
Środowisko programowe do obliczenia
poziomów energetycznych
studni kwantowych typu III-V
Praca dyplomowa inżynierska
Jarosław Zawojski
Opiekun:
dr hab. inż. Włodzimierz Salejda, prof. PWr.
Wrocław, marzec 2007
Wydział Podstawowych Problemów Techniki

3
Serdecznie dziękuję opiekunowi pracy
za cierpliwość oraz cenne uwagi merytoryczne
przekazane mi w trakcie pisania pracy dyplomowej
.

4
Spis treści
1.
WPROWADZENIE..................................................................................................................................... 5
2.
ELEMENTY PASMOWEJ TEORII HETEROSTRUKTUR ................................................................. 6
2.1
H
ETEROSTRUKTURY
...................................................................................................................................... 6
2.2
R
ÓWNANIE MASY EFEKTYWNEJ
................................................................................................................... 10
3. METODY STRZAŁÓW ROZWIĄZYWANIA RÓWNANIA SCHRÖDINGERA ................................. 12
3.1
P
RZEGLĄD METOD
....................................................................................................................................... 12
3.2
P
ROSTA METODA STRZAŁÓW
....................................................................................................................... 12
3.3
U
LEPSZONA METODA STRZAŁÓW
................................................................................................................ 13
4. PREZENTACJA ORAZ DYSKUSJA WYNIKÓW .................................................................................... 16
4.1
W
YNIKI NUMERYCZNE DLA STUDNI KWANTOWEJ TYPU
III/V...................................................................... 16
4.2
Z
ALEśNOŚĆ WARTOŚCI ENERGII WŁASNYCH STUDNI KWANTOWEJ OD MASY EFEKTYWNEJ
......................... 16
4.4
Z
ALEśNOŚCI ENERGII WŁASNYCH W STUDNI KWANTOWEJ OD JEJ SZEROKOŚCI
........................................... 19
4.5
A
NALIZA DOKŁADNOŚCI
.............................................................................................................................. 21
4.6
C
ZAS OBLICZEŃ
........................................................................................................................................... 26
4.7
S
UPERSIECI
.................................................................................................................................................. 28
5. PODSUMOWANIE ........................................................................................................................................ 32
6. BIBLIOGRAFIA............................................................................................................................................. 33
DODATEK A. STRUKTURY KRYSTALICZNE ........................................................................................... 34
DODATEK B. ELEMENTY TEORII PASMOWEJ PÓŁPRZEWODNIKÓW........................................... 36
DODATEK C. OPIS PROGRAMU STRZAŁY ................................................................................................ 37
DODATEK D. PARAMETRY UśYTEGO W OBLICZENIACH MATERIAŁU
PÓŁPRZEWODNIKOWEGO .......................................................................................................................... 41
DODATEK E. PROGRAMY ŹRÓDŁOWE .................................................................................................... 42

5
1. Wprowadzenie
Jednym z ważnych problemów teoretycznych współczesnej fizyki układów
niskowymiarowych o dużym znaczeniu aplikacyjnym jest wyznaczenie poziomów
energetycznych nośników prądu w strukturach półprzewodnikowych typu III/V. Struktury te
są podstawowym elementem budowy między innymi laserów półprzewodnikowych [1].
W celu wyznaczania struktury energetycznej heterostruktur półprzewodnikowych
stosowanych jest wiele technik doświadczalnych oraz obliczeniowych. Techniki
doświadczalne w większości opierają się na zjawisku spektroskopii fotoodbiciowej [2].
Natomiast metody obliczeniowe polegają na rozwiązywaniu odpowiedniego równania
Schrödingera, w którym opis ilościowy właściwości elektrycznych heterostruktur jest
prowadzony między innymi w ramach jednoelektronowego i jednopasmowego przybliżenia
za pomocą równania masy efektywnej [3]. Metody i algorytmy numeryczne odgrywają dużą
rolę w początkowych etapach modelowania struktur, oraz często służą do weryfikacji
poprawności wyników doświadczalnych.
Praca poświęcona jest tzw. ulepszonej metodzie strzałów, zaproponowanej
w artykule [4], umożliwiającej rozwiązywanie pełnego (wyznaczane są wartości i wektory
własne)
równania
Schrödingera
opisującego
stany
własne
nośników
prądu
w heterostrukturach półprzewodnikowych zawierających studnie kwantowe typu III/V.
Dyskutowana metoda jest specyficznym algorytmem numerycznym służącym do
rozwiązywania jednowymiarowego stacjonarnego równania masy efektywnej [3].
Celem pracy jest zaprojektowanie i opracowanie programu komputerowego
pozwalającego, w ramach ulepszonej metody strzałów, rozwiązywać jednowymiarowe
stacjonarne równanie masy efektywnej dla heterostruktur półprzewodnikowych typu III/V.
Zanim przejdziemy do prezentacji wyżej wspomnianej metody przedstawiamy krótkie
streszczenie pracy. Rozdział 2 zawiera zwięzłe przedstawienie najważniejszych wiadomości
o heterostrukturach półprzewodnikowych [5, 6, 7] (rozdz. 2.1) a rozdział 2.2 poświęcony jest
scharakteryzowaniu równania masy efektywnej. W rozdziale 3 dokonujemy przeglądu
wybranych algorytmów oraz omawiamy szczegółowo ulepszoną metodę strzałów. Rozdział 4
zawiera prezentację wybranych wyników obliczeń numerycznych wykonanych omawianą
metodą dla różnych rodzajów heterostruktur i supersieci półprzewodnikowych. W rozdziale
tym przedstawiona jest także analiza dokładności oraz czasu obliczeń dla trzech wybranych
metod – prostej metody strzałów, ulepszonej metody strzałów oraz metody macierzowej [3].
Kolejny rozdział stanowi podsumowanie najważniejszych wyników. Pracę zamykają spis
literatury oraz dodatki.
6
2. Elementy pasmowej teorii heterostruktur
2.1 Heterostruktury
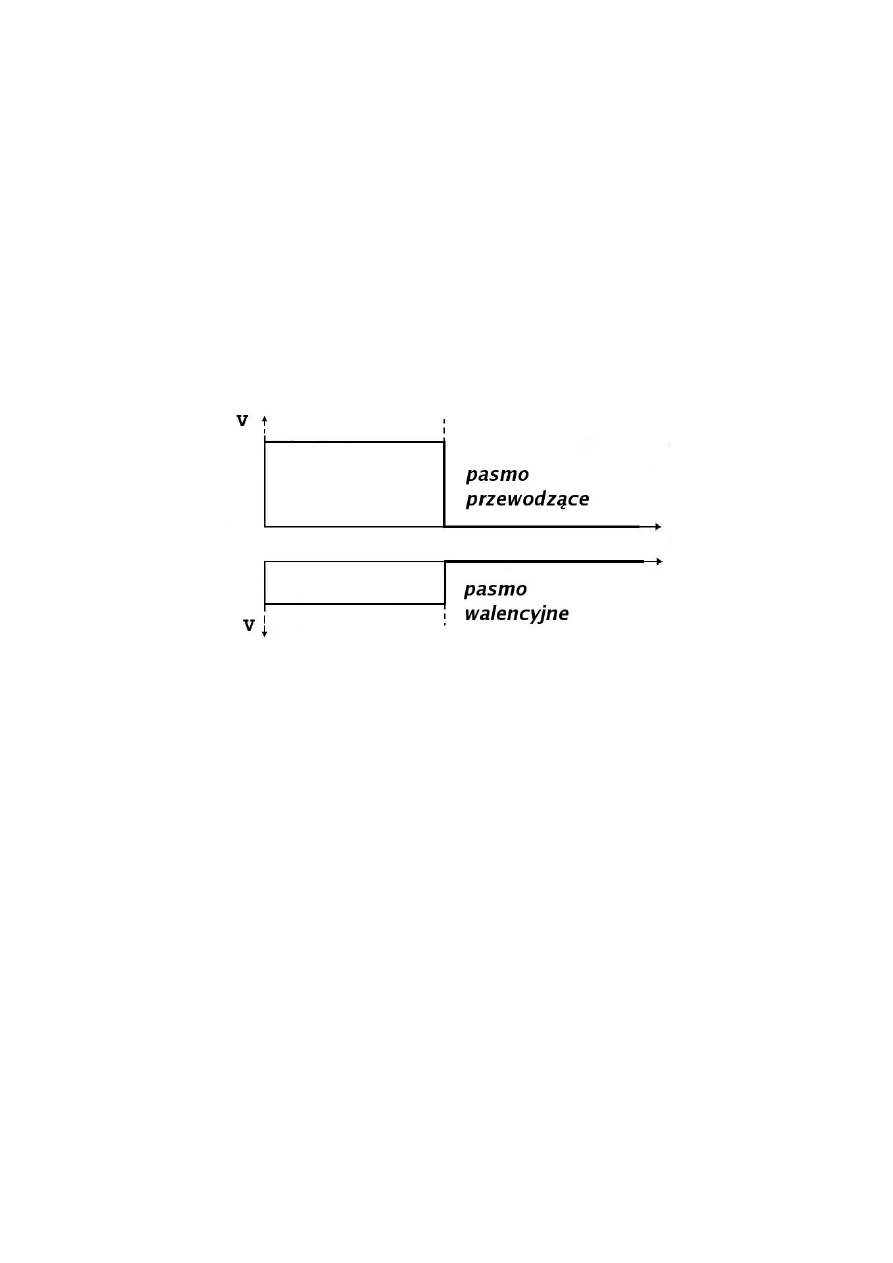
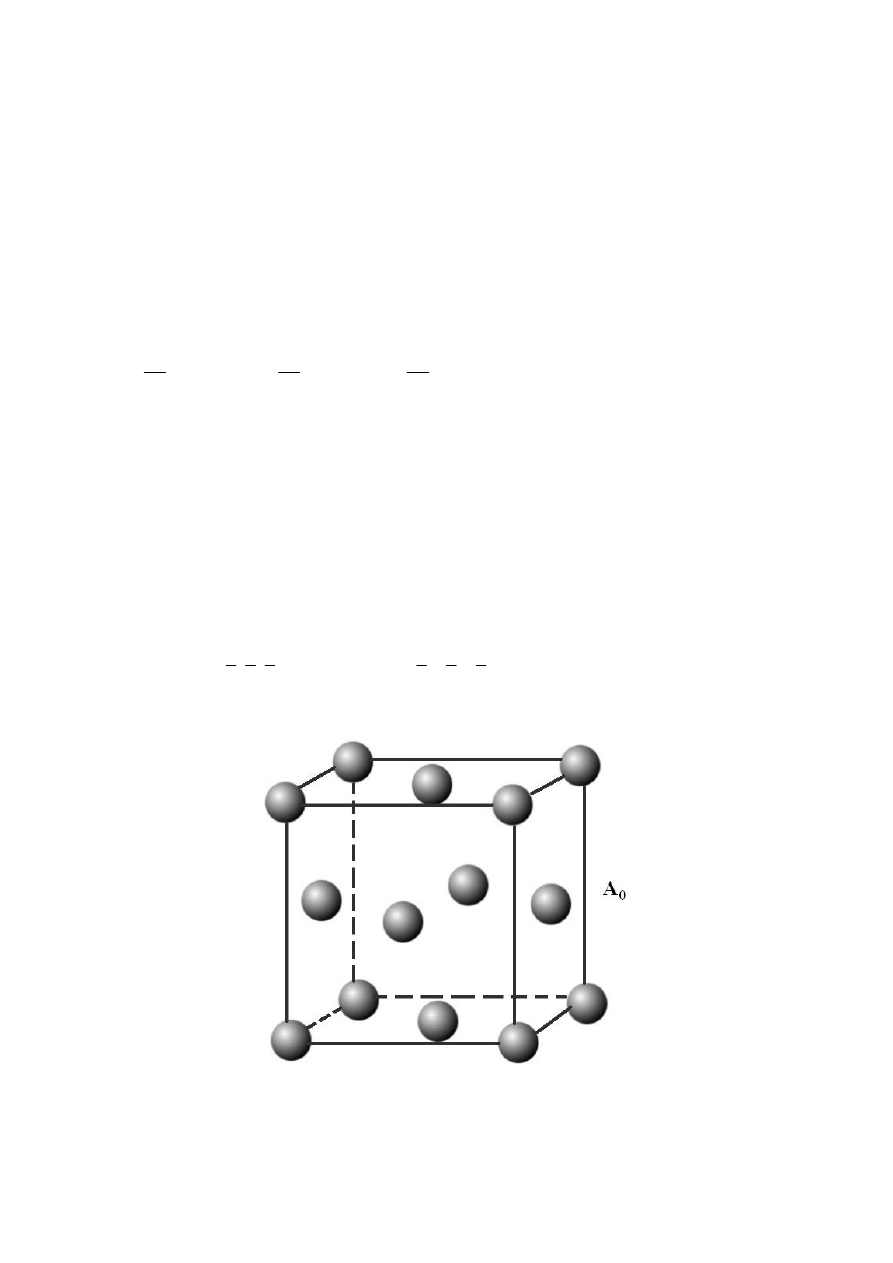
Jedną z najprostszych struktur kwantowych jest heterozłącze, które powstaje na skutek
zetknięcia ze sobą dwóch różnych materiałów półprzewodnikowych (A i B)
charakteryzujących się różnymi masami efektywnymi oraz struktura pasmową (patrz rys. 1).
Więcej informacji o strukturach krystalicznych oraz półprzewodnikach można znaleźć
w dodatkach A i B oraz w licznych książkach takich jak [5, 7].
Rys. 1. Struktura energetyczna w obszarze heterozłącza zaznaczonego linią przerywaną [3].
Należy wspomnieć, że przedstawiony model heterozłącza (rys. 1) jest daleko idącą
idealizacją rzeczywistości. Pokazana struktura pasmowa opisuje wyizolowane złącze,
w którym nie występują żadne lub występuje względnie mała ilość nośników elektrycznych.
Ponadto na układ nie działają pole elektromagnetyczne i ciśnienie a jego temperatura jest
równa 0 K. W rzeczywistym świecie takie warunki nie istnieją i cała struktura energetyczna
ma odmienną od rys. 1 postać. Więcej na ten temat można znaleźć w [2, 5, 6]. Niemniej
jednak dyskutowane tutaj przybliżenie jest dostatecznie dobre do zastosowań numerycznych,
o czym
jest mowa w książkach i publikacjach między innymi w [3 ,4, 5, 7].
Heterostruktury [2,3,4] zawierają wiele hetorozłącz, co oznacza, że istnieje
nieskończona liczba możliwych heterostruktur. Jeśli umieścimy cienką warstwę jednego
półprzewodnika o mniejszej przerwie energetycznej – materiał A – pomiędzy warstwami
półprzewodnikowymi o większej przerwie energetycznej – materiał B – (rys. 2), to wtedy
tworzą one strukturę zwaną podwójnym heterozłączem.
Jeśli warstwa materiału A jest odpowiednio wąska, dla zastosowań kwantowych
(szerokości to od kilku do kilkudziesięciu nanometrów), to wtedy takie ułożenie warstw jest
nazywane pojedynczą studnią kwantową (ang. single quantum well). Jeśli istnieją
jakiekolwiek nośniki ładunku (elektrony bądź dziury), to w takim układzie będą one zawsze
dążyły do obniżenia swojej energii. W takim przypadku elektrony i dziury będą gromadzić się
wewnątrz odpowiednich studni kwantowych.
7
Rys. 2 Schematyczne przedstawienie jednowymiarowej studni kwantowej [5].
Dodatkowe warstwy półprzewodnikowe mogą być dołączane do opisanej wcześniej
heterostruktury tworząc np. schodkowe bądź asymetryczne studnie kwantowe, co przedstawia
rys. 3.
Rys. 3 Schematyczne przedstawienie jednowymiarowych studni kwantowych typu schodkowego [5].
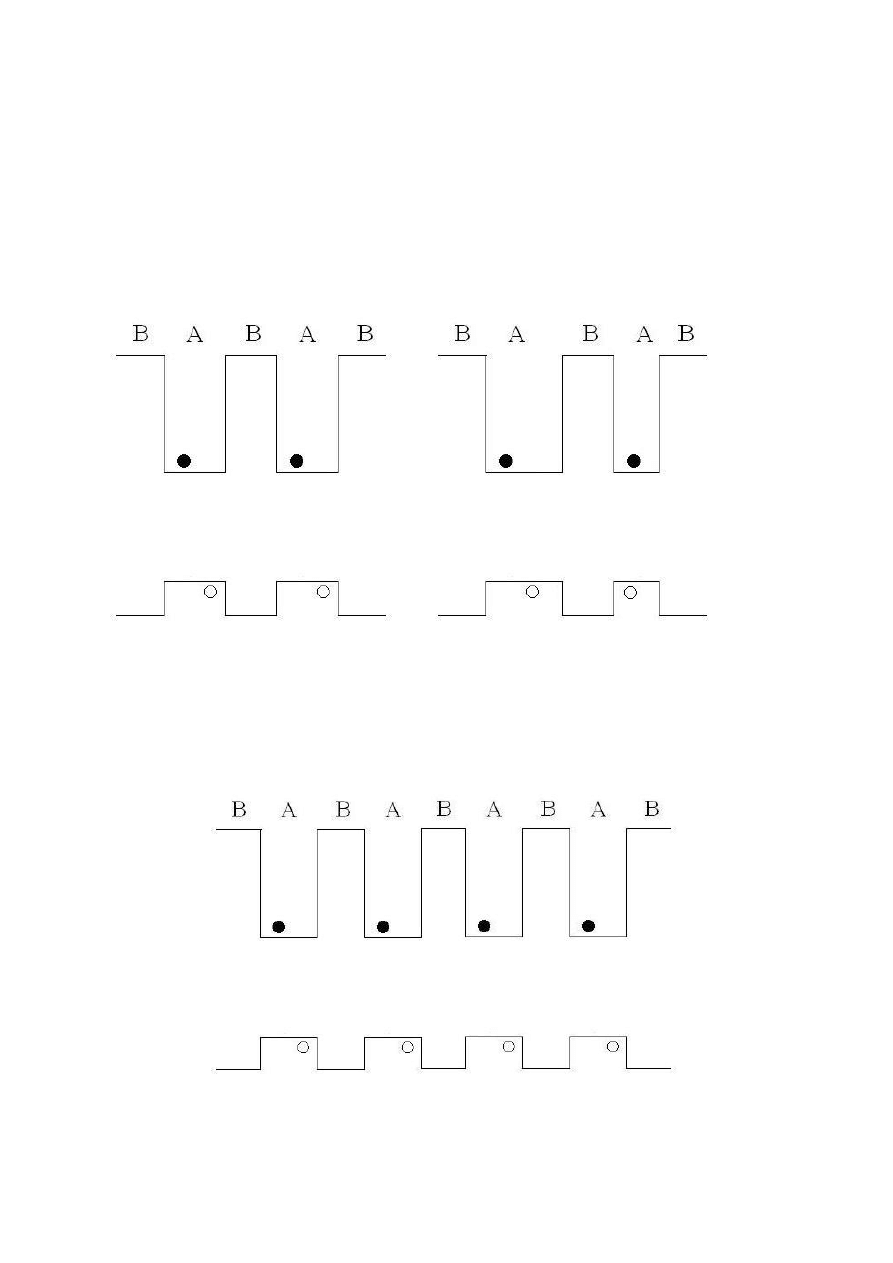
8
W ten sposób może być uformowana niezliczona ilość skomplikowanych
heterostruktur, takich jak symetryczne lub niesymetryczne podwójne studnie kwantowe (patrz
rys. 4), wielokrotne studnie bądź tzw. supersieci – ang. superlattice. Różnica pomiędzy
pojedynczą studnią bądź kombinacją wielu studni czy supersieci tkwi w oddziaływaniu
pomiędzy pojedynczymi studniami. Zestawienie wielu jam potencjałów w zależności od
odległości pomiędzy pojedynczymi jamami może zachowywać się jak skupisko pojedynczych
studni, (gdy odległość pomiędzy poszczególnymi studniami jest dostatecznie duża) bądź jak
supersieć, gdy obszar oddzielający studnie jest dostatecznie wąski, co prowadzi do
oddziaływania nośników prądu w sąsiednich studniach
.
Rys. 4. Struktura energetyczna symetrycznej (lewa strona) i asymetrycznej (prawa strona) podwójnej studni
kwantowej [5].
W supersieciach może zachodzić zjawisko tunelowania polegające na tym, że elektron
bądź dziura może przedostać się przez obszar zabroniony klasycznie do sąsiadującej studni
kwantowej.
Rys. 5. Struktura pasmowa supersieci [5].
9
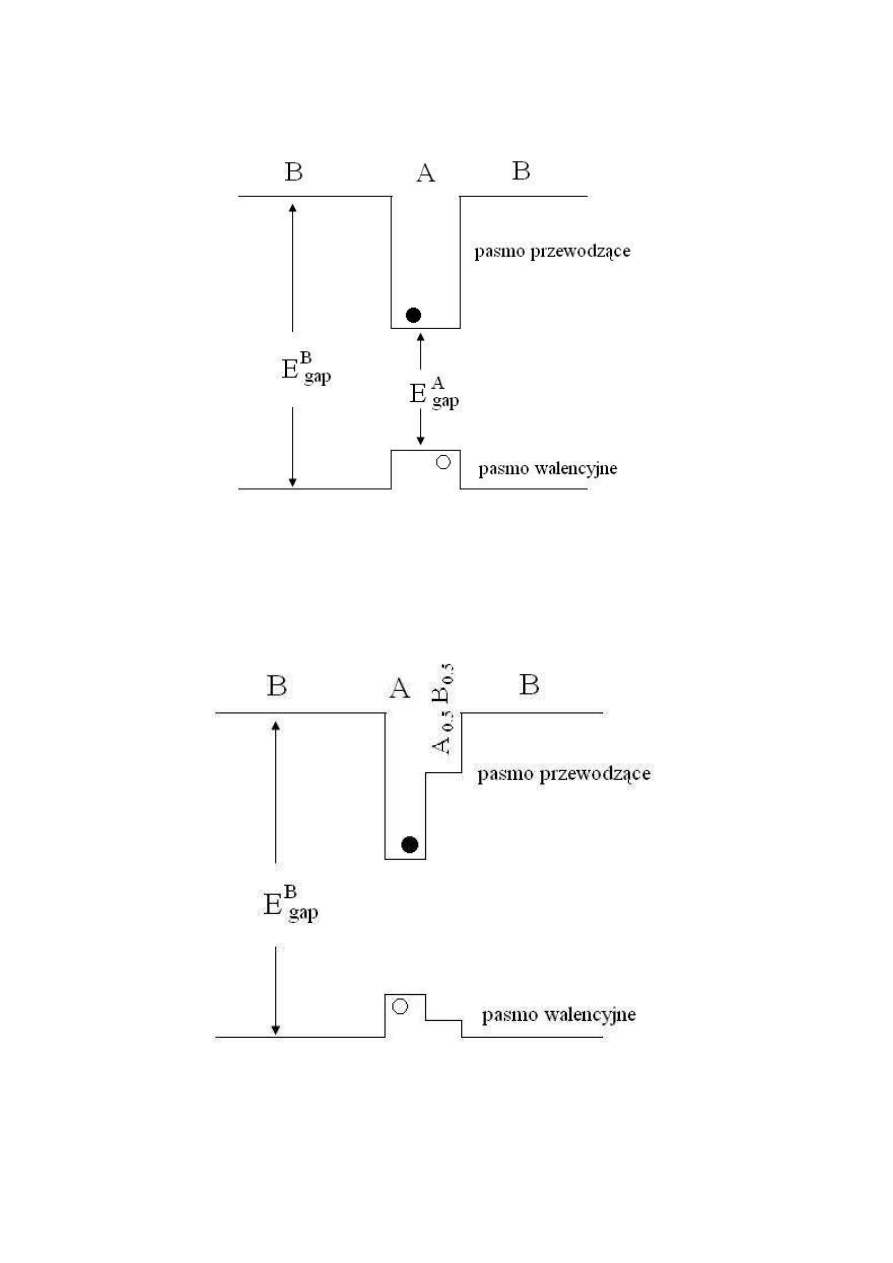
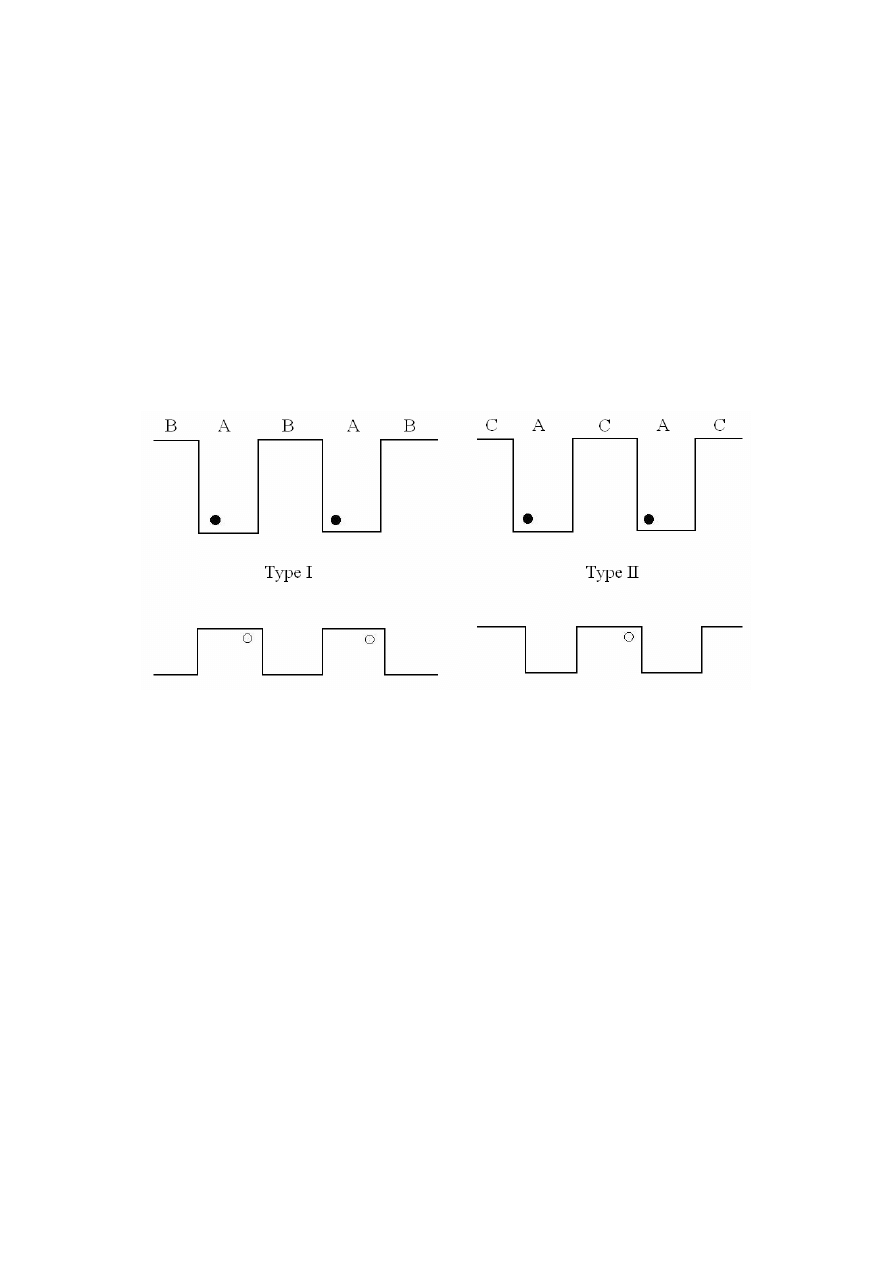
Wszystkie zaprezentowane powyżej układy półprzewodnikowe są przykładami tzw.
struktur pierwszego typu (ang. type-I systems). Są to układy, w których, przerwa
energetyczna jednego z materiałów półprzewodnikowych jest całkowicie zagnieżdżona
pomiędzy materiałem o innej przerwie energetycznej. Skutkiem takiego ułożenia jest to, że
każdy elektron czy dziura wpada w studnię kwantową wewnątrz tego samego materiału (np.
pobudzony elektron przez wzrost temperatury przeskakuje z warstwy walencyjnej do warstwy
przewodzącej wewnątrz tego samego materiału). W ten sposób dwa typy nosicieli ładunku
(elektrony i dziury) są zlokalizowane w tym samym obszarze przestrzeni, co z kolei zwiększa
szybkość rekombinacji dziura-elektron.
Istnieją jednak inne możliwości rozkładu warstw walencyjnych w strukturach
drugiego typu (ang. type-II systems), w którym przerwy energetyczne materiałów (A i C jak
zaznaczono na rys. 6) są tak rozłożone, że uformowane w ten sposób studnie kwantowe
w obydwu warstwach znajdują się w różnych materiałach.
Rys. 6. Struktura energetyczna układów pierwszego i drugiego typu [5].
Ustawienie takie powoduje rozłożenie elektronów i dziur wewnątrz warstw różnych
półprzewodników, co znacznie zwiększa czas rekombinacji par dziura-elektron.
Wiele z wyżej wymienionych struktur i układów znalazły zastosowania
w urządzeniach elektronicznych takich jak diody, tranzystory, termopary, diody
elektroluminescencyjne, lasery półprzewodnikowe [1].

