkowego. Skrócenie si´ telomerów
poni˝ej pewnej wartoÊci progowej jest
sygna∏em nakazujàcym komórkom we-
jÊcie w okres starzenia. JeÊli komórki
wcià˝ b´dà si´ dzieliç, dalsze skracanie
si´ telomerów mo˝e w koƒcu wywo∏aç
kryzys; po nadmiernym ich skróceniu
chromosomy w komórce zacznà si´ ∏à-
czyç ze sobà albo p´kaç, wprowadzajàc
chaos genetyczny majàcy dla komórki
Êmiertelne skutki.
JeÊli w komórkach nowotworowych
telomerowy system liczenia podzia∏ów
funkcjonuje prawid∏owo, ich namna˝a-
nie zostaje wstrzymane na d∏ugo, zanim
guz osiàgnie du˝e rozmiary. Niebez-
piecznej ekspansji guza zapobiega pro-
gram starzenia lub, jeÊli komórka omi-
nie t´ blokad´, zamieszanie w chro-
mosomalnym porzàdku wyst´pujàce
w fazie kryzysu. Jednak podczas rozwo-
ju wi´kszoÊci komórek nowotworowych
aktywacja genu kodujàcego enzym zwa-
ny telomerazà niweczy nadzieje na
skutecznoÊç tej ostatniej linii obrony.
Telomeraza, praktycznie nieobecna
w wi´kszoÊci zdrowych komórek, ale
wyst´pujàca prawie we wszystkich ko-
mórkach nowotworowych, systema-
tycznie odbudowuje segmenty telome-
rowe, które podczas ka˝dego cyklu ko-
mórkowego sà zwykle przycinane
i odrzucane. W ten sposób enzym ten
zapewnia zachowanie telomerów w ich
nienaruszonej postaci, dzi´ki czemu ko-
mórki mogà dzieliç si´ w nieskoƒczo-
noÊç. NieÊmiertelnoÊç komórek – kon-
sekwencja tego procesu – mo˝e byç
groêna z kilku powodów. Przede wszy-
stkim pozwala guzom osiàgaç du˝e roz-
miary. Daje tak˝e komórkom przedra-
kowym i rakowym czas na akumulacj´
dodatkowych mutacji, co zwi´ksza ich
zdolnoÊç do namna˝ania si´, inwazyj-
noÊci i w koƒcu do przerzutowania.
Z punktu widzenia komórki rakowej
produkcja jednego enzymu jest racjo-
nalnym sposobem obalenia bariery
ÊmiertelnoÊci. Ale zale˝noÊç od jedne-
go enzymu mo˝e byç tak˝e jej pi´tà
Achillesowà. Gdyby uda∏o si´ zabloko-
waç telomeraz´ w komórkach rako-
wych, ich telomery mog∏yby znowu
skracaç si´ przy ka˝dym podziale, po-
pychajàc je w faz´ kryzysu i Êmierci.
Z tego powodu liczne firmy farmaceu-
tyczne usi∏ujà opracowaç lek skierowa-
ny przeciw telomerazie.
Dlaczego pojawiajà si´ wczeÊniej
Zwykle potrzeba kilkudziesi´ciu lat,
aby powstajàcy guz zdo∏a∏ zgromadziç
wszystkie mutacje niezb´dne do jego
nowotworowego, z∏oÊliwego wzrostu.
Jednak˝e u niektórych ludzi czas roz-
woju nowotworu jest wyraênie skróco-
ny; zapadajà oni na pewne rodzaje no-
wotworów z∏oÊliwych kilkadziesiàt lat
wczeÊniej ni˝ wi´kszoÊç populacji. W ja-
ki sposób dochodzi do przyspieszenia
procesu nowotworowego?