10
2.2 Równanie masy efektywnej
W reprezentacji położeń stacjonarne równanie Schrödingera [3] dla cząstki swobodnej
ma postać zagadnienia własnego dla operatora energii (hamiltonianu) H
Ψ
Ψ
E
H
=
,
(2.1)
gdzie liczbę E nazywamy wartością własną operatora H (energią własną), a funkcję falową
Ψ
– funkcją własną.
W układach jednowymiarowych równanie (2.1) przybiera postać
( )
( )
x
EΨ
x
Ψ
dx
d
2m
2
2
2
=
−
h
,
(2.2)
gdzie m oznacza masę cząstki,
( )
π
2
/
h
=
h
– stałą Diraca, x – położenie cząstki [3].
Równania (2.2) nie można jednak stosować do układów zawierających studnie
kwantowe lub elektrony/dziury wewnątrz ciała stałego. Cząstka wewnątrz kryształu poddana
jest różnym oddziaływaniom pochodzącym od cząstek budujących kryształ bądź
przyłożonego pola elektromagnetycznego. Jak wynika z badań fizyki ciała stałego rozkład
potencjału wewnątrz kryształu jest bardzo skomplikowany (więcej informacji w załącznikach
A i B).
Zostało jednak dowiedzione w licznych pracach (takich jak [3], [5],[8]), że potencjał
ten może być dostatecznie dobrze przybliżony (dla potrzeb numerycznych) stałym
parametrem zwanym masą efektywną – m
*
. Jest to najczęściej stosowana wielkość w fizyce
niskowymiarowych układów półprzewodnikowych, takich jak druty i studnie kwantowe czy
supersieci [3,5,6]. Badania wykazały, że masa efektywna jest anizotropowa, jak również, że
jest dobrym przybliżeniem tylko dla relatywnie niskich energii elektronu. Dla przykładu
w materiale GaAs, elektrony wykazują masę równą 0,067
0
m , gdzie
0
m to spoczynkowa masa
elektronu w próżni.
Równanie (2.1) przybiera wówczas postać
ψ
ψ
E
m
=
∇
−
2
*
2
2
h
,
(2.3)
gdzie
2
2
2
dx
d
=
∇
,
∗
m
– masa efektywna a wartości własne dane są wzorem
*
2
2
2m
k
E
h
=
(2.4)
Równanie (2.3) nadal jednak nie pozwala nam przeprowadzać obliczeń dla systemów
bardziej skomplikowanych takich jak heterostruktury.

11
Dotychczas rozpatrywaliśmy układ z zerowym lub stałym na całym przedziale
potencjałem. W przypadku złącz półprzewodnikowych niezbędne jest uwzględnienie różnic
potencjału. Równanie (2.3) z uwzględnieniem potencjału jako funkcji zależnej od położenia
V(x) przybiera postać
( ) ( )
)
(
2
2
2
*
2
x
E
x
x
V
dx
d
m
Ψ
Ψ
=
+
−
h
.
(2.5)
Należy pamiętać, że równanie (2.5) nie daje nam możliwości traktowania masy jako
funkcji położenia. W celu obliczenia wartości własnej układu przedstawionego na rys. 2 za
pomocą równania (2.5) należałoby je rozwiązać na dwóch przedziałach, dla których masa
efektywna wynosi (zakładając najprostsze przybliżenie stałych lecz różnych mas efektywnych
dla różnych materiałów)
(
)
(
)
>
<
=
0
0
*
dla
dla
x
x
m
x
x
m
m
B
A
,
gdzie x
0
to punkt zetknięcia się materiałów A i B, a następnie zszyć rozwiązania w punkcie
styku x
0
, nakładając odpowiednie warunki na funkcję
Ψ
oraz jej pochodną
dx
d
Ψ
. Więcej o tej
metodzie można znaleźć w [3].
Strukturę energetyczną bardziej skomplikowanych układów, którymi są np.
heterostruktury półprzewodnikowe wyznaczamy za pomocą jednowymiarowego równania o
postaci
( )
( ) ( ) ( )
( )
x
E
x
x
V
x
dx
d
x
m
dx
d
Ψ
=
Ψ
+
Ψ
−
1
2
*
2
h
.
(2.6)
Równanie (2.6) – zwane dalej równaniem masy efektywnej – uwzględnia zarówno
zależność masy jak i potencjału od położenia.
Taka matematyczna reprezentacja nosi także nazwę przybliżenia funkcją obwiedni
(ang. the envelope function approximation). Nazwa ta pochodzi od możliwości przybliżenia
fizycznych właściwości układu za pomocą wolno zmieniającej się funkcji obwiedni
( )
x
Ψ
,
a niżeli całkowitą funkcją falową o rozmiarach atomowych
( ) ( )
x
u
x
Ψ
, która gwałtownie
zmienia swoje wartości na odległościach międzyatomowych.
Funkcja obwiedni ma swoje ograniczenia. Zastosowanie jej do np. bardzo cienkich
warstw nie daje pozytywnych rezultatów. Niemniej jednak dla większości stosowanych
struktur przybliżenie to działa dobrze, co zostanie przedstawione w dalszej części pracy.
Nad poprawnością i dokładnością zaprezentowanego powyżej przybliżenia
prowadzonych jest nadal wiele badań. Należy pamiętać, że przybliżenie to stosowane jest
w kontekście aproksymacji materiałowej, a nie jako przybliżenie mechaniki kwantowej.
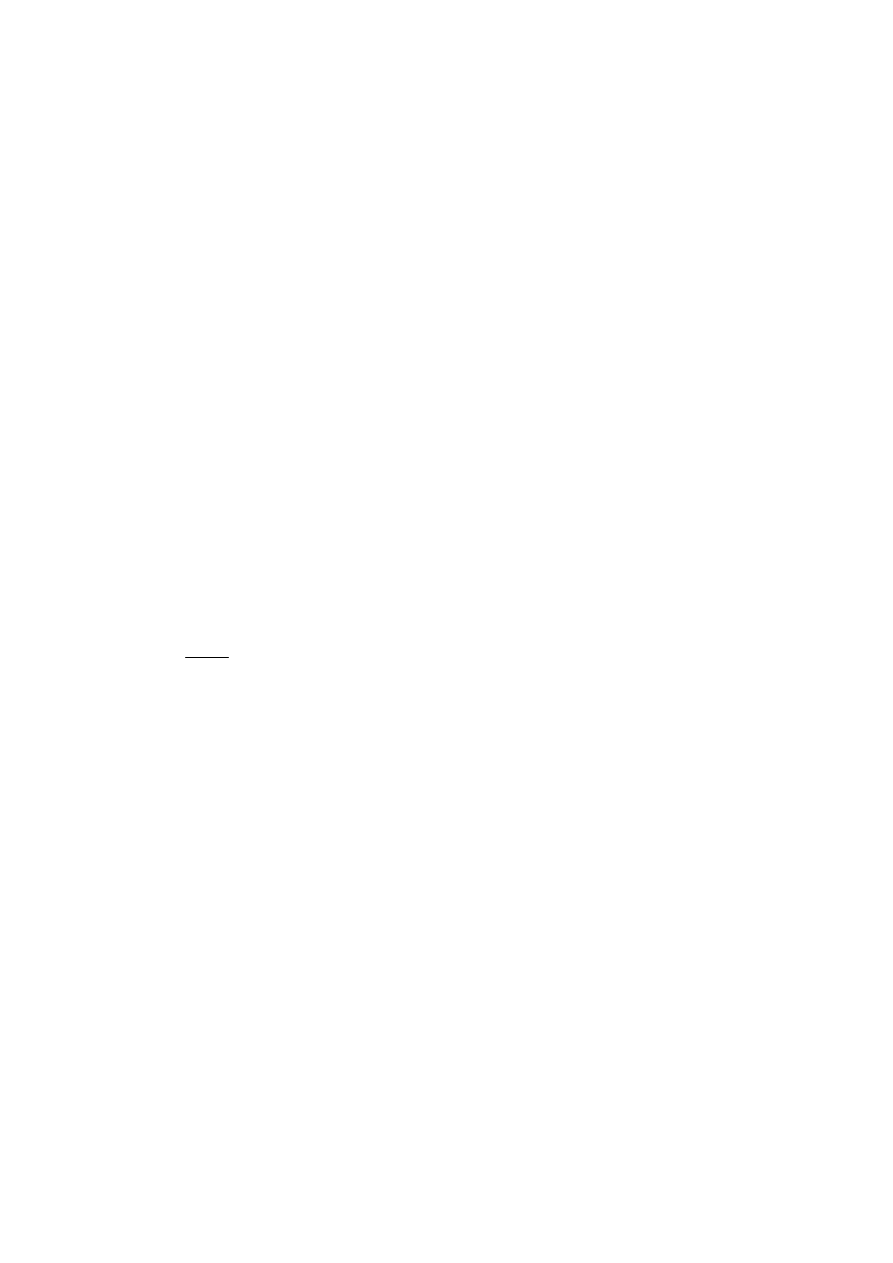
12
3. Metody strzałów rozwi
ą
zywania równania Schrödingera
3.1 Przegl
ą
d metod
Równanie (2.1) jest, jak zostało wcześniej wspomniane, zagadnieniem własnym.
Analiza numeryczna zagadnienia własnego jest prowadzona, wieloma metodami takimi jak:
•
Metodami różnic skończonych, zwanymi tez metodami siatkowymi, wśród nich wyróżnia
się:
♣
Metody macierzowe, zwane także metodami globalnymi,
♣
Metody strzałów,
♣
Metodą wariacyjną,
•
Metodą elementów skończonych [3].
3.2 Prosta metoda strzałów
Przedstawiony tu algorytm wzięty jest z [3]. Korzystając z aproksymacji drugiej
pochodnej za pomocą różnic skończonych na dyskretnym zbiorze punktów, na przedziale
< A, B >, których współrzędne określają związki
(
)
1
n
0,...,
i
,
1
+
=
+
=
+
−
+
=
x
i
A
n
A
B
i
A
X
i
δ
,
(3.1)
gdzie x
δ
jest długością kroku całkowania na zadanym przedziale. Na tak zadanej siatce
formułujemy jednowymiarowe odwzorowania (w ogólnym przypadku nieliniowe)
(
)
,
,
,
,
,...,
1
E
F
m
i
i
i
P
C
−
−
→
Ψ
Ψ
=
Ψ
(3.2)
(
)
,
,
,
,
,...,
1
E
F
m
i
i
i
P
C
+
+
←
Ψ
Ψ
=
Ψ
(3.3)
gdzie
C = (C
1
,…,C
c
) i
P = (P
1
,…,P
p
) oznaczają stałe i parametry modelu, a E to szukana
wartość własna.
Obliczenia prowadzimy zazwyczaj według następującego schematu:
1.
Dokonujemy próbnego wyboru wartości własnej E = E
0
.
2.
Obliczmy wartości
i
Ψ
funkcji falowej na zadanej siatce (3.1) za pomocą dwóch
różnych procedur. Jeden ciąg
(
)
n
n
Ψ
Ψ
Ψ
Ψ
=
Ψ
−
→
,
,...
,
1
2
1
,
(3.4)
wyznaczmy za pomocą odwzorowania (3.2), rozpoczynając obliczenia od obszaru

13
< A, A + D
A
>, gdzie D
A
<< B – A; jest to tzw. procedura wstępująca. Drugi ciąg
(
)
1
2
1
,
,...
,
Ψ
Ψ
Ψ
Ψ
=
Ψ
−
←
n
n
,
(3.5)
wyznaczmy za pomocą odwzorowania (3.3), startując z obszaru < B – D
B
, B >, gdzie
D
B
<< B – A (procedura zstępująca) Zauważamy, że do uruchomienia procedur (3.4)
i (3.5) wymagana jest znajomość dokładnego lub przybliżonego rozwiązania równania
Schrödingera na brzegach przedziału całkowania < A, B >.
3.
Sprawdzamy poprawność wyboru wartości własnej E
0
oraz otrzymanych wartości
funkcji falowej za pomocą warunku zszycia w wybranym punkcie siatki X
m
.
Zazwyczaj żąda się, aby pochodne logarytmiczne funkcji falowej otrzymane
za pomocą obu procedur obliczeniowych były w punkcie X
m
równe z zadaną
dokładnością.
4.
Kończymy obliczenia, jeśli warunek zszycia jest spełniony. Za wartość własną
przyjmujemy wtedy E
0
, a za wartość odpowiadającej jej funkcji własnej w punktach
siatki – obliczone w procesach odwzorowania wartości
i
Ψ
(3.4) i (3.5).
W przeciwnym razie obliczenia powtarzamy dla nowej próbnej wartości własnej E
0
.
3.3 Ulepszona metoda strzałów
Przedstawiony tu algorytm wzięty jest z [4]. Rozpatrując najprostszą formę
jednowymiarowego niezależnego od czasu równania Schrödingera
Ψ
=
Ψ
+
Ψ
′′
E
V
ϕ
(3.6)
stosując najprostsze przybliżenie drugiej pochodnej, na zadanej siatce punktów (3.1), dane
wzorem
( )
2
2
1
1
O
2
x
x
i
i
i
i
δ
δ
ϕ
ϕ
ϕ
ϕ
+
+
−
=
′′
−
+
,
(3.7)
możemy zapisać równanie (3.6) w postaci
(
)
( ) ( ) ( ) (
) ( )
( )
x
E
x
x
x
x
x
V
x
x
x
Ψ
Ψ
Ψ
Ψ
2
2
2
δ
δ
δ
δ
=
+
−
+
+
−
−
,
(3.8)
i
(
)
( ) ( )
[
]
{
}
( ) (
)
x
x
x
E
x
V
x
x
x
δ
δ
δ
−
−
−
+
=
+
Ψ
Ψ
Ψ
2
2
,
(3.9)
Równanie (3.8) można rozwiązywać metodami macierzowymi [3] a (3.9) za pomocą
metody strzałów korzystając z algorytmu opisanego w rozdziale 3.2.
Jak zostało pokazane wcześniej równanie masy efektywnej dla półprzewodnikowych
struktur przybiera postać daną wzorem (2.5). Stosując przybliżenie pochodnej (3.7) do
równania (2.5) otrzymujemy równania analogiczne do (3.8 i 3.9)

14
(
) (
)
( ) ( )
(
)
(
) ( )
(
) (
) ( )
E
x
x
x
x
x
m
x
x
x
m
x
x
m
x
x
x
x
x
x
m
2
2
*
*
*
2
2
*
2
2
/
2
/
1
2
/
1
2
/
1
2
2
/
1
h
h
δ
δ
δ
δ
δ
δ
δ
δ
=
+
+
−
−
+
+
+
+
−
−
−
Ψ
Ψ
V
Ψ
, (3.10)
stosowane w metodach macierzowych [2], oraz
(
) (
)
( ) ( )
[
]
(
)
(
) ( )
(
) (
)
2
/
2
/
1
2
/
1
2
/
1
2
2
/
1
*
*
*
2
2
*
x
x
x
x
m
x
x
x
m
x
x
m
E
x
x
x
x
x
x
m
δ
δ
δ
δ
δ
δ
δ
+
+
−
−
+
+
+
−
=
−
−
−
Ψ
Ψ
V
Ψ
h
,
(3.11)
stosowane w prostej metodzie strzałów.
W odróżnieniu od prostej metody strzałów, w ulepszonej metodzie nie stosuje się
przybliżenia drugiej pochodnej (3.7). W tym przypadku równanie różniczkowe drugiego
rzędu zapisujemy jako parę równań pierwszego rzędu
( )
( ) ( )
,
~
*
x
x
m
x
x
Ψ
Ψ
=
δ
δ
(3.12)
( )
( )
[
]
( )
x
E
x
x
x
Ψ
V
Ψ
−
=
2
2
~
h
δ
δ
,
(3.13)
z nową pomocniczą funkcją
( )
( )
( )
x
x
x
m
x
Ψ
Ψ
δ
δ
*
1
~
=
,
(3.14)
Stosując przybliżenie pochodnych, różnicami skończonymi z krokiem siatki
x
δ
,
równania (3.12) i (3.13) sprowadzamy do układu równań
(
)
( )
( ) ( )
,
~
*
x
x
x
m
x
x
x
δ
δ
Ψ
Ψ
Ψ
+
=
+
(3.15)
(
)
( )
( )
[
]
( )
,
2
~
~
2
x
x
E
x
x
x
x
δ
δ
Ψ
V
Ψ
Ψ
−
+
=
+
h
(3.16)
Równania (3.15) i (3.16) pozwalają obliczyć kolejne wartości funkcji falowej, jeśli
znane są wartości początkowe, tak samo jak i równanie (3.11) w prostej metodzie strzałów.
W odróżnieniu jednak od niej równania (3.15) i (3.16) są znacznie prostsze, zawierają
znacznie mniej obliczeń zmiennoprzecinkowych.
Początkowa wartość funkcji obwiedni na brzegu przedziału całkowania w naszych
obliczeniach numerycznych jest wybrana jako
.
0
0
=
=
x
Ψ
(3.17)

15
W związku z wykładniczą naturą zanikania funkcji obwiedni Ψ na zewnątrz badanej
heterostruktury funkcja Ψ
~
nie może przyjmować wartości zerowej. W pracy przyjęto
,
1
~
0
=
=
x
Ψ
(3.18)
Mając do dyspozycji równania (3.15 i 3.16) z warunkami początkowymi (3.17 i 3.18)
przeprowadziliśmy obliczenia dla różnych wartości energii próbnej E. Za wartość własną
układu przyjmowano tę, dla której wartość funkcji falowej (3.15) dla końcowego punktu
przedziału całkowania przyjmuje zero z zadaną dokładnością numeryczną.