W wielu przypadkach mo˝na to wy-
t∏umaczyç odziedziczeniem od które-
goÊ z rodziców zmutowanego genu po-
wodujàcego raka. Gdy zap∏odniona
komórka jajowa zaczyna swe podzia∏y,
zestawy genów z plemnika i komórki
jajowej sà kopiowane i rozdzielane do
wszystkich komórek nowego organi-
zmu. Wtedy rzadkie na ogó∏ zdarzenie
– mutacja w kluczowym genie kontrolu-
jàcym wzrost – staje si´ powszechna,
poniewa˝ zostaje wszczepiona we
wszystkie komórki cia∏a, a nie tylko
w niektóre, przypadkowo dotkni´te
takim defektem. Innymi s∏owy, proces
tworzenia si´ nowotworu przeskakuje
jedno ze swych wczesnych, powoli za-
chodzàcych stadiów i w rezultacie jako
ca∏oÊç ulega przyspieszeniu. W konse-
kwencji rozwój guza, który zwykle trwa
Â
WIAT
N
AUKI
Listopad 1996 39
16
17
18
18
17
16

30–40 lat, mo˝e si´ zakoƒczyç w ciàgu
jednego lub dwóch dziesi´cioleci. JeÊli
takie zmutowane geny b´dà przekazy-
wane z pokolenia na pokolenie, wielu
cz∏onkom rodziny zagrozi ryzyko wcze-
snego rozwoju nowotworu.
Dramatycznym przyk∏adem jest tu
dziedziczna postaç raka jelita grubego.
Wi´kszoÊç przypadków tej choroby po-
jawia si´ sporadycznie, w wyniku
przypadkowych zdarzeƒ genetycznych
zachodzàcych w ciàgu ˝ycia danego
cz∏owieka. Jednak w niektórych rodzi-
nach wiele osób zapada na wczeÊnie
wyst´pujàce raki jelita grubego, które
sà zaprogramowane dziedziczonym ge-
nem. W sporadycznych przypadkach
raka rzadko wyst´pujàca mutacja w na-
b∏onkowej komórce jelita wycisza (czy-
li unieczynnia) gen supresorowy APC.
W wyniku podzia∏ów zmutowanej ko-
mórki powstaje ∏agodny polip, który
mo˝e w koƒcu przekszta∏ciç si´ w z∏oÊli-
wego raka. Jednak w niektórych rodzi-
nach defektywne formy genu APC mo-
gà przechodziç z rodziców na dzieci.
U cz∏onków takich rodzin setki lub na-
wet tysiàce polipów jelitowych rozwi-
jajà si´ w ciàgu pierwszych 10 lat ˝ycia
i istnieje du˝e prawdopodobieƒstwo,
˝e niektóre z nich ulegnà transformacji
do raków.
Lista nowotworów wyst´pujàcych w
obr´bie rodzin, wiàzanych dziÊ bezpo-
Êrednio z dziedziczeniem zmutowa-
nych genów supresorowych, stale ro-
Ênie. Na przyk∏ad wrodzona defek-
tywna wersja genu kodujàcego bia∏ko
pRB cz´sto prowadzi u dzieci do roz-
woju raka oka – siatkówczaka (retino-
blastoma); w póêniejszym okresie ˝y-
cia mutacje te sà odpowiedzialne za
podwy˝szone ryzyko zachorowania na
mi´saka koÊciopochodnego (nowotwo-
ru z∏oÊliwego koÊci). Odziedziczenie
zmutowanej wersji genu supresorowe-
go p53 prowadzi do zachorowania na
nowotwory o ró˝nym umiejscowieniu;
chorob´ t´ nazwano zespo∏em Li-Frau-
meni (cz´Êciowo od nazwiska Frederi-
cka Li, wspó∏autora artyku∏u „Co po-
woduje raka?”, strona 50). Wyizolowa-
ne ostatnio geny BRCA1 i BRCA2 od-
powiadajà – jak si´ wydaje – za wi´k-
szoÊç tzw. rodzinnych raków gruczo∏u
mlecznego
2
, w tym a˝ za 20% wszyst-
kich przedmenopauzalnych raków pier-
si w USA, a tak˝e znacznà cz´Êç rodzin-
nych raków jajnika
3
.