16
4. Prezentacja oraz dyskusja wyników
Ulepszona metoda strzałów dana wzorami (3.15) i (3.16) pozwala na zaprojektowanie
oraz zaprogramowanie stabilnego numerycznie algorytmu. Do zaimplementowania
algorytmu został użyty język wyższego poziomu FORTRAN 77 oraz darmowy kompilator
GNU fortran g77 dostępny wraz z edytorem Force 2.0 w Internecie pod adresem [9].
Dysponując dobrze dobranymi procedurami (zawartymi w dodatku E oraz na
dołączonej płycie CD) można prześledzić zachowanie się energii własnych oraz
odpowiadającym im funkcjom falowym dla struktur o różnych parametrach. Wszystkie
przedstawione w tym rozdziale testy przeprowadzone były programami napisanymi w języku
FORTRAN 77, których kody źródłowe zawarte są we wspomnianym dodatku.
W ramach tej pracy został napisany także program w środowisku Delphi 5 – Strzały –
z interfejsem graficznym (zawartym także na dołączonej płycie CD), dzięki któremu można
uzyskać podobne wyniki. Program ten jest uboższy od programów napisanych w języku
FORTRAN 77 i dlatego służyć on może jedynie do prostych obliczeń. W celu uzyskania
dokładnych wyników z użyciem małych wartości kroku całkowania (program Strzały
umożliwia obliczenia z maksymalną liczbą punktów siatki równą 30000) należy używać
programów zaimplementowanych w języku FORTRAN 77. Dokładniejszy opis programu
Strzały
zawarty jest w dodatku C.
4.1 Wyniki numeryczne dla studni kwantowej typu III/V
Rysunek 7 przedstawia najprostsze przybliżenie jednowymiarowego potencjału dla
pojedynczej studni kwantowej wraz z uwzględnieniem skokowego rozkładu masy efektywnej.
Na wykresie zaprezentowany jest rozkład potencjału na zadanym przedziale całkowania
<
−
10,10>, oraz funkcje falowe odpowiadające kolejnym wartościom własnym energii.
Obliczenia przeprowadzone dla studni
As
Al
Ga
GaAs
4
.
0
6
.
0
/
o szerokości 10 nm (zastosowane
wartości parametrów znajdują się w załączniku D).
4.2 Zale
ż
no
ść
warto
ś
ci energii własnych studni kwantowej od masy
efektywnej
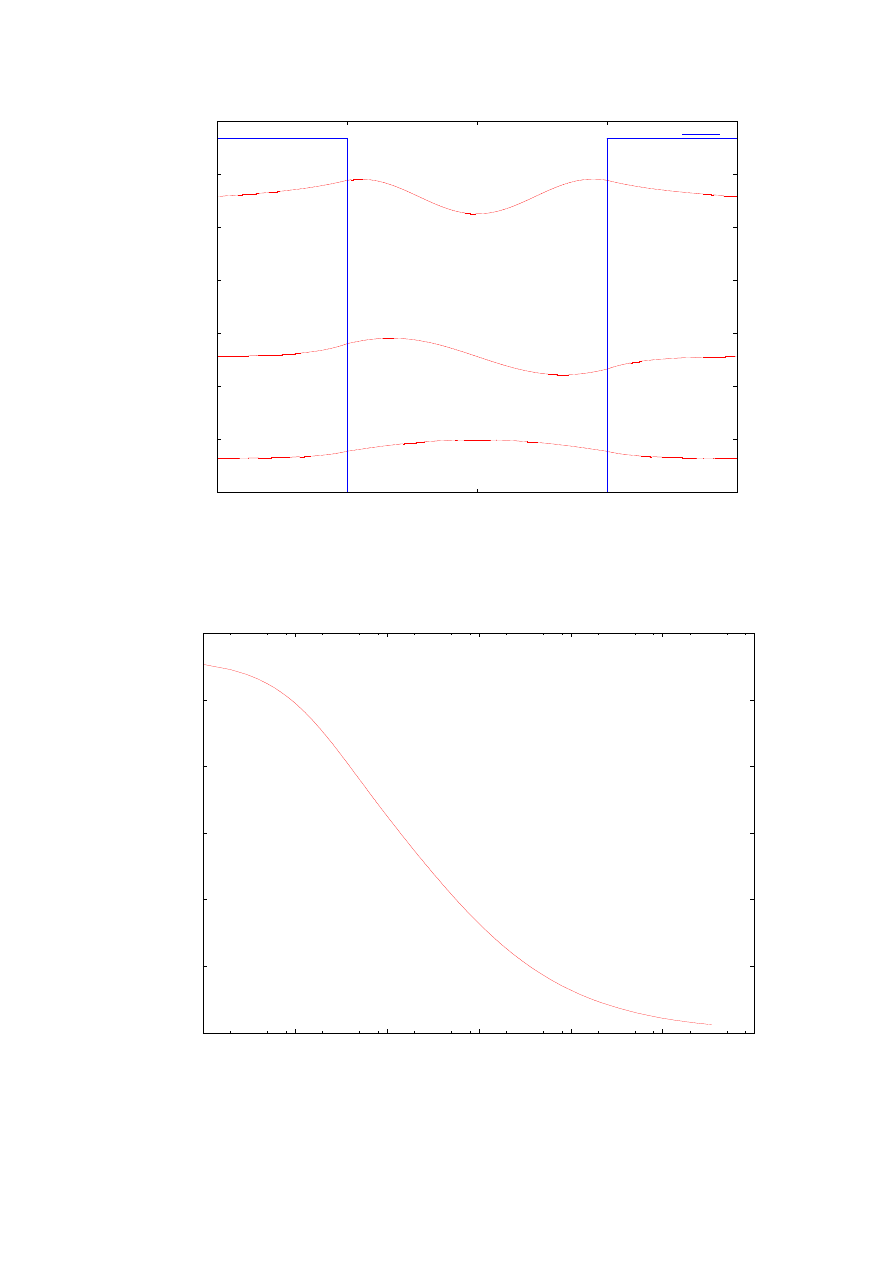
Na rysunku 8 przedstawiono zależność wartości energii poziomu podstawowego od
zmieniającej się masy efektywnej elektronu na zewnątrz studni w jednostkach masy
spoczynkowej elektronu. Wykres ten ilustruje tendencję obniżania się energii stanu
podstawowego wraz ze wzrostem masy efektywnej elektronu wewnątrz bariery. Dla bardzo
dużych wartości masy efektywnej elektronu wartość energii dąży do zera. Warto wspomnieć
w tym punkcie, że wraz ze wzrostem masy efektywnej elektronu wewnątrz bariery głębokość
przenikania funkcji falowej w obszar zabroniony (obszar bariery) staje się mniejsza, dlatego
w tych przypadkach dla przeprowadzenia obliczeń numerycznych należy koniecznie zawęzić
przedział całkowania. Obliczenia zostały przeprowadzone dla struktury z rozdziału 4.1.
17
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
-10
-5
0
5
10
E
[
e
V
]
x [nm]
V(x)
Rys.7. Prostokątna studnia potencjału oraz funkcje falowe odpowiadające trzem wartościom energii
własnych. Obliczenia przeprowadzone dla struktury opisanej w rozdziale 4.1.
0
0.01
0.02
0.03
0.04
0.05
0.06
0.001
0.01
0.1
1
10
100
1000
E
[
e
V
]
masa efektywna elektronu wewnatrz bariery [m0]
Rys. 8. Zależność wartości pierwszej energii własnej od masy efektywnej elektronu w barierze Obliczenia
przeprowadzone dla struktury opisanej w rozdziale 4.1.
18
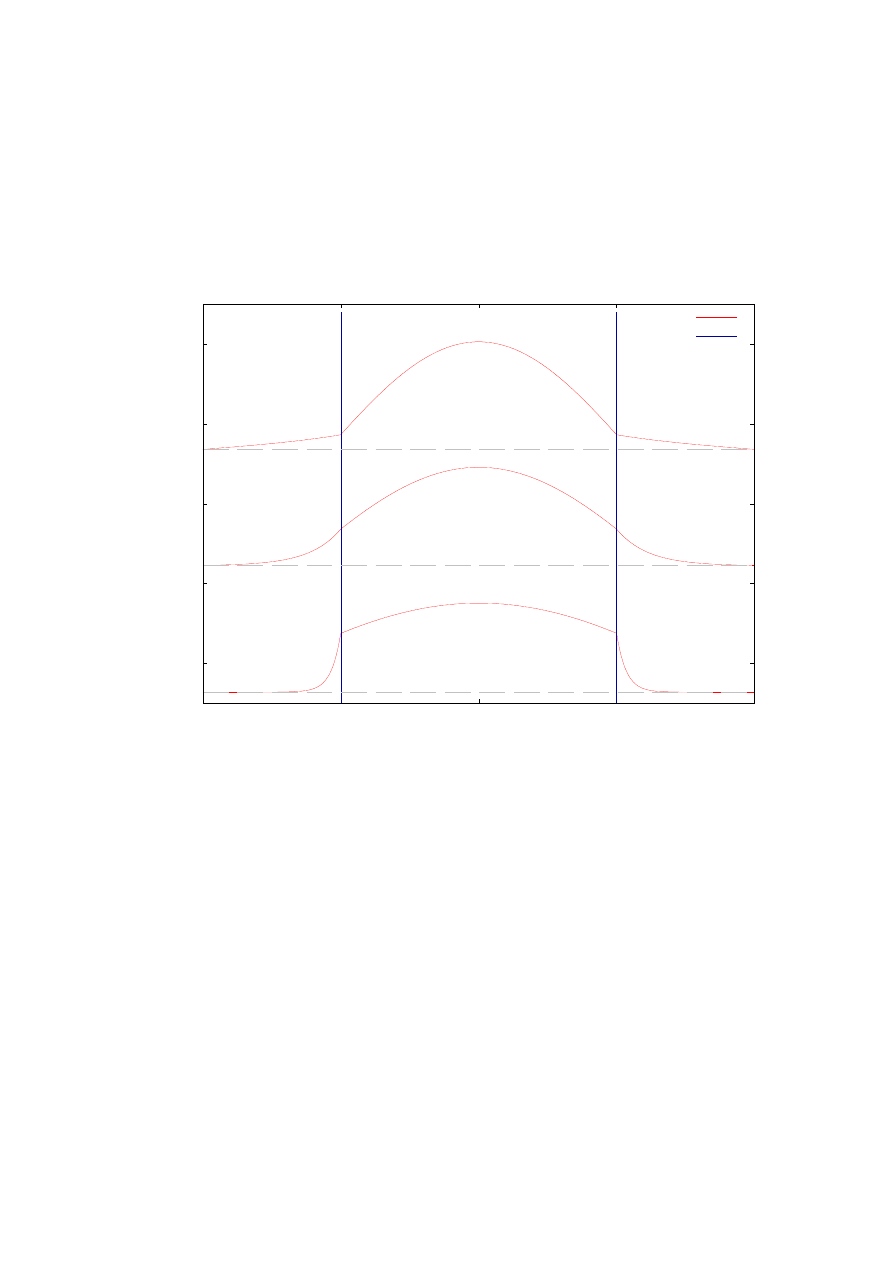
Wykres z rys. 9 przedstawia funkcje falowe dla wybranych wartości masy efektywnej
elektronu wewnątrz bariery. Wartości znormalizowanych funkcji falowych są powiększone
o wartość odpowiadającej im bezwymiarowej energii własnej (zaznaczone na wykresie
liniami poziomymi). Na wykresie zaznaczone są również bariery potencjału, dzięki czemu
dokładnie widać zmniejszającą się głębokość wnikania funkcji falowej w obszar zabroniony
bariery wraz ze zwiększającą się masą efektywna elektronu na zewnątrz studni.
0.02
0.03
0.04
0.05
0.06
-10
-5
0
5
10
E
[
e
V
]
x [nm]
V(x)
Rys. 9. Funkcje falowe pierwszego poziomu energetycznego dla różnych wartości masy własnej elektronu
wewnątrz bariery. Obliczenia przeprowadzone dla struktury opisanej w rozdziale 4.1.
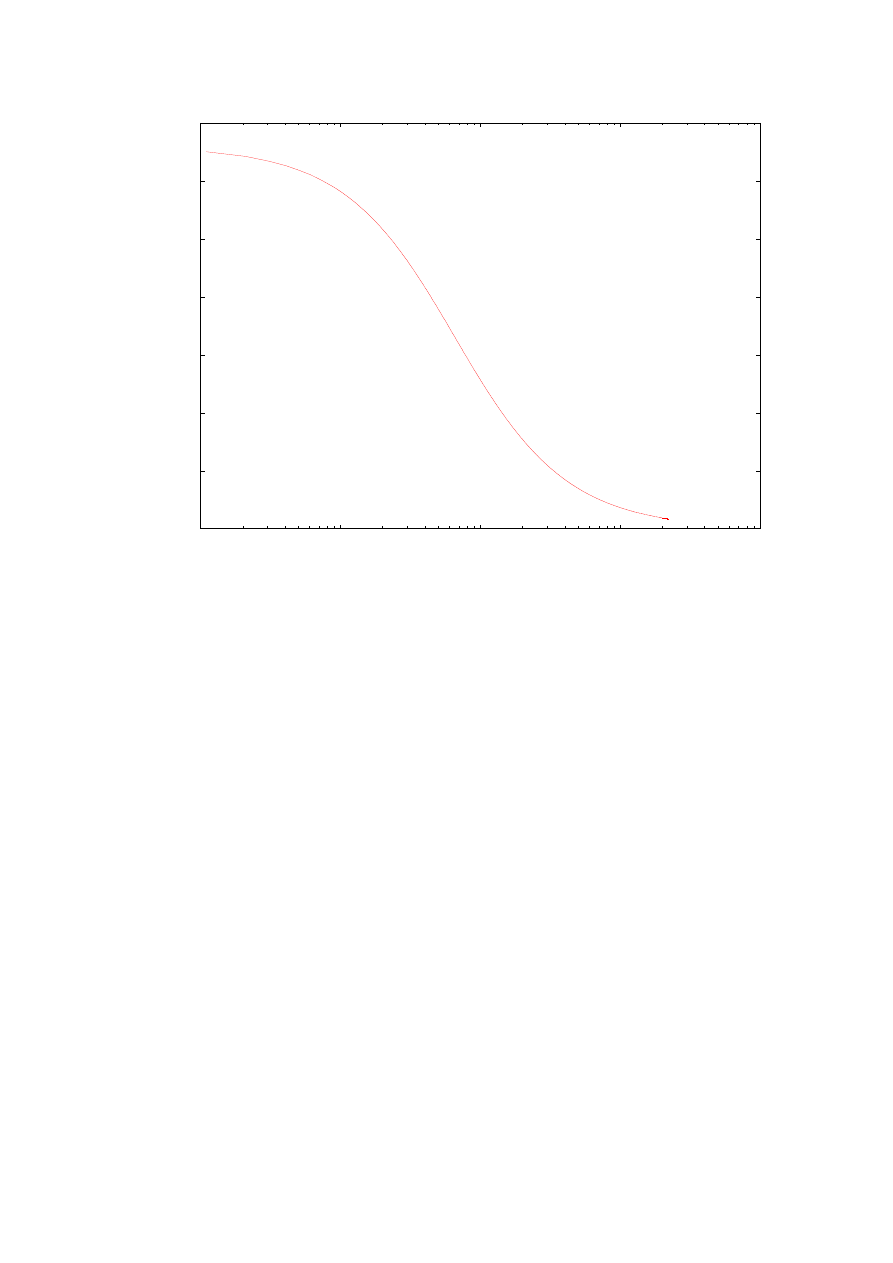
Wykresy z rys. 10 i 11 przedstawiają zależności odpowiednio energii oraz funkcji
falowych od zmieniającej się masy efektywnej wewnątrz studni (nadal dla tej samej struktury
z rozdziału 4.1). Jak można się było spodziewać wyniki są bardzo zbliżone do poprzednich.
Energia pierwszego poziomu energetycznego (oraz wyższych poziomów) także maleje wraz
ze wzrostem masy elektronu wewnątrz studni. W porównaniu z przypadkiem zwiększającej
się masy elektronu wewnątrz bariery, tutaj zmniejszenie wartości energii zachodzi znacznie
gwałtowniej. Można zaobserwować także zanikanie głębokości wnikania elektronu do
warstwy zabronionej wraz ze wzrostem masy elektronu wewnątrz studni.
m
b
= 0,1
m
b
= 1
m
b
= 0,01
ψ
19
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.001
0.01
0.1
1
10
E
[
e
V
]
masa efektywna elektronu wewnatrz studni [m0]
Rys. 10. Zależność wartości pierwszej energii własnej od wartości masy efektywnej elektronu wewnątrz
studni. Obliczenia przeprowadzone dla struktury opisanej w rozdziale 4.1
4.4 Zale
ż
no
ś
ci energii własnych w studni kwantowej od jej szeroko
ś
ci
Rysunek 12 przedstawia wartości pierwszych trzech poziomów energetycznych
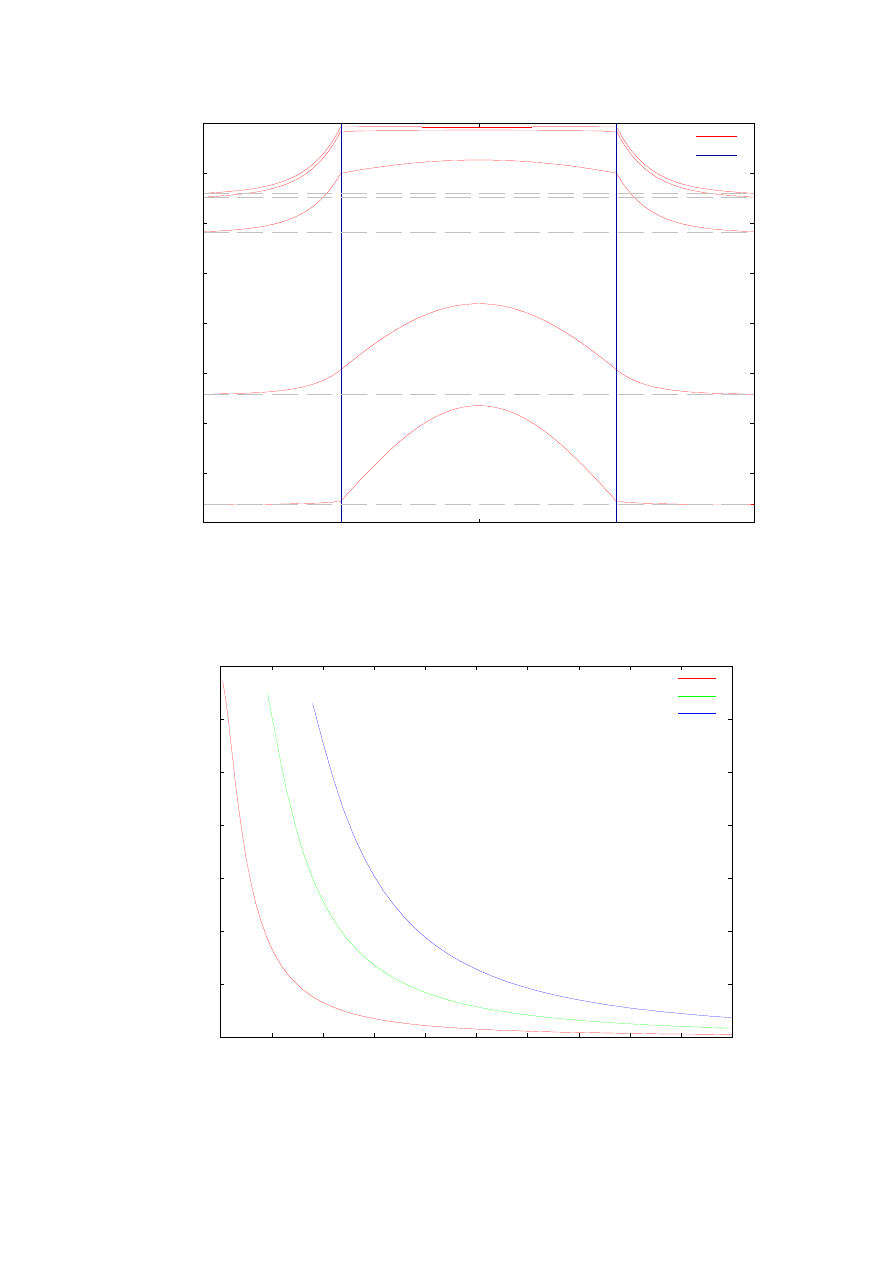
omawianej (rozdział 4.1) studni kwantowej w zależności od jej szerokości. Jak widać
szerokość studni ma bardzo duże znaczenie w projektowaniu struktur. Choć możemy także
dobierać różne wartości masy efektywnej elektronów czy dziur wewnątrz studni jak i bariery,
są to jednak wartości określone dla różnych materiałów i nie można ich zmieniać dowolnie.
Natomiast szerokość studni jest parametrem, który może być zmieniany w dość szerokim
zakresie wartości (w nanonometrach). Jak widzimy dla pojedynczej studni kwantowej
w zależności od jej szerokości możemy nie tylko dobierać wartości energii własnych
(w dużym przedziale do około 300 meV), ale również liczbę energii własnych.
20
0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
-10
-5
0
5
10
E
[
e
V
]
x [nm]
V(x)
Rys. 11 Funkcje falowe pierwszego poziomu energetycznego dla różnych wartości masy efektywnej
elektronu wewnątrz studni. Obliczenia przeprowadzone dla struktury opisanej w rozdziale 4.1
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0
5
10
15
20
25
30
35
40
45
50
E
[
e
v
]
szeroko
ść
studni [nm]
E1
E2
E2
Rys. 12. Zależność wartości pierwszych trzech energii własnych od szerokości studni. Obliczenia
przeprowadzone dla pojedynczej studni kwantowej typu GaAs/Ga
0,6
Al
0,4
As.
1,0 m
0
0,1 m
0
0,01 m
0
0,001 m
0
0,0001 m
0
ψ
E3
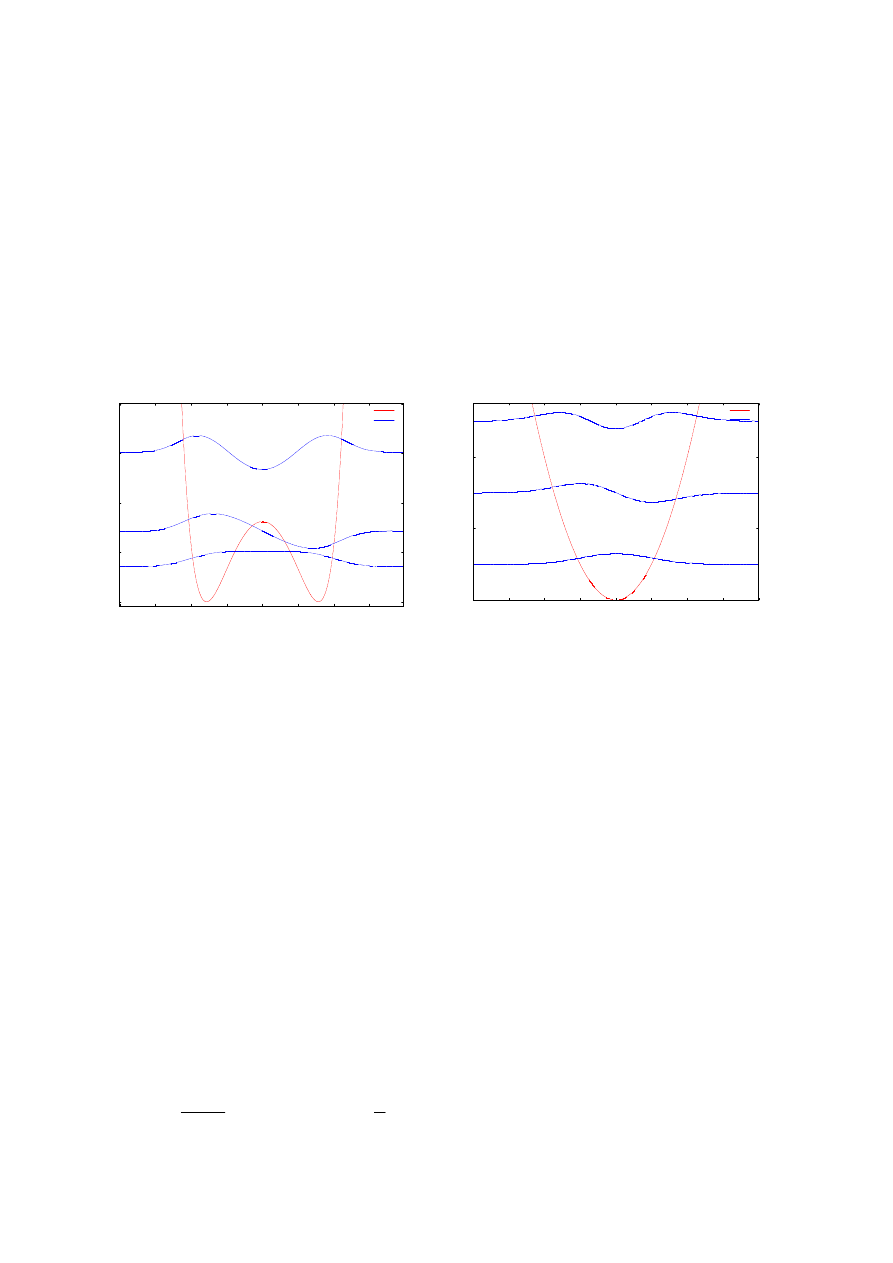
21
4.5 Analiza dokładno
ś
ci
W celu przeprowadzenia analizy błędów niezbędna jest znajomość dokładnego
rozwiązania. W podręczniku [2] zaprezentowanych zostało kilka potencjałów wiążących,
które posiadają rozwiązania analityczne, co umożliwia weryfikację poprawności wyników
numerycznych. Ponieważ porównywanie wyników numerycznych i analitycznych jest
dogodniejsze w postaci bezwymiarowej (analityczne wartości energii własnych są zwykle
prostymi ułamkami), zaprezentowane są tylko postacie bezwymiarowe (dołączone programy
w dodatku E oraz zawarte na płycie CD operują na wartościach bezwymiarowych).
Więcej informacji można znaleźć w książce [3], skąd zaczerpnięte są poniższe rozwiązania.
Oscylator harmoniczny (rysunek 13 lewy) to jeden z potencjałów wybranych do
przetestowania wyników. Jest to najprostszy z możliwych potencjałów, którego
bezwymiarowa zależność od położenia dana jest wzorem
( )
2
x
x
V
=
,
(4.1)
a energie kolejnych stanów własnych dane są zależnością:
1
2
−
=
i
E
i
,
(4.2)
gdzie
(
)
...
2
,
1
=
i
Następnym potencjałem, jaki został przetestowany jest potencjał Konwenta (rysunek
13 prawy). Jest to przykład potencjału zawierającego dwie oddziaływujące ze sobą studnie,
opisany jest zależnością
( )
( )
2
1
cosh
1
2
−
+
=
x
k
b
x
V
α
,
(
)
2
1
2
4
1
+
=
k
α
,
(4.3)
0
0.5
1
1.5
2
-4
-3
-2
-1
0
1
2
3
E
[
e
V
]
x [nm]
V(x)
0
1
2
3
4
5
-3
-2
-1
0
1
2
3
4
E
[
e
V
]
x [nm]
V(x)
Rys. 13. Poglądowe wykresy potencjałów wraz z pierwszymi trzema funkcjami falowymi wybranych do testowania
procedur numeryczny: potencjał oscylatora harmonicznego (po lewej), potencjał Konwenta (po prawej).
ψ
ψ
22
który dla współczynnika
)
4
9
(
,
1
=
=
α
k
posiada rozwiązania analityczne dane wzorami
(
)
.
4
1
4
7
9
1
,
5
9
1
,
4
1
4
7
9
1
2
2
3
2
2
2
2
1
+
+
+
=
+
+
−
+
=
b
b
E
b
E
b
b
E
(4.4)
do obliczeń przyjęte została wartość
2
,
1
=
b
.
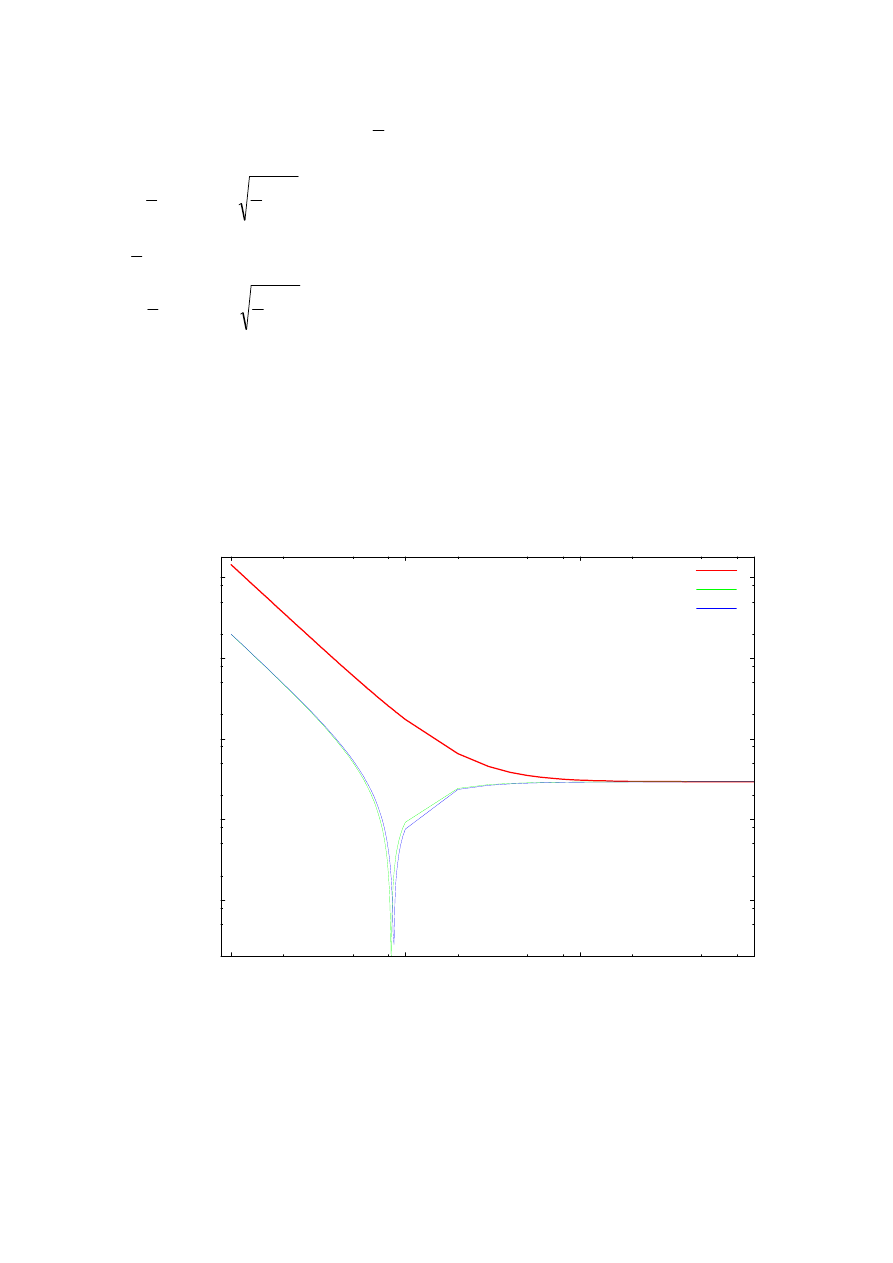
Rys. 14 przedstawia zależność dokładności obliczeń dla drugiej energii własnej
(a dokładniej różnicę pomiędzy otrzymanymi numerycznie wynikami a wartościami
analitycznymi danymi wzorem (4.2)) oscylatora harmonicznego (4.1) od ilości punktów siatki
na zadanym przedziale całkowania
1e-006
1e-005
0.0001
0.001
0.01
100
1000
10000
100000
E
[
e
V
]
ilo
ść
punktów siatki
ulepszona ms
prosta ms
M-D
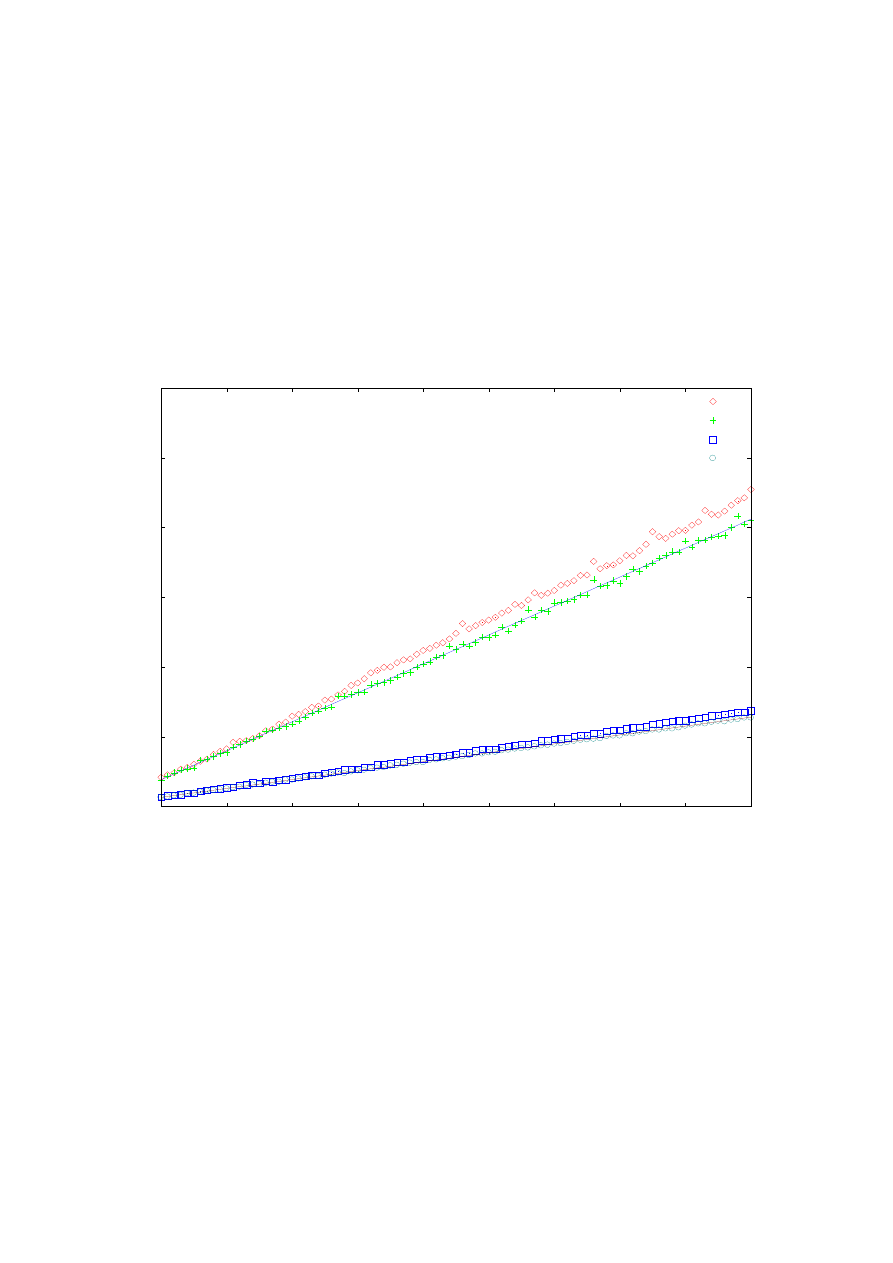
Rys. 14. Zależność dokładności obliczeń drugiej energii własnej od ilości punktów siatki. Zestawienie
obliczeń dla ulepszonej metody strzałów, prostej metody strzałów oraz metody Martina-Deana dla potencjału
oscylatora harmonicznego na przedziale <-4, 4>.
<
−
4, 4>. Na wykresie przedstawione są wyniki dla trzech metod – prostej metody strzałów
opisanej wzorem (3.11), metody macierzowej Martina-Deana, która oparta jest na wzorze
liczba punktów siatki
23
(3.10) a algorytm opisany jest w [3], oraz ulepszonej metody strzałów opisanej wzorami
(3.15) i (3.16). Wykres ilustruje tendencję osiągania lepszej dokładności wraz ze wzrostem
ilości punktów siatki a tym samym pomniejszania się kroku całkowania, aż do momentu
osiągnięcia granicy dokładności. Zależność ta jednak nie jest liniowa. Zbieżność metod
Marena-Deana oraz prostej metody strzałów nie dziwi ze względu na zastosowanie tego
samego przybliżenia drugiej pochodnej (3.7) Widoczny jest fakt polegający na tym że dla
pewnej wartości kroku siatki prosta metoda strzałów oraz metoda Marena-Deana osiągają
maksimum dokładności, po czym stabilizują się na pewnej wartości, do której zbiega także
ulepszona metoda strzałów. Aby uzyskać większą dokładność należałoby lepiej dobrać
przedział całkowania.
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
6
7
8
9
10
11
12
13
14
ulepszona ms
prosta ms
M-D
Rys. 15. Zależność dokładności obliczeń pierwszej energii własnej oscylatora harmonicznego od szerokości
przedziału całkowania dla trzech wybranych metod, ulepszonej i prostej metody strzałów, metody
Martina-Deana.
Kolejny wykres (rys. 15) to zestawienie dokładności obliczeń numerycznych
podstawowego stanu energetycznego dla oscylatora harmonicznego (4.1) ze zmieniającym się
przedziałem całkowania dla ustalonej liczby punktów siatki równej 10
4
dla trzech
dyskutowanych tutaj algorytmów. Rys. 15. przedstawia podobne zachowanie się algorytmów
do sytuacji z rys. 14. Widoczne jest pokrywanie się wyników prostej metody strzałów
z wynikami metody Marena-Deana oraz osiągania przez te metody maksymalnej dokładności
dla jednej z wartości długości przedziału całkowania. W tym przypadku możemy także i dla
algorytmu ulepszonej metody strzałów zaobserwować osiągnięcie maksimum dokładności –
nie jest ono jednak tak wyraźne. Wykres przedstawia również tendencję spadku dokładności
wraz z nadmiernym wzrostem długości przedziału całkowania, aż do momentu, dla którego,
otrzymanie poprawnych wyników numerycznych jest niemożliwe. Widoczną różnicę
w dokładnościach można zmniejszyć zwiększając odpowiednio liczbę punktów całkowania.
szeroko
ść
przedziału całkowania [nm]
E
[
e
V
]
24
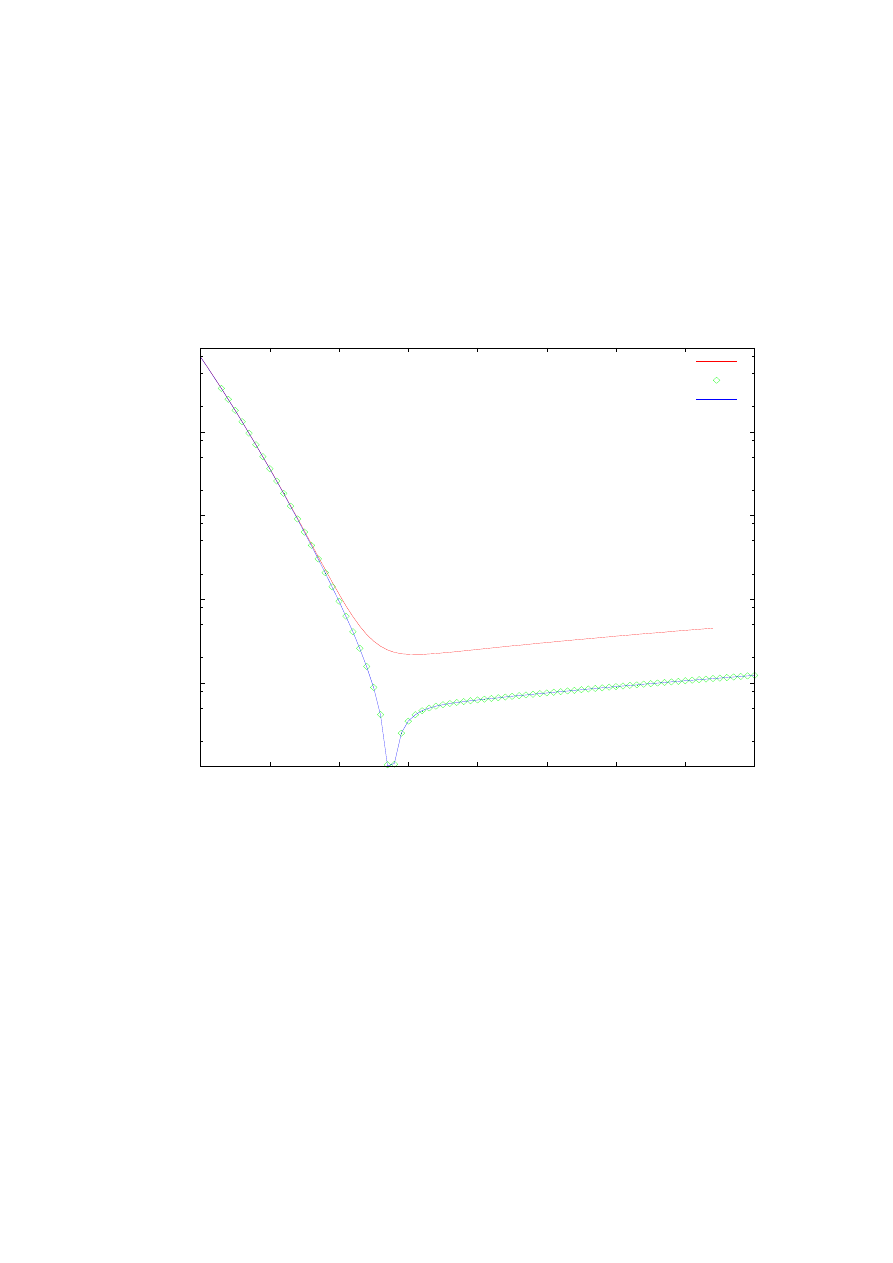
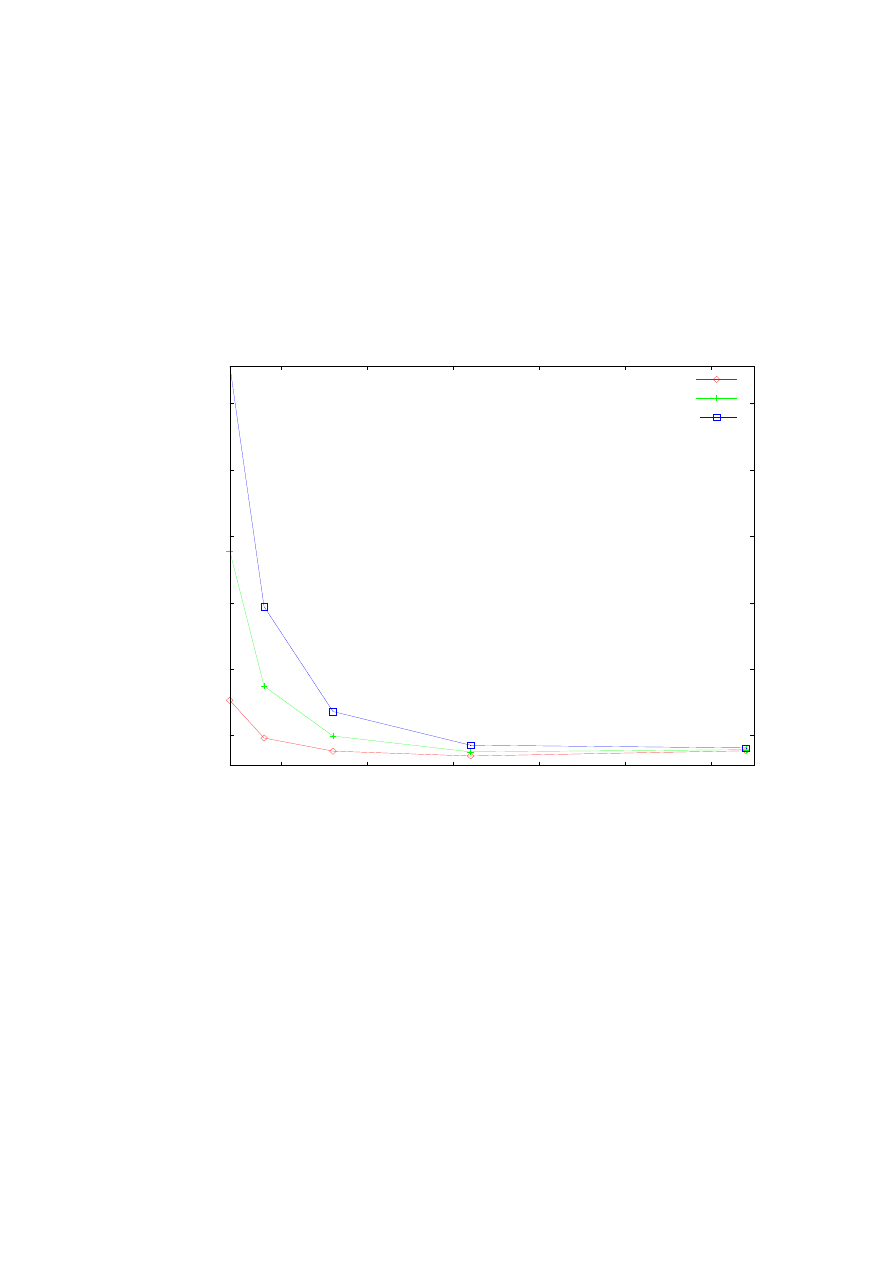
Rys. 16 zawiera zależności dokładności wyznaczenia (różnic pomiędzy wynikami
numerycznymi i wynikami analitycznymi danymi wzorami (4.4)) pierwszych trzech wartości
własnych energii potencjału Konwenta (4.3) od długości przedziału całkowania.
Przedstawiono wyniki dla metody ulepszonej strzałów oraz macierzowej Martina-Deana.
Wyniki dla najniższych 3 poziomów energii zaznaczone są odpowiednimi kolorowymi
symbolami, co wyjaśnia opis rysunku. Zauważmy jedynie, że wypełnione kolorami symbole
reprezentują wyniki otrzymane metodą macierzową, natomiast niewypełnione („puste”)
symbole ilustrują rezultaty wyznaczone ulepszoną metodą strzałów. Otrzymane wyniki nie
odbiegają wiele od wyników prezentowanych na rys. 15. Widoczna jest tendencja polepszania
się dokładności aż do momentu osiągnięcia największej dokładności dla obydwu metod dla
określonej długości przedziału całkowania, po czym daje się zauważyć niewielki jej spadek
wraz ze wzrostem długości przedziału całkowania.
1e-009
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
0.01
0.1
1
4
6
8
10
12
14
16
E
[
e
V
]
x [nm]
E1 ulepszona
E2 ulepszona
E3 ulepszona
E1 M-D
E2 M-D
E3 M-D
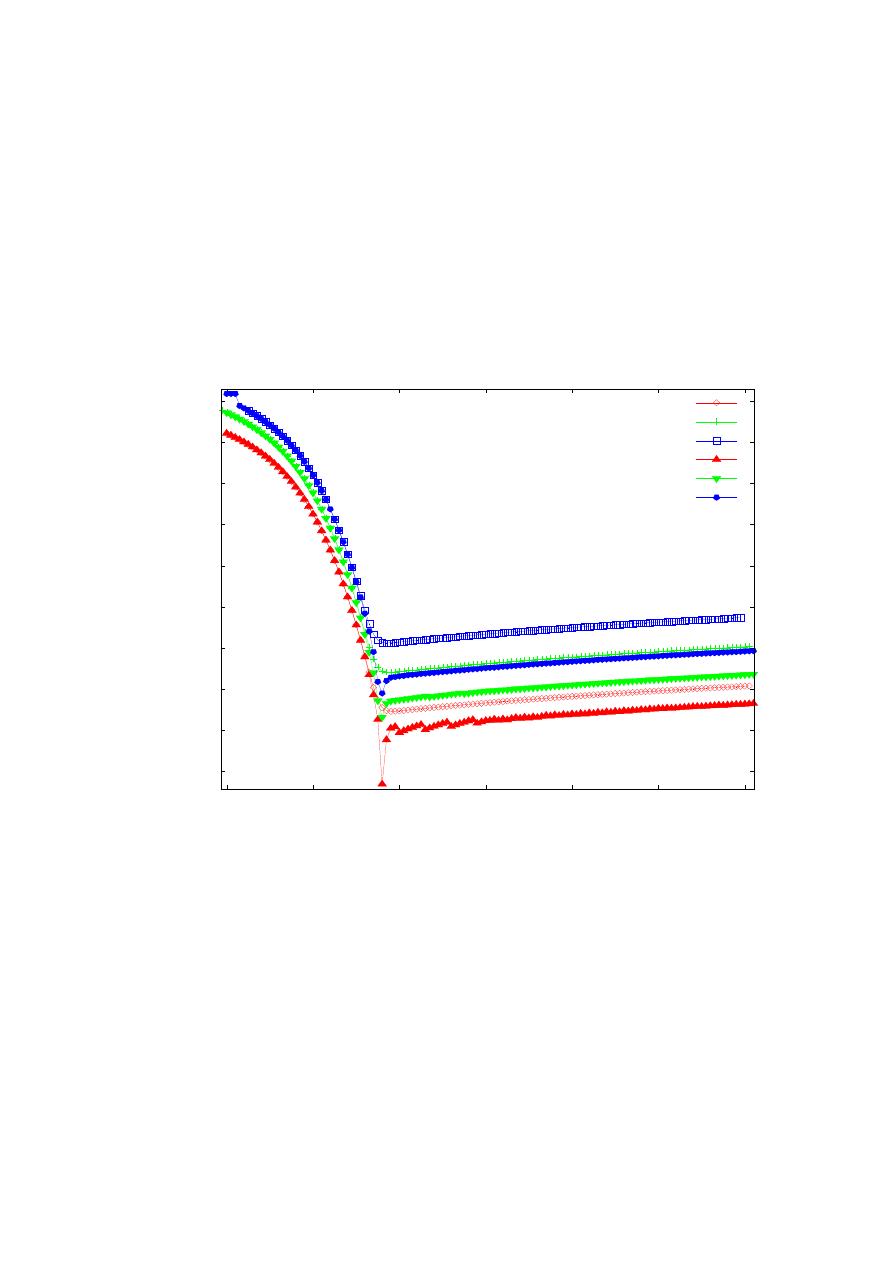
Rys. 16. Zależność dokładności obliczanych energii własnych od szerokości przedziału całkowania.
Obliczenia przeprowadzone ulepszona metodą strzałów (symbole niewypełnione) oraz metodą macierzową
Martina-Deana (symbole wypełnione) dla potencjału Konwenta z parametrami k=1, b=1,2. Obliczenia
przeprowadzone dla stałej ilości punktów siatki równej 10
4
.
Następny wykres (rys. 17) jest analogiczny do wykresu z rys. 14. Przedstawiono
zależność dokładności wyznaczenia energii stanu podstawowego cząstki kwantowej
w potencjale Konwenta (4.3) od ilości punktów siatki na zadanym przedziale całkowania
o długości równej 8 nm (przedział wynosił <
−
4 nm,4 nm>) dla omawianych tutaj trzech
metod numerycznych. Tym razem dzięki właściwemu doborowi długości przedziału
całkowania algorytmy mogły osiągnąć znacznie większe dokładności. Maksima osiągane
przez prostą metodę strzałów oraz metodę macierzową Marena-Deana są słabo widoczne, bo
w tym przypadku przypadają one na granicy wykresu odpowiadającej liczbie punktów siatki
rzędu 10
5
. Ze względu na ograniczenia sprzętowe, jakimi dysponowaliśmy, dalsza
25
kontynuacja wykresu jest bardzo czasochłonna lub wręcz niemożliwa ze wzglądu na objętość
potrzebnej pamięci do zapisania wartości potencjału oraz masy w macierzy (dla prostej
metody strzałów jak i M-D niezbędna jest znajomość masy w dwukrotnie większej liczbie
punktów siatki aniżeli potencjału).
1e-011
1e-010
1e-009
1e-008
1e-007
1e-006
1e-005
0.0001
0.001
100
1000
10000
100000
E
[
e
V
]
ilo
ść
punktów siatki
M-D
Ulepszona strzałów
prosta strzałów
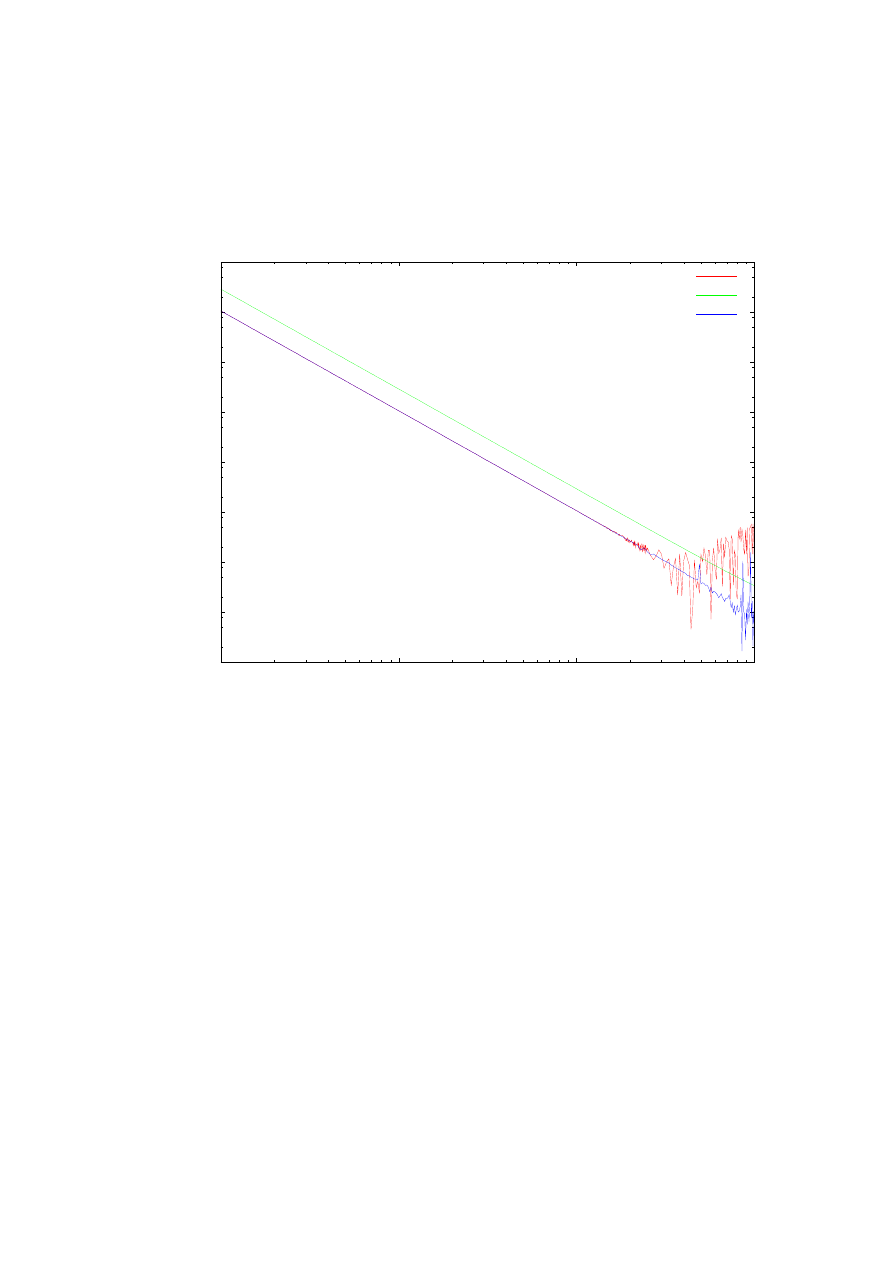
Rys. 17. Zależność dokładności obliczeń pierwszej energii własnej od ilości punktów siatki. Zestawienie
obliczeń dla ulepszonej metody strzałów, prostej metody strzałów oraz metody Matrena-Deana dla potencjału
Konwenta z parametrami k=1, b=1,2 na przedziale całkowania <-4, 4>.
liczba punktów siatki
26
4.6 Czas oblicze
ń
Kolejną ważną cechą każdego algorytmu numerycznego jest czas potrzebny do
wykonania określonego zadania. Rozdział ten zawiera krótkie przedstawienie czasu obliczeń
dla omawianych metod. Obliczenia zostały przeprowadzone, (jeśli nie zostało to zaznaczone
inaczej) na komputerze przenośnym typu PC z pamięcią dynamiczną 256 Mb wyposażonego
w procesor Intel® Pentium® Mobile III o częstotliwości taktowania 1GHz.
Rys. 19 przedstawia zależność czasu obliczeń drugiej energii własnej dla potencjału
oscylatora harmonicznego od liczby punktów siatki. Omawiany wykres przedstawia czas
0
0.5
1
1.5
2
2.5
1000
10000
100000
c
z
a
s
[
s
]
ilo
ść
punktów siatki
prosta ms
ulepszona ms
M-D
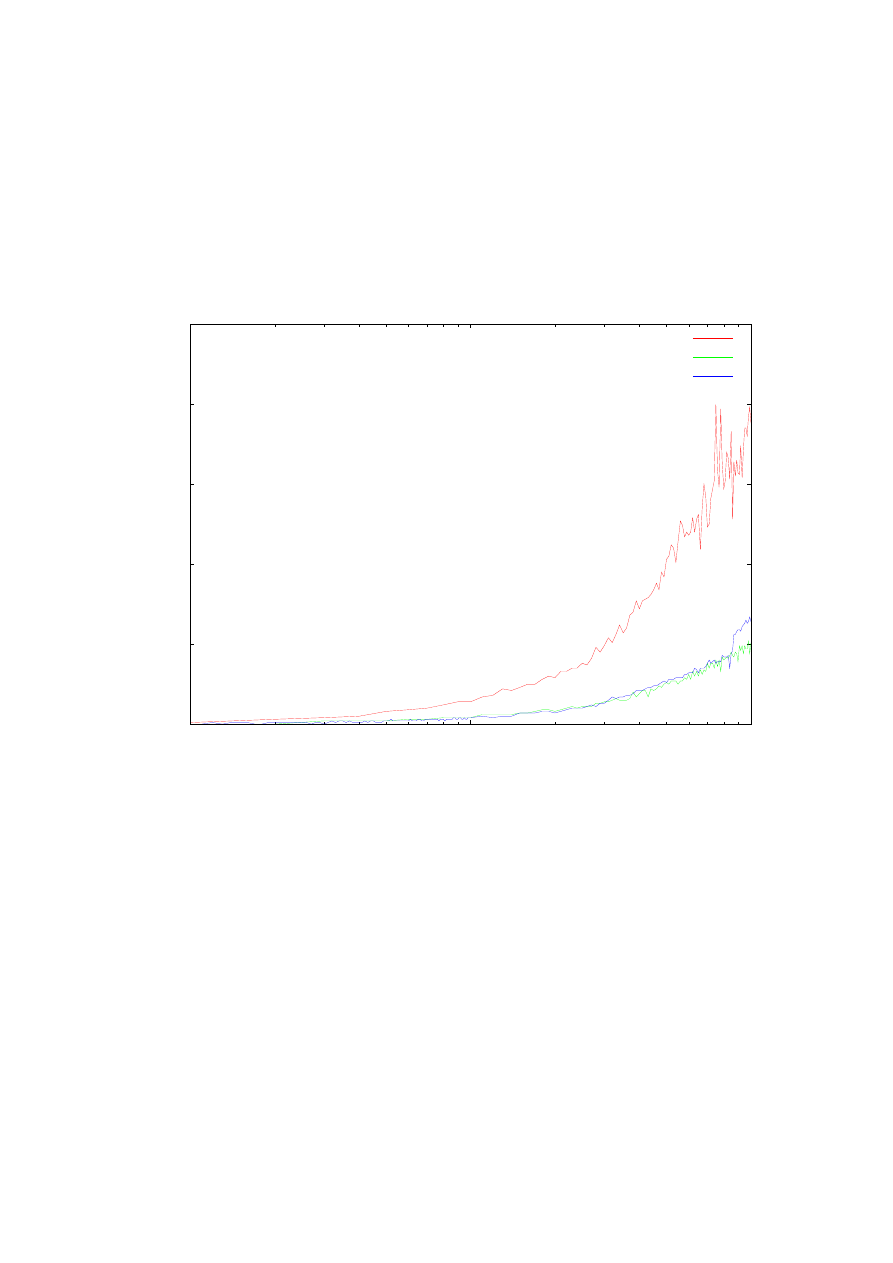
Rys. 19. Zależność czasu obliczeń drugiej energii własnej od ilości punktów siatki. Zestawienie obliczeń dla
ulepszonej metody (ms) strzałów, prostej metody strzałów oraz metody Matrena-Deana dla potencjału
oscylatora harmonicznego na przedziale <-4, 4>.
obliczeń, które zostały przeprowadzone w celu zbadania dokładności algorytmów, a których
wyniki zostały przedstawione na rys.14.
Jak widać na rys. 19 najgorzej wypada prosta metoda strzałów, która dla dużych ilości
punktów siatki potrzebuje około trzy razy więcej czasu do przeprowadzenia obliczeń
w porównaniu z pozostałymi dwiema metodami. Metoda Martina-Deana oraz ulepszona
metoda strzałów utrzymują się na tym samym poziome niezbędnego czasu dla dostatecznie
małych wartości liczby punktów siatki, choć wraz z ich wzrostem obserwujemy tendencję
rozbiegania się wykresów reprezentujących wyniki otrzymane dyskutowanymi metody na
korzyść ulepszonej metody strzałów.
Kolejny wykres (rys. 20) przedstawia zestawienie czasów obliczeń tym razem
wszystkich wartości własnych energii wraz z wyznaczeniem odpowiadających im wektorów
własnych dla podanych liczb punktów siatki. Obliczenia zostały przeprowadzone dla
liczba punktów siatki
27
struktury GaAs/Ga
0.67
Al
0.33
As zawierającej pojedynczą studnię potencjału o szerokości 15
nanometrów, na przedziale całkowania <
−
15 nm, 15 nm>. Przedstawiono wyniki tylko dla
metody macierzowej Martina-Deana z zastosowaniem metody DWSZ do obliczania
wektorów własnych [2] oraz ulepszonej metody strzałów. Obliczenia zostały przeprowadzone
również
na
dodatkowym
komputerze
nowszej
generacji
(komputer
typu
PC
z dwurdzeniowym procesorem firmy Intel® Core2Duo® taktowany zegarem 2,6GHz,
posiadający 1Gb RAM). Rys. 20 pokazuje, że ulepszona metoda jest w niewielkim stopniu
szybsza, powiększając swoją przewagę wraz z rosnącą liczbą punktów siatki. Potwierdzają to
wyniki otrzymane na dwóch PC. Możemy również zaobserwować około cztery razy szybsze
obliczenia na nowszym komputerze.
0
2
4
6
8
10
12
10000
20000
30000
40000
50000
60000
70000
80000
90000
100000
M-D PIII
ulepszona ms PIII
M-D Core2Duo
ulepszona ms COre2Duo
Rys.20. Zależność czasu obliczeń wszystkich energii własnych wraz z wyznaczeniem wektorów własnych dla
studni GaAs/Ga
0.67
Al
0.33
As o szerokości 15 nm. Obliczenia dla metody Martina-Deana oraz ulepszonej
metody strzałów przeprowadzone na dwóch różnych komputerach – jeden z procesorem PIII, drugi
z Core2Duo. Zamieszczone zostały także aproksymacje liniowe wraz z podanymi wzorami.
Punkty wykresu (patrz rys. 20) pokrywają się z aproksymującą je prostą (linie ciągłe
na wykresie wraz z odpowiadającymi im wzorami). Takie zachowanie się badanych
algorytmów świadczy o liniowej zależności czasu obliczeń od ilości punktów siatki.
y = 8,299E-05x - 5,785E-02
y = 2,554E-05x +
9,860E-03
liczba punktów siatki
c
z
a
s
[
s
]
28
4.7 Supersieci
W rozdziale tym przedstawiamy zastosowanie ulepszonego algorytmu strzałów do
wyznaczania poziomów energetycznych nośników prądu w wybranych supersieciach.
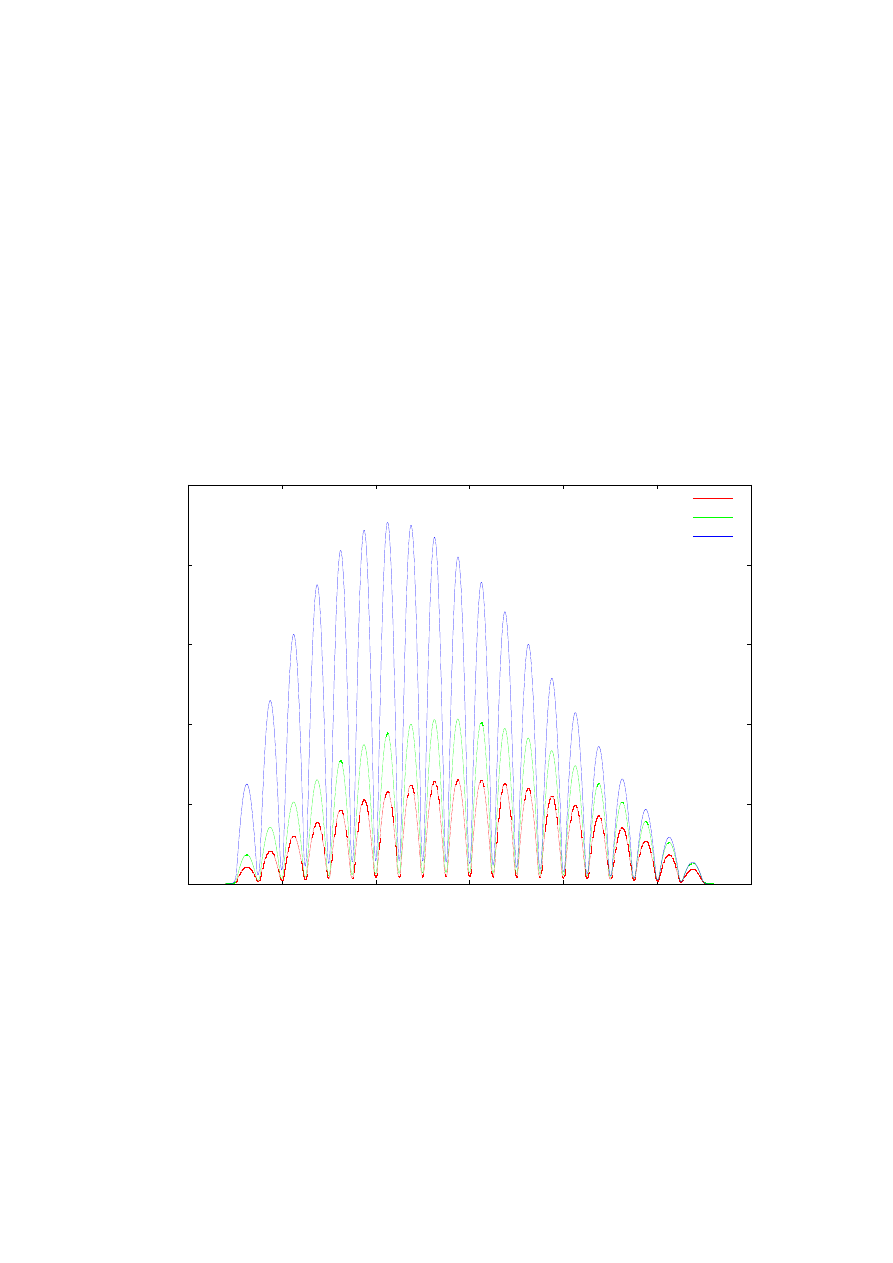
Rys. 20 przedstawia unormowaną funkcję falową stanu podstawowego elektronu
w supersieci zawierającej dwadzieścia studni typu GaAs/Ga
0,67
Al
0,33
As o szerokości 20 nm
oddalonych od siebie o 5 nm. Prezentowane wektory własne powiększone są
o odpowiadającą im bezwymiarową wartość energii. Wykres ilustruje tę samą funkcję falową
obliczaną z różną długością kroku całkowania. Jak łatwo jest zauważyć potrzeba dużej liczby
punktów siatki, aby otrzymany wynik był dostatecznie dobry (dokładność wektora można
oszacować np. przez porównanie symetryczności funkcji falowej względem środka osi X).
Uzasadnia to zastosowanie ulepszonej metody strzałów, która nie tylko jest szybsza, ale
przede wszystkim potrzebuje mniej pamięci na zapisanie wartości funkcji rozkładu masy
efektywnej.
0.01
0.015
0.02
0.025
0.03
0.035
-300
-200
-100
0
100
200
300
x [nm]
10e5 punktów siatki
4e4 punktów siatki
9e3 punktów siatki
Rys. 20. Wykresy funkcji falowej podstawowego stanu energetycznego dla supersieci zawierającej
dwadzieścia studni typu GaAs/Ga
0,67
Al
0,33
As o szerokości 20 nm. oddalonych od siebie o 5 nm. Wynik
obliczeń dla różnej liczby punktów siatki, ulepszoną metodą strzałów.
E
[
e
V
]
29
Rys. 21 przedstawia zależność wartości trzech pierwszych stanów energetycznych dla
supersieci od liczby zawartych w niej studni kwantowych typu GaAs/Ga
0,67
Al
0,33
As
o szerokości 15 nm oddalonych od siebie o 5 nm. Na wykresie tym uwidacznia się tendencja
dążenia poziomów energetycznych do określonej wartości. Im więcej studni składających się
na supersieć, tym bardziej kolejne poziomy energetyczne zbliżają się do siebie, co jest
związane z tworzeniem się pasm energetycznych w naszej heterostrukturze będącej
jednowymiarowym kryształem zbudowanym ze skończonej liczby studni kwantowych.
Zauważmy, że zbieżność wartości własnych wraz ze wzrostem liczby studni sprawia
znaczne
utrudnienia
w
obliczaniach
numerycznych
poszczególnych
poziomów
energetycznych oraz wydłuża czas obliczeń.
0.01625
0.0163
0.01635
0.0164
0.01645
0.0165
10
20
30
40
50
60
E
[
e
v
]
ilo
ść
studni
E1
E2
E2
Rys. 21. Zależność wartości pierwszych trzech energii własnych od ilości oddziaływających ze sobą studni.
Obliczenia przeprowadzone dla studni typu GaAs/Ga
0,67
Al
0,33
As o szerokości 15 nm., oddalonych od siebie
o 5nm.
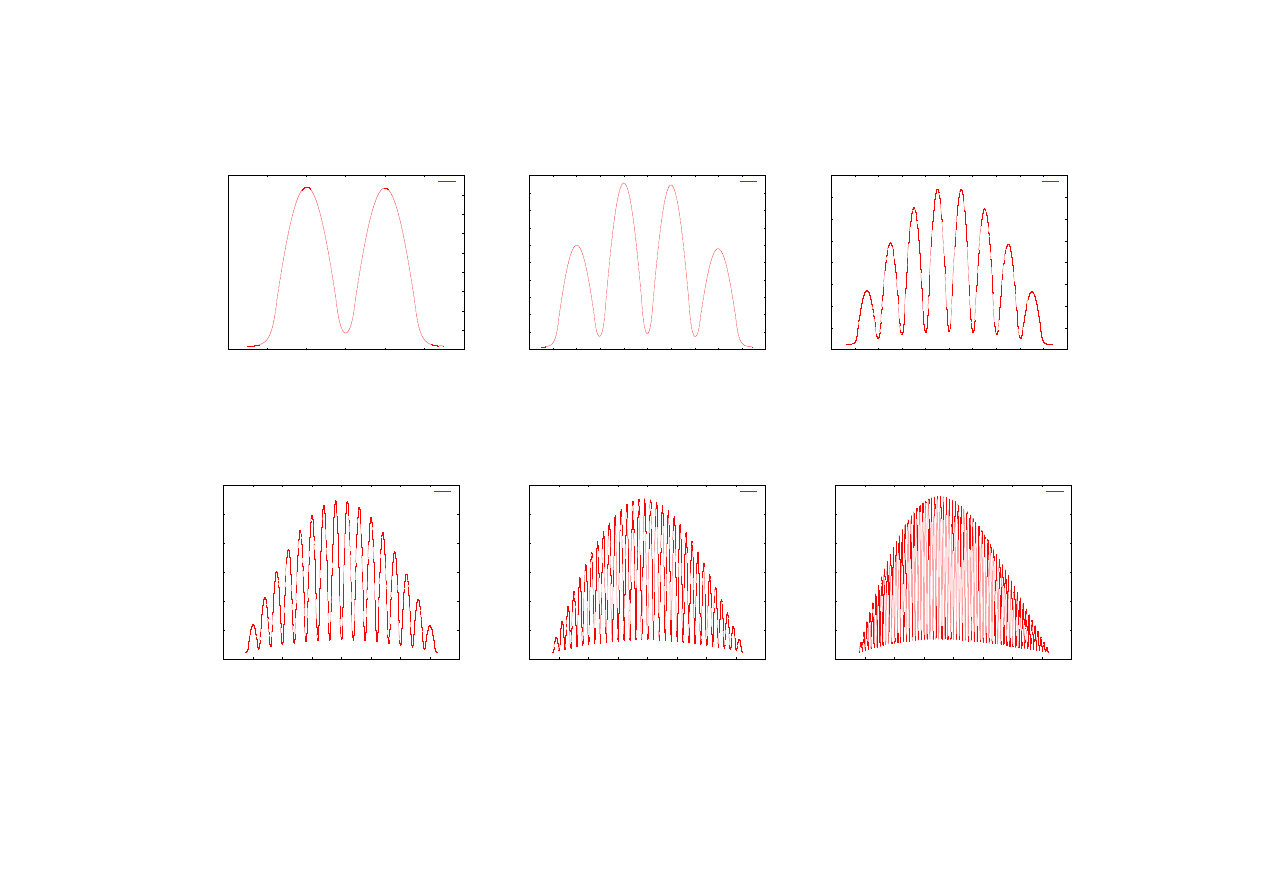
Kolejne wykresy (rys. 22-33) przedstawiają unormowane funkcje falowe odpowiednio
stanu podstawowego oraz pierwszego stanu wzbudzonego dla supersieci zawierających różne
liczby studni kwantowych. Prezentowane wektory własne zostały powiększone
o odpowiadającą im energię własną, która podana jest w opisach wykresów. Wyznaczenie
wektorów funkcji falowych jest bardzo ważnym elementem pozwalającym na określenie
liczby porządkowej L (numeru poziomu energetycznego) obliczonej wartości energii. Dla
supersieci składających się z dużej liczby studni kwantowych analiza kształtu funkcji falowej
może być jedynym sposobem na określenie numeru poziomu energetycznego, ponieważ
liczba węzłów funkcji falowej wynosi L
−
1.
E3
30
0.016
0.018
0.02
0.022
0.024
0.026
0.028
0.03
0.032
0.034
-30
-20
-10
0
10
20
30
E
[
e
v
]
x [nm]
E1
0.016
0.018
0.02
0.022
0.024
0.026
0.028
0.03
0.032
0.034
0.036
-50
-40
-30
-20
-10
0
10
20
30
40
50
E
[
e
v
]
x [nm]
E1
0.016
0.017
0.018
0.019
0.02
0.021
0.022
0.023
0.024
-100
-80
-60
-40
-20
0
20
40
60
80
100
E
[
e
v
]
x [nm]
E1
0.016
0.017
0.018
0.019
0.02
0.021
0.022
-200
-150
-100
-50
0
50
100
150
200
E
[
e
v
]
x [nm]
E1
0.016
0.017
0.018
0.019
0.02
0.021
0.022
-400
-300
-200
-100
0
100
200
300
400
E
[
e
v
]
x [nm]
E1
0.016
0.017
0.018
0.019
0.02
0.021
0.022
-800
-600
-400
-200
0
200
400
600
800
E
[
e
v
]
x [nm]
E1
Rys.22. Funkcja falowa stanu podstawowego
dla supersieci zawierającej dwie studnie.
E1 = 0,0163429614 [eV].
Rys.23. Funkcja falowa stanu podstawowego
dla supersieci zawierającej cztery studnie.
E1 =0,016276492 [eV].
Rys.24. Funkcja falowa stanu podstawowego
dla supersieci zawierającej osiem studni.
E1 = 0,0162480602 [eV].
Rys.25. Funkcja falowa stanu podstawowego
dla supersieci zawierającej szesnaście studni.
E1 = 0,0162352162 [eV].
Rys.26. Funkcja falowa stanu podstawowego
dla supersieci zawierającej trzydzieści dwie
studnie. E1 = 0,0162345508 [eV].
Rys.27. Funkcja falowa stanu podstawowego
dla supersieci zawierającej sześćdziesiąt
cztery studnie. E1 = 0,0162384657 [eV].
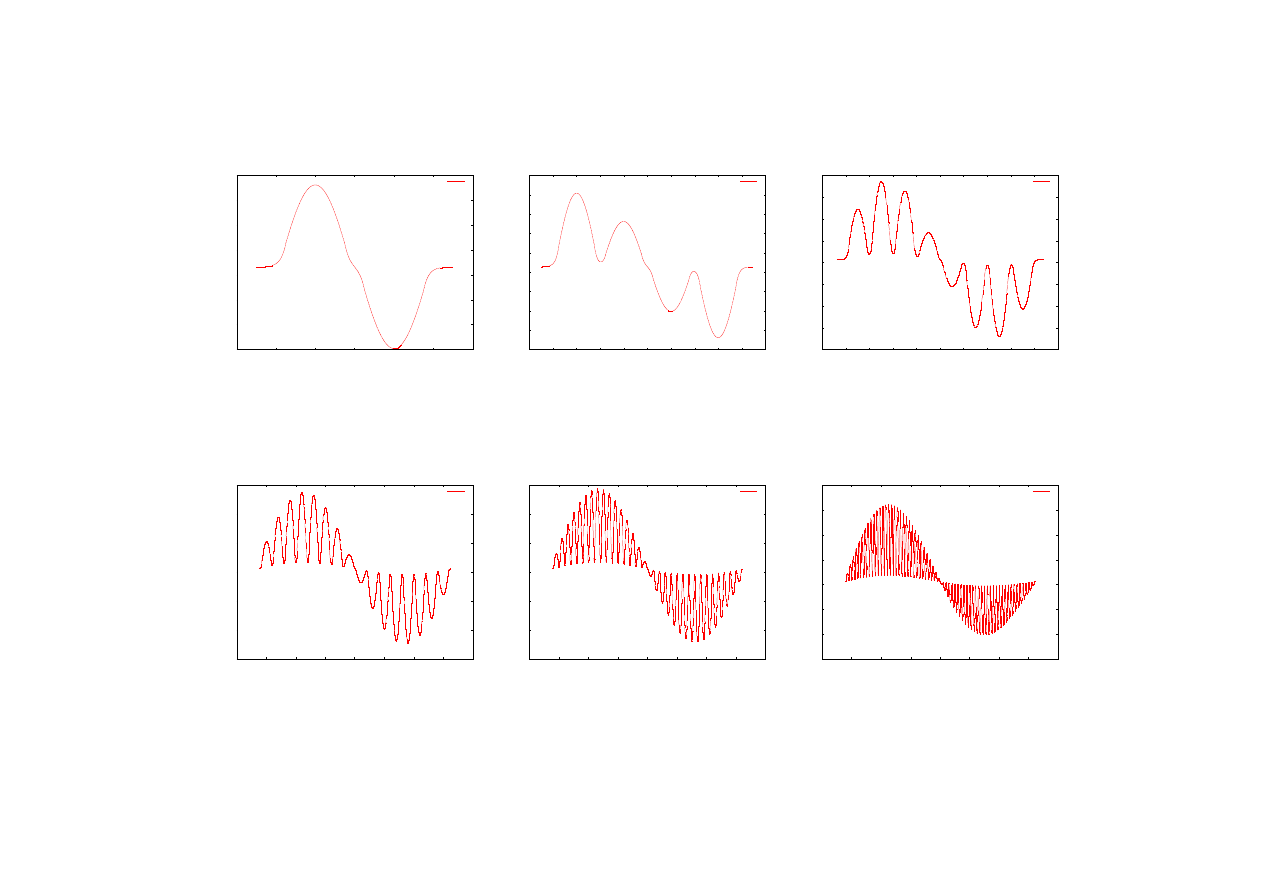
31
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
-30
-20
-10
0
10
20
30
E
[
e
v
]
x [nm]
E2
-0.005
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0.04
-50
-40
-30
-20
-10
0
10
20
30
40
50
E
[
e
v
]
x [nm]
E2
0.008
0.01
0.012
0.014
0.016
0.018
0.02
0.022
0.024
-100
-80
-60
-40
-20
0
20
40
60
80
100
E
[
e
v
]
x [nm]
E2
0.01
0.012
0.014
0.016
0.018
0.02
0.022
-200
-150
-100
-50
0
50
100
150
200
E
[
e
v
]
x [nm]
E2
0.01
0.012
0.014
0.016
0.018
0.02
0.022
-400
-300
-200
-100
0
100
200
300
400
E
[
e
v
]
x [nm]
E2
0.01
0.012
0.014
0.016
0.018
0.02
0.022
0.024
-800
-600
-400
-200
0
200
400
600
800
E
[
e
v
]
x [nm]
E2
Rys.28. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
dwie studnie. E2 = 0,0165702687 [eV].
Rys.29. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
cztery studnie. E2 = 0,0163892205 [eV].
Rys.30. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
osiem studni. E2 = 0,0162870794 [eV].
Rys.31. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
szesnaście studni. E2 = 0,016289791 [eV].
Rys.32. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
trzydzieści
dwie
studnie
E2 = 0,0162375734 [eV].
Rys.33. Funkcji falowa drugiego poziomu
energetycznego dla supersieci zawierającej
sześćdziesiąt
cztery
studnie.
E2 = 0,0162392443 [eV].

32
5. Podsumowanie
W pracy przedstawiono i przetestowano opracowane programy komputerowe napisane
w języku FORTRAN 77 (spis w tabeli E.1 Dodatku E; kody źródłowe programów są zapisane
na CD dołączonym do pracy) przy zastosowaniu algorytmów opisanych w rozdziale 3.
Przeprowadzono szereg testów numerycznych dotyczących m.in. dokładności oraz czasu
trwania obliczeń numerycznych. Pokazano, że dokładność obliczeń wartości i wektorów
własnych zależy od kroku całkowania, szerokości przedziału całkowania i wybranego
algorytmu. Opracowane programy komputerowe – wymienione w tabeli E.1 i oparte
o ulepszoną metodę strzałów – bardzo dobrze nadają się do rozwiązywania równań masy
efektywnej opisujących struktury energetyczne prostych (pojedyncza studnia kwantowa) jak
również bardziej skomplikowanych (supersieci) heterostruktur półprzewodnikowych typu
III/V. Warto dodać, że stosując te programy można analizować i badać heterostruktury
o zaprogramowanych przez użytkownika kształtach energii potencjalnej oraz zależności masy
efektywnej od położenia.
Ś
rodowisko programowe pt. Strzały (zapisane na CD) wyposażone w interfejs
graficzny pozwala w przyjazny dla użytkownika sposób wyznaczać poziomy i wektory
własne dla pojedynczych studni kwantowych różnych typów (szczegółowy opis w Dodatku
C).
Najważniejszym wynikiem pracy jest stwierdzenie, że czasy trwania obliczeń
numerycznych opracowanych programów komputerowych opartych o ulepszoną metodę
strzałów wykazują liniową zależność czasu ich trwania od liczby kroków całkowania.
Otrzymane wyniki numeryczne (patrz rys. 14
−
33) możemy podsumować w następujący
sposób:
– uzyskanie poprawnych i dokładnych wyników numerycznych dla wartości energii
własnych w ramach każdej z przedstawionych metod jest trudne i wymaga odpowiednio
właściwego wyboru zarówno przedziału całkowania jak również liczby punktów siatki;
– numeryczne wartości najniższych poziomów energetycznych otrzymane prostą
metodą strzałów (3.11) oraz metodą Martina-Deana (3.10) są praktycznie zbieżne, co jest
konsekwencją zastosowania takiego samego przybliżenia drugiej pochodnej wzorem (3.7);
– ulepszona metoda strzałów w niewielkim stopniu ustępuje dwóm pozostałym
omawianym metodom pod względem osiąganych dokładności i w przeciwieństwie do nich
zachowuje się bardziej stabilnie – polega to na tym, ze wraz ze wzrostem liczby punktów
siatki wartości numeryczne energii monotonicznie dążą do wartości rzeczywistych;
– przewaga rozpatrywanej w pracy ulepszonej metody strzałów nad dwoma
pozostałymi ujawnia się zauważalnie dla bardzo dużej liczby punktów siatki, (tj. dla małych
wartości kroków całkowania), zarówno pod względem dokładności jak i czasu obliczeń;
– pomimo niewielkiej liczby zaprezentowanych testów (a do których zapraszamy
czytelników) wiemy, że ulepszona metoda strzałów jest znacznie szybsza od prostej metody
strzałów oraz w niewielkim stopniu od metody Martina-Deana;
– ulepszona metoda strzałów idealnie nadaje się do wyznaczania energii oraz
wektorów własnych dla supersieci, ze względu na oszczędność pamięci potrzebnej do
przechowywania punktów rozkładu masy.