Niekiedy wczesne wyst´powanie no-
wotworów mo˝na wyjaÊniç odziedzi-
czeniem mutacji, które znajdujà si´ w in-
nych klasach genów. Jak zaznaczy∏em
wczeÊniej, wi´kszoÊç ludzi nie zapada
na nowotwór do póênego okresu ˝ycia
albo nie choruje nigdy, poniewa˝ przy-
chodzi na Êwiat z nienaruszonym zesta-
wem genów. W ciàgu ˝ycia jednak na-
sze geny sà atakowane przez czynniki
rakotwórcze pochodzàce ze Êrodowi-
ska, a tak˝e przez substancje chemicz-
ne produkowane w naszych w∏asnych
komórkach. B∏´dy genetyczne mogà po-
wstawaç te˝ wtedy, gdy enzymy re-
plikujàce DNA podczas cyklu komór-
kowego mylà si´ w kopiowaniu. W
wi´kszoÊci przypadków sà one szybko
korygowane przez z∏o˝ony system na-
prawczy, który dzia∏a w ka˝dej komór-
ce. JeÊli jednak system naprawczy si´
myli i nie potrafi ich usunàç, uszkodze-
nie staje si´ trwa∏à mutacjà w jednym
z genów tej komórki oraz wszystkich jej
komórek potomnych.
Wysoka sprawnoÊç systemu napraw-
czego jest jednym z powodów, dla któ-
rego up∏ywa zwykle wiele dziesi´ciole-
ci, zanim wszystkie mutacje niezb´dne
do nowotworzenia przypadkowo zgro-
madzà si´ w pojedynczej komórce. Nie-
które defekty dziedziczne sà jednak
zdolne przyspieszyç rozwój nowotwo-
ru w szczególnie podst´pny sposób: za-
burzajà one dzia∏anie bia∏ek naprawia-
jàcych uszkodzenia DNA. W wyniku
tego mutacje, które normalnie groma-
dzà si´ powoli, zaczynajà wyst´powaç
z alarmujàcà cz´stoÊcià w ca∏ym DNA
komórek. WÊród takich uszkodzonych
genów nieuchronnie sà i te, które kon-
trolujà namna˝anie si´ komórek.
Tak dzieje si´ na przyk∏ad w przy-
padku innego wrodzonego raka jelita
grubego: dziedzicznego, niepolipowa-
tego raka jelita. Dotkni´te nim osoby
produkujà defektywnà form´ bia∏ka
odpowiedzialnego za napraw´ uszko-
dzeƒ pope∏nionych podczas kopiowa-
nia przez aparat replikacyjny DNA.
Wskutek tego komórki jelita grubego
nie mogà skutecznie usuwaç uszkodzeƒ
DNA, dlatego te˝ gromadzà one muta-
cje znaczenie szybciej, co przyspiesza
o dziesi´ciolecie lub dwa rozwój nowo-
tworu. Ludzie dotkni´ci innym rakiem
rodzinnym – zespo∏em tzw. skóry per-
gaminowatej barwnikowej (Xeroderma
pigmentosum) – odziedziczyli defektyw-
nà kopi´ genu, który steruje naprawà
uszkodzeƒ DNA indukowanych pro-
mieniami ultrafioletowymi. Tacy pa-
cjenci podatni sà na liczne rodzaje raka
skóry wywo∏anego promieniowaniem
s∏onecznym.
Podobnie komórki ludzi urodzonych
z defektywnym genem ATM majà trud-
noÊci z rozpoznawaniem obecnoÊci
pewnych uszkodzeƒ DNA i z urucha-
40 Â
WIAT
N
AUKI
Listopad 1996
PODSTAWOWE MECHANIZMY
FUNDACJA NA RZECZ
CHORYCH
Z NOWOTWORAMI
KRWI
PRZY
INSTYTUCIE
HEMATOLOGII
I TRANSFUZJOLOGII
I TY MO˚ESZ
Wyobraê sobie nast´pujàce zdarzenie:
TONIE CZ¸OWIEK!
JesteÊ jedynym Êwiadkiem wypadku.
Co robisz?
Ryzykujàc ˝ycie rzucasz si´ do wody!
URATOWAå
A teraz wyobraê sobie
sal´ szpitalnà.
Le˝y w niej cz∏owiek chory
na bia∏aczk´. On te˝ ginie!
Szansà dla niego jest bardzo
kosztowne leczenie.
Tylko ono mo˝e uratowaç
mu ˝ycie.
Czy ofiarowujàc swój dar
zechcesz daç mu szans´?
Chorych z nowotworami krwi
jest coraz wi´cej!
Ofiarowujàc swój dar
– ofiarowujesz komuÊ ˝ycie!
˚YCIE
ul. Chocimska 4;
00-957 Warszawa
tel./fax: 0-22-6460417
Numer konta:
PKO BP XIII O/Warszawa
1632-21018-132-3

mianiem odpowiedniej reakcji napraw-
czej. U takich osób ∏atwiej dochodzi do
chorób degeneracyjnych uk∏adu nerwo-
wego, zaburzeƒ w tworzeniu naczyƒ
krwionoÊnych, cz´Êciej te˝ wyst´pujà
u nich nowotwory. Niektórzy naukow-
cy sàdzà, ˝e a˝ 10% zachorowaƒ na
wrodzonego raka piersi mo˝e byç
spowodowane dziedziczeniem uszko-
dzonych kopii tego genu.
W ciàgu nast´pnego dziesi´ciolecia
lista genów warunkujàcych podatnoÊç
na nowotwory zapewne gwa∏townie
wzroÊnie za przyczynà programu Hu-
man Genome Project – Projektu Pozna-
nia Ludzkiego Genomu (którego celem
jest zidentyfikowanie ka˝dego genu
w komórce ludzkiej). Wraz z rozwojem
coraz pot´˝niejszych narz´dzi do ana-
lizy DNA wiedza o tych genach pozwo-
li nam przewidzieç, którym cz∏onkom
rodzin podatnych na nowotwory grozi
wysokie ryzyko, a którzy szcz´Êliwym
trafem odziedziczyli nietkni´te ich
kopie.
Co oprócz proliferacji
Chocia˝ posiedliÊmy ju˝ ogromnà
wiedz´ o genetycznych uwarunkowa-
niach nie kontrolowanej proliferacji
komórek, wcià˝ bardzo ma∏o wiemy o
mutacjach genów uczestniczàcych w
póêniejszych stadiach rozwoju nowo-
tworu. Chodzi szczególnie o te geny,
które umo˝liwiajà komórkom nowo-
tworowym wp∏yw na rozwój od˝ywia-
jàcych je naczyƒ krwionoÊnych, inwa-
zj´ przyleg∏ych tkanek i przerzutowanie
do odleg∏ych miejsc cia∏a. Jednak˝e ba-
dania w tych dziedzinach posuwajà si´
szybko naprzód. (Judah Folkman w ar-
tykule „Atak na uk∏ad krwionoÊny gu-
za” na stronie 122 przedstawia genialnà
zdolnoÊç komórek nowotworowych do
pobudzania rozwoju w∏asnej sieci do-
starczajàcej krew; Erkki Ruoslahti opi-
suje proces powstawania przerzutów
w artykule „Jak rozsiewa si´ rak?” na
stronie 42.)
Daleko nam jeszcze do przedstawie-
nia w szczegó∏ach „historii naturalnej”
wielu ludzkich nowotworów od ich
powstania do gro˝àcego cz∏owiekowi
Êmiercià koƒca. Biografie te b´dà pisane
w j´zyku genów i czàsteczek. W ciàgu
najbli˝szych 10 lat poznamy z niezwy-
k∏à precyzjà nast´pstwo zdarzeƒ, które
sk∏adajà si´ na ewolucj´ normalnych ko-
mórek do ich wysoce z∏oÊliwych, inwa-
zyjnych potomków.