33
6. Bibliografia
1. Toshiaki Suhara „Semiconductor Laser Fundamentals”, Marcel Dekker, Inc., 270 Madison
Avenue, NY 10016; R. Diehl „High-Power Diode Lasers Fundamentals, Technology,
Applications”, Springer-Verlag Berlin Heidelberg 2000; http://photoni-x.net/
−
strona
poświęcona laserom półprzewodnikowym.
2.
J.
Misiewicz,
G.
Sęk,
P. Sitarek,
„Spektroskopia
fotoodbiciowa
struktur
półprzewodnikowych”, Oficyna Wydawnicza Politechniki Wrocławskiej, Wrocław 1999.
3. W. Salejda, M. H. Tyc, M. Just; „Algebraiczne metody rozwiązywania równania
Schrödingera”, Wydawnictwo Naukowe PWN S.A., Warszawa 2002.
4. Sphen F.-P. Paul, Henning Fouckhardt; „An improved shooting approach for solving the
time-independent Schrödinger equation for III/V QW structures", Physics Letters A 286,
1999-2004 (2001).
5. Paul Harrison, „Quantum Wells, Wires and Dots”, John Wiley and Sons Ltd, Chichester,
2001.
6. P. Butcher, N. H. March, M. P. Tosi; “Physics of Low-Dimensional Semiconductor
Structures”, Plenum Press New York, 1993.
7. K. Sierański, M Kubisa, J. Szatkowski, J. Misiewicz, „Półprzewodniki i struktury
półprzewodnikowe”, Oficyna Wydawnicza Politechniki Wrocławskiej, Wrocław 2002.
8. N.W Ashcroft, N.D Mermin, “Fizyka ciała stałego”, Wydawnictwo Naukowe PWN SA,
Warszawa 1986.
9. http://www.thefreecountry.com/compilers/fortran.shtml
−
strona zawierająca m.in.
darmowe kompilatory języka Fortran 77.
10.
http://people.deas.harvard.edu/~jones/ap216/images/bandgap_engineering/bandgap_engineering.html
−
strona poświęcona teorii pasmowej półprzewodników.
11. http://www.gnuplot.info/
−
oficjalna strona darmowego programu Gnuplot, którym
zostały stworzone wszystkie wykresy zawarte w tej pracy.
34
Dodatek A. Struktury krystaliczne
Podstawowe informacje na temat struktury kryształu są niezbędne do zrozumienia
budowy i złożoności materiałów półprzewodnikowych [3].
Większość popularnych półprzewodników posiada regularny, ściennie centrowany
rozkład atomów (ang. face-centered cubic Bravis lattice), jak pokazano na rys. A.1.
Struktura krystaliczna powstaje poprzez umieszczenie atomów w punktach tak zwanej
sieci Bravais. Punkty siatki Bravais są zdefiniowane jako liniowa kombinacja wektorów
translacji, które mają następująca postać [3]:
( )
( )
( )
j
i
a
i
k
a
k
j
a
ˆ
ˆ
2
,
ˆ
ˆ
2
,
ˆ
ˆ
2
0
3
0
2
0
1
+
=
+
=
+
=
A
A
A
, (A.1)
gdzie A
0
to odległość pomiędzy atomami wewnątrz kryształu, jak pokazano na rys.A.1.
Czasem wektory (A.1) zapisywane są jako rozkład
3
3
2
2
1
1
a
a
a
α
α
α
+
+
=
R
,
(A.2)
gdzie
3
2
1
,
,
α
α
α
to liczby całkowite.
Materiały takie jak Si, Ge, GaAs, AlAs, InP itd., składają się z dwóch atomów, jeden
w położeniu
8
1
,
8
1
,
8
1
i drugi w
−
−
−
8
1
,
8
1
,
8
1
, w jednostkowej komórce
0
A (jak
zaznaczono na rys. A.1).
Rys. A.1. Ściennie centrowany rozkład atomów [3].
35
Dla półprzewodników typu III-V i II-IV takich jak GaAs, AlAs, InP, HgTe oraz CdTe,
kation znajduje się w położeniu
−
−
−
8
1
,
8
1
,
8
1
natomiast anion w
+
+
+
8
1
,
8
1
,
8
1
– takie typy
kryształów nazywane są strukturami siarczku cynku ZnS (ang. zinc blende). Jedynym
wyjątkiem jest tutaj GaN, oraz jego stop,
N
Ga
In
x
1
x
−
, które w ostatnich latach stały się
bardzo ważne z powodu zastosowań w produkcji „zielonych” i „niebieskich” diod
elektroluminescencyjnych oraz laserów – te materiały mają tzw. strukturę wurcytu (ang.
wurtzite strukture).
Z elektrostatycznego punkt widzenia, potencjał wewnątrz kryształu składa się
z trójwymiarowej siatki sferycznie symetrycznych potencjałów jąder atomowych oraz
elektronów (patrz rys. A.2), które są połączone wiązaniami kowalencyjnymi utrzymującymi
wszystko razem [3].
Rys. A.2. Schematyczna ilustracja potencjałów składających się na skomplikowaną strukturę wewnątrz
kryształu [3]. Ilustracja przedstawia trójwymiarową macierz sferycznie symetrycznych potencjałów na
płaszczyźnie [001].