Byç mo˝e zaczniemy wtedy rozu-
mieç, dlaczego niektóre umiejscowio-
ne masy komórek nowotworowych ni-
gdy nie zmieniajà swojego charakteru
nowotworu ∏agodnego, nieinwazyjne-
go, w przeciwieƒstwie do nowotworów
z∏oÊliwych, agresywnych. Takie ∏agod-
ne nowotwory mo˝na znaleêç prawie
w ka˝dym narzàdzie organizmu. Praw-
dopodobnie b´dziemy mogli równie˝
zrozumieç, dlaczego pewne zmutowa-
ne geny przyczyniajà si´ do powsta-
wania tylko okreÊlonych rodzajów no-
wotworów. Na przyk∏ad zmutowana
wersja genu supresorowego RB (który
koduje inhibitorowe bia∏ko pRB) poja-
wia si´ cz´sto w siatkówczaku, raku p´-
cherza moczowego i drobnokomórko-
wym raku p∏uca, ale rzadko wyst´puje
w rakach piersi i rakach jelita grubego.
Jest wysoce prawdopodobne, ˝e wiele
tych tajemnic rozwià˝à badania w dzie-
dzinie biologii rozwoju (embriologii).
Przecie˝ to w∏aÊnie geny kierujàce roz-
wojem zarodkowym sà póêniej, w na-
szym doros∏ym ˝yciu, êród∏em nowo-
tworów z∏oÊliwych.
Jedno jest ju˝ pewne: iloÊç informacji
o przyczynach powstawania nowotwo-
rów zebranych w ciàgu ostatnich dwóch
dziesi´cioleci nie ma sobie równej w hi-
storii badaƒ biomedycznych. Niektóre
elementy tej wiedzy zosta∏y ju˝ wyko-
rzystane do stworzenia molekularnych
narz´dzi wykrywania i okreÊlania agre-
sywnoÊci niektórych typów nowotwo-
rów [patrz: David Sidransky, „Post´p
w wykrywaniu raka”, strona 74]. Mimo
doÊç dobrej wiedzy na temat procesu
nowotworowego nowe podejÊcia lecz-
nicze wcià˝ jeszcze sà zawodne. Jednà z
przyczyn tego jest fakt, ˝e komórki no-
wotworowe tylko nieznacznie ró˝nià
si´ od prawid∏owych; w czasie transfor-
macji nowotworowej zaledwie niewiel-
ka cz´Êç z dziesiàtków tysi´cy genów w
komórce ulega uszkodzeniu. Tak wi´c
sprzymierzeniec i z∏oÊliwy przeciwnik
sà utkani z podobnego materia∏u i ka˝-
dy atak skierowany przeciwko wrogo-
wi mo˝e w takim samym stopniu uszko-
dziç wartoÊciowe, normalne tkanki.
Jednak˝e nasze szanse na wygranà
rosnà. Ró˝nice mi´dzy komórkami nor-
malnymi a nowotworowymi sà mo˝e
subtelne, ale naprawd´ istniejà. A te uni-
kalne cechy guzów nowotworowych
stanowià doskona∏y obiekt interwencji
za pomocà nowo opracowanych leków
[patrz: Terapie jutra, strona 107]. Obec-
nie takie metody sà wcià˝ w stadium
poczàtkowym, lecz wkrótce odejdzie-
my od metody prób i b∏´dów i zamiast
dokonywaç przypadkowych odkryç za-
czniemy racjonalnie opracowywaç
leki, projektujàc je tak, by trafia∏y w
precyzyjnie wybrane cele. Mam nadzie-
j´, ˝e pierwsze dziesi´ciolecie nowego
wieku przyniesie takie metody leczenia
raka, jakie nawet nie Êni∏y si´ wczeÊniej-
szym pokoleniom. Wytrwa∏e inwesto-
wanie narodu amerykaƒskiego w pod-
stawowe badania nad rakiem zostanie
hojnie wynagrodzone.