36
Dodatek B. Elementy teorii pasmowej półprzewodników
Jak zostało wykazane doświadczalnie w strukturze energetycznej półprzewodników
wyróżnić można dwa zasadniczo różne pasma energii. Niższe pasmo, zwane walencyjnym,
jest całkowicie wypełnione elektronami. W paśmie tym związane ze sobą kowalencyjnie
atomy tworzą chmurę elektronów walencyjnych, które po dostarczeniu im dodatkowej energii
mogą przedostać się do wyższych stanów, z tzw. pasma przewodnictwa, pozostawiając po
sobie tzw. dziurę – ładunek dodatni. Pasmo walencyjne może przewodzić prąd przez ruch
tychże stanów pustych [3].
Wyższe pasmo jest natomiast całkowicie pozbawione elektronów w stanie
podstawowym (w idealnym modelu w temperaturze zera bezwzględnego). W warstwie tej
mogą
znaleźć
się
wzbudzone
elektrony,
które
poprzez
dodatkową
energie
(np. elektromagnetyczną lub cieplną) przedostały się z niższego pasma. Pasmo to nosi nazwę
pasma przewodnictwa.
W półprzewodnikach (w odróżnieniu od metali) pasma walencyjne i przewodnictwa
są oddzielone od siebie energią zwana przerwą energetyczną (ang. band gap). Dlatego
w niskich temperaturach wykazują bardzo dużą rezystancję.
Przedstawiony
tutaj
opis
struktury
energetycznej
niedomieszkowanych
półprzewodników jest wyidealizowanym modelem. W praktyce okazuje się, że w typowych
półprzewodnikach znajduje się więcej niż tylko jedno pasmo walencyjne, co powoduje, że
dziury wykazują różne wartości masy efektywnej w zależności od położenia (przybliżenie
masy efektywnej zostało przedstawione wcześniej w tej pracy).
37
Dodatek C. Opis programu Strzały
Program Strzały jest prostą w obsłudze aplikacją środowiska Windows®
umożliwiającą przeprowadzenie obliczeń energii oraz wektorów własnych dla wielu rodzajów
pojedynczych studni kwantowych. Dzięki przejrzystemu układowi graficznemu stanowi on
dobre narzędzie obliczeniowe umożliwiające natychmiastową prezentację wyników. Dodatek
ten stanowi opis oraz instrukcję obsługi programu. Program Strzały znajduję się na dołączonej
płytce CD.
Aby przeprowadzić obliczenia dla danej struktury, należy wykonać następujące kroki
opisane poniżej.
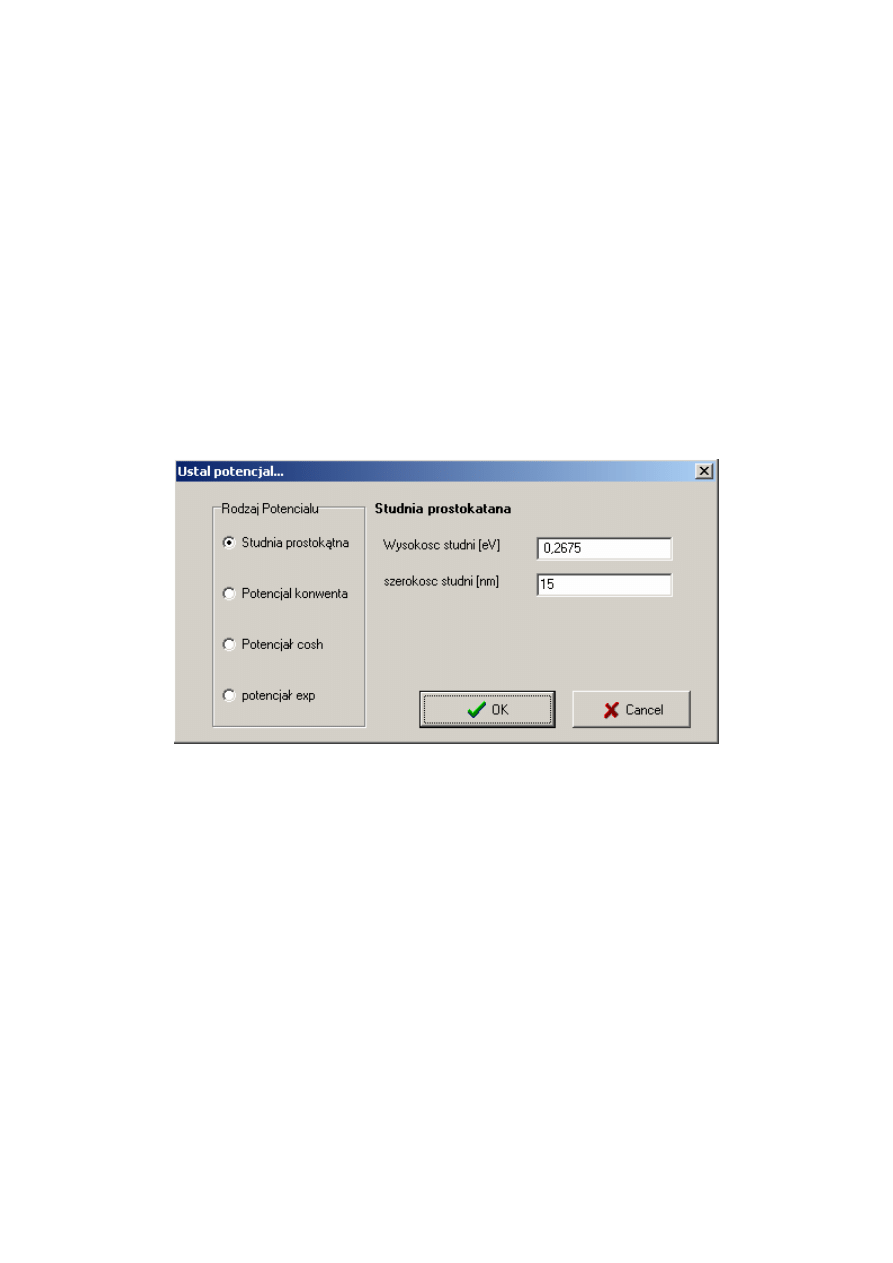
1. Wprowadzić postać rozkładu potencjału
Rozkład potencjału ustala się poprzez naciśnięcie przycisku ‘1 Ustal Potencjał’ oraz
odpowiednim wyborze dostępnych rozkładów potencjału.
Rys. C. 1. Okno dialogowe programu Strzały pozwalające na określenie rozkładu potencjału.
Jak widzimy na rys. C.1 mamy możliwość wyboru z pośród czterech dostępnych
rodzajów rozkładu potencjału. Po wyborze jednego z nich ukazuje się nam dokładniejszy jego
opis oraz miejsca, w które musimy wpisać odpowiednie parametry.
2. Wprowadzić postać rozkładu masy
Podobnie jak do ustalenia potencjału okno dialogowe pozwalające nam na określenie
rozkładu masy wywołuje się przyciskiem ‘2 Ustal Masę’.

38
Rys. C.2 Okno dialogowe programu Strzały pozwalające na określenie rozkładu masy.
Program oferuje nam (patrz rys C.2) cztery możliwe rozkłady masy. Po wyborze
odpowiedniego z nich należy wpisać niezbędne dla niego parametry.
3. Ustalić parametry obliczeniowe dla badanego zagadnienia
Rys. C.3. Część głównego okna programu Strzały pozwalająca wprowadzenie parametrów obliczeniowych.
Parametry takie jak: całkowita długość przedziału całkowania, ilość punktów siatki,
dokładność przeprowadzonych obliczeń oraz maksymalna energia (Emax), dla której mają
być wyszukiwane stany własne, wprowadza się w przypisane im pola (patrz rys. C.3).
Odpowiedni dobór parametrów jest bardzo ważną częścią każdego z obliczeń, bowiem
nieodpowiedni ich dobór jest częstą przyczyną błędnych obliczeń.
4. Zweryfikować rozkład masy oraz potencjału na zadanym przedziale całkowania
Krok ten nie jest konieczny do przeprowadzenia obliczeń, jednakże czynności
związane ze sprawdzeniem poprawności wprowadzonego potencjału oraz masy pomagają
w uniknięciu błędów obliczeniowych.

39
Rys. C. 4. Okno głównego menu wykresu programu Strzały.
W celu sprawdzenia wybranych rozkładów należy wybrać odpowiednie polecenie
z głównego menu wykresu (patrz rys. C.4) znajdującego się w górnej części okna programu.
Kształt potencjału zostanie przedstawiony na głównym wykresie programu (który można
usunąć wybierając odpowiednie polecenie w menu wykres – rys. C.4), co sprzyja wykreślaniu
obliczonych wektorów własnych, oraz jest niezbędne dla manualnej metody strzałów (opis
manualnej metody strzałów znajduje się w dalszych punktach). Wykres masy zostanie
przedstawiony w dodatkowym oknie. Sprawdzając wybrane rozkłady należy zwrócić
szczególną uwagę na to czy ich wartości nie są ujemne (dotyczy to potencjałów różnych od
prostokątnej studni potencjału – rys. C.1). Program jest tak skonstruowany, że prowadzi
obliczenia tylko dla potencjałów o wartościach dodatnich.
5. Rozpocząć obliczenia poprzedzone wyborem odpowiedniego algorytmu – domyślnym
jest algorytm ulepszonej metody strzałów
Rys. C.5. Część głównego okna programu Strzały pozwalająca określenie algorytmu obliczeń.
Jak pokazano na rys. C.5 mamy do wyboru trzy metody: ulepszona metoda strzałów,
metoda macierzowa Martina-Deana oraz tzw. manualną metodę strzałów. Pierwsze dwie
służą do obliczeń wszystkich stanów własnych rozpatrywanej jamy potencjału niższych od
zadanej energii maksymalnej (Emax). Wyniki tych metod zostają wypisane w miejscu
przedstawionym na rys. C.7. Metoda manualna służy do samodzielnego wyznaczenia energii
własnej poprzez „strzelanie” wartościami energii, dla których wykreślony zostaje wektor
własny. Wartości te można wybrać na dwa sposoby. Pierwszy z nich to wpisanie
odpowiedniej wartości w pole przypisane energii próbnej oraz zatwierdzenie jej przyciskiem
‘5 LICZ’ (patrz rys. C.6). Drugim sposobem jest pobranie wartości z wykresu klikając
w wybrany jego punkt (na wykresie tym musi znajdować się wykres potencjału). „Strzelana”
przez nas w ten sposób wartość energii zostaje również wpisana w pole poświęcone
manualnej metodzie strzałów (patrz rys. C. 6). Jak zostało powiedziane w 3 rozdziale pracy
szukana wartość energii to ta, dla której wartości wektora własnego dążą do zera na granicy
przedziału.
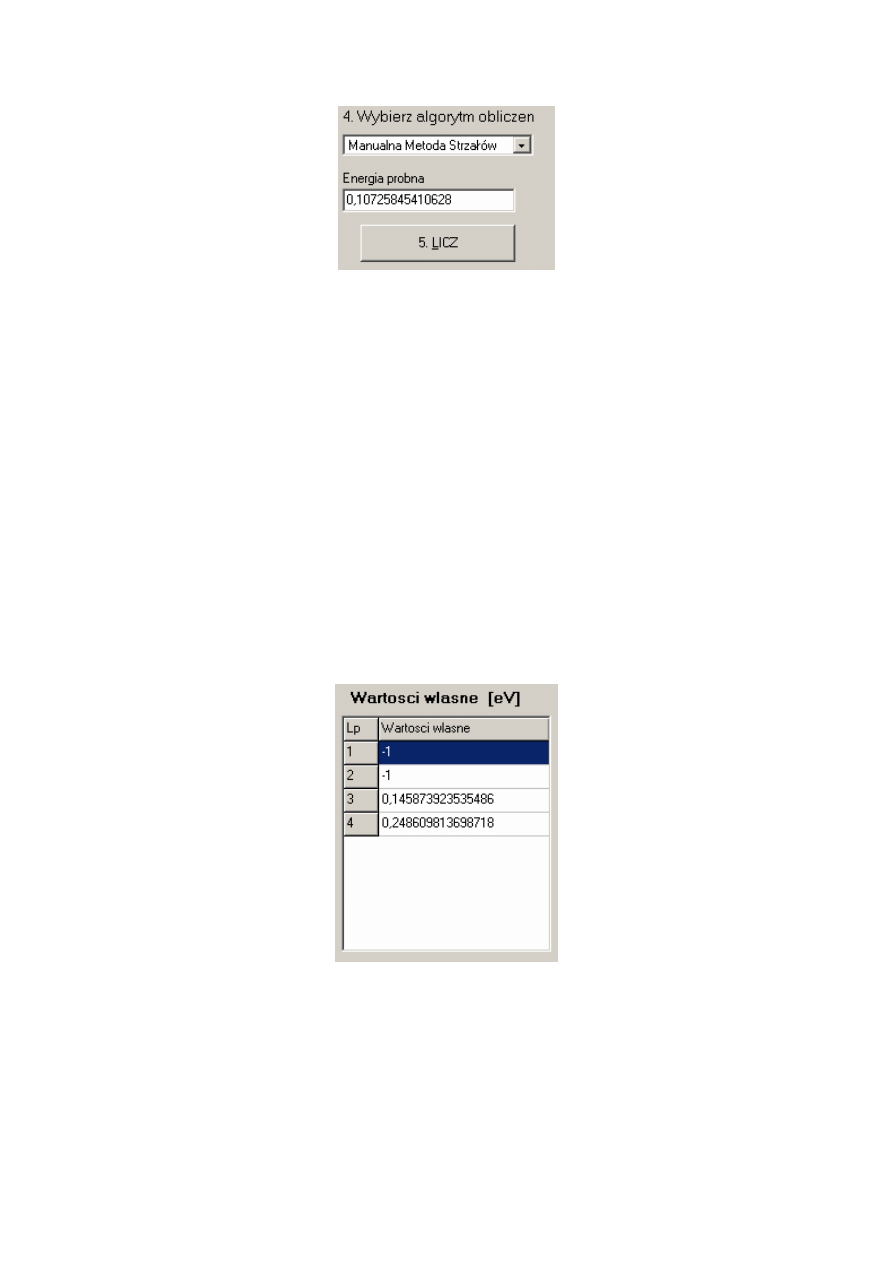
40
Rys. C. 6. Wygląd części głównego okna programu Strzały poświęconej manualnej metodzie strzałów.
6. Wyznaczyć wektory własne
Po przeprowadzeniu obliczeń metodą inną niż manualna metoda strzałów obliczone
energie własne zostają wypisane w okienku poniżej jak pokazano na rys. C.7. Aby wyznaczyć
wektor własny odpowiadający wybranej przez nas wartości energii wystarczy wybrać ją
myszką. Po kliknięciu na interesującą nas wartość energii zostanie wykreślony na głównym
wykresie odpowiadający jej wektor własny (może on zostać usunięty z wykresu przez wybór
odpowiedniego polecenia z głównego menu wykresu – patrz rys.C.4).
Wektory własne w metodzie macierzowej wyznaczane są algorytmem rekurencyjnym
DWSZ opisanym w [2].
Jak zostało pokazane na rys. C.7. program zwraca czasem energie własne równe
wartości –1. Oznacza to, że pomimo znalezienia przedziału zawierającego energię, nie została
ona dokładnie obliczona. Dzieje się tak z powodu złego doboru danych obliczeniowych (patrz
rys. C.3). Najczęściej przyczyną jest zadanie zbyt dużej dokładności obliczeń, za mała liczba
punktów siatki bądź nieodpowiednio dobrany przedział całkowania [3].
Rys. C.7. Część głównego okna programu Strzały poświęcona prezentacji wyników.

41
Dodatek
D.
Parametry
u
ż
ytego
w
obliczeniach
materiału
półprzewodnikowego
Poniżej
przedstawione
są
wszystkie
niezbędne
parametry
struktury
półprzewodnikowej
As
Al
Ga
GaAs
x
x
−
1
/
jakie potrzebne są do przeprowadzenia obliczeń
numerycznych (zarówno programem opisanym w Dodatku C jak i programami opisanymi
w Dodatku E). Parametry te zostały zaczerpnięte z [3].
••••
Przerwa energetyczna,
(
)
]
[
247
,
1
426
,
1
eV
x
E
g
+
=
••••
Przyjęte rozłożenie przerwy energetycznej przypadającej na pasmo przewodnictwa to
67% – co oznacza, że jeśli przerwa energetyczna wynosi 1 eV, to głębokość studni
w paśmie przewodnictwa wynosić będzie 0,67 eV natomiast głębokość studni pasma
walencyjnego wynosić będzie odpowiednio 0,33 eV.
••••
Masa efektywna elektronu,
(
)
0
*
083
,
0
067
,
0
m
x
m
+
=
••••
Masa efektywna dziury,
(
)
0
*
14
,
0
62
,
0
m
x
m
+
=
42
Dodatek E. Programy
ź
ródłowe
Dołączona do pracy płytka CD zawiera kody źródłowe wszystkich użytych w pracy
podprogramów napisanych w języku FORTRAN 77, realizujących algorytmy rozwiązywania
równania Schrödingera omówione w rozdziale 3 oraz dodatkowo metodę macierzową
Martina-Deana opisaną w [3]. Na płycie znajdują się również przykładowe kody programów
umożliwiające przetestowanie poszczególnych algorytmów.
Tabela E.1 zawiera opis wszystkich podprogramów oraz programów z krótkim opisem
ich działania.
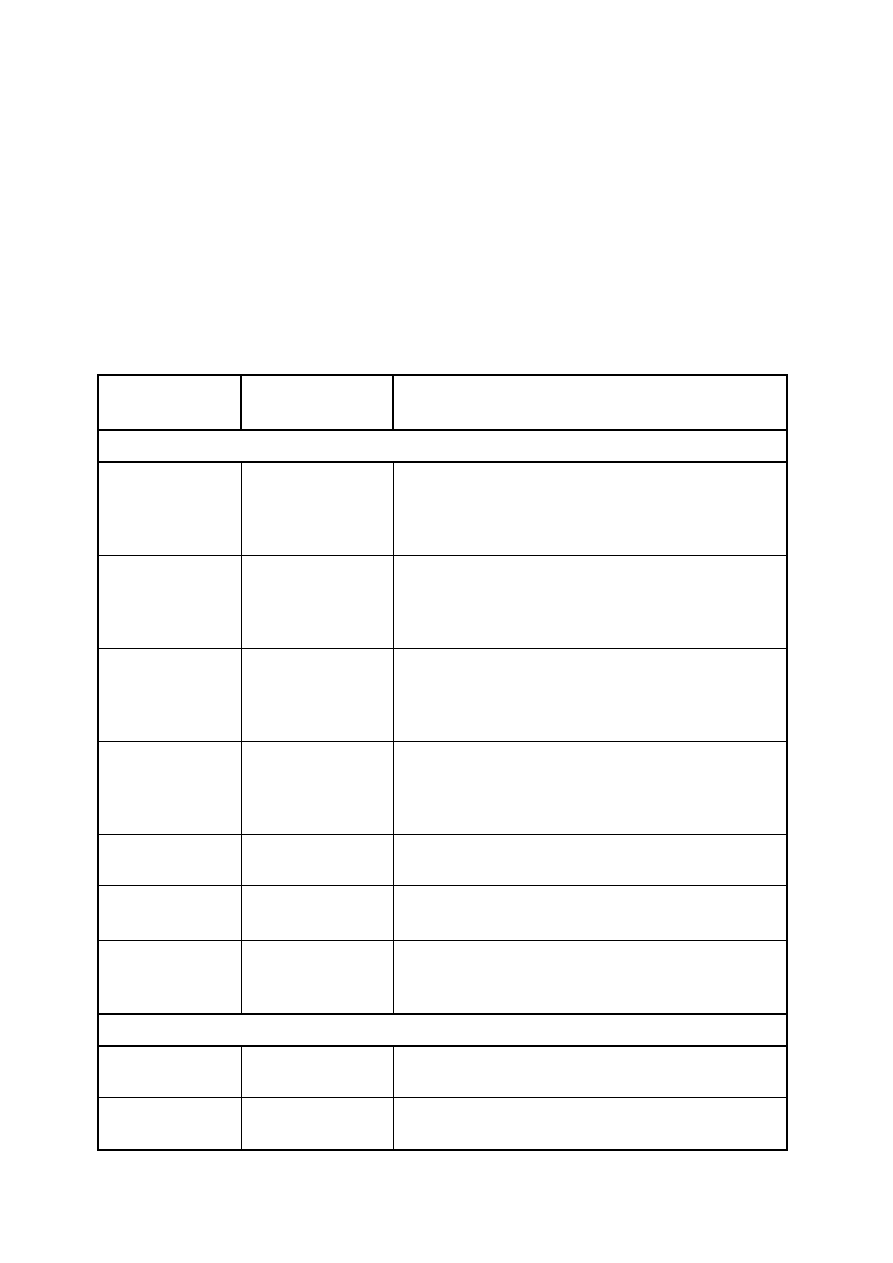
Tabela E.1 Podprogramy dołączone na płycie CD
Nazwa
podprogramu
Plik zawierający
Opis
Ulepszona Metoda Strzałów
shootP
fStrzalow.f
funkcja zwracająca wartość wektora własnego na
końcu przedziału całkowania dla zadanej energii
odwzorowaniem wstępującym – używana w
podprogramie solution
shootL
fStrzalow.f
funkcja zwracająca wartość wektora własnego na
końcu przedziału całkowania dla zadanej energii
odwzorowaniem
zstępującym
–
używana
w
podprogramie solution
shootPV
fStrzalowV.f
procedura
obliczająca
wszystkie
wartości
współrzędnych wektora własnego dla zadanej
energii odwzorowaniem wstępującym – używana do
wyznaczania wektorów własnych
shootLV
fStrzalowV.f
procedura
obliczająca
wszystkie
wartości
współrzędnych wektora własnego dla zadanej
energii odwzorowaniem zstępującym – używana do
wyznaczania wektorów własnych
IleP
fIleP.f
funkcja
wyznaczająca
przedziały
zawierające
pojedynczą energię własną
solution
fbisec.f
funkcja znajdująca wartości własne w zadanym
przedziale (metoda bisekcji)
UlepszonaMS
UlepszonaMS.f
przykładowy program wyznaczający energie własne
oraz wektory własne dla pojedynczej studni
kwantowej lub supersieci ulepszoną metodą strzałów
Metoda Martina-Deana [3]
IlePMD
fIlePMD.f
funkcja
wyznaczająca
przedziały
zawierające
pojedynczą energię własną
M_D
fMD.f
funkcja znajdująca wartości własne w zadanym
przedziale (metoda bisekcji)
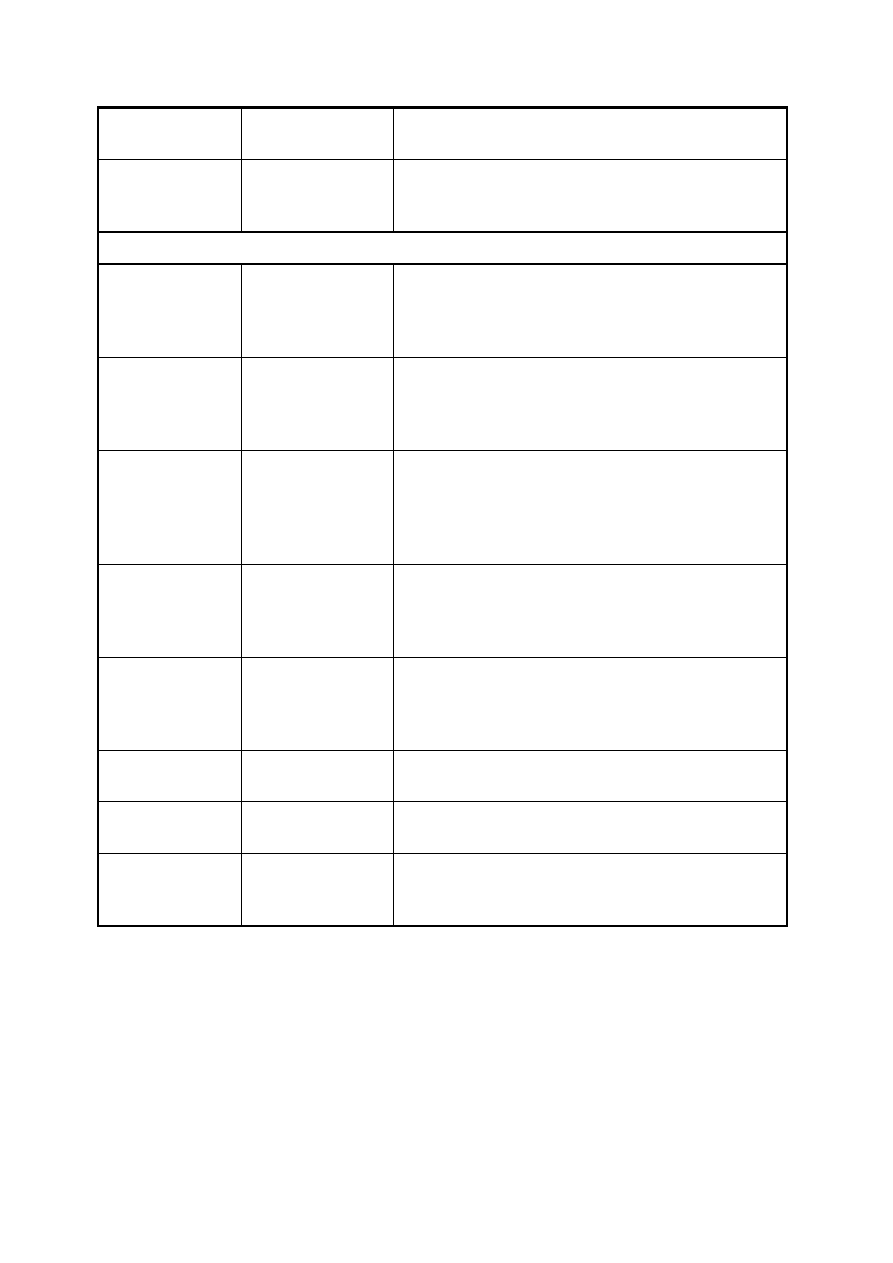
43
DWSZ
DWSZ.f
procedura wyznaczająca wektory własne metodą
DWSZ
Przykład_MD
MD.f
przykładowy program wyznaczający energie własne
oraz wektory falowe dla pojedynczej studni
kwantowej metodą Martina-Deana
Prosta Metoda Strzałów (PMS)
strzalPK
fstrzalow_PMS.f
funkcja zwracająca wartość współrzędnej wektora
własnego dla zadanej energii w zadanym punkcie
odwzorowaniem wstępującym – używana w
podprogramie solution_PMS
strzalLK
fstrzalow_PMS.f
funkcja zwracająca wartość współrzędnej wektora
własnego dla zadanej energii w zadanym punkcie
odwzorowaniem zstępującym – używana w
podprogramie solution_PMS
strzalLKP
fstrzalow_PMS.f
funkcja zwracająca wartość współrzędnej wektora
własnego dla zadanej energii w zadanym punkcie
odwzorowaniem
zstępującym
dla
stanów
parzystych
–
używana
w
podprogramie
solution_PMS
strzalP
fstrzalowV_PMS.f
procedura
obliczająca
wszystkie
współrzędne
wektora
własnego
dla
zadanej
energii
odwzorowaniem wstępującym – używana do
wyznaczania wektorów własnych
strzalL
fstrzalowV_PMS.f
procedura
obliczająca
wszystkie
współrzędne
wektora
własnego
dla
zadanej
energii
odwzorowaniem zstępującym – używana do
wyznaczania wektorów własnych
IleP_PMS
fIleP_PMS.f
funkcja
wyznaczająca
przedziały
zawierające
pojedynczą energię własną
wynik_PMS
Fbisec_PMS.f
funkcja znajdująca wartości własne w zadanym
przedziale (metoda bisekcji)
PMS
PMS.f
przykładowy program wyznaczający energie własne
oraz wektory falowe pojedynczej studni kwantowej
prostą metodą strzałów
Szczegółowe opisy przedstawionych programów oraz podprogramów zawarte są
w komentarzach wewnątrz kodów źródłowych.
Poniżej zamieszczone zostały kody źródłowe najważniejszych podprogramów wraz
z opisem, realizujących opisany w rozdziale 3 algorytm ulepszonej metody strzałów [4].
Podprogram shootP jest funkcją zwracającą ostatni obliczony punkt funkcji falowej na
zadanym przedziale, dla podanej wartości energii. Jest to funkcja napisana w celu
przyspieszenia obliczeń – nie potrzebuje rezerwacji miejsca na wszystkie punkty funkcji