T∏umaczy∏
Mieczys∏aw Chorà˝y
Przypisy t∏umacza:
1
Rak (cancer) w znaczeniu ogólnym jest synoni-
mem nowotworu z∏oÊliwego. W Êcis∏ym, medycz-
nym znaczeniu termin „rak” (∏ac. carcinoma) okre-
Êla nowotwory z∏oÊliwe pochodzenia nab∏on-
kowego (nab∏oniaki) w odró˝nieniu od mi´saków,
którym to terminem okreÊla si´ nowotwory z∏oÊli-
we pochodzenia ∏àcznotkankowego. Guz (tumor;
po grecku onkos, stàd „onkologia”) oznacza ogól-
nie „nowotwór” bez bli˝szego precyzowania po-
chodzenia i z∏oÊliwoÊci. W j´zyku polskim wiele
nazw poszczególnych rodzajów nowotworów zo-
sta∏o utworzonych od nazw ∏aciƒskich, np. „glio-
ma”, „feochromocytoma” itd.
2
Gruczo∏ mleczny kobiety (i innych ssaków) ma
termin anatomiczny „sutek”. Ogólnie stosuje si´
termin „pierÊ”, stàd rak piersi i sutka u˝ywane sà
wymiennie.
3
W Êwietle ostatnich badaƒ wysoki odsetek
dziedzicznych mutacji genu BRCA1 odpowiedzial-
nych za podwy˝szone ryzyko raka piersi stwier-
dza si´ w populacji ˚ydówek aszkenazyjskich.
WÊród kobiet nie-˚ydówek odsetek ten jest znacz-
nie mniejszy.
Â
WIAT
N
AUKI
Listopad 1996 41
Informacje o autorze
ROBERT A. WEINBERG jest cz∏onkiem zespo∏u Whitehead Institute for
Biomedical Research i profesorem biologii w Massachusetts Institute
of Technology, w którym uzyska∏ doktorat z biologii w 1969 roku. Je-
go laboratorium przyczyni∏o si´ do wyizolowania pierwszego ludz-
kiego onkogenu i pierwszego ludzkiego genu supresorowego nowo-
tworów. Weinberg jest cz∏onkiem National Academy of Sciences.
Zdoby∏ wiele nagród za wyjaÊnienie genetyki raka, ostatnio G.H.A.
Clowes Memorial Award nadanà przez American Association for Can-
cer Research. Jest to jego czwarty artyku∏ w Scientific American.
Literatura uzupe∏niajàca
CANCER: SCIENCE AND SOCIETY.
J. Cairns; W.H. Freeman, 1978.
GENES AND THE BIOLOGY OF CANCER.
H. Varmus i R. A. Weinberg; Scien-
tific American Library (dystrybucja: W. H. Freeman), 1993.
THE MULTISTEP NATURE OF CANCER.
B. Vogelstein i K. W. Kinzler, Trends
in Genetics, vol. 9, nr 4, ss. 138-141, IV/1993.
CANCER: THE RISE OF THE GENETICS PARADIGM.
J. M. Bishop, Genes and
Development, vol. 9, nr 11, ss. 1309-1315, 1 VI 1995.
ONCOGENES.
Wyd. II. G. M. Cooper; Jones and Bartlett Publishers,
Boston, 1995.
Wyszukiwarka
Podobne podstrony:
jak powstaje rak
Jak powstaje rak
jak powstaje rak
Jak powstaje dziecięca agresja, Materiały niezbędne w pracy nauczyciela przedszkola
2012 11 17 Jak powstaje ustawa str 1
Jak powstają ergonomiczne narzędzia dla elektroników 1 cz
Jak powstaje film A4
4 JAK POWSTAJĄ LUDZKIE POSTAWY
jak powstaje dziecięca agresja
Jak powstał Black Tiger
Jak powstaje film A3
Jak Powstaly Tatry
ADOLF WARSKI, JAK POWSTAŁA KPRP
anomalia PS napisac jak powstaje pytania, GIG, semestr 6, Geofizyka górnicza
2 Jak powstaje dziura ozonowa
jak powstaje ustawa action(1)
jak powstają opady, scenariusze zajęć-edukacja zdrowotna
Jak powstaje książka, Informacje i Ciekawostki
więcej podobnych podstron