44
falowej. Posługując się dwiema lokalnymi zmiennymi wyznacza tylko potrzebny nam ostatni
punkt w celu odnalezienia energii własnej. Wykorzystywana jest ona w podprogramie
solution, który wyznacza energię dla której końcowa wartość funkcji falowej (na końcu
przedziału całkowania) przyjmuję wartość zero z zadaną dokładnością.
C funkcja strzalu, wstepujaca zwracajaca ostatni obliczony punkt
C wektora falowego - przydatna tylko do wyznaczenia energi wlasnej
Double precision Function shootP(krok,xca,k1,k2,m,V,E,wym)
C DP - krok -dlugosc kroku calkowania
C DP - xca - szerokosc przedzialu calkownaia (symetryczny)
C DP - k1 - wspolczynniki
C DP - k2
C Dp - m - tablica o rozmiarze wym - zawierajaca rozklad masy
C DP - V - tablica o rozmiarze wym - zawierajaca rozklad potencjalu
C DP - E - energia probna
C Integer - wym - rozmiar tablicy wym
Implicit None
C zmienne robocze i lokalne
Integer i, wym
Double precision krok,E,k1,k2,x
Double precision xca, V(wym), m(wym)
Double precision Psi2,PsiD
! warunki poczatkowe
shootP=0d0
PsiD= 1d0
do i=2, wym+1
Psi2 = shootP ! potrzbujemy wartosci Psi 2 razy
shootP = shootP +(k1*krok*m(i)*PsiD)
PsiD = PsiD +(k2*krok*(V(i-1)-E) * Psi2)
end do
end ! koniec funkcji shootP!
Na płycie znajduje się również analogiczna do przedstawionej funkcja zstępująca (shootL),
która przeprowadza obliczenia od końcowego punktu przedziału całkowania do
początkowego.
Następnym ważnym podprogramem jest wspomniany wcześniej solution. Jest to
funkcja, która poszukuje (na zadanym przedziale) takiej wartości energii dla której
przekazana jej funkcja (shootP lub shootL) zwróci punkt równy zeru z zdaną dokładnością.
Funkcja ta wykorzystuje algorytm bisekcji w celu wyznaczenia energii własnej.
Double precision Function solution (fShoot,
!wzsystko niezbedne do funkcji strzalu
& krok,xca,k1,k2,m,V,E,wym,
!---------------------------------------------
& Ep,Ek,dok)
C DP funkcja - fShoot ktora funkcja strzalu - prawa lub lewa
C-------wszystko potrzebne do fShoot
C DP - krok -dlugosc kroku
C DP - xca - szerokosc przedzialu calkownaia (symetryczny)
C DP - k1 - wspolczynnik
C DP - k2 - wspolczynnik
C Dp - m - tablica o rozmiarze wym - zawierajaca rozklad masyC
C DP - V - tablica z potencjalem.
C DP - E - energia probna
C Integer - wym - rozmiar tablicy V oraz m
C--------------------------------------------
C DP - Ep,Ek - poczatek, koniec przedzilau energi
C DP - dok - dokladnosc szukania miejsca zerowego
Implicit none
C zmienne robocze i lokalne
character PlubL
integer wym,i,j
Double precision krok,E,k1,k2,x ,fShoot
Double precision xca, A,Ep,Ek,dok,tempP,tempK,tempS
Double precision EtempS ,EtempK,EtempP, V(wym), m(wym)

45
External fShoot
EtempK=Ek
EtempP=Ep
j=0
343 continue
EtempS = ((EtempK+EtempP)*5d-1)
tempP =fShoot(krok,xca,k1,k2,m,V,EtempP,wym)
tempK = fShoot(krok,xca,k1,k2,m,V,EtempK,wym)
tempS =fShoot(krok,xca,k1,k2,m,V,EtempS,wym)
j=j+1 ! obliczenie ilosci wywolan
if (j.EQ.2000) then ! kontrola przed zapetleniem sie programu
Solution = EtempS
write(*,*) 'zadana dokladnosc nie zostala osiagnieta'
write(*,*) 'aby osiagnac zadana dokladnosc nalezy zwiekszys
& ilosc punktow siatki,badz zmniejszyc dokladnosc'
elseif (ABS(tempS).LE.dok) then
solution = EtempS
elseif((tempP*tempS).LT.0d0) then
EtempK = EtempS
goto 343
elseif((tempS*tempK).LT.0d0) then
EtempP = EtempS
goto 343
else
write(*,*) 'nie ma rozwiazan na zadanym przedziale'
solution = 0d0
endif
END
! koniec funkcji solution
Funkcja ta realizuję przypisane jej zadanie pod warunkiem, że zadany jej przedział
zawiera dokładnie jedno rozwiązanie. Dlatego konieczny jest dodatkowy podprogram
wyszukujący przedziały zawierające dokładnie jedną energię własną.
W tym celu napisany został podprogram IleP, który zadany mu przedział dzieli
dodatkowo (z podanym krokiem) na mniejsze przedziały i wyszukuje takich dla których
wykorzystywana funkcja (shootP lub shootL) posiada miejsce zerowe. Przedział zawiera
energię własną jeśli dla dwóch wartości końców badanego przedziału ostatni punkt wektora
falowego przyjmuje różne znaki. Podprogram ten jest funkcją, która pod swą nazwa zwraca
ilość znalezionych przedziałów a w przekazanej tablicy (przedzialy) zwraca znalezione
przedziały (dwie następujące po sobie liczby w tablicy określają odpowiednio początek
i koniec przedziału).
Integer function IleP (fShoot,tempKr,
!wzsystko niezbedne do funkcji fShoot
& krok,xca,k1,k2,m,V,Ep,Ek,wym,
!---------------------------------------------
& przedzialy)
C DP funkcja - fShoot - funkcjia obliczjaca ostatni punkt wektora falowego
C (moze byc przekazana zarowno funkcjia shooP jak i shooL
C-------wszystko potrzebne do fShoot
C DP - krok -dlugosc kroku
C DP - xca - szerokosc przedzialu calkownaia (symetryczny)
C DP - k1 - wspolczynnik
C DP - k2 - wspolczynnik
C Dp - m - tablica o rozmiarze wym - zawierajaca rozklad masy
C DP - V - tablica o rozmiarze wym - zawierajaca rozklad potencjalu
C DP - Ep - pocz
Ą
tek przeszukiwanego przedzialu energi
C DP - Ep - pocz
Ą
tek przeszukiwanego przedzialu energi
C Integer - wym - rozmiar tablicy V i m, oraz przedzialy
C--------------------------------------------

46
C przedzialy - tablica ktora zawiera obliczone przedzialy
C kolejno - xp1,xk1,xp2,xk2......
Implicit none
C zmienne robocze i lokalne
integer wym,pk,i,j
Double precision krok,Ep,Ek,k1,k2,wynik(wym+1),x, przedzialy(wym)
Double precision xca, TempKr, TempP,TempK, V(wym),m(wym)
Double precision fShoot
external fShoot
!jesli nie jest zadeklarowane przyjmij warotsc domyslna
if(TempKr.Eq.0) then
TempKr = (Ek-Ep)/1d2 !krok
endif
x=Ep
i=1
!jesli funkcjia fShoot wywolana dla wartosc x zwraca wynik o przeciwnym znaku do
!wyniku gdy funkcja fShoot wywolana jest dla (x+TempKr)
! wtedy przedzial stanowia liczby x i x+TempKr
678 CONTINUE
if(x.LT.Ek) then
TempP=fShoot(krok,xca,k1,k2,m,V,x,wym)
TempK=fShoot(krok,xca,k1,k2,m,V,x+TempKr,wym)
if(TempP*TempK.LT.0d0) then
przedzialy(i) = x
przedzialy(i+1)= x+TempKr
i=i+2
C mamy przedzial (x, x+TempKr)
endif
x=x+TempKr
goto 678
endif
IleP = int(i/2) ! zwrocenie ilosci znalezionych przedzialow
END ! koniec funkci IleP
Do wyznaczenia wektorów własnych służą funkcje shootPV oraz shootLV, które
obliczają wszystkie punkty wektora odpowiadające przekazanej im energii. Funkcje shootPV
oraz shootLV to odpowiednio funkcja wstępująca i zstępująca. Poniżej przedstawiamy kod
ź
ródłowy tylko jednej z nich – wszystkie użyte, lecz nie zamieszczone tutaj kody źródłowe
znajdują się na dołączonej płycie CD.
C procedura strzalow (wstepujaca) do wyznaczenia wektora falowego dla zadanej Energi
Subroutine shootPV(krok,xca,k1,k2,m,V,E,wynik,wym)
C DP - krok -dlugosc kroku
C DP - xca - szerokosc przedzialu calkownaia (symetryczny)
C DP - k1 - wspolczynnik
C DP - k2 - wspolczynnik
C Dp - m - tablica o rozmiarze wym - zawierajaca rozklad masy
C DP - V - tablica o rozmiarze wym - zawierajaca rozklad potencjalu
C DP - E - energia probna
C tablica DP - wynik - tablica o wymiarze wym - zawiera punkty wektora wlasnego
C Integer - wym - rozmiar tablicy
Implicit None
C zmienne robocze i lokalne
Integer i, wym
Double precision krok,E,k1,k2,wynik(wym),x
Double precision xca, V(wym), m(wym)
Double precision PsiD ,suma
Dimension PsiD(wym)
! warunki poczatkowe
wynik(1)=0d0
PsiD(1)= 1d0
suma=0d0

47
do i=2, wym+1
x=(-xca*5d-1)+((i-1)*krok)
wynik(i) = wynik(i-1) +(k1*krok*m(i)*PsiD(i-1))
PsiD(i)= PsiD(i-1) +(k2*krok*(V(i-1)-E)*wynik(i-1))
suma = suma +(Wynik(i)*wynik(i))
end do
!normowanie wektora
suma = sqrt(suma)
do i=1, wym+1
wynik(i) = wynik(i)/suma
enddo
end ! koniec procedury shootPV!
end ! koniec procedury shootPV!
Procedura ta zwraca znormalizowaną funkcję falową wewnątrz przekazanej tablicy
wynik.
Wykorzystując przedstawione wyżej procedury numeryczne możemy wyznaczyć
zarówno energie własne jak również odpowiadające im wektory własne dla dowolnie
skonstruowanej heterostruktury przedstawionej jako rozkład potencjału oraz masy (tablice
zawierające wartości potencjału oraz masy na zadanym przedziale są parametrami
wejściowymi funkcji shootP oraz shootL). Efekt wykorzystania opisanych procedur został
krótko przedstawiony w treści pracy (rozdział 4).
Poniżej znajduje się kod źródłowy przykładowego programu (znajdującego się w pliku
UlepszonaMs.f) wykorzystującego wszystkie z zaprezentowanych podprogramów. Program
umożliwia przeprowadzenie obliczeń wartości własnych energii oraz wektorów własnych jak
również pomiar czasu obliczeń, dla pojedynczej studni kwantowej lub supersieci,
ograniczając się tylko to skokowego rozkładu potencjału jak i masy (co może być zmienione
na rozkład ciągły przez wprowadzenie niewielkich zmian w programie). Wynikiem
omawianego programu, oprócz wyświetlenia wyznaczonych energii własnych oraz
potrzebnego czasu obliczeń na ekranie są cztery pliki zawierające bezwymiarowe wartości:
••••
v.txt – zawiera rozkład potencjału na zadanym przedziale całkowania – plik zawiera
dwie kolumny, pierwsza z nich to współrzędna położenia (x), druga to wartość
potencjału w danym punkcie ( V(x) ),
••••
m.txt – zawiera rozkład masy na zadanym przedziale całkowania – plik zawiera dwie
kolumny, pierwsza z nich to współrzędna położenia (x), druga to wartość masy
w danym punkcie ( m(x) ),
••••
E.txt – zawiera obliczone energie własne – plik zawiera trzy kolumny, pierwsza to
liczba porządkowa wyznaczonej energii, druga to wartość energii obliczona procedurą
wstępującą, trzecia to wartość energii obliczona procedurą zstępującą,
••••
wektor.txt – zawiera punkty wektorów falowych rozpatrywanej struktury – plik
zawiera trzy kolumny, pierwsza to współrzędna położenia (x), druga to wartość
wektora falowego w danym punkcie obliczona procedurą wstępującą
( )
x
ψ
r
, trzecia to
wartość wektora falowego w danym punkcie obliczona procedurą zstępującą
( )
x
ψ
s
.
Pliki skonstruowane są w sposób łatwy do sporządzenia wykresów np. darmowym
programem Gnuplot, który można bezpłatnie pobrać z [11], bądź do importu przez wiele
innych programów takich jak np. Microsoft Exel.

48
C Przykladowy program umozliwjajacy obliczenie wartosci wlasnych energii
C oraz odpowiadajacych im wektorom falowym dla pojedynczych studni kwantowych
C badz supersieci
include 'fstrzalow.f'
include 'fstrzalowV.f'
include 'fbisec.f'
include 'fIleP.f'
Program Program_shoot
Implicit none
C zmienne
Integer wym,N, i,j,temp,l, IleP,ileS
Double precision V,Vdane,V0,x,xca,szerokosc,krok ,przedzialy
Double precision m, Eprob,hkr,L0,Ep,Ek,krokE,dok
Double precision VfalP,VfalL, fm,k1,k2, solution
double precision TempDPP,TempDPL, VTab, MTab ,mz,mw,d
double precision shootP,shootL
************ Potrzebne do pomiaru czasu********************************
real Atime, time ,etime
dimension Atime(2)
external etime
C Parametry
Parameter (N=200000) ! maksymalny rozmiar tablicy
Parameter (hkr = 1.0545726663d-34)! stala Diraca
Parameter (m = 9.1093d-31, V0=1.602176d-19)! jednostka masy oraz jednostka energi [1eV]
Parameter (L0 = 1d-9) !jednostka dlugosci [1nm]
C Rozmiary tablic
Dimension VfalP(N+1),VfalL(N+1), przedzialy(N), Vtab(N+1)
Dimension MTab(N+1)
External V, fm ,solution,IleP , shootP,shootL,shootPV,shootLV
C stworzenie plikow wyjsciowych
Open(10,File='v.txt') !plik zawierajacy rozklad potencjalu
Open(20,File='m.txt') !plik zawirejacy rozklad masy
Open(30,File='wektor.txt')!plik zawierajacy obliczone wektory wlasne
OPEN(50,File='E.txt') !plik zawierajacy obliczone energie wlasne
C oblicznie wspolczynnikow dla bezwymiarowego row. Schrodingera
k1=L0*m *1d40
k2=L0*2*(1/(hkr**2))*V0 *1d-40
!write(*,*) 'k1*k2= ', k1*k2
Write(*,*)'******************************************************'
Write(*,*)'* Przykladowy program umozliwjajacy obliczenie *'
Write(*,*)'* wartosci wlasne energii oraz odpowiadajace im *'
Write(*,*)'* wektory falowe dla pojedynczych studni kwantowych *'
Write(*,*)'* badz supersieci *'
Write(*,*)'*----------------------------------------------------*'
Write(*,*)'* wynikiem progrmu sa cztery pliki: *'
Write(*,*)'* v.txt - zawiera rozklad potencjalu *'
Write(*,*)'* m.txt - zawiera rozklad masy *'
Write(*,*)'* wektor.txt - zawiera obliczone wektory wlasne *'
Write(*,*)'* powiekszone o odpowiadajaca im energie *'
Write(*,*)'* E.txt - zawieraja obliczone energie wlasnie *'
Write(*,*)'******************************************************'
Write(*,*)
C Wprowadzenie danych
write(*,*) 'Podaj Wysokosc studni [eV]'
read(*,*) Vdane
write(*,*) 'Podaj calkowita szerokosc studni [nm]'
read(*,*) szerokosc
write(*,*) 'Podaj mase efektywna na zewnatrz studni [m0]'
read(*,*) mz
write(*,*) 'Podaj mase efektywna wewnatrz studni [m0]'
read(*,*) mw
write(*,*) 'Podaj ilosc studni '
read (*,*) ileS
if(ileS.GT.1)then
write(*,*)'Podaj odstepy miedzy studniami [nm]'
read(*,*) d
else
d=0d0
endif
write(*,*) 'Podaj szerokosc calkowania [nm] - min ',
& (szerokosc*ileS)+(d*(ileS-1))
read(*,*) xca
write(*,*)'Podaj dolna granice wyszukiwania wartosci wlasnych [eV
&]'
read(*,*) Ep
write(*,*)'Podaj gorna granice wyszukiwania wartosci wlasnych [eV
&]'
read(*,*) Ek
write(*,*) 'Podaj krok wyszukiwania przedzialow energii [eV]'
read (*,*) krokE
write(*,*) 'Podaj ilosc punktow siatki'
read(*,*) wym
write(*,*) 'Podaj dokladnosc wyszukiwania wartosci wlasnych'
read(*,*) dok
C Wywolanie funckji implementujacej tablice z potencjalem jak i masa

49
call ImplementVM(V,Vdane,szerokosc,fM,mz,mw,d,ileS,xca,VTab,MTab,
& wym)
C Obliczenie kroku calkowania
krok=xca/wym
C Zapis do pliku rozkladu masy jak i potencjalu
do i=1,wym+1
x=(-xca/2d0)+((i-1)*krok) !przesuniecie o 1
write(10,*)x, VTab(i)
write(20,*) x, Mtab(i)
enddo
C wywolanie procedury obliczajacej przedzialy warosc energi wlasnej
temp = IleP(shootP,krokE,krok,xca,
& k1,k2,MTab,VTab,Ep,Ek,wym, przedzialy)
write(*,*) 'ilosc znalezionych przdzialow = ', temp
C Petala w ktorej wyznaczone beda doklade wartosci enrgii wlasnej oraz wektory wlasne
do i =1, temp !znajdz wszystkie energie
j=2*i-1
time=etime(Atime) ! pomiar czasu
C Wywolanie funckji znajdujacej energie wlasna z zadana dokladnoscia na
C przedziale zawierajacym tylko jedna energie wlasna, z wykorzystaniem
C funkcji wstepujacej
TempDPP = solution(shootP,krok,xca,
& k1,k2,MTab,VTab,Eprob,wym,
& przedzialy(j) ,przedzialy(j+1),dok)
time = ABS(time - etime(Atime)) ! pomiar czasu
WRITE(*,*)'obliczona energia z uzyciem fuckji wstepujacej = ',
& TempDPP, ' potrzebny czas = ', time
time=etime(Atime) ! pomiar czasu
C Wywolanie funckji znajdujacej energie wlasna z zadana dokladnoscia na
C przedziale zawierajacym tylko jedna energie wlasna, z wykorzystaniem
C funkcji zstepujacej
TempDPL = solution(shootL,krok,xca,
& k1,k2,Mtab,VTab,Eprob,wym,
& przedzialy(j) ,przedzialy(j+1),dok)
time = ABS(time - etime(Atime)) ! pomiar czasu
WRITE(*,*)'obliczona energia z uzyciem fuckji zstepujacej = ',
& TempDPL, ' potrzebny czas = ', time
C zapis wartosci do pliku
write(50,*) i,TempDPP, TempDPL
write(*,*)'************************************'
C Wywolanie procedury obliczajacej vektory wlasne dla obliczonej energi wlasnej
C Z uzyciem procedury wstepujacej
Call shootPV(krok,xca,k1,k2,MTab,VTab,TempDPP,VfalP,wym)
C Z uzyciem procedury zstepujacej
Call shootLV(krok,xca,k1,k2,MTab,VTab,TempDPL,VfalL,wym)
C Zapis wektorow do pliku
write(30,*)
do l=1,wym +1
x=(-xca/2d0)+((l-1)*krok)
write(30,*) x, VfalP(l) +TempDPP,VfalL(l) +TempDPL
enddo
enddo
C Zamkniecie pliku
close(10)
close(20)
close(30)
close(50)
Write(*,*)'KONIEC PROGRAMU!!!'
END ! Koniec programu
*************FUNKCJIE I PROCEDURY***************
C Funkcjia tabliconujaca potencjal oraz mase na zadanym przedziale
C dla zadanej ilosci kwadratowych studni
Subroutine ImplementVM(fV,V0,a,fM,mz,mw,d,N,xca,TabV,TabM,wym)
C fV - funkcja DP - funkcja zwracjaca potencjal w damym punkcjie
C V0 - DP - wysokosc studni
C a - DP - szerokosc pojedynczej studni
C fV - funkcjia DP -funkcja zwracjaca mase w damym punkcjie
C mz,mw -DP - odpowiednio masa efektywna na zewnatrz i wewnatrz studni
C d -DP - odleglosci pomiedzy odpowiednimi studniami
C N - integer - ilosc studni
C xca -DP- dlugosc symetrycznego przedzialu na ktorym maja byc zaimplementowane masa i potencjal
C TabV - Tablica DP - tablica zwracajaca punkty rozkaldu potencjalu
C TabM - Tablica DP - tablica zwracajaca punkty rozkaldu masy
C wym - integer - wymiar tablic
Implicit none
integer N,j,k,wym,i,index
Double precision V0, x,xg,srodek,a, d,mz,mw,xca
Double precision TabV(wym+1),TabM(wym+1), krok,fV,fM
external fV, fM
srodek= ((N*a) + ((N-1)*d))/2d0;

50
krok = xca/wym
xg =-Xca*0.5 !//- srodek;
i=0
index =1
123 if (xG .le.( Xca*0.5 ) ) then
if (xg .lt. -srodek) then
!poza pierwsza studnia
TabV(index)=fV(-a,V0,a)
TabM(index) = fM(-a,mz,mw,a)
else if (( xg .ge. -srodek).AND.(xg.le. srodek)) then !//jestesmy na paczatku pierwszej studni
do j= 1,N !//ile studni
do k = 0 ,((a+ d)/krok)
x = (-a*5d-1) + k*krok
TabV(index)=fV(x,V0,a)
tabM(index) = fM(x,mz,mw,a)
index=index+1
xG= xG+ krok
enddo
enddo
else if (xg.gt. srodek ) then
!//poza ostatnia studnia
TabV(index-1)= v0!fV(a,V0,a)
tabM(index-1)=mz!fM(a,mz,mw,a)
endif !koniec ifa z 343
xG = xG + krok
index=index+1
goto 123
endif
TabV(index-1)=v0
tabM(index-1)=mz
TabV(index)=v0
tabM(index)=mz
end !koniec procedury implement
C pomocnicza Funkcja zwracajaca potencjal w danym punkcie - potrzebana w ImplementVM
Double precision Function V(x,V0,a)
C x - DP - miejsce w ktorym ma byc zwracany potencjal
C V0 - DP - glebokosc studni
C a - DP - szerokosc studni - symetrycznie
Implicit none
Double precision V0, x, a !a- szerokosc studni
If(x.LT.(-a/2)) then
V = V0
else if ( (x.GE.(-a/2)).AND.(x.LE.(a/2))) then
V = 0d0
else if (x.GT.(a/2)) then
V = V0
endif
end !koniec funkcji V
C pomocnicza Funkcja zwracajaca mase w danym punkcie - potrzebana w ImplementVM
Double precision Function fm (x,mz,mw,a)
C x - DP - miejsce w ktorym ma byc zwracany potencjal
C xw - DP - stosunek do masy wewnatrz studni
C mz - DP - stosunek do masy elektronu poza studnia
C a - DP - szerokosc studni - symetrycznie
Double precision x,mw,mz,a
If ( (x.LT.(-a/2)) .OR. (x.GT.(a/2))) then
fm = mz
else if ( (x.GE.(-a/2)).AND.(x.LE.(a/2))) then
fm = mw
endif
end ! koniec funkcji fm
.
Wyszukiwarka
Podobne podstrony:
Środowisko programowe do symulacji zjawiska tunelowania
AOL2, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń
A4, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń P
tab lam, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do oblic
Rysunek 1wykres do obliczenia wydajności eksploatacyjnej studni
tabsworzen, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do ob
w7, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń P
tabsr1, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do oblicz
w5, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń P
w10, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń
tab cp, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do oblicz
AOL4, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obliczeń
podkładka, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do obl
tabpodkladka, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do
tab f, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do oblicze
tab cv, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do oblicz
tabzawleczka, Akademia Morska -materiały mechaniczne, szkoła, Mega Szkoła, PODSTAWY KON, Program do
więcej podobnych podstron